
Effects of peripheral substitutions on the singlet oxygen quantum yields of monophthalocyaninato ytterbium(III) complexes†
Quanquan Zhanga,
Guoe Chenga,
Hanzhong Ke*a,
Xunjin Zhu*b,
Nianyong Zhub,
Wai-Yeung Wongb and
Wai-Kwok Wongb
aFaculty of Material Science and Chemistry, China University of Geosciences, Wuhan, Hubei 430074, P. R. China. E-mail: kehanz@aliyun.com; Fax: +86-27-6788-3731
bDepartment of Chemistry, Hong Kong Baptist University, Waterloo Road, Hong Kong, P. R. China. E-mail: xjzhu@hkbu.edu.hk; Fax: +86-852-3411-7348
First published on 20th February 2015
Abstract
A new monophthalocyaninato ytterbium(III) complex 5 with eight 3,5-di-t-butylphenoxyl substituents at peripheral positions is synthesized. X-ray structural analysis of 5·CHCl3·MeOH reveals that the Yb3+ ion is seven-coordinate, surrounded by four nitrogen atoms from the phthalocyaninate dianion and three oxygen atoms from the anionic tripodal LOMe− ligand {LOMe− = [(cyclopentadienyl)tris(dimethylphosphito) cobaltate(III)]}. The effects of substituents on the relative singlet oxygen (1O2) quantum yields of monophthalocyaninato ytterbium(III) complexes are investigated through comparison. It is found that the monophthalocyaninato ytterbium(III) complex has higher 1O2 quantum yield than its corresponding phthalocyaninate ligand. The introduction of a 3,5-di-t-butylphenoxy substituent on the microcycle can enhance the yield of singlet oxygen. Due to the heavy-atoms of both iodine and lanthanide ions, the ytterbium(III) complex 4 based on tert-butyl and iodine substituted phthalocyanine ligands has the highest 1O2 quantum yield (0.82).
Introduction
Photodynamic therapy is a noninvasive methodology used for the treatment of a variety of premalignant and malignant diseases.1–4 This therapy requires a photosensitizer, visible light, and molecular oxygen. After irradiation of light with an appropriate wavelength in the presence of oxygen, the photosensitizer will induce cellular and tissue damage by generating reactive oxygen species (ROS), such as singlet oxygen and other radical species.5 Evidence favors that singlet oxygen (1O2) is the key cytotoxic agent for photobiological activity.6,7 The 1O2 is produced by energy transfer from the photoexcited sensitizer to the ground-state oxygen. This is known as the Type II photosensitization mechanism.8,9 As the first generation photosensitizers, porphyrin derivatives have been studied extensively and used in clinical work.2,10 However, the efficiency of the treatment is limited by their small extinction coefficient at the body therapeutic window (650–900 nm).11 Phthalocyanines have been proposed as the second-generation photosensitizers because of their many advantages such as the intense absorption in the red visible region, high efficiency to generate singlet oxygen, ease of chemical modification, and low dark toxicity.11–14 However, one major drawback of phthalocyanines in PDT is their aggregation tendency,15–17 which severely diminishes their photosensitizing ability. We have previously reported two novel monophthalocyaninato ytterbium(III) complexes.18 These Yb(III) complexes with an organometallic tripodal ligand at axial position show no aggregation in solution. As the energy of the emissive 2F5/2 state of Yb(III) is about the same as that of the phthalocyanine T1 state, there is a significant back-energy transfer, only 1O2 phosphorescence, not Yb(III) emission, was observed in the NIR region. Due to the heavy-atom effect of the Yb3+ ion enhances the intersystem crossing from the ligand-centered singlet state (1LC) to the ligand-centered triplet state (3LC), the monophthalocyaninato ytterbium(III) complexes have high ability to generate singlet oxygen than H2(t-Bu)4Pc. However, the singlet oxygen quantum yields of these Yb(III) complexes were not reported.It has been found that peripheral substituents can influence the electron delocalization, reduce aggregation, and enhance intersystem crossing in the case of heavy-atom substitution.19–22 It is necessary to evaluate the structure–activity relationship of photosensitizers via variedly substituted monophthalocyaninato ytterbium(III) complexes. The main aim of this work is to explore the influences of peripheral substituents at the monophthalocyaninato ytterbium(III) complexes on the singlet oxygen quantum yields.
