
Supramolecular assembly of isomeric SN-38 prodrugs regulated by conjugation sites†
Zhenhai
Tang
,
Jianhua
Zhang
,
Wenting
Li
,
Kaiying
Wen
,
Zhipeng
Gu
 ,
Dongdong
Zhou
* and
Hao
Su
*
,
Dongdong
Zhou
* and
Hao
Su
*
College of Polymer Science and Engineering, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, China. E-mail: hsu@scu.edu.cn; zhoudd@scu.edu.cn
First published on 22nd May 2024
Abstract
Supramolecular polymers (SPs) are an emerging class of drug transporters employed to improve drug therapy. Through the rational design of self-assembling monomers, one can optimize the properties of the resulting supramolecular nanostructures, such as size, shape, surface chemistry, release, and, therefore, biological fates. This study highlights the design of isomeric SN38 prodrugs through the conjugation of hydrophilic oligo(ethylene glycol) (OEG) with hydroxyls at positions 10 and 20 on hydrophobic SN-38. Self-assembling prodrug (SAPD) isomers 10-OEG-SN38 and 20-OEG-SN38 can self-assemble into giant nanotubes and filamentous assemblies, respectively, via aromatic associations that dominate self-assembly. Our study reveales the influence of modification sites on the assembly behavior and ability of the SN38 SAPDs, as well as drug release and subsequent in vitro and in vivo antitumor effects. The SAPD modified at position 20 exhibits stronger π–π interactions among SN38 units, leading to more compact packing and enhanced assembly capability, whereas OEG at position 10 poses steric hindrance for aromatic associations. Importantly, owing to its higher chemical and supramolecular stability, 20-OEG-SN38 outperforms 10-OEG-SN38 and irinotecan, a clinically used prodrug of SN38, in a CT26 tumor model, demonstrating enhanced tumor growth inhibition and prolonged animal survival. This study presents a new strategy of using interactions among drug molecules as dominating features to create supramolecular assemblies. It also brings some insights into creating effective supramolecular drug assemblies via the engineering of self-assembling building blocks, which could contribute to the optimization of design principles for supramolecular drug delivery systems.
 Hao Su | Hao Su received his bachelor's degree in polymer science and engineering from Sichuan University in 2013, a master's degree in polymer science from the University of Akron in 2014 under the supervision of Prof Stephen Z. D. Cheng, and PhD in chemical and biomolecular engineering from the Johns Hopkins University in 2019 under the supervision of Prof. Honggang Cui. He was Postdoctoral Fellow with Prof. Bert Meijer at the Institute for Complex Molecular Systems at the Eindhoven University of Technology from 2019 to 2021. He is currently Assistant Professor in the Department of Polymer Science and Engineering at Sichuan University. He is a member of the Early Career Board of Biomacromolecules. His research interests include the design, synthesis and self-assembly of prodrugs, peptides and polypeptides for applications in drug delivery and tissue engineering. |
Introduction
Supramolecular polymers (SPs) constructed via the non-covalent linkage of monomeric units offer great advantages in improving the delivery of drugs.1–3 One unique feature is that the physiochemical properties of supramolecular nanostructures can be facilely controlled via the molecular-level design of self-assembling building blocks. For example, the morphologies of peptide assemblies are highly sequence-specific4–6 and sensitive to the amino acid position in isomeric peptide sequences.7–9 In other examples, the self-assembly behavior of monomeric building blocks is controlled by the attachment and removal of enzyme cleavable pendant functional groups, enabling the sol–gel transition of supramolecular materials.10,11 In addition, the composition and functionality of SPs can be tuned via the supramolecular copolymerization of multiple monomers.12,13 Among these monomeric units, self-assembling prodrugs (SAPDs) have been recently recognized as a class of novel monomers of particular interest that are capable of spontaneously associating into SPs in aqueous solutions, achieving self-formulation and self-delivery of active drugs.14,15 Conjugation of hydrophobic drugs to hydrophilic auxiliaries affords amphiphilic prodrugs, which subsequently self-assemble into discrete drug nanostructures in aqueous environments. Compared with traditional nanomedicines prepared via drug encapsulation, this strategy avoids external carriers, which could potentially overcome the heterogeneity between drug molecules and carriers, thereby significantly increasing drug loading and reducing the toxicity risk from drug leakage and external carrier materials. The physicochemical properties, such as size, shape, surface chemistry, internal ordering, and stability, of the resulting drug assemblies can be meticulously regulated through the design and refinement of the prodrug molecular structure, thereby offering high levels of operability.16,17 For example, block copolymer-drug conjugates consisting of poly(ethylene glycol)-poly(α,β-aspartic acid) (PEG-b-PAsp) and doxorubicin can assemble into drug-containing micelles.18 The Chikoti group synthesized a class of chimeric polypeptide–drug conjugates that can self-assemble into sub-100-nm-sized nanoparticles.19 The modification of small molecule drugs with short peptides created a library of peptide–drug conjugates that enable the formation of filamentous assemblies in aqueous solutions.20,21Irinotecan (CPT-11), a derivative of the prototypical anticancer drug camptothecin (CPT), manifests cytotoxicity by arresting the DNA replication process through the entrapment of topoisomerase I and remains a prevalent treatment modality for advanced colorectal cancer in the clinic.22–24 Nonetheless, the metabolic conversion rate of CPT-11 is low and highly depends on the esterase level in the patients, causing drawbacks such as rapid clearance from the bloodstream and nonspecific cytotoxicity.25 Studies have indicated that the irinotecan needs to hydrolyze to the parent drug SN-38 to exert its therapeutic efficacy, and the in vitro cytotoxicity of the parent drug, SN38, is 100–1000 times greater than that of CPT-11.26 Structurally, SN38 incorporates an additional hydroxyl compared to CPT; however, similar to CPT, it suffers from exceedingly poor solubility in water, ranging from 2 to 5 μg ml−1, instability at physiological pH, and strong binding with plasma proteins, which hinders its direct use in the clinic.27 Despite these problems, the strong potency of SN-38 attracts great interest in making it a great candidate for more effective cancer treatment.28,29 Considering the molecular structure of SN-38, the inherent strong hydrophobicity of the SN-38 molecules could serve as the driving force in self-assembly, and the π–π stacking interaction among the aromatics could provide directional forces to aggregate one-dimensionally. Therefore, chemical modification to make SN-38 amphiphilic is feasible for creating SAPDs that can self-assemble into supramolecular nanostructures. Simultaneously, the two hydroxyl modification sites on the SN38 molecule afford more possibilities and flexibility in the amphiphilic prodrug design.30 For example, the Cui group synthesized a nonionic self-assembling prodrug hydrogelator (SAPHs) by conjugating SN38 on the 20-site with a peptide auxiliary decorated with hydrophilic oligo(ethylene glycol) (OEG) on the lysine side chain.31 Remarkably, this prodrug molecule spontaneously assembles into stable nanofibers in water and, interestingly, under physiological conditions, which can respond to the Hofmeister effect to transition into a gel, thereby enabling the sustained release of SN38. Yang and colleagues synthesized a negatively charged prodrug, HCPT (10-site)-peptide (FFERGD), and co-assembled it with cisplatin.32 The resultant dual-drug assembly, aided by the positive charge of cisplatin on its outer surface, efficiently penetrates the cell nucleus, achieving promising therapeutic outcomes both in vitro and in vivo. Li, Zhang and colleagues conjugated SN38 with RGDR and HKD peptides through β-sheet peptide sequences to form co-assembled nanofibers. These fibrous nano-prodrugs effectively suppressed primary tumors and lung metastasis by upregulating integrin expression, providing a synergistic approach to chemotherapy and immunotherapy.33 To create SN-38 filamentous assemblies, current strategies predominantly involve the use of peptides as the hydrophilic auxiliaries, which could provide hydrogen bonding interactions to enhance the self-assembly capability.17,34 However, in some cases, the hydrogen bonding and π–π associations could also compete with each other, reducing the orderliness of molecular arrangements.35 Considering that the supramolecular self-assembly could occur in a π–π association-controlled manner, the utilization of peptide segments as structural domains is unnecessary. Therefore, we envision that solely using the interactions from SN-38 could also direct ordered molecular arrangements. The conjugation of short hydrophilic segments could simplify the molecular design, in which SN38 plays both therapeutic and structural roles. In addition, because SN38 has two conjugation sites, self-assembly behavior could be affected by modifying different hydroxyl positions.
