
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEstablishing ultraporous permanently polarized hydroxyapatite as a green and highly efficient catalyst for carbon dioxide conversion in continuous flow under mild conditions†
Marc
Arnau
 a,
Jordi
Sans
*a,
Pau
Turon
*b and
Carlos
Alemán
*ac
a,
Jordi
Sans
*a,
Pau
Turon
*b and
Carlos
Alemán
*ac
aDepartament d’Enginyeria Química, EEBE and Barcelona Research Center in Multiscale Science and Engineering, Universitat Politècnica de Catalunya, Planta I-2, C/ Eduard Maristany, 10-14, Ed. I2, 08019, Barcelona, Spain. E-mail: jordi.sans.mila@upc.edu; carlos.aleman@upc.edu; Tel: (+34) 93 401 08 83
bB. Braun Surgical, S. A. U. Carretera de Terrassa 121, 08191, Rubí, Barcelona, Spain. E-mail: pau.turon@bbraun.com
cInstitute for Bioengineering of Catalonia (IBEC), The Barcelona Institute of Science and Technology, Baldiri Reixac, 10-12, 08028, Barcelona, Spain
First published on 13th August 2024
Abstract
We present the use of an ultraporous permanently polarized hydroxyapatite (upp-HAp) catalyst for continuous and highly efficient production of formic acid (predominant) and acetic acid using wet CO2 (i.e. CO2 bubbled into liquid water) as a reagent. In all cases, reactions were conducted at temperatures ranging from 95 to 150 °C, using a CO2 constant flow of 100 mL s−1, and without applying any external electric field and/or UV radiation. Herein, we study how to transfer such a catalytic system from batch to continuous reactions, focusing on the water supply (proton source): (1) wet CO2 or (2) liquid water in small amounts is introduced in the reactor. In general, the reduction of CO2 to formic acid predominates over the C–C bond formation reaction. On the other hand, when liquid water is added, two interesting outcomes are observed: (1) the yield of products is higher than in the first scenario (>2 mmol gc−1·min−1) while the initial liquid water remains largely available due to the mild reaction temperature (95 °C); and (2) a high yield of ethanol (>0.5 mmol gc−1·min−1) is observed at 120 °C, as a result of the increased efficiency of the C–C bond formation. Analysis of kinetic studies through temporal and temperature dependence shows that CO2 fixation is the rate limiting step, ruling out the competing effect of proton adsorption on the binding sites and confirming the crucial role of water. The activation energy for the CO2 fixation reaction has been determined to be 66 ± 1 kJ mol−1, which is within the range of conventional electro-assisted catalysts. Finally, mechanistic insights on the CO2 activation and role of the binding sites of upp-HAp are provided through isotopic-labeling (13CO2) and near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) studies.
Sustainability spotlightThe catalytic conversion of carbon dioxide (CO2) into value-added chemical products (CO2-revalorization) is interesting because of its potential to close the carbon cycle while generating green circular economies. However, truly sustainable CO2-revalorization processes are normally restricted by the use of noble metal-based catalysts and/or intensive reaction conditions. Accordingly, we explore the use of ultraporous permanently polarized hydroxyapatite (upp-HAp) as a catalyst for green CO2-revalorization. upp-HAp is abundant and biocompatible (main inorganic component of biological hard tissues). Besides, upp-HAp is capable of performing successful reactions under mild conditions (achievable through renewable sources) and in batch/continuous configurations, representing a viable alternative to conventional catalysts. This work is aligned with the SDG7 (affordable and clean energy) and SDG13 (climate action) UN sustainable development goals. |
Introduction
The concentration of carbon dioxide (CO2), the most significant greenhouse gas in the atmosphere, has significantly increased in recent years mainly due to the combustion of fossil fuels.1–4 This environmental impact has accelerated research activities to capture CO2, and thus avoid or minimize CO2 release into the atmosphere. Two research approaches have been prioritized looking for net zero emissions: (1) CO2 capture and long term geological storage or reutilization in several innovative ways;5–10 and (2) catalytic conversion of CO2 into value-added compounds of chemical and industrial interest using efficient catalysts and low energy consuming processes (i.e. CO2 reutilization and valorization).11–19Although many noble metal-based catalysts have been investigated for CO2 conversion into chemicals of industrial interest (e.g. Rh, Ru, Pt, Au and Pd),15,20–31 simple systems based on non-precious metal catalysts are preferred due to their lower cost and higher sustainability (e.g. Fe, Cu, Zn, and Co among others).32–40 Similarly, although efficient catalytic processes have been developed to transform CO2 into valuable chemicals using harsh reaction conditions (i.e. high temperature and/or pressure), processes involving mild reaction conditions are the most desired to avoid contradictions related to energy/mass balance inefficiencies, for example that the energy consumption and CO2 emission associated with the elimination of this greenhouse gas is higher than the CO2 transformed.41–45 A large number of studies have been devoted to the catalytic conversion of CO2 into organic products (e.g. alcohols and acids with one or more carbon atoms), with the main results being compiled in recent reviews.28,46–54
In a recent study we proposed a polarized bioceramic, a sustainable metal-free catalyst, for the conversion of CO2 into ethanol using mild reaction conditions.55 This catalyst consisted of hydroxyapatite (HAp), a naturally occurring mineral with the formula Ca5(PO4)3(OH), that is non-toxic and biocompatible and, therefore, frequently used for biomedical applications.56–59 HAp was permanently polarized by applying a thermally stimulated polarization (TSP) process at high temperature. The resulting material (named pp-HAp) became catalytically active due to extraordinary charge accumulation on its surface. Thus, pp-HAp was found to exhibit not only vacancies and charge accumulation at the HAp crystal lattice but also charge accumulation at the boundaries among the microscopic HAp crystal grains.60,61 Compact pp-HAp discs, which were prepared by applying a pressure of 620 MPa to HAp powder before applying the TSP, were found to convert CO2 into a mixture of ethanol (predominant) and some other minor compounds (i.e. acetic acid, formic acid, methanol and acetone) using a starting mixture of CO2 and CH4 (3 bar each) and 1 mL of liquid water.55 However, the yield of the reaction (expressed in μmol per gram of catalyst; μmol gc−1), which was performed in a batch reactor at 140 °C, was very low after 72 h: 18.6 ± 1.9, 2.7 ± 0.6, 1.9 ± 0.6, 1.5 ± 0.3 and 0.9 ± 0.2 μmol gc−1 for ethanol, acetic acid, formic acid, methanol, and acetone, respectively.
More recently, the performance of the pp-HAp catalyst was significantly improved by modifying the synthesis process to provide ultra-porosity and printability.62 Thus, printable inks were prepared by mixing HAp powder with Pluronic® F-127 hydrogel. The latter was removed after 3D printing the scaffold (but before applying the TSP process) by calcining the samples at 1000 °C. The resulting catalyst exhibited spherical HAp nanoparticles with an average diameter of 159 ± 43 nm. More specifically, this printed catalyst consisted of a 3D network forming branches made of coalesced nanoparticles separated by nanometric pores (average size: 167 ± 51 nm). The performance of the ultraporous pp-HAp catalyst (hereafter named upp-HAp) for the production of ethanol was studied by loading a CO2°:°CH4 mixture (3 bar each) and 1 mL of liquid water into the batch reactor and applying 140 °C for 48 h. The ethanol yield increased up to 55.0 ± 4.9 μmol gc−1, which represented an outstanding increment of the catalytic performance (by a factor of 2.9).
Despite that, most of the studies reported focus on the enhanced electrical properties of the material, rather than elucidating the catalytic mechanisms involved. In this work, we go one step further by transferring the use of the upp-HAp catalyst from the batch reactor to a continuous process where the kinetics of the conversion of CO2 into ethanol have been studied. Because of the extremely high yields obtained, we have focused on the catalytic activity of upp-HAp to clarify, for the first time, the fundaments of upp-HAp as a highly efficient and sustainable catalyst. For this purpose, first we have examined the formation of products in the different phases (i.e. adsorbed on the catalyst surface, liquid water and gas phase, as this is a heterogeneous catalytic process) as a function of the time using a batch process. Next, the influence of water, which is rapidly consumed in the continuous process, and how it is supplied have been examined (i.e. wet CO2 or liquid water). Third, the catalytic performance of upp-HAp has been studied by varying the temperature and reaction time to determine the kinetics of the reaction. Finally, isotope-labeling 13CO2 reactions and near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) studies have been performed to support the results presented. Accordingly, the catalytic mechanism of CO2 activation has been experimentally determined further establishing upp-HAp as a green catalyst for CO2 fixation.
Experimental methods
Synthesis of the ultraporous pp-HAp catalyst
The calcined HAp porous structures were thermally and electrically activated to achieve the catalytic properties by placing the samples between two stainless steel plates (AISI 304), which acted as electrodes. The HAp sample was left in control with the negative electrode, while the positive electrode was separated 4 cm from the calcined sample. A constant DC voltage of 500 V was applied for 1 h with a GAMMA power supply, while the temperature was kept at 1000 °C. Samples were allowed to cool down maintaining the applied electric potential for 30 minutes, and finally, the whole system was powered off and left to cool down overnight.
Characterization of the upp-HAp catalyst
Raman analyses were performed by means of an inVia Qontor confocal Raman microscope (Renishaw) equipped with a Renishaw Centrus 2957T2 detector and a 532 nm laser. In order to obtain representative data, 32 single point spectra were averaged.X-ray diffraction (XRD) studies were conducted using a Bruker D8 Advance model with Bragg–Brentano 2θ configuration and Cu Kα radiation (λ = 0.1542 nm). Measurements were performed in a 2θ range of 20°–60° in steps of 0.02°, and a scan speed of 2 s using a one-dimensional Lynx Eye detector.
Scanning electron microscopy (SEM) images were obtained using a Zeiss Neon40 microscope equipped with a SEM GEMINI column.
High resolution transmission electron microscopy (HRTEM) was performed using a JEOL 2010F microscope equipped with a field emission electron source and operated at an accelerating voltage of 200 kV. The point-to-point resolution was 0.19 nm, and the resolution between lines was 0.14 nm. Samples were dispersed in an aqueous (Milli-Q) suspension in an ultrasonic bath, and a drop of the suspension was placed over a grid with holey-carbon film. Images were not filtered or treated by means of digital processing and they correspond to raw data.
Near-Ambient Pressure X-ray Photoelectron Spectroscopy (NAP-XPS) experiments were performed with a SPECS Surface Nano Analysis GmbH (Berlin Germany) system. The spectrometer was equipped with an Al (XR50) anode operating at 400 W and with a Phoibos 225 (SPECS) detector (hemispherical energy analyzer, HAS). The pass energy of the hemispherical analyzer was set to 20 eV and the energy step of high resolution spectra was set at 0.1 eV. The temperature of the analysis was controlled using an Electron Beam Heater (EBH-150, SPECS Surface Nano Analysis GmbH) which has been monitored during the whole experiment through a thermocouple sensor. The C 1s peak was used as an internal reference with a binding energy of 284.5 eV. The obtained experimental spectra were fitted using a Gaussian–Lorentzian curve.
Batch reactions
The reactor consisted of an inert reaction chamber (120 mL) coated with a perfluorinated polymer where both the catalyst and water were placed. All surfaces were coated with a thin film of perfluorinated polymer in order to avoid any contact between the reactants, catalyst and the stainless steel reactor surfaces, in order to rule out other catalytic effects. The reactor was equipped with an inlet valve for the entry of gases and an outlet valve to recover the gaseous reaction products.Both the upp-HAp catalyst and deionized liquid water (20 mL) were introduced into the reactor. After exhaustive purging with CO2, the chamber pressure was increased up to 6 bar of CO2 (measured at room temperature). The reaction was conducted for 1, 4, 8 and 18 h at 120 °C. All processes were performed in triplicate. The isotope-labeling experiment was carried out using 13CO2 purchased from Cambridge Isotope Laboratories INC (purity: 13C 99%). 1 bar of 13CO2 and 5 bar of 12CO2 were introduced in the reactor. The reaction temperature was set at 120 °C for 72 h.
