Tailored graphene systems for unconventional applications in energy conversion and storage devices
Received
16th August 2014
, Accepted 30th September 2014
First published on 30th September 2014
Abstract
Graphene-based materials have shown great potential in various fields across physics, chemistry, biology, and electronics, due to their unique electronic properties, facile synthesis, and ease of functionalization. In this review, we summarize the significant advances in tailored graphene systems for the recently developed unconventional energy conversion and storage devices reported by our group and others, namely focused on their tunable and controllable preparation and remarkable applications in new types of supercapacitors, lithium ion batteries, photovoltaic cells, and other emerging generators. This featured article also highlights the working principles and outlines the problems hindering the practical applications of graphene-based materials in these energy-related devices. Future research trends towards new methodologies in the design and synthesis of graphene-based systems with unique properties for emerging energy storage and energy conversion devices are also proposed.
Broader context
Energy and environmental issues, including the depletion of fossil fuels, waste pollution, and global warming, pose great threats to the sustainable development of human society. Consequently, finding reliable ways of efficiently producing and conveniently storing clean energy is of paramount importance. A practical solution to these problems lies in two aspects. First, energy should be efficiently converted from inexhaustible or renewable sources, such as solar, wind, water, and geothermal energy, into applicable forms such as electricity or fuels; and in this consideration, photovoltaic cells, photoelectrochemical cells, and fuel cells are the prevailing devices currently under intense investigation. Second, effective, low-cost, and environmentally benign energy storage devices are needed to complement the new power generating systems, due to the sporadic and off-grid features of most renewable energy sources, which is furthermore creating a high demand for supercapacitors and lithium-based rechargeable batteries. Recently, due to graphene's unique properties of large surface area, high electric/thermal conductivity, and excellent stability, there has been an ongoing trend to employ graphene materials to replace/reduce the usage of noble and rare metals in the present energy conversion and energy storage devices with materials incurring lower costs and having a higher environmental benignity. For example, nitrogen-containing graphene sheets, instead of Pt, could be used as catalysts for the oxygen reduction reaction (ORR) in fuel cells. Moreover, specially designed graphene structures also enable the fabrication of high-efficiency miniaturized devices with high flexibility and a light weight for use in wearable electronics, as shown in several recent paradigm products. In this review, we summarize the recent progress in tailoring graphene structures for unconventional applications in energy conversion and storage devices to meet the various requirements of modern devices and gadgets.
|
1. Introduction
Graphene, a two-dimensional (2D) form of carbon atoms with a hexagonal lattice structure has attracted enormous research interest since its first use in 2004.1 As the first true 2D material, graphene has demonstrated many intriguing properties, such as its intrinsically superior electrical conductivity, excellent mechanical flexibility, remarkable thermal conductivity, high surface area, etc.2,3 For instance, graphene shows an ambipolar electric field effect with high charge carrier mobilities of ca. 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1 at room temperature,1 while suspended graphene exhibits a strikingly high mobility approaching 200000 cm2 V−1 s−1 at low temperatures.4 Optically, the light absorption of a graphene monolayer is only 2.3% with a negligible reflectance of <0.1%.5 Thermally, graphene is highly stable and conductive (thermal conductivity of 3000–5000 W m−1 K−1).6 In addition, graphene has a large theoretical specific surface area (ca. 2600 m2 g−1)7,8 and a high chemical diffusivity of Li+ of 10−7 to 10−6 cm2 s−1.9,10 Its unique structure and these outstanding properties qualify graphene as one of the most important materials for a wide range of applications in electronics,11,12 high-strength materials,13 chemical and biosensors,14–16 catalysts,17 and various high-power-density/high-energy-density energy storage and conversion devices, such as lithium ion batteries,18–21 capacitors,22–25 fuel cells,26–33 and solar cells.34–37 Recently, ongoing efforts in the development of electronics has led to the emergence of various types of future electronics in unconventional forms, including smart skins, human friendly devices, and flexible/stretchable circuitries and energy devices.38–53 These advanced devices can perform sophisticated motions and/or deform into desirable shapes under external stimuli of bending, stretching, compressing, and twisting without sacrificing their device performance and reliability, thus attracting broad interest in many diverse applications, ranging from robots to memory chips to integrations into stretchable and wearable electronics.54–61
000 cm2 V−1 s−1 at room temperature,1 while suspended graphene exhibits a strikingly high mobility approaching 200000 cm2 V−1 s−1 at low temperatures.4 Optically, the light absorption of a graphene monolayer is only 2.3% with a negligible reflectance of <0.1%.5 Thermally, graphene is highly stable and conductive (thermal conductivity of 3000–5000 W m−1 K−1).6 In addition, graphene has a large theoretical specific surface area (ca. 2600 m2 g−1)7,8 and a high chemical diffusivity of Li+ of 10−7 to 10−6 cm2 s−1.9,10 Its unique structure and these outstanding properties qualify graphene as one of the most important materials for a wide range of applications in electronics,11,12 high-strength materials,13 chemical and biosensors,14–16 catalysts,17 and various high-power-density/high-energy-density energy storage and conversion devices, such as lithium ion batteries,18–21 capacitors,22–25 fuel cells,26–33 and solar cells.34–37 Recently, ongoing efforts in the development of electronics has led to the emergence of various types of future electronics in unconventional forms, including smart skins, human friendly devices, and flexible/stretchable circuitries and energy devices.38–53 These advanced devices can perform sophisticated motions and/or deform into desirable shapes under external stimuli of bending, stretching, compressing, and twisting without sacrificing their device performance and reliability, thus attracting broad interest in many diverse applications, ranging from robots to memory chips to integrations into stretchable and wearable electronics.54–61
In this review, we aim to summarize the very recent developments, contributed by us and other groups, of graphene-based systems in emerging and unconventional forms of energy conversion and storage devices, mainly in supercapacitors, lithium ion batteries, photovoltaic cells, and other power generators. As graphene may play different roles in these devices, the current work is accordingly structured to discuss its property-motivated applications in each type of device. Finally, future trends in designing and improving smart devices for energy conversion and storage are also envisaged.
2. Tailored graphene systems for energy storage
2.1 Graphene-based supercapacitors
With the recent boom in studies on powering electric vehicles and portable electronics, electrochemical capacitors, also known as supercapacitors, have attracted increasing attention, due to their high power density, high rates of charge–discharge, long cycling life, and their ability to complement secondary batteries in reducing the cycling duty and, therefore, in extending the battery life.62–66 However, conventional supercapacitors are usually heavy and bulky, whereas there is currently a strong demand for highly efficient, flexible, miniaturized supercapacitor devices compatible with flexible and wearable electronic energy storage devices to cater for the power requirements of modern gadgets.67–71 Furthermore, the simple and low-cost assembly of these flexible and lightweight supercapacitors would enable the easy fabrication of capacitor arrays via a convenient design and integration that is compatible with the existing electronic industry, making them industrially viable for powering a range of small/micro devices in different scenarios.
2.1.1 Flexible and compressible graphene supercapacitors.
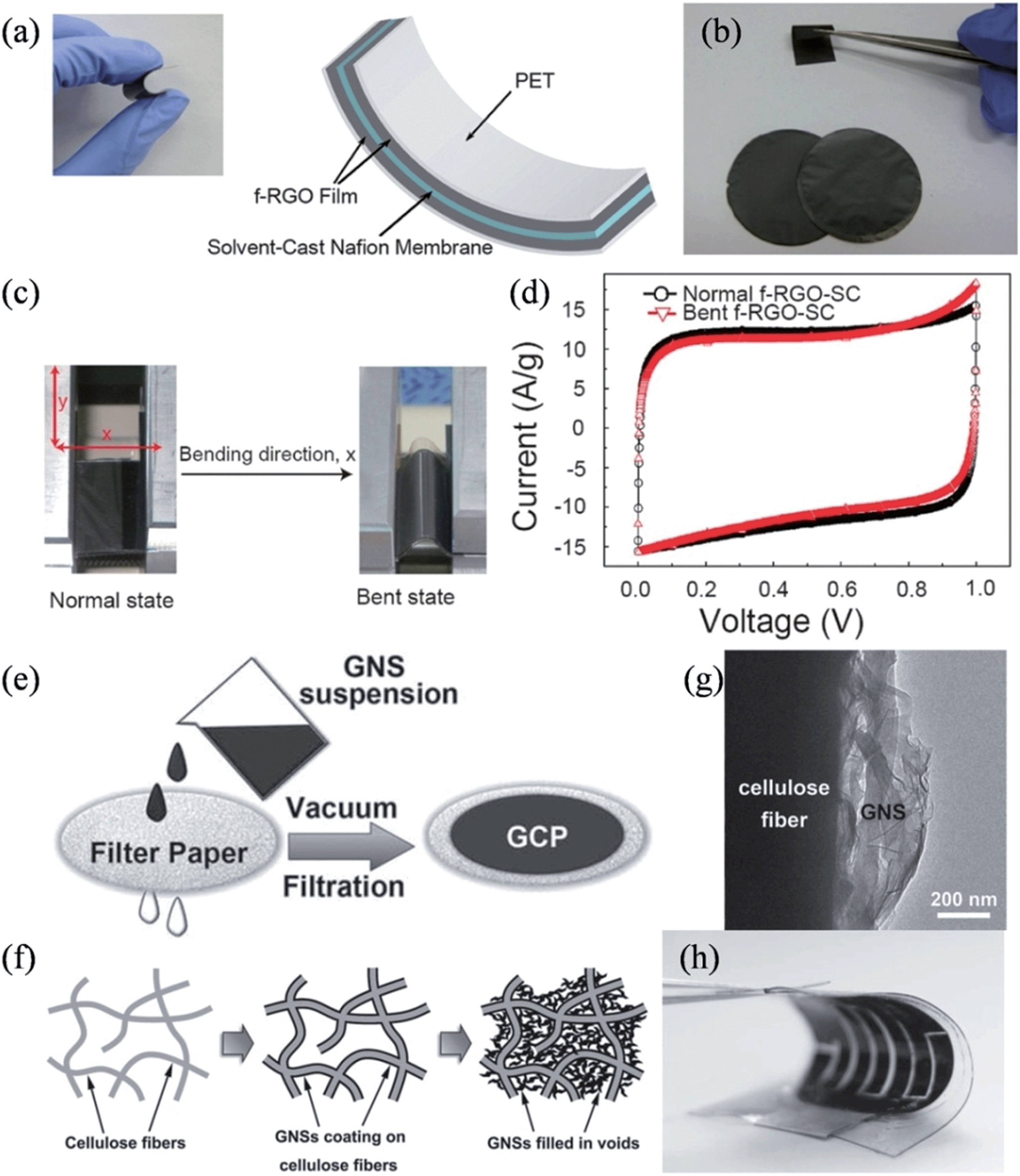
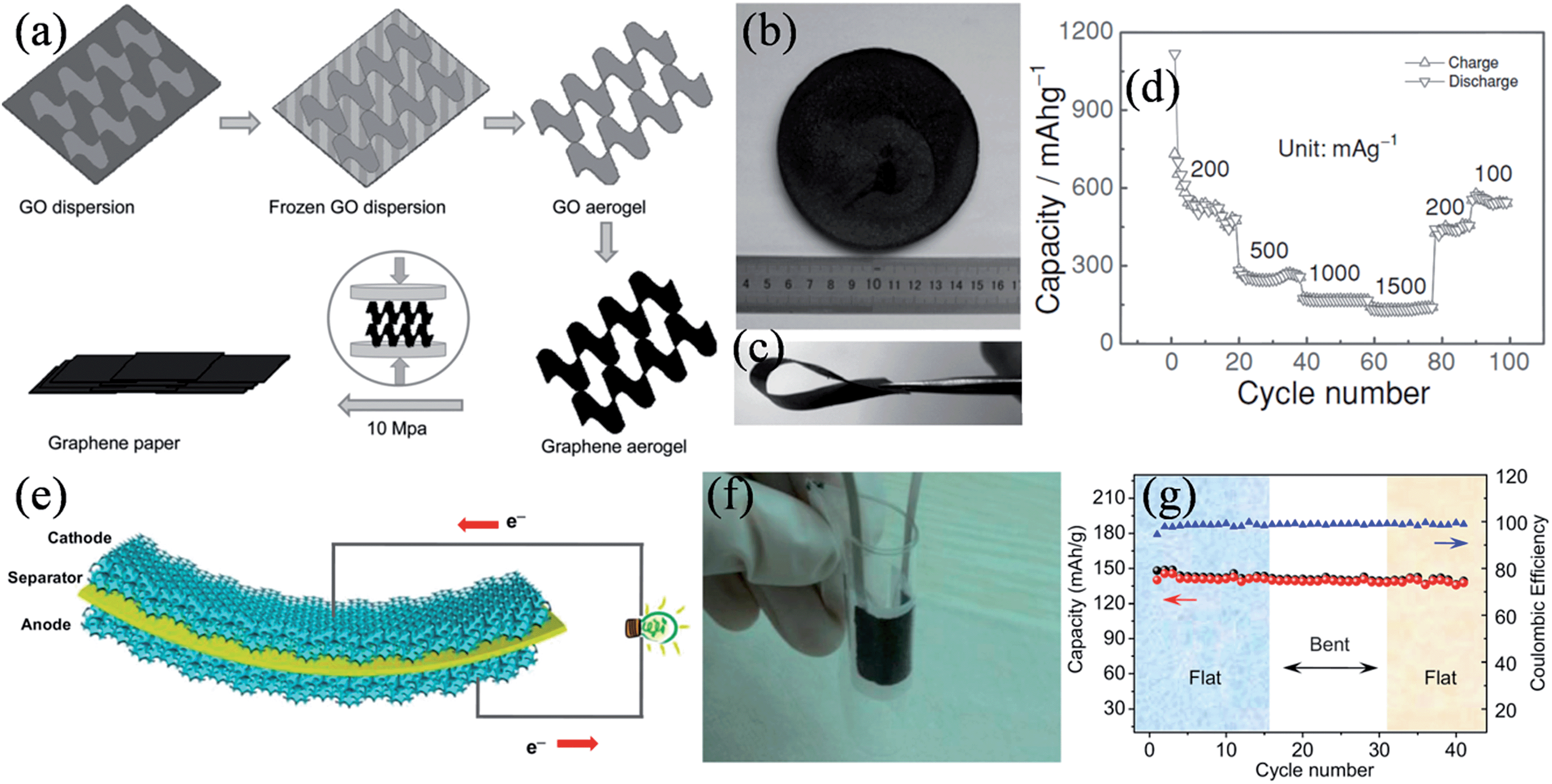
Graphene-assembled three-dimensional (3D) macroscopic materials, such as foam and aerogel, have a tunable hierarchical morphology with high surface areas and can form large-sized monolithic materials. These properties provide a new design platform, characterized by a light weight, high porosity, high mechanical stability, and electrical conductivity, thus offering great technological promise for supercapacitors.72–74 Graphene-based porous bulk materials are usually fabricated by chemical vapor deposition over a porous template,75,76 dip-coating on a porous framework,77,78 or sol–gel reactions.79–83 However, these materials are usually brittle, and have low compression resilience ratios.75,81,84 Hence, skeletons of elastomeric polymers are introduced to the graphene materials to enhance their flexibility and compressibility for practical applications. Choi et al. demonstrated an all-solid-state flexible supercapacitor by assembling a film electrode of Nafion-functionalized reduced graphene oxide (f-RGO) and solvent-cast Nafion electrolyte membranes as the electrolyte and separator (Fig. 1a).85 The functionalized-RGO film showed high flexibility (Fig. 1b), and the obtained supercapacitor was demonstrated to have a high capacitance of 118.5 F g−1 at 1 A g−1 and a good performance durability under 1000 cycles of bending and relaxing, owing to the improved adhesion and the facilitated charge/ion transport at the electrode/electrolyte interfaces (Fig. 1c and d). Alternatively, Cheng et al. developed a graphene–cellulose paper (GCP) membrane by simply filtering a suspension of graphene nanosheets (GNSs) through a slice of filter paper (Fig. 1e and f).86 Due to the strong interactions between the abundant functional groups on the cellulose fibers (CFs) and the negatively charged GNSs, GNSs uniformly covered the CFs to form an interconnected conductive framework (Fig. 1g), thereby combining the porous and ductile nature of the cellulose filter paper with the good electrical conductivity and electroactivity of GNSs. As a result, the strain on the GCP membrane was 3% elongation, about three times higher than that of the graphene paper (0.76%). The excellent flexibility and strength of the GCP membrane enables the construction of more sophisticated devices and, as shown in Fig. 1h, a flexible interdigital supercapacitor was conveniently fabricated by solidifying two interdigital GCP electrodes on a polydimethylsiloxane (PDMS) substrate with a polymer gel electrolyte. Since the interdigital supercapacitor can be very thin, it is deemed ideal for powering ultrathin electronic devices and micro-electromechanical systems.
