DOI:
10.1039/C4RA03384A
(Paper)
RSC Adv., 2014,
4, 23779-23789
Facile cyclization in the synthesis of highly fused diaza cyclooctanoid compounds using retrievable nano magnetite-supported sulfonic acid catalyst†
Received
14th April 2014
, Accepted 20th May 2014
First published on 20th May 2014
Abstract
Magnetically separable Fe3O4–SO3H nano particles have been prepared and established as an efficient catalyst for the synthesis of dihydrofuran fused cyclooctanoids via the dehydration reaction of dihydroxy cyclooctanoid heterocylces. Nanomagnetite, Fe3O4–SO3H was prepared using an impregnation protocol and fully characterized by XRD, FT-IR, TEM, and FE-SEM-EDS. The catalyst was recycled for five consecutive cycles with almost unaltered catalytic activity. The dehydration step was performed keeping the acid sensitive amide linkage intact under ambient temperature and milder reaction conditions. The efficacy of the methodology lies in the excellent yield of product formation within a short span of reaction time avoiding undesirable side products due to the substrate decomposition by ring rupture and tedious purification and work-up procedure.
Introduction
The functionalized cyclooctanes are frequently found in pharmacologically significant compounds and natural products. Because of their intriguing geometrical features and potential biological activities, the construction of cyclooctanoid frameworks has drawn significant attention from synthetic organic chemists for a long time.1,2 In many natural products eight-membered carbo and heterocycles are fused with one or more five- or six-membered rings. For examples, the cyclooctanoid fusicoauritone (1),3 aleurodiscal (2),4 Kopsia alkaloid specially lapidilectam (3),5 ophiobolin A (4),6 serotobenine (5)7 and deoxyisoaustamide (6)8 are natural compounds containing fused penta- and hexacyclic rings (Fig. 1). Extensive investigation on the above mentioned eight membered hetero or carbocycle family shows that they have broad spectrum of bioactivity against fungi, nematodes, bacteria, the Plasmodium falciparum and cultured B-16 melanoma cells.6,9,10 Marine-products cyclooctanoid (6) containing indole ring are presently considered as the affluent sources of active secondary metabolites (Fig. 1).8 Therefore, synthesis of cyclooctanoids fused with penta- and hexacyclic ring is very important. In recent years a variety of synthetic methodologies have been developed for the synthesis of fused cyclooctanoid rings which include olefin metathesis,11 cycloadditions,12 nucleophilic and electrophilic substitution reactions,13 a Mizoroki–Heck reaction,14 ring expansion reactions15 and other reactions.16 In our continuing endeavor to synthesize biologically potent heterocyclic compounds from ninhydrin,17 we wish to report herein a cyclization method for the synthesis of isoindole and benzofuran fused eight-membered spiro heterocycles following the Green Chemistry principles.
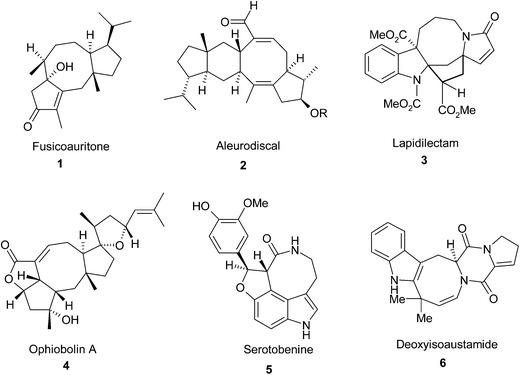 |
| | Fig. 1 Some important naturally occurring eight-membered heterocycles. | |
Catalysis is becoming a strategic field of science because it represents a new way to meet the challenges of energy and sustainability. In order to explore environmentally benign synthetic pathways, we need a catalyst system that not only shows high activity and selectivity (like a homogeneous system) but also can be separated and recovered easily after the reactions (like a heterogeneous system). In this respect nanocatalysts have recently been emerged as highly promising catalysts which mimic both homogeneous (high surface area, easily accessible) and heterogeneous (stable, easy to handle) catalyst systems. The main difficulty in the nanocatalyst mediated organic reaction is the separation and recovery of the catalyst through conventional filtration as it blocks the pores and valves of the filter papers. In this respect, the use of magnetically separable nanoparticles (MSNPs) such as iron oxide supported nano materials attributes excellent isolation and separation procedure from the reaction mixture by means of an external magnet because of their insolubility and paramagnetic nature. Moreover, they are inexpensive, easy to prepare, and most importantly easily recyclable for many times.18
Chlorosulfonic acid is a well-known strong organic acid, which is used as an active catalyst in organic syntheses. However, its direct application in the reaction mixture may cause the decomposition of the reactants and also derivation of several impurities. The best way to avoid the harmful effect of chlorosulfonic acid is by supporting it on a solid surface.19 In this regard, magnetically retrievable Fe3O4@SO3H emerges as an efficient and sustainable acid catalyst for many organic reactions.19,20 Here, we have exploited this acid catalyst for the construction of dihydrofuran ring in cyclooctanoid compounds (±)-2a–r/2a′–r′ starting from dihydroxy cyclooctanoid heterocylces (±)-1a–r/1a′–r′ (Scheme 1).
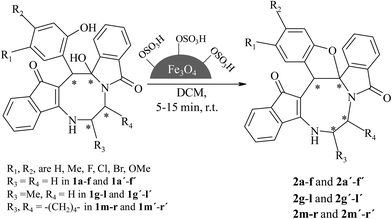 |
| | Scheme 1 Optimized reaction condition for the formation of fused cyclooctanoids. | |
Results and discussion
In our initial study, we attempted to optimize the reaction conditions for the formation of (±)-2a/2a′ (R1 = Me, R2 = R3 = R4 = H) from (±)-1a/1a′ (R1 = Me, R2 = R3 = R4 = H) in presence of different solvents, catalysts, solid supports at different temperature and time (Scheme 1 and Table 1). From literature survey, it is known that, to carry out dehydration reaction in Dean–Stark apparatus, benzene is the most commonly used solvent. So at first, we tried to carry out the dehydration in (±)-1a/1a′ using benzene as the solvent. Unfortunately, no product was isolated after prolonged reflux (Table 1, entry 1). Then we tried to carry out the dehydration and subsequent intra-molecular etheration reaction under acid catalyzed condition in presence of several organo-acid catalysts such as lactic acid, formic acid, citric acid and acetic acid in benzene (Table 1, entries 2–5). In all the cases, trace amount of desired product was isolated through column chromatography and the structure of the product 2a and 2a′ was confirmed by IR, 1H and 13C NMR spectroscopy, elemental analysis, and X-ray diffraction studies. To obtain the desired product with satisfactory yield, we employed higher acidic condition in the reaction, using acetic acid as solvent (Table 1, entry 6). But yield of the product (±)-2a/2a′ was still very low (30%) and unsatisfactory. When stronger Brønsted acid catalyst PTSA was used in benzene, moderate yield (48%) of the desired product was obtained (Table 1, entry 7), which increased further (65–68%) in dichloromethane (DCM) and dimethylformamide (DMF) solvent because of better solubility of the starting material (Table 1, entries 8 and 9). Since the yields are comparable in these two solvents, DCM was chosen as the best solvent for our reaction because of operational simplicity and easier work-up procedure. It was also clear from these studies that, –SO3H functional group is acting as a better activating agent compared to –COOH group. So next, we employed several solid supports bearing –SO3H group such as homogeneous polymeric acid–surfactant combined catalyst PEG–SO3H and heterogeneous solid acid supports MSA and SSA as catalyst in DCM (Table 1, entries 11–13). Moderate yield of the product (±)-2a/2a′ under reduced reaction time and temperature in presence of SSA (Table 1, entry 13) incited us for further investigation on the optimization of the reaction condition using magnetic nano-catalysts bearing –SO3H group on the surface. In this regard, two nano-catalysts Fe3O4–SO3H and γ-Fe3O4@SiO2–SO3H (ref. 21 and 22) were considered since they have interesting structural features, high level of catalytic activity for large surface area and facile separation procedure using external magnets (Table 1, entries 14–20). Gratifyingly, significant increase in the product yield (92%) was obtained with drastic diminution of required temperature (30 °C) and reaction time (10 min) on employing nano-magnetite Fe3O4–SO3H (50 mg) in DCM (Table 1, entry 18) after careful tuning of catalyst load (20–150 mg) under same reaction condition (Table 1, entries 14–19). Interestingly, comparable yield of (±)-2a/2a′ was observed using magnetically retrievable γ-Fe3O4@SiO2–SO3H as catalyst (Table 1, entry 20). Since nano-magnetite Fe3O4–SO3H is easier to prepare compared to γ-Fe3O4@SiO2–SO3H, the former was chosen as the most suitable catalyst for the present reaction.
