
A photo-induced C–C bond formation methodology to construct tetrahydrofluorenones and their related structures†
Shujun
Cai
,
Zheming
Xiao
,
Jinjie
Ou
,
Yingbo
Shi
and
Shuanhu
Gao
*
Shanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, 3663N Zhongshan Road, Shanghai 200062, China. E-mail: shgao@chem.ecnu.edu.cn
First published on 14th January 2016
Abstract
A metal-free, photo-induced C–C bond formation methodology was developed to construct tetrahydrofluorenones and their related structures. This mild and efficient method proceeds either through electrocyclization or radical cyclization from β-Cl or β-Br aryl vinyl ketone. We believe that this method is particularly useful for the synthesis of related tetrahydrofluorenone containing natural products.
 Shuanhu Gao | Shuanhu Gao received his B.S. degree from the Lanzhou University in 2001. In 2006 he earned his Ph.D. in organic chemistry from Lanzhou University under the direction of Professor Yongqiang Tu. From 2006 to 2010, he was a postdoctoral fellow at the UT Southwestern Medical Center at Dallas. He started his independent career in the Department of Chemistry at East China Normal University in October 2010. His current research interests are primarily focused on the synthesis of complex natural products and medicinal chemistry. |
The naturally occurring fluorenones were discovered from various natural sources including plants, fungi and bacteria. This family of natural products contain similar core units of fluorenone,1 azafluorenone2 or hydrofluorenone. Regarding the core structures of hydrofluorenone, this group of natural molecules could be further divided into the subgroups of tetrahydro-, hexahydro- and decahydro-fluorenones. As shown in Fig. 1, the representatives of fluorenone, azafluorenone or hydrofluorenone containing natural products demonstrate diverse and challenging chemical structures. Together with potential bioactivities, this family of natural products aroused considerable attention from synthetic chemists. For instance, the core skeleton of kinamycins3 and lomaiviticins4 could be considered as the derivatives of tetrahydrofluorenones. The chemical synthesis and biological studies of these bacterial metabolites were carried out by several research groups over the past few years.5 Selective and efficient construction of the core hydrofluorenone rings is the key to synthesizing