Urea as an organic solvent and reagent for the addition/cyclization/fragmentation cascades leading to 2-R-7H-dibenzo[de,h]quinolin-7-one analogues of Aporphinoid alkaloids†
Denis S.
Baranov
a,
Sergei F.
Vasilevsky
*a,
Brian
Gold
b and
Igor V.
Alabugin
*b
aInstitute of Chemical Kinetics and Combustion, Siberian Branch of the Russian Academy of Science, 630090, Novosibirsk, Russian Federation. E-mail: vasilev@ns.kinetics.nsc.ru; Fax: +7(383)3307350
bDepartment of Chemistry and Biochemistry, Florida State University, Tallahassee, Florida 32306, USA.. E-mail: alabugin@chem.fsu.edu
First published on 25th October 2011
Abstract
Molten urea serves both as a solvent and a reagent for the one-pot addition/cyclizations/fragmentation cascade which converts peri-alkynyl-9,10-anthraquinones into 2-R-7H-dibenzo[de,h]quinolin-7-ones – substituted analogues of Aporphinoid alkaloids.
Alkynes are indispensable building blocks in organic synthesis, medicinal chemistry and materials science.1 The activated alkyne moiety in peri-substituted acetylenic anthraquinones provides a very convenient platform for probing fundamental electronic factors involved in alkyne reactivity using polyfunctional hemiaminal groups derived from the addition of a polyfunctional nucleophile to the adjacent carbonyl moiety (Scheme 1).
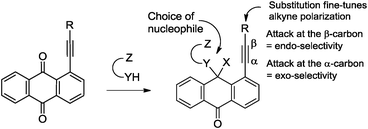 | ||
| Scheme 1 The delicate balance between endo and exo ring closure is controlled by the choice of nucleophile as well as electronic character of substituents on the alkyne. | ||
The multichannel reactivity of peri-substituted ethynyl-9,10-anthraquinones towards bifunctional nucleophiles depends on the nature of the attacking species (Scheme 2). For example, the reaction of arylethynyl-9,10-anthraquinones with hydrazine leads to the mixture of 2-R-7H-dibenzo[de,h]quinolin-7-one (I) and 3-R-4H-anthra[9,1-cd]-1,2-diazepin-8-one (II),2 whereas interaction with guanidine leads to three condensed heterocycles: 2-R-7H-dibenzo[de,h]quinolin-7-one (I), 1-R-2H-dibenzo[de,h]isoquinoline-3,7-dione (III) and 2-amino-3-aroyl-7H-dibenzo[de,h]quinolin-7-one (IV).3
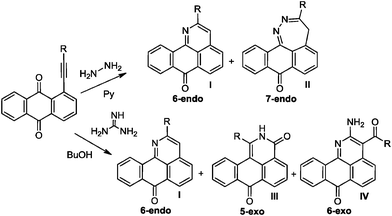 | ||
| Scheme 2 Observed products of reactions of anthraquinones with hydrazine and guanine. | ||
Not only do the above mentioned products provide examples of new heterocyclizations, but in the case of 1-R-2H-dibenzo-[de,h]isoquinoline-3,7-dione, this study led to the discovery of an unusual reaction of alkynes leading to the formal complete disassembly of a triple bond with the insertion of a nitrogen atom at the break point and the creation of six new bonds at the expense of the alkyne moiety. From a fundamental point of view, the ratio of the products in the reaction with guanine reflected a competition between three types of alkyne cyclizations: nucleophilic 6-endo-dig and 5-exo-dig attacks at an electrophilic alkyne moiety and 6-exo-dig closure in which the alkyne formally acts as a nucleophile (see Scheme 3).
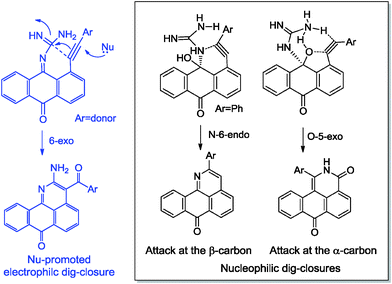 | ||
| Scheme 3 Effect of alkyne electronic properties on the selectivity of ring closure in the reactions of anthraquinones and guanine. | ||
The cascade nature of these transformations is potentially valuable for green chemistry because the combination of several steps in a one-pot procedure eliminates the need for the isolation and purification of the intermediates. This work will describe an efficient addition/cyclization/fragmentation cascade which will illustrate the unusual properties of urea—an environmentally friendly but under utilized polar solvent. We will show that this solvent/reagent allows us to bypass the need for the toxic reagents (hydrazine hydrate and pyridine) and/or strong bases (guanidine) which have been used in the previously described approaches to condensed anthraquinones. The new procedure adheres closer to the green chemistry practices because it minimizes the use and generation of such hazardous substances.
These considerations led to expansion of our studies to urea. Urea is not only the very first organic compound prepared by a chemist but also a common organic metabolite which is used in many body functions such as nitrogen excretion. It is solid, colorless, odorless, highly soluble in water, non-toxic and widely used in fertilizers. Its solutions in water are neither acidic nor alkaline.
