
A copper(I)-catalyzed three-component reaction of triethoxysilanes, sulfur dioxide, and alkyl halides†
Danqing
Zheng
a,
Runyu
Mao
a,
Zhiming
Li
*a and
Jie
Wu
*ab
aDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai 200433, China. E-mail: jie_wu@fudan.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 354 Fenglin Road, Shanghai 200032, China
First published on 18th January 2016
Abstract
A copper(I)-catalyzed three-component reaction of triethoxysilanes, DABCO·(SO2)2, and alkyl halides is reported for the first time. This transformation provides a convenient and facile route to sulfones under ligand-free conditions catalyzed by copper(I) oxide. The insertion of sulfur dioxide is efficient, and both triethoxyarylsilanes and triethoxyalkylsilanes are practicable during the coupling process. A plausible mechanism is proposed, supported by theoretical calculations.
Introduction
Sulfones have attracted considerable attention over the past few decades due to their widespread presence in pharmaceutical and agrochemical molecules.1,2 They have also been used as essential intermediates in organic synthesis because of their versatile reactivities.3 Traditional methods to access sulfones include alkylation of sulfonate salts and oxidation of the corresponding sulfides or sulfoxides.4 However, these methods usually suffer from significant limitations and shortcomings. For example, the oxidative method requires foul-smelling thiols as starting materials and has restricted substrate scope due to the oxidizing conditions. On the other hand, sulfinate salts have limited commercial availability and their preparation usually requires procedures with harsh reaction conditions when sulfonyl chlorides are utilized as the starting materials.5Recent efforts have been directed to the fixation of sulfur dioxide into small molecules since the pioneer work reported by Willis and co-workers, which described a palladium-catalyzed aminosulfonylation using innoxious and bench-stable DABCO·(SO2)2 as the SO2 source.6,7 For example, addition of organometallic reagents to sulfur dioxide would generate metal sulfinates, which could then react with electrophiles leading to sulfones (Scheme 1, a).8 So far, Grignard reagents, organolithium reagents, and organozinc reagents have been proven to be efficient for this transformation. Willis and co-workers also reported a palladium-catalyzed formation of ammonium sulfinates from aryl halides in the presence of DABCO·(SO2)2, triethylamine and isopropyl alcohols.9 Shavnya and co-workers developed a similar palladium-catalyzed sulfinate generation using K2S2O5 as the sulfur dioxide source.10 Additionally, a gold-catalyzed sulfination of arylboronic acids utilizing K2S2O5 as the sulfur dioxide source was reported by Toste and co-workers.11a Further conversion of the sulfinate intermediates would lead to the corresponding sulfones and sulfonamides. Very recently, Shavnya and co-workers utilized arylboronic acids as substrates to generate sulfones via a palladium-catalyzed coupling reaction (Scheme 1, b).11b However, the above transformations could not proceed without the use of air-sensitive organometallic reagents, expensive metal catalysts or specialized ligands. Therefore, it is still necessary to develop efficient and environmentally benign methods for the synthesis of sulfones. Organosilanes have been paid continuous attention in cross-coupling reactions due to their advantages of low toxicity, high stability, ease of preparation and wide functional group tolerance.12 Recently Wang and co-workers developed an aminosulfonylation process catalyzed by a copper catalyst utilizing triethoxysilanes as the reaction substrates.7f It would be also attractive and highly desirable if organosilanes are utilized in sulfone synthesis. Herein, we would like to report a copper(I)-catalyzed three-component reaction of triethoxysilanes, DABCO·(SO2)2, and alkyl halides for the first time. This transformation provides a convenient and facile route to sulfones under ligand-free conditions catalyzed by copper(I) oxide. The insertion of sulfur dioxide is efficient, and both triethoxyarylsilanes and triethoxyalkylsilanes are practicable during the coupling process (Scheme 1, c).
