A bis(maleonitriledithiolato)nickelate charge-transfer salt with mixed stacks exhibiting novel non-ferroelectric-type dielectric phase transition and bistability†
Yao Chena,
Shun-Ping Zhaoa,
Jian-Lan Liu*a,
Wei-Hua Ninga,
Xiao-Mei Suna and
Xiao-Ming Ren*ab
aState Key Laboratory of Materials-Oriented Chemical Engineering and College of Science, Nanjing University of Technology, Nanjing 210009, P. R. China. E-mail: xmren@njut.edu.cn; Fax: +86 25 58139481; Tel: +86 25 58139476
bCollege of Material Science and Engineering, Nanjing University of Technology, Nanjing 210009, P. R. China
First published on 10th December 2013
Abstract
A charge-transfer salt consisting of 1,1′-dioctyl-4,4′-bipyridinium and bis(maleonitriledithiolato)nickelate is described. Its crystal is built from mixed stacks of the anion and cation, with the anions (A) and cations (C) adopting an …ACAC… fashion within a stack. This salt shows a novel non-ferroelectric-type dielectric phase transition at around 324 K and bistability.
The physical properties of matter may change massively in a solid-to-solid phase transition,1–6 and as a result, materials undergoing fast, reversible, especially hysteretic phase transitions probably show diverse applications.7
The dielectric property is a technologically important functional characteristic of matter, and special dielectric solids have been widely used in microelectronic and electrical engineering applications recently. For example, a dielectric resonator oscillator that exhibits resonance for a narrow range of frequencies can be used as a dielectric resonator antenna,8 and a high-κ dielectric material can be used in semiconductor manufacturing processes where it is usually utilized to replace a silicon dioxide gate dielectric or another dielectric layer of a device.9 A low-κ dielectric material is generally used to separate the conducting parts (such as the wire interconnects and transistors) from one another in digital circuits, and reduces parasitic capacitance, enabling faster switching speeds and lower heat dissipation for a microelectronic device.10
Recently, dielectric properties have been widely studied for inorganic materials and polymers, but rarely investigated for metal coordination compounds11,12 and small molecule organic compounds.13 Herein, we report a charge-transfer salt (1), consisting of bis(maleonitriledithiolato)nickelate and 1,1′-dioctyl-4,4′-bipyridinium (abbr. as [C8-4,4′-Bipy][Ni(mnt)2]), which shows a mixed stack of alternating anions and cations in the crystal and a novel non-ferroelectric-type dielectric phase transition.‡
Disodium maleonitriledithiolate (Na2mnt) was synthesized according to a previously published procedure,14 and a similar process as for the preparation of 4-amino-1-hexylpyridinium bromide was used for the synthesis of 1,1′-dioctyl-4,4′-bipyridinium dibromide.15 Charge-transfer salt 1 was prepared according to the similar approach for the preparation of [TBA]2[Ni(mnt)2]14 where TBA+ = tetrabutylammonium, and was characterized by elemental analysis for C, H and N elements, IR spectroscopy in the range of 4000–400 cm−1, powder X-ray diffraction and thermogravimetric analysis (see ESI†). The single crystals suitable for X-ray structure analyses were grown by evaporation of a solution of 1 in DMF at ambient temperature after ∼30 days.
Single crystal X-ray structural analysis‡ revealed that 1 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , as shown in Fig. 1a, and an asymmetric unit is comprised of one half [Ni(mnt)2]2− anion together with one half C8-4,4′-Bipy2+ cation, with both the anion and cation possessing Ci symmetry. In the planar [Ni(mnt)2]2− moiety, the Ni2+ ion occupies a crystallographic inversion center. The Ni–S bond lengths and the S–Ni–S bite angle, summarized in Table S2,† are comparable to those in the reported [Ni(mnt)2]2− salts.16
, as shown in Fig. 1a, and an asymmetric unit is comprised of one half [Ni(mnt)2]2− anion together with one half C8-4,4′-Bipy2+ cation, with both the anion and cation possessing Ci symmetry. In the planar [Ni(mnt)2]2− moiety, the Ni2+ ion occupies a crystallographic inversion center. The Ni–S bond lengths and the S–Ni–S bite angle, summarized in Table S2,† are comparable to those in the reported [Ni(mnt)2]2− salts.16
 | ||
| Fig. 1 (a) ORTEP view with displacement ellipsoids at the 20% probability level and hydrogen atoms omitted for clarity, (b) packing diagram projected approximately along the crystallographic a-b direction, (c) the mixed stack and (d) arrangement of anions and cations along their short molecular axes for 1. | ||
The C8-4,4′-Bipy2+ cation adopts a chair-shaped conformation, in which an inversion center coincides with the midpoint of the C15–C15#1 bond (the symmetric code #1 = 1−x, −y, 1−z). Two hydrocarbon chains show a completely trans-planar conformation and two pyridyl rings are coplanar owing to the constraint of Ci-symmetry. The bond length and bond angles in the cation fall within the expected values.
