Functionalized graphene oxide modified polysebacic anhydride as drug carrier for levofloxacin controlled release
Jie
Gao
a,
Feng
Bao
*a,
Lingling
Feng
a,
Kaiyan
Shen
a,
Qiandong
Zhu
a,
Dongfang
Wang
a,
Tao
Chen
b,
Rui
Ma
*b and
Chunjie
Yan
b
aCollege of Chemistry, Central China Normal University, Wuhan, 430079, China. E-mail: polymerbaofeng@yahoo.com.cn
bEngineering Research Center of Nano-Geomaterials of Ministry of Education, China University of Geosciences, Wuhan, 430074, China. E-mail: marui.cug@gmail.com
First published on 20th October 2011
Abstract
Functionalized graphene oxide modified polysebacic anhydride composites were synthesized by Steglich esterification and characterized by various techniques such as Fourier transform-infrared spectrometry, differential scanning calorimetry, gel permeation chromatography, X-ray powder diffraction, dynamic light scattering, and scanning electron microscopy. In the release study using these composites, the release kinetics of drug loaded composites were studied in phosphate buffer saline, when the pH value was 7.4, at 37 °C using levofloxacin as a model antibacterial drug. In contrast to the pure polysebacic anhydride, different feeding ratio of graphene oxide modified polysebacic anhydride provides much longer drug release duration, and the drug release duration could be controlled by the molecular weight of the composites. In addition, the 2% graphene oxide modified polysebacic anhydride (Mw = 5000 D) exhibits a nearly perfect linear release behavior with an extended release time as long as 80 days, and the effective drug release amount exceeding 95%. The results of XRD indicated that the prepared graphene oxide contains adsorbing water, and it led to an obvious decrease in the molecular weight of polysebacic anhydride grafted on graphene oxide. This has affected the release behaviors to a certain extent. High molecular weight polysebacic anhydride grafted on dried graphene oxide and their release behaviors will be provided in our future work.
Introduction
Graphene oxide (GO) offers interesting electronic, mechanical, and novel biocompatible properties that are currently being explored for advanced sensors,1–4 electronic devices,5–8 and composites.9–12Graphene oxide has a one-atom-thick two-dimensional layered carbon system, which contains several kinds of oxygen functional groups on their basal planes and edges. Its excellent physical and chemical properties have attracted great attention from scientists of materials, chemistry, physics and biology. Potential applications of graphene oxide have been widely investigated, and the biosafety of graphene oxide in cells and biosystems has been reported.13,14Graphene oxide under a low dose (0.1 mg) and medium dose (0.25 mg) would not exhibit obvious toxicity to mice and under a high dose (0.4 mg) exhibit chronic toxicity. Further studies15–17 indicated that graphene oxide can be used for cellular imaging and cancer drug delivery, such as SN38 and doxorubicin. However, up to now, no report is closely associated with the applications of graphene oxide loaded with antibiotic in the treatment of chronic osteomyelitis.Once acute haematogenous osteomyelitis or infection of open fracture becomes chronic, systemic administration of antibiotics usually cannot attain an effective level at the site of bone infection. Further more, long-term use of intravenous antibiotics may increase toxicity and side effect of the antibiotics. Though PMMA beads containing gentamycin have been locally used to treat osteomyelitis for years, the drawbacks are obvious, an additional surgical procedure is needed to remove the beads.18 To overcome this obstacle, a series of biodegradable materials, such as polyanhydrides,19–21 and polylactide acid22–24 have been explored to deliver antibiotics.
In view of the facts mentioned above, the improvement of the controlled release properties of polysebacic anhydride (PSA) were realized by using the novel filler of graphene oxide, a prolonged and controlled release time was expected with a different ratio of the graphene oxide modified polysebacic anhydride (GO/PSA). In this article, high-grade graphene oxide and high molecular weight polysebacic anhydride were synthesized through modified Hummers method and melt condensation polymerization respectively. GO/PSA composites were obtained when the GO and PSA undergo Steglich esterification, which was catalyzed by the dicyclohexyl carbodiimide and 4-dimethylaminopyridine. After being characterized using various techniques, the mixture of modified polysebacic anhydride and polylactic acid was chosen as the matrix, and levofloxacin (LOF) as a model antibacterial drug to investigate its release properties. The release kinetics of the pill were studied in phosphate-buffered saline (PBS), when the pH value was 7.4, at 37 °C.