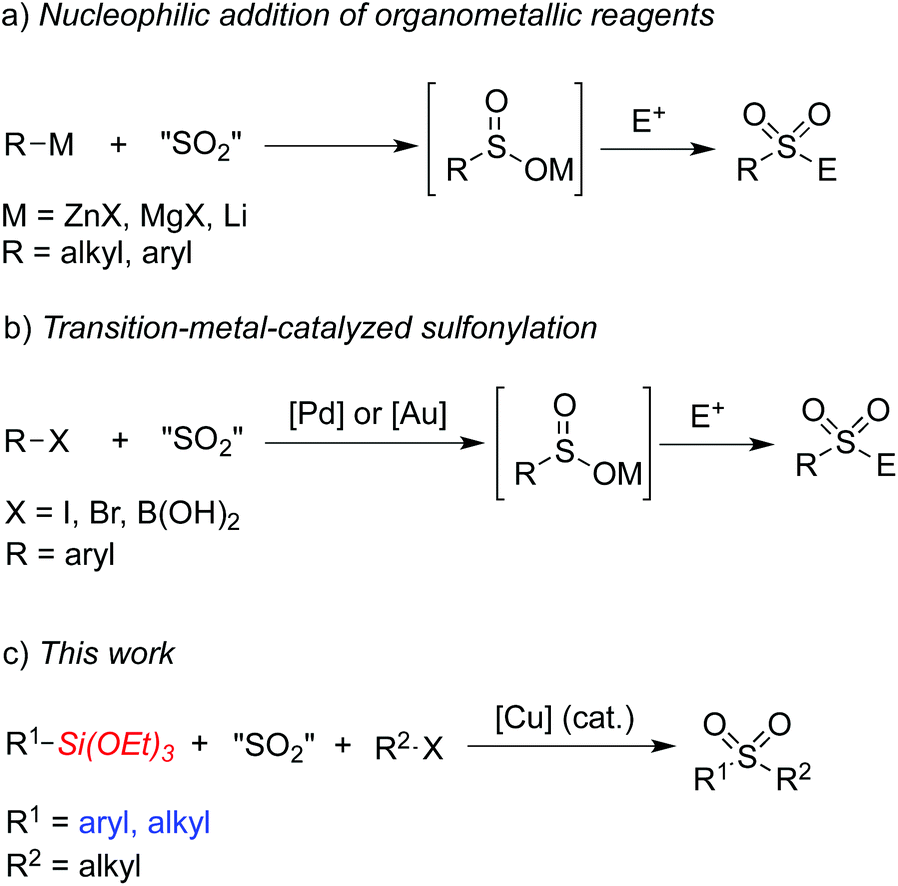 | ||
| Scheme 1 Synthesis of sulfones via the insertion of sulfur dioxide. | ||
Results and discussion
We began our investigation with the reaction of triethoxy-phenysilane 1a, DABCO·(SO2)2 and benzyl bromide 2a. Initially, the reaction was catalyzed by Cu(OAc)2 in the presence of Xphos and CsF in toluene under N2 at 100 °C (Table 1, entry 1). To our delight, the desired sulfonylation product 3a was obtained in 20% yield. Further screening of solvents indicated that DMF is the best choice, affording the expected product 3a in 43% yield (Table 1, entries 2–5). When NaF was used as a replacement of CsF, only a trace amount of product 3a was detected (Table 1, entry 6). The yield was decreased to 20% when TBAF was used as the fluoride source. No better results were obtained when other ligands were employed (Table 1, entries 8–10). To our surprise, the reaction took place smoothly in the absence of ligands, leading to the product 3a in 45% yield (Table 1, entry 11). Considering the low solubility of the sulfinate salts in organic solvents, water was added to this reaction as a co-solvent. Gratifyingly, the yield was improved to 55% with the addition of water (Table 1, entry 12). Further exploration of copper catalysts showed that the reaction worked efficiently in the presence of Cu2O, leading to the corresponding sulfone 3a in 71% yield (Table 1, entries 13–15). This transformation took place smoothly at 80 °C as well, while the yield was decreased to 62% (Table 1, entry 16). However, the yield was much lower when the reaction occurred at 120 °C (Table 1, entry 17). No desired product was detected when K2S2O5 was utilized as the sulfur dioxide source.|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | “F” | [Cu] | Ligand | Yieldb (%) |
| a Reaction conditions: copper salt (0.03 mmol), ligand (0.06 mmol), triethoxyphenylsilane 1a (0.3 mmol), DABCO·(SO2)2 (0.3 mmol), benzyl bromide 2a (0.6 mmol), “F” (0.6 mmol), solvent (2.0 mL), 100 °C, 12 h. b Isolated yield based on triethoxyphenylsilane 1a. c H2O (0.1 mL), DMF (2.0 mL). d The reaction was performed at 80 °C. e The reaction occurred at 120 °C. f K2S2O5 was used instead of DABCO·(SO2)2. | |||||
