
Bioinspired highly electrically conductive graphene–epoxy layered composites†
Peng Ming‡
a,
Yuanyuan Zhang‡a,
Jianwen Baob,
Gang Liub,
Zhou Lic,
Lei Jianga and
Qunfeng Cheng*a
aKey Laboratory of Bio-inspired Smart Interfacial Science and Technology of Ministry of Education, School of Chemistry and Environment, BeiHang University, Beijing, P. R. China. E-mail: cheng@buaa.edu.cn; Fax: +86-10-82627566
bScience and Technology on Advanced Composites Laboratory, Beijing Institute of Aeronautical Materials, AVIC Composites Technology Center, Beijing 100095, P. R. China
cKey Laboratory for Biomechanics and Mechanobiology of Ministry of Education, School of Biological Science and Medical Engineering, Beihang University, Beijing, P. R. China
First published on 23rd February 2015
Abstract
Inspired by the nano/micro-scale hierarchical structure of nacre, we developed a new method for fabricating highly electrically conductive graphene–epoxy layered composites. In this new method, the graphene loading can be easily controlled, and the intrinsic three-dimensional network of graphene in the composites results in high electrical conductivity. Through effective surface modification, the interface strength between graphene and epoxy matrix was dramatically improved, leading to the 23-fold improvement in tensile strength, 136-fold in Young's modulus, and 8-fold in electrical conductivity compared with the pure graphene foam. These high performance bioinspired graphene–epoxy layered composites have a great potential for applications in electromagnetic interference (EMI) shielding, aerospace, and other electrical devices.
Introduction
Highly electrically conductive graphene–polymer composites are desired in many practical applications,1 including, electromagnetic interference (EMI) shielding,2–4 electrode materials for a range of electrochromic devices, flexible electrode in LEDs and many others. Recently, bioinspired graphene-based thermoplastic polymer layered composites have been fabricated with high strength and electrical conductivity, such as graphene oxide (GO)–poly(vinylalcohol) (PVA),5 GO–thermoplastic polyurethane (TPU),6 etc. However, few work has reported the preparation of graphene-based thermosetting polymer layered composites. Epoxy is a typical thermosetting polymer, which is widely used as the matrix in the composites due to its outstanding mechanical and heat resistance properties. Several traditional approaches for fabricating the graphene–epoxy composites have been attempted, such as in situ polymerization,7–9 and solution intercalation.10–14 However, these methods usually require large amounts of organic solvents and are not environment-friendly.9 On the other hand, it is very difficult to achieve high graphene loading and electrical conductivity in the graphene–epoxy composites. Recently, the graphene foam or three-dimensional (3D) graphene scaffolds were developed and were infiltrated with epoxy resin in attempt to solve this problem.4,15,16 For example, Tang et al. demonstrated the graphene aerogel–epoxy composites with compressive properties but low electrical conductivity (below 1 S m−1).17 Jia et al. demonstrated the graphene foam–epoxy composites with high electrical conductivity (6.4 S cm−1) with only 0.16 wt% graphene.15 However, the graphene loading is still low due to the large voids in the 3D graphene scaffold, resulting in low electrical conductivity. Thus, a great challenge still remains to develop new approaches to fabricate highly electrically conductive graphene–epoxy composites.After millions of years of evolution, natural nacre shows outstanding mechanical properties through assembling the organic–inorganic components into layered nano/micro-scale hierarchical structure.18–22 The inorganic phase of aragonite platelets reaches 95 vol% and the organic phase is only 5 vol% in the nacre.23 Inspired by the structure of nacre, we developed a new method to fabricate the graphene–epoxy layered composite with high graphene loading and electrical conductivity, simultaneously. In our approach, the graphene oxide nanosheets were assembled into the 3D foam structures. Then the epoxy resin was impregnated into the 3D graphene foam (GF) to obtain the GF–epoxy preform. Finally, the GF–epoxy preform was cured by hot-press into the layered structure. In this method, the graphene loading can be easily controlled, and the intrinsic 3D network of GF in the composites results in highly electrical conductivity. Through effective surface modification, the interface strength between graphene and epoxy matrix was dramatically improved, leading to the 23-fold improvement in tensile strength, 136-fold in Young's modulus, and 8-fold in electrical conductivity compared with the pure graphene foam. These high performance bioinspired graphene–epoxy layered composites have a great potential for applications in electromagnetic interference (EMI) shielding, aerospace, and other electrical devices.