Table 1 Optimization studies for etheration reaction in (±)-1a/1a′ to form (±)-2a/2a′
| Entry |
Catalyst |
Solvent |
Catalyst load |
Temperature (°C) |
Time |
Yielda (%) |
| Isolated yields. |
| 1 |
__ |
Benzene |
__ |
80 |
24 h |
__ |
| 2 |
Lactic acid |
Benzene |
20 mol% |
80 |
24 h |
10 |
| 3 |
Formic acid |
Benzene |
20 mol% |
80 |
24 h |
8 |
| 4 |
Citric acid |
Benzene |
20 mol% |
80 |
24 h |
10 |
| 5 |
Acetic acid |
Benzene |
20 mol% |
80 |
24 h |
10 |
| 6 |
__ |
Acetic acid |
__ |
80 |
24 h |
30 |
| 7 |
PTSA |
Benzene |
20 mol% |
80 |
24 h |
48 |
| 8 |
PTSA |
DCM |
20 mol% |
40 |
24 h |
65 |
| 9 |
PTSA |
DMF |
20 mol% |
80 |
24 h |
68 |
| 10 |
PTSA |
H2O |
20 mol% |
80 |
24 h |
45 |
| 11 |
PEG–SO3H |
DCM |
500 mg |
40 |
24 h |
35 |
| 12 |
Melamine sulphonic acid (MSA) |
DCM |
500 mg |
40 |
24 h |
27 |
| 13 |
Silica sulfuric acid (SSA) |
DCM |
500 mg |
40 |
3 h |
47 |
| 14 |
Fe3O4–SO3H |
DCM |
100 mg |
40 |
10 min |
72 |
| 15 |
Fe3O4–SO3H |
DCM |
100 mg |
30 |
10 min |
72 |
| 16 |
Fe3O4–SO3H |
DCM |
150 mg |
30 |
10 min |
71 |
| 17 |
Fe3O4–SO3H |
DCM |
75 mg |
30 |
10 min |
80 |
| 18 |
Fe3O4–SO3H |
DCM |
50 mg |
30 |
10 min |
92 |
| 19 |
Fe3O4–SO3H |
DCM |
20 mg |
30 |
10 min |
85 |
| 20 |
γ-Fe3O4@SiO2–SO3H |
DCM |
50 mg |
30 |
10 min |
92 |
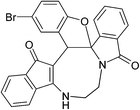
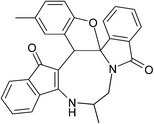
Having prepared (±)-2a/2a′ successfully, we decided to explore the scope and generality of this reaction in the synthesis of other analogues. At first, racemic mixtures of diaza-cyclooctanoid derivatives (±)-1a–r/1a′–r′ have been prepared from ninhydrin following the reported procedure.17b The diaza-cyclooctanoid compounds (±)-1a–r/1a′–r′ derived from different aliphatic 1,2-diamines (1,2-ethylenediamine, (±)-1,2-propylenediamine and (±)-trans-1,2-cyclohexanediamine) were treated with Fe3O4–SO3H under optimized reaction condition (Table 1, entry 18) to get fused eight membered heterocycles. As evident from Table 2, etheration reaction proceeded well with various electron-withdrawing and donating phenols and 1,2-diamine derivatives (Scheme 1 and Table 2). In all the cases reaction time is short (10 min) and yield of the product formation is high (Table 2). The isolated yellow colored solid products (±)-2a–r/2a′–r′ are fully characterized by 1H and 13C NMR spectroscopy. When diaza-cyclooctanoids (±)-(R,S)-1a–f/(S,R)-1a′–f′ cyclized in presence of Fe3O4–SO3H (±)-(R,S)-2a–f/(S,R)-2a′–f′ were formed which is established by X-ray crystal data analysis (Fig. 2). The other possible diastereomer (R,R or S,S) is not formed in the reactions. Therefore, retention of configuration takes place at the chiral centre adjacent to amide nitrogen (Scheme 2). The same reaction was also carried out for (±)-(R,S,S)-1g–l/(S,R,R)-1g′–l′, as predicted from the above results reaction passed through retention of configuration to furnish (±)-(R,S,S)-2g–l/(S,R,R)-2g′–l′ (Scheme 2 and Fig. 3). Then etheration reaction was carried out with compounds having four stereo-centre (±)-(R,S,S,S)-1m–r/(S,R,R,R)-1m′–r′. As expected, products (±)-(R,S,S,S)-2m–r/(S,R,R,R)-2m′–r′ were isolated from the reaction mixture which is established by X-ray crystal structure analysis (Fig. 4). So it is clear from the above information that the cyclization reaction is enantioselective.
Table 2 Scope of highly fused cyclooctanoid rings syntheses
 |
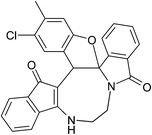 |