| 1 | Toluene | CsF | Cu(OAc)2 | Xphos | 20 |
| 2 | 1,4-Dioxane | CsF | Cu(OAc)2 | Xphos | 34 |
| 3 | DMA | CsF | Cu(OAc)2 | Xphos | 40 |
| 4 | NMP | CsF | Cu(OAc)2 | Xphos | 42 |
| 5 | DMF | CsF | Cu(OAc)2 | Xphos | 43 |
| 6 | DMF | NaF | Cu(OAc)2 | Xphos | Trace |
| 7 | DMF | TBAF | Cu(OAc)2 | Xphos | 23 |
| 8 | DMF | CsF | Cu(OAc)2 | PPh3 | 36 |
| 9 | DMF | CsF | Cu(OAc)2 | L-Proline | 29 |
| 10 | DMF | CsF | Cu(OAc)2 | PtBu3 | 35 |
| 11 | DMF | CsF | Cu(OAc)2 | — | 45 |
| 12c | H2O/DMF | CsF | Cu(OAc)2 | — | 55 |
| 13c | H2O/DMF | CsF | CuOAc | — | 43 |
| 14c | H2O/DMF | CsF | Cu2O | — | 71 |
| 15c | H2O/DMF | CsF | Cu(CF3COO)2 | — | 54 |
| 16c,d | H2O/DMF | CsF | Cu2O | — | 62 |
| 17c,e | H2O/DMF | CsF | Cu2O | — | 50 |
| 18c,f | H2O/DMF | CsF | Cu2O | — | ND |
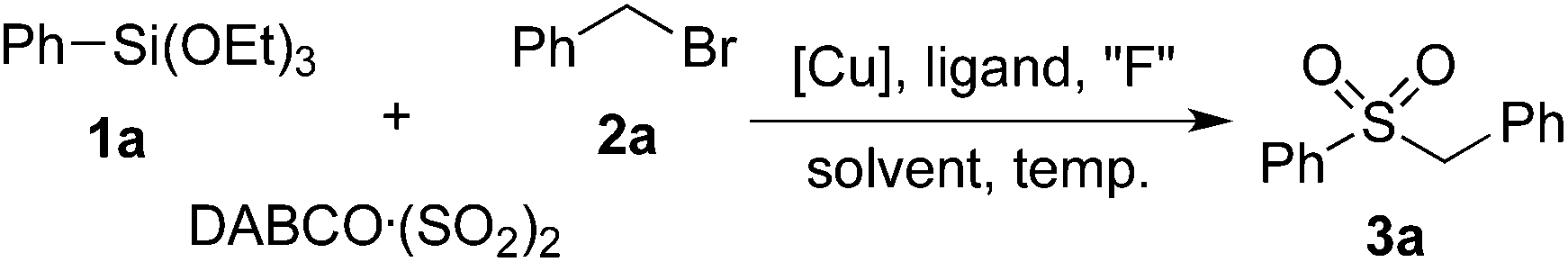
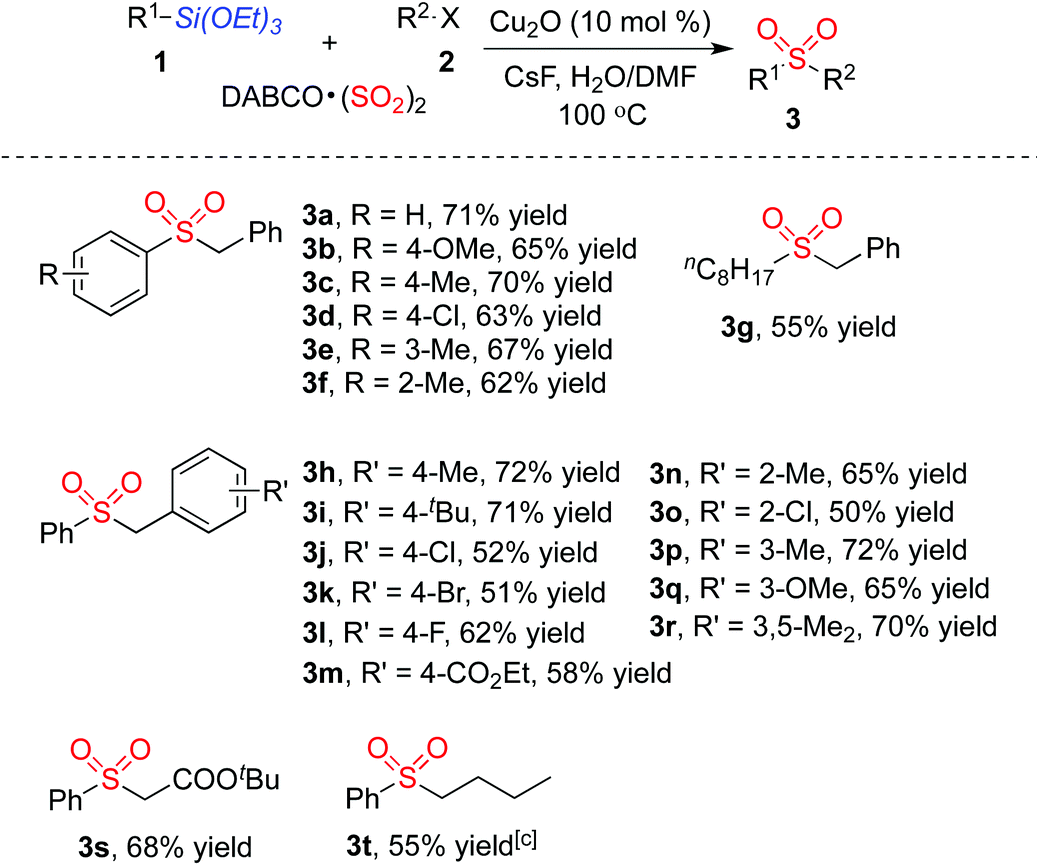
With the optimized reaction conditions in hand, we next explored the scope of the sulfonylation reaction of triethoxysilanes 1, DABCO·(SO2)2 and alkyl halides 2. Various triethoxysilanes 1 reacted with DABCO·(SO2)2 and alkyl halides 2 efficiently, giving rise to the corresponding sulfones 3 in moderate to good yields (Table 2). A range of triethoxyarylsilanes was firstly examined. Triethoxyarylsilanes with electron-withdrawing or electron-donating groups on the aromatic ring were all compatible under the standard conditions, delivering the desired products 3a–3f. It is noteworthy that the triethoxyarylsilane bearing ortho-substituent also proceeded well to afford the desired products 3f in 62% yield. Interestingly, triethoxyalkylsilanes could be utilized in this transformation as well. For instance, the corresponding product 3g was obtained in 55% yield when triethoxy(octyl)silane was employed in the reaction. Various benzyl bromides were next investigated for this process. Although both electron-withdrawing and electron-donating groups on the aromatic ring of benzyl bromides were tolerated in this transformation affording the corresponding sulfones 3h–3r, the reactions of benzyl bromides with electron-withdrawing substituents resulted in only moderate yields. The substrates featuring various functional groups, including ether, ester