Recently, we reported four monophthalocyaninato ytterbium(III) complexes 1–4 shown in Fig. 1.18,23 As nonsubstituted phthalocyanine ligand is insoluble in organic solvent, identically structured nonsubstituted PcYb 1 was obtained in less than 10% yield. 2–4 were obtained in about 65% yield with the disadvantage of a mixture of place isomers. We hereby describe the synthesis, spectroscopic and photochemical properties of a new unique structured monophthalocyaninato ytterbium(III) complex 5 with eight 3,5-di-t-butylphenoxyl substituents at peripheral positions of the microcycle ring. The influences of peripheral substituents on the singlet oxygen quantum yields are discussed through the comparison.
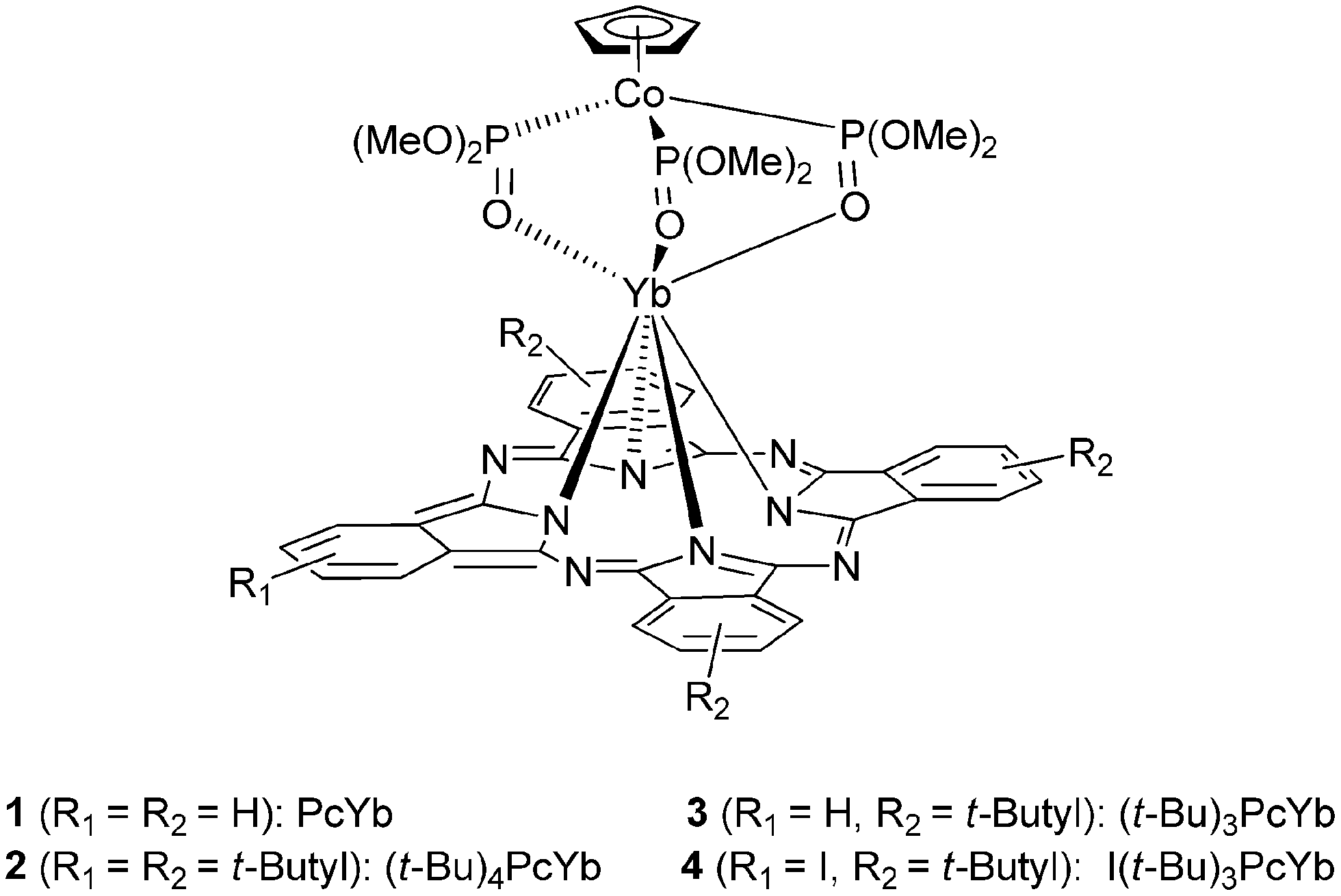 | ||
| Fig. 1 Structures of monophthalocyaninato ytterbium(III) complexes 1–4. | ||
Results and discussion
The synthetic route for the monophthalocyaninato ytterbium(III) complex 5 is shown in Scheme 1. Following the procedure described in the literature,24 the intermediate 4,5-dichloro-1,2-dicyanobenzene was prepared in three steps from 4,5-dichloro-1,2-benzenedicarboxylic acid via the anhydride, imide and diamide with an overall yield of 50%. The precursor 4,5-bis(3,5-di-t-butylphenoxy)phthalonitrile was prepared in 90% yield by the reaction of 3,5-di-t-butylphenol with 4,5-dichloro-1,2-dicyanobenzene in dry DMSO at 90 °C in the presence of solid K2CO3. The phthalocyanine free-base (3,5-di-t-butylphenoxy)8Pc 6 was synthesized in 43% yield by condensation of 4,5-bis(3,5-di-t-butyl-phenoxy)phthalonitrile in 1-pentanol in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) under reflux for 24 h. Then the reaction of (3,5-di-t-butylphenoxy)8Pc with Yb[N(SiMe3)2]3·[LiCl(THF)3]x followed by addition of Na(LOMe−) gave (3,5-di-t-butylphenoxy)8PcYb 5 in 61% yield. | ||
| Scheme 1 Synthesis of (3,5-di-t-butylphenoxy)8PcYb 5: (a) 3,5-di-t-butylphenol, DMSO/K2CO3, 90 °C; (b) DBU/1-pentanol, reflux, 48 h; (c) Yb[N(SiMe3)2]3·[LiCl(THF)3]x, toluene, reflux; (d) NaLOMe−, room temperature. | ||