This work describes a new approach to the synthesis of pharmacologically important 2-R-7H-dibenzo-[de,h]quinolin-7-ones via the interaction of 9,10-anthraquinones and urea in the absence of solvent. This approach avoids the use and generation of toxic substances and solvents.
The requisite 1-alkynyl-9,10-anthraquinones 1a–e,i were prepared from 1-iodo-9,10-anthraquinone and the respective terminal alkynes under Sonogashira conditions4 in an 80–98% yield. For the preparation of alkynes 1f,g,h, we have found that a more convenient alternative is provided by the Castro method5 based on the use of Cu(I) acetylides as cross-coupling partners (obtaining yields of 70–83%).
Taking into account the very low solubility of urea in the majority of organic solvents, we have carried out this transformation using molten urea itself as solvent.6 The results were intriguing. We have found that interaction of 1-alkynyl-9,10-anthraquinones 1a–i with an excess of urea in the melt at 135–140 °C is a regiospecific process which leads to the formation of 2-R-7H-dibenzo-[de,h]quinolin-7-ones 2a–i in 50–87% after 4–30 h (Scheme 4).
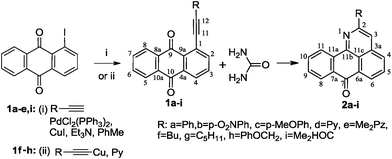 | ||
| Scheme 4 Preparation of 1-alkynyl-9,10-anthraquinones 1a–i and their reaction with urea. | ||
It is worth noting that our attempts to use common solvents for this transformation were unsuccessful. In particular, the reaction with an excess of urea in refluxing pyridine provided only 25% of heterocycle 2a with an incomplete conversion of alkyne 1a (∼60%) even after 86 h. For comparison, the reaction of alkyne 1a in molten urea is complete after 25 h, providing 2a in a 70% yield. In alcohols, the unfavorable solvent effect is pronounced even further. For example, the only reaction observed in butan-1-ol was the slow decomposition of the alkyne reactant (Table 1).
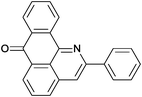
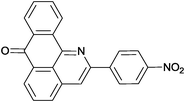
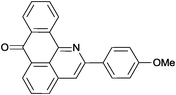
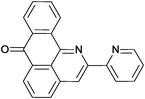
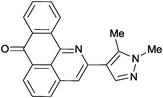
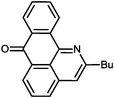
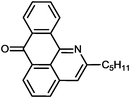
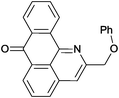
Reaction times and product yields depend on the structure of the alkyne. We found that faster reaction times and higher yields are observed for alkynes 1f–i, which have a better solubility and lower melting point. The triple bond polarization which was found to be very important for the reaction of 1-arylethynyl-9,10-anthraquinones with guanidine,3 seems to be of minor importance for the urea-promoted transformations.
The successful use of alkynes with substituents of different natures and properties demonstrates the universal character of the newly discovered process. It seems that the only likely limitation is provided by steric factors as illustrated by the lack of reactivity in the reaction with 1-mesitylethynyl-9,10-anthraquinone. A similar effect has been observed earlier in the reaction of 1-mesitylethynyl-9,10-anthraquinone with guanidine.3c
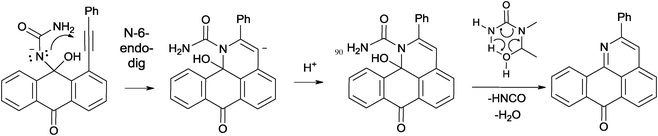
The plausible cyclization mechanism involves nucleophilic 6-endo-dig closure8 leading to the pyridine ring formation with subsequent or concomitant loss of water and isocyanic acid (Scheme 5). In order to gain an insight into the feasibility of this mechanism, we have calculated the full potential energy surface for the addition/cyclization/fragmentation cascade at the B3LYP/6-31G(d,p) level of theory.
![Suggested mechanism and B3LYP/6-31G(d,p) energies of suggested intermediates for the formation of 2-R-7H-dibenzo-[de,h]quinolin-7-ones 2a–i in the reaction of 1-alkynyl-9,10-anthraquinones 1a–i with urea. aDielectric = 3.5,7 volume calculations were performed in order to define radii.](/image/article/2011/RA/c1ra00622c/c1ra00622c-s5.gif) | ||
| Scheme 5 Suggested mechanism and B3LYP/6-31G(d,p) energies of suggested intermediates for the formation of 2-R-7H-dibenzo-[de,h]quinolin-7-ones 2a–i in the reaction of 1-alkynyl-9,10-anthraquinones 1a–i with urea. aDielectric = 3.5,7 volume calculations were performed in order to define radii. | ||
 | ||
| Scheme 6 Key calculated steps in the cascade reaction of anthraquinone 1a which correspond to reaction and activation energies given in Table 2 | ||
The initial step, formation of hemiaminal adduct via addition to the carbonyl group, is predicted to be thermodynamically unfavorable. However, the subsequent steps in the cascade are sufficiently exergonic for the overall cascade to be thermodynamically feasible (Scheme 5). Overall, the transformation of anthraquinone and urea into the 6-endo product is ∼34 kcal mol−1 exergonic (Table 2).