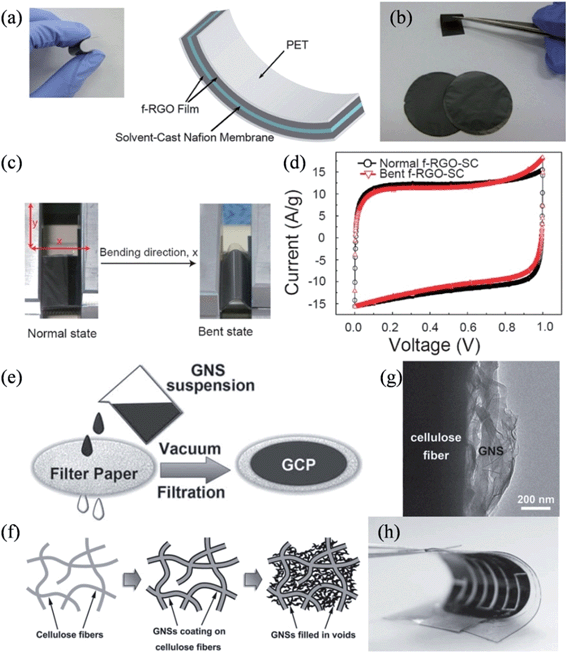 |
| | Fig. 1 (a) Image and schematic presentation of the all-solid-state flexible supercapacitor. (b) Image of the f-RGO films showing the flexibility. Images (c) and cyclic voltammograms (d) of the flexible supercapacitor in the normal and bent states (reprinted with permission from ref. 85. Copyright 2011 American Chemical Society). Illustrations of (e) fabrication of the GCP membrane and (f) structural evolution of GCP as the GNS loading increases. (g) TEM image of a cellulose fiber in a GCP membrane, showing GNSs anchored on the fiber surface. (h) Image of a flexible interdigital GCP supercapacitor (reprinted with permission from ref. 86. Copyright 2011 Wiley-VCH). | |
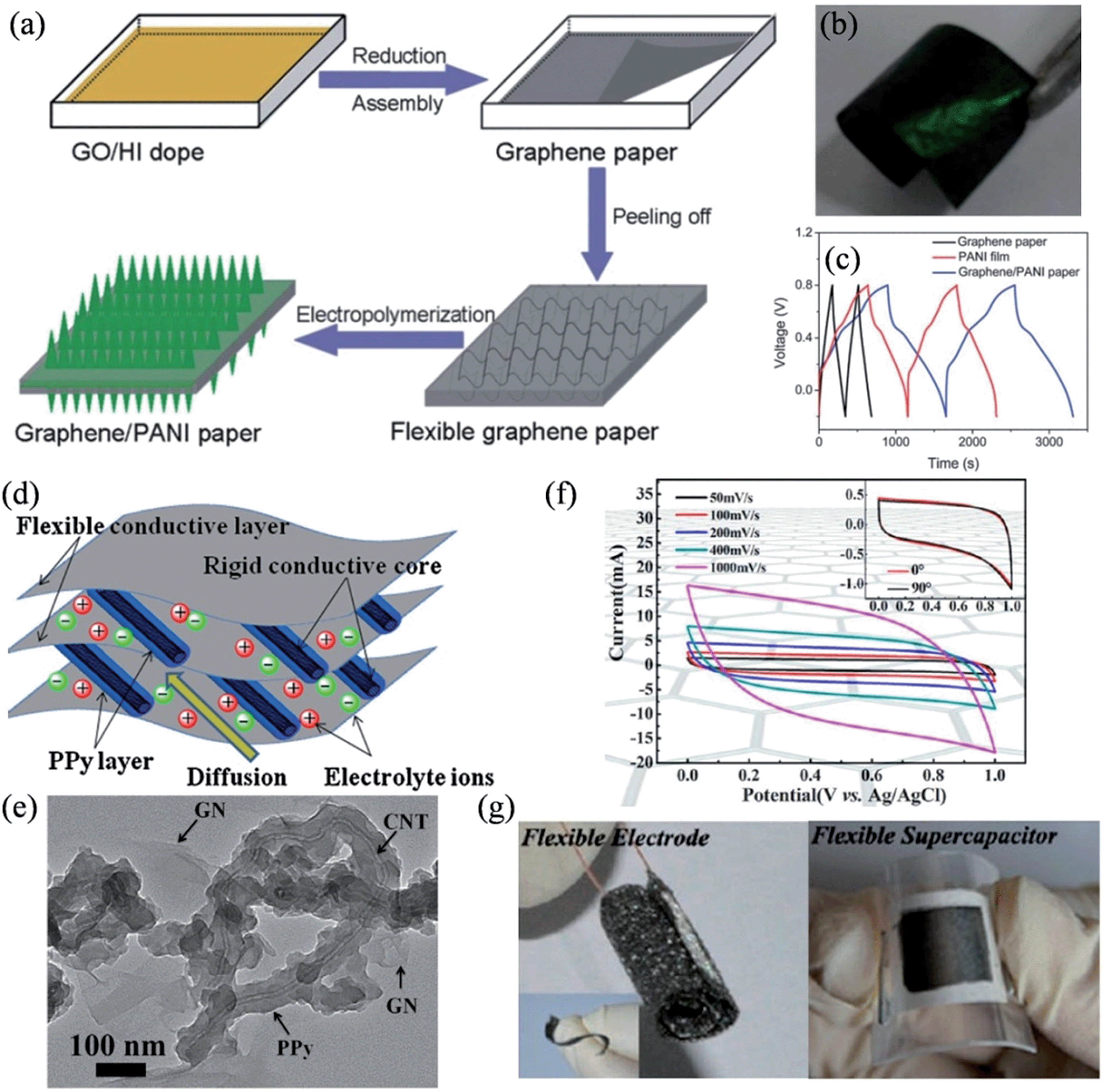
Polyaniline (PANI) and polypyrrole (PPy) are well-studied conducting polymers that are widely used in energy conversions and storage systems, mostly because of their good conductivity, easy-to-handle synthesis, the low cost of their monomers, environmental stability, and the high redox pseudocapacitive charge storage associated with the multiple redox states.73,87–90 To further improve the electrochemical performance of the flexible supercapacitor, PANI was included to fully exploit the unique potential of graphene paper as a free-standing electrode by the in situ anodic electropolymerization of aniline monomers on the graphene paper (Fig. 2a–c).91 The obtained electrode contained graphene/polyaniline composite sheets as the building blocks, and showed a favorable tensile strength of 12.6 MPa and a stable large electrochemical capacitance of 233 F g−1 over 1500 cycles. In another approach, Chen et al. deployed a “dipping and drying” strategy to coat the cellulose fibers (CFs) of the paper with a thin and uniform surface layer of graphene oxide (GO), and then assembled the GO sheets into reduced graphene oxide (RGO) networks to form a RGO/CF composite paper rich in micro-sized pores (Fig. 2d).92 Such a composite structure could simultaneously alleviate the agglomeration of RGO in the pores of the paper and promote the diffusion of ions in the electrolyte, fully utilizing the interconnected CF network and the porous nature of the paper (Fig. 2e). PANI can be further deposited onto the RGO surface to achieve a high specific capacitance of 464 F g−1. The excellent flexibility and conductivity of the PANI-RGO/CF composite papers enable their direct application as electrodes for flexible supercapacitors with a narrow Pt wire as the current collector, eliminating the need to resort to heavy and bulky metallic foils or foams that are generally used as current collectors in conventional supercapacitors (Fig. 2f). An all-solid-state supercapacitor based on the PANI-RGO/CF paper attained 89% of the maximum capacity after 1000 cycles and lost only about 3.4% and 5% of its maximum capacity when bent to 10% of its original length and folded by 180° (Fig. 2g), respectively, proving its candidacy as a novel kind of flexible electrode for wearable and roll-up devices.
 |
| | Fig. 2 (a) Illustration of the anodic electropolymerization of aniline on the graphene paper. Digital camera images of (b) two free-standing graphene papers (30 mm × 10 mm) and (c) a flexible graphene paper (reprinted with permission from ref. 91. Copyright 2009 American Chemical Society). (d) Schematic diagram of preparing PANI-RGO/CF composite paper. (e) Illustration of the nanostructure of the RGO/CF composite paper. (f) Schematic diagram of a compact supercapacitor based on PANI-RGO/CF paper, where the inset is an optical image of four compact supercapacitors lighting an LED. (g) Variation of the specific capacitance of the all-solid-state supercapacitor in different folding states, with the inset showing an LED illuminated using four integrated supercapacitors in folding states (reprinted with permission from ref. 92. Copyright 2014 Wiley-VCH). | |
Recently, Yu and coworkers reported a convenient one-step method to fabricate free-standing, low-density graphene paper with good electrical conductivity, where PANI nanorod arrays could electrochemically grow in situ to form flexible graphene–PANI composite paper (Fig. 3a).93 The composite electrode was highly flexible (Fig. 3b), exhibiting an enhanced capacitive performance of 763 F g−1 at a discharge current of 1 A g−1, and a good cycling stability with 82% capacity retention after 1000 cycles (Fig. 3c). The pronounced improvements both in the specific capacitance and stability were ascribed to the unique structure of the PANI–graphene composite. On the one hand, the PANI nanorods render a high surface area, thus facilitating ion diffusion at the electrolyte/nanorod interface, and the high conductivity of the graphene paper favors the quick transport of electrons across the device. On the other hand, the vertically aligned PANI nanorods make graphene paper a perfect base for relaxing the mechanical strain and deformation that occur during the charge/discharge process, mitigating the swelling and shrinking of the electrode materials against serious collapses by the insertion/extraction process of the counter ions.
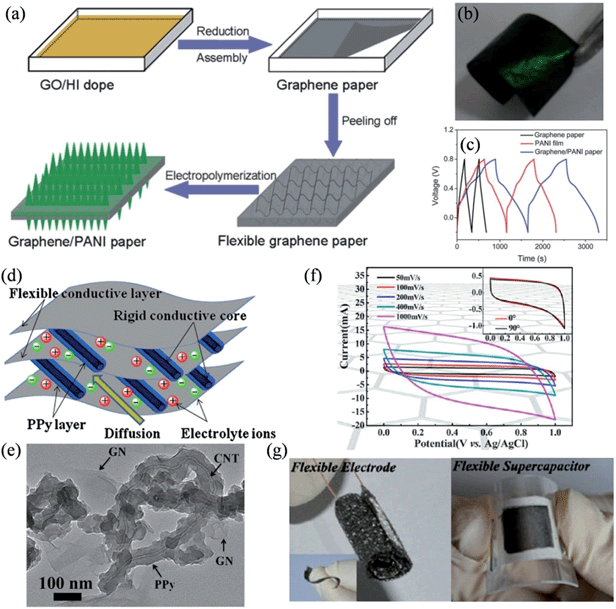 |
| | Fig. 3 (a) Schematic illustrations of the formation process of graphene–PANI paper. (b) Photograph of the flexible graphene–PANI paper (3 × 1.5 cm) with an electrochemical deposition time of 10 min. (c) Galvanostatic charge–discharge curves of graphene paper, PANI film on the Pt electrode, and graphene–PANI paper at a current density of 1 A g−1 (reprinted with permission from ref. 93. Copyright 2013 Royal Society of Chemistry). (d) Schematic representation of the microstructure and energy storage characteristics of the GN-PPy/CNT film. (e) TEM image of GN-PPy/CNT (reprinted with permission from ref. 94. Copyright 2011 Elsevier B.V.). (f) Cyclic voltammograms of the flexible supercapacitors at scan rates of 50, 100, 200, 400, and 1000 mV s−1. Inset shows the CVs of the flexible supercapacitors with bending angles of 0° and 90° at a fixed scan rate of 10 mV s−1. (g) Digital photographs showing the curled 3D graphene network and the flexible supercapacitor in the bent state (reprinted with permission from ref. 95. Copyright 2013 American Chemical Society). | |
Besides PANI, PPy was also introduced to enhance the specific capacitance of the flexible graphene electrodes. Lu et al. proposed a novel flexible graphene-polypyrrole/carbon nanotube (G-PPy/CNT) ternary composite film through a flow-assembly method (Fig. 3d), where coaxial PPy/CNT nanocables were uniformly sandwiched between the graphene sheets, so that the mechanical stability of the PPy chains was significantly improved by the flexible graphene sheets and the rigid CNT core (Fig. 3e).94 The specific capacitance of the GN-PPy/CNT composite reached 211 F g−1 at a discharge current of 0.2 A g−1, and an excellent cycling stability of 5% capacity loss after 5000 cycles. Additionally, He et al. reported a type of MnO2-coated freestanding, flexible, lightweight, and highly conductive three-dimensional (3D) graphene network, on which a remarkably high mass loading of 92.9% MnO2 was achieved.95 These hybrid flexible electrodes exhibit a high areal capacitance of 1.42 F cm−2 and a high specific capacitance of 130 F g−1 (Fig. 3f), and more importantly, they could be twisted or bent to a large extent without suffering any performance loss (Fig. 3g). To this extent, we have designed a unique strategy for the in situ formation of PPy–graphene (PPy–G) foam, and have demonstrated a highly compression-tolerant graphene-based supercapacitor.29,96 The PPy–G matrix could sustain large-strain deformations (e.g., ε = 50%) under manual compression and could recover most of the material volume without structural fatigue within seconds. Consequently, the fabricated deformation-tolerant supercapacitors showed no significant change with a 50% applied strain over 1000 measured cycles, highlighting the excellent electrochemical stability of the compressible supercapacitor based on PPy–graphene foam electrodes.
Graphene nanoribbons (GNRs) elegantly combine the structure and properties of carbon nanotubes (CNTs) and graphene sheets.97–99 GNRs feature a flat crystal surface that extends into a one-dimensional structure with high length-to-width aspect ratios compared to exfoliated graphene sheets of irregular sizes and shapes. Measurements and simulations on individual GNRs have revealed fascinating electrical and thermal properties.99,100 Tour et al. demonstrated the splitting of vertically aligned CNTs with preservation of their vertical integrity to produce a mixture of split multi-walled carbon nanotubes (MWNTs) and GNRs.101 The largely opened spaces and the aligned vertical structure not only increased the effective surface area for the ions, but also provided facilely accessible paths for fast ion transport. The energy density and power density are in the range of 5.2–9.4 W h kg−1 and 1.1–103 kW kg−1, respectively, superseding those of the prevailing electrochemical capacitor technology102 and some single-walled carbon nanotube (SWNT) electrode materials.103 When a pristine vertically aligned CNT carpet was used as the active material, a maximum energy density of ∼3.1 W h kg−1 was measured, and the maximum power density was ∼35 kW kg−1 at an energy density of 0.95 W h kg−1. Also, Baughman et al. fabricated long and narrow GNRs that could be assembled and aligned into the first large GNR sheets, and then into macroscopic neat GNR fibers with high conductivity and good mechanical performance.104 The gravimetric capacitance of the graphene yarns was much higher than that of conventional twist-spun MWNT yarns, reaching a maximum value of 99 F g−1, which compares favorably to a value of 20 F g−1 obtained by the starting MWNT yarns. This novel conversion process was scalable and could be employed for the industrial production of GNR yarns that would be useful as woven electrodes for flexible supercapacitors.
2.1.2 Graphene micro-supercapacitors.
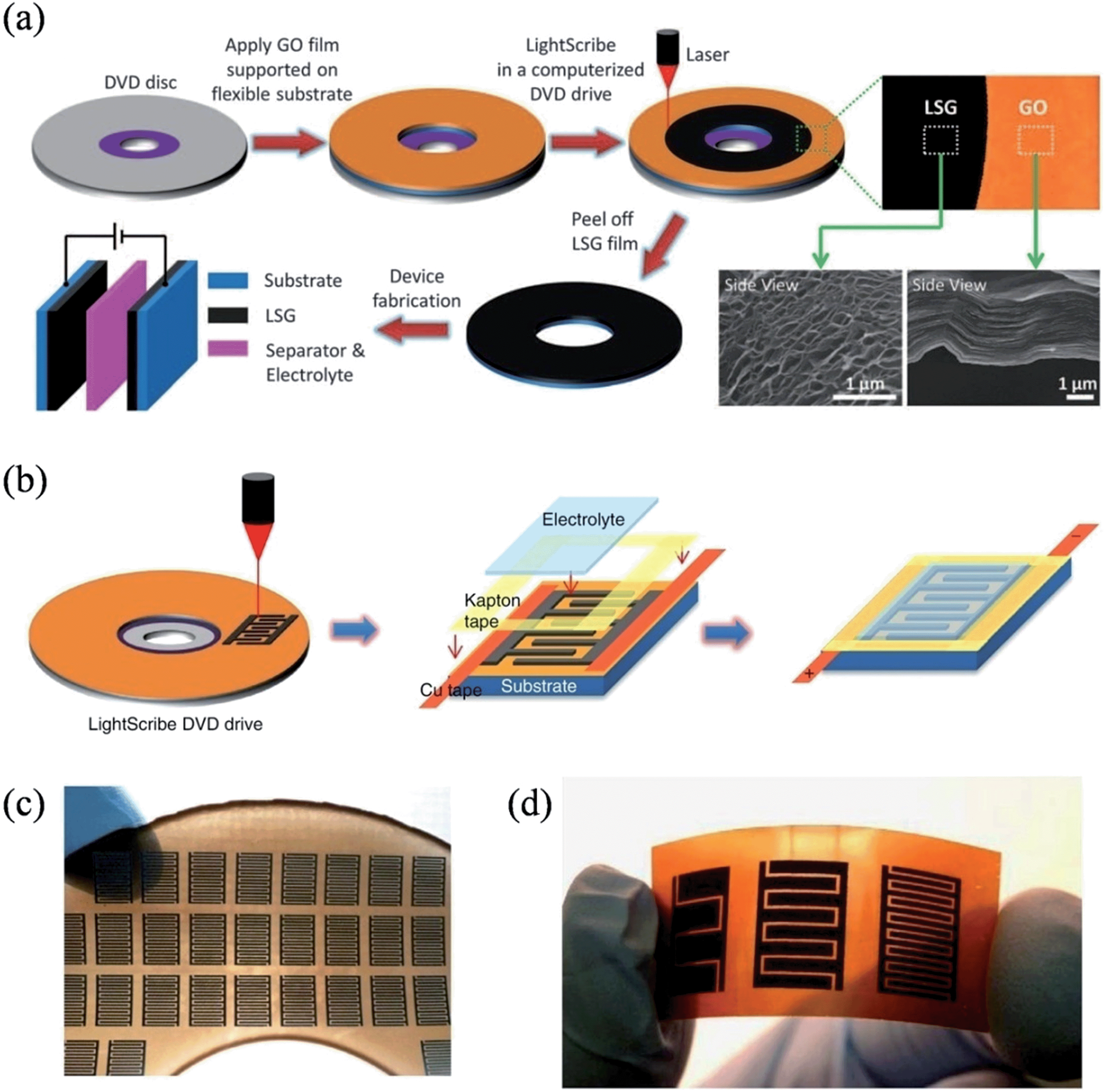
The recent soaring demand in portable electronics has triggered the urgent need to miniaturize energy-storage units. In 2012, El-Kady et al. employed a standard LightScribe DVD optical drive to perform the direct laser reduction of graphite oxide films to laser-scribed graphene (LSG),105 an approach that effectively prevents the aggregation of graphene sheets and enables the LSG to feature an open network structure with a large specific surface area of 1520 m2 g−1 and a high electrical conductivity of 1738 S m−1 (Fig. 4a). Superthin symmetrical capacitors with a total thickness of less than 100 μm could be achieved by the assembly of two identical LSG electrodes, qualifying them as potential power systems for micro-device applications. The same group further reported the direct fabrication of interdigitated graphene micro-supercapacitors by etching a computer-designed circuit onto the film (Fig. 4b).106 In contrast to conventional micro-fabrication methods, this mask-free method is able to work under ambient conditions to facilely build supercapacitor arrays with a high areal density, where 112 micro-supercapacitors can be produced on a DVD disc in a single run (Fig. 4c). As the LightScribe has a lateral spatial resolution of ∼20 microns, micro-devices with different numbers of interdigitated electrodes could be easily fabricated, commensurate with the working domain of the conventional micro-fabrication techniques in the order of ∼100 μm. The LSG produced in this method had an even higher conductivity of 2350 S m−1, and the micro-capacitors with 16 interdigitated microelectrodes demonstrated a power density of ∼200 W cm−3, representing one of the highest values for any supercapacitor ever reported. Again, the LSG supercapacitors were highly stable, and maintained 96% of their initial performance after 10000 consecutive charge–discharge cycles.
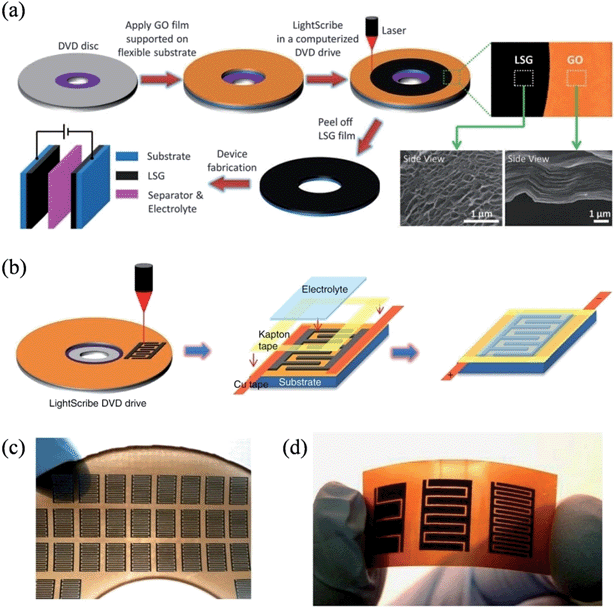 |
| | Fig. 4 (a) Schematic illustration of the fabrication of laser-scribed graphene-based electrochemical capacitors (reprinted with permission from ref. 105. Copyright 2012 the American Association for the Advancement of Science). (b) Schematic diagram showing the fabrication process for laser-scribed graphene micro-supercapacitors. (c) Demonstration of more than 100 micro-devices produced on a single run. (d) A digital photograph of the laser-scribed micro-devices with 4, 8, and 16 interdigitated electrodes (reprinted with permission from ref. 106. Copyright 2013 Nature Publishing Group). | |
Following a similar procedure, Ajayan and coworkers demonstrated the application of hydrated GO as an ionic conductor as well as an electrical insulator, making it suitable to work simultaneously as a viable electrolyte and electrode separator in the micro-supercapacitor.107 In-plane and sandwiched supercapacitors, with different geometries in the RGO-GO-RGO configuration, were built by layer writing and patterning without the use of any external electrolyte (Fig. 5a). The device with the in-plane concentric circular pattern had the highest specific capacitance of about 0.51 mF cm−2 and a power density of 1.7 W cm−3 (Fig. 5b), while scanning electron microscopy (SEM) revealed the porous nature of the laser-reduced RGO, which was due to gases released from decomposing the functional groups and the water vapor under localized laser heating (Fig. 5c). The performance of the micro-supercapacitor could be significantly improved if an external electrolyte, even a drop of deionized water, was used to improve the ion transport. Nevertheless, the hydrated GO films represented a completely different type of porous solid electrolyte, and capable of making the current micro-capacitors potentially much lighter and more compact. To further improve the capacitive performance of the micro-supercapacitor, Xue et al. adopted a solution-based method to electrochemically grow a uniform layer of ordered PANI nanorod arrays vertically on patterned RGO thin films (Fig. 5d).108 The PANI nanorods were shown to be densely packed on the surface of RGO, with a diameter of about 20 nm and a height of around 100 nm to 200 nm, leading to a synergistic effect in supercapacitive storage that decreased the ionic diffusion resistance and the charge-transfer resistance. The all-solid-state RGO/PANI micro-supercapacitor exhibited a specific capacitance as high as 970 F g−1 at a discharge current density of 2.5 A g−1, with 90% performance retention after 1700 consecutive cycles, making it ideal for powering flexible miniaturized electronic devices.