and halo, proceeded well in this process. Other alkyl halides were examined as well, which were proved to be suitable under the standard conditions. tert-Butyl 2-bromoacetate reacted with triethoxyphenylsilane 1a and DABCO·(SO2)2 leading to the desired product 3s in 68% yield. Reaction of 1-iodobutane, triethoxyphenylsilane 1a and DABCO·(SO2)2 was carried out in the meantime, providing the corresponding butylsulfonylbenzene 3t in 55% yield. We also examined heterocyclic siloxanes such as 2-(triethoxysilyl)pyridine and 3-(triethoxysilyl)pyridine; however, these reactions did not afford the desired products.
According to the experimental observations and the previous reports, we proposed a plausible mechanism shown in Scheme 2. We reasoned that a transmetallation of Cu(I) with triethoxysilanes 1 would occur first to generate a Cu(I) species, which would coordinate with the DABCO·(SO2)2 to form intermediate A. This followed by the attack of the aryl to the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond would generate the intermediate B. Release of DABCO would produce the sulfinate salt C, which would subsequently be trapped by the alkyl halide 2 to afford the final product 3.
O bond would generate the intermediate B. Release of DABCO would produce the sulfinate salt C, which would subsequently be trapped by the alkyl halide 2 to afford the final product 3.
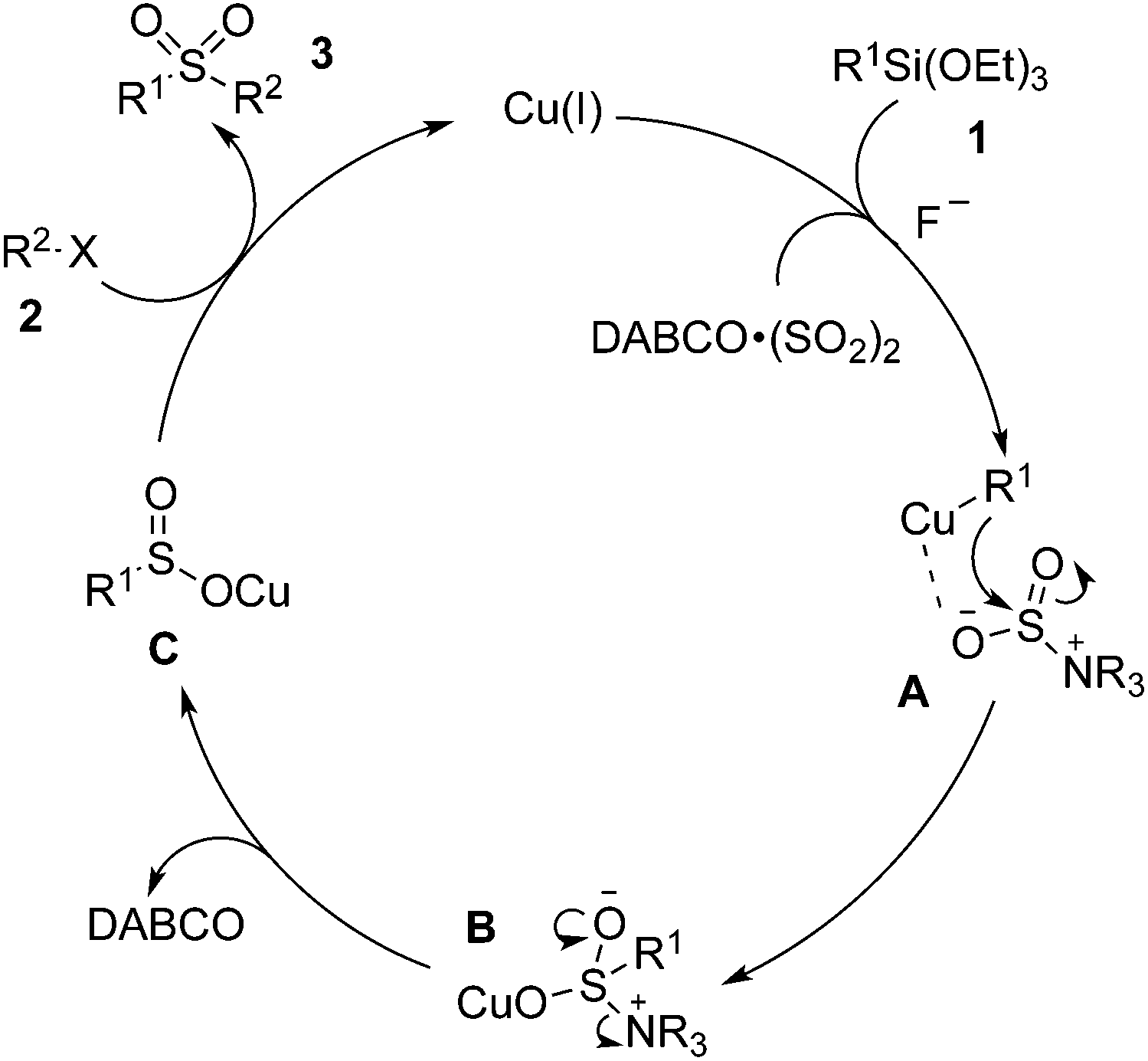 | ||
| Scheme 2 A proposed mechanism for the sulfonylation reaction of triethoxysilane 1 with DABCO·(SO2)2 and alkyl halide 2. | ||
In order to shed more light on the reaction mechanism, especially on the role of the copper reagent, B3LYP density functional theory (DFT) calculations were performed with the Gaussian09 software package.13 In view of the reaction conditions, DMF would be the main ligand for Cu, and the coordinated atom should be O instead of N.14 Though copper has many kinds of coordination modes, the simplest linear one was initially studied. The computation results are shown in Fig. 1 (in black). In the catalytic cycle, the first transmetallation step is thermodynamically favoured to give organic copper reagent L-CuPh by 10.2 kcal mol−1. Instant static interaction between