these molecules. The frequently used strategy to install these hydrofluorenone units may be the cyclopentanone ring formation, wherein a series of Friedel–Crafts reactions,3i,j,6 and metal-catalyzed coupling reactions3k,l were applied to execute the intramolecular cyclization. However, these reactions require harsh conditions that cannot tolerate the sensitive functional groups in most cases. Therefore, it's still necessary to develop new methodologies to achieve the same goal in mild reaction conditions.
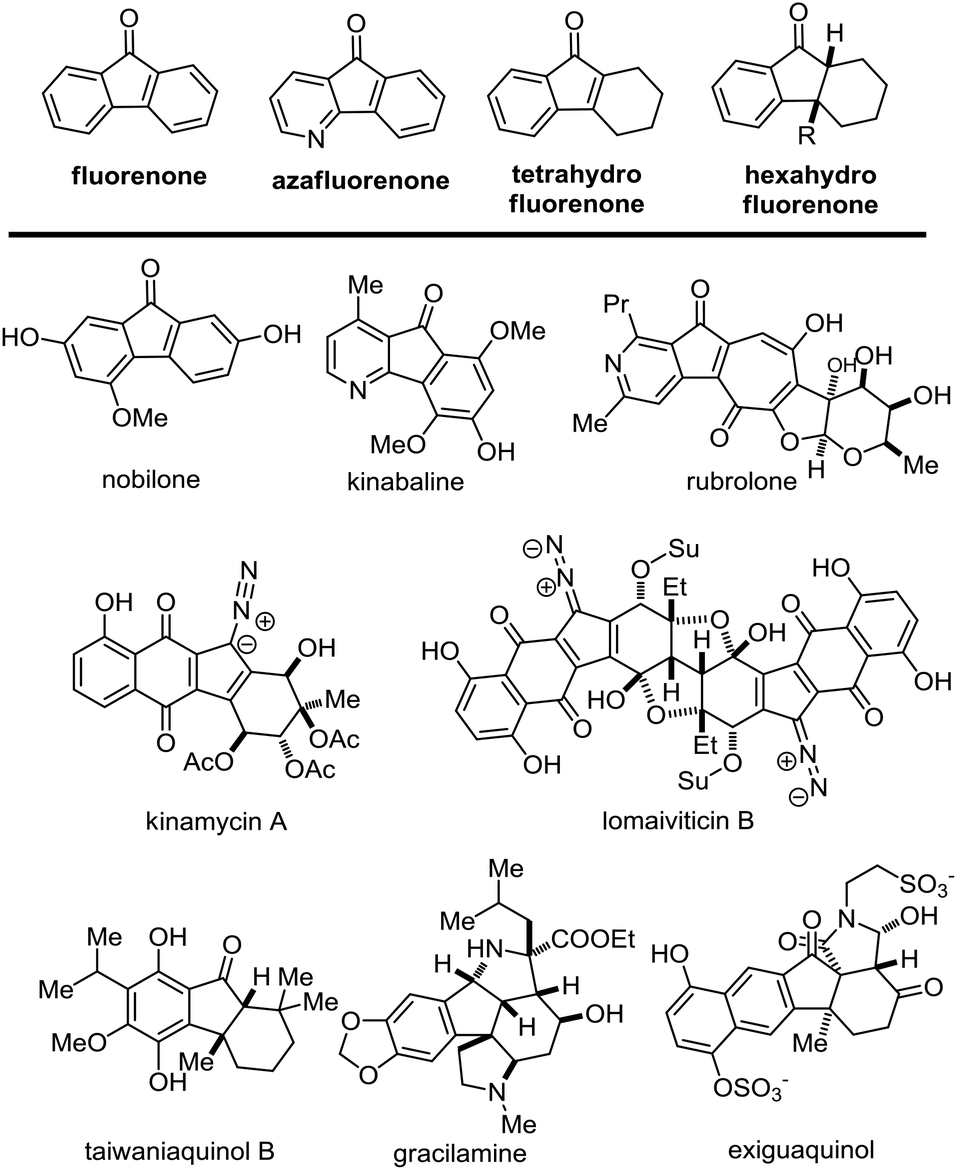 | ||
| Fig. 1 Representatives of fluorenone, azafluorenone or hydrofluorenone containing natural products. | ||
The Nazarov reaction is one of the most effective methods for building the cyclopentenone rings through 4-π-electron cyclization followed by protonation.7 However, the acid promoted electrocyclization of aromatic vinyl ketones to construct indanones, hexahydrofluorenones and related skeletons always requires the use of strong acids and high temperature. In contrast to the conventional acid mediated Nazarov reaction, the photo-Nazarov reaction of vinyl aryl ketones provides a mild and reliable method to prepare hydrofluorenones. The pioneering studies of the photo-Nazarov reaction were reported by Smith and Agosta in 1973.8 Mechanistic studies of this reaction were investigated by the group of Leitich and Schaffner.9 During our synthesis of hydrofluorenone containing natural products, we systematically studied the photo-Nazarov reaction of various aryl vinyl ketones (Scheme 1B).10 We found that this mild photolytic electrocyclization proceeds under neutral or basic conditions to give hexahydrofluorenones and the corresponding polycyclic rings efficiently. The photo-Nazarov reaction is particularly useful for preparing the core hexahydrofluorenones that are not easily accessible by the traditional acid-promoted methods. We have successfully applied this mild photo-Nazarov reaction in the synthesis of taiwaniaquinol B10 and gracilamine.11a On further investigation of the synthesis of this family of natural molecules, we hope to construct the tetrahydrofluorenones and their related structures through the same electrocyclization process by adding a leaving group (R2 = Cl) at the β position of the aryl vinyl ketones (Scheme 1B). We assumed that the photolysis of these substrates may undergo electrocyclization followed by a rapid regioselective β elimination to form the desired tetrahydrofluorenones. This rational design is inspired by the elegant work developed by Denmark and co-workers to efficiently construct the decahydrofluorenones through the silicon-directed Nazarov reaction (Scheme 1A).12