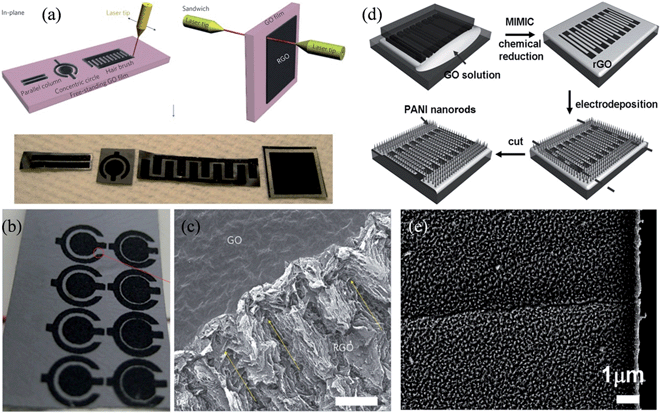 |
| | Fig. 5 (a) Schematics of the laser-patterning of free-standing hydrated GO films to fabricate RGO-GO-RGO devices with in-plane and sandwich geometries. The bottom row shows images of the patterned films. (b) Image of an array of concentric circular patterns fabricated on a free-standing hydrated GO film. (c) SEM image of the interface between GO and RGO (scale bar, 100 μm), with yellow arrows indicating a long-range pseudo-ordered structure generated by laser-beam scanning (reprinted with permission from ref. 107. Copyright 2011 Nature Publishing Group). (d) Fabrication of RGO/PANI microelectrodes. (e) SEM image of PANI nanorods on the RGO pattern (reprinted with permission from ref. 108. Copyright 2012 Wiley-VCH). | |
2.1.3 Graphene fiber supercapacitors.
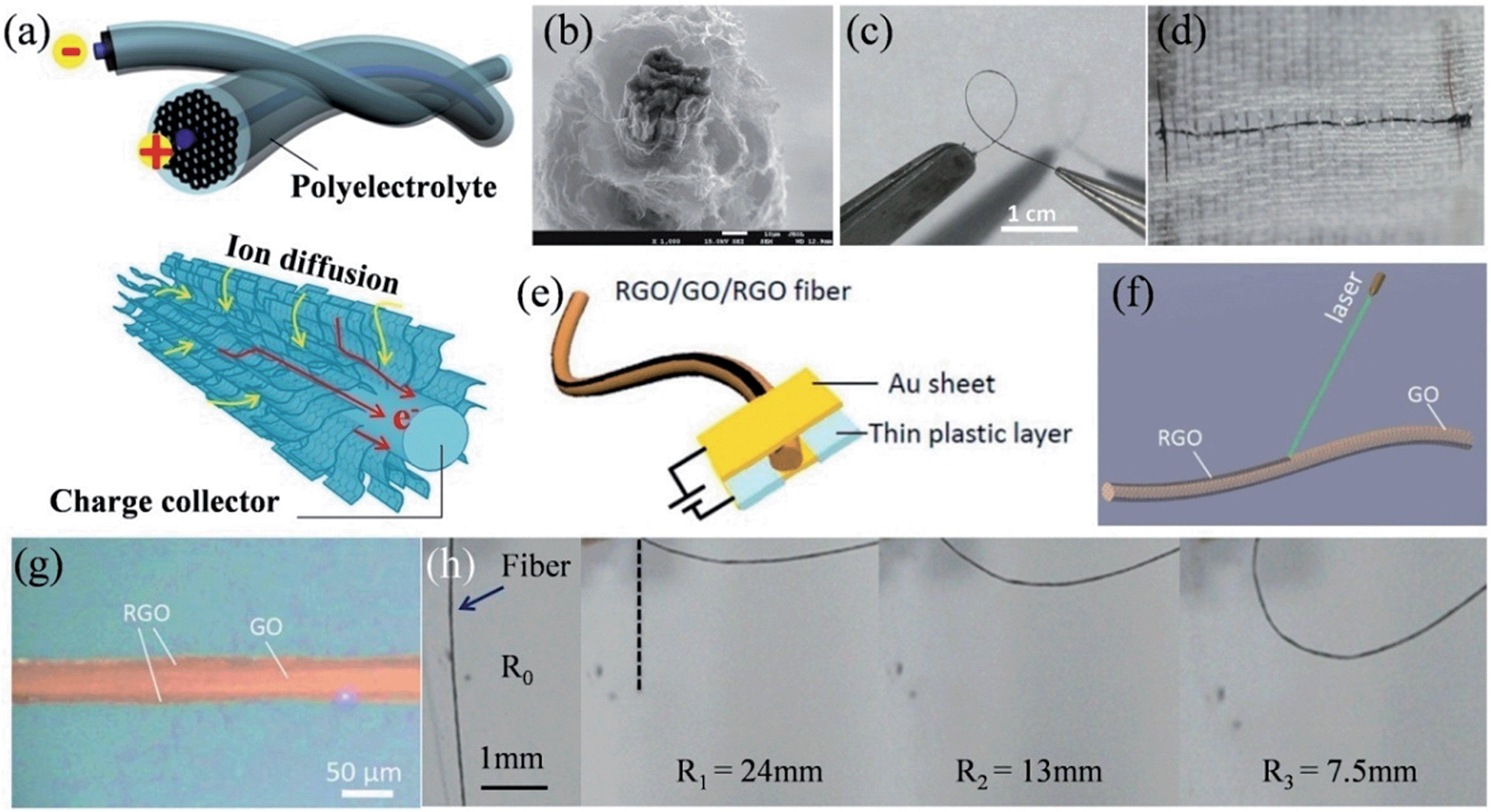
The recent successful demonstration of the assembly of 2D graphene sheets into macroscopic graphene fibers (GFs) has now made the construction of GF supercapacitors possible.109–111 We have thus designed a series of unique hierarchical structures to fabricate different GF supercapacitors. A sheath of a 3D porous graphene framework was deposited onto a GF core to form an all graphene core-sheath fiber supercapacitor (Fig. 6a), where the high electronic conduction of the core graphene fiber and the highly exposed surface of the 3D graphene network were well combined (Fig. 6b).112 The assembled fiber supercapacitor was highly flexible (Fig. 6c) and could be conveniently woven into a textile for wearable electronics (Fig. 6d). In a further effort, an all-in-one fiber graphene supercapacitor was fabricated by rationally integrating the two electrodes and the separator on a single GF, without involving any extra bonding agent or assembly process (Fig. 6e).113 The controllable laser reduction of the GO fiber could facilely realize a RGO-GO-RGO configuration (Fig. 6f), where the RGO parts had an approximate width of 1/4 diameter of the fiber along the surface, acting as the active electrode materials and current collectors, and the remaining GO part between the RGO layers worked as the separator (Fig. 6g). This all-in-one fiber supercapacitor inherited the mechanical flexibility of GF and could be deliberately bent to different radii of curvature, while maintaining its original high capacitance (Fig. 6h).
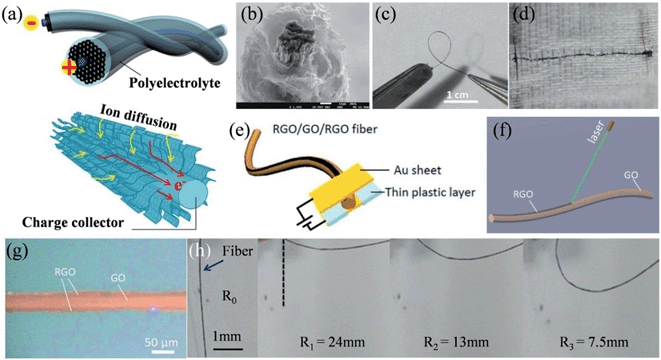 |
| | Fig. 6 (a) Schematic illustration of a graphene core–sheath fiber supercapacitor with a polyelectrolyte. The lower part shows the advantage of the graphene core–sheath fiber in charge collection and ion diffusion. (b) Cross-section SEM image showing the core GF surrounded by standing graphene sheets. Images of the fiber supercapacitor (c) in the bending state and (d) embedded in the textile (reprinted with permission from ref. 112. Copyright 2013 Wiley-VCH). (e) Representation of the structure of the all-in-one fiber supercapacitor. (f) Schematic of the laser reduction of GO fiber for the formation of RGO-GO-RGO fiber. (g) Optical image of the RGO-GO-RGO fiber. (h) Scheme of the bent state of the RGO-GO-RGO fiber capacitor with a certain radius of curvature (R1 to R3), where R0 stands for the initial state (reprinted with permission from ref. 113. Copyright 2014 Royal Society of Chemistry). | |
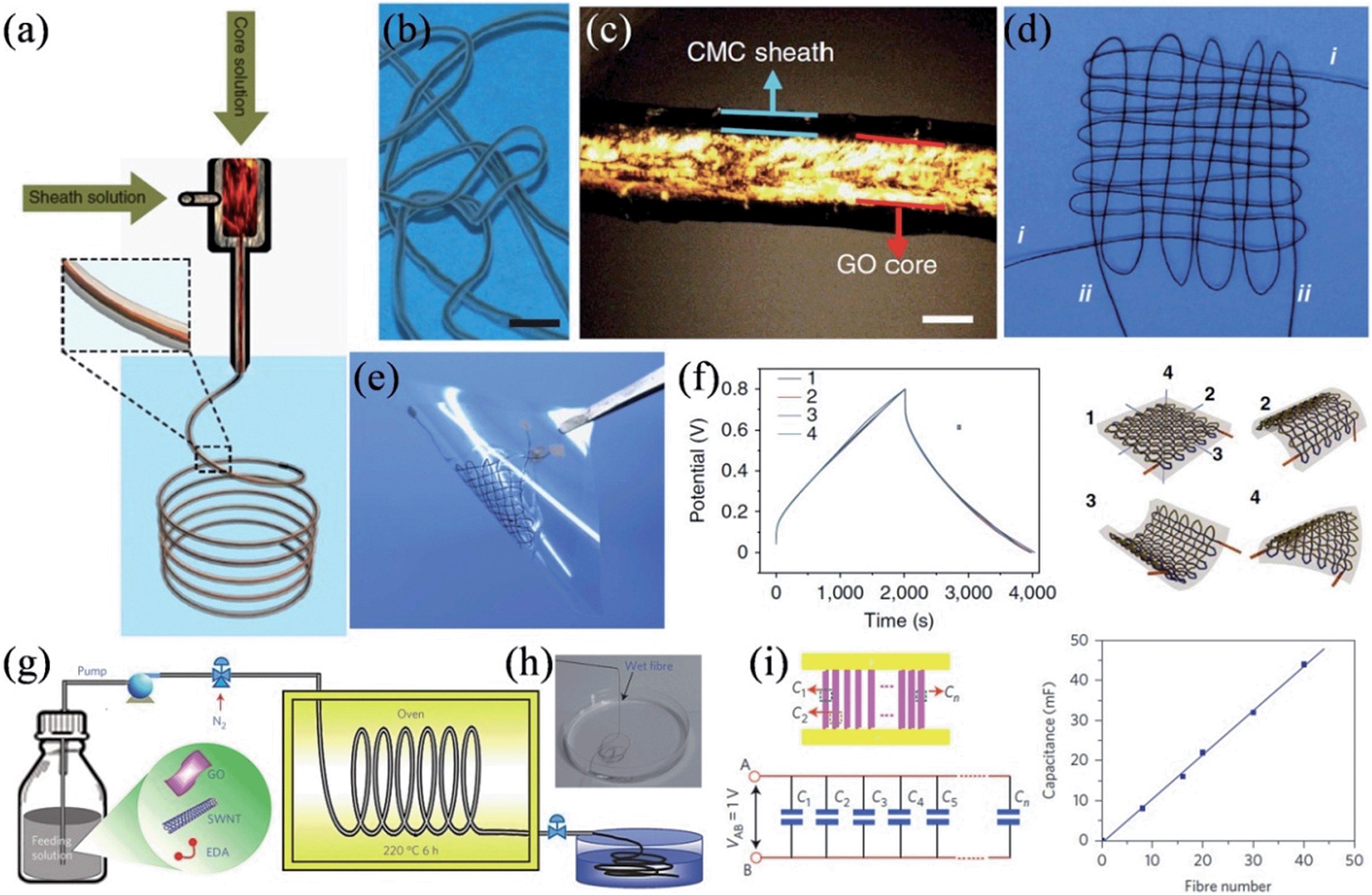
In a recent work, Gao and coworkers presented a coaxial wet-spinning assembly strategy to prepare sodium carboxymethyl cellulose (CMC) coated graphene core-sheath fibers (Fig. 7a–c).114 As CMC is an ionic conducting but electrically insulating polyelectrolyte, two individual 40 cm long coaxial CMC-coated graphene core–sheath fibers could be directly employed to interweave a cloth supercapacitor (Fig. 7d and e). The cloth supercapacitor demonstrated a capacitance of 28 mF at a current of 10 mA, and the corresponding charge–discharge curves remained undistorted under the bending angle of 180° along all three directions (Fig. 7f), confirming its excellent tolerance to arbitrary bending actions. Also, by wet-spinning, Yu et al. described the scalable synthesis of hierarchically structured carbon microfibers of a single-walled carbon nanotube (SWNT)/nitrogen-doped RGO sheet interconnected network architecture (Fig. 7g and h).115 As the RGO renders a large surface area for ion adsorption and the aligned SWNT network reduces the RGO interlayer resistance, the as-prepared dry hybrid fibers showed a good tensile strength of 84 MPa to 165 MPa, excellent electrical conductivity of 20 S cm−1 to 102 S cm−1, and a large specific surface area up to 396 m2 g−1. Consequently, micro-supercapacitors fabricated with these fibers featured volumetric energy densities of up to ∼300 F cm−3, with no loss in the cyclic stability or rate capability. The hybrid fibers were robust enough to be assembled in parallel by incorporating up to 40 fibers, and the overall capacitance of the array followed a linear increase with the number of fibers (Fig. 7i), showing the good scalability of micro-supercapacitors based on the hybrid fiber.
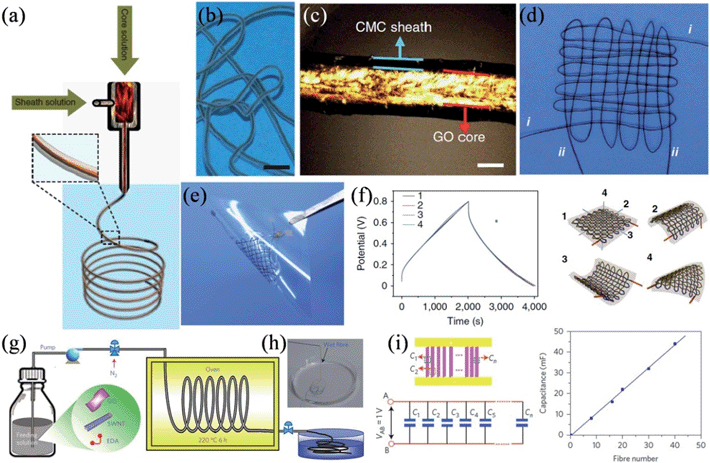 |
| | Fig. 7 (a) Schematic showing the coaxial spinning process. (b) A single intact wet CMC-coated graphene core-sheath fiber. (c) Polarized-light optical microscopy image of the wet fiber, indicating the core-sheath structure and the well-aligned GO sheets in the core part. (d) Cloth woven by two individual coaxial fibers. (e) Supercapacitor device based on the cloth fabricated by two coaxial fibers. (f) Charge–discharge curves of the cloth supercapacitor, where 1 represents the initial cloth supercapacitor without bending, and 2, 3, and 4 show the cloth supercapacitor with bending angles of 180° along three directions. Scale bars are 5 mm (b) and 100 μm (c) (reprinted with permission from ref. 114. Copyright 2014 Nature Publishing Group). (g) Synthesis of the carbon hybrid microfibers. (h) Image of the as-prepared fibers collected in water.(i) Schematic and equivalent circuit for the scalable integration of multiple fibers in micro-supercapacitors in parallel (left) and the relation between the total device capacitance and number of fibers integrated (right) (reprinted with permission from ref. 115. Copyright 2014 Nature Publishing Group). | |
We also reported a simple and facile method for continuously producing graphene/PPy fibers via a direct wet spinning strategy.116 A roll of initially produced GO/PPy fiber with a length of 0.5 m was collected in FeCl3 solution within only 5 s and the subsequent HI treatment yielded graphene/PPy fibers, with diameters ranging from 15 to 80 μm that could be tuned by controlling the diameter of the needle of a single-capillary spinneret and the concentration of GO. The graphene/PPy fibers had a tensile strength of up to 80 MPa and thus led to highly flexible all-solid-state fiber supercapacitors with high electrochemical capacitances of up to 100 mF cm−2 under repetitive deformations. In other attempts, we designed other interesting hybrid structures toward flexible fiber capacitor applications. One-dimensional (1D) macroscopic CNT/graphene hybrids with a large surface area and high flexibility were prepared by directly growing CNTs along graphene fibers with embedded Fe3O4 nanoparticles as catalysts for chemical vapor deposition (CVD) of the nanotubes (Fig. 8a).117 The as-formed textile of the CNT/graphene fibers was still very flexible (Fig. 8b) and the supercapacitor using the CNT/graphene fiber textiles as electrodes could light a commercial LED either in the flat or bended state, though the charge voltage was only 0.6 V (Fig. 8c–e). Meanwhile, a novel hybrid fiber of MnO2-modified graphene on GF (MnO2/graphene/GF) was developed by forming the core-sheath graphene on GF first and then depositing MnO2 nanostructures.118 The SEM images clearly show the morphology change from the pristine GF (Fig. 8f) to the coating of the interpenetrating networks of graphene sheets on the GF (Fig. 8g), and finally to the formation of a densely deposited layer of MnO2 along the whole graphene/GF scaffold (Fig. 8h). Due to the incorporation of the electrochemically active MnO2 component, the symmetrical solid supercapacitors of two intertwined MnO2/graphene/GFs showed an area-specific capacitance of 9.1 mF cm−2 to 9.6 mF cm−2, far exceeding that of all graphene core-sheath fiber supercapacitors (ca. 1.2 mF cm−2 to 1.7 mF cm−2). Additionally, the deposition of MnO2 did not compromise the mechanical property of the fiber supercapacitor, and the performance of the MnO2/graphene/GF fiber capacitor remained unchanged during 1000 repetitive straight-bending cycles, showing the exceptional tolerance of the MnO2/graphene/GF supercapacitor to mechanical deformations and its great potential to be woven into a textile for a range of wearable electronics (Table 1).