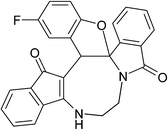 |
| (±)-2a/2a′ (92%) |
(±)-2b/2b′ (90%) |
(±)-2c/2c′ (89%) |
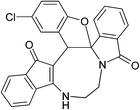 |
 |
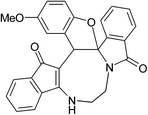 |
| (±)-2d/2d′ (91%) |
(±)-2e/2e′ (92%) |
(±)-2f/2f′ (94%) |
 |
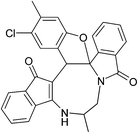 |
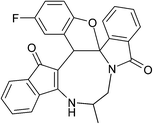 |
| (±)-2g/2g′ (89%) |
(±)-2h/2h′ (86%) |
(±)-2i/2i′ (84%) |
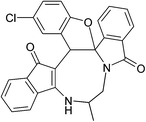 |
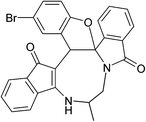 |
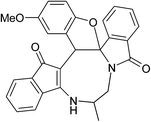 |
| (±)-2j/2j′ (85%) |
(±)-2k/2k′ (88%) |
(±)-2l/2l′ (92%) |
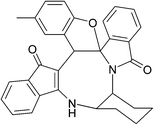 |
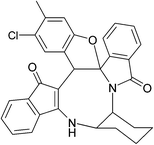 |
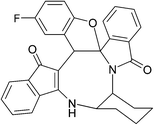 |
| (±)-2m/2m′ (85%) |
(±)-2n/2n′ (81%) |
(±)-2o/2o′ (78%) |
 |
 |
 |
| (±)-2p/2p′ (80%) |
(±)-2q/2q′ (83%) |
(±)-2r/2r′ (89%) |
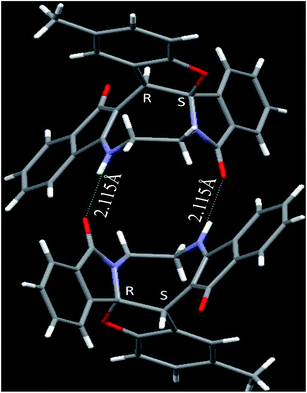 |
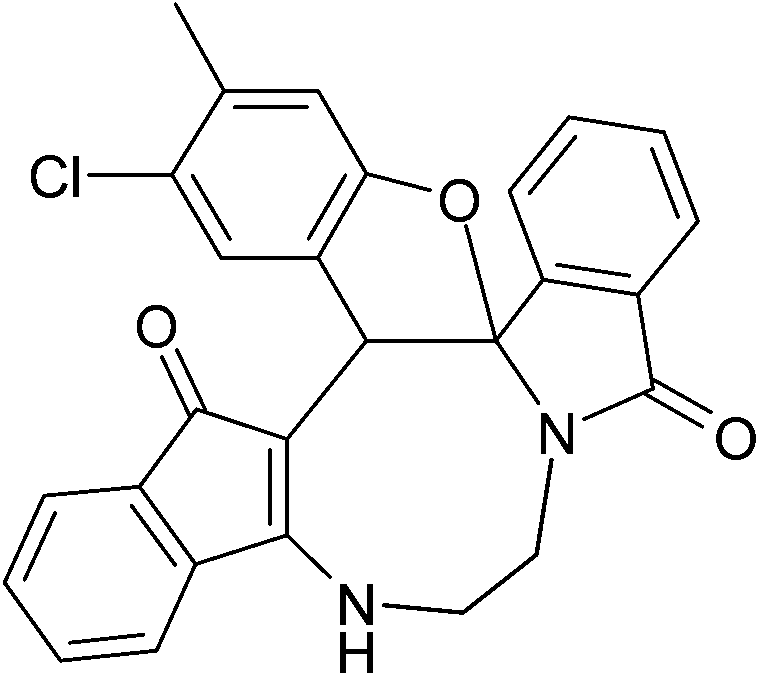
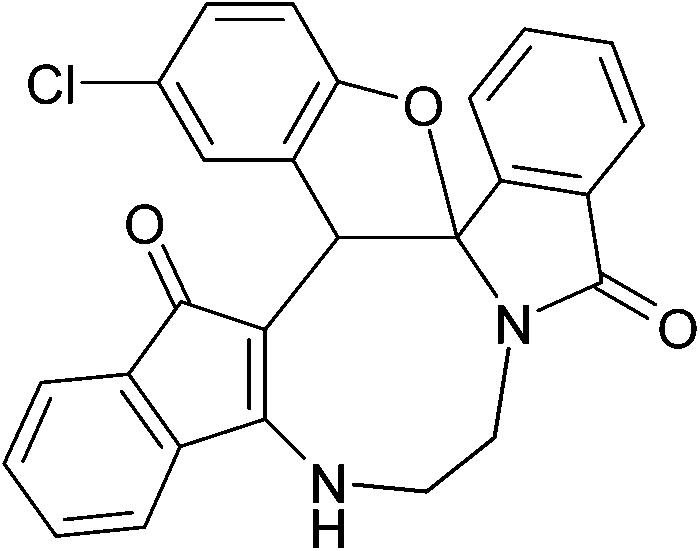
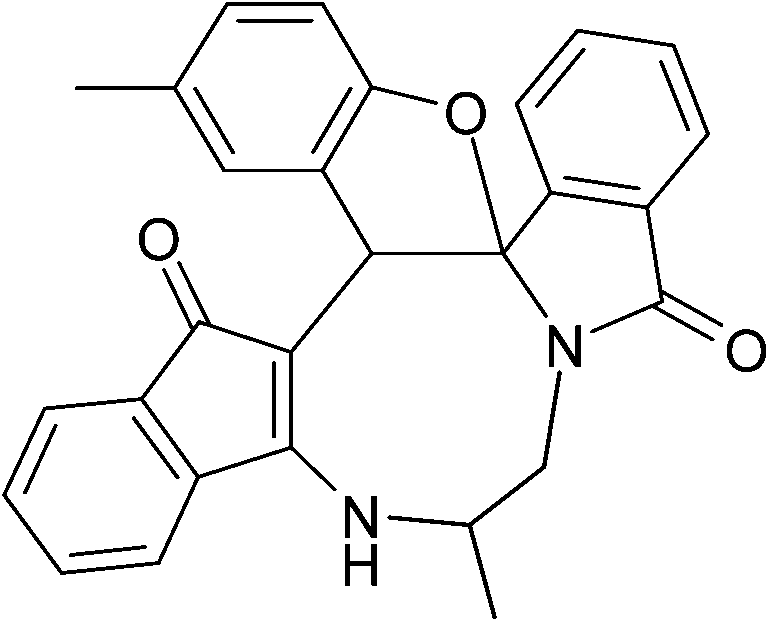
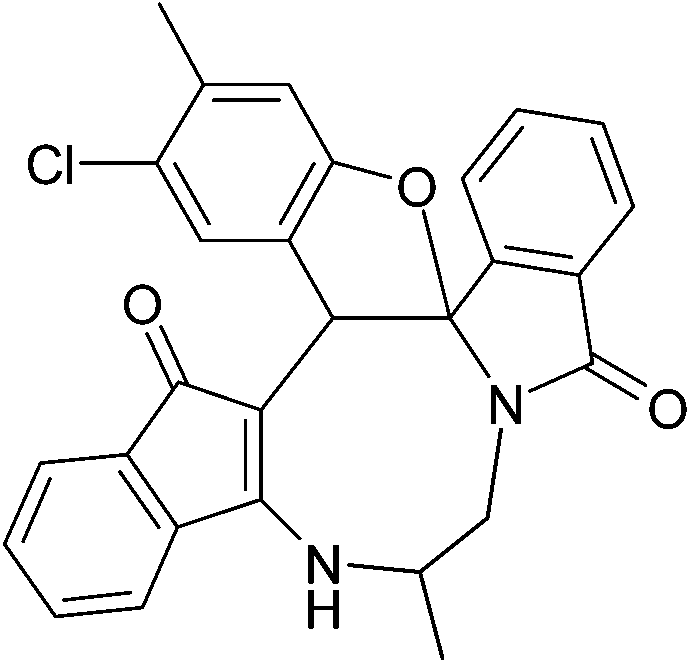
| | Fig. 2 Crystal structure of compound 2a and 2a′. Color code: red, oxygen; blue, nitrogen; grey, carbon; white, hydrogen.† | |
 |
| | Scheme 2 Synthesis of isoindole fused cyclooctanoid rings as a racemic mixture. | |
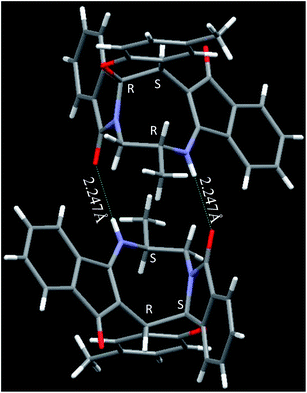 |
| | Fig. 3 Crystal structure of compound 2g and 2g′. Color code: red, oxygen; blue, nitrogen; green, chlorine; large white, carbon; small white, hydrogen.† | |
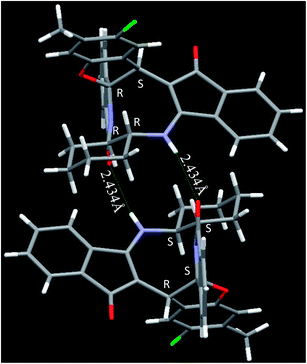 |
| | Fig. 4 Crystal structure of compound 2n and 2n′. Color code: red, oxygen; blue, nitrogen; green, chlorine; large white, carbon; small white, hydrogen.† | |
A possible mechanism for the formation of spirofuran fused eight-membered heterocycles 2 from dihydroxy eight-membered heterocycles 1 is depicted in Scheme 3. The initially formed hydrogen bond between aliphatic hydroxyl and –SO3H group provokes the breaking of C–O bond to generate dehydrated cationic intermediate 3. Finally, intramolecular nucleophilic attack of hydroxyl group of phenol to the imine carbon produces the oxonium intermediate 4 which subsequently loses proton to form furan ring fused eight-membered heterocycles 2. During the intramolecular nucleophilic attack via S1N process, the phenolic hydroxyl group attacks from the same face of the double bond where the aromatic phenols are aligned, as a result retention of configuration takes place at the stereogenic centre.
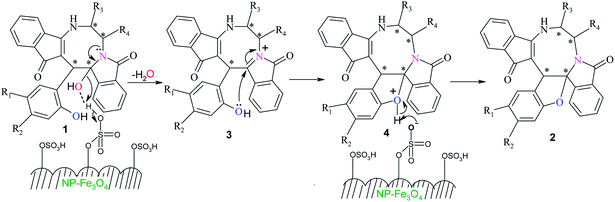 |
| | Scheme 3 Plausible mechanism for the formation of furan ring using NP Fe3O4–SO3H as catalyst. | |
The crystal structure analysis of (±)-2a/2a′, (±)-2g/2g′ and (±)-2n/2n′ reveals that in the crystalline state two enantiomeric molecules are locked in antiparallel fashion stabilized by two intermolecular hydrogen bonds between –NH– and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups (Fig. 2–4). The hydrogen bonding distance increases with the increase of the bulkiness of the substituents on 1,2-diamine moieties. In all the cases the central diazacyclooctane rings adopt a tub or boat like conformation rather than crown or boat–chair conformation (Fig. 2–4). The substituents around the diazacyclooctane rings are alternatively in up and down orientation due to steric reasons. In (R,S,S,S)-2n and (S,R,R,R)-2n′ the central boat diazacyclooctane ring is further fused with chair cyclohexane ring (Fig. 4).