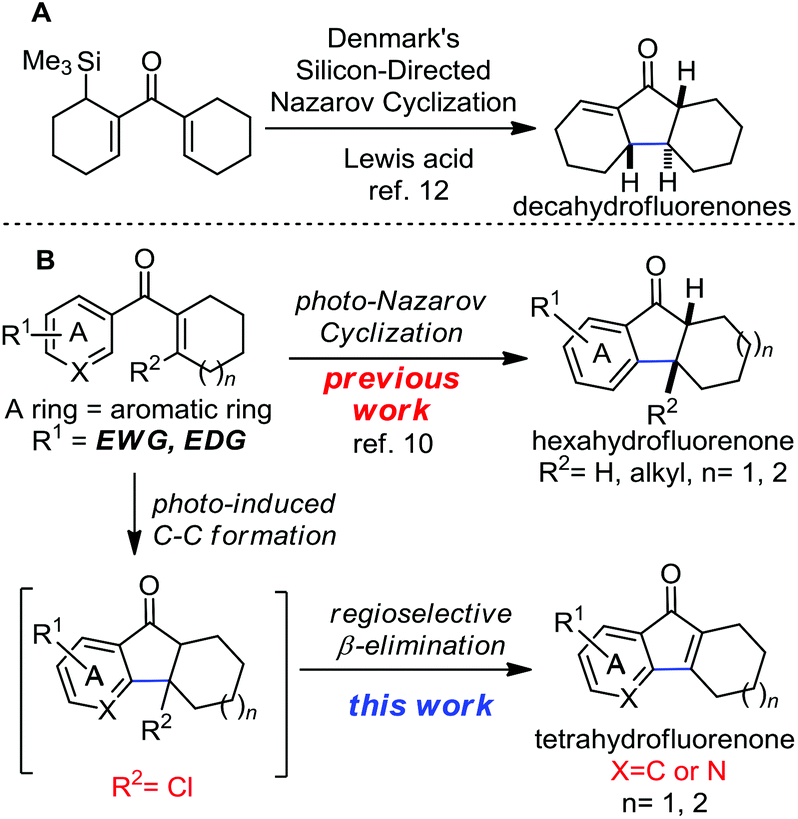 | ||
| Scheme 1 Photo-Nazarov reaction and proposed photo-induced C–C formation reaction. | ||
To test this synthetic hypothesis, we prepared substrate 1 with a β-Cl group on the aryl vinyl ketone as a leaving group, which is compatible to the model substrate used in the condition screening of the photo-Nazarov reaction. Irradiating a solution of 1 (2.0 mg mL−1) in degassed 1,2-dichloroethane with UV-light at 254 nm for 2 h gave the desired product 2 in 45% yield and its regioisomer 3 in 29% yield (Table 1, entry 1). We postulated that the photo-induced electrocyclization of 1 led the formation of an unstable intermediate which underwent a fast elimination to give 2 and release volatile HCl. This tetrahydrofluorenone 2 may be photoactive and was further activated by UV light to undergo isomerization and yielded 3 under acidic conditions. In order to restrain the photo-isomerization of 2, we explored the photolysis under basic conditions by adding a stoichiometric amount (2 μL mL−1) of Et3N, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), N,N,N′,N′-tetramethylethylenediamine (TMEDA) diisopropylamine (iPr2NH) and 2,2,6,6-tetramethylpiperidine (TMP) to neutralize the in situ formed HCl (Table 1, entries 2–6). We observed that the only desired product 2 was detected under these basic photolysis conditions except the condition with TMP as the base. Notably, the photolysis of 1 in the presence of iPr2NH gave 64% isolated yield at room temperature. We envisioned that hydrogen bonding between iPr2NH and the carbonyl group of 2 may stabilize the enone group and effectively restrain the photo-isomerization. Encouraged by this result, we explored the photolysis of 1 with iPr2NH in various solvents including acetonitrile, methanol, acetone, ether, dichloromethane and chloroform (Table 1, entries 8–13). It was found that performing this reaction in dichloromethane and chloroform gave similar results with 1,2-dichloroethane, and other solvent systems either led to the decomposition of substrate 1 or the formation of the product in very low yields. The reaction can also be done by irradiating with a longer wavelength (300 or 366 nm) UV-light (Table 1, entries 14–17) at the expense of reaction time. The reaction yield was improved slightly by using the degassed solvent; increasing the concentration to 4 mg mL−1 gave 2 in 79% yield (entry 18).