 |
| | Fig. 8 (a) Fabrication scheme of CNT/graphene fibers. (b) Image of CNT/graphene fiber textile, and the inset showing the bent state. (c) Schematic illustration of a flexible supercapacitor using the textile of CNT/G fibers as electrodes. Images of the fabricated textile supercapacitor in the (d) flat and (e) bending state, respectively, where the insets show a light-emitting-diode (LED) lighted by the textile supercapacitor in the corresponding state. The scale bars 0.5, 1, and 1 cm for (b), (d), and (e), respectively (reprinted with permission from ref. 117. Copyright 2014 Royal Society of Chemistry). SEM images of (f) GF, (g) graphene/GF, and (h) MnO2/graphene/GF. (i) The stability of the MnO2/graphene/GF fiber supercapacitor undergoing 1000 repetitive straight-bending cycles (reprinted with permission from ref. 118. Copyright 2013 Elsevier B.V.). | |
Table 1 Comparison of the materials, preparation methods, specific performance, and characteristics of the reported supercapacitors
| Samples |
Preparation methods |
Performance |
Characteristics |
Ref. |
| Nafion-functionalized RGO film |
Filtration and hydrazine reduction |
Capacitance: 118.5 F g−1 at 1 A g−1 |
All-solid-state; flexible |
85
|
| Graphene-cellulose paper |
Filtration |
Capacitance: 81 mF cm−2 |
Binder-free; free-standing; flexible |
86
|
| Graphene/polyaniline composite sheets |
Filtration and anodic electropolymerization |
Gravimetric capacitance: 233 F g−1; volumetric capacitance: 135 F cm−3 |
Free-standing; flexible |
91
|
| PANI-RGO/cellulose fibers |
“Dipping and drying” |
Capacitance: 464 F g−1 |
Flexible; wearable; rolling-up |
92
|
| Graphene–PANI composite paper |
HI treatment and in situ electrochemically growth |
Capacitance: 763 F g−1 at a 1 A g−1 |
Low-density; free-standing; flexible |
93
|
| G-PPy/CNT composite |
Flow-assembly method |
Capacitance: 211 F g−1 at 0.2 A g−1 |
Uniformly sandwiched; flexible |
94
|
| MnO2-coated 3D graphene network |
Electrodeposition |
Gravimetric capacitance: 130 F g−1; volumetric capacitance: 1.42 F cm−3 |
Freestanding; flexible; lightweight |
95
|
| PPy–graphene foam |
Hydrothermal process and electrodeposition |
Gravimetric capacitance: 350 F g−1; volumetric capacitance: 14 F cm−3 |
Lightweight; compressible |
96
|
| Laser-scribed graphene |
Laser reduction |
Capacitance: 3.67 mF cm−2 at 1A/gLSG in 1.0 M H3PO4; 4.04 mF cm−2 in 1.0 M H2SO4 |
All solid-state; flexibility; lightweight; microdevice |
105
|
| Interdigitated graphene |
Etching circuit onto a film |
Power density: ∼200 W cm−3 |
High areal density; superthin |
106
|
| RGO-GO-RGO patterns |
Layer writing and patterning |
Capacitance: 0.51 mF cm−2; power density: 1.7 W cm−3 |
In-plane; sandwiched; lightweight |
107
|
| PANI nanorod arrays on patterned RGO films |
Electrochemically grow |
Capacitance: 970 F g−1 at 2.5 A g−1 |
All-solid-state; flexible; miniaturized |
108
|
| G@3D G core-sheath fiber |
Hydrothermal process and electrodeposition |
Capacitance: 1.2–1.7 mF cm−2 |
Flexible; wearable |
109
|
| RGO-GO-RGO fiber |
Laser reduction |
Capacitance: 1.2 mF cm−2 at 80 mA cm−2; 0.45 mF cm−2 at 200 mA cm−2 |
All-in-one; flexible |
110
|
| CMC coated graphene core-sheath fibers |
Wet-spinning assembly and HI treatment |
Capacitance: 28 mF at 10 mA |
Cloth supercapacitor; flexible; wearable |
111
|
| SWNT/N-doped RGO sheet network |
Wet-spinning and hydrothermal treatment |
Volumetric energy densities: ∼300 F cm−3 |
Tensile; flexible |
112
|
| Graphene/PPy fibers |
Wet-spinning and HI treatment |
Capacitance: 100 mF cm−2 at 0.24 mA cm−2 |
All-solid-state; tensile; flexible |
113
|
| CNT/graphene hybrid |
Hydrothermal process chemical vapor deposition |
Capacitance: 0.74 mF cm−2 at 10 mA cm−2 |
Flexible; wearable |
114
|
| MnO2/graphene/GF |
Hydrothermal process and electrodeposition |
Capacitance: 9.1–9.6 mF cm−2 |
All-solid-state; flexible; wearable |
115
|
2.2 Graphene-based flexible and unconventional lithium ion batteries
Lithium ion batteries have emerged as a main power source for portable electronic devices, due to their high energy density, large output voltage, appreciable life, and environmentally benign operation.119,120 To match the rapid development of flexible electronics, the current trend in portable battery research is focused on making lighter, thinner, and smaller flexible batteries to drive various bendable, deformable, and wearable electronic devices. As graphene-based flexible lithium ion batteries have been well summarized lately,52,61,121 we hereby focus on the recent progress in this field.
As graphene paper is mechanically strong and electrically conductive, it is deemed to be an ideal binder-free electrode material in lithium ion batteries. Liu et al. mechanically pressed a graphene aerogel to prepare a unique graphene paper with a folded structure of graphene sheets (Fig. 9a and b).122 The graphene paper was free-standing and very flexible (Fig. 9c), with a high electronic conductivity of 18 S cm−1. Impressively, the folded structure of the graphene sheets with fewer layers provided more lithium insertion active sites with an easy access of the electrolyte, leading to improved coulombic efficiencies of up to 98% and a reversible capacity of 568 mA h g−1 at 100 mA g−1 after 100 charge–discharge cycles (Fig. 9d). In another approach, Cheng and coworkers developed a graphene foam as a 3D conductive network for simultaneously collecting electrons and transporting ions, and based on this platform, they demonstrated thin, lightweight, and a flexible LiFePO4/graphene foam cathode and Li4Ti5O12/graphene foam anode capable of charging and discharging at rates of up to 200 C (Fig. 9e).123 The total thickness of the flexible full battery was less than 800 μm, and it was highly tolerant to the mechanical deformations of repeatedly bending to a radius of <5 mm (Fig. 9f). The flexible battery showed an excellent stability in a cyclic bending test, retaining ∼97% and ∼95% of its original capacity in the first 15 cycles under the flat state and the following 15 cycles under the bent state with a bend radius of 5 mm (Fig. 9g).
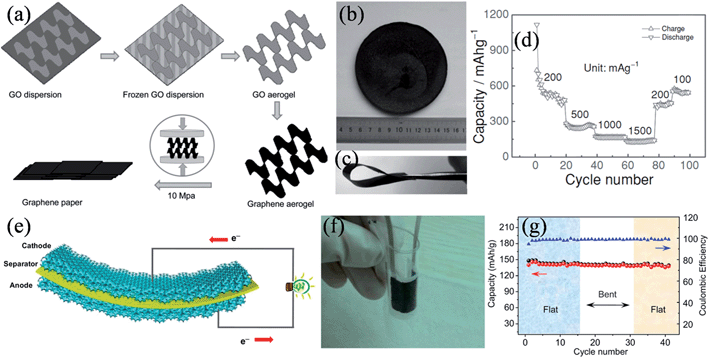 |
| | Fig. 9 (a) Illustration of the formation process of graphene paper. Digital images of (b) a graphene aerogel and (c) graphene paper. (d) Cycle performance of the graphene paper as a lithium ion battery anode at different current densities (reprinted with permission from ref. 122. Copyright 2012 Wiley-VCH). (e) Schematic of a free-standing flexible battery containing a 3D interconnected graphene foam anode and cathode. (f) Photograph of a bent battery encapsulated by PDMS, showing its good flexibility. (g) Cyclic performance of the battery under flat and bent states (reprinted with permission from ref. 123. Copyright 2012 National Academy of Sciences). | |
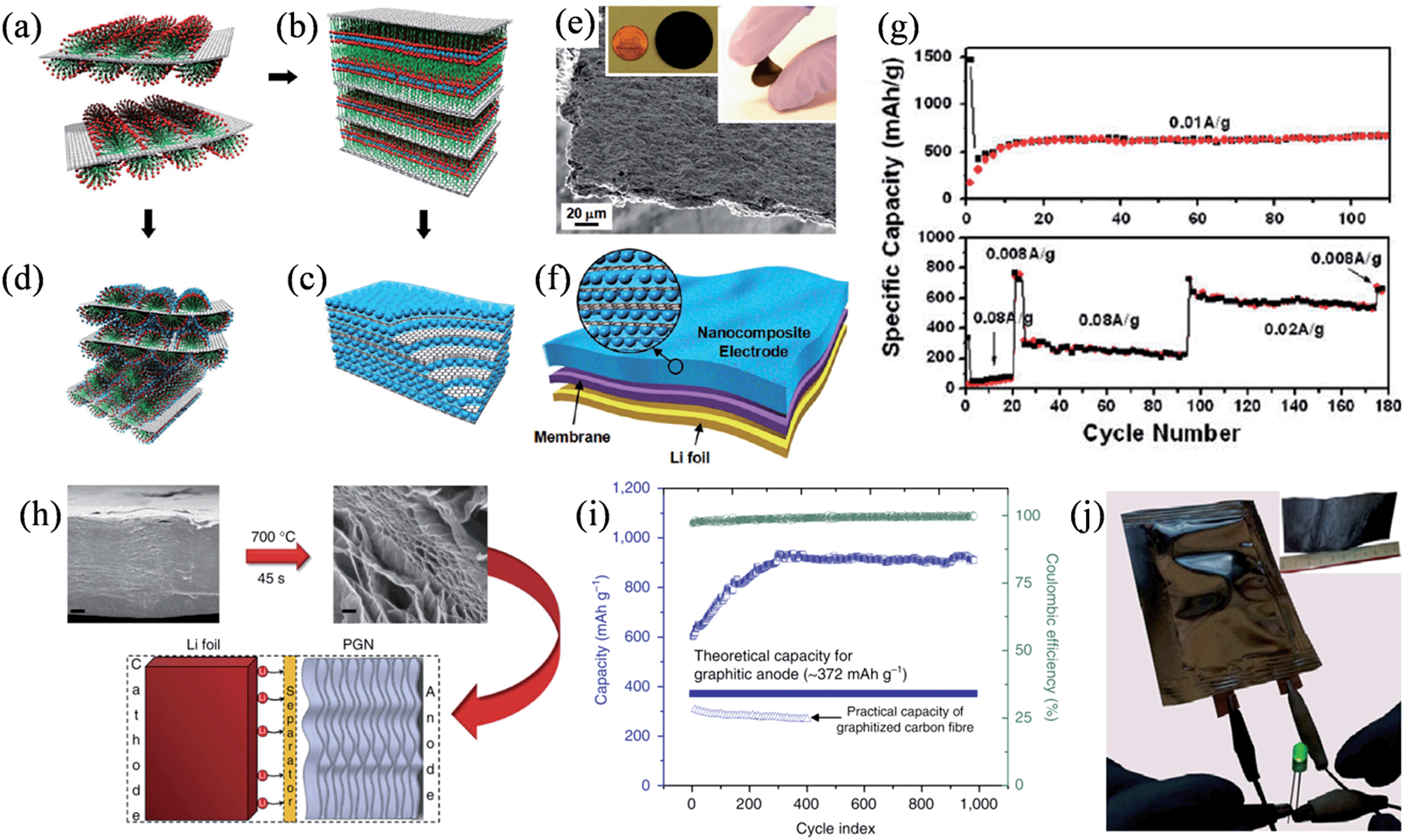
To obtain electrode materials with a high capacity, Wang et al. designed a ternary self-assembly of metal oxides, surfactants, and graphene to synthesize ordered nanocomposites composed of metal oxide and graphene stacks that could be used as an additive-free and binder-free electrode in the battery.124 Anionic surfactants were first absorbed on the surface of graphene stacks (Fig. 10a), and then they functioned as the fundamental building blocks for binding to the metal cations, thereby forming an ordered nanocomposite (Fig. 10b). Finally, alternating layers of graphene stacks and metal oxide nanocrystals could assemble into layered nanostructures after crystallization and the removal of surfactant (Fig. 10c), while hexagonal mesoporous metal oxide–graphene nanocomposites could also be obtained by the direct assembly of metal oxides with the surfactant into a hexagonal mesophase on the surface of the stacked graphene (Fig. 10d). The layered metal oxide–graphene nanocomposites featured as free-standing flexible films of 5 to 20 μm thickness (Fig. 10e). The SnO2–graphene nanocomposite electrodes showed a steady specific capacity of 625 mA h g−1 after 10 cycles at a current density of 0.01 A g−1 and 760 mA h g−1 at a current density of 0.008 A g−1 over 100 cycles (Fig. 10f and g), confirming the good adhesion between SnO2 and the sandwiched graphene layers resistant to the phase transformation and volume change upon lithiation/delithiation. In another attempt to boost the capacity of the battery, Mukherjee et al. found that defects in the graphene lattice could work as seed points to trap lithium metal within the interior of a thermally reduced, free-standing porous graphene network (PGN) (Fig. 10h).125 The PGN effectively prevented dendritic growth and exhibited exceptional specific capacities higher than 850 mA h g−1, also with an excellent reversibility and coulombic efficiencies above 99% in 1000 charge–discharge cycles (Fig. 10i). A full-cell composed of PGN anodes and lithiated PGN cathodes could be easily fabricated from a large-scale GO paper with an area of ∼30 cm2 in a similar process compatible with the conventional Li-ion battery, demonstrating the versatility and scalability of the PGNs in developing low-weight, all-carbon lithium batteries.
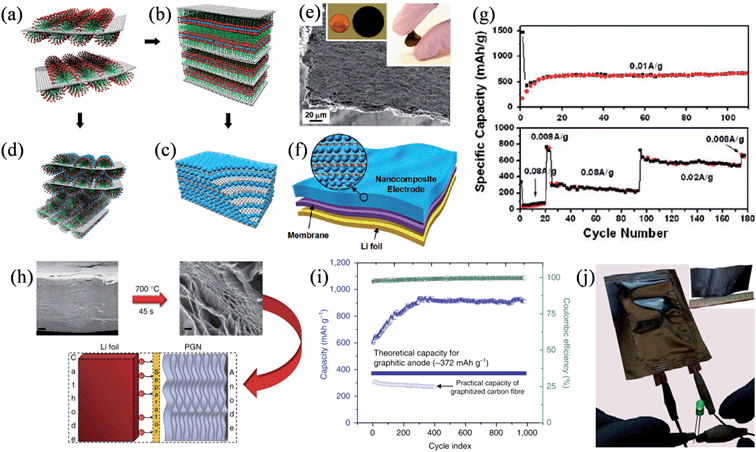 |
| | Fig. 10 (a–d) The ternary self-assembly approach to ordered metal oxide–graphene composites. (a) Graphene stacks with surfactant hemimicelles. (b) The transition into the lamella mesophase toward the formation of metal-oxide–graphene nanocomposites. (c) Metal oxide–graphene layered nanocomposites. (d) Self-assembled metal oxide precursor with nonionic surfactants on graphene stacks. (e) Side-view SEM image of a self-assembled SnO2–graphene nanocomposite, where the inset shows a disk-like 3 cm diameter SnO2–graphene nanocomposite electrode on the left and a folded electrode on the right. (f) A Li-ion battery configuration directly using the metal oxide–graphene nanocomposite. (g) Specific capacity of SnO2 as a function of the charge–discharge cycles in the SnO2–graphene nanocomposite electrode at a current density of 0.01 A g−1 (top) and at different charge–discharge current densities (bottom) (reprinted with permission from ref. 124. Copyright 2010 American Chemical Society). (h) Reduction of GO paper to produce a free-standing PGN, tested as an anode material in lithium ion batteries. Scale bars are 2 μm (left) and 200 nm (right). (i) Capacity and coulombic efficiency versus cycle index of the PGN anode. (j) Macroscopic pouch cell powering an LED device, where the inset is an image of a large-scale GO paper (reprinted with permission from ref. 125. Copyright 2014 Nature Publishing Group). | |
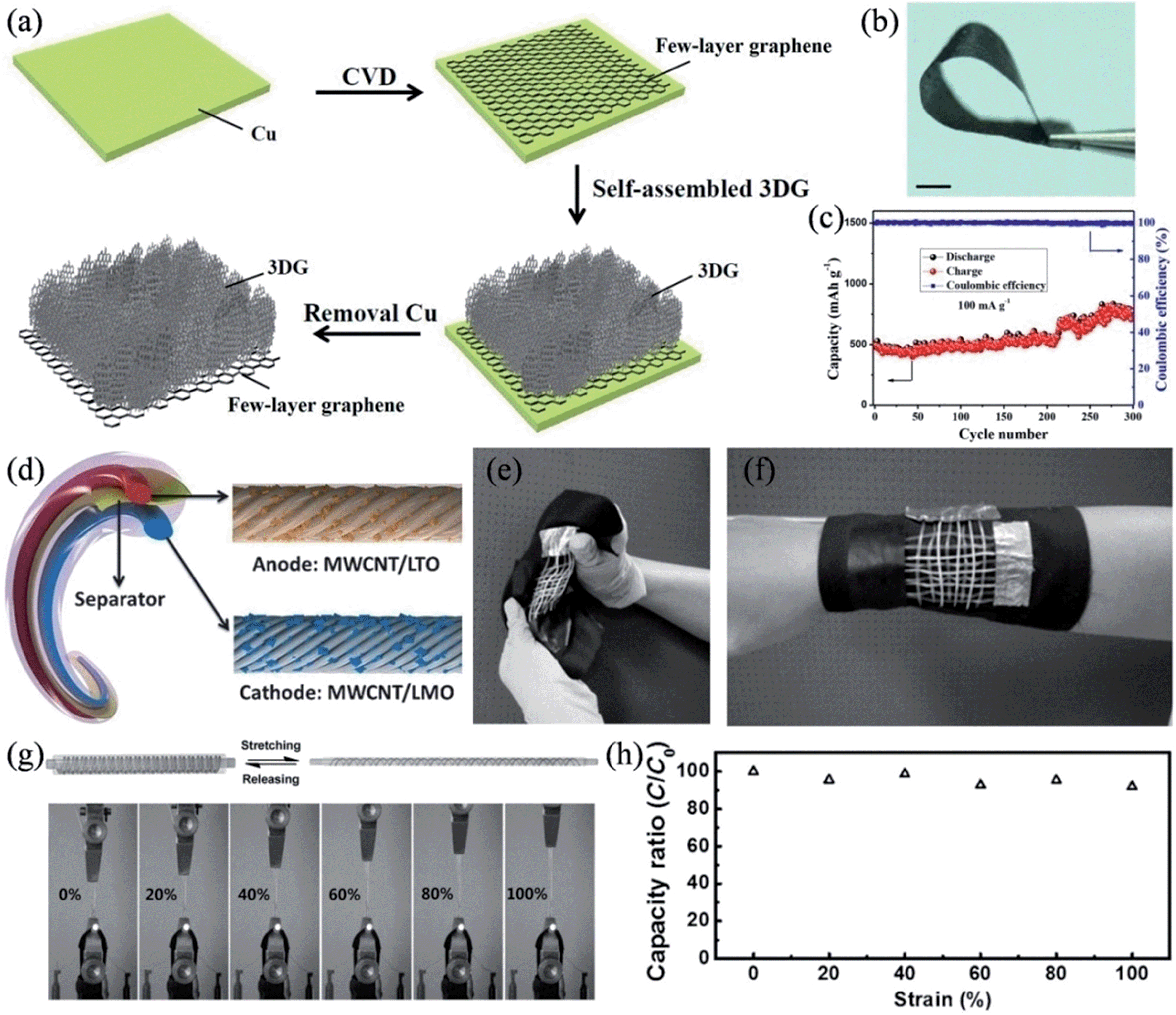
We also prepared a hierarchical graphene-on-graphene hybrid structure as a flexible, binder-free, all-graphene anode of the lithium ion battery.126 3D graphene (3DG) interconnected frameworks were homogenously connected with the few-layer graphene film, where the pore-rich 3D graphene networks promoted the Li+ diffusion and the few-layer graphene worked as a non-metallic current collector to provide a potent scaffold to support the 3DG and extract the charge from/to the 3D graphene networks during the charge and discharge processes (Fig. 11a and b). Compared to the metallic current collector, the few-layer graphene possesses advantageous characteristics of light weight, chemical stability, excellent electrical conductivity and tolerance to corrosion and oxidation. As a result, the hybrid structure showed an excellent specific capacity of ∼770 mA h g−1 with an energy density of ∼388 W h kg−1, superseding those of commercial graphitic anodes.