| 6-endocyclization | Fragmentation | Overall from 1a | |||
|---|---|---|---|---|---|
| ΔEa (ΔGa) | ΔErxn (ΔGrxn) | ΔEa (ΔGa) | ΔErxn (ΔGrxn) | ΔErxn (ΔGrxn) | |
| a Dielectric = 3.5,7 volume calculations were performed in order to define radii | |||||
| Gas Phase | 16.0 (18.4) | 8.7 (12.6) | 23.0 (19.9) | −6.1 (−34.8) | −24.1 (−34.4) |
| SCRF a | 17.0 (19.4) | 8.9 (12.7) | 24.0 (20.9) | −7.6 (−36.4) | −24.8 (−35.1) |
A more detailed analysis of the cyclization step revealed that direct formation of the 6-endo product via nucleophilic attack of the N-atom in the neutral hemiaminal provides the zwitter-ionic cyclized product which is not an energy minimum and opens without an energy barrier to give the starting material. This result suggested that deprotonation of the hemiaminal9 is needed to assist the cyclization. Indeed, although the cyclizations of the N-anion is still predicted to be endergonic, the activation barrier for this process is sufficiently low (∼18 kcal mol−1, Table 2) to be accessible under the reaction conditions and the initially formed vinyl anion resides in a sufficiently deep energy minimum to be trapped by the highly exergonic protonation step. The relatively low basicity of urea suggests that a concerted “proton wire” mechanism may be operational where several urea molecules participate in accepting the proton from the nucleophilic nitrogen and delivering it to the developing carbanion (Scheme 6).
As far as the fragmentation is concerned, the DFT analysis supports the concerted nature of this process which proceeds via a six-membered transition state of relatively low (∼20 kcal mol−1) energy (Fig. 1). The process is exergonic and provides the driving force for the overall transformation.10
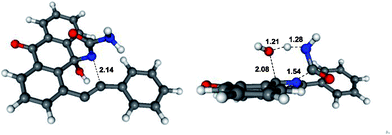 | ||
| Fig. 1 Optimized geometries and selected bond lengths (Angstroms) for the 6-endo-dig (left) and fragmentation (right) transition states. | ||
The relatively mild reaction conditions and high efficiency render this reaction an attractive alternative to the known approaches to the 7H-dibenzo[de,h]quinoline skeleton because our method does not require use of strongly acidic dehydrating agents (H2SO4, SO2Cl2, HSO3Cl, P2O5, oleum), high temperatures (180–240 °C) and a multistep synthesis.11 7H-Dibenzo[de,h]quinolin-7-ones are structurally related to natural alkaloids of the Aporphinoid family.12
The new synthesis of 2-R-7H-dibenzo[de,h]quinolin-7-ones developed in this work can be of significant interest to medicinal chemistry because their analogues display anticancer activity13 and increased acetylcholine esterase inhibition activity.14 In addition, several 7H-dibenzo[de,h]quinolin-7-ones were used as building blocks for the preparation of more complex polycyclic compounds15 and dyes.16
Conclusions
In summary, we have developed a new one-step synthesis of 2-R-7H-dibenzo[de,h]quinolin-7-ones from urea and 1-R-ethynyl-9,10-anthraquinones with a wide selection of functional groups. The new process described in this work follows many practices of green chemistry, which minimize the use of toxic substances and generation of large amounts of solvent waste. Not only is urea a non-toxic compound which is commercially available at a low price but it is also an odorless, common metabolite and neither acidic nor alkaline. High solubility in water greatly facilitates the work-up procedure – the crystalline crude reaction product is provided simply by pouring the reaction mixture onto ice.Experimental
Melting points were determined with a Kofler apparatus. The IR-spectra were recorded in KBr pellets on a ‘Bruker Vector 22’ instrument. Combustion analysis was performed with a CHN-analyzer (Model 1106, ‘Carlo Erba’, Italy). NMR spectra were recorded on a ‘Bruker AV-400’ 400.13 MHz (1H) and 100.61 MHz (13C) at 25 °C. Column chromatography was performed on 60 (Merck) and the Silufol UV-254 plates were used for TLC analysis.Preparation of 1-(R-ethynyl)-9,10-anthraquinones. Method A