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | λ [nm] | Solvent | Degasseda | Base (2 μL mL−1) | Con. (mg mL−1) | Time | Yield of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
Yield of 3b |
| a The solvents was degassed by bubbling nitrogen through the solution for 0.5 h. b All were determined by 1H NMR crude analysis using CH2Br2 as an internal standard, unless noted. c All were purified by silica gel column chromatography to give the product. d Not detected. e The concentration is 4 μl mL−1. | ||||||||
| 1 | 254 | ClCH2CH2Cl | Yes | — | 2 | 2 h | 45% | 29% |
| 2 | 254 | ClCH2CH2Cl | Yes | Et3N | 2 | 1.5 h | 38% | — |
| 3 | 254 | ClCH2CH2Cl | Yes | DBU | 2 | 1.5 h | 38% | — |
| 4 | 254 | ClCH2CH2Cl | Yes | TMEDA | 2 | 1.5 h | 13% | — |
| 5 | 254 | ClCH2CH2Cl | Yes | i Pr2NH | 2 | 1.5 h | 67% (64%)c | — |
| 6 | 254 | ClCH2CH2Cl | Yes | TMP | 2 | 1.5 h | 48% | 10% |
| 7 | 254 | ClCH2CH2Cl | No | i Pr2NH | 2 | 1 h | 61% (62%)c | — |
| 8 | 254 | CH3CN | No | i Pr2NH | 2 | 1 h | 6% | — |
| 9 | 254 | CH3OH | No | i Pr2NH | 2 | 1 h | NDd | |
| 10 | 254 | CH3COCH3 | No | i Pr2NH | 2 | 1 h | Decomposed | |
| 11 | 254 | Et2O | No | i Pr2NH | 2 | 1 h | Decomposed | |
| 12 | 254 | CH2Cl2 | No | i Pr2NH | 2 | 1 h | 56% | — |
| 13 | 254 | CHCl3 | No | i Pr2NH | 2 | 1 h | 51% | — |
| 14 | 300 | ClCH2CH2Cl | Yes | — | 2 | 2.5 h | 13% | 35% |
| 15 | 300 | ClCH2CH2Cl | Yes | i Pr2NH | 2 | 2.5 h | 45% | — |
| 16 | 366 | ClCH2CH2Cl | Yes | — | 2 | 3.0 h | 44% | 21% |
| 17 | 366 | ClCH2CH2Cl | Yes | i Pr2NH | 2 | 3.0 h | 82% (72%)c | — |
| 18 | 254 | ClCH2CH2Cl | Yes | i Pr2NHe | 4 | 2.0 h | 79% | — |
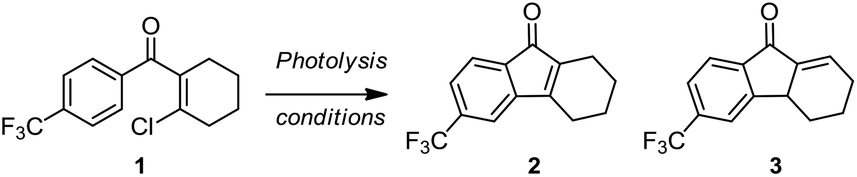
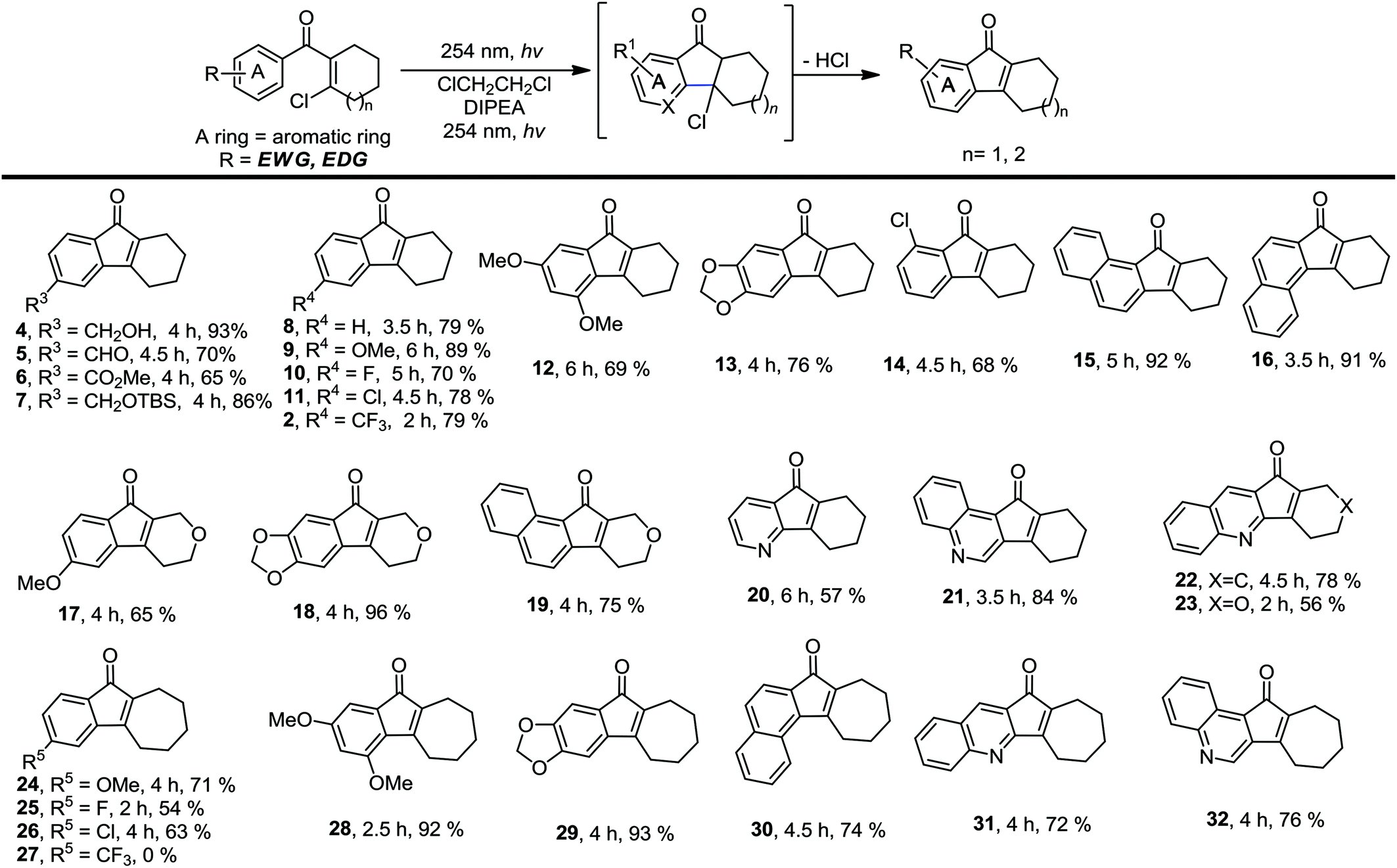
We then investigated the scope of this photo-induced C–C bond formation to construct tetrahydrofluorenone under the optimized conditions with respect to the functional-group tolerance, aromatic ring compatibility, and cycloalkene ring size. To our delight, this mild photolysis tolerated substrates with hydroxyl (4, Table 2), aldehyde (5) and ester functional