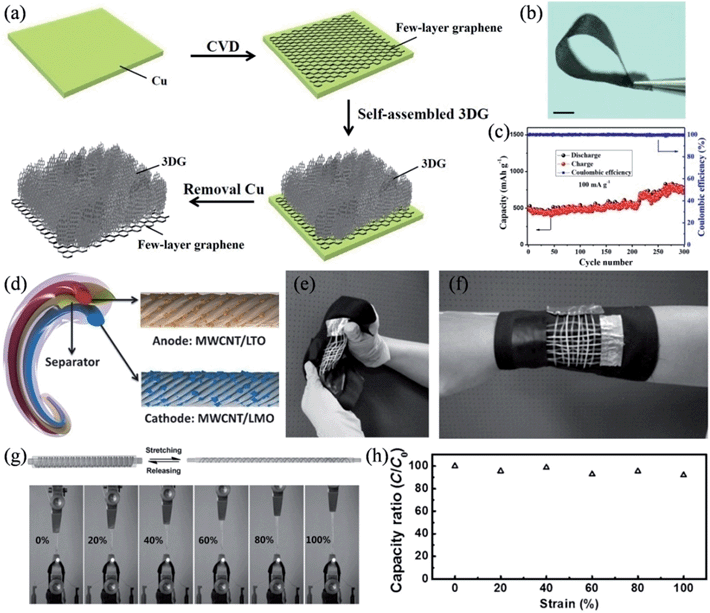 |
| | Fig. 11 (a) Schematic procedure and mechanism for the formation of a 3DG/few-layer graphene hybrid structure. (b) Image of the 3DG/few-layer graphene with good flexibility. Scale bar is 1 cm. (c) Cycling test of the 3DG/few-layer graphene electrode at a current density of 50 mA g−1. (d) Structure of the flexible wire-shaped lithium-ion battery with the aligned MWNT/LTO anode and MWNT/LMO cathode. (e and f) Wire-shaped batteries woven into flexible textiles. (g) Images of an elastic battery used to power a light-emitting diode at increasing strains, with stretching and releasing process illustrated. (h) Dependence of the specific capacity on the strain (reprinted with permission from ref. 127. Copyright 2014 Wiley-VCH). | |
Very recently, Peng and coworkers demonstrated safe wire-shaped lithium-ion batteries with low weight, high flexibility, and high energy density.127 Li4Ti5O12 (LTO) and LiMn2O4 (LMO) nanoparticles were well-incorporated into two aligned MCNT yarns that functioned as the anode and cathode, respectively (Fig. 11d). A thin layer of GO coating was applied on the surface of the anode to stabilize the structure with an improved capacity retention. The aligned nanostructure and high electrical conductivity of the MCNT yarns made the current collector and binders in conventional lithium ion batteries redundant, and therefore, the wire-shaped battery could be conveniently woven into various flexible structures, such as electronic textiles (Fig. 11e and f). The wire-shaped battery could be placed around an elastic substrate into a spring structure to give a very elastic device, where the elastic battery was able to uninterruptedly illuminate an LED whilst it was continuously stretched by 100% (Fig. 11g). The stretchable lithium ion battery only suffered a less-than-10% decrease in its specific capacity when its length was doubled, and it retained over 80% of its capacity performance after 200 cycles of stretching by 100% (Fig. 11h), due to the unique spring-like structure and the excellent elasticity of the substrate and gel electrolyte, thereby, qualifying these materials as highly desirable for diverse applications in wearable and portable electronics (Table 2).
Table 2 Comparison of the materials, preparation methods, specific performance, and characteristics of the reported lithium ion batteries
| Samples |
Preparation methods |
Performance |
Characteristics |
Ref. |
| Graphene paper |
Pressed a graphene aerogel |
Capacitance: 568 mA h g−1 at 100 mA g−1 |
Binder-free free-standing; flexible |
122
|
| LiFePO4/graphene and Li4Ti5O12/graphene |
Hydrothermal deposition |
Capacitance: 170 mA h g−1 and 164 mA h g−1 at 744 mA g−1 |
Thin; lightweight; flexible |
123
|
| SnO2–graphene nanocomposite |
Self-assembly |
Capacitance: 625 mA h g−1 at 10 mA g−1; 760 mA h g−1 at 8 mA g−1 |
Additive-free; binder-free; flexible |
124
|
| Porous graphene network |
Filtration and rapid heating |
Capacitance: 850 mA h g−1 at 372 mA g−1; energy density: 547 W h kg−1 |
Free-standing; binder-free |
125
|
| Graphene-on-graphene hybrid film |
Chemical vapor deposition and spontaneous reduction of GO |
Capacitance: 770 mA h g−1 at 372 mA g−1; energy density: 388 W h kg−1 |
All-graphene; flexible; binder-free |
126
|
| GO-coated LTO and LMO/MWNT yarns |
Hydrothermal process and rolled method |
Energy densities: 27 W h kg−1; power densities: 880 W kg−1 |
Stretchable; flexible; wearable; low weight |
127
|
3. Tailored graphene systems for power generation
3.1 Graphene-based photovoltaic devices
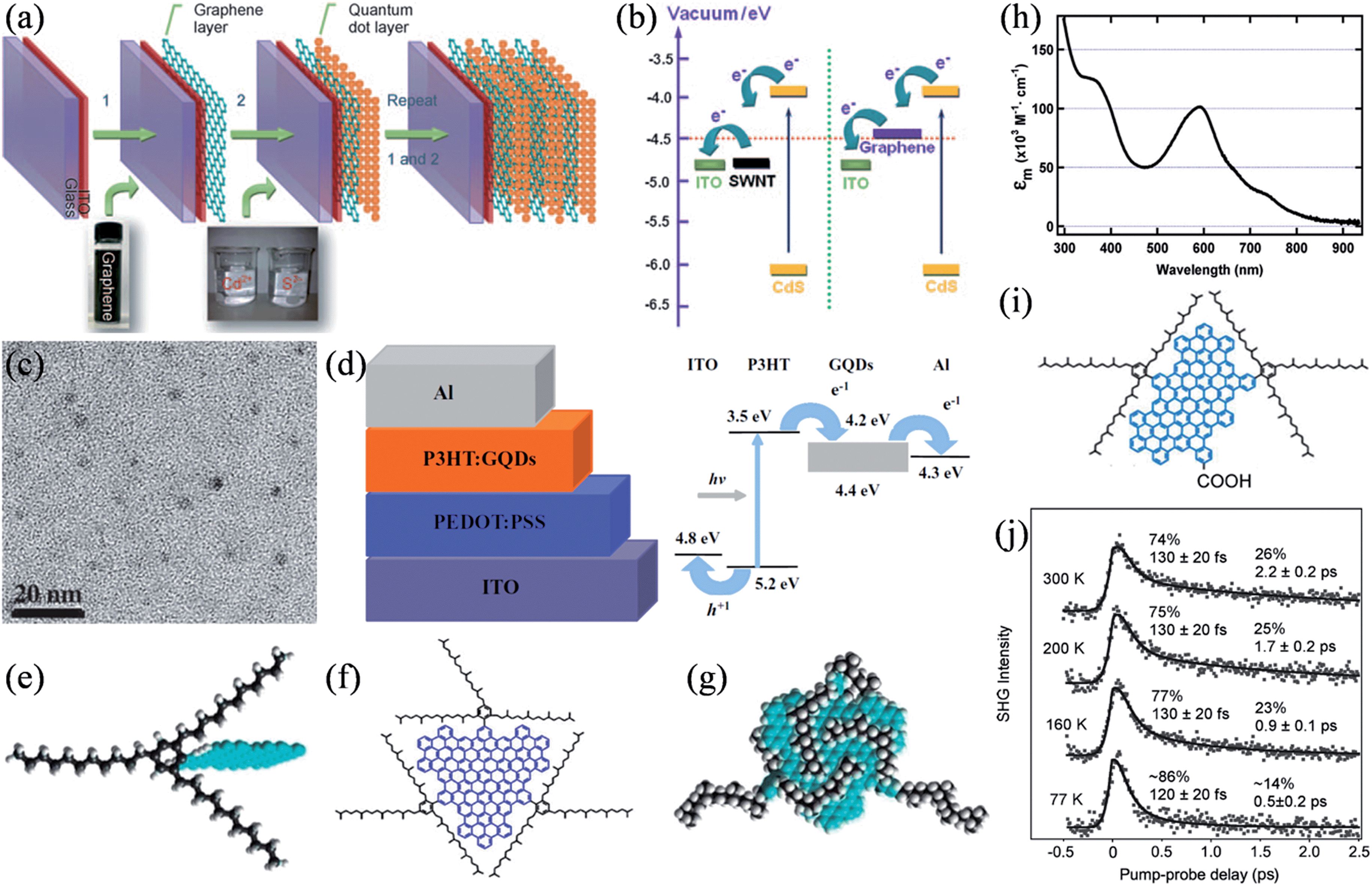
Due to the high transparency and in-plane conductivity of single-layer graphene and the tunable catalytic activity of doped graphene frameworks, tailored graphene structures have been intensively studied as transparent conducting films for charge collection29,128,129 and as catalytic counter electrodes (CEs)130–132 in a range of photovoltaic devices. Besides these applications, Guo et al. demonstrated that graphene could effectively work as the electron acceptor in CdS quantum dot (QD) solar cells.133 The nanoscale layer-by-layer assembly of graphene and CdS (Fig. 12a) seemed to alleviate the inherent problems of charge collection and transport in QD solar cells. Energy-level diagrams indicated that, compared to that of SWNTs, the work function of graphene is better positioned for a more balanced charge transfer from the QDs and the following electron collection by the collection electrode (Fig. 12b). Meanwhile, Liu et al. reported the application of solution-processable isocyanate-functionalized graphene as the electron acceptor in the bulk heterojunction (BHJ) solar cell.134 In a different attempt, we studied the viability of using electrochemically synthesized graphene quantum dots (GQDs) as a better electron acceptor (Fig. 12c and d).135 The large surface areas of GQDs can conveniently afford ample interfaces for exciton dissociation, while the high electron mobility of GQDs could be expected to increase the conductivity of the active layer and greatly facilitate the charge transport through the active layer. Without any device optimization in this primary study, a power conversion efficiency of 1.28% was achieved. Later, aniline-functionalized GQDs (ANI-GQDs) were further employed as the electron acceptor in poly(3-hexylthiophene) (P3HT) based on an optimized cell structure, where P3HT/ANI-GQD films showed a heterogeneous nanoscale phase separation commensurate with the diffusion length of excitons (∼10 nm), which leads to an enhanced performance.136
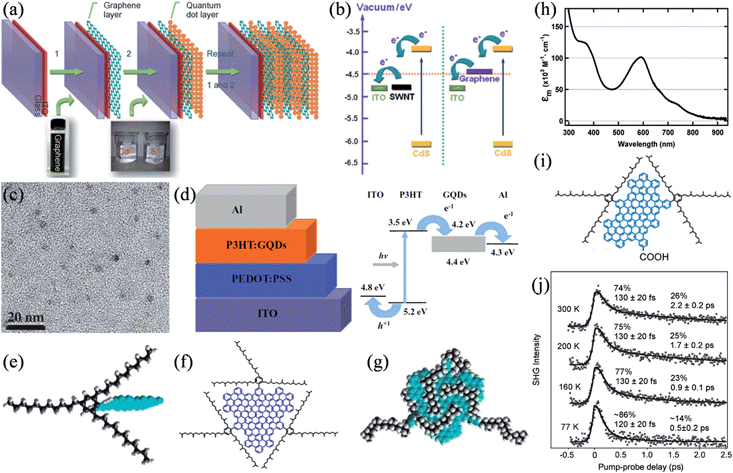 |
| | Fig. 12 (a) Fabrication of the layered graphene/QDs on the indium tin oxide (ITO) glass. (b) Energy-level diagram of the bilayer system (reprinted with permission from ref. 133. Copyright 2010 Wiley-VCH). (c) TEM image of the as-prepared GQDs. (d) Schematic (left) and energy levels (right) of the BHJ solar cell based on GQDs (reprinted with permission from ref. 135. Copyright 2011 Wiley-VCH). (e) The shielding of graphenes (blue) from one another in three dimensions by covalently attaching 1,3,5-trialkyl phenyl moieties. (f) Molecular structure of GQD containing the graphene moiety with 168 conjugated carbon atoms (blue) and the three solubilizing groups (black). (g) A theoretically energy-minimized configuration of the GQD, showing the three-dimensional enclosure of the graphene core by the alkyl chains (black). (h) Absorption spectrum of the GQD in dichloromethane (reprinted with permission from ref. 138. Copyright 2010 American Chemical Society). (i) Structure of the GQD molecule of a 132-C graphene core and functionalized with two 2,4,6-trialkylsubstituted phenyl groups and a carboxylic acid group. (j) Time-resolved second harmonic generation (TR-SHG) spectra (dots) of GQD/TiO2 at a pump photon energy of 2.41 eV and at different sample temperatures, with biexponential fits for the solid curves (reprinted with permission from ref. 141. Copyright 2013 American Chemical Society). | |
Additionally, graphene can also function actively as the exciton generator in sensitized solar cells. Li and coworkers synthesized a series of well-defined GQDs with substituted phenyl groups functionalized at the edge of the graphene core.137 The introduction of multiple 1,3,5-trialkyl-substituted phenyl moieties effectively shields the GQDs from one another by wrapping them three-dimensionally, thus separating the GQDs in all three dimensions and reducing their tendency to form insoluble aggregates (Fig. 12e–g).138 By this strategy, GQDs with a 168-carbon core were highly soluble in various common organic solvents, with solubilities up to 30 mg mL−1 at room temperature and an absorbance maximum at 591 nm with a molar extinction coefficient of 1.0 × 105 M−1 cm−1, about an order of magnitude larger than those of the widely used sensitizers based on metal complexes (Fig. 12h). Unfortunately, the lack of any anchoring group led to a low affinity of the GQDs to the TiO2 film in a pure physical adsorption, and consequently resulted in a slightly colored film despite its long time sensitization and a limited photocurrent in μA cm−2. Nevertheless, it was found that these defect-free, colloidal GQDs featured an exceptionally slow hot-carrier cooling process, where the lifetimes of the hot-carriers were about 2 orders of magnitude longer than those in bulk graphene materials.139 As the hot-carriers survived for up to hundreds of picoseconds, intersystem crossing could efficiently compete with internal conversion, thereby, potentially increasing the access to triplet states with notably longer lifetimes and triplet excitons with larger diffusion lengths.140 Recently, the dynamics of the hot-carrier injection and subsequent charge recombination was studied by monitoring the photoexcitation of a similar GQD molecule of a 132-C graphene core anchored to the TiO2 (110) surface via a carboxylic acid anchoring group (Fig. 12i).141 The ultrafast electron injection from GQDs to the TiO2 conduction band took place in less than 15 fs, followed by charge recombination in a fast channel (80–130 fs) associated with the instantaneous recombination of the bound electron–hole pair right at the interface and a slow one (0.5–2 ps), where the hot electrons underwent a process of injection, cooling down, drifting back to the interface, and ultimately recombining. Besides the excitation photon energy, the slow channel was also strongly dependent on the sample temperature, whereby it slowed down with increasing temperatures, presumably due to the decreased mobility of the electron polarons (Fig. 12j). Based on these results, it was proposed that graphene absorbers are needed to be at least hundreds of nanometers to micrometers in size to ensure multiple excitations in each molecule under normal solar radiation, which necessitates more effort to be devoted to judiciously design the corresponding graphene structure and device configuration for harvesting hot-carriers in a real device.
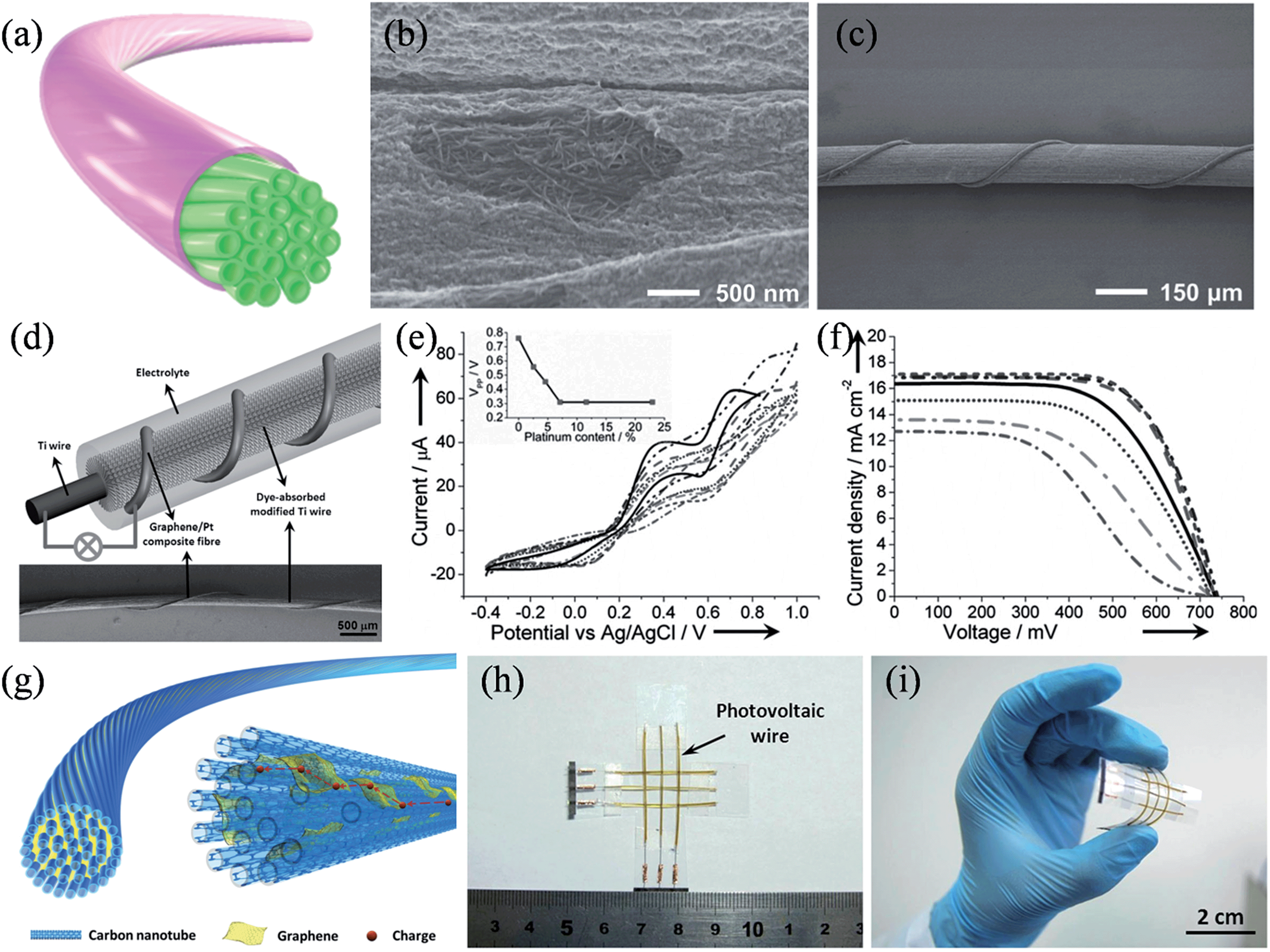
For flexible electronics oriented applications, Peng and coworkers developed a range of flexible graphene-based fibers as efficient CEs in wire-shaped dye-sensitized solar cells. A CNT fiber was first treated with an oxidizing solution, and then reduced to form a core-sheath structure with the surface covered by graphene nanoribbons and other carbonaceous residues (Fig. 13a).142 As shown in the SEM image in Fig. 13b, the dense CNT core was clearly visible and the carbonaceous sheath was postulated to be 20 nm in thickness. Such a core-sheath structure integrated the high tensile strength and electrical conductivity of aligned CNTs with the high electrocatalytic activity of the edge-rich sheath materials, and as a result, wire-shaped dye-sensitized solar cells with energy conversion efficiencies of up to 6.83% could be achieved (Fig. 13c). In another work, they spun graphene into flexible fibers and electrodeposited platinum nanoparticles on to them to serve as the CE (Fig. 13d).143 Cyclic voltammetry results showed that with increased Pt loadings up to 7.1%, graphene/Pt composite fibers produced higher current densities and lower peak-to-peak voltage separations (VPP), which are indicative of the improved catalytic activities for CEs (Fig. 13e). The J–V characteristics revealed that the open-circuit voltage (Voc) remained nearly the same at 0.73 V, while the short-circuit current density (Jsc) and the fill factor (FF) improved from 12.67 mA cm−2 to 17.11 mA cm−2 and from 0.42 to 0.67, respectively as the Pt content increased from 0% to 7.1%, eventually reaching a maximized photo-to-electricity conversion efficiency of 8.41% at a Pt loading of 7.1% (Fig. 13f). Recently, the same group further synthesized a new range of graphene/CNT composite fibers with graphene sheets incorporated among neighboring CNTs to work as effective bridges to facilitate the charge transport, because of the strong π–π interactions between CNT and graphene sheet (Fig. 13g).144 With the deposition of Pt nanoparticles, a wired dye-sensitized solar cell with the graphene/CNT/Pt composite fiber CE showed conversion efficiencies of 8.36% ± 0.10%, which compares favorably to those of its counterpart with a bare Pt wire (6.66% ± 0.15%). Furthermore, the wire-shaped solar cells could be facilely integrated into a photovoltaic textile, with stable and high energy conversion efficiencies under bending (Fig. 13h and i). Additionally, symmetrical wire-shaped supercapacitors of two intertwined graphene oxide/CNT composite fibers showed specific capacitances up to ca. 31.50 F g−1 (4.97 mF cm−2 or 27.1 μF cm−1) with no notable decay in capacitance after 5000 charge–discharge cycles, validating the concept of using these miniaturized wire-shaped devices in flexible and portable electronic facilities (Table 3).
