Fosmidomycin analogues with N-hydroxyimidazole and N-hydroxyimidazolone as a chelating unit†
Camille Midriera,
Sonia Montela,
Ralf Braunb,
Klaus Haafb,
Lothar Willmsb,
Arie van der Leec,
Jean-Noël Vollea,
Jean-Luc Pirat*a and
David Virieux*a
aInstitut Charles Gerhardt, UMR5253, AM2N, ENSCM, 8, Rue de l'Ecole Normale, F34296 Montpellier Cedex 5,, France
bBayer CropScience AG, Chemistry Frankfurt, G 836, Industriepark Höchst, 65926 Frankfurt am Main, Germany
cInstitut Européen des membranes, cc047 Université de Montpellier 2, 34095, Montpellier, France
First published on 14th May 2014
Abstract
Fosmidomycin has been reported to have many biological activities as an antibacterial and antimalarial, along with being a herbicidal agent. Its unique mode of action involves the inhibition of a key step of the non mevalonate pathway by blockade of a crucial enzyme, the 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR), whose expression is present in bacteria, plasmodium parasites and higher plants, but not in mammals. Herein we report the development of fosmidomycin and of FR-900098 constrained analogues belonging to an unusual heterocyclic based complexing subunit involving N-hydroxyimidazoles and cyclic N-hydroxyureas.
Introduction
During the late 1970s, fosmidomycin 1a, also known under the acronym FR-31564, was isolated from Streptomyces lavendulae and first evaluated as a natural antibiotic in an early phase II study for the management of bacterial infections.1 In an in vitro assay with the purified recombinant E. coli DXR, Seto et al. showed that a crucial enzyme of the initial step of the mevalonate-independant pathway of isoprenoid biosynthesis,2 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR), was inhibited by fosmidomycin in a dose-dependent manner with an IC50 value of 8.2 nM.3 Later, the natural antibiotic 1a and its methyl derivative FR-900098 1b have been reported for their in vitro antimalarial activity against Plasmodium falciparum and in vivo in mice against Plasmodium vinckei.4 The efficiency of both compounds against Plasmodium falciparum on human has been proved after the latter cured uncomplicated malaria.5 Nevertheless, in human a high rate of recrudescence has been observed, which coupled to a moderate gastrointestinal absorption rate (20 to 40% after administration of an oral dose of 7.5 mg kg−1 of 1a) and a short half-life per os (∼2 h) has probably precluded fosmidomycin as a monotherapeutic agent towards malaria.6 On the other hand, two other clinical trials using a combination of fosmidomycin–clindamycin gave promising results for a possible new treatment against malaria.7 But at the best of our knowledge, no therapy using these two compounds has been commercialized yet and this prompted the search for better effective inhibitors.8 This phosphonic acid antibiotic, also proved to be effective against the DXR enzyme of higher plants,9 and its herbicide activity has even been patented.10 The combined low mammalian toxicity (LD50 rats, oral > 8 g kg−1)4 and high hydrophilic properties of the fosmidomycin 1a and its acetyl analogue 1b present a great interest to discover a novel class of compounds for herbicide applications. Consequently, both structures have been seen as valuable leads for the preparation of new inhibitors of DXR in higher plants. Due to considerable conformational rearrangement of DXR upon formation of the DXR–fosmidomycin complex, two sites are of a crucial importance for tight binding, selectivity and for the development of effective molecules.11 The positively charged pocket binds the phosphonate group of fosmidomycin with a high specificity whereas an amphipathic region binds the hydroxamic acid through complexation with a divalent cation, generally magnesium. The hydrophobic region of the carbon backbone is considered as a modulatory region and is exploited by different classes of fosmidomycin-like inhibitors. Then, modulation of the cation-complexing unit along with the modulatory region offers fine-tuning possibilities.12In this context, the 5-acyl-N-hydroxyimidazoles 2 were perceived as molecules having a highly potent complexing unit by comparison with the parent molecules 1a–b (Fig. 1). Chelation based on acyl-N-hydroxyimidazoles 2 has without ambiguity interesting features.