groups (6) and acid-sensitive protecting groups such as tert-butyldimethylsilyl ether (OTBS) (7) producing the corresponding tetrahydrofluorenone in good yields. We then surveyed the electron density of aromatic rings and observed that both electron-donating (OMe−, −OCH2O−) and -withdrawing substituted groups (F−, Cl−, CF3−) on the phenyl rings (9–14) worked well in the photolysis. The desired electrocyclized products were obtained in acceptable yields. Photolysis of substrates with naphthyl groups smoothly yielded the desired cyclized products in excellent yields (15 and 16). When changing the cyclohexenyl ring to the unsaturated pyran ring, electrocyclization was also applicable and the desired polycyclic rings (17–19) were achieved in acceptable yields. We also investigated substrates containing hetero-aromatic rings including pyridine and quinoline in this photolysis. We were pleased to find that these substrates reacted efficiently and installed tetrahydroazafluorenone (20) and the corresponding heteroaromatic cycles (21–23) in good yields. We also studied the ring size of the cycloalkene ring. In accordance with the photo-Nazarov reaction, the cyclohexene could only be replaced by the cycloheptene ring and the corresponding cyclopentenyl and cyclooctenyl phenyl ketones failed to give the electrocyclization products under the optimized photolytic conditions. Aromatic rings with electron-donating groups such as −OMe (24 and 28), −OCH2O− (29), naphthyl (30), and electron-rich heteroaromatic rings (31 and 32) facilitated electrocyclization and furnished the desired cyclized products in good yields. Substrates with substituted halogens like −F (25) and −Cl (26) also worked and formed the corresponding products in moderate yields. Notably, the strong electron-withdrawing group trifluoromethyl on the phenyl ring (27) made the substrate inert under photolysis. The structures of the photo reaction products were determined unambiguously by using X-ray crystallographic analysis of 2, 7, 13 and 25 (Fig. 2). Based on the reaction scope study, we concluded that the reactivity of the substrates with a β-Cl group on aryl vinyl ketones totally agree with those used in the photo-Nazarov reactions.10 We considered that these reactions further support our originally proposed reaction process and also provide a platform for preparing related tetrahydrofluorenones.