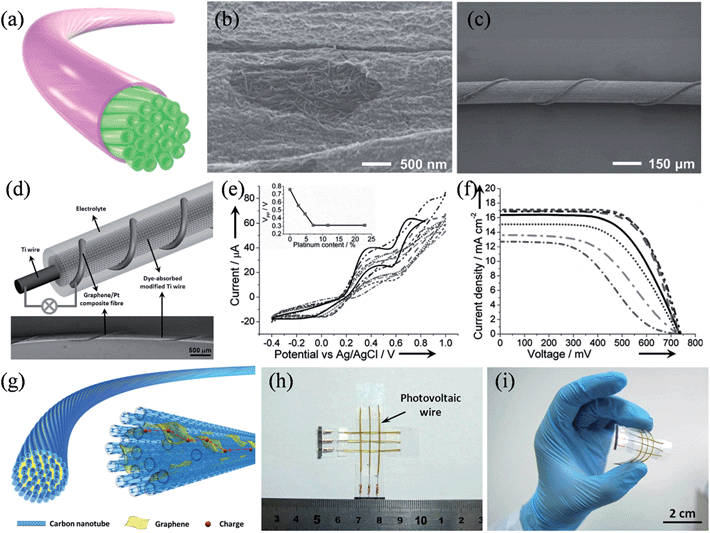 |
| | Fig. 13 (a) Schematic illustration of the core-sheath nanostructured fiber. (b) SEM image of a core-sheath fiber with the sheath part peeled off to unveil the CNTs. (c) SEM image of a wire-shaped dye-sensitized solar cell (reprinted with permission from ref. 142. Copyright 2014 Wiley-VCH). (d) Illustration (top) and SEM image (bottom) of a wired solar cell prepared by using a graphene/Pt composite fiber as the CE and a Ti wire impregnated with TiO2 nanotubes as the working electrode. (e) Electrocatalytic characterization of graphene/Pt composite fibers and (f) J–V characteristics of the wired cell with increasing Pt content. The inset of (e) shows the peak-to-peak voltage separation (VPP) as a function of Pt content. (reprinted with permission from ref. 143. Copyright 2013 Wiley-VCH). (g) Schematic illustration of the structure of the graphene/CNT composite fiber. Photographs of the photovoltaic textile woven from six wire-shaped solar cells (h) before and (i) after bending, respectively (reprinted with permission from ref. 144. Copyright 2014 Wiley-VCH). | |
Table 3 Comparison of the materials, preparation methods, specific performance, and characteristics of the reported photovoltaic devices
| Samples |
Preparation methods |
Performance |
Characteristics |
Ref. |
| Graphene and CdS composite |
Layer-by-layer assembly |
Quantum efficiency at 400 nm: 16%; photoresponse: 1.08 mA cm−2 under light illumination of 100 mW cm−2 |
Graphene as electron acceptor; balanced charge transfer |
133
|
| Graphene and P3HT composite |
Spin-coating |
Power conversion efficiency: 0.88%; open-circuit voltage: 0.77 V; short-circuit current density: 3.72 mA cm−2 |
Electron acceptor; balanced charge transfer |
134
|
| Graphene quantum dots |
Electrochemical corrosion approach |
Power conversion efficiency: 1.28%; open-circuit voltage: 0.67 V; short-circuit current density: 6.33 mA cm−2 |
Electron acceptor; high electron mobility |
135
|
| Aniline-functionalized GQDs and P3HT |
Spin-casting |
Power conversion efficiency: 1.14%; open-circuit voltage: 0.67 V; short-circuit current density: 6.33 mA cm−2; fill factor: 0.53 |
Electron acceptor; heterogeneous nanoscale phase |
136
|
| Phenyl-functionalized GQDs |
Suzuki coupling reactions |
As a new type of light harvesting media for photovoltaics |
Exciton generator; large extinction coefficients |
137
|
| 1,3,5-Trialkyl-substituted phenyl moieties functionalized GODs |
Solution synthesis method |
An absorbance maximum at 591 nm with a molar extinction coefficient of 1.0 × 105 M−1 cm−1 |
Exciton generator; large extinction coefficients |
138
|
| Colloidal graphene quantum dots |
Solution synthesis method |
Faster component of 18 ± 3 ps and slower one ranging from approximately 100 ps to nanoseconds |
Exciton generator; increased efficiency of hot electron extraction or multiexciton generation |
139
|
| Monodispersed graphene quantum dots |
Solution synthesis method |
Relaxing into triplet states and emitting both phosphorescence and fluorescence |
Exciton generator; long lifetimes and triplet excitons |
140
|
| GQD and TiO2 composite |
Chemically anchored method |
Electron injection from GQDs to the TiO2 conduction band: less than 15 fs; charge recombination: 80–130 fs; instantaneous recombination of the bound electron–hole pair: 0.5–2 ps |
Hot carrier chromophores; ultrafast electron injection |
141
|
| CNT@ graphene nanoribbons |
Oxidation-reduction method |
Energy conversion efficiencies: 6.83% |
Counter electrode; tensile; flexible |
142
|
| Graphene/Pt composite fibers |
Spinning and electrodeposition |
Open-circuit voltage: ca. 0.73 V; short-circuit current density: from 12.67 to 17.11 mA cm−2; fill factor: from 0.42 to 0.67 |
Counter electrode; flexible; wearable; lightweight |
143
|
| Graphene/CNT/Pt composite fiber |
Biscrolling method and electrodeposition |
Conversion efficiencies: 8.36% ± 0.10%; capacitances of two intertwined fibers: ca. 31.50 F g−1 (4.97 mF cm−2 or 27.1 μF cm−1) |
Counter electrode; flexible; wearable; miniature devices |
144
|
3.2 Graphene-based nanogenerators harvesting mechanical energy
When graphene is seamlessly coupled with a layer of piezoelectric material, the device is able to harvest acoustic or impact energy through the deformation-induced piezoelectric effect. Earlier studies on graphene-based piezoelectric nanogenerators involved sandwiching a layer of ZnO nanostructures145,146 or Pb(ZrxTi1−x)O3 (PZT) ribbons147 between the upper and lower graphene layers, as well summarized in a recent review,148 while recent research has focused on developing flexible, lightweight and/or transparent nanogenerators. For this purpose, Lee et al. developed a stretchable transparent piezoelectric nanogenerator using a polymeric piezoelectric material of poly(vinylidene fluoride trifluoroethylene) [P(VDF-TrFE)] sandwiched between mobility-modified graphene electrodes grown by chemical vapor deposition (CVD) (Fig. 14a).149 With excellent mechanical properties, the PDMS-supported, completely flexible device exhibited high sensitivity to any external mechanical energy, such as a small bending or even exposure to sound. The measured peak to peak output voltage varied from 50 mV to 600 mV when the exposure of the device to sound changed from 82 decibels (dB) to 110 dB at 100 Hz, a response significantly higher than that of a similar nanogenerator constructed on polyethylene naphthalate (PEN) film (10 mV to 22 mV). Alternatively, upon its exposure to an air flow, the device could generate piezoelectric voltages of up to 3.9 V with the air flow speed varying from 0.5 ms−1 to 3 ms−1, and maintained a continuous output voltage, owing to the vibration of the PDMS, which served as a stretchable mechanical spring. In another work, Bae et al. demonstrated a graphene/P(VDF-TrFE)/graphene acoustic actuator and its application as a nanogenerator during the bending/unbending process.150 When the device was subjected to a bending test with an applied strain of ∼0.3% at a rate of 60 mm s−1, it showed an average output voltage of ∼2.5 V (maximum value ∼3 V), a current density of over ∼0.2 μA cm−2 (maximum value ∼0.37 μA cm−2), and an energy conversion efficiency of 8%, with a reasonably stable performance for >500 cycles.
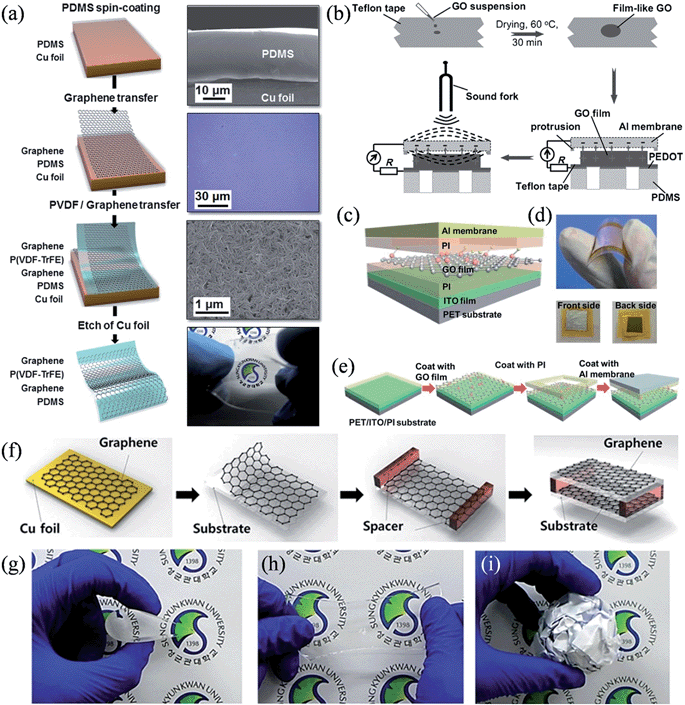 |
| | Fig. 14 (a) Fabrication of a graphene-based stretchable transparent piezoelectric nanogenerator, where the right panel shows the corresponding SEM or optical images at each step (reprinted with permission from ref. 149. Copyright 2013 Royal Society of Chemistry). (b) Schematic of the fabrication of a GO-based acoustic energy harvesting device (reprinted with permission from ref. 151. Copyright 2012 Wiley-VCH). (c) Schematic view of the flexible GO-based nanogenerator. (d) Images showing the device's flexibility, front side, and back side. (e) Schematic representation of the device fabrication (reprinted with permission from ref. 153. Copyright 2013 Royal Society of Chemistry). (f) Schematic diagrams of graphene triboelectric device fabrication. (g–i) The flexibility, stretchability, and adjustability of graphene with the crumpled substrate are shown (reprinted with permission from ref. 155. Copyright 2014 Wiley-VCH). | |
Interestingly, without the incorporation of any piezoelectric materials, graphene-based electrostatic nanogenerators were able to work on the asymmetrical variation in the charge amount of the adjacent top and bottom electrodes during the mechanical deformation if a GO layer was sandwiched in a Al/GO/PEDOT:PSS configuration151 or embedded in the poly(methyl methacrylate) matrix.152 The Al/GO/PEDOT:PSS device fabricated by Que et al. was highly sensitive and even responded to a vibrating sound fork with a power output of 0.424 nW and a conversion efficiency of acoustic to electrical energy of 12.1% (Fig. 14b). A later work by Tian et al. adopted GO films with a release structure, and reported a flexible GO nanogenerator that could generate an output power as high as 60 nW with a peak voltage of 2 V upon a 15 N force applied at 1 Hz frequency (Fig. 14c–e),153 providing another viable solution to effectively harvest repetitive acoustic and impact energy.
Very recently, and following the successful demonstration of the flexible triboelectric nanogenerator,154 stretchable and transparent graphene nanogenerators working on triboelectricity have been realized by separating two wrinkled and rippled graphene electrodes with a plastic spacer (Fig. 14f).155 Owing to its excellent mechanical property, graphene was found to be compatible with an arbitrarily shaped substrate with high flexibility and stretchability (Fig. 14g–i). The output performance of the device diminished with the growing number of randomly stacked graphene layers but improved when regularly stacked few-layer graphene was employed, an effect mainly related to the difference in the work function and in the accessible amount of friction. A device constituting single-layer graphene electrodes showed an output voltage of 5 V and a current density of 0.5 μA cm−2 under a vertical compressive force of 9.88 N, qualifying it as an attractive candidate to drive low-power portable devices and self-powered flexible electronic systems (Table 4).
Table 4 Comparison of the materials, preparation methods, specific performance, and characteristics of the reported nanogenerators
| Samples |
Preparation methods |
Performance |
Characteristics |
Ref. |
| P(VDF-TrFE) sandwiched between mobility-modified graphene |
Chemical vapor deposition |
Output voltage varied from 50 mV to 600 mV when the sound changed from 82 dB to 110 dB at 100 Hz |
Flexible; high sensitivity; stretchable |
149
|
| Graphene/P(VDF-TrFE)/graphene |
Doping layer method |
Under a bending test with an applied strain of ∼0.3% at a rate of 60 mm s−1, an output voltage of ∼2.5 V, a current density of ∼0.2 μA cm−2, and an energy conversion efficiency of 8% |
Acoustic actuator; bendable; flexible |
150
|
| Sandwiched Al/GO/PEDOT:PSS |
Assembly |
Vibrating sound fork with a power output of 0.424 nW and a conversion efficiency of 12.1% |
Highly sensitive; flexible |
151
|
| Poly(methyl methacrylate)/GO |
Soaking |
Maximum output electrical power from the device was 6 × 10−9 W |
Flexible |
152
|
| GO films |
Filtration and assembly |
Output power of as high as 60 nW with a peak voltage of 2 V upon a 15 N force at 1 Hz frequency |
Flexible |
153
|
| Wrinkled and rippled graphene electrodes |
Chemical vapor deposition and transferred method |
Output voltage of 5 V and a current density of 0.5 μA cm−2 under a vertical compressive force of 9.88 N |
Stretchable; transparent; flexible |
155
|
3.3 Moisture-triggered motor, fluid-induced generator, and beyond
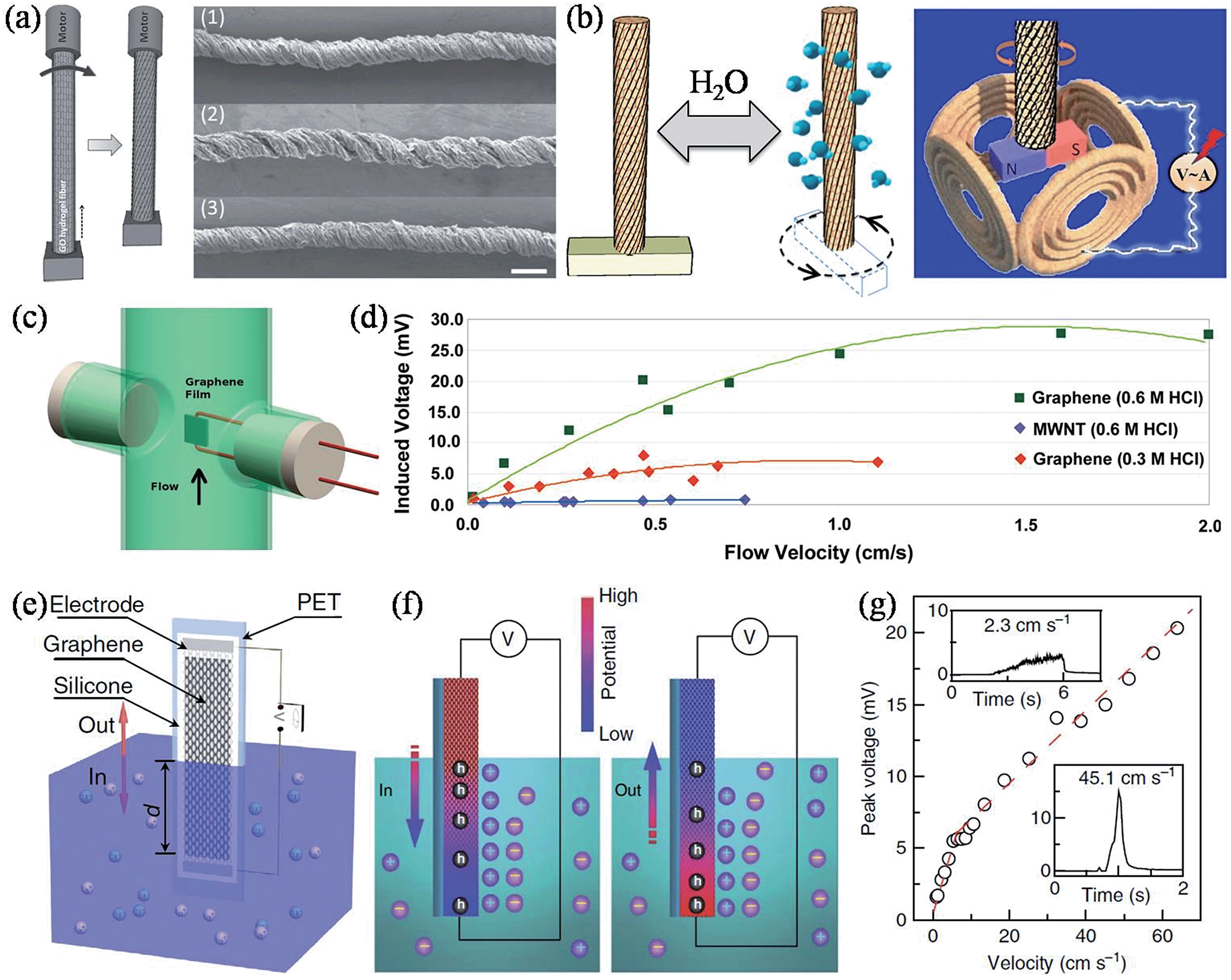
Apart from the above-mentioned power generators, there are other graphene-based devices that could harvest other types of energy and convert them to electricity. Continuing our effort in developing unconventional applications and exploring the full potential of GFs, we reconstructed the intrinsic configuration of graphene within the fiber body and achieved a new type of moisture-driven rotational motor by rationally designing the rotary processing of the freshly spun GO fiber hydrogel (Fig. 15a).156 Due to the presence of oxygen-rich functional groups in GO, a relatively fast and reversible expansion/contraction of GO layers took place through the adsorption/desorption of water molecules, and accordingly, caused a large deformation of 5% upon increasing/decreasing the relative humidity (RH) of the environment between 20% and 85%. As a result, the twisted GO fiber (TGF) served as a superb reversible rotary motor, with a rotary speed of up to 5190 revolutions per minute, presenting us with a prototype of a moisture-triggered electric generator when a magnet bar was attached to the end of the TFG and put at the center of several copper coils (Fig. 15b). When the whole device was exposed to an environment with varied humidity, the TGF drove the magnet in reversible rotations, and according to Faraday's law, the periodic change of the magnetic flux in the copper coils induced an electromotive force (EMF) of up to 1 mV, without any systematic device optimization in this preliminary study.