 | ||
| Fig. 1 Fosmidomycin 1a, FR-900098 1b and a weak inhibitor of DXR. Sterically strained analogues 2 and 3. Isoxaflutole and its active metabolite (DKN). | ||
First of all, the complexing moiety can be seen as a close pharmacophore of the bioactive metabolite, diketonitrile (DKN) which is referred to be an iron complexing agent and an inhibitor of the 4-hydroxyphenylpyruvate dioxygenase (HPPD), a ferrous iron metalloenzyme. DKN is generated by conversion in plants from isoxaflutole herbicide,13 and the presence of a β-(1,3)-diketone moiety permits the creation of a stable ion–dipole interaction in the active enzyme active site (Fig. 1). Secondly, conformational restriction due to imidazole ring, constituted another feature justifying the preparation of acyl-N-hydroxyimidazoles 2. In the same way, N′-benzyl-N-hydroxyurea was already reported as a modest of DXR (Fig. 1).14 Nevertheless, the targeted cyclic N-hydroxyureas 3 can be considered as closer and conformationally modified fosmidomycin 1a analogues. These original structures are fully different from those previously tested and could be present interesting features for the development of new herbicides. N-Hydroxyimidazoles 2 were obtained from the phosphonate 4. The key step of the chemical pathway was a three-component cyclisation of substituted α-ketooximes 6 (ref. 15 and 16) with the 3-phosphonopronionaldehyde 5 (ref. 17) and ammonia (Fig. 2). In the other side, N-hydroxyurea derivatives 3 were intended to be synthesized by a 5-exo-trig cyclization according to the Baldwin rules of compound 8 coming from the phosphonoallylamine 9 by reaction with an isocyanate. The O-benzylhydroxyamino group should be accessible from reductive oximination of the β-phosphonoacrolein 10. The last steps of the synthesis of 3 are the cleavage of the phosphonic ester functions and a critical hydrogenolysis of the O-benzyl protective group without cleavage of the N–O bond.
 | ||
| Fig. 2 Retrosynthetic pathways for targeted hydroxyimidazoles 2 and hydroxyimidazolones 3. | ||
Results and discussion
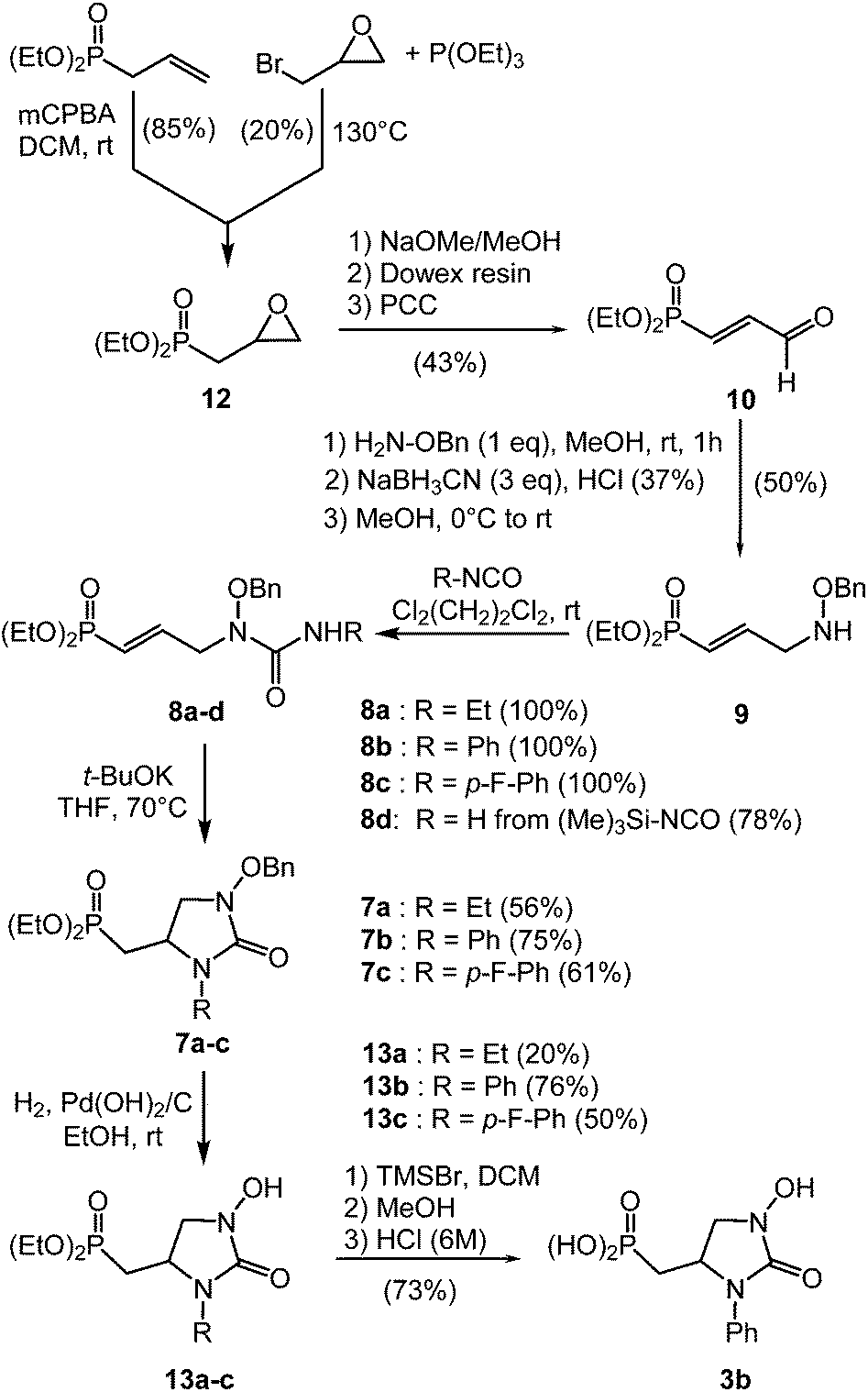
The 4-dibenzylphosphono-propionaldehyde 5 was an expected intermediate for the preparation of the N-hydroxyimidazolyl derivatives 2 (Scheme 1). It was accessible by adapting a two-step sequence which started from the Michaelis–Becker reaction of dibenzyl phosphite with bromoethyldioxolane promoted by caesium carbonate (Cs2CO3) instead of sodium hydride. The resulting dibenzyl (2-(1,3-dioxolane-2-yl)ethylidene)phosphonate 11 was obtained in 74% yield. An acidic work-up using acetic acid in water gave almost quantitatively the aldehyde 5 without hydrolysis of the phosphonic ester function (Scheme 1).17 In parallel, substituted ketooximes 6a–d were prepared according to the reaction of the corresponding ketone with a hydrochloric solution of sodium nitrite in 41% to 86% yields.15,16 The reaction of cyclisation between aldehyde 5 and α-hydroxyiminocarbonyl derivatives 6a–d was performed in mild conditions using ammonium acetate as a source of ammonia in acetic acid at room temperature.18 The desired products 4a–d were obtained after flash chromatography in isolated yield ranging from 56% to 94%. Interestingly, when unsymmetrical diketone such as 2-hydroxyimino-1-phenyl-butane-1,3-dione 6c was used, a full