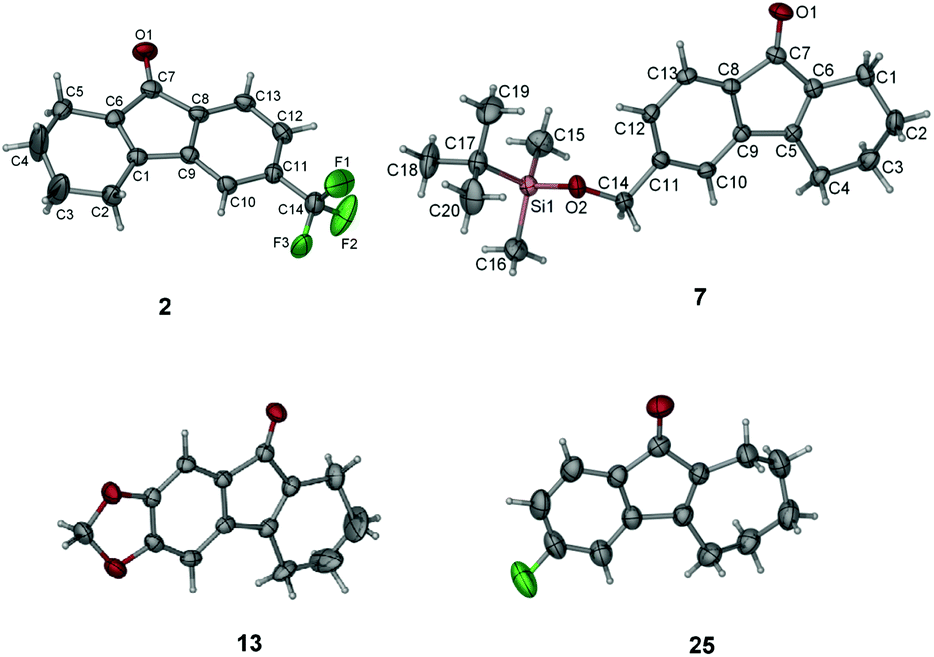 | ||
| Fig. 2 X-ray structures of 2, 7, 13 and 25. | ||
|
In order to solve the limitation of this photo-induced electrocyclization and expand the reaction scope, we decided to examine photolysis using the substrates bearing a β-Br group on aryl vinyl ketones. Instead of the photolytic electrocyclization, we realized that a photo-induced homolysis of the C–Br bond may occur on these substrates to produce vinyl radicals which may undergo fast radical cyclization to furnish the desired tetrahydrofluorenones.13,14 To test this hypothesis, we selectively prepared some substrates related to those shown in Table 2 and investigated their photolysis. To our delight, irradiation of these substrates under the optimized conditions smoothly gave the same cyclized products in even better yields (Table 3). We observed that this photolysis proceeds with a faster reaction rate in most cases which demonstrates that the intramolecular radical cyclizations are favourable for the ring closure. We also prepared the β-Br substrate with the trifluoromethyl group on the phenyl ring. Remarkably, we found that photolysis of this substrate efficiently gave the desired product 27 in 69% yield which was not achieved using the related β-Cl substrate. We believe that this photo-induced intramolecular radical cyclization is a beneficial supplement for photo-induced electrocyclization. An effective combination of these two photolytic strategies may provide a reliable solution for synthesizing a variety of natural products and their derivatives.

|
In conclusion, we developed a metal-free, photo-induced C–C bond formation methodology to construct tetrahydrofluorenones and their related structures based on our previously reported photo-Nazarov reaction. This mild photolysis proceeds either through electrocyclization or radical cyclization from a β-Cl or β-Br aryl vinyl ketone. We systematically studied the reaction conditions and scope with respect to the functional group tolerance and properties of aromatic rings. Based on these results, we believe this method is particularly useful for synthesizing tetrahydrofluorenones and their related structures. We are currently studying the applications of this methodology in natural product synthesis.
Acknowledgements
We thank the National Natural Science Foundation of China (21272076, 21422203), the Qi Ming Xing Foundation of Shanghai Ministry of Science and Technology (14QA1401400), the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning, Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) and Program for New Century Excellent Talents in University (NCET-13-0199) for generous financial support.References
- For isolation of nobilone, see: X. Zhang, J. Xu, J. Wang, N. Wang, H. Kurihara, S. Kitanaka and X. Yao, J. Nat. Prod., 2007, 70, 24–28 CrossRef CAS PubMed.