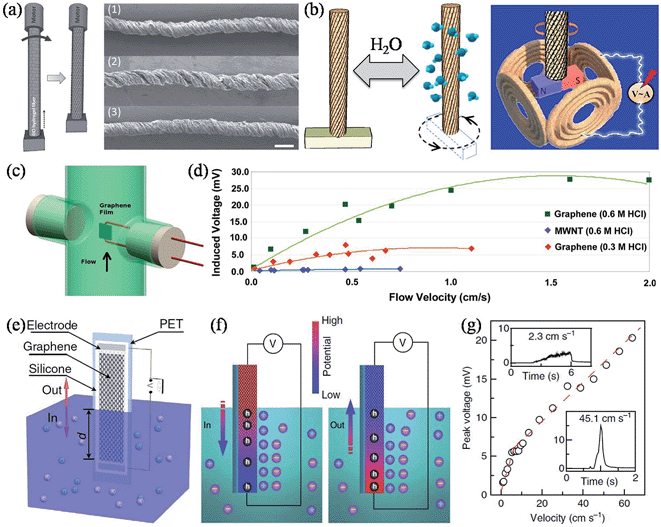 |
| | Fig. 15 (a) Scheme of the TGF fabrication, with the arrow indicating the direction of rotation. The right panel shows SEM images of the initial TGF at RH = 20% (1), on exposure to a high RH of 85% (2), and returning to a RH of 20% (3). The scale bars are 100 μm. (b) Schematic rotation of a TGF with a paddle driven by the increased humidity. The paddle can be reversed to the initial state when the moisture decreases. The right panel shows an illustration of the alternating current generator based on the humidity-responsive TGFs (reprinted with permission from ref. 156. Copyright 2014 Wiley-VCH). (c) Scheme of the fluid flowing parallel to the graphene film. (d) The induced voltage as a function of the fluid flow velocity for the graphene film and for a MWNT film (reprinted with permission from ref. 157. Copyright 2011 American Chemical Society). (e) Schematic illustration of the wave generating potential in graphene moving across the surface of seawater. (f) Schematic of the electric double layer and its boundaries by ion adsorption (left) and desorption (right) on the surface of graphene during the insertion and pulling out processes, respectively. The induced potential difference and hole concentration gradient in the graphene sheet are also illustrated. (g) Peak voltage as a function of the insertion velocity of graphene into 0.6 M NaCl solution. The insets zoom in on the signals at velocities of 2.25 cm s−1 and 45.1 cm s−1 (reprinted with permission from ref. 161. Copyright 2014 Nature Publishing Group). | |
Alternatively, an inducted voltage could also be generated by fluid flowing over the graphene sheets. Dhiman et al. observed induced voltages for few-layer graphene of a few orders of magnitude higher than for MWNTs with hydrochloric acid (HCl) flowing over the surface (Fig. 15c and d).157 The power generation was believed to originate from the coupled movement of free charges in the flow direction induced by a physical drift of the adsorbed Cl− ions on the continuous graphene film surface, and the larger induced voltage of graphene over MWNT could be attributed to the high electron mobility, structure integrity, and the flat nature of the graphene sheets. The power generated was ∼85 nW for a ∼30 × 16 μm graphene film with a flow of 0.6 M HCl solution at 0.01 ms−1, equivalent to a power density of ∼175 W m−2. Lee et al. further studied the flow-induced voltage generation in graphene perpendicularly aligned to the flow of the non-ionic liquids.158 It was found that a notable voltage of tens of μV could still be generated when the non-ionic liquids flowed in a perpendicular direction, and also that the polarity of the fluid was influential in determining the magnitude of the voltage generated. Due to the absence of salt ions, the observed voltage was understood to be associated with instantaneous potential differences induced in the graphene by interacting with polar liquids, and also from the momentum transferred from the flowing media to the graphene. Similarly, Sun and coworkers studied the flow-induced current in 3D graphene foam, and demonstrated that the value of the induced current was closely related to the flow velocity and the polarity of the liquid, but was independent of the flow direction and the external bias voltage.159,160 Lately, in contrast to the previous reports with liquids streaming over immersed graphene surfaces, Yin et al. found that a waving potential of up to 0.1 V could be induced if a liquid–gas boundary was moving along apiece of monolayer graphene sheet (Fig. 15e), and, furthermore, that the phenomenon could not be understood by any mechanism used to explain flow-induced voltage.161 Theoretical calculations showed that Na+ was more favorably adsorbed onto graphene rather than Cl−, thus forming an electric double layer comprising the compact layer of adsorbed Na+ and the diffuse layer of attracted Cl− under equilibrium, hence making the graphene p-doped. During the insertion/extraction of the graphene electrode from the liquid, the moving boundary of the electric double layer caused a local surplus/deficiency of Na+, driving a hole current from/to the boundary (Fig. 15f). The induced voltage was proportional to the moving velocity and was even capable of stimulating a sciatic nerve of a frog. Additionally, the induced voltage and current at lower velocities were multipliable simply via series/parallel connections of multiple graphene sheets, demonstrating the viability and scalability of building self-powered sensors and monitors in remote areas with sea wave energy (Table 5).
Table 5 Comparison of the materials, preparation methods, specific performance, and characteristics of the other generators
| Samples |
Preparation methods |
Performance |
Characteristics |
Ref. |
| Spun GO fiber hydrogel |
Wet-spinning |
GO fiber served as a superb reversible rotary motor with a rotary speed of up to 5190 revolutions per minute; the periodical change of magnetic flux induced an electromotive force (EMF) up to 1 mV |
Moisture-driven motor; flexible; screwable |
156
|
| Few-layer graphene sheets |
Chemical vapor deposition and transferred method |
The power generated was ∼85 nW for a ∼30 × 16 μm graphene film by the flow of 0.6 M HCl solution at 0.01 ms−1, equivalent to a power density of ∼175 W m−2 |
Voltage driven from fluid flow with ionic liquids |
157
|
| Graphene |
Chemical vapor deposition |
Tens of μV could still be generated when non-ionic liquids flowed |
Voltage driven from fluid flow with non-ionic liquids |
158
|
| 3D graphene foam |
Chemical vapor deposition |
10 μA was induced when the water flowing velocity was 10 cm s−1 |
The value of induced current is related to the flow velocity |
159
|
| 3D graphene foam |
Chemical vapor deposition |
20 μA was induced when the volume mixing ratio of ethanol:water = 75:25 |
Current induced when the mixtures of ethanol and water flowed |
160
|
| Monolayer graphene sheet |
Chemical vapor deposition |
0.1 V could be induced if a liquid-gas boundary was moving along the piece of monolayer graphene sheet |
High voltage; high sensitivity |
161
|
4. Summary and future outlook
Ten years has elapsed since the discovery of graphene from the mechanical cleavage of graphite, and scientists have now developed various methods to synthesize functionalized graphene materials of desirable dimensions to fully exploit their outstanding electronic, optical, thermal, and mechanical properties for application in a range of scientific fields, ranging from material science to biology, chemistry, and physics. Recently, with the advent of portable and wearable electronics, the ongoing search for low-cost, lightweight, and robustly deformable material has firmly focused on graphene as one of the most promising candidates in device fabrication, integration, optimization, and energy provision. In this featured article, we have discussed tailored graphene materials for unconventional energy conversion and storage applications, particularly in flexible and miniaturized forms, while unconventional applications of graphene in other types of energy devices, such as fuel cells are also emerging and are envisaged to grow significantly in the next few years.162–164
To date, the developments of these emerging devices are still in their infancy, with more than 80% of our references coming from the past three years. However, the current quest for energy devices compatible with flexible and wearable electronics has provided a golden opportunity and the much needed resources for academia to scrutinize on what graphene could potentially offer. Despite all the current and recent research, at least the following challenges or issues still remain. First, unconventional devices require high-quality tailored graphene materials. Unfortunately, there are no cost-efficient and eco-friendly techniques for the large-scale preparation of tailored graphene materials. Second, such a demand-driven scenario poses rigid standards for the stability, compatibility, feasibility, and stretchability of graphene-based devices. However, still only limited examples can be found in the design of flexible electronics and their application in recycling schemes. Third, several important intrinsic properties of tailored graphene materials, such as swelling, diffusion of molecules and ions within the gels, and the rheological and mechanical properties, have only rarely been studied. An in-depth understanding of these properties will not only facilitate the elucidation of the intrinsic structures of tailored graphene materials, but also provide an important clue on how to develop high-performance unconventional devices for energy conversion and storage. Finally, the tailored graphene materials for unconventional energy conversion and storage that we have discussed in this paper are mainly in the miniaturized form, but it demands additional effort to increase the capacitance and power density of the devices to approach practical scenarios.
To meet these requirements, we believe the following issues deserve special attention. First, it is important to have simple and inexpensive methods to controllably assemble the graphene building blocks into desired structures with reasonable reproducibility, thus rendering the material as a basis for further device evolution. Second, a continuous emphasis is needed on increasing the device performance (energy density, power density, conversion efficiency, etc.), and also the accessible lifetime should be highly considered, as this could make the technology a practical and viable solution to power real devices efficiently to compete with existing technologies. Third, developing multifunctional integrated devices with the ability to harvest and store different types of energy would be advantageous. For instance, a supercapacitor or lithium ion battery that could harvest thermal energy has the potential to turn the unwanted heat produced by the working portable electronics back into desired electricity, and thus could extend the discharge cycle for powering. Last, the integration of appropriate smart compositions, such as the recently developed self-healing materials, into energy conversion and storage devices could possibly help to repair the structural micro-fractures of electrodes, retain the electrical properties of the active components, and restore the configuration integrity of the devices in cases of longtime, repetitive cycling with loads or from unpredicted mechanical damage. With the increasing amount of effort devoted to this dynamic research field, we believe these emerging graphene-based energy conversion and storage devices will become more durable and more powerful and find greater application in flexible, wearable electronics and in small/micro gadgets.
Acknowledgements
We thank the financial support from the 973 program (2011CB013000, 2011CB605802) of China and NSFC (21325415, 21301018, 21174019 and 51161120361), Fok Ying Tong Education Foundation (no. 131043), the 111 Project 807012. ZZ thanks Excellent Young Scholars Research Fund of Beijing Institute of Technology for the financial support.
Notes and references
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 Search PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 Search PubMed.
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 Search PubMed.
- X. Du, I. Skachko, A. Barker and E. Y. Andrei, Nat. Nanotechnol., 2008, 3, 491–495 Search PubMed.
- R. R. Nair, P. Blake, A. N. Grigorenko, K. S. Novoselov, T. J. Booth, T. Stauber, N. M. R. Peres and A. K. Geim, Science, 2008, 320, 1308 Search PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 Search PubMed.
- B. Luo, B. Wang, X. L. Li, Y. Y. Jia, M. H. Liang and L. J. Zhi, Adv. Mater., 2012, 24, 3538–3543 Search PubMed.
- C. H. Xu, B. H. Xu, Y. Gu, Z. G. Xiong, J. Sun and X. S. Zhao, Energy Environ. Sci., 2013, 6, 1388–1414 CAS.
- K. Persson, V. A. Sethuraman, L. J. Hardwick, Y. Hinuma, Y. S. Meng and A. van der Ven, J. Phys. Chem. Lett., 2010, 1, 1176–1180 CrossRef CAS.
- C. Uthaisar and V. Barone, Nano Lett., 2010, 10, 2838–2842 Search PubMed.
- G. Eda and M. Chhowalla, Adv. Mater., 2010, 22, 2392–2415 Search PubMed.
- X. Huang, Z. Zeng, Z. Fan, J. Liu and H. Zhang, Adv. Mater., 2012, 24, 5979–6004 Search PubMed.
- H. Bai, C. Li and G. Q. Shi, Adv. Mater., 2011, 23, 1089–1115 Search PubMed.
- Q. Y. He, S. X. Wu, Z. Y. Yin and H. Zhang, Chem. Sci., 2012, 3, 1764–1772 Search PubMed.
- S. Wu, Q. He, C. Tan, Y. Wang and H. Zhang, Small, 2013, 9, 1160–1172 Search PubMed.
- W. J. Yuan and G. Q. Shi, J. Mater. Chem. A, 2013, 1, 10078–10091 CAS.
- N. Zhang, Y. H. Zhang and Y. J. Xu, Nanoscale, 2012, 4, 5792–5813 RSC.
- D. Q. Wu, F. Zhang, H. W. Liang and X. L. Feng, Chem. Soc. Rev., 2012, 41, 6160–6177 Search PubMed.
- C. Li and G. Q. Shi, Adv. Mater., 2014, 26, 3992–4012 Search PubMed.
- A. Zhamu, G. R. Chen, C. G. Liu, D. Neff, Q. Fang, Z. N. Yu, W. Xiong, Y. B. Wang, X. Q. Wang and B. Z. Jang, Energy Environ. Sci., 2012, 5, 5701–5707 CAS.
- L. W. Ji, Z. Lin, M. Alcoutlabi and X. W. Zhang, Energy Environ. Sci., 2011, 4, 2682–2699 CAS.
- G. P. Wang, L. Zhang and J. J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 Search PubMed.
- J. B. Hou, Y. Y. Shao, M. W. Ellis, R. B. Moored and B. L. Yi, Phys. Chem. Chem. Phys., 2011, 13, 15384–15402 Search PubMed.
- Y. Q. Sun, Q. Wu and G. Q. Shi, Energy Environ. Sci., 2011, 4, 1113–1132 CAS.
- Q. F. Zhang, E. Uchaker, S. L. Candelaria and G. Z. Cao, Chem. Soc. Rev., 2013, 42, 3127–3171 Search PubMed.
- J. Guo and S. J. Dong, Chem. Soc. Rev., 2011, 40, 2644–2672 Search PubMed.
- D. Chen, L. H. Tang and J. H. Li, Chem. Soc. Rev., 2010, 39, 3157–3180 Search PubMed.
- C. C. Huang, C. Li and G. Q. Shi, Energy Environ. Sci., 2012, 5, 8848–8868 CAS.
- J. Zhang, F. Zhao, Z. P. Zhang, N. Chen and L. T. Qu, Nanoscale, 2013, 5, 3112–3126 Search PubMed.
- M. M. Liu, R. Z. Zhang and W. Chen, Chem. Rev., 2014, 114, 5117–5160 Search PubMed.
- C. G. Hu, Y. Zhao, H. H. Cheng, Y. Hu, G. Q. Shi, L. M. Dai and L. T. Qu, Chem. Commun., 2012, 48, 11865–11867 Search PubMed.
- C. G. Hu, H. H. Cheng, Y. Zhao, Y. Hu, Y. Liu, L. M. Dai and L. T. Qu, Adv. Mater., 2012, 24, 5493–5498 Search PubMed.
- C. G. Hu, X. Q. Zhai, Y. Zhao, K. Bian, J. Zhang, L. T. Qu, H. M. Zhang and H. X. Luo, Nanoscale, 2014, 6, 2768–2775 Search PubMed.
- X. Huang, X. Y. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 Search PubMed.
- D. Chen, H. Zhang, Y. Liu and J. H. Li, Energy Environ. Sci., 2013, 6, 1362–1387 CAS.
- H. J. Tang, C. M. Hessel, J. Y. Wang, N. L. Yang, R. B. Yu, H. J. Zhao and D. Wang, Chem. Soc. Rev., 2014, 43, 4281–4299 Search PubMed.
- J. D. Roy-Mayhew and I. A. Aksay, Chem. Rev., 2014, 114, 6323–6348 Search PubMed.
- C. G. Hu, Y. Zhao, H. H. Cheng, Y. H. Wang, Z. L. Dong, C. C. Jiang, X. Q. Zhai, L. Jiang and L. T. Qu, Nano Lett., 2012, 12, 5879–5884 Search PubMed.
- R. F. Servic, Science, 2003, 301, 909–911 Search PubMed.
- D. J. Lipomi, M. Vosgueritchian, B. C. Tee, S. L. Hellstrom, J. A. Lee, C. H. Fox and Z. Bao, Nat. Nanotechnol., 2011, 6, 788–792 Search PubMed.
- S. H. Ko, H. Pan, C. P. Grigoropoulos, C. K. Luscombe, J. M. J. Fréchet and D. Poulikakos, Nanotechnology, 2007, 18, 345202–345209 Search PubMed.
- Y. Ahn, E. B. Duoss, M. J. Motala, X. Guo, S. I. Park, Y. Xiong, J. Yoon, R. G. Nuzzo, J. A. Rogers and J. A. Lewis, Science, 2009, 323, 1590–1593 Search PubMed.
- Y. Son, J. Yeo, H. Moon, T. W. Lim, K. H. Nam, C. P. Grigoropoulos, S. Yoo, D. Y. Yang and S. H. Ko, Adv. Mater., 2011, 23, 3176–3181 Search PubMed.
- T. Sekitani, Y. Nouguchi, K. Hata, T. Fukushima, T. Aida and T. Someya, Science, 2008, 32, 1468–1472 Search PubMed.
- K. Takei, T. Takahashi, J. C. Ho, H. Ko, A. G. Gillies, P. W. Leu, R. S. Fearing and A. Javey, Nat. Mater., 2010, 9, 821–826 Search PubMed.
- V. Chabot, D. Higgins, A. P. Yu, X. C. Xiao, Z. W. Chen and J. J. Zhang, Energy Environ. Sci., 2014, 7, 1564–1596 CAS.
- H. Gwon, H. S. Kim, K. U. Lee, D. H. Seo, Y. C. Park, Y. S. Lee, B. T. Ahna and K. Kang, Energy Environ. Sci., 2011, 4, 1277–1283 CAS.
- L. Li, Z. Wu, S. Yuan and X. B. Zhang, Energy Environ. Sci., 2014, 7, 2101–2122 CAS.
- N. Mahmood, C. Z. Zhang, H. Yin and Y. L. Hou, J. Mater. Chem. A, 2014, 2, 15–32 CAS.
- H. Gwon, J. Hong, H. Kim, D. H. Seo, S. Jeon and K. Kang, Energy Environ. Sci., 2014, 7, 538–551 CAS.
- X. F. Wang, X. H. Lu, B. Liu, D. Chen, Y. X. Tong and G. Z. Shen, Adv. Mater., 2014, 26, 4763–4782 Search PubMed.
- G. M. Zhou, F. Li and H. M. Cheng, Energy Environ. Sci., 2014, 7, 1307–1338 CAS.
- X. Peng, L. L. Peng, C. Z. Wu and Y. Xie, Chem. Soc. Rev., 2014, 43, 3303–3323 RSC.
- J. Liu, M. A. G. Namboothiry and D. L. Carroll, Appl. Phys. Lett., 2007, 90, 063501–063503 Search PubMed.
- J. Ramier, C. J. G. Plummer, Y. Leterrier, J. A. E. Manson, B. Echkert and R. Gaudiana, Renewable Energy, 2008, 33, 314–319 Search PubMed.
- B. O. Connor, K. P. Pipe and M. Shtein, Appl. Phys. Lett., 2008, 92, 193306 CrossRef PubMed.