Experimental section
Materials
Graphene oxide (GO) was prepared by the modified Hummers' method. The epoxy resin system was supplied by the Beijing Institute of Aeronautical Materials (BIAM). Silane coupling agent (3-glycidyloxypropyl)trimethoxysilane ≥98% (KH-560) was obtained from Sigma-Aldrich. Concentrated sulfuric acid (98%), sodium nitrate, hydrochloric acid, potassium permanganate, 30% H2O2 solution, hydrazine hydrate (N2H4·H2O) (85%), acetone, ethanol, acetic acid were of reagent grade and purchased from Beijing Chemical Works Co., Ltd.,. All the reagents were used without further purification.Fabrication of KH-560 treated GO
100 mg GO was dissolved in 100 ml ethanol and then ultrasound for 1 hour to obtain the suspension. 10 ml deionized water, 5 ml acetic acid (36%) and different amount of (1 mg, 5 mg, 10 mg) silane coupling agent (3-glycidyloxypropyl)trimethoxysilane ≥98% (KH-560) were mixed uniformly and then added into GO suspension and ultrasound for 0.5 h. Afterwards, the solution was condensed at 80 °C for 4 hours, and the KH-560 treated GO was obtained by washing with 100 ml ethanol and 100 ml water for two times under vacuum filtration respectively.Fabrication of bioinspired graphene–epoxy layered composites
The graphene foam (GF) was obtained according to the report,24 as following: 60 mg GO was dispersed in 60 ml deionized water and then sonicated for 1 hour to obtain the GO colloidal suspension. Then the GO film, obtained through vacuum filtration, was coated with 40 μl N2H4·H2O. Finally, the GO film was covered by Tin foil and placed into the autoclave at 90 °C for 10 h. The epoxy resin was diluted with acetone into 5 wt% solution. Then the GF–epoxy preform was obtained by immersing the GF into epoxy resin solution with assistance of vacuum for 30 minutes. The residual acetone was removed at 80 °C in the vacuum oven for 2 h. The GF–epoxy preform was cured by the hot-press process at a pressure of 2 MPa following the curing cycle at 94 °C for 10 minutes plus 177 °C for 4 h. The exact weight percentage of graphene was determined by the thermogravimetric analysis (TGA), as shown in Fig. S1.†Characterization
Mechanical properties were tested using the Shimadzu AGS-X at a loading speed of 1 mm min−1 with a gauge length of 5 mm. All the samples tested were cut into 20 mm in length and 3 mm in width. The thickness and the fracture surface morphology of each specimen were obtained by the environmental scanning electron microscope (Quanta 250 FEG ESEM). The results for each kind of sample are based on the average value of 3–5 specimens. Fourier transform infrared (FTIR) spectra were obtained using the iN10MX FTIR instrument. The electrical conductivity was measured by a standard two-probe method using a source meter (Agilent E4980A). The thermogravimetric analysis (TGA) was performed on NETZSCH TG 209F1 Libra TGA209F1D-0173-L under argon with a temperature rising rate of 20 °C min−1. X-ray photoelectron spectroscopy (XPS) was performed using ESCALab220i-XL (Thermo Scientific) with the X-ray source of a monochromatic Al Kα. Raman spectroscopy was conducted using a LabRAM HR800 with 633 nm laser excitation.Results and discussion
The fabrication process of bioinspired graphene–epoxy layered composite is shown in Fig. 1. The GO sheets were first dispersed into deionized (DI) water. Then GO film was obtained by vacuum assisted filtration. Herein the GF was fabricated according to the previous report.24 The porous structure of the GF is similar to the previous report,24 and the pore size shows a little of big, as shown in Fig. 2a. Several characterizations were performed to confirm the formation of reduced GF, as shown in Fig. 2. The characteristic peaks of carboxyl groups at 1720 cm−1 and carboxy C–O at 1396 cm−1 on GO nanosheets almost disappears in GF after reduction, indicating that oxygen functional groups on GO nanosheets were dramatically removed during the reduction. Raman spectra was also utilized to confirm the reduction of GO. After the hydrazine reduction, the ID/IG ratio increased from 1.8 for GO film to 3.4 for GF, indicating that the GO films were chemically converted to GF after hydrazine treatment and well consistent with previous report.25 Furthermore, XPS spectrum shows that peak intensity of C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C(O)O groups dramatically decreased after hydrazine reduction. The ratio of O1s to C1s is decreased from 0.32 for the GO to 0.13 for GF after hydrazine reduction. Moreover, a new peak at 285.8 eV corresponding to C–N, further confirmed the reduction reaction happened according to the previous report.26 Finally, TGA curves of GO and GF indicated that there was the 38% mass loss during the “leavening” stage of hydrazine reduction,27 which further confirmed that the GO nanosheets in the film were reduced. Then the GF was impregnated with the epoxy resin solution to make GF–epoxy preform, and the GF–epoxy preform was cured by hot-press. The amount of graphene content in the resultant bioinspired layered composites was 10%, 20%, 40 wt%, 45 wt%, 60 wt%, 80 wt%, and the specimens were designated as Composite-I, II, III, IV, V, and VI, respectively. The exact graphene loading was determined by thermogravimetric analysis (TGA), as shown in Fig. S1,† and the detail values are listed in Table 1.