regioselectivity was observed, only affording the phenylketo-hydroxyimidazole 4c. The benzyl protected phosphonic esters were not stable on storage for a long period of time giving products resulting from an internal nucleophilic substitution and forming a N-benzyloxyimidazolyl phosphonic acid monoester. Then reaction of 4b and 4c by addition of one equivalent of lithium hydroxide in a mixture water–THF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) gave the stable lithium salts. An acid-base titration with a diluted aqueous solution of hydrogen chloride permitted to determine a pKa around 5.5 for these products 4b-Li and 4c-Li. Several catalysts were tested for the cleavage of benzyl group of phosphonate 4a–d (Pd/C, Pd(OH)2/C and Pd/BaSO4) keeping in mind to minimize the cleavage of the sensitive N–O bond.19,20 The best results were observed with Pd(OH)2/C in methanol at room temperature with hydrogen at 1.0 bar pressure (Scheme 2). Then, phosphonic acid 2b was obtained after recrystallization in 50% yield. In the same conditions, the reaction on 4c gave the alcohol 2e in 51% yield by reduction of the ketone concomitantly with the cleavage of the benzyl group. To circumvent this side-reaction, bromo trimethylsilane has been used for 4a, 4c and 4d, affording after solvolysis in methanol the desired phosphonic acids 2a, 2c and 2d.
:1) gave the stable lithium salts. An acid-base titration with a diluted aqueous solution of hydrogen chloride permitted to determine a pKa around 5.5 for these products 4b-Li and 4c-Li. Several catalysts were tested for the cleavage of benzyl group of phosphonate 4a–d (Pd/C, Pd(OH)2/C and Pd/BaSO4) keeping in mind to minimize the cleavage of the sensitive N–O bond.19,20 The best results were observed with Pd(OH)2/C in methanol at room temperature with hydrogen at 1.0 bar pressure (Scheme 2). Then, phosphonic acid 2b was obtained after recrystallization in 50% yield. In the same conditions, the reaction on 4c gave the alcohol 2e in 51% yield by reduction of the ketone concomitantly with the cleavage of the benzyl group. To circumvent this side-reaction, bromo trimethylsilane has been used for 4a, 4c and 4d, affording after solvolysis in methanol the desired phosphonic acids 2a, 2c and 2d.
 | ||
| Scheme 1 Synthesis of hydroxyimidazoles 4a–d and 2a–e. | ||
 | ||
| Scheme 2 Synthesis of protected N-benzyloxyimidazolones 7a–c and 3c. | ||
Slow crystallisation of the three final products into water gave single crystals which were analysed by X-ray experiments, confirming thus each structure (Fig. 3).21 These compounds 2a–b and 2d existed as zwitterionic imidazoliums by protonation of nitrogen in position 3.
 | ||
| Fig. 3 Conformations adopted in the crystal for phosphinic acids 2a, 2b and 2d and their crystal packing. | ||
The molecular packing in the single crystals showed intermolecular hydrogen bonding between the N-hydroxy group and an oxygen atom of the phosphonic acid group. Surprisingly, no intramolecular hydrogen bonding between the N-hydroxy group and heteroatom on the substituent in position 5 was highlighted. The N-hydroxyimidazole 2b crystallized with one molecule of water in a P21/n crystal cell. On the contrary N-hydroxyimidazole 2a and 2d crystallized without water and presented a crystal cell with Pn and P21/n space group symmetry, respectively.