Results and discussion
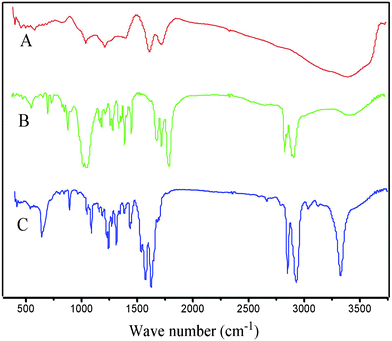
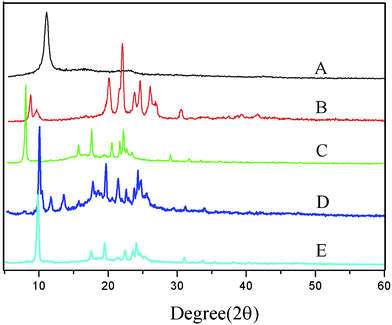
In a typical preparation, GO was synthesized from graphite powder by a modified Hummers method.25,26FT-IR spectra of the GO (Fig. 1, A) clearly marked the presence of GO as evident from the observed bands at 3407 cm−1 (O–H stretching), 1735 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1408 cm−1 (O–H bending), 1054 cm−1 (C–O bending).27,28Differential scanning calorimetry (DSC) (Fig. 2, A) curves suggested the GO decomposed from 189.4 °C, and the structure of GO was completely destructed at 243.1 °C, with an intense exothermic peak at this point.29X-Ray powder diffraction (XRD) patterns of the GO (Fig. 3, A) implied the presence of graphene oxide for the most intensive peak at around 2θ = 10.6°, which corresponds to an interlayer spacing of 0.75 nm. This is much larger than that of pristine graphite (0.34 nm) and pure graphene oxide (0.61 nm), due to the introduction of the oxygen-containing functional groups and adsorbed water on the graphite sheets.29,30 This was a clear indication of the complete transformation from graphite to GO.15,31Dynamic light scattering (DLS) results indicated that the size of the GO made via the modified method was 538.3 nm, with a polydispersity index of 0.582. Scanning electron microscopy (SEM) of the GO (Fig. 4a, 4b) revealed that the exfoliated graphene oxide material consisted of layered structures.32,33 The wrinkled and folded surface resulted in the graphene oxide having a large aspect ratio and specific area, which is beneficial for filler like graphene oxide.
O stretching), 1408 cm−1 (O–H bending), 1054 cm−1 (C–O bending).27,28Differential scanning calorimetry (DSC) (Fig. 2, A) curves suggested the GO decomposed from 189.4 °C, and the structure of GO was completely destructed at 243.1 °C, with an intense exothermic peak at this point.29X-Ray powder diffraction (XRD) patterns of the GO (Fig. 3, A) implied the presence of graphene oxide for the most intensive peak at around 2θ = 10.6°, which corresponds to an interlayer spacing of 0.75 nm. This is much larger than that of pristine graphite (0.34 nm) and pure graphene oxide (0.61 nm), due to the introduction of the oxygen-containing functional groups and adsorbed water on the graphite sheets.29,30 This was a clear indication of the complete transformation from graphite to GO.15,31Dynamic light scattering (DLS) results indicated that the size of the GO made via the modified method was 538.3 nm, with a polydispersity index of 0.582. Scanning electron microscopy (SEM) of the GO (Fig. 4a, 4b) revealed that the exfoliated graphene oxide material consisted of layered structures.32,33 The wrinkled and folded surface resulted in the graphene oxide having a large aspect ratio and specific area, which is beneficial for filler like graphene oxide.
 | ||
| Fig. 1 FT-IR spectra of GO (A), pure PSA (B), 1% GO/PSA (C). | ||
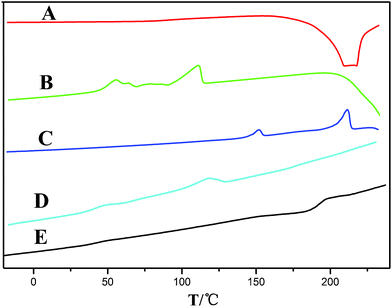 | ||
Fig. 2
DSC curves of GO (A), PSA (B), 1% GO/PSA (C), PSA (Mw = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 D) + PLA + LOF (D), 1% GO/PSA (Mw = 11000 D) + PLA + LOF(E). 000 D) + PLA + LOF (D), 1% GO/PSA (Mw = 11000 D) + PLA + LOF(E). | ||
 | ||
| Fig. 3
XRD patterns of GO (a), PSA (Mw = 30000 D) (b), 1% GO/PSA (c), 2% GO/PSA (d), 4% GO/PSA (e). | ||
 | ||
| Fig. 4 SEM images of GO at low-solution (a) and high-solution (b); image of 1% GO/PSA (c); image of 2% GO/PSA (d); image of 4% GO/PSA (e). | ||
High molecular weight polysebacic anhydride was synthesized through melt condensation polymerization. The FT-IR spectra (Fig. 1, B) marked the presence of PSA, as evident from the observed bands at 3434 cm−1 (O–H stretching), 2931 cm−1 (–CH2– stretching), 1812 cm−1, 1742 cm−1 (CO stretching), 1071 cm−1 (C–O bending).34 The DSC curves of PSA (Fig. 2, B) showed that two endothermic peaks due to PSA were located at 55.6 °C and 113.0 °C, and decomposed from 231.3 °C to 283.8 °C. PSA is a semi-crystalline polymer with the strong characteristic peaks observed in the XRD pattern (Fig. 3, B). The peaks for pure PSA were at 2θ values of 8.3°, 19.7°, 21.7°, 23.4°, 24.3° and 25.7°, respectively. The characteristic peaks at 2θ values of 9.2°, 23.4° and 30.2° indicated that a few sebacic acid monomers exist, which was reported by Gong et al.35 Three kinds of polysebacic anhydride (Mw = 30000 D; Mw = 16000 D; Mw = 11000 D) were synthesized by changing the ratio of acetic anhydride and sebacic acid. The data in Table 1 indicated that an increase in the molecular weight (PSA) related to the ratio of acetic anhydride and sebacic acid, with an increase in molecular weight achieved an increase in ratio.