In this study, we directly modify the 10- and 20-site hydroxyl groups of SN38 with pentaethylene glycol monomethyl ether (OEG5-OH) by carbonate bond, giving two prodrugs (10-OEG-SN38 and 20-OEG-SN38) that exist as isomers. The use of OEG as hydrophilic segments does not introduce extra intermolecular interactions to ensure the dominant role of π–π associations; simultaneously, it also provides neutral surface chemistry and steric effect, which could potentially reduce protein absorption and improve pharmacokinetics in circulation. The incorporation of the carbonate bond enables the degradation of the prodrugs through either chemical or enzymatic hydrolysis to release the parent drug SN-38. Luckily, both SN-38 prodrugs can self-assemble into one-dimensional nanostructures in aqueous solutions. This straightforward molecular design also enables a more intuitive understanding of how different modification sites influence the assembly behavior and capability of SN38. The findings of our study indicate a strong correlation between the π–π interactions among SN38 molecules and the sites of modification. Specifically, when the hydroxyl group at position 20 is modified, it tends to form filamentous structures with a regular chiral arrangement. This tendency leads to more compact packing and enhanced assembly capabilities. Conversely, the chiral stacking of SN38 is disrupted when modification occurs at the 10-site, resulting in compromised assembly capacity. These disparities directly affect the drug release, cytotoxicity and therapeutic efficacy of the SAPDs both in vitro and in vivo. Our results provide an example of using interactions among drug molecules as dominating features to create supramolecular assemblies and the strategic selection of the most appropriate hydroxyl modification sites for designing SN38 SAPD to maximize its performance in self-assembly and therapy.
Experimental section
1 Synthesis of OEG-NO2
Pentaethylene glycol monomethyl ether (OEG5-OH, 1.00 g, 3.9 mmol), 4-dimethylamino pyridine (DMAP, 1.65 g, 13.5 mmol) and 4-nitrobenzoyl chloride were mixed together into dry DCM (30 ml) and then stirred at room temperature overnight. The reaction mixture was evaporated to remove the solvent, and the residue was further purified by chromatography on silica gel with EA/PE (1/9 to 1/1) and then EA/MeOH (19/1) as the eluent to obtain the OEG-NO2 (colorless oil, 1.2 g, 73%). 1H NMR (300 MHz; CDCl3; ppm): δ 3.38 (s, 3H), 3.50–3.85 (m, 18H), 4.40–4.47 (m, 2H), 7.34–7.46 (m, 2H), 8.22–8.34 (m, 2H). MS (ESI-MS) m/z: [M + H]+ calculated for C18H27NO10, 418.16; found, 418.05.2 Synthesis of 10-OEG-SN38
SN38 (0.34 g, 0.86 mmol) was suspended in a DCM solution (20 ml) of OEG-NO2 (0.3 g, 0.72 mmol) and triethylamine (0.36 g, 3.59 mmol). The reaction mixture was stirred for 24 hours. After removing the solvent under a vacuum, the residue was first purified by flash chromatography on silica gel with DCM/MeOH (10/1) and then by RP-HPLC (10-SN38-OEG, white solid, 0.3 g, 62%). 1H NMR (300 MHz; CDCl3; ppm): δ 1.0–1.08 (t, 3H), 1.36–1.44 (s, 3H), 1.81–1.98 (m, 2H), 3.10–3.22 (q, 2H), 3.34–3.41 (s, 3H), 3.49–3.91 (m, 20H), 4.44–4.51 (m, 2H), 5.22–5.36 (m, 3H), 5.71–5.80 (d, 1H), 7.61–7.70 (t, 2H), 7.90–7.96 (s, 1H), 8.21–8.29 (d, 1H). MS (ESI-MS) m/z: [M + H]+ calculated for C34H42N2O12, 670.27; found, 671.12.3 Synthesis of 20-OEG-SN38
SN38 (1 g, 2.55 mmol) was suspended in a solution of di-tert-butyl dicarbonate (0.72 g, 3.3 mmol) and triethylamine (0.52 g, 5 mmol) in DCM (40 ml). After being stirred at room temperature for 48 h, the resulting mixture was washed three times with 0.5 M HCl (40 ml) and brine (40 ml) and then dried using anhydrous sodium sulfate to obtain 10-BOC-SN38 (pale-yellow solid, 1.02 g, 92%). 1H NMR (300 MHz; CDCl3; ppm): δ 0.97–1.10 (t, 3H), 1.34–1.46 (s, 3H), 1.52–1.68 (s 9H), 1.81–1.99 (m, 2H), 3.08–3.24 (q, 2H), 5.21–5.37 (m, 3H), 5.68–5.82 (d, 1H), 7.59–7.72 (t, 2H), 7.85–7.94 (s, 1H), 8.19–8.27 (d, 1H).10-BOC-SN38 (0.4 g, 0.9 mmol), OEG-NO2 (0.46 g, 1 mmol) and DMAP (0.25 g, 2 mmol) were dissolved in DCM (30 ml). The reaction mixture was heated reflux at 55 °C and stirred overnight. The yellow solution was concentrated in vacuo, and the residue was further purified using RP-HPLC. The product fractions were combined and lyophilized to give 10-Boc-20-OEG-SN38 (pale-yellow solid, 0.3 g, 43%). 1H NMR (300 MHz; CDCl3; ppm): δ 0.95–1.04 (t, 3H), 1.35–1.43 (s, 3H), 1.61–1.64 (s, 9H), 2.08–2.34 (m, 2H), 3.12–3.21 (q, 2H), 3.35–3.40 (s, 3H), 3.5–3.75 (m, 17H), 4.19–4.32 (m, 2H), 5.22–5.30 (s, 2H), 5.35–5.74 (q, 2H) 7.28–7.33 (s, 1H), 7.64–7.72 (d 1H), 7.88–7.93 (s, 1H), 8.20–8.26 (d, 1H).