For the second series of fosmidomycin analogues 3, the 3-phosphonoacrolein intermediate 10 was obtained from oxiranylmethylphosphonate 12 (Scheme 2). For that purpose, two different methodologies have been tested. The first one was an Arbuzov reaction between epichlorhydrin and triethyl phosphite, nevertheless the reaction revealed unsuccessful forming diethyl methylphosphonate. When using epibromohydrin, oxiranylmethyl phosphonate 12 was isolated only in a low yield (20%, litt.22 61.9%). Therefore, another way using oxidation of commercial allylphosphonate with m-CPBA in dichloromethane has been preferred and furnished the expected epoxide 12 in multigram scale (Scheme 2).23 The 3-phosphonoacrolein 10 synthesis was achieved according to a three-step sequence involving a treatment by sodium methoxide, followed by an elimination mediated by Dowex resin and subsequent oxidation of the resulting alcohol with PCC.24 A reductive amination was finally performed using O-benzyl hydroxylamine in methanol and sodium cyanoborohydride. After flash chromatography, the phosphonoallyl benzyloxyamine 9 was isolated in 50% yield. The chemical diversity was introduced at the following step, by reaction of different isocyanates affording N-hydroxybenzylureas 8a–d (78–100%). A favourable 5-exo-trig intramolecular Michael addition has been led with success, using potassium tert-butoxide (20 mol%) in tetrahydrofuran at 70 °C overnight. Cyclic imidazolones 7a, 7b and 7c were isolated in 56 to 75% yields. For 7d (R = H), two unidentified products were observed by 31P NMR with an identical ratio, but none of them was separable by column chromatography on silica or by preparative HPLC, and therefore imidazolone 7d characterization revealed unsuccessful. The deprotection of the benzyl group of 7a–c was first tested by hydrogenolysis with palladium hydroxide in ethanol at room temperature and overnight. For all the reactions, we observed the cleavage of the N–O bond. For phenyl 7b and p-fluorophenyl 7c derivatives, the expected imidazolones 13b–c were obtained in 76% and 50% yields, respectively. Compound 13a was successfully obtained by modification of the reaction conditions, using palladium hydroxide on charcoal in ethanol for only three hours. Nevertheless, only low yield (20%) of deprotected imidazolone 13a was obtained after purification. Then, a final removal of both ethyl groups was performed on 13b by treatment with trimethylsilyl bromide, followed by methanolysis. Nevertheless, despite a large excess of trimethylsilyl bromide (12 eq.) the reaction never reached the completion. Then a solution of hydrochloric acid (6 M) was added and the mixture was refluxed. After one night, fully deprotected phosphinic acid 3b was obtained in 73% yield.
Experimental
General information
All air and/or water sensitive reactions were carried out under a nitrogen atmosphere. The solvents were dried using standard methods, distilled and stored under nitrogen. Reactions were monitored by 31P NMR using DMSO-d6 as internal references. Chromatography columns were performed on silica gel (Merck 60 AC. 35–70 μm). All NMR spectra were recorded on a BRUKER Ultra shield 400 plus instrument at 161.99 MHz for 31P, 376.50 MHz for 19F, 400.13 MHz for 1H and 100.61 MHz for 13C. The spectrometer used for low and high mass resolution spectra was electrospray ionization (ESI) WATERS Micromass Q-Tof spectrometer with as internal reference H3PO4 (0.1% in water–acetonitrile, 1:1).
Preparation of precursor
Preparation of precursors
3-Hydroxyimino-pentane-2,4-dione (6a) (ref. 15a and b). At −5 °C, a solution of nitrite sodium (7.1 g, 0.10 mol) in water (20 mL) was added dropwise to a solution of acetyl acetone (10 g, 0.10 mol) in hydrochloric acid (50 mL, 2 M), and the mixture was allowed to stand for 20 min. The mixture was extracted with ethyl acetate (3 × 40 mL), then combined organic layers were dried over magnesium sulfate and concentrated under vacuum. The crude was purified by recrystallization from chloroform affording the expected compound as a white solid. Yield: 86% (11.1 g).