| Sample | SA (g) | AA (ml) | Yield (%) | M w (D) | M w/Mn |
|---|---|---|---|---|---|
| 1 | 8 | 80 | 82.4 | 30000 |
1.314 |
| 2 | 8 | 40 | 79.1 | 16000 |
1.036 |
| 3 | 8 | 30 | 70.2 | 11000 |
1.040 |
GO/PSA composites were fabricated through Steglich esterification reaction catalyzed by DCC and DMAP. Centrifugation was used to remove the non-reacted GO, and various techniques were carried out to ensure that the esterification reaction was successfully completed.36,37
FT-IR experiments were carried out to investigate the interaction between the GO and PSA in 1% GO/PSA (Fig. 1, C). It was found that PSA had reacted with suspended hydroxyl in GO to yield esters, as evidenced by the almost disappearance of the peaks at 1735 cm−1 of GO and 1812 cm−1, 1742 cm−1 of PSA, and the new bands at 1626 cm−1 and 1575 cm−1, respectively; this is consistent with the results published before.30,36,37
1% GO/PSA (Mw = 14000 D) was studied by DSC under a nitrogen atmosphere (Fig. 2, C). Regarding the heating scan, it can be observed that Tm and the melting enthalpy (ΔHm) are dramatically influenced by the presence of graphene oxide, which also demonstrated the presence of PSA and the filler GO in the covalent linkage. This effect can be explained as the effective covalent attachment of the polymer to the graphene oxide, which prevents the segment motions of the polymer chains. Conspicuously, the presence of the huge rigid graphene oxide structure in the polymer chains restricts the movements intensively.12,35,36,38Fig. 2, D–E show the thermostability of the composites PSA (Mw 11000 D) + PLA + LOF and 1% GO/PSA (Mw 11000 D) + PLA + LOF, it indicates that the introduction of GO increases the thermostability dramatically. This could be due to the formation of π–π stacking between GO and LOF, similar to the appearance of red-shifts which has been reported.15–17 The spectral features of the composites with other feeding ratios were similar.
Different feeding ratios (1%, 2%, 3%, 4%, 5%) of graphene oxide modified with two kinds of polysebacic anhydride (Mw = 30000 D; Mw = 16000 D) are shown in Table 2 and Table 3 respectively. By comparing the molecular weight of the GO/PSA and the pure polysebacic anhydride used for synthesizing GO/PSA, we know that the molecular weight of all the composites decreased after esterification due to the GO adsorbing water,29 as no water was produced in the course of Steglich esterification.37 Moreover, all the polymers prepared had a narrow molecular distribution.
Fig. 2, C–E, show the XRD patterns of 1% GO/PSA, 2% GO/PSA and 4% GO/PSA respectively. After the GO reacted with PSA, the XRD pattern of the GO/PSA composites showed only the PSA diffraction peaks with the diffraction peaks of GO having disappeared.12 It is noticed that for all three GO/PSA mentioned above, an obvious increase in the intensity of the 8.3° 2θ peak was observed, with no obvious decrease in the intensity of the peaks at 19.7°, 21.7°, 23.4°, 24.3° and 25.7° 2θ. The first peak indicates the existence of the crystalline structure, while the rest of the peaks correspond to an amorphous structure.39 This could be due to the increased crystallinity which inhibited the PSA phase introduced by the graphene oxide, and the molecular weight decreasing process40 is similar with carbon nanotubes.41
Fig. 4 c–e, shows SEM images of GO/PSA particles with different feeding ratios (1%, 2% and 4%) and shows that the GO is well-dispersed in the PSA matrix. The SEM images of the three GO modified PSA particles are similar.12 The slight difference observed among them can be explained by the difference in their molecular weights. With a decrease in the molecular weight, the surface of the particles becames more rough, which can affect the performances in the drug release dramatically.42
In Table 4, the size of the GO, and the three kinds GO/PSA were listed, their sizes are smaller than 1 μm. The size of GO was 538.3 nm, and GO modified PSA are larger than that of GO; this also indicates the attachment of PSA to GO. The size of composites and the geometry of the pill has a great influence on the release kinetics.42,43
Polysebacic anhydride has been considered to be a useful medical polymer as a carrier for drugs, with its excellent biocompatibility and unique degradability. When it was used in the release system, polyanhydrides can exhibit fine surface corrosion and the effect of zero order release,44 which means that the drug release rate was not affected by the time and drug concentration. However, it’s very difficult to obtain high molecular weight polyanhydrides and it is hard to process, so it cannot be used as a matrix for drug delivery solely. To solve this problem, we chose the mixture of polylactic acid and pure or modified polysebacic anhydride as carrier for the antibacterial drug levofloxacin in our study.45–47 Upon blending the two together, the combined advantages of the drug releasing properties of the two polymers was expected, and by changing the molecular weight of the polysebacic anhydride in the blend, we can achieve a long release time. The images and structure profiles of the pills were shown in Fig. 5, a–d. The pill containing pure PSA and GO/PSA were white and black respectively. From the structure profiles of the component in the pill, we know that the difference of the pill's color was caused by the addition of graphene oxide.