Continuous flow reactions
The continuous CO2 conversion into valuable chemicals was carried out using a commercial borosilicate glass continuous-flow reactor (novaLight CUBE 100, Peschl Ultraviolet GmbH, Germany). All reactions were conducted using a constant CO2 flow of 100 mL s−1. Despite the reactor being designed to allow UV irradiation, all reactions were conducted without UV light since previous studies using a batch reactor showed that such energy supply is not required when pp-HAp or upp-HAp catalysts are used.55,62 Reactions were performed introducing 1 mL of liquid water into the reactor. When performed without liquid water this is explicitly indicated for each case below. All the studied processes were performed in triplicate.Analysis of the reaction products
The reaction products dissolved in the liquid water (batch reactions), the reaction products condensed by the cold trap (continuous reaction), and the reaction products adsorbed on the catalyst were analyzed by 1H NMR spectroscopy. In order to desorb the reaction products from the catalyst, samples were dissolved in a solution containing 100 mM HCl and 50 mM NaCl with the final addition of deuterated water. All 1H NMR spectra were acquired with a Bruker Avance III-400 spectrometer operating at 400.1 MHz. The chemical shift was calibrated using tetramethylsilane as the internal standard. 256 scans were recorded in all cases to obtain an optimum signal-to-noise-ratio for proper area integration. In order to compare the different products obtained from the studied reactions, the areas associated with the proton contribution were normalized and regularly calibrated through external references to ensure a high accuracy of the measurements.After the batch reactions, the gas was collected by means of an MSHA portable pump and stored in a Tedlar® gas sampling bag (LB-2 Septa) and analyzed on a Trace1300 gas chromatograph equipped with a valve for gas injection fitted with a thermal conductivity detector (ThermoFisher Scientific). Prior to the measurements, the samples were transferred from the Tedlar® bags to multi-sorbent bed tubes (Carbotrap, Carbopack X and Carboxen 569) to ensure proper storage and manipulation.
Results and discussion
Design and characterization of the catalyst
The upp-HAp catalyst was prepared as is described in the Experimental methods section. The new properties of this catalytically active ceramic rely on the enhanced charge accumulation and boosted electron transport properties achieved through intensive lattice refinement. As depicted in Scheme 1, while vacancy engineering (i.e. generation of OH− vacancies) is considered to increase the quantity of available charges (Fig. S1 and S2†), and thus, the activity of the naturally occurring acid binding sites (Ca2+ as Lewis and P–OH as Brønsted acidic sites) and basic Lewis binding sites (O2− atoms of PO43− and OH−) of HAp,63 the TSP treatment imposes a specific global orientation to the hydroxyl groups oriented in columns along the c-axis that boosts the electron transport (Fig. S3†), and consequently, favors the catalytic conversion of carbon dioxide to new products. Note that although upp-HAp could be strictly referred to as a thermal catalyst (i.e. neither external electric currents nor UV irradiation are needed for the reaction) the nature of the catalyst strongly relies on its refined electrical properties. Hence, upp-HAp finds more similarities to the classical electroreduction mechanisms and formulations reported in the literature. In this sense, this dual approach proposes upp-HAp as a sustainable green candidate to replace the current metal-based thermocatalysts, further competing directly with the efficiencies obtained by means of conventional electrocatalysis. Such features have been widely studied in the literature in terms of the customization of the enhanced electrical properties (material characterization approach), reporting exhaustive structural analyses to ensure its proper catalytic activation.55,60–62,64–69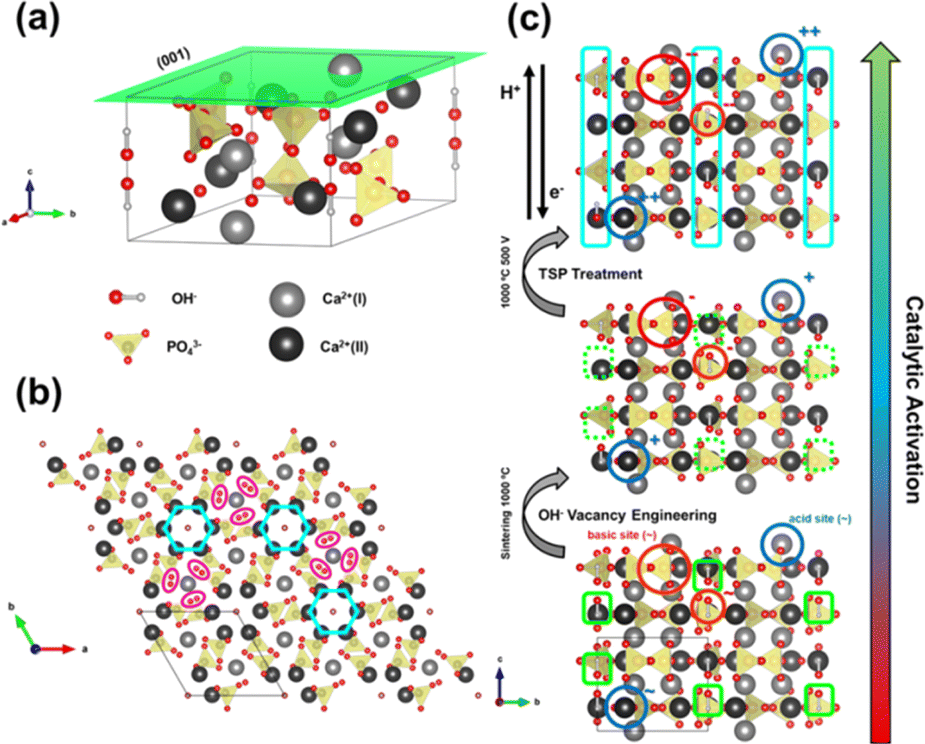 | ||
| Scheme 1 Structure of HAp as a catalyst. (a) HAp unit cell P63/m. (b) (001) crystallographic plane observed from the c-axis direction. The OH− channels and the Ca(I) 6-fold coordination are highlighted. (c) Structural changes and lattice refinement strategies adopted to achieve the catalytic activation of HAp. | ||
Accordingly, the Raman spectrum of upp-HAp (Fig. 1a) displays the characteristic active modes of HAp with ν1 = 962 cm−1 (P–O symmetric stretching), ν2 = 400–480 cm−1 (doubly degenerated O–P–O bending), ν3 = 570–625 cm−1 (P–O triply degenerated asymmetric stretching), ν4 = 1020–1095 cm−1 (triply degenerated O–P–O bending mode) and ν5 = 3574 cm−1 (O–H stretching).63 Besides, the peaks at 878, 844 and 794 cm−1 are attributed to the formation of brushite (CaHPO4·2H2O) due to the polarization process, thus proving the correct catalytic activation of HAp.66 Consistently, the XRD diffractogram of upp-HAp (Fig. 1b) shows the typical HAp reflections at 2θ = 25.9° (002), 31.8° (211), 32.3° (112) and 33.0° (300), while the presence of brushite is confirmed by the peak at 31.1° (121).66 Also, comparison with the Raman spectrum and XRD diffractogram of calcined HAp samples (i.e. just before applying the TSP process), which are shown in Fig. S4,† confirms the success of the treatment used to induce permanent polarization.
 | ||
| Fig. 1 (a) Raman spectrum, (b) XRD diffractogram, (c) SEM and (d) HRTEM micrographs, and (e) fast Fourier transform of the HRTEM micrograph for upp-HAp. In (a) the characteristic HAp (ν1–ν5) and brushite (diamonds) Raman vibrational modes are displayed. In (b), the characteristic HAp (spots) and brushite (diamonds) reflections are indicated. | ||
SEM and HRTEM micrographs of upp-HAp are shown in Fig. 1c–e. As can be seen, the SEM image reflects the ultraporous structure of the catalyst with an average pore size of 156 ± 32 nm. The porosity, which was estimated considering five independent SEM images, was 17% ± 2%. The crystallinity of the samples was confirmed by the lattice fringes observed in HRTEM images (Fig. 1d). Fast Fourier transform of the upp-HAp fringes (Fig. 1e) allowed not only the distinction between the (002) and (112) crystallographic planes of HAp at 3.40 Å and 2.75 Å, respectively, but also the identification of the characteristic polarized superstructure (001) at 6.87 Å d-spacing.
From batch to continuous reactions
In previous studies, the efficiency of pp-HAp and upp-HAp catalysts to convert CO2 alone67 or mixed with CH4 (ref. 55 and 62) into ethanol (predominant) and other valuable chemicals was examined, quantifying by 1H NMR the amount of reaction products dissolved in the liquid water (hereafter named “supernatant”) and adsorbed on the catalyst. Thus, we assumed that the content of products in the gas phase was negligible in comparison to those in the supernatant and on the catalyst. In this work, as a first step and before transferring the process to a continuous-flow reactor, we experimentally examined the time evolution of the gas composition compared to the supernatant and the catalyst of the batch reactions. The results presented in Fig. 2 are expected to provide better understanding about the distribution of products among the different phases in the continuous-flow process described in the next section.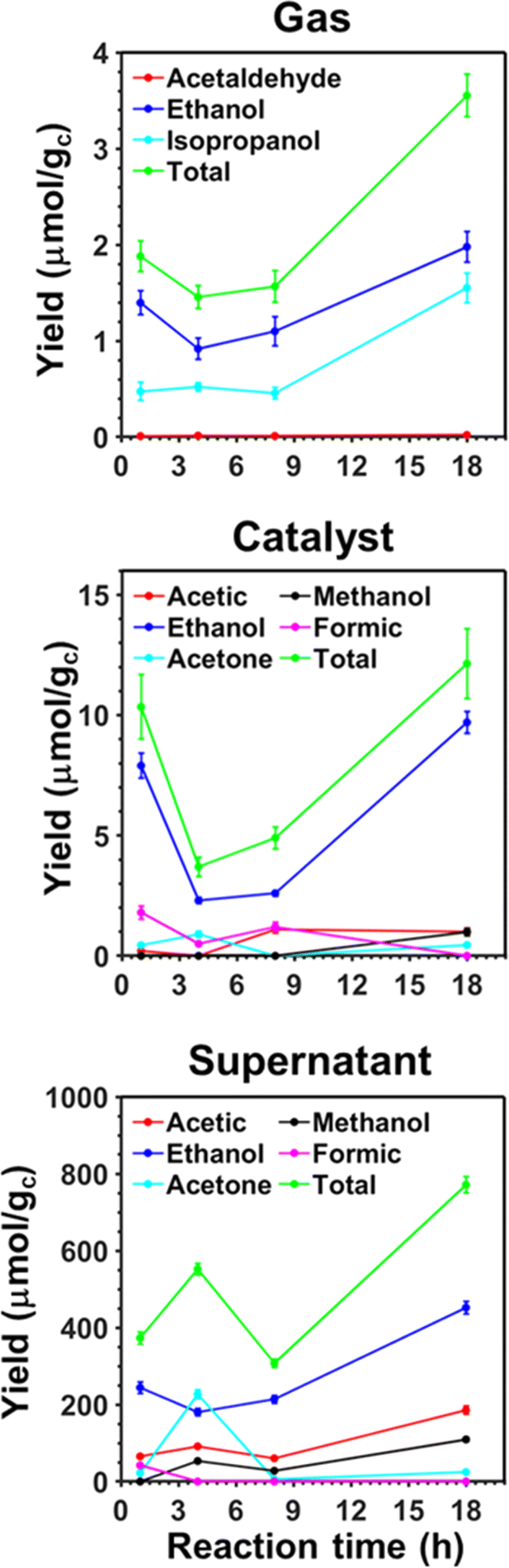 | ||
| Fig. 2 Variation of the different product yields in the gas, adsorbed on the catalyst and dissolved in the supernatant vs. the reaction time obtained from batch reactions catalyzed by upp-HAp. Reactions were conducted at 120 °C using 6 bar of CO2 and 20 mL of liquid water. The total yield, which corresponds to the sum of the yields of all the individual products, is presented as well. The yields are expressed in μmol of product per gram of catalyst (μmol gc−1). | ||
The variations of the product yields over time confirm that the resulting species are partially adsorbed on the solid catalyst and mainly dissolved in the supernatant. Thus, the yields of the different products are one and two orders of magnitude smaller in the gas than in the catalyst and the supernatant, respectively. Furthermore, the three analyzed phases present reaction products containing two or three carbon atoms (C2 and C3, respectively), while products with one carbon atom (C1) are only detected in the catalyst and supernatant (i.e. methanol and formic acid). This feature suggests that C1 products evolve into C2 and C3 before migrating to the gas phase. The thermodynamics of water splitting and CO2 reduction into formic acid, which is an exhaustively studied reaction,68–76 have been discussed in a recent review.77 Besides, all C2 products seem to be inter-related in the sense that acetic acid and acetaldehyde probably come from the complete or partial oxidation of ethanol, respectively. Similarly, acetone is catalytically hydrogenated to isopropanol at low pressure and temperature when the mass transfer between the liquid and the gas is high enough.78 Further analysis of the results is provided in the ESI.†
On the other hand, the water content available in the reactor also merits special attention when moving from batch to continuous reactions. That is because water not only provides a medium to collect the products desorbed from the catalyst (i.e. supernatant) but also acts as the proton source. Accordingly, in Scheme 2 and Fig. S5† the batch and continuous reactor systems used are compared. Thus, for the continuous reactions two different scenarios were considered: (1) liquid water is directly introduced into the reactor (1 mL) and (2) CO2 is bubbled into liquid water to produce wet CO2 (see Scheme 2b). Both reaction approaches were carried out at 95 °C and 120 °C analyzing the products obtained in the cold trap (Scheme 2b) after 15 minutes. Representative 1H NMR spectra are presented in Fig. 3a (complete spectra are shown in Fig. S6†), while the yields of the reaction are provided in Fig. 3b and c for scenarios (1) and (2), respectively. Moreover, a blank (i.e. reaction without the upp-HAp catalyst at 120 °C using the same experimental conditions as those in the upp-HAp catalyzed reactions) was also conducted (Fig. 3a) to rule out other carbon sources.