The newly synthesized compound 5 was fully characterized by high resolution mass spectrometry, 1H and 31P NMR, UV/Vis absorption and fluorescence spectroscopy. The MALDI-TOF high-resolution mass spectrum (HRMS) of 5 showed the [M]+ peak at 2770.2902, giving mass accuracy Δm of 0.5 ppm relative to their corresponding calculated m/z values of 2770.2916. The isotopic distribution pattern also matched the expected profile. 5 displayed a singlet at δ = 65.36 ppm (vs. 85% H3PO4) in their 31P NMR spectra for the phosphito group of the anionic LOMe− ligand, The 1H NMR spectrum (in CDCl3) of 5 showed a singlet at δ = −5.90 ppm for the five cyclopentadienyl protons.
The solid-state structure of 5·CHCl3·MeOH was ascertained by X-ray crystallography. Complex 5·CHCl3·MeOH crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , in which the asymmetric unit is composed of one neutral molecular 5 and two solvates CHCl3 and MeOH. As shown in Fig. 2, it is evident that the Yb3+ ion is sandwiched between the phthalocyaninato ring and the anionic tripodal ligand and its seven-coordinate environment with a slightly distorted tricapped polygon prism is surrounded by four nitrogen atoms (N1A, N1C, N1E and N1G) from the phthalocyaninate dianion and three oxygen atoms (O2A, O2B and O2C) from the LOMe− anion as a cap. The bond lengths between Yb3+ and four N atoms of the phthalocyaninato ring and three O atoms of LOMe− are in the range of 2.286(6)–2.314(6) Å and 2.240(6)–2.255(6) Å, respectively, where the higher affinity of the Yb3+ ion to O atoms than that to N atoms endows relatively longer Yb–N distances than those of Yb–O bonds. The three mean planes (C5 of the cyclopentadienyl ring, N4 of the phthalocyaninate ligand, and O3 of the phosphito groups) are almost parallel to one another. The dihedral angles formed between the C5 and O3 mean planes, O3 and N4 mean planes, and C5 and N4 mean planes are, 2.345(3)°, 3.531(2)°, and 5.587(3)°, respectively. The Yb3+ center is 1.186(9) Å above the plane formed by the four phthalocyaninato N atoms and 1.489(3) Å below the plane formed by the three phosphito O atoms. The solvates CHCl3 and MeOH exhibit no observed interaction with the host structure.