O, C(O)O groups dramatically decreased after hydrazine reduction. The ratio of O1s to C1s is decreased from 0.32 for the GO to 0.13 for GF after hydrazine reduction. Moreover, a new peak at 285.8 eV corresponding to C–N, further confirmed the reduction reaction happened according to the previous report.26 Finally, TGA curves of GO and GF indicated that there was the 38% mass loss during the “leavening” stage of hydrazine reduction,27 which further confirmed that the GO nanosheets in the film were reduced. Then the GF was impregnated with the epoxy resin solution to make GF–epoxy preform, and the GF–epoxy preform was cured by hot-press. The amount of graphene content in the resultant bioinspired layered composites was 10%, 20%, 40 wt%, 45 wt%, 60 wt%, 80 wt%, and the specimens were designated as Composite-I, II, III, IV, V, and VI, respectively. The exact graphene loading was determined by thermogravimetric analysis (TGA), as shown in Fig. S1,† and the detail values are listed in Table 1.
 | ||
| Fig. 1 Illustration of the preparation process of bioinspired graphene–epoxy layered composite. The GO nanosheets were first dispersed into deionized (DI) water. Then the GO solution was subjected to vacuum-assisted filtration into the GO film. After N2H4·H2O (85%) leavening strategy treatment, the graphene foam (GF) was obtained. The GF was impregnated with epoxy/acetone solution (5 wt%) into the preform. Finally the GF–epoxy preform was cured by hot-press, and the bioinspired graphene–epoxy layered composites were obtained. | ||
 | ||
| Fig. 2 (a) Porous structure of GF, (b) FITR spectra of GO film and the reduced GF: the characteristic peaks of carboxyl groups at 1720 cm−1 and carboxy C–O at 1396 cm−1 on GO nanosheets almost disappears in GF after reduction, indicating that oxygen functional groups on GO nanosheets were dramatically removed during the reduction. (c) Raman spectra of GO film and GF shows that the ID/IG ratio increased from 1.8 for GO film to 3.4 for GF, indicating that the GO films were chemically converted to GF after reduction, (d) and (e) XPS spectrum shows that peak intensity of C–O, CO, C(O)O groups dramatically decreased after hydrazine reduction, and the ratio of O1s to C1s is decreased from 0.32 for the GO film to 0.13 for GF after hydrazine reduction. (f) TGA curves show that there was the 38% mass loss during the “leavening” stage of hydrazine reduction, further confirming that most of the GO sheets are reduced. | ||
| Sample | Input graphene loading [wt%] | Graphene loading by TGA | Electrical conductivity (S cm−1) |
|---|---|---|---|
| GF | — | — | 2.9 ± 0.2 |
| Control GF | — | — | 53.2 ± 0.7 |
| Composite-I | 10 | 12 | 2.3 ± 0.4 |
| Composite-II | 20 | 24 | 5.6 ± 0.7 |
| Composite-III | 40 | 42 | 21.2 ± 1.0 |
| Composite-IV | 45 | 46 | 23.2 ± 1.5 |
| Composite-V | 60 | 59 | 33.9 ± 1.5 |
| Composite-VI | 80 | 83 | 51.1 ± 1.3 |
| Treated I | 60 | 63 | 33.0 ± 7.4 |
| Treated II | 60 | 56 | 26.8 ± 2.0 |
| Treated III | 60 | 57 | 25.3 ± 2.3 |
Mechanical properties
The typical stress–strain curves of bioinspired graphene–epoxy layered composites are shown in Fig. 3a and the details values are listed in Table S1.† The home-made GF shows the tensile strength of 2.9 ± 0.5 MPa, and Young's modulus of 0.04 ± 0.01 GPa (curve 1), respectively, which are comparable to previous reported value of 3.2 MPa.24 To make a comparison with composites, the GF is treated with the same hot-pressing processing and designated as control GF. The mechanical properties show the tensile strength of 19.2 ± 1.2 MPa, and Young's modulus of 0.57 ± 0.14 GPa (curve 2). The mechanical properties of bioinspired graphene–epoxy layered composites show dramatical improvement in tensile strength, and Young's modulus (curve 4) as compared to control GF. With increased graphene loading, the tensile strength and the Young's modulus of Composite-V reached 55.1 MPa, and 5.1 GPa, corresponding to 18-fold and 126-fold improvement over the pure GF. However, with further increase of GF loading, the mechanical properties of resultant composites decrease, which may be the lack of resin in the composite and leads to the dry spot similar to carbon fiber–reinforced epoxy composites.28 Thus the mechanical properties show an optimized maximum value with graphene loading (Fig. 3b). | ||