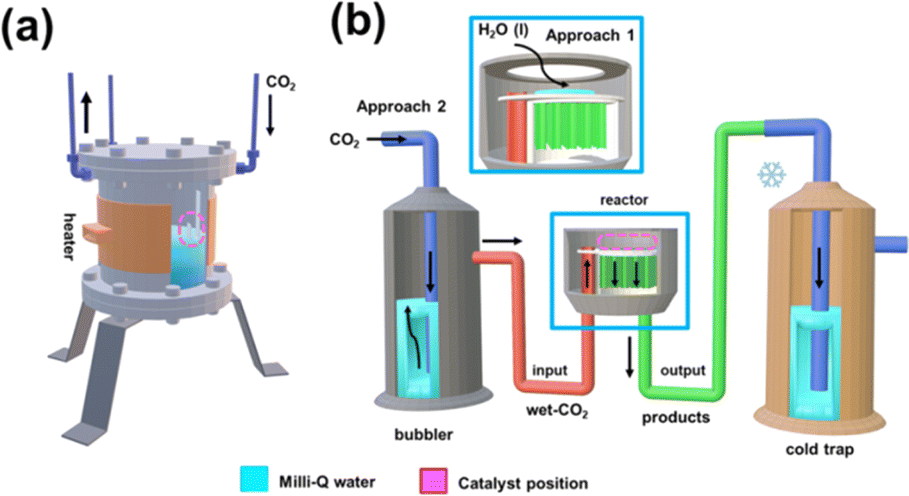 | ||
| Scheme 2 (a) Scheme of the batch reactor used. (b) Scheme of the continuous reactor used. The continuous reactor was filled with four upp-HAp catalysts (3.60 × 5.00 × 0.44 mm3 each) and 1 mL of deionized water which was homogeneously distributed (approach 1). The outlet valve of the reactor was connected to a cold trap to condense the reaction products obtained in the gas phase, while the inlet valve was connected to a CO2 cylinder to achieve a constant flow of 100 mL s−1, which was controlled using a back-pressure regulator. This high flow was selected to mimic the conditions at the pre-pilot scale. To have the required moisture, as water is the proton source, CO2 was bubbled into liquid water to produce wet CO2, the bubbler being located between the CO2 cylinder and the inlet valve (approach 2). | ||
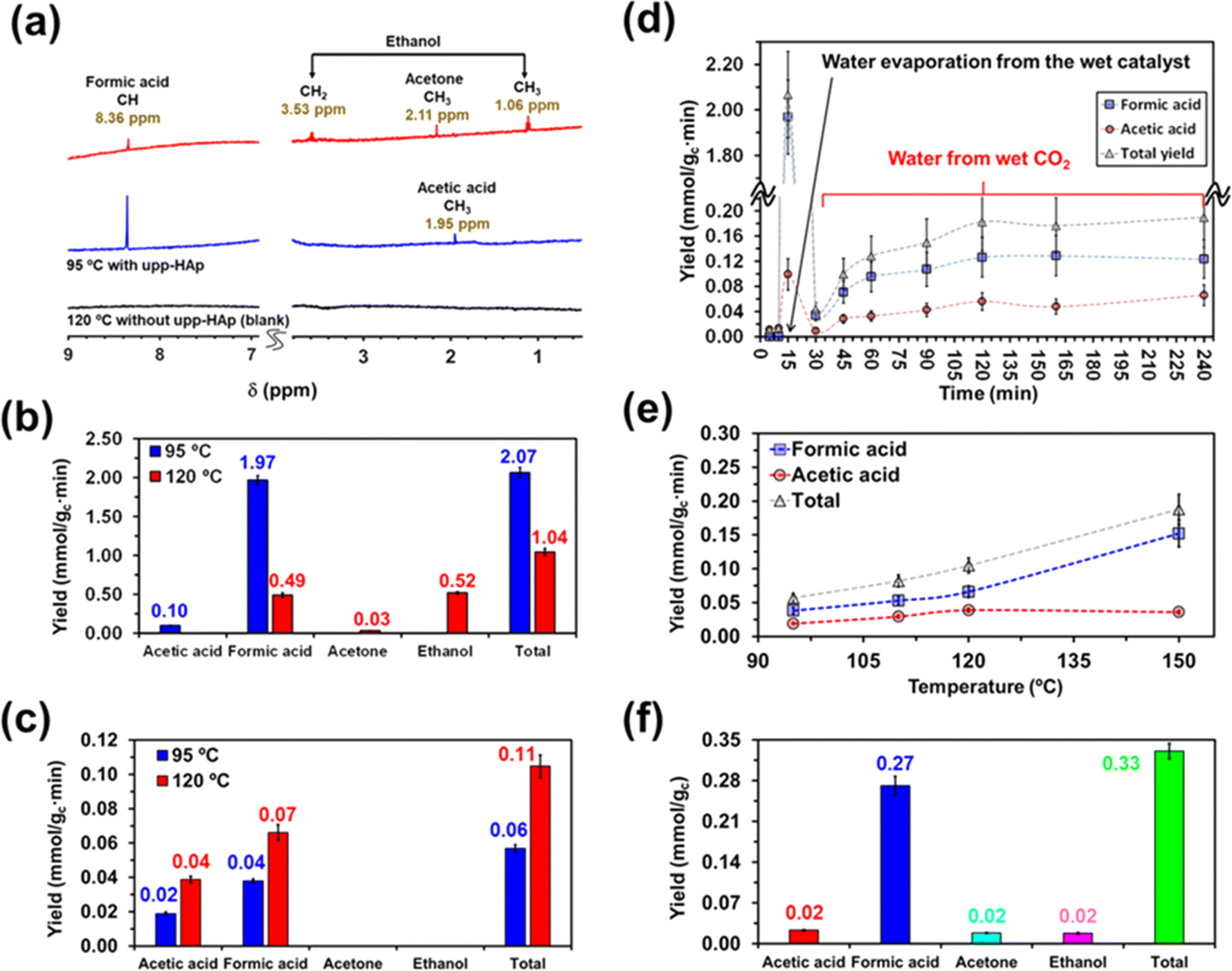 | ||
| Fig. 3 Continuous reaction studies. (a) Representative 1H NMR spectra used to identify the reaction products for the continuous processes catalyzed by upp-HAp and performed at 95 and 120 °C using a CO2 flow of 100 mL s−1 and 1 mL of liquid water. The spectrum of a blank (i.e. without catalyst) at 120 °C is also displayed. Each spectrum corresponds to the liquid collected in the cold trap after 15 min of reaction. (b) Yield of the reaction products for the continuous process described in (a). The yields were calculated from the 1H NMR spectra of the liquids collected in the cold trap after 15 min of reaction. The total reaction yield (i.e. sum of the yields of the individual products) is also displayed. (c) Yield of the reaction products for the continuous process catalyzed by upp-HAp at 95 °C and 120 °C but without including liquid water in the reactor. The yields were derived from the 1H NMR spectra of the liquids collected in the cold trap after 15 min of reaction. (d) Evolution of the non-accumulated yields of the reaction products identified by 1H NMR against the reaction time for the continuous processes conducted including 1 mL of liquid water in the reactor and catalyzed by upp-HAp at 95 °C. (e) Variation of the temperature for a process conducted up to 30 min. In all cases, the reactions were catalyzed by upp-HAp and using wet CO2 but without including liquid water in the reactor. (f) Reaction products adsorbed on the upp-HAp catalyst after several continuous-flow catalytic processes at 120 °C without including liquid water in the reactor. The amount of product was calculated from the 1H NMR spectrum collected after dissolving the catalyst using an aqueous solution containing 100 mM HCl and 50 mM NaCl. The total yield, which is the sum of the yields of all the individual products, is also presented. The yields are expressed as mmol of product per gram of catalyst per min (mmol gc−1·min−1). | ||
As can be observed, the collected spectra showed some important differences, which suggested that the evaporation rate of water plays a crucial role in the reaction mechanisms. The reaction at 95 °C led to the formation of formic acid identified by the singlet at 8.36 ppm as the predominant product and acetic acid denoted by the CH3 singlet at 1.95 ppm as the residual product. Thus, the reaction exhibited not only a high yield of formic acid (expressed as mmol of product per gram of catalyst per minute), 1.97 ± 0.06 mmol gc−1·min−1, but also a very high selectivity (∼95%, comparable to the photocatalysts)79 towards such C1 product, the ratio of formic acid/acetic acid yields being around 20 (Fig. 3b). Compared to the literature, the results reported here represent a compelling step forward in the field of heterogeneous catalysis which is extremely competitive not only when considering the total yield obtained (124 mmol gc−1·h−1 for upp-HAp compared with the standard range of 2–20 mmol gc−1·h−1)79 but also because of the greener and more sustainable reaction conditions achieved (i.e. mild conditions, use of water instead of H2, avoiding the use of electrical supply or UV irradiation, use of a green and abundant catalyst). Table 1 compares all the features obtained with respect the conventional heterogeneous catalysts used (more detailed information can be also found in Table S1†). Surprisingly, the total yield of products at 95 °C (2.07 ± 0.07 mmol gc−1·min−1) decreased by half at 120 °C (1.04 ± 0.05 mmol gc−1·min−1) and the reaction became much less selective. Thus, in addition to formic acid (0.49 ± 0.03 mmol gc−1·min−1), both ethanol (C2) and acetone (C3) were clearly detected at 120 °C. The yield of ethanol (0.52 ± 0.02 mmol gc−1·min−1), which was identified by the quartet from CH2 at 3.53 ppm and the triplet from CH3 at 1.06 ppm (Fig. 3a), was comparable to that of formic acid. Conversely, acetone, with the CH3 singlet at 2.11 ppm (Fig. 3a), remained as a residual product (0.03 ± 0.01 mmol gc−1·min−1). These observations indicate that such conditions (i.e. temperature high enough to promote the rapid evaporation of liquid water) favor the catalytic formation of C–C bonds, although they are detrimental to the efficiency of the overall CO2 conversion process (Fig. 3b).
| Catalyst | Proton source | T (°C) | Maximum selectivity (%) | Productivity (mmol gc−1·h−1) | |
|---|---|---|---|---|---|
| Heterogeneous catalysis79 | Pd, Au, Cu–ZnO, M–ZrOx,… | H2 | 220–300 | >70 (Methanol) | 2–29 (Total products) |
| This work | upp-HAp | H2O | 95 | ∼95 (Formic acid) | 124 (Total products) |
In the batch reactions, which were conducted using 20 mL of liquid water, ethanol was the predominant product in both the supernatant and the catalyst, while the content of formic acid was residual (Fig. 2). Instead, ethanol and formic acid were formed with similar yields when only 1 mL of liquid water was introduced in the continuous reactor. This feature suggests that, in the batch process, the excess evaporated water favors the formation of ethanol (i.e. generation of a C–C bond and hydrogenation) over the reduction of CO2 to formic acid (C1), while the continuous flow of CO2 and the decrease of available water molecules promote the conversion of CO2 to formic acid instead of producing ethanol.