, in which the asymmetric unit is composed of one neutral molecular 5 and two solvates CHCl3 and MeOH. As shown in Fig. 2, it is evident that the Yb3+ ion is sandwiched between the phthalocyaninato ring and the anionic tripodal ligand and its seven-coordinate environment with a slightly distorted tricapped polygon prism is surrounded by four nitrogen atoms (N1A, N1C, N1E and N1G) from the phthalocyaninate dianion and three oxygen atoms (O2A, O2B and O2C) from the LOMe− anion as a cap. The bond lengths between Yb3+ and four N atoms of the phthalocyaninato ring and three O atoms of LOMe− are in the range of 2.286(6)–2.314(6) Å and 2.240(6)–2.255(6) Å, respectively, where the higher affinity of the Yb3+ ion to O atoms than that to N atoms endows relatively longer Yb–N distances than those of Yb–O bonds. The three mean planes (C5 of the cyclopentadienyl ring, N4 of the phthalocyaninate ligand, and O3 of the phosphito groups) are almost parallel to one another. The dihedral angles formed between the C5 and O3 mean planes, O3 and N4 mean planes, and C5 and N4 mean planes are, 2.345(3)°, 3.531(2)°, and 5.587(3)°, respectively. The Yb3+ center is 1.186(9) Å above the plane formed by the four phthalocyaninato N atoms and 1.489(3) Å below the plane formed by the three phosphito O atoms. The solvates CHCl3 and MeOH exhibit no observed interaction with the host structure.
 | ||
| Fig. 2 A perspective view of the host structure of compound 5·CHCl3·MeOH. Hydrogen atoms, solvates and parts of substituted groups were omitted for clarity. Thermal ellipsoids are drawn at the 25% probability level. Selected bond lengths [Å] and bond angles [°]: Yb1–N1A 2.313(7), Yb1–N1C 2.314(6), Yb1–N1E 2.295(6), Yb1–N1G 2.286(6), Yb1–O2A 2.255(6), Yb1–O2B 2.242(6), Yb1–O2C 2.240(6); O2A–Yb1–O2B 78.4(2), O2A–Yb1–O2C 80.2(2), O2B–Yb1–O2C 83.6(2), N1A–Yb1–N1C 73.4(2), N1A–Yb1–N1E 118.4(2), N1C–Yb1–N1E 75.3(2), N1E–Yb1–N1G 74.9(2), N1C–Yb1–N1G 117.4(2), N1G–Yb1–N1A 74.7(2). The labels of A, B, C, D, E and G for N, O or P atoms with the same symmetric code as Co1 and Yb1 are used to discriminate different repeating portions of the phthalocyaninato ring and the tripodal ligand. | ||
The photophysical properties of 5 and 6 are summarized in Table 1. Fig. 3 shows the absorption spectra of 5 and 6. The UV/Vis spectrum of 6 shows two strong Q bands at 667 and 703 nm and two additional weak bands at 613 and 649 nm, and a strong B band at 353 nm. The Yb(III) complex 5 shows a strong Q band at 680 nm and two additional weak bands at 605 and 638 nm, and a strong B band at 361 nm. The absorption can be assigned to the π–π* transitions of the phthalocyaninate ligand. Fig. 4 shows the room-temperature emission and excitation spectra of 5 in toluene. Complex 5 displays visible emission peak at 688 nm with lifetime (τ) of 6.23 ns and quantum yields (Φem) of 0.010. However, ligand-centered (LC) long-lived phosphorescence was not observed. The excitation spectrum of complex 5 monitored at 688 nm matched well with its absorption spectrum. Thus, the visible fluorescence of compounds can be assigned to the intraligand emission.