A mixture of terminal acetylene (4.5 mmol), 1-iodo-9,10-anthraquinone (4.5 mmol), CuI (0.027 mmol), PdCl2(PPh3)2 (0.013 mmol) and Et3N (12.7 mmol) in 50 mL of toluene was stirred under an argon atmosphere at 65 °C (2–4 h) The reaction mixture was cooled and filtered through SiO2 (25 × 30 mm) using toluene as the eluent. The solvents were evaporated in vacuo. The residue was recrystallized from toluene to give compounds 1a–e,i.1-Phenylethynyl-9,10-anthraquinone (1a). The yield is 1.25 g (90%), m.p. 160–161 °C (from toluene), m.p.(lit) 159–160 °C.17
1-[2-(4-Nitrophenyl)ethynyl]-9,10-anthraquinone (1b). The yield is 1.52 g (95.6%), m.p. 214–215 °C (from toluene), m.p.(lit) 214–215 °C.17
1-[2-(4-Methoxyphenyl)ethynyl]-9,10-anthraquinone (1c). The yield is 1.49 g (98%), m.p. 194–195 °C (from toluene), m.p.(lit) 193–194 °C.17
1-(Pyridin-2-ylethynyl)-9,10-anthraquinone (1d). The yield of 1.25 g (90%), m.p. 186–187 °C (from toluene). IR (KBr): 1673 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 2220 (C
O), 2220 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) cm−1.1H NMR (CDCl3) δ (ppm): 7.25–7.30 (1H, m, H–3), 7.71–7.81 (5H, m, H–6, H–7, Py), 8.04 (1H, dd, J = 1.4, 7.72 Hz, H–2), 8.25–8.27 (1H, m, H–4), 8.32–8.36 (2H, m, H–5, H–8), 8.66–8.67 (1H, m, H–14). 13C NMR (CDCl3) δ (ppm): 88.39 (C–11), 94.14 (C–12), 122.47 (C–1), 123.05 (C–15), 126.79 (C–17), 127.46 (C–8), 127.87 (C–5), 127.89 (C–4), 132.58 (C–10a), 132.84 (C–16), 133.60 (C–9a), 133.75 (C–7), 133.88 (C–8a), 134.29 (C–6), 134.35 (C–4a), 136.09 (C–3), 140.41 (C–2), 143.29 (C–13), 150.10 (C–14), 181.66 (C–10), 182.49 (C–9). Anal. calcd (%) for C21H11NO2: C (81.54%), H (3.58%), N (4.53%). Found: C (81.69%), H (3.49%), N (4.24%).
C) cm−1.1H NMR (CDCl3) δ (ppm): 7.25–7.30 (1H, m, H–3), 7.71–7.81 (5H, m, H–6, H–7, Py), 8.04 (1H, dd, J = 1.4, 7.72 Hz, H–2), 8.25–8.27 (1H, m, H–4), 8.32–8.36 (2H, m, H–5, H–8), 8.66–8.67 (1H, m, H–14). 13C NMR (CDCl3) δ (ppm): 88.39 (C–11), 94.14 (C–12), 122.47 (C–1), 123.05 (C–15), 126.79 (C–17), 127.46 (C–8), 127.87 (C–5), 127.89 (C–4), 132.58 (C–10a), 132.84 (C–16), 133.60 (C–9a), 133.75 (C–7), 133.88 (C–8a), 134.29 (C–6), 134.35 (C–4a), 136.09 (C–3), 140.41 (C–2), 143.29 (C–13), 150.10 (C–14), 181.66 (C–10), 182.49 (C–9). Anal. calcd (%) for C21H11NO2: C (81.54%), H (3.58%), N (4.53%). Found: C (81.69%), H (3.49%), N (4.24%).
1-(1,5-Dimethylpyrazol-4-ylethynyl)-9,10-anthraquinone (1e). The yield is 1.34 g (91%), m.p. 177–178 °C (from toluene), m.p.(lit) 177–178 °C.3c
1-(3′-Hydroxy-3′-methylbutynyl)-9,10-anthraquinone (1i). The yield is 1.05 g (80%), m.p. 157–158 °C (from toluene), m.p.(lit) 157.5–158.5 °C.18
Preparation of 1-(R-ethynyl)-9,10-anthraquinones. Method B
A mixture of 1-iodo-9,10-anthraquinone (6 mmol) and copper acetylides (9 mmol) in 60 mL of pyridine was stirred under a stream of argon at 60 °C for 40 min. Then CH2Cl2 (250 mL) was added, the organic layers were washed with 25% aqueous NH3 (2 × 100 mL) and water (250 mL), then dried over Na2SO4. The crude product was purified by column chromatography on Al2O3 (elution with toluene). Subsequent recrystallization gave pure compounds 1f–h.1-(Hexyn-1-yl)-9,10-anthraquinone (1f). The yield is 1.44 g (80%), m.p. 75–76 °C (from hexane), m.p.(lit) 92–93 °C.2
1-(Heptyn-1-yl)-9,10-anthraquinone (1g). The yield is 1.5 g (83%), m.p. 62–63 °C (from hexane). IR (KBr): 1675 (CO), 216 (CC), 2949–2846 (Alk) cm−1. 1H NMR (CDCl3) δ (ppm): 0.95 (3H, t, J = 7.2 Hz, –CH3), 1.36–1.45 (2H, m, –CH2–Me), 1.49–1.57 (2H, m, –CH2–Et), 1.74 (2H, p, J = 7.1, 14.4 Hz, –CH2–Pr), 2.60 (2H, t, J = 7.1 Hz, –CH2–Bu), 7.66 (1H, t, J = 7.7 Hz, H–3), 7.72–7.81 (2H, m, H–6, H–7), 7.84 (1H, dd, J = 1.3, 7.7 Hz, H–2), 8.26 (2H, dd, J = 1.4, 7.7 Hz, H–5, H–8), 8.31–8.33 (1H, m, H–4). 13C NMR (CDCl3) δ (ppm): 14.19 (–CH3), 20.27 (–CH2–Me), 22.43 (–CH2–Et), 28.52 (–CH2–Pr), 31.39 (–CH2–Bu), 80.34 (C–11), 98.46 (C–12), 124.86 (C–1), 126.85 (C–8), 126.97 (C–5), 127.64 (C–4), 132.82 (C–7), 132.92 (C–10a), 133.33 (C–9a), 133.80 (C–6), 134.40 (C–8a), 134.44 (C–3), 134.55 (C–4a), 140.96 (C–2), 182.24 (C–10), 183.08 (C–9). Anal. calcd (%) for C21H18O2: C (83.42%), H (6.00%). Found: C (83.95%), H (5.96%).