This hypothesis can be further corroborated by analyzing the yields of the products obtained using wet CO2 (approach 2). As seen in Fig. 3c, both formic and acetic acid are obtained, whereas ethanol and acetone were not detected. Comparison of the yields, which are shown in Fig. 3c, with Fig. 3b reveals a reduction in the total yield of two and one order of magnitude at 95 and 120 °C, respectively. Furthermore, in the absence of liquid water, the yields of the two reaction products increased with temperature, as expected. Overall, these results demonstrate that liquid water is responsible for both the variety of products and the large yields reflected in Fig. 3b.
All these features can be clearly observed when merging both approaches (i.e. wet CO2 and addition of 1 mL of liquid water in the reactor) in a single long term (4 hours) continuous reaction depicted in Fig. 3d and exhaustively described in the ESI.† Considering a CO2 flow of 100 mL s−1 (0.24 mol min−1), the yields reported at 15 min in Fig. 3d in the lab scale indicate that conversion of CO2 into products is around 1%. The yields at 95 °C (Fig. 3d) stabilized at 7.4 ± 0.4 and 4.0 ± 0.4 mmol gc−1·h−1 for formic acid and acetic acid, respectively. These values were significantly higher than those reported in the literature for the photocatalytic and the electrocatalytic reduction and hydrogenation of CO2.80,81
To better understand the reaction sequence, the yields of the individual products and the total yield were analyzed as a function of the temperature after 30 min of reaction. Results for reactions performed at 95, 110, 120 and 150 °C, which are plotted in Fig. 3e, showed that all yields increased with the temperature, even though two regimes were observed. At temperatures ≤120 °C, the yields of formic acid and acetic acid, as well as the total yield, increased linearly with the temperature. However, at 150 °C, the yield of acetic acid remained unchanged while that of formic acid increased significantly, suggesting an enhancement in the dominance of the CO2 reduction to formic acid with respect to the formation of C–C bonds. After these assays, the products adsorbed in the catalyst were analyzed by 1H NMR. Accordingly, results presented in Fig. 3f have been further discussed in the ESI.†
Another important observation refers to the catalytic stability of upp-HAp for both batch and continuous processes, which was evidenced by the fact that the yields were maintained in all repetitions carried out (i.e. the same catalyst samples were always used). Fig. S7,† which compares the Raman spectra of the catalyst before and after reaction, confirms such stability. As can be observed, the main vibration (ν1 peak at 962 cm−1), which was assigned to the main symmetric stretching P–O mode, does not show any significant difference. It is worth mentioning that any loss of electrical and, consequently, catalytic activity would be detected by a widening and/or splitting of the ν1 band in the Raman spectrum. In this sense, this study is in agreement with the results reported in a recent work from where the complete stability of the upp-HAp catalysts is studied through different characterization techniques and under a wide range of temperatures.82
Mechanistic insights on CO2 activation: NAP-XPS studies and isotope-labeling reaction
Due to the novelty of the catalyst and the relevance of converting the thermodynamically stable CO2 molecule in value-added molecules, significant effort has been made to experimentally explore the initial steps of CO2 adsorption and activation on the upp-HAp surface. Firstly, we performed isotope-labeling experiments by loading 13CO2 and 5 bar of 12CO2 in a batch reactor (20 mL liquid water and 120 °C for 72 hours) to isotopically confirm the CO2 fixation reaction. Fig. 4a compares the 1H NMR spectra collected from the supernatant of a standard (i.e. 6 bar of 12CO2) and the isotope-labeling reaction, focusing on the region of acetic acid (1.6–2.4 ppm). As can be seen, the distinctive doublet of 13CH3COOH can be observed to be shifted ∼0.3 ppm from the 12CH3COOH peak at 1.95 ppm.83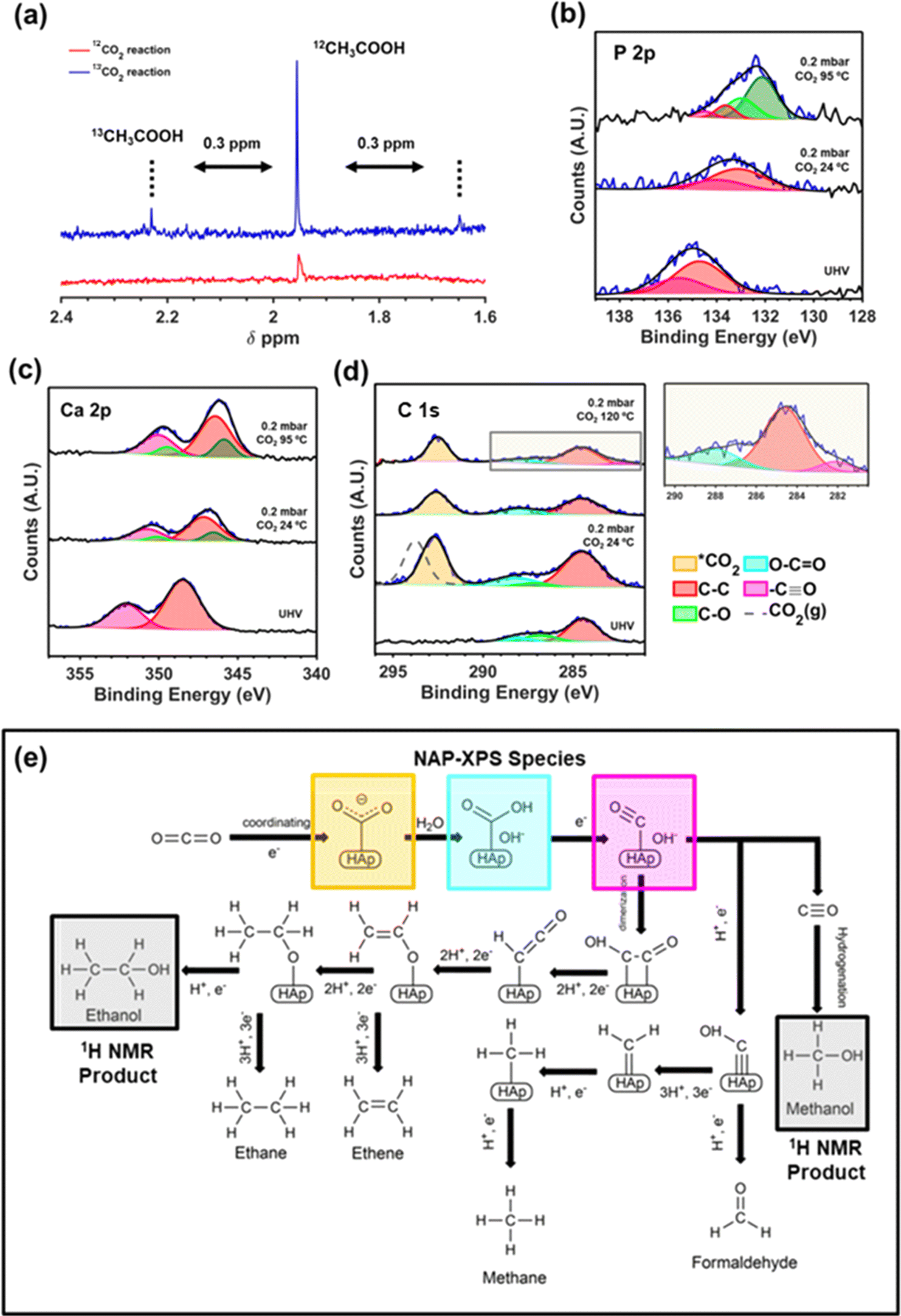 | ||
| Fig. 4 (a) Isotope-labeling experiments. High resolution NAP-XPS studies using 12CO2 of the P 2p (b), Ca 2p (c) and C 1s (d). (e) Fixation mechanism derived from 1H NMR and NAP XPS results. | ||
Additionally, near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) studies using 0.2 mbar of CO2 and at different reaction temperatures has been further performed to better understand the role of the binding sites of upp-HAp. Accordingly, Fig. 4b and c show the high-resolution spectra of the P 2p and Ca 2p regions, respectively. As soon as the CO2 is introduced in the analysis chamber, the characteristic peaks of P 2p at 134.7 eV (P 2p3/2) and 135.5 eV (P 2p1/2) experience a clear shift of 1.6 eV attributed to the adsorption of CO2 on the PO43− basic sites. Moreover, at 95 °C the new species at 132.1 eV (P 2p3/2) and 132.9 eV (P 2p1/2) shifted an additional 1 eV, suggesting the subsequent fixation steps of the adsorbed CO2 molecules. On the other hand, Ca 2p (Fig. 4c) is also affected by the CO2 atmosphere, shifting from the original binding energy under ultra-high vacuum (UHV) conditions of 348.5 eV (Ca 2p3/2) and 352.1 eV (Ca 2p1/2) by 1.3 eV and 2 eV.
As expected, due to its acidic nature, the Ca species remains stable (compared to P) when temperature is increased. Thus, the shift observed is attributed to the 6-fold coordination with the oxygen atoms of the lattice. Moreover, the fact that two different species appear is in agreement with the fact that two distinctive crystallographic sites exist for calcium cations. More specifically, Ca(I) is strictly coordinated with the oxygens from the PO43− tetrahedra, while Ca(II) is also coordinated with a hydroxyl group.84
The C 1s region has also been examined to further confirm the results obtained. Several interesting insights can be derived from the acquired spectra presented in Fig. 4d. Firstly, the shift of 1.1 eV observed when comparing the CO2 gas measured far from the surface of upp-HAp (dashed grey line; characteristic peak at 293.7 eV for C 1s which is in agreement with the literature)85 confirms the adsorption and activation of the CO2 molecule (*CO2) on the surface of upp-HAp, showing an outstanding atomic *CO2/P ratio of ∼3. These results suggest that apart from the PO43− sites, the CO2 adsorption might also occur on the OH− binding sites. This preliminary experimental evidence of the active role of several binding sites might support the strong affinity towards the catalytic production of C2 and C3 products obtained. This conclusion merits special attention due to the possibility of developing upp-HAp dual site catalysts.