Trifluoroacetic acid (TFA, 0.88 g, 7.8 mmol) was added dropwise to a solution of 10-BOC-20-OEG-SN38 (0.3 g, 0.39 mmol) in DCM (10 ml). The resulting mixture was washed with a saturated solution of NaHCO3 and dried over anhydrous sodium sulfate. The mixture was concentrated in vacuo to obtain the 20-OEG-SN38 (bright yellow powder, 0.22 g, 84.6%). 1H NMR (300 MHz; CDCl3; ppm): δ 1.01–1.05 (t, 3H), 1.25–1.38 (s, 3H), 2.10–2.30 (m, 2H), 2.96–3.10 (q, 2H), 3.35–3.45 (s, 3H), 3.46–3.78 (m, 19H), 4.13–4.33 (m, 2H), 5.02–5.18 (s, 2H), 5.32–5.77 (q, 2H) 7.28–7.30 (s, 1H), 7.31–7.38 (s 1H), 7.44–7.55 (d, 1H), 8.01–8.12 (d, 1H). MS (ESI-MS) m/z: [M + H]+ calculated for C34H42N2O12, 670.27; found, 671.13.
4 Synthesis of di-OEG-SN38
SN38 (0.5 g, 1.274 mmol) was suspended in the solution of triphosgene (0.2647 g, 0.892 mmol) in dry DCM (50 ml). DMAP (0.7165 g, 5.86 mmol) was dissolved in dry DCM (20 ml) and added dropwise to the mixture. After one hour, OEG (0.7073 g, 2.80 mmol) was then added to the mixture and stirred overnight. The mixture was washed with 0.5 M HCl (50 ml × 3) and brine (50 ml × 3). Then, the extracts were dried with anhydrous sodium sulfate. The reaction mixture was concentrated under a vacuum, and the residue was purified using RP-HPLC. The product fractions were combined and lyophilized to produce di-OEG-SN38 (pale-yellow solid, 0.6 g, 50%). 1H NMR (300 MHz; CDCl3; ppm): δ 0.99–1.05 (t, 3H), 1.25–1.39 (s, 3H), 2.10–2.30 (m, 2H), 2.96–3.10 (q, 2H), 3.08–3.78 (m, 46H), 4.17–4.32 (m, 2H), 4.44–4.51 (m, 2H), 5.26 (d, 2H), 5.39–5.69 (q, 2H), 7.30 (s, 1H), 7.67 (dd 1H), 7.94 (d, 1H), 8.24 (d, 1H). [M + H]+ calculated for C46H64N2O19, 948.41; found, 949.13.Results and discussions
Synthesis of self-assembling SN38 prodrugs
Fig. 1A and Scheme S1 (ESI†) show the molecular design of the studied self-assembling prodrugs (SAPDs), consisting of the hydrophobic SN38 and hydrophilic OEG5-OH. The commercially available SN38 possesses two different hydroxyl groups located at positions 10 and 20, exhibiting varied reactivity when coupled with OEG5-OH via carbonate bonds. By carefully regulating the molar ratio between SN38 and OEG5-OH, only phenolic (C10) or both hydroxyl groups were conjugated, yielding mono- and di-functional SN38 (labeled as 10-OEG-SN38 and di-OEG-SN38), respectively. Furthermore, the phenolic hydroxyl group could be protected by tert-butyloxycarbonyl (Boc), and the residual C20-hydroxyl group was then capped by OEG5-OH. Another mono-substituted prodrug (20-OEG-SN38) was obtained after deprotecting and served as the regio-isomer of 10-OEG-SN38. Details of the synthesis of these self-assembling prodrugs can be found in Section 1–4 and Fig. S1–S6 (ESI†). | ||
| Fig. 1 Schematic illustration of the design and synthesis of SN38 SAPDs (A). Comparison of the RP-HPLC chromatograms of 10-OEG-SN38 and 20-OEG-SN38 (B) and comparison of 1H NMR spectra of 10-OEG-SN38 and 20-OEG-SN38 (C). | ||
The HPLC trace of each compound, as shown in Fig. 1B, displays a narrow and unimodal peak, confirming the high purity of the obtained molecules. The chemical accuracy of each sample was further confirmed by LC-MS, with a series of single m/z peaks in excellent accordance with the calculated values (Fig. S1–S6, ESI†). All these characterizations clearly demonstrate the successful synthesis of the prodrugs. The resultant prodrugs were purified using reversed-phase high-performance liquid chromatography (RP-HPLC), and their chemical structures and purity were fully characterized by nuclear magnetic resonance (NMR), analytical HPLC and liquid chromatography-mass spectrometry (LC-MS). The characteristic resonances of both precursory motifs (SN38 and OEG) could be captured, and the integration agrees well with the proposed chemical structures.
Interestingly, the subtle variations between samples 10-OEG-SN38 and 20-OEG-SN38, a pair of regio-isomers with the same composition but different architectures, can be clearly identified by HPLC and 1H NMR. The eluting time of sample 20-OEG-SN38 is substantially longer than that of 10-OEG-SN38, which can be well resolved due to the appreciable difference (Fig. 1B). This may indicate that sample 20-OEG-SN38 possesses a stronger hydrophobic interaction. As depicted in Fig. 1C, obvious disparities emerge within the 4–8 ppm range corresponding to various modifications at distinct positions of SN38. Specifically, the 20-OEG-SN38 spectrum exhibits noteworthy variations. The peaks corresponding to OEG proximal to carbonate bonds undergo fragmentation into multiple peaks, accompanied by chemical shift variations from 4.5 ppm to 4.2 ppm due to the influence of the chiral carbon. In contrast, both 10-OEG-SN38 and OEG-NO2 (Fig. 1C and Fig. S1 (ESI†)) exhibit conventional doublet splitting at the same position. Another obvious transformation manifests within the 6–8 ppm regime, indicating the modifications at the 10-site hydroxyl moiety. Evidently, both 10-OEG-SN38 and 10-Boc-SN38 (Fig. 1C and Fig. S3, ESI†) show similar peaks within the 6–8 ppm range, conspicuously distinguishing them from 20-OEG-SN38.