| Absorption, λmax/nm [log(ε/dm3 mol−1 cm−1)] | Excitation λex/nm | Emission at 298 K λem/nm (τ, Φem × 102)b | |
|---|---|---|---|
| a Measurements were done in 1 × 10−6 M solution in toluene.b Quantum yield was measured relative to a 1 × 10−6 M solution of H2(t-Bu)4Pc in toluene (Φem = 0.67).c Not detected. | |||
| 5 | 361 (5.04), 613 (4.70), 649 (4.65), 680 (5.52) | 361 | 688 (6.23 ns, 1.0) |
| 6 | 353 (4.84), 605 (4.52), 638 (4.69), 667 (5.22), 703 (5.30) | 667 | 707 (—c, 63), 741, 790 |
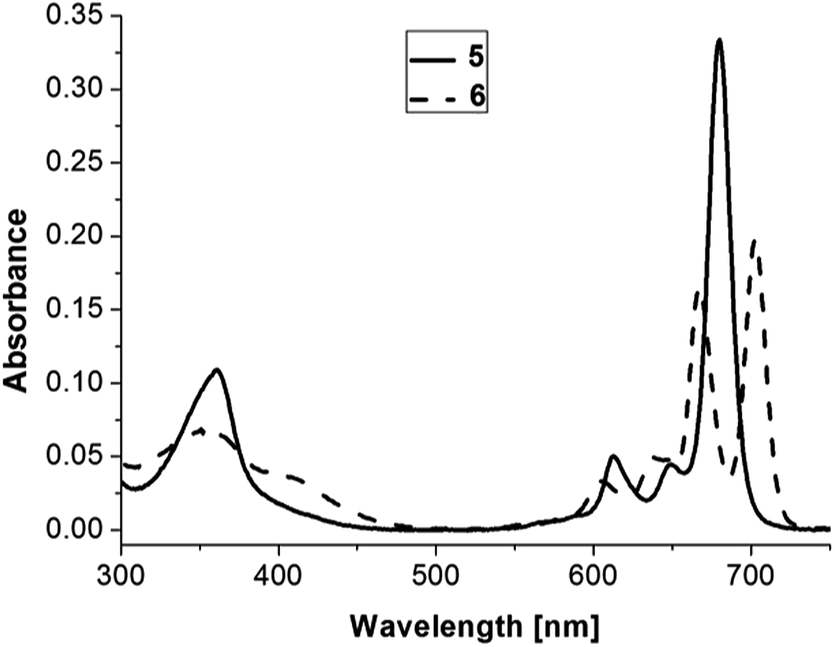 | ||
| Fig. 3 Absorption spectra of 5 and 6 solutions in toluene (1 × 10−6 M). | ||
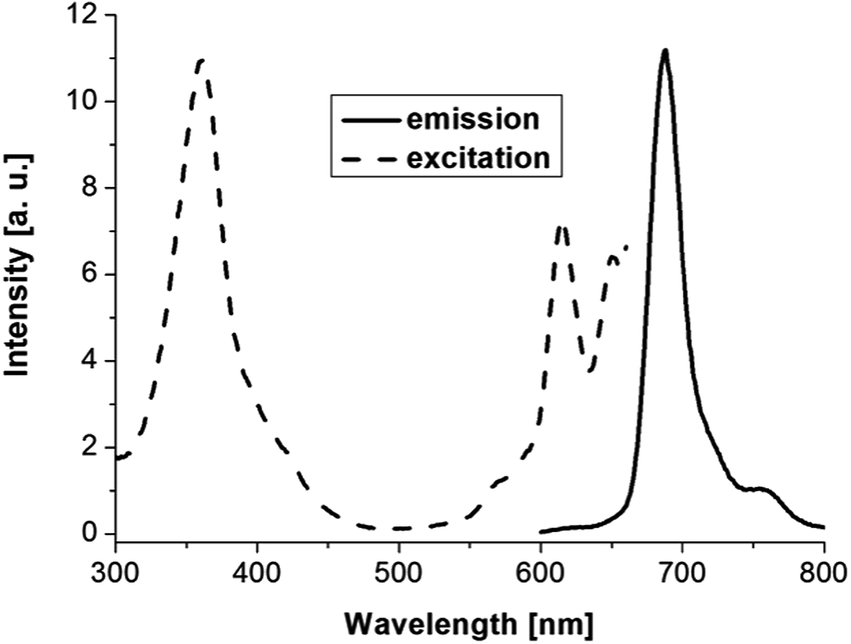 | ||
| Fig. 4 Room-temperature visible emission (excited at 361 nm) and excitation (monitored at 688 nm) spectra of complex 5 in toluene (1 × 10−6 M). | ||
In order to evaluate the photosensitizing efficiency of monophthalocyaninato ytterbium(III) complexes 1–5, the 1O2 quantum yields (ΦΔ) were determined by measuring the near-infrared (NIR) phosphorescence intensity of 1O2 (at 1270 nm) produced from these compounds at irradiation at 675 nm, using unsubstituted zinc(II) phthalocyanine (ZnPc) as the reference (ΦΔ = 0.58).25 The relative 1O2 quantum yields of these compounds and the reference ligands (3,5-di-t-butylphenoxy)8Pc 6 are given in Table 2. The error in the determination of ΦΔ was ∼10% (determined from several ΦΔ values).