As suggested in Fig. 4d, the initial steps of CO2 activation can be initiated at 95 °C. Accordingly, an increment of the peak at 288.1 eV, attributed to the O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species, can be observed showing a ratio with adventitious carbon (284.5 eV) of 0.31 (24 °C) and 0.41 (95 °C). Note that during all the experiments samples were kept inside the analysis chamber avoiding the presence and absorption of other carbon-based products on the upp-HAp surface. In contrast, desorption of C–O (contamination during sample preparation) due to temperature can be also observed. To obtain more insights on the fixation mechanism, the temperature of the experiment was finally raised to 120 °C. As can be observed in the inset of Fig. 4d, a small peak appears at 282.1 eV which has been attributed to a partially dissociated C
O species, can be observed showing a ratio with adventitious carbon (284.5 eV) of 0.31 (24 °C) and 0.41 (95 °C). Note that during all the experiments samples were kept inside the analysis chamber avoiding the presence and absorption of other carbon-based products on the upp-HAp surface. In contrast, desorption of C–O (contamination during sample preparation) due to temperature can be also observed. To obtain more insights on the fixation mechanism, the temperature of the experiment was finally raised to 120 °C. As can be observed in the inset of Fig. 4d, a small peak appears at 282.1 eV which has been attributed to a partially dissociated C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O species anchored on the catalyst (resulting in carbidic species). This result supports the unique structural and electronic properties derived from the vacancy engineering and TSP treatments (i.e. superior crystallinity and high charge delocalization) favoring the fast evolution of the generated carbon intermediates. Overall, the combined 1H NMR and NAP-XPS results allow the first catalytic CO2 fixation mechanism on upp-HAp (see Fig. 4e) to be proposed which not only supports experimentally the theoretical pathways proposed for HAp67 and/or other classical electro-mechanisms28 but also proves the feasibility of performing extremely efficient CO2 fixation reactions under strictly mild reaction conditions using only upp-HAp as a sustainable and green catalyst. It is worth highlighting the fact that the mechanism proposed involves the C–C coupling by means of a dimerization process, supported by the literature.67 This mechanistic pathway requires a high abundance of HCOO− species simultaneously adsorbed on the surface of the catalyst. According to Fig. 4e the presence of water is crucial to do so, which further corroborates the experimental results discussed above.
O species anchored on the catalyst (resulting in carbidic species). This result supports the unique structural and electronic properties derived from the vacancy engineering and TSP treatments (i.e. superior crystallinity and high charge delocalization) favoring the fast evolution of the generated carbon intermediates. Overall, the combined 1H NMR and NAP-XPS results allow the first catalytic CO2 fixation mechanism on upp-HAp (see Fig. 4e) to be proposed which not only supports experimentally the theoretical pathways proposed for HAp67 and/or other classical electro-mechanisms28 but also proves the feasibility of performing extremely efficient CO2 fixation reactions under strictly mild reaction conditions using only upp-HAp as a sustainable and green catalyst. It is worth highlighting the fact that the mechanism proposed involves the C–C coupling by means of a dimerization process, supported by the literature.67 This mechanistic pathway requires a high abundance of HCOO− species simultaneously adsorbed on the surface of the catalyst. According to Fig. 4e the presence of water is crucial to do so, which further corroborates the experimental results discussed above.
Kinetics of the CO2 fixation reaction
Herein, we present a kinetic model for the CO2 fixation reaction over upp-HAp, which has been developed combining the fixation mechanisms described in the literature66 with the experimental results obtained in this work. The main challenge resides in discerning the crucial reaction steps governing the kinetics of the CO2 fixation, which is essential to discern among the complex known network of intermediate pathways.67Fig. 3d shows that both the total yield and the yields for the individual products present an initial linear behavior (first-order reaction) followed by a stabilization regime (zero-order reaction). This fact clearly indicates a competing phenomenon between the adsorption of reactants and desorption of products. For this reason, the Langmuir–Hinshelwood–Hougen–Watson (LHHW) and the Eley–Rideal (ER) approaches, which are frequently used to model heterogeneous catalysis,86,87 have been considered to derive the expression of the reaction rate, according to a classical electroreduction mechanism, as follows:Water splitting:
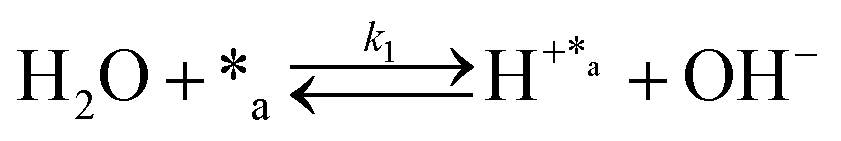 | (1) |
CO2 adsorption:
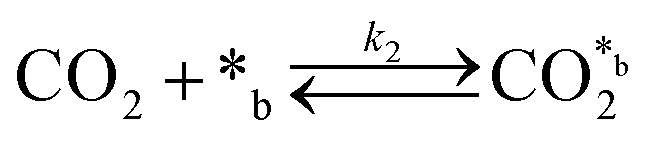 | (2) |
CO2 fixation:
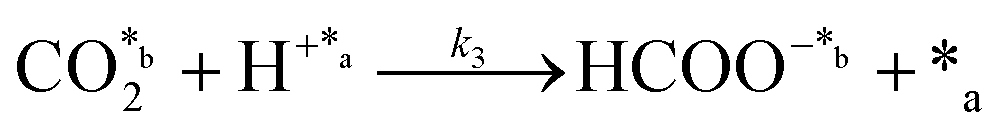 | (3) |
Chain reaction:
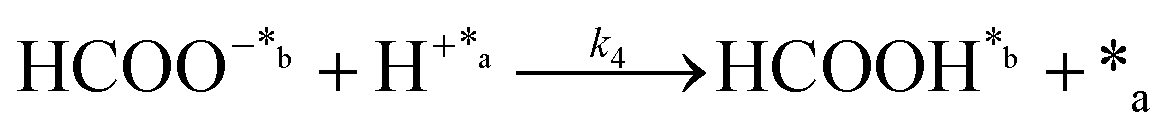 | (4) |
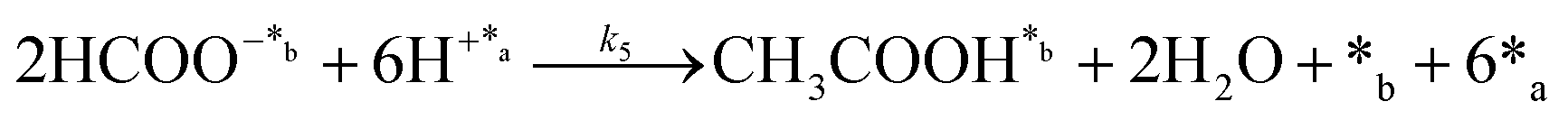 | (5) |
Product desorption:
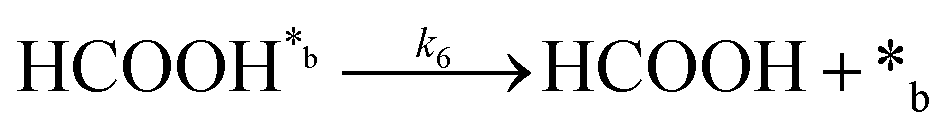 | (6) |
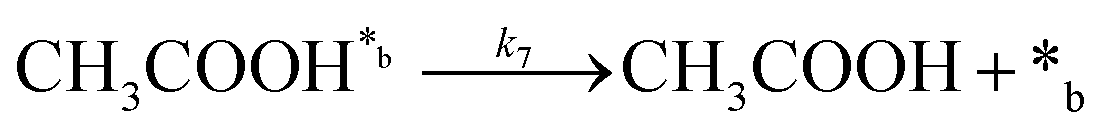 | (7) |
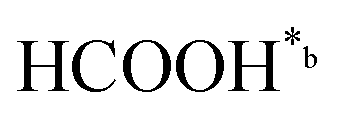 ). It should be emphasized that the already reported unique electrical and charge storage properties of upp-HAp are, in general, consistent with this classical reduction mechanism.51,52
). It should be emphasized that the already reported unique electrical and charge storage properties of upp-HAp are, in general, consistent with this classical reduction mechanism.51,52
In eqn (1)–(7), we propose a four step chain mechanism consisting of:53,54,67 (1) an initial water splitting reaction in the acid sites (*a) corresponding to the Ca2+ sites of upp-HAp (eqn (1)), occurring simultaneously with CO2 adsorption in the basic sites (*b) mainly attributed to the OH− and PO43− sites (eqn (2));87,88 (2) CO2 fixation into HCOO− due to the addition of a neighboring proton (eqn (3)), which is going to be initially considered as the rate determining step (RDS); (3) chain reaction with adsorbed formic acid (eqn (4)) or less likely, with adsorbed acetic acid (eqn (5)) due to the C–C coupling of two HCOO− adsorbed species through a dimerization process;53 and (4) product desorption (eqn (6) and (7)). Foremost and derived from point (1), the fraction of vacant sites (θ) defined according to the LHHW model has to be described separately for both acid and basic binding sites:
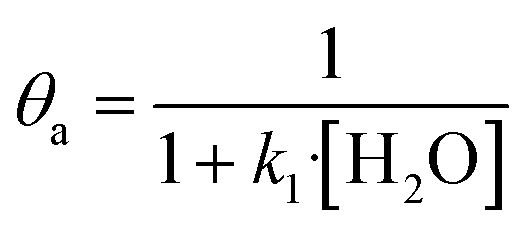 | (8) |
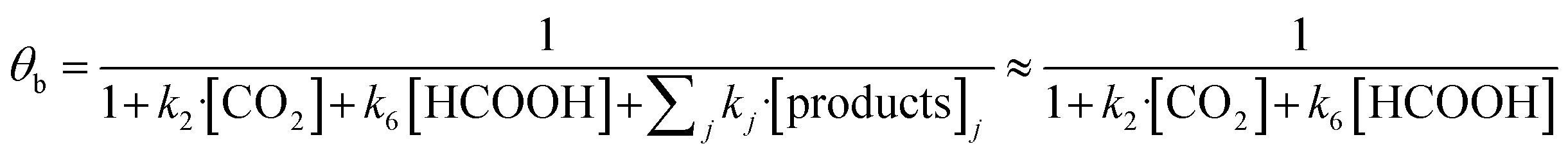 | (9) |
According to the results displayed in Fig. 3f, the term  in eqn (9), which accounts for the contribution of the rest of the products adsorbed in the catalyst (including acetic acid), has been assumed to be negligible in light of the adsorbed formic acid content.
in eqn (9), which accounts for the contribution of the rest of the products adsorbed in the catalyst (including acetic acid), has been assumed to be negligible in light of the adsorbed formic acid content.
Due to the complexity of the system, interesting questions arise related to the contribution of the different chain reactions to the overall kinetics (which might regulate the RDS) and to the competing nature of the water splitting reaction (as a result of the huge proton demand needed to generate the reported products). In order to shed some light, we modeled the kinetics of the CO2 fixation reaction under different hypotheses and compared the resulting statistical parameters. For this purpose, the minimum value for the sum of squared errors (SSE) was chosen. Also, the variance of the error and the Model Selection Criterion (MSC, eqn S1†),89 commonly used in kinetic analyses, were calculated to further confirm the results. The minimum value for variance and higher value for MSC were chosen. A comparison of the different assumptions (hereafter denoted A#) is summarized in Table 2.