1-(3-Phenoxypropyn-1-yl)-9,10-anthraquinone (1h). The yield is 1.4 g (70%), m.p. 141–142 °C (from toluene), m.p.(lit) 140–141 °C.2
Reaction of 1-(R-ethynyl)-9,10-anthraquinones with urea
A mixture of 1-(R-ethynyl)-9,10-anthraquinone (3 mmol) and urea 30 g (0.5 mol) was stirred at 135–140 °C for 2–36 h. Then the hot melt was added to ice and the precipitated product was filtered and purified by column chromatography on Al2O3 (elution with toluene). Subsequent recrystallization gave pure compounds 2a–i.2-Phenyl-7H-dibenzo[de,h]quinolin-7-one (2a). M.p. 204–205 °C (from toluene-hexane), m.p.(lit) 207–208 °C.2
2-(4-Nitrophenyl)-7H-dibenzo[de,h]quinolin-7-one (2b). M.p. 294–295 °C (from 1,4-dioxane). IR (KBr): 1346, 1518 (NO2),1666 (CO) cm−1. 1H NMR (CDCl3) δ (ppm): 7.72 (1H, td, J = 1.2, 7.49 Hz, H–9), 7.89 (1H, dt, J = 1.2, 7.4 Hz, H–10), 7.99 (1H, t, J = 7.8 Hz, H–5), 8.27 (1H, d, J = 7.8 Hz, H–4), 8.30 (1H, s, H–3), 8.41–8.52 (5H, m, H–8, Ho, Hm), 8.71 (1H, dd, J = 0.9, 7.4 Hz, H–6), 9.09 (1H, d, J = 7.8 Hz, H–11). 13C NMR (CDCl3) δ (ppm): 118.14 (C–3), 122.79 (C–11c), 124.33 (Co), 125.72 (C–11), 125.90 (Cm), 127.91 (C–8), 129.45 (C–6a), 130.59 (C–6), 131.03 (C–9), 131.39 (C–5), 132.77 (C–7a), 134.02 (C–4), 134.30 (C–10), 136.13 (C–3a), 136.62 (Ci), 145.27 (C–11a), 148.35 (C–11b), 149.24 (C–2), 149.37 (Cp), 183.36 (C–7). Anal. calcd (%) for C22H12N2O3: C (74.99%), H (3.43%), N (7.95%). Found: C (74.93%), H (3.39%), N (7.83%).
2-(4-Methoxyphenyl)-7H-dibenzo[de,h]quinolin-7-one (2c). M.p. 206–207 °C (from toluene). IR (KBr): 1660 (CO) cm−1. 1H NMR (CDCl3) δ (ppm): 3.92 (3H, s, –CH3), 7.07 (2H, dt, J = 2.9, 2.1, 8.9 Hz, Hm), 7.64 (1H, td, J = 1.4, 7.8 Hz, H–5), 7.79–7.85 (2H, m, H–10, H–9), 8.02 (1H, s, H–3), 8.11 (1H, dd, J = 1.1, 8.2 Hz, H–4), 8.21 (2H, dt, J = 2.9, 2.1, 8.9 Hz, Ho), 8.40 (1H, dd, J = 1.1, 7.8 Hz, H–8), 8.55 (1H, dd, J = 1.1, 7.3 Hz, H–6), 9.03 (1H, dd, J = 1.1, 7.8 Hz, H–11). 13C NMR (CDCl3) δ (ppm): 55.57 (–CH3), 114.37 (Cm), 115.44 (C–3), 121.70 (C–11c), 125.63 (C–11), 127.61 (C–8), 128.44 (Co), 129.18 (C–6), 129.26 (C–6a), 130.38 (C–9), 130.64 (C–5), 131.81 (Ci), 132.64 (C–7a), 133.68 (C–4), 133.98 (C–10), 136.38 (C–3a), 137.07 (C–11a), 148.20 (C–11b), 151.63 (Cp), 160.68 (C–2), 183.67 (C–7). Anal. calcd (%) for C23H15NO2: C (81.88%), H (4.48%), N (4.15%). Found: C (81.86%), H (4.36%), N (4.03%).