Compared to the phthalocyanine ligand 6, those monophthalocyaninato ytterbium(III) complexes 1–5 show higher 1O2 quantum yield (ΦΔ) in the range of 0.63–0.82. This can be ascribed to the heavy-atom effect of the Yb3+ ion which, upon coordination with the phthalocyaninate ligand, enhances the intersystem crossing from the 1LC to the 3LC state. And the presence of heavy atom iodine at the phthalocyanice skeleton can also enhances the intersystem crossing from the ground state to the triplet state of phthalocyanine ligand, resulting in the highest 1O2 quantum yield. Complex 5 shows the second highest 1O2 quantum yield (0.79), which might be explained that the introduction of eight bulky 3,5-di-t-butylphenoxy substituents on the macrocycle provide a good shield from solvent induced nonradiative quenching and hence enhance the yield of singlet oxygen. This also indicates that singlet oxygen is produced by the energy transfer from the triple states of the photosensitizers and the ground state of molecular oxygen. Fig. 5 shows the phosphorescence spectra of 1O2 generated upon photoirradiation of an aerated solution of 1–6 in toluene (1.0 × 10−5 M) at 671, 678, 676, 682, 680 and 703 nm, respectively.
 | ||
| Fig. 5 Phosphorescence spectra of 1O2 generated from photoirradiation of complexes 1–6 in toluene (1.0 × 10−5 M) excited at the λmax 671, 678, 676, 682, 680 and 703 nm, respectively. | ||
Experimental section
General
All reactions were carried out under dry nitrogen atmosphere unless specified otherwise. Solvents were dried by standard procedures, distilled, and deaerated prior to use. All chemicals were obtained from Sigma-Aldrich Chemical Company and used without further purification. 4,5-Dichloro-1,2-dicyanobenzene was prepared according to the literature methods.25 Yb[N(SiMe3)2]3·[LiCl(THF)3]x was synthesized according to procedures in the literature.26 Electronic absorption spectra in the UV/Vis region were recorded with a Varian Cary 100 UV/Vis spectrophotometer, steady-state visible fluorescence and photo-luminescence excitation spectra were recorded with a Photon Technology International (PTI) Alphascan spectrofluorimeter, and quantum yields of the visible emissions were determined according to the literature method27 using H2(t-Bu)4Pc as reference standard (Φem = 0.67 in toluene).28 NMR spectra were recorded with a Varian Unity Inova 400 MHz spectrometer. 1H NMR chemical shifts were referenced to internal CDCl3 and then re-referenced to TMS (δ = 0.00 ppm). High-resolution mass spectra, reported as m/z, were obtained with a Bruker Autoflex MALDI-TOF mass spectrometer.Singlet oxygen quantum yield
Singlet oxygen was detected directly by its phosphorescence emission at 1270 nm using an InGaAs detector on the PTI QM4 luminescence spectrometer. The singlet oxygen quantum yields (ϕΔ) of all compounds were determined in toluene by comparing the singlet oxygen emission intensity of the sample solution to that of a reference material according to eqn (1).29
 | (1) |
Preparations of [(3,5-di-t-butylphenoxy)8PcYb] (5)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent. The yield was 43% (230 mg). MALDI-TOF HRMS: calcd for [M]+ 2148.3795; found 2148.3726. 1H NMR (CDCl3): δ = −0.38 (s, 2H), 1.32 (s, 144H), 7.13 (s, 16H), 7.24 (t, 8H), 9.02 (s, 8H) ppm.:1) as the eluent. The yield was 61% (85 mg). MALDI-TOF HRMS: calcd for [M]+ 2770.2916; found 2770.2902. 1H NMR (CDCl3): δ = −5.90 (s, 5H), 1.86 (s, 144H), 7.61 (s, 18H), 7.92 (s, 8H), 8.66 (s, 16H), 14.12 (s, 8H) ppm. 31P NMR (CDCl3): δ = 65.36 ppm.