| Approach | Assumption | SSE (×10−7) | MSC | Variance error (×10−7) |
|---|---|---|---|---|
| A1 |
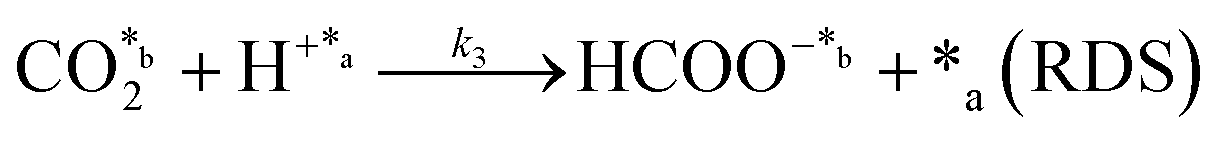
|
6.38 | 2.953 | 1.06 |
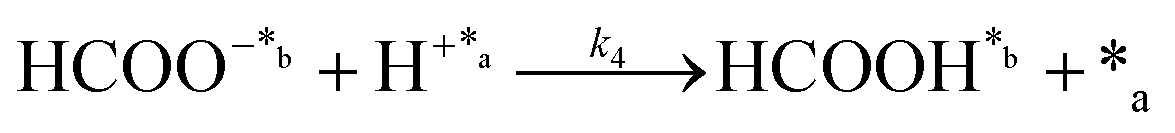
|
||||
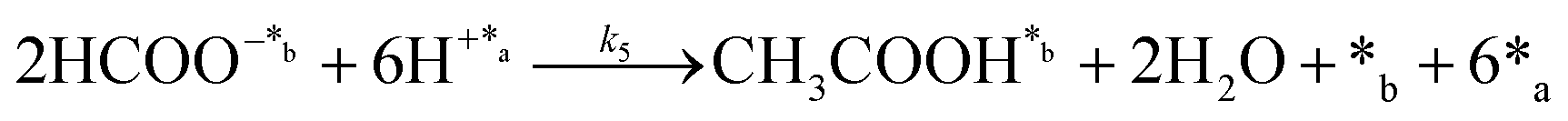
|
||||
| A2 |

|
2.30 | 4.196 | 0.37 |
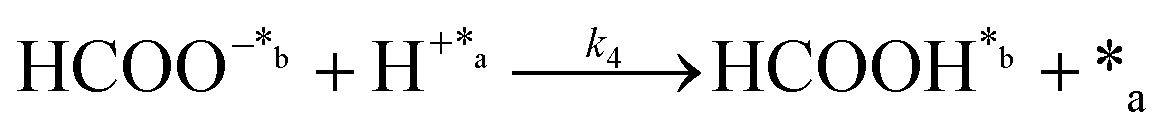
|
||||
| A3 |
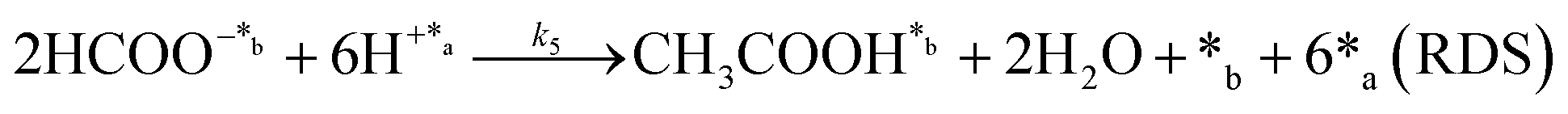
|
3.56 | 3.759 | 0.56 |
| (Modeling the kinetics of total CO2 fixation) | ||||
| A3* |
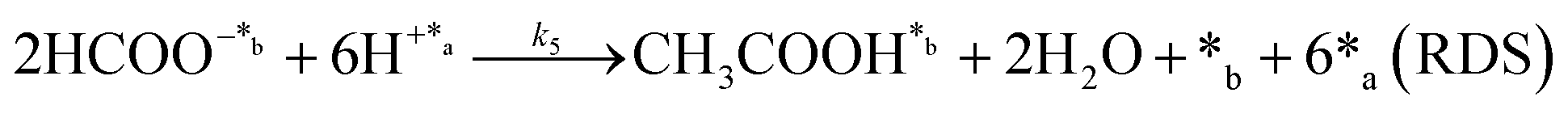
|
0.20 | 6.380 | 0.03 |
| (Modeling the kinetics of CH3COOH alone) | ||||
| A4 |
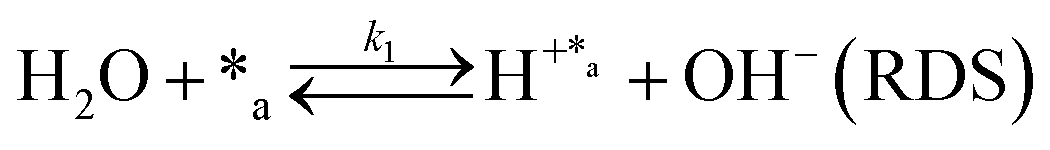 – experimentally ruled out – experimentally ruled out |
— | — | — |
| A5 |
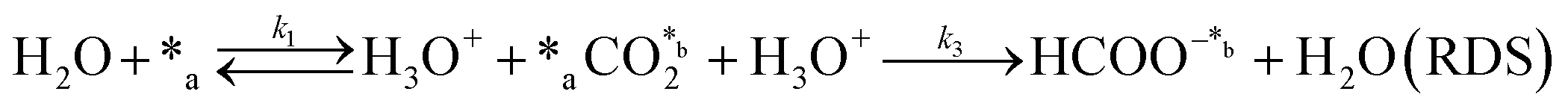
|
2.27 | 4.194 | 0.37 |
As a starting point, assumption A1 was modeled considering that the RDS was eqn (6) and assuming chain reaction with both formic and acetic acids.53 The results showed the worst data correlation (Table 2) and were ruled out. In scenarios A2 and A3, the chain reaction with formic acid alone and the dimerization process (eqn (5)), respectively, were considered as RDS.53 Although both assumptions provided much better statistical data than A1, A2 showed lower SSE and higher MSC values, indicating that the kinetics of the CO2 fixation is primarily governed by the first fixation step (eqn (3)) and its evolution to formic acid alone. This conclusion is supported by recently reported density functional theory (DFT) calculations used to study the conversion of CO2 into formic acid.90 Moreover, ruling out the A1 approach is also in agreement with other CO2 fixation mechanisms proposed for both electrocatalysts and thermocatalysts.53,54,90,91
On the other hand, the synthesis of acetic acid remains extremely limited to eqn (5) (C–C coupling), which acts as the RDS for the latter product in agreement with the experimental results. Accordingly, the expression of the rate from A3 was used to fit the experimental data obtained for acetic acid only (instead of the total CO2 fixation, A3*), showing excellent statistical results. Despite the fact that the aforementioned conclusion explains the small presence of C2 and C3 adsorbed products (Fig. 3f), the non-negligible amount of acetic acid detected might be contradictory. While remarkable differences in the desorption constants of C2 products (i.e. acetic acid vs. ethanol) have been ruled out, the possibility of re-adsorption of formic acid and its subsequent reaction with adsorbed HCOO− is more plausible. In order to demonstrate this hypothesis, a batch reaction consisting on an inert N2 atmosphere, with 1 mL of formic acid and 20 mL of water, at 95 °C and for 2 h was carried out. The recorded 1H NMR showed the unequivocal existence of acetic acid both adsorbed in the catalyst and dissolved in the supernatant (Fig. S8†). Hence, although A3* provides good statistical results, it has to be considered as the very first approach to the specific kinetics of C2 and C3 products, as it has been demonstrated that the re-adsorption and subsequent reaction of C1 products play a considerable role in the final model, increasing remarkably the complexity of the system. This mechanism is currently under study under in situ and operando conditions to support this hypothesis with experimental evidence.
Finally, the nature of the water splitting reaction has been explored. It was experimentally ruled out as the RDS since a first-order reaction regime was found by changing the water content from 1 mL to 80 mL in the batch reactor (Fig. S9†), which is inconsistent with the yields reported in Fig. 3d. Despite this fact, the extensive proton demand suggests that k1 ≫ kj. Thus, A5 assumes an almost instantaneous desorption of protons, which results in a non-competing behavior, resembling the ER approach. Surprisingly, statistical data showed slightly better results than A2, implying that the generation of products linearly depends on the initial water concentration without expecting any stagnation. This result is in total agreement with the experimental measurements presented in Fig. 3d. The high proton reactivity and mobility were previously explained from a structural perspective since upp-HAp showed enhanced proton conductivity along different crystalline domains.60 Therefore, protons might not be chemisorbed in the acid sites or completely desorbed in the reaction atmosphere but electrostatically attracted to the surface of the catalyst due to the reported charge surface accumulation. Consistently, the null catalytic CO2 fixation activity found for non-polarized HAp should be related with the lack of protons supplied to the adsorbed CO2, clearly limiting the RDS.
Due to the resemblance among some results (Table 2) and the mathematical restrictions, the final expression for the rate of total CO2 fixation reaction was expressed as follows considering A2:
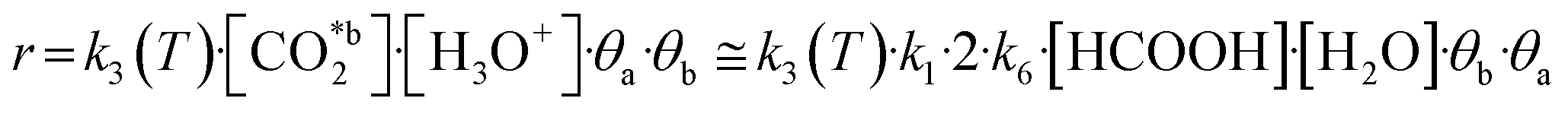 | (10) |
The activation energy Ea was obtained using the temperature studies reported in Fig. 3e and the Arrhenius relation (eqn S2†). Analysis of the kinetic models supports the conclusions derived from the behavior observed in Fig. 3e. Thus, the linear increase of formic acid with temperature, while acetic acid remains stable, is in agreement with assumptions A2 and A3.
Fig. 5a displays the fittings obtained from both total carbon fixation and acetic acid models in conjunction with the experimental results. For the sake of completeness and due to their intrinsic similarities, the same model (eqn (10)) was used to fit the synthesis of formic acid alone. In order to show the good correlation between the experimental data and the points estimated with the kinetic model, a parity plot is presented in Fig. 5b. As can be observed, the predicted model shows an excellent correlation with the experimental results without showing any systematic deviations. Thus, almost all of the points are found inside the ± 10% interval range, with more than 50% of the points located in the ± 5% interval range. Finally, Fig. 5c proves the good correlation (R2 = 0.989) between the experimental data and the Arrhenius relation.
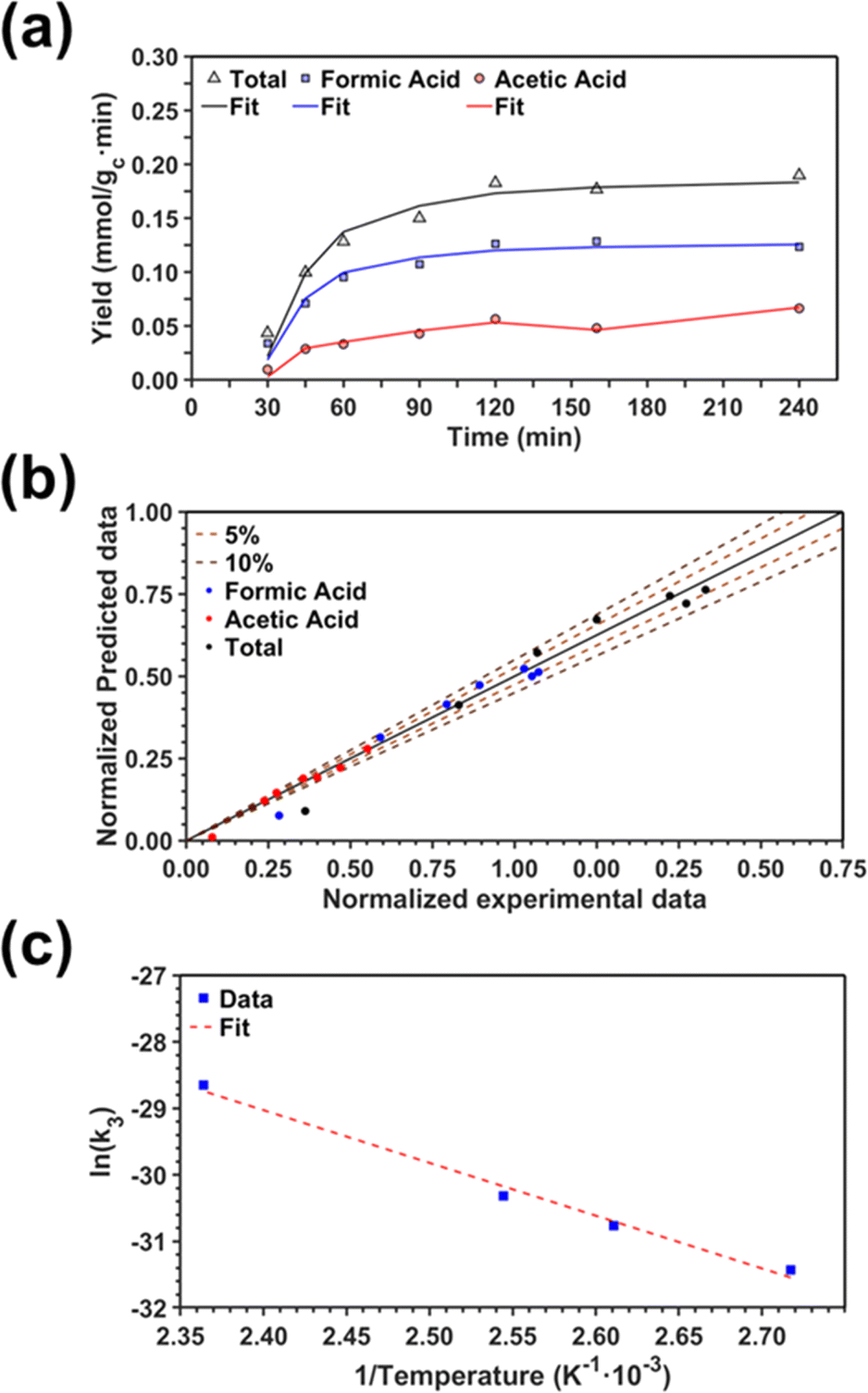 | ||
| Fig. 5 Comparison between the experimental data and the fitting model: (a) fitting of the different experimental yields obtained from the kinetic models proposed; (b) parity plot; and (c) linearization of the Arrhenius expression. | ||
The resulting kinetic parameters are reported in Table 3. k3(T = 95 °C) presents the smallest value (supporting the RDS A2 assumption), while k1 (related with the water splitting reaction) is two orders of magnitude higher than the RDS. The value for the activation energy (Ea = 66 ± 1 kJ mol−1) falls within the range of values reported in the literature (40–100 kJ mol−1).92–95 This can be considered as an outstanding result, as upp-HAp (a biocompatible and cheap ceramic) shows the same catalytic potential than conventional metal-based catalysts (e.g. Cu- and Ni-based)93,94 with the great advantage that no electric potential (which has been demonstrated to clearly affect the Ea) is applied.