Self-assembly of SN38 prodrugs
After dissolving these samples in an aqueous solution at a concentration of 1 mM and aging overnight at room temperature, transmission electron microscopy (TEM) was utilized to investigate their morphologies. With the alternation of the regio architecture and the number of OEG, different supramolecular nanostructures were observed. 10-OEG-SN38 self-assembled into a unique giant nanotube with 1.842 ± 0.985 μm (n > 20) in length and an approximate 64.3 ± 8.9 nm (n > 50) in diameter (Fig. 2A and Fig. S7, ESI†). The wall thickness approximates twice the extended length of molecule 10-OEG-SN38 (Fig. S8A, ESI†), indicating that the packing follows a bilayer packing model. The 20-OEG-SN38 assembled into ordered nanofibers with ca. 9.0 ± 1.1 nm (n > 50) in diameter and several micrometers in length (Fig. 2C and Fig. S9, ESI†). The varying morphologies reveal the critical importance of regio architecture on the self-assembly behaviors. Moreover, the molecular packing in supramolecular filaments generally follows the core–shell cylindrical fashion, and the diameter is close to the double size as the motif extended length.36,37 The size of the 20-OEG-SN38 assembly in diameter, however, is more than twice the molecular extended length (Fig. S8B, ESI†), indicating looser molecular packing. With an increased number of OEG, no ordered structures were observed for di-OEG-SN38, indicating that the increasing hydrophilic region may impede molecular aggregation. For convenience, this sample, which cannot form an ordered nanostructure, is not discussed further. | ||
| Fig. 2 Supramolecular polymers formed by SAPDs in water. TEM images of supramolecular assemblies of 10-OEG-SN38 (A) and 20-OEG-SN38 (C) at a concentration of 1 mM. CD of 10-OEG-SN38 (1 mM) (B) and 20-OEG-SN38 (200 μM). (D). | ||
To further illustrate the molecular arrangement of the regio-isomeric prodrugs in the assemblies, we next performed circular dichroism (CD) spectroscopy measurements. 20-OEG-SN38 exhibits two CD negative signals centered at 366 nm and a strong positive signal at 389 nm (Fig. 2D). This type of peak position has been reported in previous studies,38 resulting from exciton coupling between two adjacent chromophores in a chiral orientation, which indicates a strong exciton coupling between SN38 units. The positive peak at 389 nm suggests a chiral and right-handed helical arrangement of the SN38 molecules within the nanofibers. The CD spectrum of 10-OEG-SN38 shows only a mainly sharp peak at 240 nm (Fig. 2B), which could be attributed to chromophore interactions (n–π*). Nevertheless, there is no 389 nm positive peak in the CD signal, indicating that the modification by OEG at the 10-position disrupts the ordered aromatic packing and chiral orientation of SN38 in the assembly as the hydroxyl is on the aromatic ring. The hydroxyl at the 20-site is far from the aromatic region; therefore, the modification does not disturb the π–π stacking. The results of the CD analysis suggest that various modifications of SN38 at different sites can result in distinct interactions between SN38 SAPDs. Specifically, for 20-OEG-SN38, the regular chiral stacking and closer molecular arrangement may potentially reduce the relative volume of the hydrophobic domain, leading to the formation of core–shell structured nanofibers, while the 10-OEG-SN38 without the chiral stacking feature tends to form a giant nanotube with a bilayer structure.
Self-assembly capability and stability
To investigate the self-assembly capability of these prodrugs, their critical micelle concentrations (CMCs) were determined using dynamic light scattering (DLS). The count rate values of the aqueous solutions at various concentrations were recorded, which underwent a transition accompanying the formation of molecular aggregates. The CMC values of these prodrugs were assessed by plotting the count rate value against the concentration of the sample.17 As shown in Fig. 3A and B, the mutations were identified when the concentrations were above the CMCs. The critical values are ca. 764 μM for 10-OEG-SN38 and ca. 13 μM for 20-OEG-SN38. It is also noteworthy that the CMC value sometimes differs from the critical assembly concentration (CAC) value. The CAC, above which the molecules start to form well-defined structures, is another key parameter associated with the dynamic feature and stability of supramolecular assemblies.35 To further illustrate the self-assembly capability of the prodrugs, the CD measurements of 10-OEG-SN38 and 20-OEG-SN38 at different concentrations were carried out. As illustrated in Fig. 3C and D, characteristic peaks inherited from the equilibrium assemblies appear as the solution concentration exceeds the CACs. For example, the distinctive peaks associated with 20-OEG-SN38 at 389 nm disappeared below 20 μM, suggesting the disassembly of nanofibers (Fig. 3C). Meanwhile, concentrations surpassing 900 μM yielded a notable positive peak at 240 nm, indicative of assembly formation for 10-OEG-SN38 (Fig. 3D). The estimated values are 900–1000 μM for 10-OEG-SN38 and 10–20 μM for 20-OEG- SN38. Both CMC and CAC values suggest that the self-assembly capability of 10-OEG-SN38 significantly decreases compared with that of 20-OEG-SN38, which may be due to the reduction of aromatic interactions resulting from the steric hindrance by the attached OEG on the benzene ring. These results indicate that various modification sites of SN38 significantly affect their associations and molecular arrangement, further affecting the assembly capability.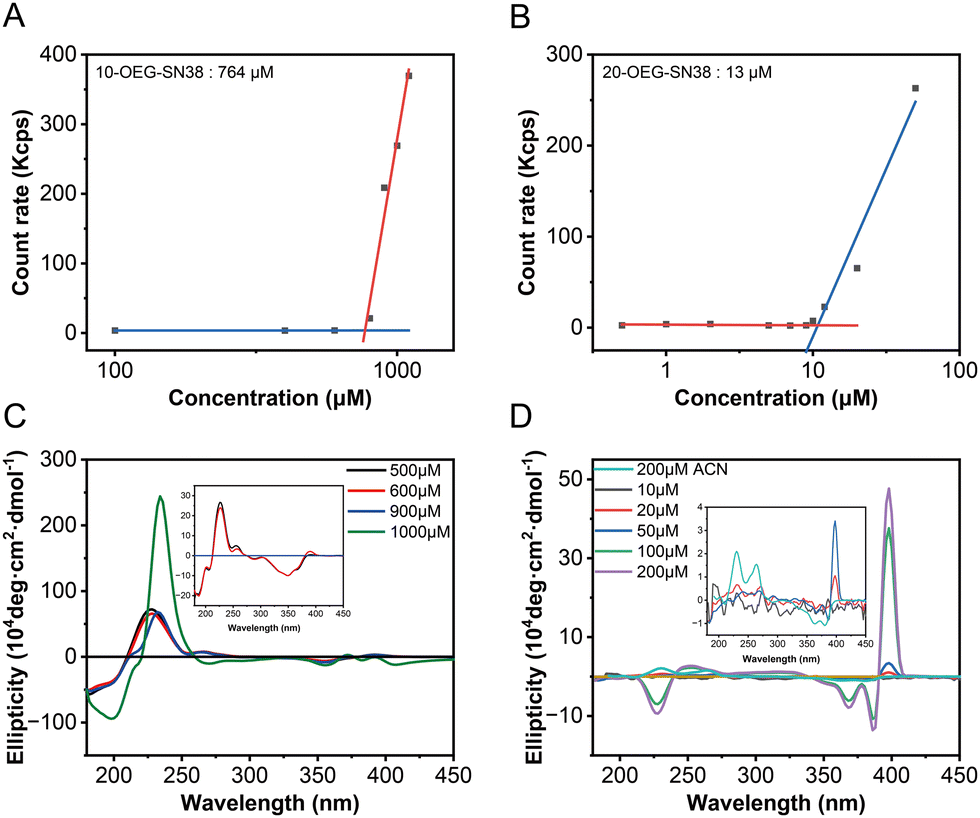 | ||