:1) as the eluent. The yield was 43% (230 mg). MALDI-TOF HRMS: calcd for [M]+ 2148.3795; found 2148.3726. 1H NMR (CDCl3): δ = −0.38 (s, 2H), 1.32 (s, 144H), 7.13 (s, 16H), 7.24 (t, 8H), 9.02 (s, 8H) ppm.:1) as the eluent. The yield was 61% (85 mg). MALDI-TOF HRMS: calcd for [M]+ 2770.2916; found 2770.2902. 1H NMR (CDCl3): δ = −5.90 (s, 5H), 1.86 (s, 144H), 7.61 (s, 18H), 7.92 (s, 8H), 8.66 (s, 16H), 14.12 (s, 8H) ppm. 31P NMR (CDCl3): δ = 65.36 ppm.X-ray crystallography
Single crystals of 5·CHCl3·MeOH suitable for X-ray diffraction study were grown by slow evaporation of solutions of the compound in chloroform/methanol at room temperature. X-ray intensity data were collected with a Bruker Axs SMART 1000 CCD area-detector diffractometer using graphite-monochromated MoKα radiation (λ = 0.71073 Å). The collected frames were processed with the SAINT software30 and an absorption correction was applied (SADABS31) to the collected reflections. The structures of these molecules were solved by direct methods and expanded by standard difference Fourier syntheses using the SHELXTL software.32 Structure refinements were made on F2 using the full-matrix least-squares technique. All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were placed in their idealized positions and allowed to ride on the respective carbon atoms. Since there are disorders of tert-butyl groups in this compound, they were located and refined with restraints: DFIX, ISOR, SADI, SAME and SIMU. And some tert-butyl groups are split to two parts. Crystal data: C157H204Cl3CoN8O18P3Yb, MW = 2922.51, triclinic, space group = P, a = 19.6111(18) Å, b = 19.6932(18) Å, c = 24.446(2) Å, α = 81.59(2)°, β = 70.14(2)°, γ = 60.83(2)°, V = 7751.4(12) Å3, Z = 2, ρcalcd = 1.252 g cm−3.†
Conclusions
In summary, a new unique structured monophthalocyaninato ytterbium(III) complex 5 has been synthesized with high yield. The photophysical properties and the crystal structure of 5 have been examined. Compared to the phthalocyanine ligand 6, those monophthalocyaninato ytterbium(III) complexes 1–5 show higher 1O2 quantum yield (ΦΔ) in the range of 0.63–0.82. This can be ascribed to the heavy-atom effect of the centered Yb3+ ion or heavy atom substitution iodine, which enhances the intersystem crossing from the 1LC to the 3LC state. In addition, the introduction of eight bulky 3,5-di-t-butylphenoxy substituents on the microcycle can provide a good shield from solvent induced nonradiative quenching and hence enhance the yield of singlet oxygen. The results offer new strategies for the design of efficient photosensitizer of phthalocyanine with high singlet oxygen quantum yield.Acknowledgements
The work described in this paper was partially supported by a grant from the Research Grants Council of the Hong Kong Special Administrative Region, P.R. China (HKBU 202509) and a grant from the Hong Kong Baptist University (FRG2/12-13/050 and FRG1/13-14/022).Notes and references
- R. Bonnett, Chemical Aspects of Photodynamic Therapy, Gordon and Breach Science Publishers, Amsterdam, 2000 Search PubMed.
- R. R. Allison, R. E. Cuenca, G. H. Downie, P. Camnitz, B. Brodish and C. H. Sibata, Photodiagn. Photodyn. Ther., 2005, 2, 205–222 CrossRef CAS.
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS PubMed.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- N. L. Oleinick, R. L. Morris and I. Belichenko, Photochem. Photobiol. Sci., 2002, 1, 1–21 CAS.
- R. Bonnett, Chem. Soc. Rev., 1995, 24, 19–33 RSC.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- M. Ochsner, J. Photochem. Photobiol., B, 1997, 39, 1–18 CrossRef CAS.