O groups (Fig. 2–4). The hydrogen bonding distance increases with the increase of the bulkiness of the substituents on 1,2-diamine moieties. In all the cases the central diazacyclooctane rings adopt a tub or boat like conformation rather than crown or boat–chair conformation (Fig. 2–4). The substituents around the diazacyclooctane rings are alternatively in up and down orientation due to steric reasons. In (R,S,S,S)-2n and (S,R,R,R)-2n′ the central boat diazacyclooctane ring is further fused with chair cyclohexane ring (Fig. 4).
The reusability test of Fe3O4–SO3H nanoparticles was carried out under optimized conditions by stirring (±)-1a/1a′ in DCM medium (Table 1, entry 18). When the magnetic stirring of the reaction mixture was discontinued, the magnetic Fe3O4–SO3H MNPs were adsorbed on the surface of the magnetic stirring bar and separated easily from the reaction mixture without filtration. Then Fe3O4–SO3H MNPs catalyst was washed three times with ethanol (3 × 5 mL), dried at room temperature under vacuum to eliminate residual solvents and used for the next cycle. The dehydration reaction of (±)-1a/1a′ was performed with recovered catalyst upto five times without any significant loss of the catalytic activity (Fig. 5).
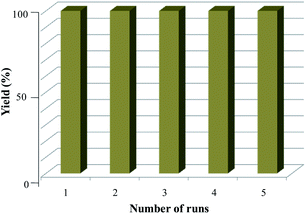 |
| | Fig. 5 Reusability of Fe3O4–SO3H MNPs for the formation of densely fused cyclooctanoid. | |
Powder XRD study was carried out to confirm the crystalline nature of as-synthesized iron oxide nanoparticles. The XRD pattern of Fe3O4 and Fe3O4–SO3H NPs are displayed in Fig. 6a and b respectively. The diffraction peaks from the (220), (311), (400), (422) and (440) lattice planes are observed at 2θ = 32.9°, 35.36°, 44.9°, 53.7° and 62.35° respectively (Fig. 6a). The peaks in the respective XRD patterns are equivalent to the Bragg reflections of face centered cubic (FCC) iron oxide and confirm both the identity and purity of the sample. Similar pattern of peaks is also obtained in Fe3O4–SO3H NPs XRD spectra (Fig. 6b), with slight alterations in the nature of peaks. The presence of sulfonic acid group coated on the surface of Fe3O4 NPs might cause slight changes in the XRD spectra of Fe3O4–SO3H NPs.19
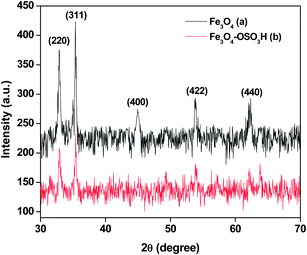 |
| | Fig. 6 XRD spectra of (a) Fe3O4 and (b) Nanocat-Fe3O4–SO3H MNPs. | |
Field emission scanning electron microscopy (FE-SEM) at operating voltage of 5.0 kV is employed to know the morphology and size of Nanocat-Fe3O4–SO3H nanoparticles (Fig. 7a). It is clear from the image in Fig. 7a that the spherical shaped Nanocat-Fe3O4–SO3H nanoparticles have diameter in the range of ∼23 to ∼43 nm. Energy dispersive spectroscopy (EDS) analysis of the prepared Nanocat-Fe3O4–SO3H nano-particle (sulfur peak) confirms the incorporation of –SO3H group on the surface which comes from the reaction of Fe3O4 NPs with chlorosulfonic acid (Fig. 7b).
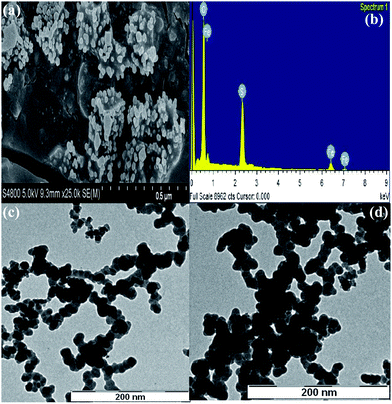 |
| | Fig. 7 (a) SEM image of Fe3O4–SO3H NPs; (b) SEM-EDS of Nanocat-Fe3O4–SO3H; and TEM images of Fe3O4–SO3H NPs (c) before use in reaction and (d) after five times applications. | |
In order to confirm the formation of Nanocat-Fe3O4–SO3H nanoparticles and to inspect its morphology, high-resolution transmission electron microscopic (HRTEM) analysis is performed (Fig. 7c and d). TEM analysis reveals that the prepared nanoparticles are uniform in size and mostly spherical in shape and the size of the nanoparticles is in the range of ∼14 to ∼23 nm. The characterization of the nano-Fe3O4–SO3H by TEM images, before (Fig. 7c) and after five times applications (Fig. 7d) shows the unaltered particle size.
Conclusions
In summary, we have successfully demonstrated a robust and magnetically recoverable Fe3O4–SO3H MNPs catalyzed dehydration reaction for the formation of spirofuran fused eight-membered heterocycles. These potentially bioactivity cyclooctanoid heterocylces have intriguing structural and geometrical features. Most importantly these alkaloids analogous are readily prepared from inexpensive starting materials in moderate to high yield under mild conditions in air.17b In the final step of the synthesis the dehydration reaction for the formation of dihydrofuran ring using Fe3O4–SO3H MNPs was very straightforward, efficient and highly economical. In addition, the employment of iron oxides as acid catalysts is also more environmentally acceptable and safer in terms of toxicity compared to other transition metal catalysts.
Experimental
Solvents and chemicals were purchased from commercial suppliers and used without further purification. Catalyst and starting materials were prepared according to reported procedure. Melting points were measured in open capillary tubes and were uncorrected. Perkin-Elmer 782 spectrophotometer was used for IR spectra. 1H (300 MHz) and 13C NMR (75 MHz) spectra were performed on Bruker instrument (300 MHz) in DMSO-d6. Elemental analyses (C, H and N) were recorded using Perkin-Elmer 240C elemental analyzer. The X-ray diffraction data for crystallized compounds were collected with MoKα radiation at 296 K using the Bruker APEX-II CCD System. The crystals were positioned at 50 mm from the CCD. Frames were measured with a counting time of 5 s. Data analyses were carried out with the Bruker APEX2 and Bruker SAINT program. The structures were solved using direct methods with the Shelxs97 program (Sheldrick, 2008). The morphological analysis of the resultant nanoparticles was confirmed by TEM. The sample suspension is drop casted on a carbon coated copper grid and the excess solution was removed by tissue paper and allowed to air dry at room temperature overnight. TEM study was monitored on a HRTEM, JEOL JEM 2010 at an accelerating voltage of 200 kV and fitted with a CCD camera. The crystallinity of synthesized Fe3O4 and Fe3O4–OSO3H nanoparticles was determined and confirmed by XRD analysis. The diffractogram was documented from PANalytical, XPERT-PRO diffractrometer using Cuα (λ= 1.54060) as X-ray source. Hitachi S-4800 Field Emission Scanning Electron Microscope (Hitachi S-4800 FE-SEM) operating voltage 5.0 kV is used for SEM.
Preparation of Fe3O4 nano particle19
FeCl3·6H2O (8.1 g) and urea (5.4 g) were stirred in 300 mL double distilled water at 85 to 90 °C for 2 h. The resultant brown colored reaction mixture was cooled to room temperature and FeSO4·7H2O (4.2 g) was added into it. The pH of the resultant solution was maintained at 10 by addition of 0.1 M NaOH solution. The molar ratio of Fe(III) to Fe(II) in the above system was nearly 2.0. The obtained hydroxides were kept under ultrasonication in the sealed flask at room temperature for 30 min. After ageing for 5 h, the obtained black powder of Fe3O4 nano particles was washed several times with distilled water, and dried under vacuum.