- For isolation of kinabaline, see: (a) D. Tadić, B. K. Cassels, M. Lcboeuf and A. Cavé, Phytochemistry, 1987, 26, 537–541 CrossRef; (b) For isolation of rubrolone, see: W. Schuep, J. F. Blount, T. H. Williams and A. Stempel, J. Antibiot., 1978, 31, 1226 CrossRef CAS PubMed.
- For isolation of kinamycins, see: (a) S. Ito, T. Matsuya, S. Ōmura, M. Otani and A. Nakagawa, J. Antibiot., 1970, 23, 315–317 CrossRef CAS PubMed; (b) T. Hata, S. Ōmura, Y. Iwai, A. Nakagawa and M. Otani, J. Antibiot., 1971, 24, 353–359 CrossRef CAS PubMed; (c) S. Ōmura, A. Nakagawa, H. Yamada, T. Hata, A. Furusaki and T. Watanabe, Chem. Pharm. Bull., 1971, 19, 2428–2430 CrossRef; (d) S. Ōmura, A. Nakagawa, H. Yamada, T. Hata and A. Furusaki, Chem. Pharm. Bull., 1973, 21, 931–940 CrossRef; (e) M. C. Cone, P. J. Seaton, K. A. Halley and S. J. Gould, J. Antibiot., 1989, 42, 179–188 CrossRef CAS PubMed; (f) K. Isshiki, T. Sawa, H. Naganawa, N. Matsuda, S. Hattori, M. Hamada, T. Takeuchi, M. Oosono, M. Ishizuka, Z. Yang, B. Zhu and W. Xu, J. Antibiot., 1989, 42, 467–469 CrossRef CAS PubMed; (g) P. J. Seaton and S. J. Gould, J. Antibiot., 1989, 42, 189–197 CrossRef CAS PubMed; (h) T. A. Smitka, R. Bonjouklian, T. J. Perun, A. H. Hunt, R. S. Foster, J. S. Mynderse and R. C. Yao, J. Antibiot., 1992, 45, 581–583 CrossRef CAS PubMed. For synthesis of kinamycins, see: (i) X. Lei and J. A. Porco, J. Am. Chem. Soc., 2006, 128, 14790–14791 CrossRef CAS PubMed; (j) S. S. Scully and J. A. Porco, Angew. Chem., Int. Ed., 2011, 50, 9722–9726 CrossRef CAS PubMed; (k) K. C. Nicolaou, H. Li, A. L. Nold, D. Pappo and A. Lenzen, J. Am. Chem. Soc., 2007, 129, 10356–10357 CrossRef CAS PubMed; (l) C. M. Woo, L. Lu, S. L. Gholap, D. R. Smith and S. B. Herzon, J. Am. Chem. Soc., 2010, 132, 2540–2541 CrossRef CAS PubMed.
- For isolation of lomivitaicins, see: (a) H. He, W. Ding, V. S. Bernan, A. D. Richardson, C. M. Ireland, M. Greenstein, G. A. Ellestad and G. T. Carter, J. Am. Chem. Soc., 2001, 123, 5362–5363 CrossRef CAS PubMed; (b) C. M. Woo, N. E. Beizer, J. E. Janso and S. B. Herzon, J. Am. Chem. Soc., 2012, 134, 15285–15288 CrossRef CAS PubMed. For synthesis of lomivitaicins, see: (c) W. Zhang, A. Baranczak and G. A. Sulikowski, Org. Lett., 2008, 10, 1939–1941 CrossRef CAS PubMed; (d) K. C. Nicolaou, A. L. Nold and H. Li, Angew. Chem., Int. Ed., 2009, 48, 5860–5863 CrossRef CAS PubMed; (e) E. S. Krygowski, K. Murphy-Benenato and M. D. Shair, Angew. Chem., Int. Ed., 2008, 47, 1680–1684 CrossRef CAS PubMed; (f) H. G. Lee, J. Y. Ahn, A. S. Lee and M. D. Shair, Chem. – Eur. J., 2010, 16, 13058–13062 CrossRef CAS PubMed; (g) S. B. Herzon, L. Lu, C. M. Woo and S. L. Gholap, J. Am. Chem. Soc., 2011, 133, 7260–7263 CrossRef CAS PubMed.