| Fig. 3 (a) Tensile stress–strain curves of GF (curve 1), controlled GF (curve 2), epoxy (curve 3), Composite-V (curve 4), and Treated-II (curve 5). (b) The ultimate strength of the composite with different graphene loading (wt%), indicating that the tensile strength increases with graphene loading until 60%, and then decrease with higher graphene loading. (c) Tensile strength of bioinspired graphene–epoxy layered composite with different treated methods. (d) Fracture surface morphology of Treated-II. | ||
The mechanical properties of Composite-V are lower than the pure epoxy resin (curve 4). The main reason may be the weak interface between graphene and epoxy resin, as the active functional groups on the graphene oxide were removed in the reduction process, which was confirmed by FTIR, Raman, XPS and TGA, as shown in Fig. 2. Thus it is difficult to form strong chemical bonds between GF with epoxy resin, and improve the stress transfer efficiency. To further improve the mechanical properties of the bioinspired graphene–epoxy layered composites, the GF was grafted with silane coupling agent,29 dramatically improving the interfacial interaction with epoxy matrix through chemically covalent bonding and stress transfer efficiency from epoxy to graphene nanosheets. The proposed mechanism is shown in Fig. S2.† Step 1: the (3-glycidyloxypropyl)trimethoxysilane is hydrolyzed into the (3-glycidyloxypropyl)trimethoxysilanol. Step 2: the graphene oxide reacted with (3-glycidyloxypropyl)trimethoxysilanol through condensation of water into KH-560 grafted GO (KH-560-GO). Then the KH-560-GO was reduced with N2H4·H2O to remove the other active functional groups on GO surface. Step 3: the epoxide groups on the KH-560-GF are attacked by one component of epoxy resin-benzyldimethylamine (BDMA), yielding zwitterions that contain a quaternary nitrogen cation and an active anion. This type of ester is functionalized as the initiator of the chainwise polymerization, and then reacts with epoxy resin of glycidyl ester, leading to the formation of an alkoxide species. Subsequently, the formed alkoxide species attacks the BDMA again. It should be noted that the reaction in step 3 can happen at both ends of glycidyl ester with the KH-560-GF, eventually resulting in formation of graphene–epoxy composite having chemical cross-linking bonds.
Herein, to obtain the maximum treatment effect, three different ratios between KH-560 and GF were applied: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100, 5:100 and 10:100, the corresponding GF are designated GF-I, GF-II, and GF-III, and the corresponding composites are designated as Treated-I, Treated-II, and Treated-III, respectively. FTIR was conducted to verify the grafting reaction between GF and KH-560 (Fig. S3a†). The absorption peaks vibration around 2800–2900 cm−1 of GF-I, GF-II, and GF-III corresponds to stretching vibration of –CH2– and –CH–, which demonstrates that the silane coupling agent was successfully grafted on the surface of GF. Furthermore, Raman spectrum was also conducted to confirm the grafting reaction between GF and KH-560 (Fig. S3b†). The ID/IG ratio was decreased from 3.4 for the GF to 2.7 for GF-I, 2.4 for GF-II and 2.3 for GF-III, respectively. Moreover, Fig. S3c† shows the XPS spectra of GF and GF-II samples, and the corresponding elements contents are listed in Table S2.† After grafting KH-560, the C1s and N1s peaks decrease 4.4% and 12.5%, respectively. The O1s peak increases 12.8%, because of the contribution of oxygen element in KH-560. Meanwhile, the appearance of silicon element peak on GF-II further confirms that KH-560 was successfully grafted to the surface of GF.30 Finally, TGA results show that the weight loss of GF-I, GF-II, GF-III are 20.2%, 24.5%, 28.1% respectively, which is higher than the GF with 19.46% weight loss. This is direct evidence of the successful grafting KH-560 on the surface of GF.