Preparation of Fe3O4–SO3H catalyst19
In a two neck round bottom flask, one neck was equipped with a dropping funnel and other neck is fixed with water vacuum to suck HCl gas generated during the reaction. Magnetite, Fe3O4 nano particles (3.0 g) was then poured into round bottom flask and neat chlorosulfonic acid was added (1.0 mL) drop by drop over a period of 10 min at room temperature. HCl gas generated immediately from the reaction was removed by suction. After complete addition of chlorosulfonic acid, the mixture was stirred vigorously for 30 min and solid brown magnetic sulfonic acid, Nanocat-Fe–SO3H (3.45 g) was collected.
Preparation of γ-Fe3O4@SiO2–SO3H catalyst21,22
Nano-Fe3O4@SiO2 was synthesized according to the previously published literature method.22 Magnetic Fe3O4 nano particles (1.0 g) were initially diluted via the sequential addition of water (20 mL), ethanol (60 mL), and concentrated aqueous ammonia (1.5 mL, 28 wt%). The resulting dispersion was then homogenized by ultrasonic vibration in a water bath. A solution of TEOS (tetraethyl orthosilicate) (0.45 mL) in ethanol (10 mL) was then added to the dispersion in a drop-wise manner under continuous mechanical stirring. Following a 12 h period of stirring, the resulting γ-Fe3O4@SiO2 nano particles were collected by magnetic separation and washed three times with ethanol.
Chlorosulfonic acid (0.5 g, 4.5 mmol) was added in a drop-wise manner to a cooled (ice-bath) solution of γ-Fe3O4@SiO2 (1.0 g) in n-hexane (5 mL) over a period of 2 h. Upon completion of the addition, the mixture was stirred for another 3 h until complete dissipation of HCl occurred from the reaction vessel. The resulting MNPs were separated using an external magnet and washed with methanol before being dried in an oven at 60 °C to give γ-Fe3O4@SiO2–SO3H as a brown powder.
General synthesis of compounds (±)-2a–r/2a′–r′
A mixture of dihydroxy cyclooctanoid heterocylces (±)-1a–r/1a′–r′ (2.0 mmol) and Fe3O4–SO3H (50 mg) in dichloromethane (10.0 mL) was stirred for 10 min (as mentioned in Table 1). When the magnetic stirring of the reaction mixture was discontinued, the magnetic Fe3O4–SO3H MNPs were adsorbed on the surface of the magnetic stirring bar and easily separated from the reaction mixture without filtration. Then dichloromethane was evaporated under vacuum and reaction mass was sonicated with ethylacetate (3 × 10 mL). The combined ethylacetate was evaporated to get yellow solids of (±)-2a–r/2a′–r′. Yellow solid (±)-2a–r/2a′–r′ was further purified through crystallization from DMSO solvent.
Spectral data for synthesized compounds
(±)-2a/2a′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3295, 1698 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 8.67 (t, J = 5.7 Hz, 1H), 7.80 (d, J = 7.2 Hz, 1H), 7.59 (t, J = 6.6 Hz, 1H), 7.44–7.40 (m, 3H), 7.28 (t, J = 7.2 Hz, 1H), 7.20 (t, J = 6.9 Hz, 1H), 7.07 (d, J = 6.9 Hz, 1H), 6.95 (d, J = 7.8 Hz, 1H), 6.83–6.77 (m, 2H), 5.41 (s, 1H), 4.15–4.14 (m, 1H), 3.19–3.16 (m, 1H), 2.85–2.81 (m, 2H), 2.12 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δH 191.0, 167.1, 164.4, 155.0, 145.0, 138.7, 134.3, 133.8, 133.7, 131.9, 131.5, 130.8, 130.6, 130.5, 129.3, 124.1, 123.4, 122.8, 120.1, 118.9, 110.0, 102.5, 95.2, 45.0, 40.5, 39.6, 20.8; C27H20N2O3 (420.47): calcd C 77.13, H 4.79, N 6.66; found C 77.11, H 4.74, N 6.61.
(±)-2b/2b′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3293, 1701 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 8.82 (t, J = 5.4 Hz, 1H), 7.94 (d, J = 7.5 Hz, 1H), 7.73–7.68 (m, 1H), 7.57–7.51 (m, 3H), 7.39 (t, J = 7.5 Hz, 1H), 7.30 (t, J = 7.5 Hz, 1H), 7.17 (d, J = 7.2 Hz, 1H), 7.08 (d, J = 4.8 Hz, 2H), 5.54 (s, 1H), 4.27–4.25 (m, 1H), 3.33–3.25 (m, 1H), 2.97–2.86 (m, 2H), 2.32 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δH 190.5, 166.7, 164.1, 155.5, 144.2, 138.2, 135.9, 133.9, 133.4, 133.1, 131.2, 130.6, 130.4, 130.2, 126.2, 123.4, 123.2, 122.5, 119.8, 118.6, 112.6, 102.6, 94.1, 44.3, 40.6, 39.6, 20.1; C27H19ClN2O3 (452.92): calcd C 71.29, H 4.21, N 6.16; found C 71.41, H 4.14, N 6.10.
(±)-2c/2c′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3286, 1706 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 8.76 (t, J = 5.7 Hz, 1H), 7.91 (d, J = 7.8 Hz, 1H), 7.63 (t, J = 6.9 Hz, 1H), 7.52–7.44 (m, 3H), 7.32 (t, J = 7.5 Hz, 1H), 7.23 (t, J = 6.9 Hz, 1H), 7.11 (d, J = 6.9 Hz, 1H), 7.02–6.99 (m, 2H), 6.89 (d, J = 7.8 Hz, 1H), 5.53 (s, 1H), 4.21–4.19 (m, 1H), 3.29–3.24 (m, 1H), 2.91–2.87 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δH 190.5, 166.6, 164.1, 159.6, 156.5, 152.7, 144.2, 138.2, 135.3, 135.2, 133.9, 133.3, 131.1, 130.6, 130.4, 130.2, 123.2, 122.5, 119.8, 118.6, 115.0, 114.7, 110.8, 110.6, 102.7, 94.0, 44.7, 40.6, 39.6; C26H17FN2O3 (424.44): calcd C 73.58, H 4.04, N 6.60; found C 73.72, H 3.95, N 6.53.
(±)-2d/2d′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3306, 1699 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 8.86 (br s, 1H), 7.98 (d, J = 7.5 Hz, 1H), 7.71 (t, J = 6.3 Hz, 1H), 7.59–7.54 (m, 3H), 7.41–7.30 (m, 3H), 7.19–7.06 (m, 3H), 5.59 (s, 1H), 4.32–4.25 (m, 1H), 3.36–3.32 (m, 1H), 2.97–2.93 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δH 190.9, 167.0, 164.5, 155.8, 144.4, 138.6, 136.2, 134.3, 133.8, 131.5, 131.0, 130.7, 130.6, 128.9, 126.5, 123.7, 123.6, 122.9, 120.2, 119.0, 111.9, 103.0, 94.3, 44.8, 40.6, 39.6; C26H17ClN2O3 (440.89): calcd C 70.83, H 3.89, N 6.35; found C 70.70, H 3.81, N 6.29.
(±)-2e/2e′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3298, 1704 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 8.79 (t, J = 5.7 Hz, 1H), 7.92 (d, J = 7.5 Hz, 1H), 7.66 (t, J = 6.6 Hz, 1H), 7.53–7.46 (m, 3H), 7.40–7.23 (m, 3H), 7.17–7.12 (m, 2H), 6.99 (d, J = 8.7 Hz, 1H), 5.55 (s, 1H), 4.22–4.20 (m, 1H), 3.27–3.26 (m, 1H), 2.92–2.89 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δH 190.9, 167.1, 164.5, 156.3, 144.5, 138.6, 136.7, 134.3, 133.8, 131.8, 131.5, 131.0, 130.8, 130.6, 126.5, 123.6, 122.9, 120.2, 119.0, 114.1, 112.6, 102.9, 94.4, 44.8, 40.6, 39.6; C26H17BrN2O3 (485.34): calcd C 64.34, H 3.53, N 5.77; found C 64.49, H 3.47, N 5.70.