As displayed in Fig. 1b and c, the anions (A) and cations (C) form a mixed stack in an …ACAC… fashion along the crystallographic a–b direction; the formation of the mixed stack is due to the matching molecular geometries and the electrostatic attraction between the anion and cation. The long molecular axes of both the anion and cation are approximately parallel to one another, while the chelate ring and the neighboring pyridyl ring overlap mutually with a dihedral angle of 4.62° and a plane-to-plane distance of 3.479 Å between the mean molecular plane of the anion and the pyridyl rings within a mixed stack, where the mean molecular plane of the anion is defined through four coordinated S atoms. The mixed stacks are arranged into a molecular layer along the short molecular axes of both the anion and the cation, which is parallel to the crystallographic (0 0 1) plane. The adjacent mixed stacks are mutually shifted along the stacking direction, leading to the pyridyl rings being almost coplanar with the anions in two neighboring mixed stacks. As shown in Fig. S2,† several short interatomic contacts are observed between the anions and cations of adjacent mixed stacks in a molecular layer, and these interatomic distances are obviously less than the sum of the van der Waals radii of corresponding atoms.17 The alkyl chains are interdigitated between the neighboring molecular layers (see Fig. 1b).
The frequency dependencies of the dielectric permittivity ε′, and the dielectric loss tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ, for 1, are shown in Fig. 2a and b, respectively. The dielectric permittivity is maintained at 7–10 over the frequency range of 1–107 Hz below 245 K, and then increases with rising temperature. The ε′ value rises rapidly when the temperature is above 273 K and reaches ∼120 at 405 K under a frequency of 1 Hz, indicating the existence of thermally-assisted dynamic dipole motion under an ac electrical field in 1. An explanation for this behaviour is that the molecules are easily reoriented and polarized in the crystals at a high temperature, owing to thermal expansion. The dielectric permittivity ε′ drops with increasing frequency in the region of 1–105 Hz (see Fig. 2a), demonstrating that the dynamic dipole motion cannot follow the switching of the applied ac electric field under higher frequencies (when f > 105 Hz). Corresponding to the dielectric permittivity drop, no clear broad peak appears in the tanδ–f plot in the 1–105 Hz region (see Fig. 2b), but this probably occurs in the frequency region below 1 Hz.
δ, for 1, are shown in Fig. 2a and b, respectively. The dielectric permittivity is maintained at 7–10 over the frequency range of 1–107 Hz below 245 K, and then increases with rising temperature. The ε′ value rises rapidly when the temperature is above 273 K and reaches ∼120 at 405 K under a frequency of 1 Hz, indicating the existence of thermally-assisted dynamic dipole motion under an ac electrical field in 1. An explanation for this behaviour is that the molecules are easily reoriented and polarized in the crystals at a high temperature, owing to thermal expansion. The dielectric permittivity ε′ drops with increasing frequency in the region of 1–105 Hz (see Fig. 2a), demonstrating that the dynamic dipole motion cannot follow the switching of the applied ac electric field under higher frequencies (when f > 105 Hz). Corresponding to the dielectric permittivity drop, no clear broad peak appears in the tanδ–f plot in the 1–105 Hz region (see Fig. 2b), but this probably occurs in the frequency region below 1 Hz.