- X. Fan, Z. Chu, F. Wang, C. Zhang, L. Chen, Y. Tang and D. Zou, Adv. Mater., 2008, 20, 592–595 CrossRef CAS.
- B. Weintraub, Y. Wei and Z. L. Wang, Angew. Chem., Int. Ed., 2009, 121, 9143–9147 Search PubMed.
- M. R. Lee, R. D. Eckert, K. Forberick, G. Dennler, C. J. Brabec and R. A. Gaudiana, Science, 2009, 324, 232–235 Search PubMed.
- T. Chen, L. B. Qiu, Z. B. Cai, F. Gong, Z. B. Yang, Z. S. Wang and H. S. Peng, Nano Lett., 2012, 12(5), 2568–2572 Search PubMed.
- S. Y. Lee, K. H. Choi, W. S. Choi, Y. H. Kwon, H. R. Jung, H. C. Shin and J. Y. Kim, Energy Environ. Sci., 2013, 6, 2414–2423 CAS.
- R. J. Miller and P. Simon, Science, 2008, 321, 651–652 Search PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4269 Search PubMed.
- R. Kotz and M. Carlen, Electrochim. Acta, 2000, 45, 2483–2498 Search PubMed.
- C. G. Liu, Z. N. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863–4868 Search PubMed.
- Z. J. Fan, J. Yan, T. Wei, L. J. Zhi, G. Q. Ning, T. Y. Li and F. Wei, Adv. Funct. Mater., 2011, 21, 2366–2375 CrossRef CAS.
- Z. B. Yang, J. Deng, X. L. Chen, J. Ren and H. S. Peng, Angew. Chem., Int. Ed., 2013, 52, 1–6 CrossRef.
- V. T. Le, H. Kim, A. Ghosh, J. Kim, J. Chang, Q. A. Vu, D. T. Pham, J. H. Lee, S. W. Kim and Y. H. Lee, ACS Nano, 2013, 7, 5940–5947 Search PubMed.
- G. Sun, L. Zheng, J. An, Y. Pan, J. Zhou, Z. Zhan, J. H. Pang, C. K. Chua, K. Leong and L. Li, Nanoscale, 2013, 5, 2870–2874 Search PubMed.
- J. Bae, M. K. Song, Y. J. Park, J. M. Kim, M. L. Liu and Z. L. Wang, Angew. Chem., Int. Ed., 2011, 50, 1683–1687 Search PubMed.
- C. J. Yu, C. Masarapu, J. P. Rong, B. Q. Wei and H. Q. Jiang, Adv. Mater., 2009, 21, 4793–4797 Search PubMed.
- H. Jiang, L. P. Yang, C. Z. Li, C. Y. Yan, P. S. Lee and J. Ma, Energy Environ. Sci., 2011, 4, 1813–1819 CAS.
- S. B. Ye and J. C. Feng, ACS Appl. Mater. Interfaces, 2014, 6, 9671–9679 CAS.
- H. Hu, Z. B. Zhao, W. B. Wan, Y. Gogotsi and J. S. Qiu, ACS Appl. Mater. Interfaces, 2014, 6, 3242–3249 CAS.
- F. Yavari, Z. P. Chen, A. V. Thomas, W. C. Ren, H. M. Cheng and N. Koratkar, Sci. Rep., 2011, 1, 166–170 Search PubMed.
- Z. P. Chen, W. C. Ren, L. B. Gao, B. L. Liu, S. F. Pei and H. M. Cheng, Nat. Mater., 2011, 10, 424–428 Search PubMed.
- C. Wu, X. Huang, X. Wu, R. Qian and P. Jiang, Adv. Mater., 2013, 25, 5658–5662 Search PubMed.
- H. B. Yao, J. Ge, C. F. Wang, X. Wang, W. Hu, Z. J. Zheng, Y. Ni and S. H. Yu, Adv. Mater., 2013, 25, 6692–6698 CrossRef CAS PubMed.
- Y. X. Xu and G. Q. Shi, J. Mater. Chem., 2011, 21, 3311–3323 RSC.
- H. Bai, C. Li, X. L. Wang and G. Q. Shi, J. Phys. Chem. C, 2011, 115, 5545–5551 CAS.
- Y. X. Xu, K. X. Sheng, C. Li and G. Q. Shi, ACS Nano, 2010, 4, 4324–4330 Search PubMed.
- H. Y. Sun, Z. Xu and C. Ga, Adv. Mater., 2013, 25, 2554–2560 Search PubMed.
- P. M. Sudeep, T. N. Narayanan, A. Ganesan, M. M. Shaijumon, H. Yang, S. Ozden, P. K. Patra, M. Pasquali, R. Vajtai, S. Ganguli, A. K. Roy, M. R. Anantharaman and P. M. Ajayan, ACS Nano, 2013, 7, 7034–7040 Search PubMed.
- Y. R. Li, J. Chen, L. Huang, C. Li, J. D. Hong and G. Q. Shi, Adv. Mater., 2014, 26, 4789–4793 Search PubMed.
- B. G. Choi, J. L. Hong, W. H. Hong, P. T. Hammond and H. S. Park, ACS Nano, 2011, 5, 7205–7213 Search PubMed.
- Z. Weng, Y. Su, D. W. Wang, F. Li, J. H. Du and H. M. Cheng, Adv. Energy Mater., 2011, 1, 917–922 Search PubMed.
- Y. Zhou, Z. Y. Qin, L. Li, Y. Zhang, Y. L. Wei, L. F. Wang and M. F. Zhu, Electrochim. Acta, 2010, 55, 3904–3908 Search PubMed.
- T. V. Vernitskaya and O. N. Efimov, Russ. Chem. Rev., 1997, 66, 443–457 Search PubMed.
- J. H. Kim, A. K. Sharma and Y. S. Lee, Mater. Lett., 2006, 60, 1697–1701 CrossRef CAS PubMed.
- A. R. Liu, C. Li, H. Bai and G. Q. Shi, J. Phys. Chem. C, 2010, 114, 22783–22789 CAS.
- D. W. Wang, F. Li, J. P. Zhao, W. C. Ren, Z. G. Chen, J. Tan, Z. S. Wu, I. Gentle, G. Q. Lu and H. M. Cheng, ACS Nano, 2009, 3, 1745–1752 Search PubMed.
- L. L. Liu, Z. Q. Niu, L. Zhang, W. Y. Zhou, X. D. Chen and S. S. Xie, Adv. Mater., 2014, 26, 4855–4862 Search PubMed.
- H. P. Cong, X. C. Ren, P. Wang and S. H. Yu, Energy Environ. Sci., 2013, 6, 1185–1191 CAS.
- X. J. Lu, H. Dou, C. Z. Yuan, S. D. Yang, L. Hao, F. Zhang, L. F. Shen, L. J. Zhang and X. G. Zhang, J. Power Sources, 2012, 197, 319–324 Search PubMed.
- Y. M. He, W. J. Chen, X. D. Li, Z. X. Zhang, J. C. Fu, C. H. Zhao and E. Q. Xie, ACS Nano, 2013, 7, 1174–1182 Search PubMed.
- Y. Zhao, J. Liu, Y. Hu, H. H. Cheng, C. G. Hu, C. C. Jiang, L. Jiang, A. Y. Cao and L. T. Qu, Adv. Mater., 2013, 25, 591–595 Search PubMed.
- L. Ma, J. Wang and F. Ding, ChemPhysChem, 2013, 14, 47–54 Search PubMed.
- L. Jiao, L. Zhang, X. Wang, G. Diankov and H. Dai, Nature, 2009, 458, 877–880 Search PubMed.
- D. K. James and J. M. Tour, Macromol. Chem. Phys., 2012, 213, 1033–1050 Search PubMed.
- T. Ouyang, Y. Chen, K. Yang and J. Zhong, Europhys. Lett., 2009, 88, 28002–28006 Search PubMed.
- C. G. Zhang, Z. W. Peng, J. Lin, Y. Zhu, G. D. Ruan, C. C. Hwang, W. Lu, R. H. Hauge and J. M. Tour, ACS Nano, 2013, 7, 65151–65159 Search PubMed.
- J. H. Chen, W. Z. Li, D. Z. Wang, S. X. Yang, J. G. Wen and Z. F. Ren, Carbon, 2002, 40, 1193–1197 Search PubMed.
- Z. Q. Niu, W. Y. Zhou, J. Chen, G. X. Feng, H. Li, W. J. Ma, J. Z. Li, H. B. Dong, Y. Ren, D. Zhao and S. S. Xie, Energy Environ. Sci., 2011, 4, 1440–1446 CAS.
- J. Carretero-González, E. Castillo-Martínez, M. Dias-Lima, M. Acik, D. M. Rogers, J. Sovich, C. S. Haines, X. Lepró, M. Kozlov, A. Zhakidov, Y. Chabal and R. H. Baughman, Adv. Mater., 2012, 24, 5695–5701 Search PubMed.
- M. F. El-Kady, V. Strong, S. Dubin and R. B. Kaner, Science, 2012, 335, 1326–1330 Search PubMed.
- M. F. El-Kady and R. B. Kaner, Nat. Commun., 2013, 4, 1475–1483 Search PubMed.
- W. Gao, N. Singh, L. Song, Z. Liu, A. L. M. Reddy, L. J. Ci, R. Vajtai, Q. Zhang, B. Q. Wei and P. M. Ajayan, Nat. Nanotechnol., 2011, 6, 496–500 Search PubMed.
- M. Q. Xue, F. W. Li, J. Zhu, H. Song, M. N. Zhang and T. B. Cao, Adv. Funct. Mater., 2012, 22, 1284–1290 Search PubMed.
- H. H. Cheng, C. G. Hu, Y. Zhao and L. T. Qu, NPG Asia Mater., 2014, 6, e113–e125 Search PubMed.
- W. Zeng, L. Shu, Q. Li, S. Chen, F. Wang and X. M. Tao, Adv. Mater., 2014, 26, 5310–5336 Search PubMed.
- K. Jost, G. Diona and Y. Gogotsi, J. Mater. Chem. A, 2014, 2, 10776–10787 CAS.
- Y. N. Meng, Y. Zhao, C. G. Hu, H. H. Cheng, Y. Hu, Z. P. Zhang, G. Q. Shi and L. T. Qu, Adv. Mater., 2013, 25, 2326–2331 Search PubMed.
- Y. Hu, H. H. Cheng, F. Zhao, N. Chen, L. Jiang, Z. H. Feng and L. T. Qu, Nanoscale, 2014, 6, 6448–6451 Search PubMed.
- L. Kou, T. Q. Huang, B. N. Zheng, Y. Han, X. L. Zhao, K. Gopalsamy, H. Y. Sun and C. Gao, Nat. Commun., 2014, 5, 3754–3763 Search PubMed.
- D. S. Yu, K. L. Goh, H. Wang, L. Wei, W. C. Jiang, Q. Zhang, L. M. Dai and Y. Chen, Nat. Nanotechnol., 2014, 9, 555–562 Search PubMed.
- X. T. Ding, Y. Zhao, C. G. Hu, Y. Hu, Z. L. Dong, N. Chen, Z. P. Zhang and L. T. Qu, J. Mater. Chem. A, 2014, 2, 12355–12360 CAS.
- H. H. Cheng, Z. L. Dong, C. G. Hu, Y. Zhao, Y. Hu, L. T. Qu, N. Chen and L. M. Dai, Nanoscale, 2013, 5, 3428–3434 Search PubMed.
- Q. Chen, Y. N. Meng, C. G. Hu, Y. Zhao, H. B. Shao, N. Chen and L. T. Qu, J. Power Sources, 2014, 247, 32–39 Search PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 Search PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 Search PubMed.
- Y. H. Hu and X. L. Sun, J. Mater. Chem. A, 2014, 2, 10712–10738 CAS.
- F. Liu, S. Y. Song, D. F. Xue and H. J. Zhang, Adv. Mater., 2012, 24, 1089–1094 Search PubMed.
- N. Li, Z. P. Chen, W. C. Ren, F. Li and H. M. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17360–17365 Search PubMed.
- D. H. Wang, R. Kou, D. W. Choi, Z. G. Yang, Z. M. Nie, J. Li, L. V. Saraf, D. H. Hu, J. G. Zhang, G. L. Graff, J. Liu, M. A. Pope and I. A. Aksay, ACS Nano, 2010, 4, 1587–1595 Search PubMed.
- R. Mukherjee, A. V. Thomas, D. Datta, E. Singh, J. W. Li, O. Eksik, V. B. Shenoy and N. Koratkar, Nat. Commun., 2014, 5, 3710–3719 Search PubMed.
- M. H. Ye, Z. L. Dong, C. G. Hu, H. H. Cheng, H. B. Shao, N. Chen and L. T. Qu, Small, 2014 DOI:10.1002/small.201401610.
- J. Ren, Y. Zhang, W. Y. Bai, X. L. Chen, Z. T. Zhang, X. Fang, W. Weng, Y. G. Wang and H. S. Peng, Angew. Chem., Int. Ed., 2014, 53, 7864–7869 CrossRef CAS PubMed.
- Y. H. Hu, H. Wang and B. Hu, ChemSusChem, 2010, 3, 782–796 CrossRef CAS PubMed.
- B. Luo, S. M. Liu and L. J. Zhi, Small, 2012, 8, 630–646 Search PubMed.
- L. J. Brennan, M. T. Byrne, M. Bari and Y. K. Gun'ko, Adv. Energy Mater., 2011, 1, 472–485 Search PubMed.
- X. J. Wan, G. K. Long, L. Huang and Y. S. Chen, Adv. Mater., 2011, 23, 5342–5358 Search PubMed.
- X. J. Wan, Y. Huang and Y. S. Chen, Acc. Chem. Res., 2012, 45, 598–607 Search PubMed.
- C. X. Guo, H. B. Yang, Z. M. Sheng, Z. S. Lu, Q. L. Song and C. M. Li, Angew. Chem., Int. Ed., 2010, 49, 3014–3017 Search PubMed.
- Z. Y. Liu, D. W. He, Y. S. Wang, H. P. Wu and J. G. Wang, Sol. Energy Mater. Sol. Cells, 2010, 94, 1196–1200 Search PubMed.
- Y. Li, Y. Hu, Y. Zhao, G. Q. Shi, L. E. Deng, Y. B. Hou and L. T. Qu, Adv. Mater., 2011, 23, 776–780 CrossRef CAS PubMed.
- V. Gupta, N. Chaudhary, R. Srivastava, G. D. Sharma, R. Bhardwaj and S. Chand, J. Am. Chem. Soc., 2011, 133, 9960–9963 Search PubMed.
- X. Yan, X. Cui and L. S. Li, J. Am. Chem. Soc., 2010, 132, 5944–5945 Search PubMed.
- X. Yan, X. Cui, B. Li and L. S. Li, Nano Lett., 2010, 10, 1869–1873 Search PubMed.
- M. L. Mueller, X. Yan, B. Dragnea and L. S. Li, Nano Lett., 2011, 11, 56–60 Search PubMed.
- M. L. Mueller, X. Yan, J. A. McGuire and L. S. Li, Nano Lett., 2010, 10, 2679–2682 Search PubMed.
- K. J. Williams, C. A. Nelson, X. Yan, L. S. Li and X. Y. Zhu, ACS Nano, 2013, 7, 1388–1394 Search PubMed.
- X. Fang, Z. B. Yang, L. B. Qiu, H. Sun, S. W. Pan, J. Deng, Y. F. Luo and H. S. Peng, Adv. Mater., 2014, 26, 1694–1698 Search PubMed.
- Z. B. Yang, H. Sun, T. Chen, L. B. Qiu, Y. F. Luo and H. S. Peng, Angew. Chem., Int. Ed., 2013, 52, 7545–7548 Search PubMed.
- H. Sun, X. You, J. Deng, X. L. Chen, Z. B. Yang, J. Ren and H. S. Peng, Adv. Mater., 2014, 26, 2868–2873 Search PubMed.
- H. J. Shin, W. M. Choi, D. Choi, G. H. Han, S. M. Yoon, H. K. Park, S. W. Kim, Y. W. Jin, S. Y. Lee, J. M. Kim, J. Y. Choi and Y. H. Lee, J. Am. Chem. Soc., 2010, 132, 15603–15609 Search PubMed.
- B. Kumar, K. Y. Lee, H. K. Park, S. J. Chae, Y. H. Lee and S. W. Kim, ACS Nano, 2011, 5, 4197–4204 Search PubMed.
- J. Kwon, W. Seung, B. K. Sharma, S. W. Kim and J. H. Ahn, Energy Environ. Sci., 2012, 5, 8970–8975 CAS.
- J. Kwon, B. K. Sharma and J. H. Ahn, Jpn. J. Appl. Phys., 2013, 52, 06GA02–06GA10 CrossRef.
- J. H. Lee, K. Y. Lee, B. Kumar, N. T. Tien, N. E. Lee and S. W. Kim, Energy Environ. Sci., 2013, 6, 169–175 CAS.
- S. H. Bae, O. Kahya, B. K. Sharma, J. Kwon, H. J. Cho, B. Ozyilmaz and J. H. Ahn, ACS Nano, 2013, 7, 3130–3138 Search PubMed.
- R. H. Que, Q. Shao, Q. L. Li, M. W. Shao, S. D. Cai, S. D. Wang and S. T. Lee, Angew. Chem., Int. Ed., 2012, 51, 5418–5422 Search PubMed.
- L. Valentini, S. B. Bon and J. Kenny, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1028–1032 Search PubMed.
- H. Tian, S. Ma, H. M. Zhao, C. Wu, J. Ge, D. Xie, Y. Yang and T. L. Ren, Nanoscale, 2013, 5, 8951–8957 Search PubMed.
- F. R. Fan, Z. Q. Tian and Z. L. Wang, Nano Energy, 2012, 1, 328–334 Search PubMed.
- S. Kim, M. K. Gupta, K. Y. Lee, A. Sohn, T. Y. Kim, K. S. Shin, D. Kim, S. K. Kim, K. H. Lee, H. J. Shin, D. W. Kim and S. W. Kim, Adv. Mater., 2014, 26, 3918–3925 Search PubMed.
- H. H. Cheng, Y. Hu, F. Zhao, Z. L. Dong, Y. H. Wang, N. Chen, Z. P. Zhang and L. T. Qu, Adv. Mater., 2014, 26, 2909–2913 Search PubMed.
- P. Dhiman, F. Yavari, X. Mi, H. Gullapalli, Y. Shi, P. M. Ajayan and N. Koratkar, Nano Lett., 2011, 11, 3123–3127 Search PubMed.
- S. H. Lee, Y. Jung, S. Kim and C. S. Han, Appl. Phys. Lett., 2013, 102, 063116–103120 Search PubMed.
- W. B. Huang, G. L. Wang, F. Q. Gao, Z. T. Qiao, G. Wang, L. Tao, M. J. Chen, F. Yu, H. C. Yang and L. F. Sun, Nanoscale, 2014, 6, 3921–3924 Search PubMed.
- W. B. Huang, G. L. Wang, F. Q. Gao, Z. T. Qiao, G. Wang, M. J. Chen, Y. Deng, L. Tao, Y. Zhao, X. K. Fan and L. F. Sun, J. Phys. Chem. C, 2014, 118, 8783–8787 CAS.
- J. Yin, Z. H. Zhang, X. M. Li, J. Yu, J. X. Zhou, Y. Q. Chen and W. L. Guo, Nat. Commun., 2014, 5, 3582 Search PubMed.
- H. J. Choi, S. M. Jung, J. M. Seo, D. W. Chang, L. M. Dai and J. B. Baek, Nano Energy, 2012, 1, 534–551 Search PubMed.
- Q. Wang, X. K. Wang, Z. F. Chai and W. P. Hu, Chem. Soc. Rev., 2013, 42, 8821–8834 Search PubMed.
- X. J. Zhou, J. L. Qiao, L. Yang and J. J. Zhang, Adv. Energy Mater., 2014, 4, 1301523–1301547 Search PubMed.
Footnote |
| † These authors contribute equally to the work. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.