| Fig. 3 Determination of self-assembly capacity and supramolecular stability. CMC was measured using DLS methods through the mutation of the count rate. 10-OEG-SN38 (A) and 20-OEG-SN38 (B). CD for various concentrations around CAC: 10-OEG-SN38 (C) and 20-OEG-SN38 (D). | ||
Drug release
To explore drug release properties, we performed degradation experiments at 37 °C in PBS solutions with a concentration of 200 μM. Because the solubility of SN38 is extremely poor, we chose to utilize the remaining ratio of SAPDs for detection by RP-HPLC. As shown in Fig. 4A, 20-OEG-SN38 exhibits a lower degradation rate than that of 10-OEG-SN38, indicating a higher level of stability. Fig. 4B and C show a significant difference in the stability and degradation profiles between isomers at pH 7.4 monitored via analytical HPLC. Two peaks were observed in HPLC traces in the first hour, located at 8.5 min and 12 min, with the remaining ratio of 10-OEG-SN38 rapidly weakening to 40% (Fig. 4B). Monitored by LC-MS, the additional peak at 8.5 min was assigned to the carboxylate form of the prodrug (Scheme S2, ESI†) rather than the parent drug SN38 after carbonate cleavage. To confirm the pH-dependent hydrolysis of lactone, we investigated the stability of lactone in SN38 at pH 5.4. The corresponding peak is absent, revealing that the lactone stability of 10-OEG-SN38 is promoted at a lower pH value (Fig. S10, ESI†). Conversely, in the case of 20-OEG-SN38, the carboxylate form was not captured at both pH levels, which may be attributed to the enhanced stability of lactone by chemical modification at the 20-site.39 It is well known that the release of the SN38 parent drug incorporates the disassembly of supramolecular structures and the subsequent hydrolysis of carbonate bonds. Here, benefiting from the lower CMC, 20-OEG-SN38 maintains a stable assembly status at a concentration of 200 μM, which can reduce solvent and ion contact and increase the stability of prodrugs. Our results reveal that the regio architecture can significantly influence the drug release process by altering chemical stability and self-assembly capability. | ||
| Fig. 4 Drug release of SN38 SAPDs at 200 μM in PBS at pH 7.4. Drug release plots of SAPDs (A) and comparison of HPLC spectra at different time points at pH 7.4:10-OEG-SN38 (B) and 20-OEG-SN38 (C). | ||
Cytotoxicity
We next studied the in vitro cytotoxicity against colorectal cancer cells CT26 and breast cancer cells MDA-MB-231 using SAPDs and the controls (i.e., the parent drug SN38 and clinically used prodrug irinotecan) at various concentrations. The SAPDs were incubated with tumor cells for 48 hours, and their efficacy was evaluated by applying MTT methods via a dose–response relationship assay based on SN38 concentration (Fig. 5). Fig. 5A illustrates that the IC50 values of irinotecan, a clinical medication used for colorectal cancer, reach approximately 74![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 443 nM for CT26 cancer cells. Both SN38 SAPDs exhibit lower IC50 values with 518 nM for the 10-site and 1517 nM for the 20-site, suggesting a more effective inhibition of colorectal tumor cells in comparison to irinotecan. In contrast to CT26 cells, both SAPDs exhibit a higher IC50 in MDA-MB-231 cells. This lower toxicity may arise from the reduction of the degradation rate of carbonate bonds due to the relatively lower pH value within the microenvironment of breast cancer cells in comparison to colorectal cancer cells.40 It is noteworthy that the efficacies of SAPDs are just slightly lower than those of the parent drug SN38, indicating that both SAPDs can effectively release the parent drug in the cellular environment. Moreover, there are still differences in efficacy between the isomers depending on their concentration levels.
443 nM for CT26 cancer cells. Both SN38 SAPDs exhibit lower IC50 values with 518 nM for the 10-site and 1517 nM for the 20-site, suggesting a more effective inhibition of colorectal tumor cells in comparison to irinotecan. In contrast to CT26 cells, both SAPDs exhibit a higher IC50 in MDA-MB-231 cells. This lower toxicity may arise from the reduction of the degradation rate of carbonate bonds due to the relatively lower pH value within the microenvironment of breast cancer cells in comparison to colorectal cancer cells.40 It is noteworthy that the efficacies of SAPDs are just slightly lower than those of the parent drug SN38, indicating that both SAPDs can effectively release the parent drug in the cellular environment. Moreover, there are still differences in efficacy between the isomers depending on their concentration levels.
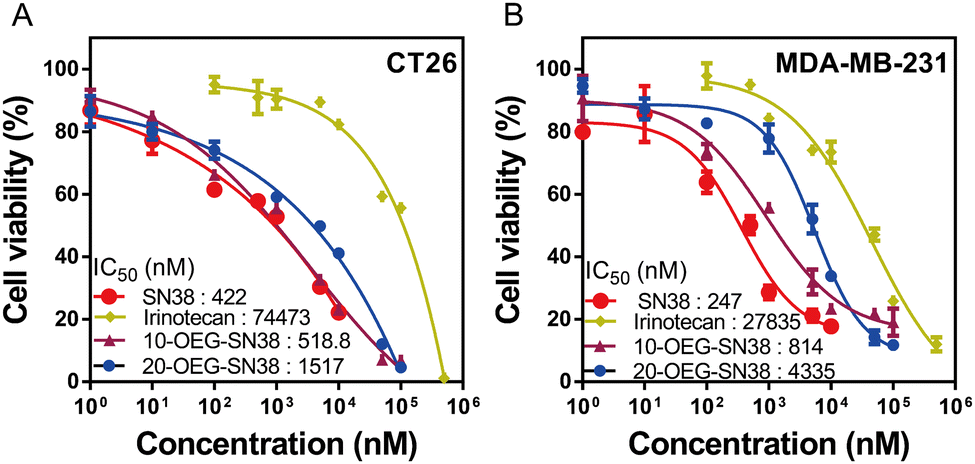 | ||
| Fig. 5 In vitro cell cytotoxicity of 10/20-OEG-SN38 against CT-26 colorectal cancer cells (A) and MDA-MB-231 breast cancer cells (B), with both free SN38 and irinotecan as controls (48-h incubation). | ||
At lower concentrations, 10-OEG-SN38 shows a higher level of cytotoxicity due to the faster hydrolysis of the carbonate bond with the phenolic hydroxyl group. Nevertheless, at concentrations ranging from 50 to 100 μM, 20-OEG-SN38 exhibits a lower cell survival rate and shows a continuous decreasing trend, especially in MDA-MB-231 cells (Fig. 5B). As mentioned above, the lactone ring of SN38 was easily opened to reduce toxicity when modified on the 10-site but remained stable on the 20-site. As the concentration increases, 20-site may release more active ingredients compared to 10-site at the same concentration, resulting in a variety of toxicity between isomers. Moreover, above its CMC (13 μM), the self-assembled nanostructures may play a certain role in cell internalization. Therefore, we can conclude that both SAPDs have excellent in vitro cytotoxicity, underscoring their substantial anticancer potential.