:100, 5:100 and 10:100, the corresponding GF are designated GF-I, GF-II, and GF-III, and the corresponding composites are designated as Treated-I, Treated-II, and Treated-III, respectively. FTIR was conducted to verify the grafting reaction between GF and KH-560 (Fig. S3a†). The absorption peaks vibration around 2800–2900 cm−1 of GF-I, GF-II, and GF-III corresponds to stretching vibration of –CH2– and –CH–, which demonstrates that the silane coupling agent was successfully grafted on the surface of GF. Furthermore, Raman spectrum was also conducted to confirm the grafting reaction between GF and KH-560 (Fig. S3b†). The ID/IG ratio was decreased from 3.4 for the GF to 2.7 for GF-I, 2.4 for GF-II and 2.3 for GF-III, respectively. Moreover, Fig. S3c† shows the XPS spectra of GF and GF-II samples, and the corresponding elements contents are listed in Table S2.† After grafting KH-560, the C1s and N1s peaks decrease 4.4% and 12.5%, respectively. The O1s peak increases 12.8%, because of the contribution of oxygen element in KH-560. Meanwhile, the appearance of silicon element peak on GF-II further confirms that KH-560 was successfully grafted to the surface of GF.30 Finally, TGA results show that the weight loss of GF-I, GF-II, GF-III are 20.2%, 24.5%, 28.1% respectively, which is higher than the GF with 19.46% weight loss. This is direct evidence of the successful grafting KH-560 on the surface of GF.
Due to the interaction between KH-560 and epoxy resin, which is confirmed by the FTIR and XPS, as shown in Fig. S4,† the interface strength is enhanced when grafting with the KH-560, resulting in improved mechanical properties. The tensile strength and Young's modulus of resultant composites increase with increased amount of KH-560. For example, the tensile strength and Young's modulus increase to 69.6 MPa, and 5.5 GPa for Treated-II from 55.4 MPa, and 3.8 GPa for Treated-I, respectively, and corresponding to 23-fold and 136-fold improvement compared with the pure GF. With further grafting by KH-560, the mechanical properties of Treated-III decrease. We believe that too much KH-560 is affecting the curing reaction of epoxy matrix.
The cross-section morphologies of GF and bioinspired graphene–epoxy layered composites are shown in Fig. S5.† With increased graphene loading, the layered structure becomes clear. When the graphene loading reaches about 60 wt%, the mechanical properties of resultant composites show maximum value, almost similar to the carbon fiber reinforced epoxy composites.28 If the epoxy loading is too low, there would be a dry spot in the resultant composites, decreasing the interface strength and mechanical properties. If the epoxy loading is too high, areas rich in resin occur, which decreases the load transfer efficiency from epoxy resin to graphene nanosheets and thus mechanical properties.
After surface modification, the interface strength of resultant composites was enhanced, resulting in the improvement of mechanical properties, especially Young's modulus. The fracture morphology of Treated-II shows typical brittle fracture mode, as shown in Fig. 3d. However, when grafting too much KH-560 on the surface of GF, the mechanical properties of resultant composites decrease. Thus there is a maximum value for treated composites. In this study, the maximum value of tensile strength was obtained for Treated-II composite. The other cross-sectional surface morphologies of treated composites are shown in Fig. S5.† Treated-II is denser than the other Treated-I and Treated-III specimens.