2-(Pyridin-2-yl)-7H-dibenzo[de,h]quinolin-7-one (2d). M.p. 236–237 °C (from 1,4-dioxane). IR (KBr): 1660 (CO) cm−1. 1H NMR (CDCl3) δ (ppm): 7.36–7.40 (1H, m, H–14), 7.67 (1H, t, J = 7.7 Hz, H–15), 7.85 (1H, t, J = 7.4 Hz, H–5), 7.88–7.95 (2H, m, H–9, H–10), 8.27 (1H, d, J = 8.2 Hz, H–4), 8.43 (1H, d, J = 7.7 Hz, H–8), 8.65 (1H, d, J = 7.2 Hz, H–13), 8.74–8.80 (2H, m, H–6, H–16), 8.89 (1H, s, H–3), 9.07 (1H, d, J = 8.0 Hz, H–11). 13C NMR (CDCl3) δ (ppm): 117.95 (C–3), 121.63 (C–14), 123.07(C–11c), 123.94 (C–16), 125.53 (C–11), 127.74 (C–8), 129.35 (C–6a), 130.16 (C–6), 130.54 (C–9), 130.86 (C–5), 132.68 (C–7a), 134.04 (C–4), 134.09 (C–10), 136.31 (C–3a), 136.96 (C–11a), 137.23 (C–15), 148.16 (C–11b), 149.49 (C–13), 150.65 (C–12), 156.15 (C–2), 183.57 (C–7). Anal. calcd (%) for C21H12N2O: C (81.80%), H (3.92%), N (9.09%). Found: C (81.28%), H (3.86%), N (9.12%).
2-(1,5-Dimethylpyrazol-4-yl)-7H-dibenzo[de,h]quinolin-7-one (2e). M.p. 238–239 °C (from toluene). IR (KBr): 1656 (CO), 2927, 3064 (CH3) cm−1. 1H NMR (CDCl3) δ (ppm): 2.77 (3H, s, C–CH3), 3.90 (3H, s, N–CH3), 7.60 (1H, td, J = 1.1, 7.6 Hz, H–5), 7.69 (1H, s, H–16), 7.70–7.78 (2H, m, H–10, H–11), 7.91 (1H, s, H–3), 7.98 (1H, d, J = 7.6 Hz, H–4), 8.35 (1H, dd, J = 0.9, 7.7 Hz, H–8), 8.46 (1H, dd, J = 0.9, 7.2 Hz, H–6), 8.76 (1H, dd, J = 7.9 Hz, H–11). 13C NMR (CDCl3) δ (ppm): 11.72 (C13–CH3), 36.59 (N–CH3), 116.03 (C–3), 119.65 (C–13), 120.86 (C–11c), 125.30 (C–11), 127.59 (C–8), 128.82 (C–6), 129.06 (C–6a), 130.22 (C–9), 130.48 (C–5), 132.56 (C–7a), 133.23 (C–4), 133.87 (C–10), 136.12 (C–3a), 137.08 (C–11a), 137.61 (C–16), 137.82 (C–12), 148.01 (C–11b), 148.40 (C–2), 183.50 (C–7). Anal. calcd (%) for C21H15N3O: C (77.52%), H (4.65%), N (12.91%). Found: C (77.44%), H (4.59%), N (12.74%).
2-Butyl-7H-dibenzo[de,h]quinolin-7-one (2f). M.p. 94–95 °C (from hexane), m.p.(lit) 97–98 °C.2
2-Pentyl-7H-dibenzo[de,h]quinolin-7-one (2g). M.p. 90–91 °C (from hexane). IR (KBr): 1666 (CO), 2854, 2921, 2954 (Alk) cm−1. 1H NMR (CDCl3) δ (ppm): 0.93 (3H, t, J = 6.5 Hz, –CH3), 1.38–1.47 (4H, m, –CH2–CH2–Me), 1.92 (2H, p, J = 7.2, 14.6 Hz, –CH2–Pr), 3.02 (2H, t, J = 7.6 Hz, –CH2–Bu), 7.51 (1H, s, H–3), 7.62 (1H, d, J = 7.6 Hz, H–5), 7.76–7.84 (2H, m, H–9, H–10), 8.03 (1H, d, J = 8.2 Hz, H–4), 8.39 (1H, d, J = 7.8 Hz, H–8), 8.55 (1H, d, J = 7.2 Hz, H–6), 8.94 (1H, d, J = 8.1 Hz, H–11). 13C NMR (CDCl3) δ (ppm): 14.22 (–CH3), 22.75 (–CH2–Me), 29.59 (–CH2–Et), 31.74 (–CH2–Pr), 38.38 (–CH2–Bu), 118.581 (C–3), 121.33 (C–11c), 125.48 (C–11), 127.55 (C–8), 128.91 (C–6), 129.14 (C–6a), 130.18 (C–9), 130.34 (C–5), 132.52 (C–7a), 133.19 (C–4), 133.94 (C–10), 136.00 (C–3a), 137.12 (C–11a), 148.03 (C–11b), 157.27 (C–2), 183.78(C–7). Anal. calcd (%) for C21H19NO: C (83.69%), H (6.35%), N (4.65%). Found: C (83.81%), H (6.23%), N (4.28%).