In vivo antitumor efficacy
To further understand how the regio architecture of SAPDs affects their in vivo performance, the antitumor efficacies of the regio-isomeric prodrugs were evaluated in a CT26 tumor model of BALB/c mice (see details in ESI†). The irinotecan dosage was set at 30 mg kg−1 every two days near its maximum tolerated dosage.41 Similarly, the dosage of SN38 in our prodrugs was 34.3 mg kg−1, matching the SN38 equivalent in irinotecan. When the tumor volume reached ∼100 mm3 after tumor inoculation, the drugs were administered once every two days for six doses (i.e., day 1, 3, 5, 7, 9, and 11) using physiological saline as a blank control. During the experiment, the weight and tumor volume of the mice were recorded as detection indicators of efficacy and safety. The body weights and tumor volumes of the mice are shown in Fig. 6A and B. After the fifth dose (day 9), the tumor volumes of the saline, irinotecan, 10-OEG-SN38, and 20-OEG-SN38 groups approached 586 mm3, 410 mm3, 124 mm3, and 98 mm3, respectively. Both SAPDs exhibited a more significant inhibitory effect on tumor growth compared with irinotecan, probably attributed to higher cytotoxicity and improved pharmacokinetics. Interestingly, although the group receiving 10-OEG-SN38 showed significant inhibition of tumor growth, we observed severe weight loss and even death in the studied animals within 11 days (Fig. 6A and C). This observation suggests that the 10-OEG-SN38 has severe toxicities in the animals studied. With a high CMC, 10-OEG-SN38 is rapidly diluted to small molecules in the bloodstream after injection, possibly leading to a fast drug release and causing toxicity to normal tissues. However, throughout the 20-OEG-SN38 treatment period, the average weight of the mice remained stable (Fig. 6A) and the 20-OEG-SN38 group showed the slowest tumor volume growth among all the treated groups (Fig. 6B). Additionally, the administration of 20-OEG-SN38 increased the median survival from 15 d (the control group) to 23 d, while the median survivals of the other groups were 11 d (10-OEG-SN38) and 17 d (irinotecan) (Fig. 6C). A reasonable explanation is that 20-OEG-SN38 can retain its nanostructure with low CMC (13 μM) upon dilution in the circulation. This could prevent rapid renal clearance and prolong circulation to improve efficacy and safety. Our results revealed the differences in toxicity and efficacy of SN38 SAPDs when modified at 10-site and 20-site, establishing the correlations among molecular design, supramolecular stability, in vitro cytotoxicity and in vivo efficacy.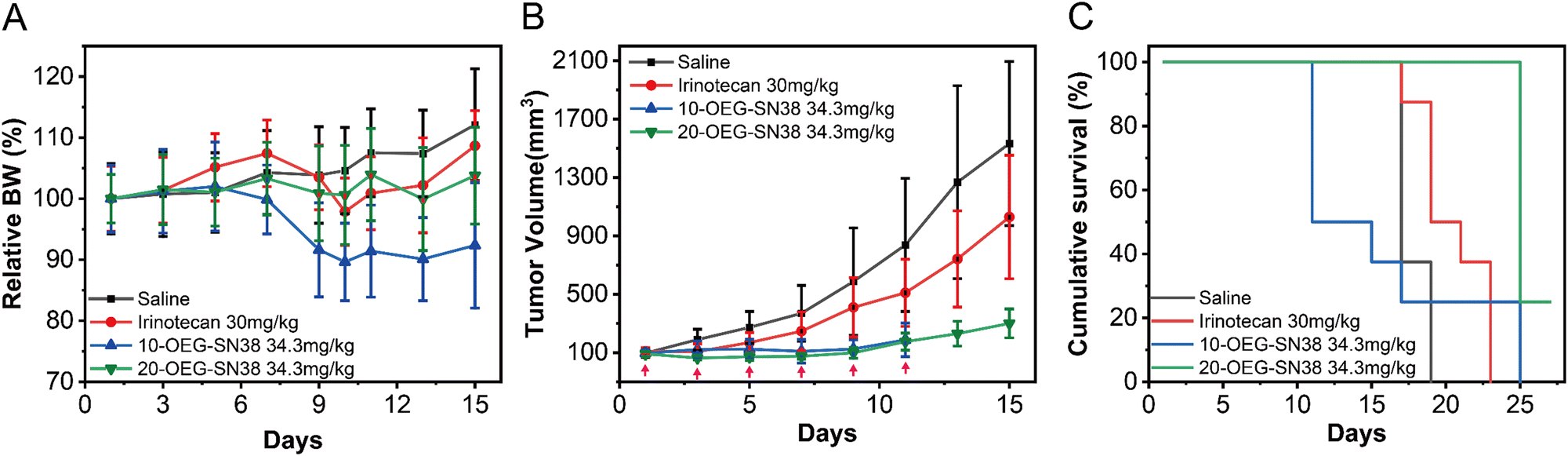 | ||
| Fig. 6 In vivo antitumor efficacy of SAPDs using irinotecan and saline as the control group with 8 mice in each group. Relative body weight (A), tumor volume (B), and percent survival (C) plots of mice between isomers. | ||
Conclusions
In this article, we synthesized a group of isomers via modification at different hydroxyl sites on SN38 to investigate their self-assembly behavior and biological functions. The SN38 SAPDs were self-assembled into giant nanotubes and nanofibers when OEG was anchored onto 10- or 20-positions. The SN38 SAPD with OEG at position 10 showed weaker aromatic associations and lower assembly capacity compared with SN-38 modified at position 20 due to the steric hindrance from OEG on the benzene ring that disrupted the close π–π stacking. The chemical and supramolecular stability of SAPDs can further influence the drug release, cytotoxicity, and therapeutic efficacy of the SAPDs both in vitro and in vivo. 20-OEG-SN38 exhibited lower cytotoxicity because of the slower hydrolysis of its carbonate bond; however, its therapeutic efficacy in tumor inhibition was better than that of commercial irinotecan. The 20-OEG-SN38 with a lower CMC retains its supramolecular integrity in the circulation, resulting in longer circulation and significantly retarding the growth of the tumor. Overall, our results indicate that the isomerization of SN38-OEG had significant effects on their associations, molecular arrangement in the assemblies, assembly capability, and, eventually, anticancer effects. Our work also presented a simplified design of amphiphilic prodrugs via direct OEGylation and gave an example of solely using the molecular interactions among drug units to create supramolecular drug assemblies without other directional intermolecular forces, such as hydrogen bonding. We hope that the current research can provide new insights into the selective modification of therapeutics into SAPDs and motivate the future design of simple SAPD systems.Author contributions
Z. T. and H. S. designed research; Z. T., J. Z., W. L., and K. W. performed research; Z. T., D. Z., and H. S. analysed data; Z. G., D. Z., and H. S. supervised the project; Z. T., D. Z., and H. S. wrote and revised the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52273136 to H.S.), the Sichuan Science and Technology Program (2023YFH0071 to H.S., 2023NSFSC0306 to D.Z.) and the Opening Project of State Key Laboratory of Polymer Materials Engineering (Sichuan University) (Grant No. sklpme2022-3-14 to H.S.). This work was also financially supported by Med-X Innovation Program of Med-X Center for Materials, Sichuan University (MCM202304 to H.S.) and the Fundamental Research Funds for the Central Universities (YJ202210 to H.S.). We would be also grateful to Minghua Zhang of the College of Polymer Science and Engineering at Sichuan University for his help of TEM analysis.Notes and references
- H. Acar, S. Srivastava, E. J. Chung, M. R. Schnorenberg, J. C. Barrett, J. L. LaBelle and M. Tirrell, Self-Assembling Peptide-Based Building Blocks in Medical Applications, Adv. Drug Delivery Rev., 2017, 110–111, 65–79 CrossRef CAS PubMed.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Supramolecular Biomaterials, Nat. Mater., 2016, 15(1), 13–26 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Functional Supramolecular Polymers, Science, 2012, 335(6070), 813–817 CrossRef CAS PubMed.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Exploring the Sequence Space for (Tri-)Peptide Self-Assembly to Design and Discover New Hydrogels, Nat. Chem., 2015, 7(1), 30–37 CrossRef CAS PubMed.