L-CuPh and SO2 of DABSO results in the formation of complex L-A, the Cu–O and S–C distances are 3.10 and 3.12 Å, respectively. The product complex L-B is formed via L-TSAB, which is apparently an early transition state from the structure and energy profile. Nucleophilic substitution with BnBr gives the final product sulfone, which is very thermodynamic stable by 43.7 kcal mol−1 compared to the starting materials. From the free energy profile, the energies of all the intermediates and transition state are below that of the starting materials, which means that the reaction can proceed at 100 °C smoothly. This is in good agreement with the experimental facts. The trigonal coordination mode was also considered, and the results are shown in Fig. 1 (in red) as well. A similar catalytic cycle was found with the linear coordination. Unexpectedly, there is no normal trigonal coordination in copper bromide T-CuBr and organic copper reagent T-CuPh, only the hydrogen bonding interaction between two DMF molecules. While in T-A, T-TSAB and T-B, the second DMF did coordinate to the Cu atom, the distances between the Cu atom and the O atom are 2.29, 2.21 and 2.16 Å, respectively (see Fig. 1 and 2). Cu39 forms 6-coordination mode with O1, S2, O3, O27, O51 and C40 in T-TSAB (see Fig. 2) and T-A and T-B. Compared to L-TSAB, the introduction of the second DMF indeed lowers the energy of TSAB by 4.6 kcal mol−1, and makes the reaction more easy. In view of that the copper has reached a very high coordination number, no more DMF molecules were tested. In addition, similar to L-TSAB, T-TSAB is also an early transition-state. The bond order of the Cu39–C40 bond is only 0.2852 from the NBO analysis.15 The bond orders for Cu39–O3 and C40–S2 are 0.0365 and 0.3889, respectively, which suggests that the C40–S2 bond formation is more advanced than C39–O3. Similar trends can be found in L-TSAB as well. Moreover, it is also noteworthy that, in A, TSAB and B, SO2s involving in the reaction are released from the DABCO part actually. For example, in T-TSAB, the distance between S2 and N7 is 5.46 Å, while the S4–N10 distance is only 2.30 Å (Fig. 2), which indicates another SO2 is still connected with DABCO firmly.