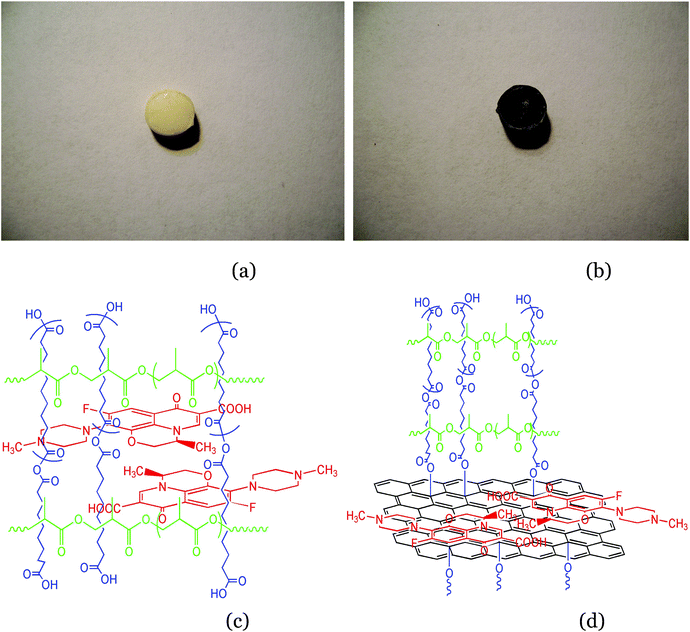 | ||
| Fig. 5 Images of the pill containing pure PSA (a), containing 1% GO/PSA (b); structure profile of the pill containing pure PSA (c), containing 1% GO/PSA (d). | ||
The drug release behavior of the pills containing four different kinds of PSA are shown in Table 5, which clearly suggested a dramatic increase in the effective release time. All of the pills containing modified PSA exhibited a longer release time than that of the pill only containing pure PSA, which also revealed that the introduction of graphene oxide and the feeding ratio play a significant role in modifying the release properties. From the dynamic release profiles shown in Fig. 6, we know that the release rate of the pill containing pure PSA is the fastest, and 2% graphene oxide modified polysebacic anhydride (Mw = 5000 D) exhibited a nearly perfect linear release behavior with an extended release time as long as 80 days. This release kinetics is is consistent with surface corrosion of polyanhydride.48 The release duration increase in the order of 1% GO/PSA < 4% GO/PSA < 2% GO/PSA. This can be explained by the sizes and surface morphologies of the modified composites. The sizes (Table 4) and SEM photos (Fig. 4, c–e) of GO/PSA indicate that the lower the molecular weight, the smaller the size and the more wrinkles on the surface; this is unfavourable for the diffusion of the LOF molecule.42 The effective release time was changed with the molecular weight range of GO/PSA composites, and all of the pills obtained a high effective drug release amount.
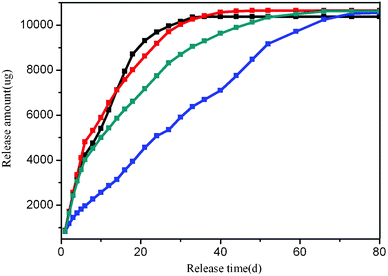 | ||
| Fig. 6 Dynamic release profiles of LOF from pure PSA–PLA (black), 1% GO/PSA–PLA (red), 2% GO/PSA–PLA (blue), 4% GO/PSA–PLA (grey). | ||
| Sample | Molecular weight (D) | First day | Effective release time (d) | Effective release amouts/% | |
|---|---|---|---|---|---|
| Weight (μg) | Ratio/% | ||||
| (1) PSA | 11000 |
878.6 | 7.9 | 36 | 93.43 |
| (2) 1wt% GO/PSA | 14000 |
870.1 | 7.8 | 48 | 95.85 |
| (3) 2wt% GO/PSA | 5000 |
823.3 | 7.4 | 80 | 95.01 |
| (4) 4wt% GO/PSA | 11000 |
844.2 | 7.6 | 66 | 95.58 |
The pill we made was Ø = 6 mm, d = 3.4 mm, with a smooth surface, and the amount of the active compound levofloxacin in the pill was 11 mg. The effective release time of the pill containing only pure PSA was 36 days, which is consistent with the report given by Chen et al.45,46 The mechanism for the increase of the effective release time may be explained, as shown in Fig. 7, A–C. The drug release kinetics of the pill contain GO/PSA (Fig. 7, A) were listed as follows, the bulk erosion22 of PLA and PSA link to GO, the strong π–π stacking between LOF and GO, so the release of GO/PSA loaded LOF from the PLA matrix should be the first step. Then, the decomposition of PSA linked on the GO, for the surface corrosion of PSA.48 The last should be the release of LOF from GO because of the strong π–π stacking interaction.49,50 While the LOF in the pill just containing pure PSA can be released into the phosphate-buffered saline directly by only one step (Fig. 7, B), the third step (Fig. 7, C) for the release of the pill containing GO/PSA play a vital role for the increase of the effective release time, as the binding of LOF onto GO/PSA was noncovalent in nature, driven by hydrophobic and π–π stacking between LOF and aromatic regions of the graphene oxide sheets.