General procedure for 6b–d
At −10 °C, a solution of nitrite sodium (1.26 g, 1.77 mmol) in water (20 mL) was added dropwise over 30 min to a solution of ketone (1.61 mmol) in acetic acid (15 mL). The mixture was allowed to stand at room temperature for 2 h. Then, the product was extracted with ethyl acetate (3 × 70 mL), combined organic layers were dried over magnesium sulfate and concentrated in vacuo. After purification, the compound was used directly in the next step because of its low stability.General procedure
:0 to 70:30)), to afford the desired product. Yield: 94% (1.26 g). 1H NMR (400.13 MHz, CDCl3) δ 2.14–2.22 (m, 2H), 2.34 (s, 3H), 2.38 (s, 3H), 2.89–2.97 (m, 2H), 4.88 (dd, 2JHH = 11.8 Hz, 3JHP = 8.0 Hz, 2H), 4.95 (dd, 2JHH = 11.8 Hz, 3JHP = 8.8 Hz, 2H), 7.19–7.27 (m, 10H). 13C NMR (100.61 MHz, CDCl3) δ 16.60 (s), 18.53 (d, 2JCP = 3.6 Hz), 22.97 (d, 1JCP = 141.3 Hz), 28.19 (s), 67.41 (d, 2JCP = 6.1 Hz), 122.27 (s), 127.87 (s), 128.47 (s), 128.61 (s), 136.04 (d, 3JCP = 6.6 Hz), 141.66 (d, 3JCP = 15.3 Hz), 190.88 (s). 31P NMR (161.97 MHz, CDCl3) δ 31.14 (s). HRMS (ESI) m/z [M + H]+ calcd for C22H26N2O5P 429.1579, found 429.1577.:0–70:30) to afford the N-hydroxyl intermediate 4b (0.85 g, 1.85 mmol). 4b was dissolved in tetrahydrofuran (10 mL) and a solution of lithium hydroxide monohydrate (0.078 g, 1.85 mmol) in water (5 mL) was slowly added. The resulting mixture was stirred for an additional 1 h. After concentration in vacuo, the lithium salts 4b-Li was obtained. Yield: 59% (0.86 g). 1H NMR (400.13 MHz, DMSO-d6) δ 1.26 (t, 3JHH = 7.0 Hz, 3H), 2.18–2.30 (m, 2H), 2.21 (s, 3H), 2.75–2.81 (m, 2H), 4.19 (q, 3JHH = 7.0 Hz, 2H), 4.98 (dd, 2JHH = 12.1 Hz, 3JHP = 7.4 Hz, 2H), 5.03 (dd, 2JHH = 12.1 Hz, 3JHP = 8.3 Hz, 2H), 7.19–7.27 (m, 10H). 13C NMR (100.61 MHz, DMSO-d6) δ 14.26 (s), 16.87 (s), 18.76 (d, 2JCP = 2.9 Hz), 21.95 (d, 1JCP = 136.8 Hz), 59.26 (s), 66.25 (d, 2JCP = 5.6 Hz), 115.42 (s), 127.60 (s), 128.04 (s), 128.40 (s), 136.71 (d, 3JCP = 6.6 Hz), 138.98 (s), 141.18 (d, 3JCP = 19.0 Hz), 163.13 (s). 31P NMR (161.97 MHz, DMSO-d6) δ 32.44 (s). HRMS (ESI) m/z [M + H]+ calcd for C23H27LiN2O6P 465.1767, found 465.1778.:0–70:30) to afford the N-hydroxyl intermediate 4c (0.87 g, 1.76 mmol). 4c was dissolved in tetrahydrofuran (10 mL) and a solution of lithium hydroxide monohydrate (0.074 g, 1.76 mmol) in water (5 mL) was slowly added. The resulting mixture was stirred for an additional 1 h. After concentration in vacuo, the lithium salts was obtained. Yield: 56% (0.88 g). 1H NMR (400.13 MHz, DMSO-d6) δ (ppm) 1.69 (s, 3H), 2.28–2.36 (m, 2H), 2.86–2.93 (m, 2H), 5.06 (dd, 2JHH = 12.1 Hz, 3JHP = 7.5 Hz, 2H), 5.1 (dd, 2JHH = 12.1 Hz, 3JHP = 8.2 Hz, 2H), 7.34–7.62 (m, 15H). 13C NMR (100.61 MHz, DMSO-d6) δ 17.42 (s), 18.74 (d, 2JCP = 2.9 Hz), 22.68 (d, 1JCP = 138.3 Hz), 66.32 (d, 2JCP = 5.8 Hz), 125.27 (s), 127.63 (s), 128.04 (s), 128.07 (s), 128.24 (s), 128.41 (s), 136.69 (d, 3JCP = 6.6 Hz), 140.45 (s), 141.41 (s), 142.72 (d, 3JCP = 18.3 Hz). 31P NMR (161.97 MHz, DMSO-d6) δ 32.37 (s). HRMS (ESI) m/z [M + H]+ calcd for C27H27LiN2O5P 497.1818, found 497.1817.:0–70:30)) to afford the desired product. Yield: 80% (1.14 g). 1H NMR (400.13 MHz, CDCl3) δ 2.11–2.19 (m, 2H), 2.30 (s, 3H), 2.89–2.98 (m, 2H), 4.91 (dd, 2JHH = 11.9 Hz, 3JHP = 8.0 Hz, 2H), 4.96 (dd, 2JHH = 11.9 Hz, 3JHP = 9.3 Hz, 2H), 6.37 (dd, 3JHH = 3.3 Hz, 3JHH = 1.76 Hz, 1H), 6.72 (d, 3JHH = 3.3 Hz, 1H), 7.19–7.27 (m, 11H). 13C NMR (100.61 MHz, CDCl3) δ 13.13 (s), 17.66 (s), 23.29 (d, 1JCP = 140.5 Hz), 67.73 (d, 2JCP = 6.6 Hz), 107.13 (s), 110.99 (s), 119.29 (s), 127.93 (s), 128.61 (s), 128.66 (s), 135.76 (d, 3JCP = 5.8 Hz), 141.13 (s), 144.18 (s). 31P NMR (161.97 MHz, CDCl3) δ 32.79 (s). HRMS (ESI) m/z [M + H]+ calcd for C24H26N2O5P 453.1579, found: 453.1582.Deprotection reaction
Method A. In a Schlenck tube was introduced palladium dihydroxyde on charcoal (10%), dibenzyl phosphonate (4b or 4c) and degassed ethanol. Three vacuum (until solvent bubbling)/nitrogen, two vacuum/hydrogen were performed on the system. After vigorously stirring at room temperature for 24 h (4b) or 40 °C for 36 h (4c), the reaction mixture was filtered on celite and the filtrate was concentrated in vacuo.