(±)-2f/2f′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3301, 1700 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 8.77 (t, J = 5.4 Hz, 1H), 7.89 (d, J = 7.5 Hz, 1H), 7.67 (t, J = 6.9 Hz, 1H), 7.55–7.47 (m, 3H), 7.36 (t, J = 7.5 Hz, 1H), 7.27 (t, J = 6.9 Hz, 1H), 7.15 (d, J = 6.9 Hz, 1H), 6.95 (d, J = 8.4 Hz, 1H), 6.78 (d, J = 8.4 Hz, 1H), 6.61 (s, 1H), 5.53 (s, 1H), 4.25–4.24 (m, 1H), 3.64 (s, 3H), 3.29–3.27 (m, 1H), 2.95–2.91 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δH 190.6, 166.7, 164.1, 155.3, 150.5, 144.5, 138.3, 134.4, 134.0, 133.2, 131.1, 130.5, 130.4, 130.2, 123.1, 122.4, 119.7, 118.6, 114.1, 110.4, 108.8, 102.4, 94.5, 55.8, 45.1, 40.6, 39.6; C27H20N2O4 (436.47): calcd C 74.30, H 4.62, N 6.42; found C 74.42, H 4.56, N 6.47.
(±)-2g/2g′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3299, 1698 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 8.23 (d, J = 6.9 Hz, 1H), 7.91 (d, J = 7.5 Hz, 1H), 7.76–7.69 (m, 2H), 7.60–7.52 (m, 2H), 7.43–7.29 (m, 2H), 7.19 (d, J = 6.9 Hz, 1H), 7.07 (d, J = 7.8 Hz, 1H), 6.95–6.89 (m, 2H), 5.55 (s, 1H), 3.96 (t, J = 13.2 Hz, 1H), 3.37–3.35 (m, 1H), 3.01–2.95 (dd, J1 = 3.9 Hz, J2 = 14.1 Hz, 1H), 2.25 (s, 3H), 1.12 (d, J = 11.7 Hz, 3H); 13C NMR (75 MHz, DMSO-d6) δH 190.9, 166.4, 163.3, 154.6, 144.5, 138.4, 133.8, 133.3, 133.0, 131.5, 131.1, 130.5, 130.4, 131.1, 128.9, 123.7, 123.0, 122.5, 119.7, 119.2, 109.5, 102.2, 95.2, 47.5, 45.6, 44.9, 20.4, 17.1; C28H22N2O3 (434.50): calcd C 77.40, H 5.10, N 6.45; found C 77.27, H 5.02, N 6.40.
(±)-2h/2h′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3280, 1694 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 8.28 (d, J = 6.3 Hz, 1H), 7.95–7.09 (m, 10H), 5.57 (s, 1H), 3.95 (t, J = 13.5 Hz, 1H), 3.40–3.37 (m, 1H), 3.02–2.98 (dd, J1 = 2.7 Hz, J2 = 13.2 Hz, 1H), 2.10 (s, 3H), 1.17 (d, J = 15.6 Hz, 3H); 13C NMR (75 MHz, DMSO-d6) δH 190.8, 166.4, 163.4, 155.5, 144.1, 138.4, 135.8, 133.8, 133.4, 132.6, 131.1, 130.6, 130.3, 130.2, 126.2, 123.3, 123.2, 122.6, 119.8, 119.3, 112.4, 102.6, 94.5, 47.7, 45.5, 44.6, 20.1, 17.1; C28H21ClN2O3 (468.94): calcd C 71.72, H 4.51, N 5.97; found C 71.87, H 4.44, N 5.91.
(±)-2i/2i′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3291, 1697 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 8.27 (d, J = 5.7 Hz, 1H), 7.95 (d, J = 5.7 Hz, 1H), 7.73–6.93 (m, 10H), 5.60 (s, 1H), 3.96 (t, J = 13.2 Hz, 1H), 3.35–3.33 (m, 1H), 2.97 (d, J = 12.9 Hz, 1H), 1.15 (d, J = 15.0, 3H); 13C NMR (75 MHz, DMSO-d6) δH 190.7, 166.3, 163.5, 159.6, 156.4, 152.7, 144.0, 138.3, 134.9, 134.8, 133.8, 133.3, 131.1, 130.6, 130.3, 130.2, 123.2, 122.5, 119.8, 119.3, 115.0, 114.7, 110.8, 110.5, 102.8, 94.3, 47.6, 45.5, 45.1, 17.0; C27H19FN2O3 (438.46): calcd C 73.96, H 4.37, N 6.39; found C 73.86, H 4.32, N 6.33.
(±)-2j/2j′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3296, 1694 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 8.26 (d, J = 5.7 Hz, 1H), 7.92 (d, J = 7.2 Hz, 1H), 7.73–7.01 (m, 10H), 5.57 (s, 1H), 3.93 (t, J = 12.9 Hz, 1H), 3.37–3.33 (m, 1H), 2.95 (d, J = 11.1 Hz, 1H), 1.12 (d, J = 15.0 Hz, 3H); 13C NMR (75 MHz, DMSO-d6) δH 190.8, 166.4, 163.5, 155.5, 144.0, 138.4, 135.4, 133.8, 133.4, 131.1, 130.7, 130.3, 130.2, 128.5, 126.1, 123.3, 123.2, 122.6, 119.8, 119.3, 111.5, 102.7, 94.3, 47.6, 45.5, 44.7, 17.1; C27H19ClN2O3 (454.92): calcd C 71.29, H 4.21, N 6.16; found C 71.18, H 4.16, N 6.10.
(±)-2k/2k′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3299, 1699 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 8.23 (d, J = 6.9 Hz, 1H), 7.91 (d, J = 7.5 Hz, 1H), 7.71–7.65 (m, 2H), 7.52–7.48 (m, 2H), 7.39–7.32 (m, 2H), 7.25 (t, J = 7.2 Hz, 1H), 7.16–7.12 (m, 2H), 6.98 (d, J = 8.4 Hz, 1H), 5.56 (s, 1H), 3.96 (t, J = 13.2 Hz, 1H), 3.30–3.28 (m, 1H), 2.99–2.94 (dd, J1 = 2.7 Hz, J2 = 12.9 Hz, 1H), 1.11 (d, J = 15.6 Hz, 3H); 13C NMR (75 MHz, DMSO-d6) δH 191.1, 166.8, 163.8, 156.3, 144.4, 138.7, 136.3, 134.1, 133.8, 131.8, 131.5, 131.1, 130.7, 130.6, 126.4, 123.6, 123.0, 120.2, 119.7, 114.0, 112.5, 103.0, 94.7, 48.0, 45.9, 45.1, 17.4; C27H19BrN2O3 (499.37): calcd C 64.94, H 3.84, N 5.61; found C 64.80, H 3.78, N 5.54.
(±)-2l/2l′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3316, 1697 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 8.24 (d, J = 6.6 Hz, 1H), 7.90 (d, J = 7.5 Hz, 1H), 7.75–7.71 (m, 2H), 7.68–7.51 (m, 2H), 7.38 (t, J = 7.2 Hz, 1H), 7.29 (t, J = 6.9 Hz, 1H), 7.17 (d, J = 6.2 Hz, 1H), 6.96 (d, J = 8.7 Hz, 1H), 6.82–6.78 (m, 1H), 6.63 (s, 1H), 5.56 (s, 1H), 3.95 (t, J = 13.5 Hz, 1H), 3.66 (s, 3H), 3.37–3.35 (m, 1H), 3.00–2.95 (dd, J1 = 3.9 Hz, J2 = 14.1 Hz, 1H), 1.12 (d, J = 15.6 Hz, 3H); 13C NMR (75 MHz, DMSO-d6) δH 190.9, 166.4, 163.5, 155.2, 150.5, 144.3, 138.4, 133.9, 133.8, 133.3, 133.2, 131.1, 130.4, 130.1, 123.1, 122.5, 119.7, 119.2, 114.1, 110.3, 108.7, 102.4, 94.8, 55.8, 47.6, 45.6, 45.3, 17.1; C28H22N2O4 (450.50): calcd C 74.65, H 4.92, N 6.22; found C 74.79, H 4.84, N 6.15.