- Y. Zhang, T. Zhou, Y. Qi, Y. Li, Y. Zhang, Y. Zhao, H. Han and Y. Wang, Engineered Assemblies from Isomeric Pentapeptides Augment Dry Eye Treatment, J. Controlled Release, 2024, 365, 521–529 CrossRef CAS PubMed.
- Y. Wang, Y. Lyu and Z. Li, Constitutionally Isomeric Peptides: Subtle Changes Leading to Dramatic Differences in Properties, ACS Appl. Eng. Mater., 2024 DOI:10.1021/acsaenm.3c00787.
- H. Cui, A. G. Cheetham, E. T. Pashuck and S. I. Stupp, Amino Acid Sequence in Constitutionally Isomeric Tetrapeptide Amphiphiles Dictates Architecture of One-Dimensional Nanostructures, J. Am. Chem. Soc., 2014, 136(35), 12461–12468 CrossRef CAS PubMed.
- A. Lampel, S. A. McPhee, H.-A. Park, G. G. Scott, S. Humagain, D. R. Hekstra, B. Yoo, P. W. J. M. Frederix, T.-D. Li, R. R. Abzalimov, S. G. Greenbaum, T. Tuttle, C. Hu, C. J. Bettinger and R. V. Ulijn, Polymeric Peptide Pigments with Sequence-Encoded Properties, Science, 2017, 356(6342), 1064–1068 CrossRef CAS PubMed.
- Y. Wang, K. Kaur, S. J. Scannelli, R. Bitton and J. B. Matson, Self-Assembled Nanostructures Regulate H2S Release from Constitutionally Isomeric Peptides, J. Am. Chem. Soc., 2018, 140(44), 14945–14951 CrossRef CAS PubMed.
- Z. Liu, J. Guo, Y. Qiao and B. Xu, Enzyme-Instructed Intracellular Peptide Assemblies, Acc. Chem. Res., 2023, 56(21), 3076–3088 CrossRef CAS PubMed.
- M. Yi, Z. Feng, H. He, D. Dinulescu and B. Xu, Evaluating Alkaline Phosphatase-Instructed Self-Assembly of d-Peptides for Selectively Inhibiting Ovarian Cancer Cells, J. Med. Chem., 2023, 66(14), 10027–10035 CrossRef CAS PubMed.
- S. I. S. Hendrikse, L. Su, T. P. Hogervorst, R. P. M. Lafleur, X. Lou, G. A. van der Marel, J. D. C. Codee and E. W. Meijer, Elucidating the Ordering in Self-Assembled Glycocalyx Mimicking Supramolecular Copolymers in Water, J. Am. Chem. Soc., 2019, 141(35), 13877–13886 CrossRef CAS PubMed.
- H. Su, W. Zhang, H. Wang, F. Wang and H. Cui, Paclitaxel-Promoted Supramolecular Polymerization of Peptide Conjugates, J. Am. Chem. Soc., 2019, 141(30), 11997–12004 CrossRef CAS PubMed.
- A. G. Cheetham, R. W. Chakroun, W. Ma and H. Cui, Self-Assembling Prodrugs, Chem. Soc. Rev., 2017, 46(21), 6638–6663 RSC.
- H. Su, J. M. Koo and H. Cui, One-Component Nanomedicine, J. Controlled Release, 2015, 219, 383–395 CrossRef CAS PubMed.
- H. Ren, X.-Z. Zeng, X.-X. Zhao, D. Hou, H. Yao, M. Yaseen, L. Zhao, W. Xu, H. Wang and L.-L. Li, A Bioactivated in Vivo Assembly Nanotechnology Fabricated NIR Probe for Small Pancreatic Tumor Intraoperative Imaging, Nat. Commun., 2022, 13(1), 418 CrossRef CAS PubMed.
- C. H. Liang, Y. X. Chen, D. B. Zheng, T. Y. Xu, G. J. Pu, Y. M. Chen, L. Wang, Z. M. Yang and H. M. Wang, Pathway-Dependent Supramolecular Polymerization of Camptothecin Derivatives into Filaments for Chemotherapy and Imaging. Appl, Mater. Today, 2020, 20, 100787 CrossRef.
- K. Kataoka, A. Harada and Y. Nagasaki, Block Copolymer Micelles for Drug Delivery: Design, Characterization and Biological Significance, Adv. Drug Delivery Rev., 2012, 64, 37–48 CrossRef.
- J. Bhattacharyya, J. J. Bellucci, I. Weitzhandler, J. R. McDaniel, I. Spasojevic, X. Li, C.-C. Lin, J.-T. A. Chi and A. Chilkoti, A Paclitaxel-Loaded Recombinant Polypeptide Nanoparticle Outperforms Abraxane in Multiple Murine Cancer Models, Nat. Commun., 2015, 6(1), 7939 CrossRef CAS PubMed.
- H. Ren, X.-Z. Zeng, X.-X. Zhao, D. Hou, H. Yao, M. Yaseen, L. Zhao, W. Xu, H. Wang and L.-L. Li, A Bioactivated in Vivo Assembly Nanotechnology Fabricated NIR Probe for Small Pancreatic Tumor Intraoperative Imaging, Nat. Commun., 2022, 13(1), 418 CrossRef CAS PubMed.