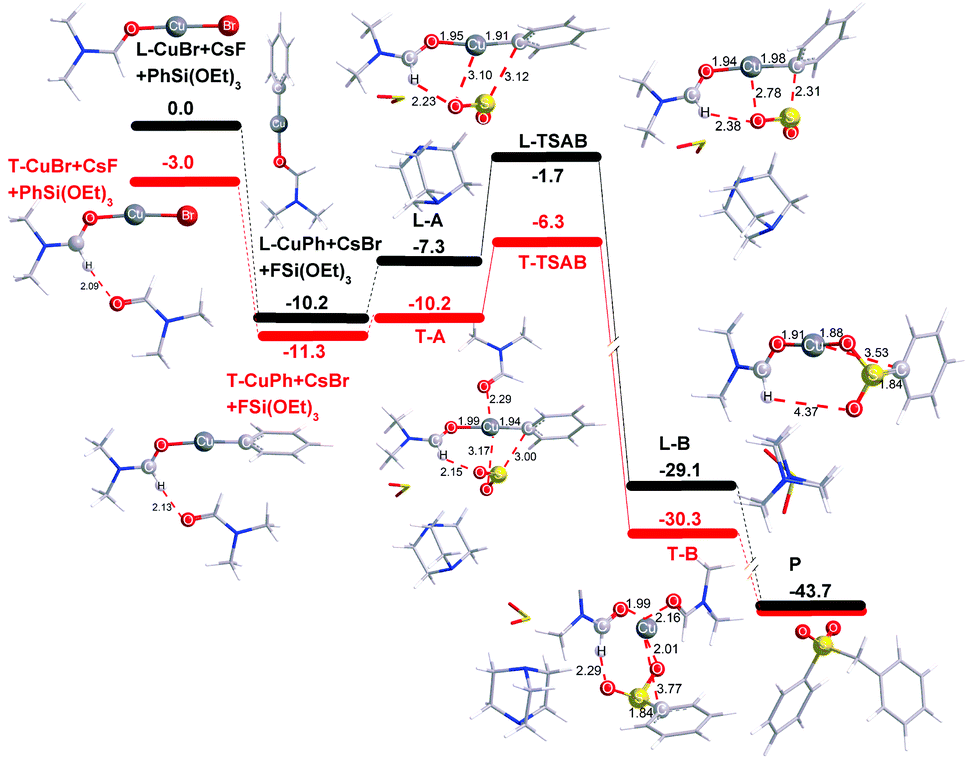 | ||
| Fig. 1 Free-energy reaction profiles (kcal mol−1) for linear (in black) and trigonal (in red) coordination modes at the PCM (DMF) B3LYP/6-311++G(d,p) with the LANL2DZ (for Cu and Cs) level at 100 °C. TS = transition state. | ||
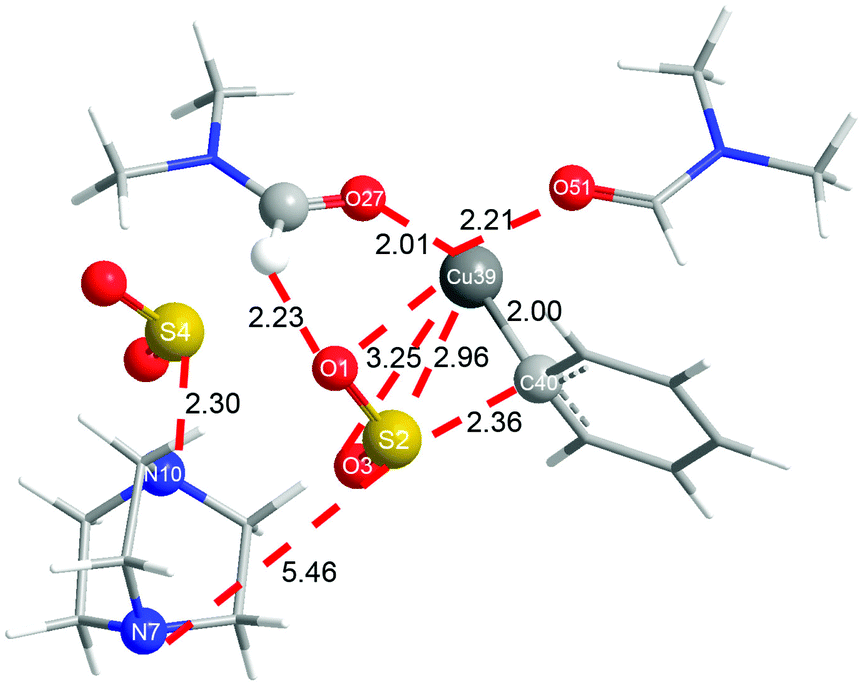 | ||
| Fig. 2 Optimized structure of T-TSAB. | ||
Conclusions
In conclusion, we have described an efficient route to sulfones via a copper(I)-catalyzed coupling of triethoxysilanes 1, DABCO·(SO2)2, and alkyl halides 2 under mild conditions. This process is attractive and practical since the copper(I) catalyst instead of a palladium or gold catalyst is utilized. Moreover, the reaction doesn't require ligands and shows broad functional group tolerance. Both triethoxyarylsilanes and triethoxyalkylsilanes work efficiently for the sulfonylation transformation. A plausible mechanism is proposed, supported by theoretical calculations. We believe that the copper(I)-catalyzed coupling reaction would broaden the reaction types of insertion of sulfur dioxide, which would open a new avenue for the fixation of sulfur dioxide.Acknowledgements
Financial support from the National Natural Science Foundation of China (no. 21372046 and 21532001) is gratefully acknowledged.Notes and references
- (a) Y. Harrak, G. Casula, J. Basset, G. Rosell, S. Plescia, D. Raffa, M. G. Cusimano, R. Pouplana and M. D. Pujol, J. Med. Chem., 2010, 53, 6560 CrossRef CAS PubMed; (b) D. A. Smith and R. M. Jones, Curr. Opin. Drug Discovery Dev., 2008, 11, 72 CAS; (c) Y. Noutoshi, M. Ikeda, T. Saito, H. Osada and K. Shirasu, Front. Plant Sci., 2012, 3, 245 CrossRef PubMed.