Electrical conductivity
One of the advantages of bioinspired graphene–epoxy layered composites is high electrical conductivity, as shown in Fig. 4. For pure GF, the electrical conductivity is 2.9 S cm−1, lower than previously reported for the graphene foam at 10 S cm−1.31 This is because the chemically reduced GO foam still has a lot of defects, after treated by the hot-press, the control GF shows an electrical conductivity of 53.23 S cm−1, the bioinspired graphene–epoxy layered composites also shows high electrical conductivity. The main reason is the intrinsic 3D network in GF. The other reason is the close contact between graphene nanosheets obtained by the hot-press curing process. For example, the Composite-I shows electrical conductivity of 21.2 S cm−1, which is almost one order of magnitude higher than the pure GF, and two times higher than the CVD-synthesized graphene foam.31 With graphene loading increasing, the electrical conductivity of bioinspired graphene–epoxy layered composites further increases to 51.1 S cm−1 for Composite-IV. | ||
| Fig. 4 (a) Electrical conductivity of bioinspired graphene–epoxy layered composites with different graphene loading. (b) Electrical conductivity of bioinspired graphene–epoxy layered composites treated with different KH-560 loadings. (c) Schematic of the circuit of bioinspired graphene–epoxy layered composites. (d) The bioinspired graphene–epoxy layered composites as a part of conductive media connected with power supply and loaded with a blue LED light bulb. | ||
After treatment, the electrical conductivity of bioinspired graphene–epoxy layered composites decrease due to the insulative KH-560. However, for the composite of Treated-II, the electrical conductivity is still as high as 26.8 S cm−1. This kind of highly conductive bioinspired graphene–epoxy layered composites can be used as the conducting wire, which is demonstrated in a circuit as shown in Fig. 4c and d. A LED blue bulb was connected with the power supply, showing that the bioinspired graphene–epoxy layered composites cable functioned well for the entire testing period. Although the electrical conductivity of the composite of Treated-II decrease when grafting with the KH-560, the improvement in tensile strength and Young's modulus is remarkable. It is thus beneficial to lower the electrical conductivity while at the same time dramatically improving mechanical properties of bioinspired graphene–epoxy layered composites.
Our bioinspired strategy for fabricating graphene–reinforced epoxy composites compared with other graphene foam reinforced thermoplastic polymer composites and graphene reinforced epoxy composites shows great advantages as shown in Fig. 5. For example, the electrical conductivity of bioinspired graphene–epoxy layered composite is far higher than that of graphene–epoxy composite fabricated from in situ polymerization,7–9 and solution intercalation,10–14 which is because of the high loading of graphene and 3D networks of graphene nanosheets. The other approaches for constructing the graphene–polymer composites only show improvement in one type of property, such as electrical conductivity or tensile strength. For example, Cheng et al. fabricated the high electrically conductive graphene foam–PDMS composites through impregnating the PDMS into graphene foam without any surface modification.31 The electrical conductivity of graphene foam–PDMS composites is as high as 10 S cm−1, which is attributed to CVD-synthesized graphene foam with perfect structure. However, this value is only half of Treated-II. On the other hand, the tensile strength and Young's modulus of bioinspired graphene–epoxy layered composites of Treated-II shows 40-fold higher than the graphene foam–PDMS, respectively. Zhu