(±)-2m/2m′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3294, 1692 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 7.84 (d, J = 6.6 Hz, 1H), 7.63 (d, J = 7.5 Hz, 1H), 7.51–7.44 (m, 2H), 7.35–7.27 (m, 2H), 7.15 (t, J = 7.5 Hz, 1H), 7.07 (t, J = 7.2 Hz, 1H), 6.95 (d, J = 6.6 Hz, 1H), 6.86 (d, J = 7.8 Hz, 1H), 6.68–6.63 (m, 2H), 5.31 (s, 1H), 4.03–4.00 (m, 1H), 2.91–2.88 (m, 1H), 2.01 (s, 3H), 1.53–1.33 (m, 6H), 0.95–0.91 (m, 1H), 0.22–0.18 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δH 190.7, 166.8, 162.4, 154.4, 145.4, 138.3, 134.0, 133.3, 133.2, 131.6, 131.0, 130.3, 130.2, 130.0, 128.7, 123.7, 122.8, 122.4, 119.7, 119.2, 110.0, 104.4, 94.8, 57.2, 53.4, 45.0, 31.0, 30.9, 25.2, 24.3, 20.4; C31H26N2O3 (474.56): calcd C 78.46, H 5.52, N 5.90; found C 78.30, H 5.45, N 5.81.
(±)-2n/2n′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3295, 1690 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 8.13 (d, J = 6.9 Hz, 1H), 7.88 (d, J = 7.5 Hz, 1H), 7.73–7.65 (m, 2H), 7.58–7.48 (m, 2H), 7.36 (t, J = 7.5 Hz, 1H), 7.28 (t, J = 7.2 Hz, 1H), 7.15 (d, J = 6.9 Hz, 1H), 7.05–7.02 (m, 2H), 5.54 (s, 1H), 4.24–4.01 (m, 1H), 3.12–3.09 (m, 1H), 2.27 (s, 3H), 1.84–1.80 (m, 1H), 1.64–1.55 (m, 5H), 1.17–1.14 (m, 1H), 0.48–0.46 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δH 190.6, 166.9, 162.5, 155.4, 145.1, 138.3, 135.9, 134.1, 133.5, 133.0, 131.1, 130.6, 130.3, 130.0, 126.3, 123.5, 123.0, 122.6, 119.9, 119.4, 112.9, 104.9, 94.3, 57.4, 53.6, 44.8, 31.1, 31.0, 25.3, 24.5, 20.1; C31H25ClN2O3 (509.01): calcd C 73.15, H 4.95, N 5.50; found C 73.05, H 4.91, N 5.45.
(±)-2o/2o′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3296, 1687 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 8.12 (d, J = 6.9 Hz, 1H), 7.93 (d, J = 6.9 Hz, 1H), 7.75–7.52 (m, 4H), 7.36 (d, J = 6.6 Hz, 1H), 7.32 (t, J = 6.9 Hz, 1H), 7.19–6.93 (m, 4H), 5.61 (s, 1H), 4.33–4.27 (m, 1H), 3.13–3.07 (m, 1H), 1.87–1.85 (m, 1H), 1.66–1.60 (m, 5H), 1.19–1.15 (m, 1H), 0.48–0.45 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δH 190.5, 166.8, 162.5, 159.7, 156.5, 152.5, 144.9, 138.2, 135.1, 133.9, 133.4, 131.0, 130.2, 129.9, 122.9, 122.4, 119.8, 119.3, 114.8, 114.5, 111.2, 111.1, 110.9, 110.5, 105.0, 94.0, 57.2, 53.5, 45.1, 31.0, 30.9, 25.2, 24.3; C30H23FN2O3 (478.53): calcd C 75.30, H 4.84, N 5.85; found C 75.14, H 4.75, N 5.78.
(±)-2p/2p′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3285, 1689 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 8.07 (d, J = 6.9 Hz, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.70–7.61 (m, 2H), 7.53–7.46 (m, 2H), 7.32–7.21 (m, 3H), 7.12 (d, J = 6.9 Hz, 1H), 7.02–6.97 (m, 2H), 5.55 (s, 1H), 4.31–4.23 (m, 1H), 3.11–3.07 (m, 1H), 1.87–1.84 (m, 1H), 1.61–1.48 (m, 5H), 1.20–1.16 (m, 1H), 0.49–0.45 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δH 190.5, 166.8, 162.5, 155.3, 144.8, 138.2, 135.6, 133.9, 133.4, 131.0, 130.5, 130.2, 129.9, 128.3, 126.2, 123.3, 123.0, 122.5, 119.8, 119.3, 111.9, 104.8, 93.9, 57.2, 53.5, 44.8, 31.0, 30.9, 25.2, 24.4; C30H23ClN2O3 (494.98): calcd C 72.80, H 4.68, N 5.66; found C 72.93, H 4.62, N 5.57.
(±)-2q/2q′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3299, 1685 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 8.14 (d, J = 6.9 Hz, 1H), 7.94 (d, J = 7.5 Hz, 1H), 7.76–7.68 (m, 2H), 7.59–7.46 (m, 3H), 7.39 (t, J = 7.5 Hz, 1H), 7.31 (t, J = 7.5 Hz, 1H), 7.21–7.17 (m, 2H), 7.02 (d, J = 8.4 Hz, 1H), 5.62 (s, 1H), 4.31–4.23 (m, 1H), 3.11–3.07 (m, 1H), 1.87–1.84 (m, 1H), 1.67–1.54 (m, 5H), 1.20–1.16 (m, 1H), 0.49–0.45 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δH 190.9, 167.2, 162.8, 156.1, 145.2, 138.6, 136.4, 134.3, 133.8, 131.6, 131.4, 130.9, 130.6, 130.2, 126.4, 123.4, 122.9, 120.2, 119.7, 114.1, 112.9, 105.1, 94.3, 57.6, 53.9, 45.1, 31.4, 31.3, 25.5, 24.7; C30H23BrN2O3 (539.43): calcd C 66.80, H 4.30, N 5.19; found C 66.95, H 4.26, N 5.12.
(±)-2r/2r′. Yellow amorphous solid, mp > 300 °C, IR (KBr) 3290, 1692 cm−1; 1H NMR (300 MHz, DMSO-d6) δH 8.09 (d, J = 7.2 Hz, 1H), 7.88 (d, J = 7.5 Hz, 1H), 7.75–7.67 (m, 2H), 7.58–7.50 (m, 2H), 7.41 (t, J = 6.6 Hz, 1H), 7.33 (t, J = 10.8 Hz, 1H), 7.18 (d, J = 6.9 Hz, 1H), 6.95 (d, J = 8.7 Hz, 1H), 6.85 (dd, J1 = 1.8 Hz, J2 = 8.7 Hz, 1H), 6.62 (s, 1H), 5.57 (s, 1H), 4.28–4.26 (m, 1H), 3.68 (m, 3H), 3.16–3.12 (m, 1H), 1.83–1.80 (m, 1H), 1.69–1.58 (m, 5H), 1.21–1.20 (m, 1H), 0.48–0.47 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δH 191.0, 167.2, 162.8, 155.8, 150.7, 145.6, 138.7, 134.5, 134.4, 133.6, 131.3, 130.7, 130.5, 130.4, 123.2, 122.8, 120.1, 119.6, 114.2, 111.1, 109.3, 105.0, 94.9, 57.6, 56.3, 53.8, 45.8, 31.4, 31.2, 25.6, 24.7; C31H26N2O4 (490.56): calcd C 75.90, H 5.34, N 5.71; found C 75.78, H 5.30, N 5.66.
Acknowledgements
S.P. and K.D. thank CSIR and UGC, New Delhi, India, for offering them Senior Research Fellowship (SRF) respectively. The author Md. M. R. Mollick gratefully acknowledge the Department of Science & Technology (DST), Govt. of India for providing fellowship under the INSPIRE program. The financial assistance of CSIR, New Delhi is gratefully acknowledged [Major Research Project, no. 02(0007)/11/EMR-II]. Crystallography was performed at the DST-FIST, India-funded Single Crystal Diffractometer Facility at the Department of Chemistry, University of Calcutta. We also acknowledge Center for Research in Nanoscience and Nanotechnology (CRNN), University of Calcutta for instrumental facilities.