Method B. At 0 °C, trimethylsilylbromide (1.27 mL, 10 mmol) was added to a solution of benzyl phosphonate (4a, 4c and 4d, 1 mmol) in dichloromethane (10 mL). The reaction mixture was allowed to stand up at room temperature and stirred for 14 h. After concentration to dryness under vacuum, methanol (10 mL) was added and the resulting mixture was stirred at room temperature for 2 h, thus concentrated in vacuo. Further purification was not required.
:40–0:100) to afford the desired product. Yield: 50% (11.8 g). 1H NMR (400.13 MHz, CDCl3) δ 1.34 (t, 3JHH = 7.1 Hz, 6H), 3.68–3.71 (m, 2H), 4.06–4.14 (m, 4H), 4.73 (s, 2H), 5.85–5.96 (ddt, 2JPH = 20.3 Hz, 3JHH = 17.2 Hz, 4JHH = 3.2 Hz, 1H), 6.77–6.89 (ddt, 3JPH = 27.8 Hz, 3JHH = 17.4 Hz, 3JHH = 5.5 Hz, 1H), 7.33–7.37 (m, 5H). 13C NMR (100.61 MHz, CDCl3) δ 16.36 (d, 3JCP = 6.4 Hz), 54.21 (d, 3JCP = 23.2 Hz), 61.77 (d, 2JCP = 5.6 Hz), 77.33 (s), 119.11 (d, 1JCP = 187.8 Hz), 127.96 (s), 128.42 (s), 137.53 (s), 148.55 (d, 2JCP = 4.9 Hz). 31P NMR (161.97 MHz, CDCl3) δ 17.84 (s).General procedure
General procedure
:50–0:100).General procedure
:18:2–0:90:10).Conclusions
N-Hydroxyimidazoles 2a–d and cyclic N-hydroxyureas 13a–c, both rigidified analogues of fosmidomycin have been successfully prepared as potential inhibitors of DXR. Compounds 2a–d have been prepared by a three-component reaction through the condensation-cyclization sequence between dibenzyl (3-oxopropyl)phosphonate 5, α-ketooximes 6 and ammonium acetate. The five-membered ring hydroxyureas derivatives have been synthesized from an unusual intramolecular Michael addition leading to the desired structures.In vivo evaluations as herbicides for compounds 4a–d, 2a–e, 13a–c and 3c were conducted by spraying on different cultures of interest belonging to monocotyledons or dicotyledons. Unfortunately, no biological activity was seen in this preliminary screening. Nevertheless other N-hydroxyurea analogues are expected and will be evaluated in due course.
Acknowledgements
Dr Camille Midrier and Dr Sonia Montel were supported in part by grants from Bayer CropScience Ag and from the Agence Nationale de la Recherche et de la Technologie (ANRT) and the Centre National de la Recherche Scientifique (CNRS).Notes and references
- (a) H. P. Kuemmerle, T. Murakawa, H. Sakamoto, N. Sato, T. Konishi and F. De Santis, Int. J. Clin. Pharmacol., Ther. Toxicol., 1985, 23, 521 CAS . For review on Isoprenoid metabolism see: ; (b) J. de Ruyck, J. Wouters and C. D. Poulter, Curr. Enzyme Inhib., 2011, 7, 79 CrossRef CAS PubMed; (c) C. Obiol-Pardo, J. Rubio-Martinez and S. Imperial, Curr. Med. Chem., 2011, 18, 1325 CrossRef CAS; (d) A. J. Wiemer, C.-H. Hsiao and D. F. Wiemer, Curr. Top. Med. Chem., 2010, 10, 1858 CrossRef CAS; (e) D. Ganjewala, S. Kumar and R. Luthra, Curr. Issues Mol. Biol., 2009, 11(suppl. 1), i35 CAS; (f) N. Singh, G. Cheve, M. A. Avery and C. R. McCurdy, Curr. Pharm. Des., 2007, 13, 1161 CrossRef CAS; (g) P. J. Proteau, Bioorg. Chem., 2004, 32, 483 CrossRef CAS PubMed.