| Parameter | |
|---|---|
| k 1 | 15.17 ± 1.5 m3 mmol−1 |
| k 6 | 0.37 ± 0.06 mmol m−1−3 |
| k 3 (T = 95 °C) | 1.43 ± 0.23 × 10−2 mmolproduct (gc−1·s−1) |
| k 0 | 2.20 ± 0.07 × 107 mmolproduct (gc−1·s−1) |
| E a | 66 ± 1 kJ mol−1 |
Conclusions
We have shown the use of metal-free upp-HAp as a catalyst for the continuous production of formic acid and acetic acid using wet CO2 as a reagent. Firstly, the distribution of reaction products in the gas, liquid and solid phases, as a function of the reaction time, was investigated for the CO2 fixation heterogeneous catalytic process using mild reaction conditions (95–120 °C) and both CO2 gas (6 bar) and liquid water (20 mL) as reagents. Results evidenced the key role of upp-HAp as a catalytic substrate able to adsorb an extremely high ratio of CO2 in the temperature regime and water during the formation of chemically valuable products that were found to be C1, C2 and C3 reflecting the competition of the two processes: the reduction of CO2 and the formation of C–C bonds.A noticeable increment of the reaction yield was observed when the CO2 conversion process was switched from batch to continuous-flow mode, using identical catalysts. Certainly, the yield obtained (>2 mmol gc−1·min−1) supposes a great step forward for the use of a competitive green heterogeneous catalyst under strictly mild conditions. Two factors were found to be crucial for the continuous-flow process: (1) the addition of liquid water at the beginning of the process, which can provide an excess of water molecules compared to the process using wet CO2 as a source of hydrogen, and therefore, enormously increasing the yield of the reaction; and (2) the reaction temperature, depending on whether it is performed above or below 100 °C, since it drastically alters the liquid–gas balance of the water molecules coming from the liquid supplied at the beginning of the process and/or from the wet CO2, affecting the distribution of the reaction products and the selectivity.
The thermodynamic and kinetic studies performed for the continuous-flow process without considering liquid water as a reactant have provided information on the relevant reaction steps of the mechanism, primarily pointing out the first CO2 fixation step as the RDS. Although the C–C bond formation is extremely challenging, the kinetic model highlights the fast and independent generation of formic acid (C1 product), boosting the total CO2 fixation yield. Besides, the competitive adsorption of species in the binding sites has exhaustively been examined, ruling out the contribution of proton adsorption to the overall kinetics, which represents a great advantage in terms of yield optimization and industrial scalability. Finally, the activation energy for the total CO2 fixation, which has been estimated to be 66 ± 1 kJ mol−1, is within the range of conventional electro-assisted catalysts. In this sense, NAP-XPS experiments further confirm the reaction pathway and mechanism for CO2 activation occurring in the basic sites of upp-HAp (PO43− and OH−). Therefore, these preliminary results suggest a synergy between the different binding sites (i.e. dual site catalysis) which would promote the generation of C2 and C3 products. Indeed, the outstanding catalytic activity of upp-HAp for CO2 fixation and to generate low organic carbon molecules has been demonstrated, representing a feasible, highly efficient, sustainable, green, cheap, and energetically low-demand alternative to conventional electro-assisted catalysts.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
Authors declare that the preparation and application of permanently polarized hydroxyapatite as a catalyst was patented by the Universitat Politècnica de Catalunya and B. Braun Surgical S.A. (EP16382381, EP16382524, P27990EP00, PCT/EP2017/069437, P58656 EP, P59205 EP, P59091 EP, P59008 EP, P59528 EP, 2021P00074 EP, P 59985 EP).Acknowledgements
This publication and other research outcomes are supported by the predoctoral program AGAUR-FI ajuts (2023 FI-100056) Joan Oró, which is backed by the Secretariat of Universities and Research of the Department of Research and Universities of the Generalitat of Catalunya. This work is part of Maria de Maeztu Units of Excellence Programme CEX2023-001300-M funded by MCIN/AEI/10.13039/501100011033. Authors also acknowledge the Agència de Gestió d'Ajuts Universitaris i de Recerca (2021 SGR 00387) and B. Braun Surgical S. A. U. for their financial support. Support for the research done by C. A. was also received through the prize “ICREA Academia” for excellence in research funded by the Generalitat de Catalunya. J. S. thanks the Spanish Ministry of Universities for the support through a Margarita Salas postdoctoral grant, funded by European Union – NextGenerationEU.Notes and references
- https://www.epa.gov/climate-indicators/greenhouse-gases(visited in november 2022).
- K. B. Karnauskas, S. L. Miller and A. C. Schapiro, Geohealth, 2020, 6, e2019GH000237 CrossRef.
- S. Paraschiv and L. S. Paraschiv, Energy Rep., 2020, 6(8), 237–242 CrossRef.
- X. Dou, J. Hong, P. Ciais, F. Chevallier, F. Yan, Y. Yu, Y. Hu, D. Huo, Y. Sun, Y. Wang, S. J. Davis, M. Crippa, G. Janssens-Maenhout, D. Guizzardi, E. Solazzo, X. Lin, X. Song, B. Zhu, D. Cui, P. Ke, H. Wang, W. Zhou, X. Huang, Z. Deng and Z. Liu, Sci. Data, 2023, 10, 69 CrossRef CAS.
- D. J. Beerling, E. P. Kantzas, M. R. Lomas, P. Wade, R. M. Eufrasio, P. Renforth, B. Sarkar, M. G. Andrews, R. H. James, C. R. Pearce, J. F. Mercure, H. Pollitt, P. B. Holden, N. R. Edwards, M. Khanna, L. Koh, S. Quegan, N. F. Pidgeon, I. A. Janssens, J. Hansen and S. Banwart, Nature, 2020, 583, 242–248 CrossRef CAS PubMed.
- E. Callagon La Plante, D. A. Simonetti, J. Wang, A. Al-Turki, X. Chen, D. Jassby and G. N. Sant, ACS Sustainable Chem. Eng., 2021, 9, 1073–1089 CrossRef.
- D. Y. C. Leung, G. Caramanna and M. M. Maroto-Valer, Renew. Sustain. Energy Rev., 2014, 39, 426–443 CrossRef CAS.
- C. Cao, H. Liu, Z. Hou, F. Mehmood, J. Liao and W. Feng, Energies, 2020, 13, 600 CrossRef CAS.
- L. Santamaría, S. A. Korili and A. Gil, Chem. Eng. J., 2023, 455, 140551 CrossRef.
- Y. Jian, Y. Wang and A. B. Farimani, ACS Sustain. Chem. Eng., 2022, 10, 16681–16691 CrossRef CAS.
- S. A. Mazari, N. Hossain, W. J. Basirun, N. M. Mubarak, R. Abro, N. Sabzoi and A. Shah, Process Saf. Environ. Prot., 2021, 149, 67–92 CrossRef CAS.
- V. Kumaravel, J. Bartlett and S. C. Pillai, ACS Energy Lett., 2020, 5, 486–519 CrossRef CAS.
- A. Ateka, P. Rodriguez-Vega, J. Ereña, A. T. Aguayo and J. Bilbao, Fuel Process. Technol., 2022, 233, 107310 CrossRef CAS.
- Y. Chen and T. Mu, Green Chem., 2019, 21, 2544–2574 RSC.
- L. Deng, Z. Wang, X. Jiang, J. Xu, Z. Zhou, X. Li, Z. You, M. Ding, T. Shishido, X. Liu and M. Xu, Appl. Catal., B, 2023, 322, 122124 CrossRef CAS.
- G. Gastelu, D. Savary, M. Hulla, D. Ortiz, J. G. Uranga and P. J. Dyson, ACS Catal., 2023, 13, 2403–2409 CrossRef CAS.
- C. Yang, Y. Hu, S. Li, Q. Huang and J. Peng, ACS Appl. Mater. Interfaces, 2023, 15, 6942–6950 CrossRef CAS PubMed.
- C. D. Koolen, W. Luo and A. Züttel, ACS Catal., 2023, 13, 948–973 CrossRef CAS.
- A. Mukhtar, S. Saqib, M. Mubashir, S. Ullah, A. Inayat, A. Mahmood, M. Ibrahim and P. L. Show, Renewable Sustainable Energy Rev., 2021, 150, 111487 CrossRef CAS.
- Y. Han, H. Xu, Y. Su, Z.-L. Xu, K. Wang and W. Wang, J. Catal., 2019, 370, 70–78 CrossRef CAS.
- P. Panagiotopoulou, Appl. Catal., A, 2017, 542, 63–70 CrossRef CAS.
- R. Xia, S. Zhang, X. Ma and F. Jiao, J. Mater. Chem. A, 2020, 8, 15884–15890 RSC.
- Z. Qi, J. Biener and M. Biener, ACS Appl. Energy Mater., 2019, 2, 7717–7721 CrossRef CAS.
- E. B. Nursanto, H. S. Jeon, C. Kim, M. S. Jee, J. H. Koh, Y. J. Hwang and B. K. Min, Catal. Today, 2016, 260, 107–111 CrossRef CAS.
- B. Nan, Q. Fu, J. Yu, M. Shu, L. L. Zhou, J. Li, W. W. Wang, C. J. Jia, C. Ma, J. X. Chen, L. Li and R. Si, Nat. Commun., 2021, 12, 3342 CrossRef CAS.
- C. Kim, S. Hyeon, J. Lee, W. D. Kim, D. C. Lee, J. Kim and H. Lee, Nat. Commun., 2018, 9, 3027 CrossRef PubMed.
- Y. Tang, Y. Li, V. Fung, D. Jian, W. Huang, S. Zhang, Y. Iwasawa, T. Sakata, L. Nguyen, X. Zhang, A. I. Frenkel and F. Tao, Nat. Commun., 2018, 9, 1231 CrossRef PubMed.
- F. Zeng, C. Mebrahtu, X. Xi, L. Liao, J. Ren, J. Xie, H. Jan Heeres and R. Palkovits, Appl. Catal., B, 2021, 291, 120073 CrossRef CAS.
- F. Zhang, R. A. Gutiérrez, P. G. Lustemberg, Z. Liu, N. Rui, T. Wu, P. J. Ramírez, W. Xu, H. Idriss, M. V. Ganduglia-Pirovano, S. D. Senanayake and J. A. Rodriguez, ACS Catal., 2021, 11, 1613–1623 CrossRef CAS PubMed.