- Y. Fang and H. Wang, Molecular Engineering of Peptide–Drug Conjugates for Therapeutics, Pharmaceutics, 2022, 14(1), 212 CrossRef CAS PubMed.
- Y. Pommier, Topoisomerase I Inhibitors: Camptothecins and Beyond, Nat. Rev. Cancer, 2006, 6(10), 789–802 CrossRef CAS PubMed.
- A. Makimoto, H. Mugishima, T. Taga, Y. Ishida, Y. Nagatoshi, K. Ida, M. Kumagai, T. Kimura, Y. Ohashi and M. Kaneko, Registration-Directed Phase 1/2 Trial of Irinotecan for Pediatric Solid Tumors, Pediatr. Int., 2019, 61(5), 453–458 CrossRef CAS PubMed.
- R. H. J. Mathijssen, R. J. van Alphen, J. Verweij, W. J. Loos, K. Nooter, G. Stoter and A. Sparreboom, Clinical Pharmacokinetics and Metabolism of Irinotecan (CPT-11), Clin. Cancer Res., 2001, 7(8), 2182–2194 CAS.
- C. Bailly, Irinotecan: 25 Years of Cancer Treatment, Pharmacol. Res., 2019, 148, 104398 CrossRef CAS PubMed.
- Y. Huang, L. Wang, Z. Cheng, B. Yang, J. Yu, Y. Chen and W. Lu, SN38-Based Albumin-Binding Prodrug for Efficient Targeted Cancer Chemotherapy, J. Controlled Release, 2021, 339, 297–306 CrossRef CAS PubMed.
- J. Zhao, S. Ma, Y. Xu, X. Si, H. Yao, Z. Huang, Y. Zhang, H. Yu, Z. Tang, W. Song and X. Chen, In Situ Activation of STING Pathway with Polymeric SN38 for Cancer Chemoimmunotherapy, Biomaterials, 2021, 268, 120542 CrossRef CAS PubMed.
- Y. Zheng, X. Yan, Y. Wang, X. Duan, X. Wang, C. Chen, D. Tian, Z. Luo, Z. Zhang and Y. Zeng, Hydrophobized SN38 to Redox-Hypersensitive Nanorods for Cancer Therapy, J. Mater. Chem. B, 2019, 7(2), 265–276 RSC.
- Y. Liu, H. Piao, Y. Gao, C. Xu, Y. Tian, L. Wang, J. Liu, B. Tang, M. Zou and G. Cheng, Comparison of Two Self-Assembled Macromolecular Prodrug Micelles with Different Conjugate Positions of SN38 for Enhancing Antitumor Activity, Int. J. Nanomed., 2015, 10, 2295–2311 CAS.
- X. Yang, H. Lu, Y. Tao, L. Zhou and H. Wang, Spatiotemporal Control over Chemical Assembly in Living Cells by Integration of Acid-Catalyzed Hydrolysis and Enzymatic Reactions, Angew. Chem., Int. Ed., 2021, 60(44), 23797–23804 CrossRef CAS PubMed.
- H. Wang, H. Su, T. Xu and H. Cui, Utilizing the Hofmeister Effect to Induce Hydrogelation of Nonionic Supramolecular Polymers into a Therapeutic Depot, Angew. Chem., Int. Ed., 2023, 62(43), e202306652 CrossRef CAS PubMed.
- Y. Cai, H. Shen, J. Zhan, M. Lin, L. Dai, C. Ren, Y. Shi, J. Liu, J. Gao and Z. Yang, Supramolecular “Trojan Horse” for Nuclear Delivery of Dual Anticancer Drugs, J. Am. Chem. Soc., 2017, 139(8), 2876–2879 CrossRef CAS PubMed.
- Y. Cai, B. Zhu, X. Shan, L. Zhou, X. Sun, A. Xia, B. Wu, Y. Yu, H. H. Zhu, P. Zhang and Y. Li, Inhibiting Endothelial Cell-Mediated T Lymphocyte Apoptosis with Integrin-Targeting Peptide–Drug Conjugate Filaments for Chemoimmunotherapy of Triple-Negative Breast Cancer, Adv. Mater., 2024, 36(3), 2306676 CrossRef CAS PubMed.
- S. Zeng, H. Ou, Z. Gao, J. Zhang, C. Li, Q. Liu and D. Ding, HCPT-Peptide Prodrug with Tumor Microenvironment -Responsive Morphology Transformable Characteristic for Boosted Bladder Tumor Chemotherapy, J. Controlled Release, 2021, 330, 715–725 CrossRef CAS PubMed.
- H. Su, F. Wang, W. Ran, W. Zhang, W. Dai, H. Wang, C. F. Anderson, Z. Wang, C. Zheng, P. Zhang, Y. Li and H. Cui, The Role of Critical Micellization Concentration in Efficacy and Toxicity of Supramolecular Polymers, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(9), 4518–4526 CrossRef CAS PubMed.
- A. G. Cheetham, P. Zhang, Y. Lin, L. L. Lock and H. Cui, Supramolecular Nanostructures Formed by Anticancer Drug Assembly, J. Am. Chem. Soc., 2013, 135(8), 2907–2910 CrossRef CAS PubMed.
- H. Su, P. Zhang, A. G. Cheetham, J. M. Koo, R. Lin, A. Masood, P. Schiapparelli, A. Quiñones-Hinojosa and H. Cui, Supramolecular Crafting of Self-Assembling Camptothecin Prodrugs with Enhanced Efficacy against Primary Cancer Cells, Theranostics, 2016, 6(7), 1065–1074 CrossRef CAS PubMed.
- A. G. Cheetham, P. Zhang, Y. Lin, L. L. Lock and H. Cui, Supramolecular Nanostructures Formed by Anticancer Drug Assembly, J. Am. Chem. Soc., 2013, 135(8), 2907–2910 CrossRef CAS PubMed.
- X. He, W. Lu, X. Jiang, J. Cai, X. Zhang and J. Ding, Synthesis and Biological Evaluation of Bis and Monocarbonate Prodrugs of 10-Hydroxycamptothecins, Bioorg. Med. Chem., 2004, 12(15), 4003–4008 CrossRef CAS PubMed.
- T. Volk, E. Jähde, H. P. Fortmeyer, K.-H. Glüsenkamp and M. F. Rajewsky, pH in Human Tumour Xenografts: Effect of Intravenous Administration of Glucose, Br. J. Cancer, 1993, 68(3), 492–500 CrossRef CAS PubMed.
- T. Schluep, J. Cheng, K. T. Khin and M. E. Davis, Pharmacokinetics and Biodistribution of the Camptothecin–Polymer Conjugate IT-101 in Rats and Tumor-Bearing Mice, Cancer Chemother. Pharmacol., 2006, 57(5), 654–662 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00717d |
| This journal is © The Royal Society of Chemistry 2024 |