2-(Phenoxymethyl)-7H-dibenzo[de,h]quinolin-7-one (2h). M.p 169–170 °C (from toluene), m.p.(lit) 174–175 °C.2
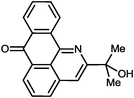
2-(2-Hydroxypropan-2-yl)-7H-dibenzo[de,h]quinolin-7-one (2i). M.p. 192–193 °C (from toluene–hexane), m.p.(lit) 196–197 °C.18
Acknowledgements
This work was supported by the Interdisciplinary Grant No.93 of SB of the Russian Academy of Sciences (2009–2011), Grant RFBR No. 10-03-00257-a; (2010–2012), Grant 5.9.3. of the Russian Academy of Sciences (2009–2011) and the Chemical Service Centre of SB RAS. Work at FSU has been supported by National Science Foundation (CHE-0848686).References
- Acetylene Chemistry: Chemistry, Biology and Material Science; C F.Diederich, P. J. Stang, R. R.Tykwinski, ed.; Wiley-VCH: Weinheim, 2005.
- (a) M. S. Shvartsberg, I. D. Ivanchikova and S. F. Vasilevsky, Tetrahedron Lett., 1994, 35, 2077–2080 CrossRef CAS; (b) M. S. Shvartsberg, I. D. Ivanchikova and S. F. Vasilevsky, Russ. Chem. Bull., Engl. Ed., 1998, 47, 1871–1974 Search PubMed.
- (a) S. F. Vasilevsky, D. S.Baranov, V. I. Mamatyuk, Y. V. Gatilov and I. V. Alabugin, J. Org. Chem., 2009, 74, 6143–6150 CrossRef CAS; (b) D. S. Baranov, S. F. Vasilevsky, V. I. Mamatyuk and Y. V. Gatilov, Mendeleev Commun., 2009, 19, 326–328 CrossRef CAS; (c) D. S. Baranov and S. F. Vasilevsky, Russ. Chem. Bull., 2010, 59, 1031–1034 CrossRef CAS; (d) Electrophilic cyclizations: A. A. Stepanov, L. M. Gornostaev, S. F. Vasilevsky, E. V. Arnold, V. I. Mamatyuk, D. S. Fadeev, B. Gold and I. V. Alabugin, J. Org. Chem. DOI:10.1021/jo2014214.
- K. Sonogashira, Y. Tohda and N. A. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- C. E. Castro and R. D. Stephens, J. Org. Chem., 1963, 28, 2163 CAS.
- (a) For the earlier use of urea and similar solvents in green chemistry processes, see: C. D. Doyle and J. M. Tour, Carbon, 2009, 47, 3215–3218 CrossRef CAS; (b) W. E. Ford, A. Jung, A. Hirsch, R. Graupner, F. Scholz and A. Yasuda, Adv Mater., 2008, 19, 1193–1197 Search PubMed; (c) J.-D. Richey, Agric. Biol. Chem., 1975, 39, 683–385 CrossRef CAS; (d) S. Gore, S. Baraskaran and B. Koenig, Green Chem., 2011, 13, 1009–1013 RSC.
- Handbook of Chemistry and Physics; R. C. Weast, M. J. Astle, W. H. Beyer, ed.; CRC Press, Inc: USA, 1984.
- (a) For earlier studies on electronic and structural factors promoting endo-dig cyclizations, see: S. F. Vasilevsky, T. F. Mikhailovskaya, V. I. Mamatyuk, G. A. Bogdanchikov, M. Manoharan and I. V. Alabugin, J. Org. Chem., 2009, 74, 8106–8117 CrossRef CAS; (b) I. V. Alabugin, V. I. Timokhin, J. N. Abrams, M. Manoharan, I. Ghiviriga and R. Abrams, J. Am. Chem. Soc., 2008, 130, 10984–10995 CrossRef CAS; (c) I. V. Alabugin and M. Manoharan, J. Am. Chem. Soc., 2005, 127, 12583–12594 CrossRef CAS; (d) I. V. Alabugin and M. Manoharan, J. Am. Chem. Soc., 2005, 127, 9534–9545 CrossRef CAS; (e) K. Gilmore and I. V. Alabugin, Chem. Rev., 2011 DOI:10.1021/cr200164y , in print; (f) I. Alabugin, K. Gilmore and M. J. Manoharan, J. Am. Chem. Soc., 2011, 133, 12608 CrossRef CAS.