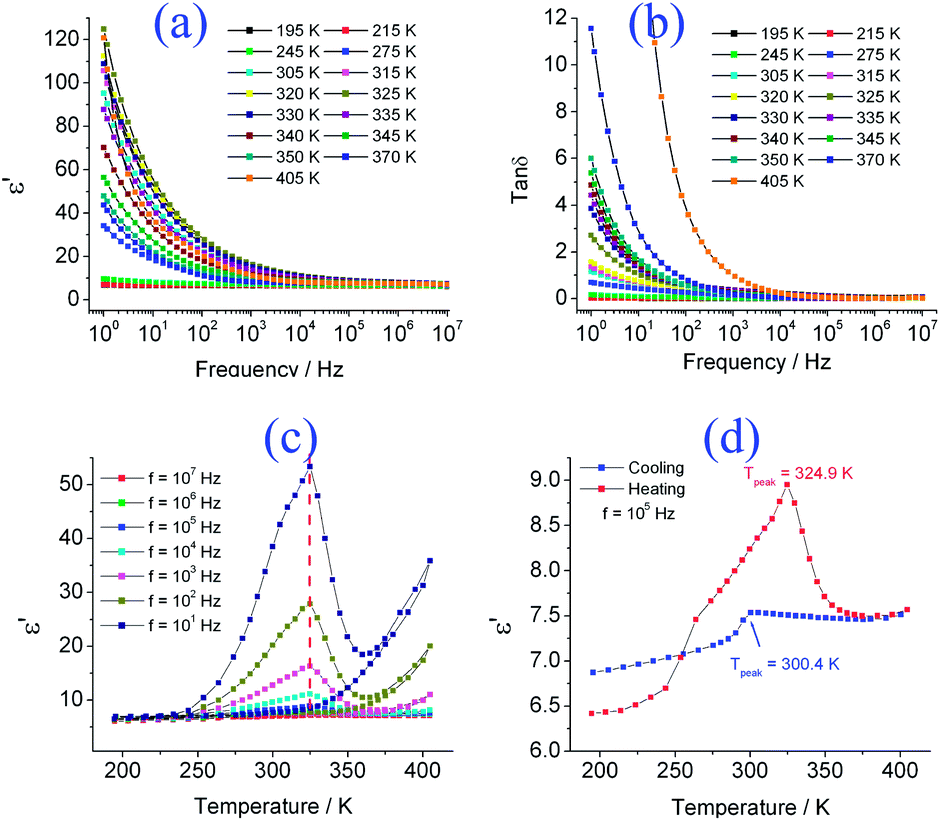 | ||
| Fig. 2 (a and b) Plots of ε′–f and tanδ–f, (c) the temperature dependency of ε′ in the frequency range of 10–107 Hz and (d) the plot of ε′–T at f = 105 Hz on a heating and cooling cycle for 1. | ||
Dynamic dipole motion arising from molecule dipole orientation or ionic displacement polarization occurs in the frequency range less than 1010 Hz. Thus, the observed dynamic dipole motion in the frequency region of 1–105 Hz can be attributed to the molecule dipole orientation or the ionic displacement polarization.
The temperature dependency of ε′ and tanδ in the frequency range of 10–107 Hz is depicted in Fig. 2c and S3,† and a dielectric anomaly appears in the ε′–T plot upon heating. Two points are worth noting for this dielectric phase transition: (1) the dielectric anomaly peak is asymmetric and the maximum is independent of the frequency of the applied ac field (Tpeak ≈ 325 K in the frequency region of 10–107 Hz). (2) A hysteresis loop is observed during the heating–cooling cycle (a hysteresis plot of ε′–T at f = 105 Hz is presented in Fig. 2d), demonstrating the existence of dielectric bistability. With bistability, crystalline materials show potential applications in sensing, switching and data storage devices, etc.1,18–20 Recently, magnetic bistability compounds have been widely studied in the molecular material field, including bistable spin-crossover (SCO) complexes,1a the bistable heterometallic valence tautomerism molecule magnets18–20 or bistable valence ordering in a mixed valence complex,21 etc. To the best of our knowledge, an example of a compound showing dielectric bistability is very rare.22 In addition, the dielectric anomaly is generally associated with a paraelectric-to-ferroelectric phase transition - obviously the dielectric anomaly observed in 1 belongs to the non-ferroelectric-type dielectric phase transition since the crystal structure in the low temperature phase is not in a polar phase.