- (a) M. Bartholow, Top 200 Drugs of 2011. Pharmacy Times. http://www.pharmacytimes.com/publications/issue/2012/July2012/Top-200-Drugs-of-2011, accessed on Jan 9, 2013; (b) For a list of topdrugs by year, see: http://cbc.arizona.edu/njardarson/group/top-pharmaceuticals-poster, accessed on Jan 9, 2013; (c) J. Drews, Science, 2000, 287, 1960 CrossRef CAS PubMed.
- (a) P. J. Crowley, J. Fawcett, B. M. Kariuki, A. C. Moralee, J. M. Percy and V. Salafia, Org. Lett., 2002, 4, 4125 CrossRef CAS PubMed; (b) N. S. Simpkins, Sulfones in Organic Synthesis, Pergamon Press, Oxford, 1993 Search PubMed.
- (a) K. Sato, M. Hyodo, M. Aoki, X.-Q. Zheng and R. Noyori, Tetrahedron, 2001, 57, 2469 CrossRef CAS; (b) B. M. Trost and R. Baslau, J. Org. Chem., 1968, 53, 532 CrossRef; (c) T. Durst, Compr. Org. Chem., 1979, 3, 171 CAS; (d) G. E. Vennstra and B. Zwanenburg, Synthesis, 1975, 519 CrossRef CAS; (e) Y. Ju, D. Kumar and R. S. Varma, J. Org. Chem., 2006, 71, 6697 CrossRef CAS PubMed.
- (a) K. Baharami, M. M. Khodaei and D. Khaledian, Tetrahedron Lett., 2012, 53, 354 CrossRef; (b) G. K. S. Prakash, T. Mathew and G. A. Olah, J. Org. Chem., 2007, 72, 5847 CrossRef CAS PubMed; (c) S. W. Wright and K. N. Hallstrom, J. Org. Chem., 2006, 71, 1080 CrossRef CAS PubMed; (d) V. Percec, T. K. Bera, B. B. De, Y. Sanai, J. Smith, M. N. Holerca, B. Barboiu, B. B. B. Grubbs and J. M. J. Fréchet, J. Org. Chem., 2001, 66, 2104 CrossRef CAS PubMed; (e) R. J. Watson, D. Batty, A. D. Baxter, D. R. Hannah, D. A. Owen and J. G. Montana, Tetrahedron Lett., 2002, 43, 683 CrossRef CAS.