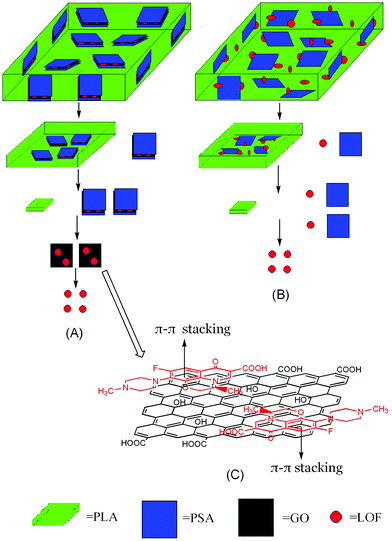 | ||
| Fig. 7 A schematic illustration of release mechanism for the pill contain GO/PSA (A), contain pure PSA (B), the interaction of π–π stacking between GO and PSA (C). | ||
Conclusion
In summary, we successfully fabricated biocompatible graphene oxide modified polysebacic anhydride composites, by Steglich esterification of high molecular weight polysebacic and high grade graphene oxide. The drug release behavior of the GO/PSA composites in vitro suggests a dramatic increase of effective release time. The smaller in size and more wrinkles on the surface, the longer the effective release time, which was dependent upon the molecular weight of GO/PSA. This study lays the foundation of graphene oxide modified biomedical polymer materials in the field of controlled release. The novel graphitic structures, combined with multi-functionalities including biocompatibility, drug loading and delivery, suggest promising applications of graphene oxide materials in biological and medical areas. Imperfectly, the prepared graphene oxide contains adsorbed water, which leads to an obvious decrease in the molecular weight of polysebacic anhydride grafted on graphene oxide. This may influence the release behaviour to a certain extent. High molecular weight polysebacic anhydride grafted on a dried graphene oxide and their release behaviour will be investigated in our future work.Experimental
Graphite powder was purchased from Sinopharm Chemical Reagent Co. Ltd. Sebacic acid was purchased from Shanghai Nanxiang Reagent Co. Ltd. Acetic anhydride was purchased from Henan Huarun Plant. N-N-Dicyclohexylcarbodiimide (DCC) was purchased from Giant Industry Group. 4-Dimethyl-aminopyridine (DMAP) was purchased from Shanghai Weifang Fine Chemical Co. Ltd. Polylactic acid was purchased from Zhongnan Hospital of Wuhan university. All chemicals were of analytical grade, and doubly distilled water was used throughout the experiment.Preparation of GO modified PSA composites
In a typical procedure, GO was made by a modified Hummers method,25,26 using purified graphite powder as starting material (Scheme 1), and the PSA was synthesized through melt condensation polymerization34,51,52 (Scheme 2). The esterification procedure36,37 for GO and PSA is shown in Scheme 3: 0.8 g of PSA and corresponding feeding ratios of GO were suspended in DMSO (20 ml), the suspension was gently stirred and maintained at 70 °C under nitrogen for 3 days. Then a solution of N-N-dicyclohexylcarbodiimide and 4-dimethylaminopyridine in DMSO were added, and the resulting mixture was stirred at room temperature for another 3 days. The coagulation of the polymer composite was accomplished by adding the suspension into 100 ml of methanol under vigorous stirring. The solid composite was filtered, washed with methanol and dried at 50 °C under vacuum. In order to eliminate non-reacted GO, the composites were redissolved in DMSO, centrifuged at a high speed, which completely removed the non-reacted GO and the supernatant coagulated with methanol, and filtered to obtain a gray powder. | ||
| Scheme 1 The procedure of synthesis of GO. | ||
 | ||
| Scheme 2 The procedure of PSA synthesis by sebacic acid and acetic anhydride. | ||
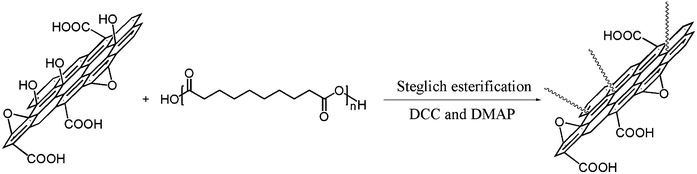 | ||
| Scheme 3 The procedure of GO/PSA synthesis by GO and PSA. | ||
Preparation of LOF–GO/PSA–PLA pill and the in vitrodrug release kinetics45,46