Differential scanning calorimetry (DSC) measurements for 1 were further carried out for the investigation of the thermal behavior of the dielectric phase transition, and the corresponding plots are shown in Fig. 3, indicating that a thermal anomaly accompanies the dielectric phase transition. The endothermic peak temperature in the DSC curve is 324 K in the first heating process, corresponding to the exothermic peak at 316 K in the first cooling process. The enthalpy and entropy changes of the phase transition are respectively estimated to be ΔH = 17.3 kJ mol−1 and ΔS = ΔH/TC = 53.2 J K−1 mol−1 (where TC = 324 K) from the first heating run. The endothermic peak temperature shifts a little towards the low temperature side during the second heating–cooling cycle, behavior which is quite analogous to that often observed in liquid crystals owing to the alkyl chain disordered motion. The large latent heat suggests that the non-ferroelectric-type dielectric phase transition is probably associated with crystal structure change, and the high entropy change value of the phase transition indicates the existence of significant structural disorder at high temperatures. Unfortunately, we failed to obtain details of the crystal structure in the high temperature phase due to the poor quality of the single crystal X-ray diffraction data above the phase transition, and we performed variable temperature powder X-ray diffraction data collection across the phase transition. The profiles at the selected temperatures in the high temperature phase are shown in Fig. S4,† and demonstrate that the powder diffraction intensities decrease dramatically above the phase transition due to significant structural disorder. Thus, the dielectric anomaly probably arises from the disorder–order transformation of alkyl chains assisting ion displacement since the alkyl chain is nearly non-polar.
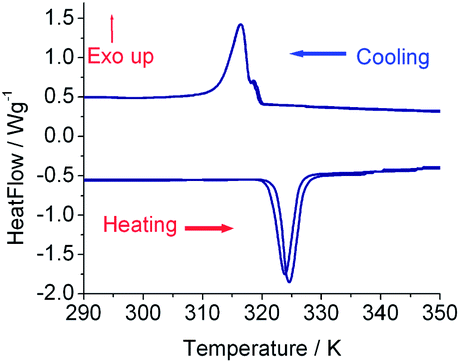 | ||
| Fig. 3 DSC plots of 1 with two heating and cooling cycles. | ||
In summary, a charge-transfer salt showing a layered crystal structure and a molecular layer built from a mixed stack of alternating anions and cations is presented. This salt experiences a reversible and non-ferroelectric-type dielectric phase transition, which is probably related to the disorder–order transformation of alkyl chains and disorder–order transformation-facilitated ion displacement. The dielectric phase transition displays bistability. This study suggests the possibility of dielectric bistability being a feature of rationally-designed non-ferroelectric systems.
The authors thank the Priority Academic Program Development of Jiangsu Higher Education Institutions and the National Nature Science Foundation of China for their financial support (grant no: 91122011 and 21071080).
Notes and references
- (a) O. Kahn and C. J. Martinez, Science, 1998, 279, 44 CrossRef CAS; (b) W. Fujita and K. Awaga, Science, 1999, 286, 261 CrossRef CAS; (c) M. E. Itkis, X. Chi, A. W. Cordes and R. C. Haddon, Science, 2002, 296, 1443 CrossRef CAS PubMed.
- S. P. Zhao, H. Gao, X. M. Ren, G. J. Yuan and Y. N. Lu, J. Mater. Chem., 2012, 22, 447 RSC.
- J. Fousek and J. Petzelt, Phys. Status Solidi A, 1979, 55, 11 CrossRef CAS.
- J. Padilla-Pantoja, C. Frontera, J. Herrero-Martín and J. Luis García-Muñoz, J. Appl. Phys., 2012, 111, 07D710 CrossRef PubMed.
- R. H. Mitchell, Z. Brkic, V. A. Sauro and D. J. Berg, J. Am. Chem. Soc., 2003, 125, 7581 CrossRef CAS PubMed; B. Scott and R. D. Willett, J. Am. Chem. Soc., 1991, 113, 5253 CrossRef; K. L. Bray, H. G. Drickamer, E. A. Schmitt and D. N. Hendrickson, J. Am. Chem. Soc., 1989, 111, 2849 CrossRef.
- Y. Garcia, F. Robert, A. D. Naik, G. Zhou, B. Tinant, K. Robeyns, S. Michotte and L. Piraux, J. Am. Chem. Soc., 2011, 133, 15850 CrossRef CAS PubMed; A. Fernández-Mato, M. D. García, C. Peinador, J. M. Quintela, M. Sánchez-Andújar, B. Pato-Doldán, M. A. Señarís-Rodríguez, D. Tordera and H. J. Bolink, Cryst. Growth Des., 2013, 13, 460 Search PubMed.