- (a) S. Rosa-Putra, A. Disch, J. M. Bravo and M. Rohmer, FEMS Microbiol. Lett., 1998, 164, 169–175 CrossRef PubMed; (b) M. Rohmer, Nat. Prod. Rep., 1999, 16, 565–574 RSC; (c) M. Rohmer, C. Grosdemange-Billiard, M. Seemann and D. Tritsch, Curr. Opin. Invest. Drugs, 2004, 5, 154 CAS; (d) M. Rohmer, Pure Appl. Chem., 2007, 79, 739 CrossRef CAS.
- T. Kuzuyama, T. Shimizu, S. Takahashi and H. Seto, Tetrahedron Lett., 1998, 39, 7913 CrossRef CAS.
- H. Jomaa, J. Wiesner, S. Sanderbrand, B. Altincicek, C. Weidemeyer, M. Hintz, I. Türbachova, M. Eberl, J. Zeidler, H. K. Lichtenthaler, D. Soldati and E. Beck, Science, 1999, 285, 1573 CrossRef CAS.
- B. Lell, R. Ruangweerayut, J. Wiesner, M. A. Missinou, A. Schindler, T. Baranek, M. Hintz, D. Hutchinson, H. Jomaa and P. G. Kremsner, Antimicrob. Agents Chemother., 2003, 47, 735 CrossRef CAS.
- J. Wiesner and H. Jomaa, Curr. Drug Targets, 2007, 8, 3 CrossRef CAS.
- (a) S. Borrmann, A. A. Adegnika, P. B. Matsiegui, S. Issifou, A. Schindler, D. P. Mawili-Mboumba, T. Baranek, J. Wiesner, H. Joomaa and P. G. Kremsner, J. Infect. Dis., 2004, 189, 901 CrossRef CAS PubMed; (b) S. Borrmann, S. Issifou, G. Esser, A. A. Adegnika, M. Ramharter, P. B. Matsiegui, S. Oyakhirome, D. P. Mawili-Mboumba, M. A. Missinou, J. F. Kun, H. Joomaa and P. G. Kremsner, J. Infect. Dis., 2004, 190, 1534 CrossRef CAS PubMed.
- For recent papers on the synthesis of fosmidomycin analogues see: (a) C. T. Behrendt, A. Kunfermann, V. Illarionova, A. Matheeussen, M. K. Pein, T. Graëwert, J. Kaiser, A. Bacher, W. Eisenreich, B. Illarionov, M. Fischer, L. Maes, M. Groll and T. Kurz, J. Med. Chem., 2011, 54, 6796 CrossRef CAS PubMed; (b) M. Andaloussi, M. Lindh, C. Björkelid, S. Suresh, A. Wieckowska, H. Iyer, A. Karlén and M. Larhed, Bioorg. Med. Chem. Lett., 2011, 21, 5403 CrossRef CAS PubMed; (c) M. Andaloussi, L. M. Henriksson, A. Wieckowska, M. Lindh, C. Björkelid, A. M. Larsson, S. Suresh, H. Iyer, B. R. Srinivasa, T. Bergfors, T. Unge, S. L. Mowbray, M. Larhed, T. A. Jones and A. Karlén, J. Med. Chem., 2011, 54, 4964 CrossRef CAS PubMed; (d) L. Deng, J. Diao, P. Chen, V. Pujari, Y. Yao, G. Cheng, D. C. Crick, B. V. V. Prasad and Y. Song, J. Med. Chem., 2011, 54, 4721 CrossRef CAS PubMed; (e) S. Ponaire, C. Zinglé, D. Tritsch, C. Grosdemange-Billiard and M. Rohmer, Eur. J. Med. Chem., 2012, 51, 277 CrossRef CAS PubMed; (f) T. Bodill, A. C. Conibear, M. K. M. Mutorwa, J. L. Goble, G. L. Blatch, K. A. Lobb, R. Klein and P. T. Kaye, Bioorg. Med. Chem., 2013, 21, 4332 CrossRef CAS PubMed; (g) A. T. Nguyen-Trung, D. Tritsch, C. Grosdemange-Billiard and M. Rohmer, Bioorg. Med. Chem. Lett., 2013, 23, 1643 CrossRef CAS PubMed; (h) G. San Jose, E. R. Jackson, R. Uh, C. Johny, A. Haymond, L. Lundberg, C. Pinkham, K. Kehn-Hall, H. I. Boshoff, R. D. Couch and C. S. Dowd, MedChemComm, 2013, 1099 RSC; (i) A. M. Jansson, A. Więckowska, C. Björkelid, S. Yahiaoui, S. Sooriyaarachchi, M. Lindh, T. Bergfors, S. Dharavath, M. Desroses, S. Suresh, M. Andaloussi, R. Nikhil, S. Sreevalli, B. R. Srinivasa, M. Larhed, T. A. Jones, A. Karlén and S. L. Mowbray, J. Med. Chem., 2013, 56, 6190 CrossRef CAS PubMed; (j) S. Montel, C. Midrier, J.-N. Volle, R. Braun, K. Haaf, L. Willms, J.-L. Pirat and D. Virieux, Eur. J. Org. Chem., 2012, 3237–3248 CrossRef CAS.