et al. realized high tensile strength of graphene–epoxy composites.32 The tensile strength reached 84 MPa, higher than the Treated-II. However, the reported electrical conductivity was only 3.28 × 10−5 S cm−1, which is six orders of magnitude lower than Treated-II. Recently, Jia et al. demonstrated the highly flexible and strong graphene foam/epoxy composite with strength of 130 MPa at low graphene content (0.16%),15 claiming that the porous graphene foam network function as crack arresters and inhibit the crack propagation. However, the electrical conductivity is only 6.4 S cm−1, which is only one fourth of Treated-II. This strategy for fabricating the graphene–reinforced epoxy composites is also superior to other approaches for constructing the graphene–polymer composites, such as graphene sponge–PDMS with strength of 8.2 MPa and electrical conductivity of only 0.027 S cm−1.33
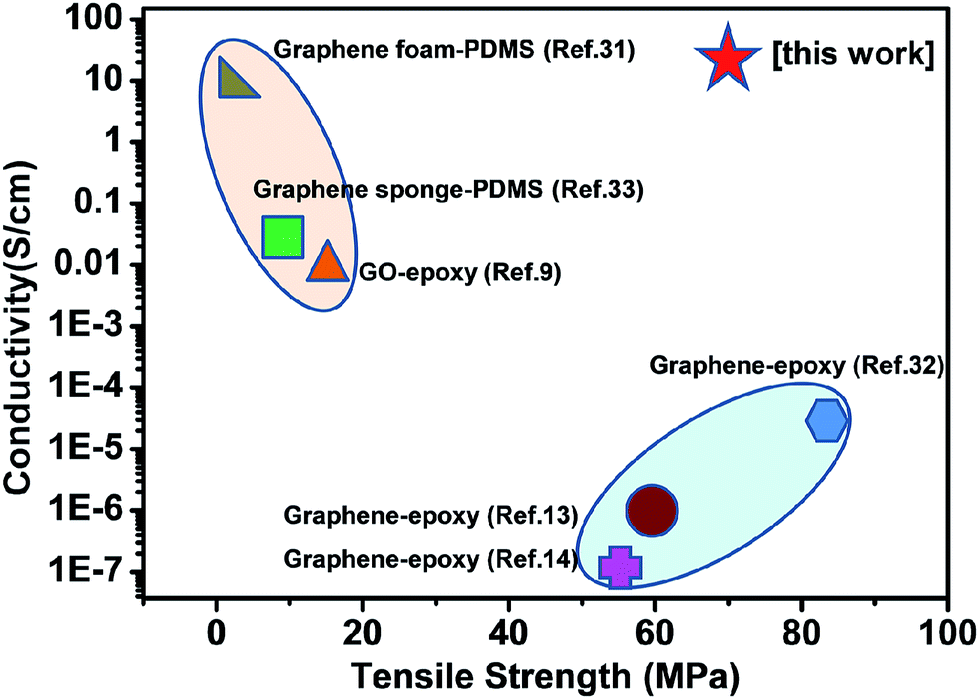 | ||
| Fig. 5 Comparison of electrical conductivity and tensile strength of the GF–epoxy layered composites with other graphene–polymer composites. The GF–epoxy layered composites show integrated high electrical conductivity and strength compared with other graphene–polymer composites, such as graphene foam–PDMS, and graphene–epoxy. | ||
Conclusions
In summary, the bioinspired graphene–epoxy layered composites were successfully fabricated. Compared with previous methods, this bioinspired strategy shows crucial advantages: (i) it dramatically enhances the graphene loading in the resultant composites, and (ii) it creates the hierarchically layered graphene–epoxy composites with high mechanical properties and electrical conductivity, simultaneously. Our study provides a novel approach for fabricating highly conductive graphene reinforced epoxy composites, and can be suitable for other thermosetting resins, such as bismaleimide, polyimide, cyanate ester and many others. These highly electrically conductive bioinspired graphene–epoxy layered composites have a great potential for applications in electromagnetic interference (EMI) shielding, aerospace, and other electrical devices.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21273017, 51103004), the National Research Fund for Fundamental Key Projects (2010CB934700), Program for New Century Excellent Talents in University (NCET-12-0034), Beijing Nova Program (Z121103002512020), Beijing Science and Technology Program (Z121100001312004), Fok Ying-Tong Education Foundation (141045), Open Project of Beijing National Laboratory for Molecular Sciences and the 111 Project (B14009), and the Fundamental Research Funds for the Central Universities (YWF-14-HXXY-018).Notes and references
- T. Kuilla, S. Bhadra, D. Yao, N. H. Kim, S. Bose and J. H. Lee, Prog. Polym. Sci., 2010, 35, 1350–1375 CrossRef CAS PubMed.
- J.-M. Thomassin, C. Jérôme, T. Pardoen, C. Bailly, I. Huynen and C. Detrembleur, Mater. Sci. Eng., R, 2013, 74, 211–232 CrossRef PubMed.
- J. Liang, Y. Wang, Y. Huang, Y. Ma, Z. Liu, J. Cai, C. Zhang, H. Gao and Y. Chen, Carbon, 2009, 47, 922–925 CrossRef CAS PubMed.
- Z. Chen, C. Xu, C. Ma, W. Ren and H. M. Cheng, Adv. Mater., 2013, 25, 1296–1300 CrossRef CAS PubMed.
- Y.-Q. Li, T. Yu, T.-Y. Yang, L.-X. Zheng and K. Liao, Adv. Mater., 2012, 24, 3426–3431 CrossRef CAS PubMed.
- S. Wan, Y. Li, J. Peng, H. Hu, Q. Cheng and L. Jiang, ACS Nano, 2015, 9, 708–714 CrossRef CAS PubMed.
- Y. Guo, C. Bao, L. Song, B. Yuan and Y. Hu, Ind. Eng. Chem. Res., 2011, 50, 7772–7783 CrossRef CAS.
- Y. Li, D. Pan, S. Chen, Q. Wang, G. Pan and T. Wang, Mater. Des., 2013, 47, 850–856 CrossRef CAS PubMed.
- N. Yousefi, X. Lin, Q. Zheng, X. Shen, J. R. Pothnis, J. Jia, E. Zussman and J.-K. Kim, Carbon, 2013, 59, 406–417 CrossRef CAS PubMed.