- M. A. Kats, D. Sharma, J. Lin, P. Genevet, R. Blanchard, Z. Yang, M. M. Qazilbash, D. N. Basov, S. Ramanathan and F. Capasso, Appl. Phys. Lett., 2012, 101, 221101 CrossRef CAS PubMed; Y. T. Wang and C. H. Chen, Inorg. Chem., 2013, 52, 2550 CrossRef PubMed.
- S.-J. Cho and N.-Y. Kim, Int. J. Electron., 2013, 100, 563 CrossRef CAS.
- J. Koch, K. Seidel, W. Weinreich, S. Riedel, J.-C. Chiang and V. Beyer, Microelectron. Eng., 2013, 109, 148 CrossRef CAS PubMed.
- J. P. Reynard, C. Verove, E. Sabouret, P. Motte, B. Descouts, C. Chaton, J. Michailos and K. Barla, Microelectron. Eng., 2002, 60, 113 CrossRef CAS.
- G. C. Xu, X. M. Ma, L. Zhang, Z. M. Wang and S. Gao, J. Am. Chem. Soc., 2010, 132, 9588 CrossRef CAS PubMed.
- W. Zhang, Y. Cai, R. G. Xiong, H. Yoshikawa and K. Awaga, Angew. Chem., Int. Ed., 2010, 49, 6608 CrossRef CAS PubMed.
- T. Akutagawa, S. Takeda, T. Hasegawa and T. Nakamura, J. Am. Chem. Soc., 2004, 126, 291 CrossRef CAS PubMed; S. Nishihara, T. Akutagawa, D. Sato, S. Takeda, S.-i. Noro and T. Nakamura, Chem.–Asian J., 2007, 2, 1083 CrossRef PubMed.
- A. Davison and H. R. Holm, Inorg. Synth., 1967, 10, 8 CrossRef CAS.
- H. B. Duan, X. M. Ren, L. J. Shen, W. Q. Jin, Q. J. Meng, Z. F. Tian and S. M. Zhou, Dalton Trans., 2011, 40, 3622 RSC.
- X. M. Ren, C. S. Lu, Y. J. Liu, H. Z. Zhu, H. F. Li, C. J. Hu and Q. J. Meng, Trans. Met. Chem., 2001, 26, 136 CrossRef CAS; W. B. Pei, J. S. Wu, J. L. Liu, X. M. Ren and L. J. Shen, Spectrochim. Acta, Part A, 2010, 75, 191 CrossRef PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- O. Sato, J. Tao and Y. Z. Zhang, Angew. Chem., Int. Ed., 2007, 46, 2152 CrossRef CAS PubMed.
- S.-i. Ohkoshi, T. Matsuda, H. Tokoro and K. Hashimoto, Chem. Mater., 2005, 17, 81 CrossRef CAS.
- T. Liu, Y. J. Zhang, S. Kanegawa and O. Sato, Angew. Chem., Int. Ed., 2010, 49, 8645 CrossRef CAS PubMed.
- M. Yamashita, D. Kawakami, H. Okamoto, H. Tanaka, K. Marumoto and S. Kuroda, Angew. Chem., Int. Ed., 2004, 43, 4763 CrossRef CAS PubMed.
- Q. Chen, P. C. Guo, S. P. Zhao, J. L. Liu and X. M. Ren, CrystEngComm, 2013, 15, 1264 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data in CIF format; the profiles of IR spectra in the 400–4000 cm−1 region, powder X-ray diffraction data, temperature-dependence of dielectric loss (tanδ) data and the details for the preparation of the charge-transfer salt (1) in PDF format. CCDC 929581. For ESI and crystallographic data in CIF or other electronic formats, see DOI: 10.1039/c3ra41607k |
| ‡ Crystallographic data in the low-temperature phase: C34H42N6NiS4 (1), Mr = 721.69, triclinic space group P, T = 296(2) K, a = 7.217(2) Å, b = 8.617(3) Å, c = 16.124(5) Å, α = 87.616(4)°, β = 87.800(5)°, γ = 72.222(4)°, V = 953.7(5) Å3, Z = 2, Dc = 1.257 g cm−3, μ(Mo-Kα) = 0.758 mm−1, F(000) = 380, number of reflections measured/number of independent reflections = 8370/4275, Rint = 0.0642; R1 = 0.0598 and wR2 = 0.1406 for I > 2σ(I) data. |
| This journal is © The Royal Society of Chemistry 2014 |