- For the reviews of kinamycins, lomivitaicins see: (a) S. J. Gould, Chem. Rev., 1997, 97, 2499–2510 CrossRef CAS PubMed; (b) J. Marco-Contelles and M. T. Molina, Curr. Org. Chem., 2003, 7, 1433–1442 CrossRef CAS; (c) C. C. Nawrat and C. J. Moody, Nat. Prod. Rep., 2011, 28, 1426–1444 RSC; (d) S. B. Herzon and A. M. Woo, Nat. Prod. Rep., 2012, 29, 87–118 RSC.
- E. Pan, S. Cao, P. J. Brodie, M. W. Callmander, R. Randrianaivo, S. Rakotonandrasana, V. E. Rasamison, K. TenDyke, Y. Shen, E. M. Suh and D. G. I. Kingston, J. Nat. Prod., 2011, 74, 1169–1174 CrossRef CAS PubMed.
- (a) I. N. Nazarov and I. I. Zaretskaya, Izv. Akad. Nauk SSSR Ser. Khim., 1941, 211–224 CAS. For reviews of the Nazarov reaction, see: (b) K. L. Habermas, S. E. Denmark and T. K. Jones, Org. React., 1994, 45, 1–158 CAS; (c) C. Santelli-Rouvier and M. Santelli, Synthesis, 1983, 429–442 CrossRef CAS; (d) Comprehensive Organic Synthesis, ed. S. E. Denmark, B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 5, p. 751 Search PubMed; (e) A. J. Frontier and C. Collison, Tetrahedron, 2005, 61, 7577–7606 CrossRef CAS; (f) H. Pellissier, Tetrahedron, 2005, 61, 6479–6517 CrossRef CAS; (g) M. A. Tius, Eur. J. Org. Chem., 2005, 2193–2206 CrossRef CAS; (h) N. Nakanishi and F. G. West, Curr. Opin. Drug Discovery Dev., 2009, 12, 732 Search PubMed; (i) N. Shimada, C. Stewart and M. A. Tius, Tetrahedron, 2011, 67, 5851–5870 CrossRef CAS PubMed; (j) T. Vaidya, R. Eisenberg and A. J. Frontier, ChemCatChem, 2011, 3, 1531–1548 CrossRef CAS.
- A. B. Smith III and W. C. Agosta, J. Am. Chem. Soc., 1973, 95, 1961–1968 CrossRef.
- (a) J. Leitich, I. Heise, S. Werner, C. Krürger and K. Schaffner, J. Photochem. Photobiol., A, 1991, 57, 127–151 CrossRef CAS; (b) J. Leitich, I. Heise, J. Rust and K. Schaffner, Eur. J. Org. Chem., 2001, 2719–2726 CrossRef CAS.
- S. Cai, Z. Xiao, Y. Shi and S. Gao, Chem. – Eur. J., 2014, 20, 8677–8681 CrossRef CAS PubMed.
- (a) Y. Shi, B. Yang, S. Cai and S. Gao, Angew. Chem., Int. Ed., 2014, 53, 9539–9543 CrossRef CAS PubMed. For other applications; see: (b) S. Gao, Q. Wang, L. Lum and C. Chen, J. Am. Chem. Soc., 2010, 132, 371–383 CrossRef CAS PubMed; (c) S. Gao, Q. Wang and C. Chen, J. Am. Chem. Soc., 2009, 131, 1410–1412 CrossRef CAS PubMed; (d) F. Churruca, M. Fousteris, Y. Ishikawa, M. von Wantoch Rekowski, C. Hounsou, T. Surrey and A. Giannis, Org. Lett., 2010, 12, 2096–2099 CrossRef CAS PubMed.
- (a) S. E. Denmark and T. K. Jones, J. Am. Chem. Soc., 1982, 104, 2642–2645 CAS; (b) T. K. Jones and S. E. Denmark, Helv. Chim. Acta, 1983, 66, 2377–2396 CrossRef CAS; (c) S. E. Denmark and R. C. Klix, Tetrahedron, 1988, 44, 4043–4060 CrossRef CAS; (d) S. E. Denmark, M. A. Wallace and C. B. Walker, J. Org. Chem., 1990, 55, 5543–5545 CrossRef CAS.
- J. N. Moorthy and S. Samanta, J. Org. Chem., 2007, 72, 9786–9789 CrossRef CAS PubMed.
- T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data. CCDC 1437982–1437985. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00392j |
| This journal is © the Partner Organisations 2016 |