The specific method was as follows: 0.9 g PLA dissolved into 20 ml THF at 60 °C, then 0.1 g pure or modified PSA and 0.125 g levofloxacin was added under stirring. When all the THF was volatilized, a white powder of the mixture was obtained. Then, 120 mg of the mixture was added into a specific mould, after heating and other processing, a white, gray or black round pill of approximately 100 mg was obtained. To obtained different drug release duration of the pill, four kinds of PSA was chosen for the release study as follows: a) pure PSA (Mw = 11000 D); b) 1% GO/PSA (Mw = 14000 D); c) 2% GO/PSA (Mw = 5000 D); d) 4% GO/PSA (Mw = 11000 D). For the in vitro test of drug release, all of the four kinds of pills were respectively put into 10 ml phosphate buffer (pH = 7.4) in a test tube which was under a homoeothermic water bath at 37 °C. The buffer was regularly replaced at different time point within 80 days of the test. The duration of effective drug release (minimal effective concentration >2 μg ml−1)53 was determined by testing the levofloxacin concentration in the regularly replaced buffer, through UV-vis spectroscopy.
Materials characterization
Fourier transform-infrared (FT-IR) and ultraviolet-vis spectroscopy (UV-vis) absorption were recorded on a Perkin- Elmer 983 and Labtech 1000 spectrophotometer, respectively. Gel permeation chromatography (GPC) were conducted with a Shimadzu LC-20AD chromatograph which use THF as mobile phase, PS as standard. Differential scanning calorimetry (DSC) was performed using a Diamond PE calorific analyzer under nitrogen atmosphere with a heating rate of 10 °C min−1 in all cases. X-Ray powder diffraction (XRD) was recorded on a Rigaku Multiflex powder diffractometer with Cu radiation between 5° and 90° with a scan rate of 0.5° min−1 and incident wavelength of 0.154056 nm (Cu Kα). Dynamic light scaterring (DLS) was conducted by a Britain Malvern zetasizer ZEN3690 at room temperature. Scanning electron microscopy (SEM) was conducted with a Cambridge S-250MK3 UK microscope with an acceleration voltage of 10 kV.Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (CUGL100407) and the National Natural Science Foundation of China (50903077, 20872044), the Natural Science Foundation of Hubei Province (2009CDB163, 2008CDZ065).References
- P. K. Ang, W. Chen, A. T. S. Wee and K. P. Loh, Solution-Gated Epitaxial Graphene as pH Sensor, J. Am. Chem. Soc., 2008, 130, 14392–14393 CrossRef CAS.
- F. Schedin, A. K. Geim and K. S. Novoselov, et al., Detection of individual gas molecules adsorbed on graphene, Nat. Mater., 2007, 6, 652–655 CrossRef CAS.
- R. S. Sundaram, C. G. Navarro and K. Kern, et al., Electrochemical Modification of Graphene, Adv. Mater., 2008, 20, 3050–3053 CrossRef CAS.
- J. T. Robinson, F. K. Perkins and P. E. Sheehan, et al., Reduced Graphene Oxide Molecular Sensors, Nano Lett., 2008, 8, 3137–3140 CrossRef CAS.
- K. Sint, B. Y. Wang and l. P. K rá., Selective Ion Passage through Functionalized Graphene Nanopores, J. Am. Chem. Soc., 2008, 130, 16448–16449 CrossRef CAS.
- C. A. Di, D. C. Wei, Y. Q. Liu and D. B. Zhu, et al., Patterned Graphene as Source/Drain Electrodes for Bottom-Contact Organic Field-Effect Transistors, Adv. Mater., 2008, 20, 3289–3293 CrossRef CAS.
- G. Eda, G. Fanchini and M. Chhowalla, Large-area ultrathin films of reduced graphene oxide as a transparent and flexible electronic material, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS.
- C. Y. Wang, D. Li, C. O. Too and G. G. Wallace, Electrochemical Properties of Graphene Paper Electrodes Used in Lithium Batteries, Chem. Mater., 2009, 21, 2604–2606 CrossRef CAS.
- S. Chen, J. W. Zhu, X. D. Wu, Q. F. Han and X. Wang, Graphene Oxide–MnO2 Nanocomposites for Supercapacitors, ACS Nano, 2010, 4, 2822–2830 CrossRef CAS.
- Z. F. Liu, Q. Liu, Y. Huang, Y. F. Ma, S. G. Yin, X. Y. Zhang, W. Sun and Y. S. Chen, Organic Photovoltaic Devies Based on A Novel Acceptor Material: Graphene, Adv. Mater., 2008, 20, 3924–3930 CrossRef CAS.