- M. Fellermeier, K. Kis, S. Sagner, U. Maier, A. Bacher and M. H. Zenk, Tetrahedron Lett., 1999, 40, 2743 CrossRef CAS.
- Y. Kamuro, T. Kawai and T. Kakiushi, Fujisawa Pharmaceutical Co. Ltd, Eur. Pat., 0256785A2, 1988.
- A. Mac Sweeney, R. Lange, A. d'Arcy, A. Douangamath, J.-P. Surivet and C. Oefner, J. Mol. Biol., 2005, 345, 115 CrossRef CAS PubMed.
- (a) J. W. Munos, X. Pu, S. O. Mansoorabadi, H. J. Kim and H.-W. Liu, J. Am. Chem. Soc., 2009, 131, 2048 CrossRef CAS PubMed; (b) U. Wong and R. J. Cox, Angew. Chem., Int. Ed., 2007, 46, 4926 CrossRef CAS PubMed; (c) A. Mac Sweeney, R. Lange, R. P. M. Fernandes, H. Schulz, G. E. Dale, A. Douangamath, P. J. Proteau and C. Oefner, J. Mol. Biol., 2005, 345, 115 CrossRef CAS PubMed; (d) A. Argyrou and J. S. Blanchard, Biochemistry, 2004, 43, 4375 CrossRef CAS PubMed; (e) K. Reuter, S. Sanderbrand, H. Jomaa, J. Wiesner, I. Steinbrecher, E. Beck, M. Hintz, G. Klebe and M. T. Stubbs, J. Biol. Chem., 2002, 277, 5378 CrossRef CAS PubMed.
- (a) K. E. Pallett, J. P. Little, M. Sheekey and P. Veerasekaran, Pestic. Biochem. Physiol., 1998, 62, 113 CrossRef CAS; (b) F. Viviani, J. P. Little and K. E. Pallett, Pestic. Biochem. Physiol., 1998, 62, 125 CrossRef CAS.
- L. Deng, S. Sundriyal, V. Rubio, Z.-Z. Shi and Y. Song, J. Med. Chem., 2009, 52, 6539 CrossRef CAS PubMed.
- (a) P. Nikitina, L. G. Kuz'mina, V. P. Perevalov and I. I. Tkach, Tetrahedron, 2013, 69, 3249 CrossRef CAS PubMed; (b) L. Wolff, Justus Liebigs Ann. Chem., 1902, 325, 129 CrossRef; (c) J. Lifschitz, Chem. Ber., 1913, 46, 3233 CrossRef.
- T. Majid, C. R. Hopkins, B. Pedgrift and N. Collar, Tetrahedron Lett., 2004, 45, 2137 CrossRef CAS PubMed.
- R. Ortmann, J. Wiesner, K. Silber, G. Klebe, H. Jomaa and M. Schlitzer, Arch. Pharm., 2007, 340, 483 CrossRef CAS PubMed.
- M. Witschel, Bioorg. Med. Chem., 2009, 17, 4221 CrossRef CAS PubMed.
- S. S. Nikam, B. E. Komberg, D. R. Johnson and A. M. Doherty, Tetrahedron Lett., 1995, 36, 197 CrossRef CAS.
- B. L. Eriksen, P. Vedso, S. Morel and M. Begtrup, J. Org. Chem., 1998, 63, 12 CrossRef CAS PubMed.
- Crystal single structures were deposited at the Cambridge Crystallographic Data Centre and referenced with numbers: CCDC 959100 (2a); CCDC 959101 (2b) and CCDC 959102 (2d).†.
- C. E. Griffin and S. K. Kundu, J. Org. Chem., 1969, 1532 CrossRef CAS.
- P. Mitula and C. Wawrzeńczyk, ARKIVOC, 2012, 4, 216 CrossRef.
- G. Just, P. Potvin and G. H. Hakimelahi, Can. J. Chem., 1980, 58, 2780 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 959100–959102. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra00757c |
| This journal is © The Royal Society of Chemistry 2014 |