References
- G. Mehta and V. Singh, Chem. Rev., 1999, 99, 881–930 CrossRef CAS PubMed.
- N. A. Petasis and M. A. Patane, Tetrahedron, 1992, 48, 5757–5821 CrossRef CAS.
-
(a) S. Huneck, G. Baxter, A. F. Cameron, J. D. Connelly and D. S. Rycroft, Tetrahedron Lett., 1983, 24, 3787–3788 CrossRef CAS;
(b) J. Zapp, G. Burkhardt and H. Becker, Phytochemistry, 1994, 37, 787–793 CrossRef CAS;
(c) H.-J. Liu, C.-L. Wu, H. Becker and J. Zapp, Phytochemistry, 2000, 53, 845–849 CrossRef CAS;
(d) F. Huang, Z.-K. Yao, Y. Wang, Y. Wang, J. Zhang and Z.-X. Yu, Chem.–Asian J., 2010, 5, 1555–1559 CrossRef CAS PubMed.
-
(a) L. Erguang, A. M. Clark, D. P. Rotella and C. D. Hufford, J. Nat. Prod., 1995, 58, 74–81 CrossRef.
- W. H. Pearson, I. Y. Lee, Y. Mi and P. Stoy, J. Org. Chem., 2004, 69, 9109–9122 CrossRef CAS PubMed.
- K. Tsuna, N. Noguchi and M. Nakada, Angew. Chem., Int. Ed., 2011, 50, 9452–9455 CrossRef CAS PubMed.
- H. Qin, Z. Xu, Y. Cui and Y. Jia, Angew. Chem., Int. Ed., 2011, 50, 4447–4449 CrossRef CAS PubMed.
- T. H. Quang, D.-S. Lee, J. H. Sohn, Y.-C. Kim and H. Oh, Bull. Korean Chem. Soc., 2013, 34, 3109–3112 CrossRef CAS.
- H. Zhang, S. Qiu, P. Tamez, G. T. Tan, Z. Aydogmus, N. Van Hung, N. M. Cuong, C. Angerhofer, D. D. Soejarto, J. M. Pezzuto and H. H. S. Fong, Pharm. Biol., 2002, 40, 221–224 CrossRef CAS.
- T.-S. Kam, K. Yoganathan, T. Koyano and K. Komiyama, Tetrahedron Lett., 1996, 37, 5765–5768 CrossRef CAS.
-
(a) L. Mitchell, J. A. Parkinson, J. M. Percy and K. Singh, J. Org. Chem., 2008, 73, 2389–2395 CrossRef CAS PubMed;
(b) C. J. Creighton and A. B. Reitz, Org. Lett., 2001, 3, 893–895 CrossRef CAS PubMed;
(c) N. Brown, B. Xie, N. Markina, D. VanderVelde, J.-P. H. Perchellet, E. M. Perchellet, K. R. Crow and K. R. Buszek, Bioorg. Med. Chem. Lett., 2008, 18, 4876–4879 CrossRef CAS PubMed;
(d) S. K. Chattopadhyay, S. Karmakar, T. Biswas, K. C. Majumdar, H. Rahaman and B. Roy, Tetrahedron, 2007, 63, 3919–3952 CrossRef CAS PubMed;
(e) K. C. Majumdar, S. Samanta, B. Chattopadhyay and R. K. Nandi, Synthesis, 2010, 863–869 CrossRef CAS PubMed.
- C. J. Roxburgh, Tetrahedron, 1993, 49, 10749–10784 CrossRef CAS.
- F. Genrich and E. Schaumann, Tetrahedron Lett., 2009, 50, 6187–6190 CrossRef CAS PubMed.
- S. L. G. Ayala, E. Stashenko, A. Palma, A. Bahsas and J. M. Amaro-Luis, Synlett, 2006, 2275–2277 Search PubMed.
- T. Zhang, X. Huang, J. Xue and S. Sun, Tetrahedron Lett., 2009, 50, 1290–1294 CrossRef CAS PubMed.
-
(a) M. Mascal, K. V. Modes and A. Durmus, Angew. Chem., Int. Ed., 2011, 50, 4445–4446 CrossRef CAS PubMed;
(b) A. Sharma, P. Appukkuttan and E. Van der Eycken, Chem. Commun., 2012, 48, 1623–1637 RSC;
(c) K. Wittstein, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 9076–9080 CrossRef CAS PubMed;
(d) M. M. Coulter, P. K. Dornan and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 6932–6933 CrossRef CAS PubMed;
(e) H. Qin, Z. Xu, Y. Cui and Y. Jia, Angew. Chem., Int. Ed., 2011, 50, 4447–4449 CrossRef CAS PubMed;
(f) A. Mishra and S. Batra, Eur. J. Org. Chem., 2010, 4832–4840 CrossRef CAS;
(g) P. G. Jones, Organometallics, 2012, 31, 3361–3372 CrossRef;
(h) R. Pflantz, J. Sluiter, M. Krička, W. Saak, C. Hoenke and J. Christoffers, Eur. J. Org. Chem., 2009, 5431–5436 CrossRef CAS;
(i) S. López-Tosco, D. Tejedor, J. González-Platas and F. García-Tellado, Eur. J. Org. Chem., 2011, 6847–6850 CrossRef;
(j) V. A. Peshkov, S. Van Hove, P. A. Donets, O. P. Pereshivko, K. Van Hecke, L. Van Meervelt and E. V. Van der Eycken, Eur. J. Org. Chem., 2011, 1837–1840 CrossRef CAS.
-
(a) S. Das, R. Fröhlich and A. Pramanik, Org. Lett., 2006, 8, 4263–4266 CrossRef CAS PubMed;
(b) S. Pathak and A. Pramanik, Eur. J. Org. Chem., 2013, 4410–4417 CrossRef CAS;
(c) S. Das, P. Koley and A. Pramanik, Tetrahedron Lett., 2011, 52, 3243–3246 CrossRef CAS PubMed;
(d) S. Pathak, A. Kundu and A. Pramanik, Tetrahedron Lett., 2011, 52, 5180–5183 CrossRef CAS PubMed;
(e) S. Pathak, K. Debnath and A. Pramanik, Beilstein J. Org. Chem., 2013, 9, 2344–2353 CrossRef PubMed;
(f) S. Das, A. Pramanik, R. Fröhlich and A. Patra, Tetrahedron, 2004, 60, 10197–10205 CrossRef CAS PubMed;
(g) S. Pathak, K. Debnath, S. k. T. Hossain, S. K. Mukherjee and A. Pramanik, Tetrahedron Lett., 2013, 54, 3137–3143 CrossRef CAS PubMed;
(h) K. Debnath, S. Pathak and A. Pramanik, Tetrahedron Lett., 2013, 54, 4110–4115 CrossRef CAS PubMed;
(i) K. Debnath, S. Pathak and A. Pramanik, Tetrahedron Lett., 2014, 55, 1743–1748 CrossRef CAS PubMed.
-
(a) V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J.-M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS PubMed;
(b) R. B. N. Baig and R. S. Varma, Chem. Commun., 2013, 49, 752–770 RSC;
(c) S. Shylesh, V. V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS PubMed;
(d) M. Shafiee, A. R. Khosropour, I. Mohammadpoor-Baltork, M. Moghadam, S. Tangestaninejad and V. Mirkhani, Catal. Sci. Technol., 2012, 2, 2440–2444 RSC;
(e) A. Wang, X. Liu, Z. Su and H. Jing, Catal. Sci. Technol., 2014, 4, 71–80 RSC.
-
(a) C. G. Piscopo, S. Bühler, G. Sartori and R. Maggi, Catal. Sci. Technol., 2012, 2, 2449–2452 CAS;
(b) M. B. Gawande, A. K. Rathi, I. D. Nogueira, R. S. Varma and P. S. Branco, Green Chem., 2013, 15, 1895 RSC.
- E. Kolvari, N. Koukabi and O. Armandpour, Tetrahedron, 2014, 70, 1383–1386 CrossRef CAS PubMed.
- F. Nemati, M. M. Heravi and R. S. Rad, Chin. J. Catal., 2012, 33, 1825–1831 CrossRef CAS.
- D. Yang, J. Hu and S. Fu, J. Phys. Chem. C, 2009, 113, 7646–7651 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 994163–994165. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra03384a |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.