- S. J. Park, D. A. Dikin, S. T. Nguyen and R. S. Ruoff, Graphene Oxide Sheets Chemically Cross-Linked by Polallyamine, J. Phys. Chem. C, 2009, 113, 15801–15804 CAS.
- X. M. Yang, Y. F. Tu, L. Li, S. M. Shang and X. M. Tao, Well-Dispersed Chitosan/Graphene Oxide Nanocomposites, ACS Appl. Mater. Interfaces, 2010, 2, 1707–1713 CAS.
- K. Wang, J. Ruan, H. Song, J. L. Zhang, Y. Wo, S. W. Guo and D.X. Cui, Biocompatibility of graphene oxide, Nanoscale. Res. Lett., 2011 DOI:10.1007/s11671-010-9751-6.
- H. Q. Chen, M. B. Müller, K. J. Gilmore, G. G. Wallace and D. Li, Mechanically Strong, Electrically Conductive, and Biocompatible Graphene Paper, Adv. Mater., 2008, 20, 3557–3561 CrossRef CAS.
- X. M. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. J. Dai, Nano-Graphene Oxide for Cellular Imaging and Drug Delivery, Nano Res., 2008, 1, 203–212 CrossRef CAS.
- Y. Yang, X. Y. Zhang, Z. F. Liu, Y. F. Ma, Y. Huang and Y. S. Chen, High-efficiency Loading and Controlled Release of Doxorubicin Hydrochloride on Graphene Oxide, J. Phys. Chem. C, 2008, 112, 17554–17558 Search PubMed.
- Z. Liu, J. T. Robins, X. M. Sun and H. J. Dai, PEGylated Nanographene Oxide for Delivery of Water-Insoluble Cancer Drug, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS.
- C. C. Chang and K. Merritt, Microbial adherence on polymthylmethacrylate (PMMA) surface, J. Biomed. Mater. Res., 1992, 26, 197–207 CrossRef CAS.
- D. S. Katti, S. Lakshmi, R. Langer and C. T. Laurencin, Toxicity, Biodegradation and Elimination of Polyanhydrides, Adv. Drug Delivery Rev., 2002, 54, 933–961 CrossRef CAS.
- K. W. Leong, P. D. Amore, M. Marletta and R. Langer, Bioerodible Poly-anhydrides as Drug Carrier Matrices II. Biocompatibilty and Chemical Reactivity, J. Biomed. Mater. Res., 1986, 20, 51–64 CrossRef CAS.
- J. P. Jain, D. Chitkara and N. Kumar, Polyanhydrides as Localized Drug Delivery Carrier: An Update, Expert Opin. Drug Delivery, 2008, 5(8), 889–907 CrossRef CAS.
- M. Miyajima, A. Koshika, O. Jun’ichi and M. Ikeda, Mechanism of drug release from poly(L-lactic acid) matrix containing acidic or neutral drugs, J. Controlled Release, 1999, 60, 199–209 CrossRef CAS.
- M. Miyajima, A. Koshika, O. Jun’ichi, A. Kusai and I. Masaru, Factors influencing the diffusion-controlled release of papaverine from poly(L-lactic acid) materix, J. Controlled Release, 1998, 56, 85–94 CrossRef CAS.
- F. Ahmed and D. E. Discher, Self-porating polymersomes of PEG–PLA and PEG–PCL: hydrolysis-triggered controlled release vesicles, J. Controlled Release, 2004, 96, 37–53 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, Preparation of Graphite Oxide, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- L. Fu, H. B. Liu, H. Yan and B. Li, Technology Research on Oxidative Degree of Graphite Oxide Prepared by Hummers Method, Carbon, 2005, 4, 10–14 Search PubMed.
- R. L. Jay, D. D. Condell and V. K. Dmitry, Diazonuim Functionalization of Surfactant Rapped Chemically Converted Graphene Sheets, J. Am. Chem. Soc., 2008, 130, 16201–16206 CrossRef.
- T. Szabo, O. Berkesi and I. Dekany, Drift Study of Deuterium Exchanged Graphite Oxide, Carbon, 2005, 43, 3186–3189 CrossRef CAS.
- Y. Z. Su, C. B. Liu, R. F. Zhang and Y. F. Xian, Research on the Synthesis and Structure of Graphite Oxide, Journal of Guang zhou Normal University., 2000, 21, 55–59 CAS.
- C. Xu, X. Wu, J. Zhu and X. Wang, Synthesis of Amphiphilic Graphite Oxide, Carbon, 2007, 46, 386–389 CrossRef.