- M. A. Rafiee, J. Rafiee, Z. Wang, H. Song, Z.-Z. Yu and N. Koratkar, ACS Nano, 2009, 3, 3884–3890 CrossRef CAS PubMed.
- D. R. Bortz, E. G. Heras and I. Martin-Gullon, Macromolecules, 2012, 45, 238–245 CrossRef CAS.
- Y.-J. Wan, L.-C. Tang, D. Yan, L. Zhao, Y.-B. Li, L.-B. Wu, J.-X. Jiang and G.-Q. Lai, Compos. Sci. Technol., 2013, 82, 60–68 CrossRef CAS PubMed.
- J. Ma, Q. S. Meng, A. Michelmore, N. Kawashima, Z. Izzuddin, C. Bengtsson and H. C. Kuan, J. Mater. Chem. A, 2013, 1, 4255–4264 CAS.
- A. S. Wajid, H. S. T. Ahmed, S. Das, F. Irin, A. F. Jankowski and M. J. Green, Macromol. Mater. Eng., 2013, 298, 339–347 CrossRef CAS.
- J. Jia, X. Sun, X. Lin, X. Shen, Y.-W. Mai and J.-K. Kim, ACS Nano, 2014, 8, 5774–5783 CrossRef CAS PubMed.
- Y. Li, Y. A. Samad, K. Polychronopoulou, S. M. Alhassan and K. Liao, Sci. Rep., 2014, 4, 4652 Search PubMed.
- G. Tang, Z.-G. Jiang, X. Li, H.-B. Zhang, A. Dasari and Z.-Z. Yu, Carbon, 2014, 77, 592–599 CrossRef CAS PubMed.
- H. D. Espinosa, J. E. Rim, F. Barthelat and M. J. Buehler, Prog. Mater. Sci., 2009, 54, 1059–1100 CrossRef CAS PubMed.
- A. R. Studart, Adv. Mater., 2012, 24, 5024–5044 CrossRef CAS PubMed.
- M. A. Meyers, J. McKittrick and P.-Y. Chen, Science, 2013, 339, 773–779 CrossRef CAS PubMed.
- H.-B. Yao, J. Ge, L.-B. Mao, Y.-X. Yan and S.-H. Yu, Adv. Mater., 2014, 26, 163–188 CrossRef CAS PubMed.
- U. G. K. Wegst, H. Bai, E. Saiz, A. P. Tomsia and R. O. Ritchie, Nat. Mater., 2015, 14, 23–36 CrossRef CAS PubMed.
- A. Jackson, J. Vincent and R. Turner, Proc. R. Soc. London, Ser. B, 1988, 234, 415–440 CrossRef.
- Z. Niu, J. Chen, H. H. Hng, J. Ma and X. Chen, Adv. Mater., 2012, 24, 4144–4150 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- R. J. Waltman, J. Pacansky and C. W. Bates, Chem. Mater., 1993, 5, 1799–1804 CrossRef CAS.
- P.-G. Ren, D.-X. Yan, X. Ji, T. Chen and Z.-M. Li, Nanotechnology, 2011, 22, 055705 CrossRef PubMed.
- B. Liu, S. Bickerton and S. G. Advani, Composites, Part A, 1996, 27, 135–141 CrossRef.
- B. Qi, Z. Yuan, S. Lu, K. Liu, S. Li, L. Yang and J. Yu, Fibers Polym., 2014, 15, 326–333 CrossRef CAS PubMed.
- J. W. Gu, J. Dang, W. C. Geng and Q. Y. Zhang, Fibers Polym., 2012, 13, 979–984 CrossRef CAS.
- Z. Chen, W. Ren, L. Gao, B. Liu, S. Pei and H. M. Cheng, Nat. Mater., 2011, 10, 424–428 CrossRef CAS PubMed.
- L. Cao, X. Liu, H. Na, Y. Wu, W. Zheng and J. Zhu, J. Mater. Chem. A, 2013, 1, 5081–5088 CAS.
- Q. Peng, Y. Li, X. He, X. Gui, Y. Shang, C. Wang, C. Wang, W. Zhao, S. Du, E. Shi, P. Li, D. Wu and A. Cao, Adv. Mater., 2014, 26, 3241–3247 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: These might include comments relevant to but not central to the matter under discussion, limited experimental and spectral data, and crystallographic data. See DOI: 10.1039/c5ra00233h |
| ‡ These two authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2015 |