- X. Cai, G. Li, W. Hu and Y. Zhu, ACS Catal., 2022, 12, 10638–10653 CrossRef CAS.
- Y. P. Du, A. M. Bahmanpour, L. Milosevic, F. Heroguel, M. D. Mensi, O. Kröcher and J. S. Luterbacher, ACS Catal., 2020, 10, 12058–12070 CrossRef CAS.
- S. Pérez-Rodríguez, E. Pastor and M. J. Lázaro, J. CO2 Util., 2017, 18, 41–52 CrossRef.
- H.-Y. Cui, Y.-X. Zhang, C.-S. Cao, T.-D. Hu and Z.-L. Wu, Chem. Eng. J., 2023, 451, 138764 CrossRef CAS.
- H. Lu, J. Tournet, K. Dastafkan, Y. Liu, Y. H. Ng, S. K. Karuturi, C. Zhao and Z. Yin, Chem. Rev., 2021, 121, 10271–10366 CrossRef CAS.
- L. Zhu, Y. Liu, X. Peng, Y. Li, Y.-L. Men, P. Liu and Y.-X. Pan, ACS Appl. Mater. Interfaces, 2020, 12, 12892–12900 CrossRef CAS PubMed.
- S.-L. Hou, J. Dong, X.-L. Jiang, Z.-H. Jiao and B. A. Zhao, Angew. Chem., Int. Ed., 2019, 58, 577–581 CrossRef CAS PubMed.
- R. Fiorenza, M. Bellardita, S. A. Balsamo, A. Gulino, M. Condorelli, G. Compagnini, S. Scirè and L. Palmisano, Catal. Today, 2023, 413–415, 113949 CrossRef CAS.
- Y. Song, J. Mao, C. Zhu, S. Li, G. Li, X. Dong, Z. Jiang, W. Chen and W. Wei, ACS Appl. Mater. Interfaces, 2023, 15, 10785–10794 CrossRef CAS.
- X. Hu, X. Liu, X. Hu, C. Zhao, Q. Guan and W. Li, Adv. Funct. Mater., 2023, 2214215 CrossRef CAS.
- H. Han, J. Im, M. Lee and D. Choo, Appl. Catal., B, 2023, 320, 121953 CrossRef CAS.
- A. P. Kulkarni, T. Hos, M. V. Landau, D. Fini, S. Giddey and M. Herskowitz, Sustain. Energy Fuels, 2021, 5, 486–500 RSC.
- M. M. F. Hasan, L. M. Rossi, D. P. Debecker, K. C. Leonard, Z. Li, B. C. E. Makhubela, C. Zhao and A. Kleij, ACS Sustain. Chem. Eng., 2021, 9, 12427–12430 CrossRef CAS.
- H. Guzmán, F. Salomone, E. Batuecas, T. Tommasi, N. Russo, S. Bensaid and S. Hernández, Chem. Eng. J., 2021, 417, 127973 CrossRef.
- T. Wang and J. Gong, Nat. Energy, 2020, 5, 642–643 CrossRef CAS.
- B. Goldsteina, D. Gounaridisa and J. P. Newell, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 19122–19130 CrossRef.
- A. Velty and A. Corma, Chem. Soc. Rev., 2023, 52, 1773–1946 RSC.
- W. Zhang, Z. Jin and Z. Chen, Adv. Sci., 2022, 9, e2105204 CrossRef PubMed.
- A. Mustafa, B. G. Lougou, Y. Shuai, Z. Wang, S. Razzaq, E. Shad¡gdar, J. Zhao and J. Shan, J. Mater. Chem. A, 2021, 9, 4558–4588 RSC.
- S. Ghosh, A. Modak, A. Samanta, K. Kole and S. Jana, Adv. Mater., 2021, 2, 3161–3187 RSC.
- Q. Wu and C. Wu, J. Mater. Chem. A, 2023, 11, 4876–4906 RSC.
- K. Wei, H. Guan, Q. Luo, J. He and S. Sun, Nanoscale, 2022, 14, 11869–11891 RSC.
- G. Wen, B. Ren, Y. Zheng, M. Li, C. Silva, S. Song, Z. Zhang, H. Dou, L. Zhao, D. Luo, A. Yu and Z. Chen, Adv. Energy Mater., 2022, 12, 2103289 CrossRef CAS.
- W. J. Zhang, Y. Hu, L. B. Ma, G. Y. Zhu, Y. R. Wang, X. L. Xue, R. P. Chen, S. Y. Yang and Z. Jin, Adv. Sci., 2018, 5, 1700275 CrossRef PubMed.
- S. Roy, A. Cherevotan and S. C. Peter, ACS Energy Lett., 2018, 3, 1938–1966 CrossRef CAS.
- J. Sans, G. Revilla-López, V. Sanz, J. Puiggalí, P. Turon and C. Alemán, Chem. Commun., 2021, 57, 5163–5166 RSC.
- S. Awasthi, S. K. Pandey, E. Arunan and C. Srisvastva, J. Mater. Chem. B, 2021, 9, 228–249 RSC.
- A. Veiga, F. Castro, F. Rocha and A. L. Oliveira, ACS Appl. Bio Mater., 2020, 3, 3441–3455 CrossRef CAS.
- M. U. Munir, S. Salman, A. Ihsan and T. Elsaman, Int. J. Nanomed., 2022, 17, 1903–1925 CrossRef PubMed.
- M. Rivas, L. J. del Valle, A. M. Rodríguez-Rivero, P. Turon, J. Puiggalí and C. Alemán, ACS Biomater. Sci. Eng., 2018, 4, 3234–3245 CrossRef CAS PubMed.
- J. Sans, M. Arnau, F. Estrany, P. Turon and C. Alemán, Adv. Mater. Interfaces, 2021, 8, 2100163 CrossRef CAS.
- J. Sans, M. Arnau, V. Sanz, P. Turon and C. Alemán, Adv. Mater. Interfaces, 2022, 9, 2101631 CrossRef CAS.
- J. Sans, M. Arnau, J. J. Roa, P. Turon and C. Alemán, ACS Appl. Nano Mater., 2022, 5, 8526–8536 CrossRef CAS PubMed.
- L. Silvester, J.-F. Lamonier, R.-N. Vannier, C. Lamonier, M. Capron, A.-S. Mamede, F. Pourpoint, A. Gervasini and F. Dumeignil, J. Mater. Chem. A, 2014, 2, 11073 RSC.
- J. Sans, M. Arnau, P. Turon and C. Alemán, Mater. Horiz., 2022, 9, 1566 RSC.
- J. Sans, J. Llorca, V. Sanz, J. Puiggalí, P. Turon and C. Alemán, Langmuir, 2019, 35, 14782–14790 CrossRef CAS PubMed.
- J. Sans, M. Arnau, V. Sanz, P. Turon and C. Alemán, Chem. Eng. J., 2022, 433, 133512 CrossRef CAS.
- J. Sans, V. Sanz, P. Turon and C. Alemán, ChemCatChem, 2021, 13, 5025 CrossRef CAS.
- H. Zhong, Y. Gao, G. Yao, X. Zeng, Q. Li, Z. Huo and F. Jin, Chem. Eng. J., 2015, 280, 215–221 CrossRef CAS.
- C. Liu, B. C. Colón, M. Ziesack, P. A. Silver and D. G. Nocera, Science, 2016, 352(6290), 1210–1213 CrossRef CAS PubMed.
- J. K. Stolarczyk, S. Bhattacharyya, L. Polavarapu and J. Feldmann, ACS Catal., 2018, 8, 3602–3635 CrossRef CAS.
- S. K. Lee, M. Kondo, G. Nakamura, M. Okamura and S. Masaoka, Chem. Commun., 2018, 54, 6915–6918 RSC.
- F. Jin, X. Zeng, J. Liu, Y. Jin, L. Wang, H. Zhong, G. Yao and Z. Huo, Sci. Rep., 2015, 4, 4503 CrossRef PubMed.
- L. Quintana-Gómez, P. Martínez-Álvarez, J. J. Segovia, A. Martín and M. D. Bermejo, J. CO2 Util., 2023, 68, 102369 CrossRef.
- Y. Wang, X. Shang, J. Shen, Z. Zhang, D. Wang, J. Lin, J. C. S. Wu, X. Fu and C. Li, Nat. Commun., 2020, 11, 3043 CrossRef CAS PubMed.
- M. SK, S. Barman, S. Paul, R. De, S. S. Sreejith, H. Reinsch, M. Grzywa, N. Stock, D. Volkmer, S. Biswas and S. Roy, Chem.–Eur. J., 2021, 27, 4098–4107 CrossRef CAS PubMed.
- D. Oluwatobi, M. Tahir and N. A. Saidina Amin, J. CO2 Util., 2017, 18, 261–274 CrossRef.
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef PubMed.
- S. Basu and N. C. Pradhan, Catal. Today, 2020, 348, 118–126 CrossRef CAS.
- Y. Wang, Y. Tian, S.-Y. Pan and S. W. Snyder, ChemSusChem, 2022, 15, e202201290 CrossRef CAS PubMed.
- V.-H. Nguyen and J. C. S. Wu, Appl. Catal., A, 2018, 550, 122–141 CrossRef CAS.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- M. Arnau, J. Sans, J. Ll. Tamarit, M. Romanini, P. Turon and C. Alemán, Adv. Mater. Interfaces, 2024, 2400422 CrossRef.
- Y. Tang, Y. Li, V. Fung, D. Jiang, W. Huang, S. Zhang, Y. Iwasawa, T. Sakata, L. Nguyen, X. Zhang, A. I. Frenkel and F. Tao, Nat. Commun., 2018, 9, 1231 CrossRef PubMed.
- E. O. López, P. L. Bernardo, N. R. Checca, A. L. Rossi, A. Mello, D. E. Ellis, A. M. Rossi and J. Terra, Appl. Surf. Sci., 2022, 571, 151310 CrossRef.
- T. G. Avval, S. Chatterjee, S. Bahr, P. Dietrich, M. Meyer, A. Thisen and M. R. Linford, Surf. Sci. Spectra, 2019, 26, 014022 CrossRef.
- E. F. de Souza, H. P. Pacheco, N. Miyake, R. J. Davis and S. F. Toniolo, ACS Catal., 2020, 10, 15162–15177 CrossRef CAS.
- C. R. Ho, S. Shylesh and A. T. Bell, ACS Catal., 2016, 6, 939–948 CrossRef CAS.
- S. C. Oh, J. Xu, D. T. Tran, B. Liu and D. Liu, ACS Catal., 2018, 8(5), 4493–4507 CrossRef CAS.
- S. Armenise, E. García-Bordejé, J. L. Valverde, E. Romeo and A. Monzón, Phys. Chem. Chem. Phys., 2013, 15, 12104–12117 RSC.
- S. Lee, H. Bae, A. Singh, T. Hussain, T. Kaewmaraya and H. Lee, ACS Omega, 2021, 6, 27045–27051 CrossRef CAS PubMed.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS PubMed.
- Y. Wu, A. Chen, X. Liu, J. Xu, Y. Wang, K. Mumford, G. W. Stevens and W. Fei, Chem. Eng. Process., 2021, 159, 108235 CrossRef CAS.
- M. Szalay, D. Buzsáki, J. Barabás, E. Faragó, E. Janssens, L. Nyulászi and T. Holtzl, Phys. Chem. Chem. Phys., 2021, 23, 21738–21747 RSC.
- C. Vogt, M. Monai, E. B. Sterk, J. Palle, A. E. M. Melcherts, B. Zijlstra, E. Groeneveld, P. H. Berben, J. M. Boereboom, E. J. M. Hensen, F. Meirer, I. A. W. Filot and B. M. Weckhuysen, Nat. Commun., 2019, 10, 5330 CrossRef PubMed.
- X. Wang, H. Shi and J. Szanyi, Nat. Commun., 2017, 8, 513 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00305e |
| This journal is © The Royal Society of Chemistry 2024 |