- For reviews: (a) G. Liu, C. Fan and J. Wu, Org. Biomol. Chem., 2015, 13, 1592 RSC; (b) P. Bisseret and N. Blanchard, Org. Biomol. Chem., 2013, 11, 5393 RSC; (c) A. S. Deeming, E. J. Emmett, C. S. Richards-Taylor and M. C. Willis, Synthesis, 2014, 2701 Search PubMed.
- (a) B. Nguyen, E. J. Emmet and M. C. Willis, J. Am. Chem. Soc., 2010, 132, 16372 CrossRef CAS PubMed; (b) E. J. Emmet, C. S. Garcia-Rubia, B. Nguyen, A. B. Garcia-Rubia, R. Hayter and M. C. Willis, Org. Biomol. Chem., 2012, 10, 4007 RSC; (c) L. Martial, Synlett, 2013, 1595 Search PubMed; (d) W. Li, H. Li, P. Langer, M. Beller and X.-F. Wu, Eur. J. Org. Chem., 2014, 3101 CrossRef; (e) W. Li, M. Beller and X.-F. Wu, Chem. Commun., 2014, 50, 9513 RSC; (f) X. Wang, L. Xue and Z. Wang, Org. Lett., 2014, 16, 4056 CrossRef CAS PubMed; (g) A. S. Deeming, C. J. Russell and M. C. Willis, Angew. Chem., Int. Ed., 2015, 54, 1168 CrossRef CAS PubMed; (h) S. Ye and J. Wu, Chem. Commun., 2012, 48, 7753 RSC; (i) S. Ye and J. Wu, Chem. Commun., 2012, 48, 10037 RSC; (j) D. Zheng, Y. An, Z. Li and J. Wu, Angew. Chem., Int. Ed., 2014, 53, 2451 CrossRef CAS PubMed; (k) S. Ye, H. Wang, Q. Xiao, Q. Ding and J. Wu, Adv. Synth. Catal., 2014, 356, 3225 CrossRef CAS; (l) Y. An, D. Zheng and J. Wu, Chem. Commun., 2014, 50, 11746 RSC; (m) Y. Luo, X. Pan, C. Chen, L. Yao and J. Wu, Chem. Commun., 2015, 51, 180 RSC; (n) D. Zheng, Y. Li, Y. An and J. Wu, Chem. Commun., 2014, 50, 8886 RSC.
- (a) C. C. Chen and J. Waser, Org. Lett., 2015, 17, 736 CrossRef CAS PubMed; (b) A. S. Deeming, C. J. Rusell, A. J. Hennessy and M. C. Willis, Org. Lett., 2014, 16, 150 CrossRef CAS PubMed; (c) B. N. Rocke, K. B. Bahnck, M. Herr, S. Lavergne, V. Mascitti, C. Perreault, J. Polivkova and A. Shavnya, Org. Lett., 2014, 16, 154 CrossRef CAS PubMed; (d) E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 12679 CrossRef CAS PubMed.
- E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2014, 38, 10204 CrossRef PubMed.
- A. Shavnya, S. B. Coffey, A. C. Smith and V. Mascitti, Org. Lett., 2013, 15, 6226 CrossRef CAS PubMed.
- (a) M. W. Johnson, S. W. Bagley, N. P. Mankad, R. G. Bergman, V. Mascitti and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 4404 CrossRef CAS PubMed; (b) A. Shavnya, K. D. Hesp, V. Mascitti and A. C. Smith, Angew. Chem., Int. Ed., 2015, 54, 13571 CrossRef CAS PubMed; (c) A. S. Deeming, C. J. Russell and M. C. Willis, Angew. Chem., Int. Ed., 2016, 55, 747 CrossRef CAS PubMed.
- (a) J. Yu, J. Liu, G. Shi, C. Shao and Y. Zhang, Angew. Chem., Int. Ed., 2015, 54, 4079 CrossRef CAS PubMed; (b) J. He, R. Takise, H. Fu and J. Yu, J. Am. Chem. Soc., 2015, 137, 4618 CrossRef CAS PubMed; (c) S. Shi and Y. Zhang, J. Org. Chem., 2007, 72, 5927 CrossRef CAS PubMed; (d) S. K. Gurung, S. Thapa, A. S. Vangala and R. Giri, Org. Lett., 2013, 15, 5378 CrossRef CAS PubMed; (e) Z. Wang and S. Chang, Org. Lett., 2013, 15, 1990 CrossRef CAS PubMed.
- For computational details and complete references, see the ESI.†.
- T. Chem, M. Yan, C. Zheng, J. Yuan, S. Xu, R. Chen and W. Huang, Chin. J. Chem., 2015, 33, 961 CrossRef.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, F. Weinhold, M. Morales and F. Weinhold, NBO 5.9, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00399g |
| This journal is © the Partner Organisations 2016 |