- (a) The hemiaminal N–H bond in the hemiaminal should be more acidic than N–H bonds in urea due to the additional stabilization of heminal anionic center by hyperconjugative nN→σ*(C–O) interaction with involves the hydroxyl group oxygen. For the many roles of negative hyperconjugation in organic structure and reactivity, see: I. V. Alabugin, K. Gilmore and P. Peterson, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 109–141 CrossRef CAS; (b) I.V. Alabugin, M. Manoharan and T. A. Zeidan, J. Am. Chem. Soc., 2003, 125, 14014–14031 CrossRef CAS.
- (a) For recent transformations where exothermic fragmentations were used to drive an unfavorable equilibrium, see: A. Baroudi, J. Alicea and I. V. Alabugin, Chem.–Eur. J., 2010, 16, 7683–7687 CrossRef CAS; (b) A. Baroudi, P. Flack and I. V. Alabugin, Chem.–Eur. J., 2010, 16, 12316–12320 CrossRef CAS; (c) A. Baroudi, J. Mauldin and I. V. Alabugin, J. Am. Chem. Soc., 2010, 132, 967–979 CrossRef CAS; (d) A. Baroudi, J. Alicea, P. Flack, J. Kirincich and I. V. Alabugin, J. Org. Chem., 2011, 76, 1521–1537 CrossRef CAS; (e) R. Pal, R. J. Clark, M. Manoharan and I. V. Alabugin, J. Org. Chem., 2010, 75, 8689–8692 CrossRef CAS; (f) S. Roy, M. P. Davydova, R. Pal, K. Gilmore, G. A. Tolstikov, S. F. Vasilevsky and I. V. Alabugin, J. Org. Chem., 2011, 76, 7482–7490 CrossRef CAS.
- (a) A. P. Krapcho and K. J. Shaw, J. Org. Chem., 1983, 48, 3341–3343 CrossRef CAS; (b) F. Ebel, Patent, DE614196, 1935 Search PubMed; (c) F. Ebel, Patent, US2086704, 1937 Search PubMed; (d) G. Boffa and G. Chiusoli, Patent, DE2010665, 1970 Search PubMed; (e) G. Goffa, G. Pieri and N. Mzzaferro, Gazz. Chim. Ital., 1972, 102, 697–708 Search PubMed; (f) M. V. Gorelik, M. V. Kazankov and M. I. Bernadskii, J. Org. Chem. USSR Engl. Ed., 1976, 12, 1988–1989 Search PubMed; (g) G. Ribaldone, G. Borsotti and F. Gonzati, Patent, DE2434466, 1974 Search PubMed; (h) M. V. Gorelik, S. P. Titova and M. A. Kanor, Russ. J. Org. Chem. Engl.Ed., 1992, 28, 1858–1864 Search PubMed; (i) J. Kunitomo and M. Satoh, Tetrahedron, 1983, 30, 3261–3265 CrossRef; (j) J. Kunitomo, S. Kaede and M. Satoh, Chem. Pharm. Bull., 1985, 33, 2778–2782 CAS.
- H. Guinaudeau, M. Lebceuf and A. Cave, J. Nat. Prod., 1994, 57, 1033–1135 CrossRef CAS.
- (a) B.-W. Yu, L.-H. Meng, J.-Y. Chen, T.-X. Zhou, K.-F. Cheng, J. Ding and G.-W. Qin, J. Nat. Prod., 2001, 64, 968–970 CrossRef CAS; (b) H. Tang, X.-D. Wang, Y.-B. Wei, S.-L. Huang, Z.-S. Huang, J.-H. Tan ;, L.-K. An, J.-Y. Wu, A. S.-C. Chan and L.-Q. Gu, Eur. J. Med. Chem., 2008, 43, 973–980 CrossRef CAS; (c) Y. D. Min, S. U. Choi and K. R. Lee, Arch. Pharmacal Res., 2006, 29, 627–632 CrossRef CAS.
- H. Tang, F.-X. Ning, Y.-B. Wei, S.-L. Huang, Z.-S. Huang, A. S.-C. Chan and L.-Q. Gu, Bioorg. Med. Chem. Lett., 2007, 17, 3765–3768 CrossRef CAS.
- (a) S. Iwashima, T. Ueda, H. Honda, T. Tsujioka, M. Ohno, J. Aoki and T. Kan, J. Chem. Soc., Perkin Trans. 1, 1984, 2177–2187 RSC; (b) J. King and G. R. Ramage, J. Chem. Soc., 1954, 936–938 RSC.
- K. Köberle and F. Ebel, Patent, DE627258, 1936 Search PubMed.
- A. A. Moroz, A. V. Piskunov and M. S. Shvartsberg, Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.), 1981, 30, 304–308 CrossRef.
- A. V. Piskunov, A. A. Moroz and M. S. Shvartsberg, Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.), 1987, 36, 755–760 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: Energies and geometries for reactants, intermediates, and transition states calculated at B3LYP/6-31G(d,p)]. See DOI: 10.1039/c1ra00622c/ |
| This journal is © The Royal Society of Chemistry 2011 |