- W. M. Sharman, C. M. Allen and J. E. van Lier, Drug Discovery Today, 1999, 4(11), 507–517 CrossRef CAS.
- K. Lang, J. Mosinger and D. M. Wagnerová, Coord. Chem. Rev., 2004, 248, 321–350 CrossRef CAS PubMed.
- M. R. Detty, S. L. Gibson and S. J. Wagner, J. Med. Chem., 2004, 47, 3897–3915 CrossRef CAS PubMed.
- C. M. Allen, W. M. Sharman and J. E. van Lier, J. Porphyrins Phthalocyanines, 2001, 5, 161–169 CrossRef CAS.
- I. J. MacDonald and T. J. Dougherty, J. Porphyrins Phthalocyanines, 2001, 5, 105–129 CrossRef CAS.
- C. C. Leznoff, M. Hu, C. R. McArthur, Y. Qin and J. E. van Lier, Can. J. Chem., 1994, 72, 1990–1998 CrossRef CAS.
- V. Iliev, V. Alexiev and L. Bilyarska, J. Mol. Catal. A: Chem., 1999, 137, 15–22 CrossRef CAS.
- P. C. Martin, M. Gouterman, B. V. Pepich, G. E. Renzeoni and D. C. Schindele, Inorg. Chem., 1991, 30, 3305–3309 CrossRef CAS.
- I. Lang, D. M. Wagnerova and J. Brodilova, J. Photochem. Photobiol., A, 1993, 72, 9–14 CrossRef.
- H. Ke, W.-K. Wong, W.-Y. Wong, H.-L. Tam, C.-T. Poon and F. Jiang, Eur. J. Inorg. Chem., 2009, 1243–1247 CrossRef CAS.
- S. Foley, G. Jones, R. Liuzzi, D. J. McGarvey, M. H. Perry and T. G. Truscott, J. Chem. Soc., Perkin Trans. 2, 1997, 1725–1730 RSC.
- A. Sastre, M. A. Diaz-Garcia, B. d. Rey, C. Dhenaut, J. Zyss, I. Lendoux, F. Agullo-Lopez and T. Torres, J. Phys. Chem. A, 1997, 101, 9773–9777 CrossRef CAS.
- I. Gürol, M. Durmuş, V. Ahsen and T. Nyokong, Dalton Trans., 2007, 3782–3791 RSC.
- S. FitzGerald, C. Farren, C. F. Stanley, A. Beeby and M. R. Bryce, Photochem. Photobiol. Sci., 2002, 1, 581–587 CAS.
- H. Ke, W. Li, T. Zhang, X. Zhu, H.-L. Tam, A. Hou, D. W. J. Kwong and W.-K. Wong, Dalton Trans., 2012, 41(15), 4536–4543 RSC.
- D. Wöhrle, M. Eskes, K. Shigehara and A. Yamada, Synthesis, 1993, 2, 194–196 CrossRef PubMed.
- A. Ogunsipe, D. Maree and T. J. Nyokong, J. Mol. Struct., 2003, 650, 131–140 CrossRef CAS.
- X. Zhu, W. K. Wong, W. K. Lo and W. Y. Wong, Chem. Commun., 2005, 1022–1024 RSC.
- V. Chauke, A. Ogunsipe, M. Durmus and T. Nyokong, Polyhedron, 2007, 26, 2663–2671 CrossRef CAS PubMed.
- D. M. Guldi, I. Zilbermann, A. Gouloumis, P. Vazquez and T. J. Torres, J. Phys. Chem. B, 2004, 108, 18485–18494 CrossRef CAS.
- Y. Li, T. M. Pritchett, J. Huang, M. Ke, P. Shao and W. Sun, J. Phys. Chem. A, 2008, 112, 7200–7207 CrossRef CAS PubMed.
- SAINT+, ver. 6.02a, Bruker Analytical X-ray System, Inc., Madison, WI, 1998 Search PubMed.
- G. M. Sheldrick, SADABS, Empirical Absorption Correction Program, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXTLTM, reference manual, ver. 5.1, Madison, WI, 1997 Search PubMed.
Footnote |
| † CCDC 1044126. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra02569a |
| This journal is © The Royal Society of Chemistry 2015 |