- T. Wei, Z. J. Fan, G. L. Luo, C. Zheng and D. S. Xie, A Rapid and Efficient Method to Prepare Exfoliated Graphite by Microwave Irradiation, Carbon, 2008, 47, 337–339 CrossRef.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. Nguyen and R. S. Ruoff, Preparation and Characterization of Graphene Oxide Paper, Nature, 2007, 448, 457–460 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Synthesis of Graphene-Based Nanosheets Via Chemical Reduction of Exfoliated Graphite Oxide, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- N. Zhang and S. G. Guo, Synthesis of High Molecular Poly-Sebacic Anhydride, Chin. Pharm. J., 2005, 11, 866–868 Search PubMed.
- K. Gong and I. U. Darr Rehman, Synthesis of Poly(sebacicanhydride)-Indomethacin Controlled Release Composites Via supercritical Carbon Dioxide Assisted Impregnation, Int. J. Pharm., 2007, 338, 191–197 CrossRef CAS.
- J. Horacio, M. A. G. Salavagione and M. Gerardo, Polymeric Modification of Graphene Esterification of Graphite Oxide by Poly(vinylalcohol), Macromolecules, 2009, 42, 6331–6334 CrossRef.
- B. Neises and W. Steglich, Simple Method for the Esterification of Carboxylic Acids, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef.
- J. L. Valentín, I. Mora-Barrantes, J. Carretero-González, M. A. López-Manchado, P. Sotta, D. R. Long and K. Saalwächter, Novel Experimental Approach to Evaluate Filler-elastomer Interactions, Macromolecules, 2010, 43, 334–346 CrossRef.
- J. W. Rhim, S. I. Hong, H. M. Park and P. K. Ng, Preparation and Characterization of Chitosan-Based Nanocomposite Films with Antimicrobial Activity, J. Agric. Food Chem., 2006, 54, 5814–5822 CrossRef CAS.
- D. A. Nguyen, Y. R. Lee, A. V. Raghu, H. M. Jrong, C. M. Shin and B. K. Kim, Morphological and physical properties of a thermoplastic polyurethane reinforced with functionalized graphene sheet, Polym. Int., 2009, 58, 412–417 CrossRef CAS.
- O. Probst, E. M. Moore, D. E. Resasco and B. P. Grady, Nucleation of polyvinyl alcohol crystallization by single-walled carbon nanotubes, Polymer, 2004, 45, 4437–4443 CrossRef CAS.
- K. Tongjit, K. Shunsaku and I. Yukio, Degradation and release profile of microcapsules made of poly[L-lactic acid-co-L-lysine (Z)], J. Controlled Release, 1998, 54, 283–292 CrossRef.
- H. Akbari, A. D. Emanuele and D. Attwood, Effect of geometry on the erosion characteristics of polyanhydride matrices, Int. J. Pharm., 1998, 160, 83–89 CrossRef CAS.
- K. A. Walter, A. C. Mitchell, A. Gur, B. Tyler, J. Hilton, O. M. Colvin, P. C. Burger, A. Domb and H. Brem, Intersitial Taxol Delivered from a Biodegradable Polymer Implant Against Experimental Malignant Gliomas, Cancer. Res., 1994, 8, 2207–2215 Search PubMed.
- L. B. Chen, M. Chen, J. Wang, D. J. He and L. Shang, Study on the Controlled Release System of Ofloxacin/PSA/PLA in Anti-Infection of Bone, Chin. J. Exp. Surg., 2006, 23, 1571 CAS.
- L. B. Chen, H. Wang, J. Wang, M. Chen and L. Shang, Ofloxacin-Delivery System of A Polyanhydride and Polylactide Blend Used in the Treatment of Bone Infection, J. Biomed. Mater. Res., Part B, 2007, 589–595 Search PubMed.
- L. X. Chen, K. Xiong, J. Fu and J. Wang, Study on the Preparation and Properties of Polysebacic Anhydride/Polylactide Blend Film, Journal of Wuhan university of technology, 2004, 7, 49–51 Search PubMed.
- A. Gopferich and J. Tessmar, Polyanhydride degradation and erosion, Adv. Drug Delivery Rev., 2002, 54, 911–931 CrossRef CAS.
- R. Chen, Y. Zhang, D. Wang and H. Dai, Noncovalent Sidewall Functionalization of Single-Walled Carbon Nanotubes for Protein Immobilization, J. Am. Chem. Soc., 2001, 123, 3838–3839 CrossRef CAS.
- Z. Liu, X. Sun, N. Nakayama and H. Dai, Supermolecular Chemistry in Water-soluble Carbon Nanotubes for Drug Loading and Delivery, ACS Nano, 2007, 1, 50–56 CrossRef CAS.
- A. J. Domb and R. Langer, Polyanhydrides. I. Preparation of High Molecular Weight Polyanhydrides, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 3373 CrossRef CAS.
- A. J. Domb, E. Mathiowitz, E. Ron, S. Giannos and R. Langer, Polyanhydrides. IV. Unsaturated and Crosslinked Polyanhydrides, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 571 CrossRef CAS.
- B. G. Katzung, Basic & Clinical Pharmacology, 8th edition: People’ hygiene publishing house, 2001, pp. 797–799 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |