
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational design of galactopyranoside-substituted N-heterocyclic carbene palladium(II) complexes. Stable and efficient catalyst for C–C coupling in aqueous media†
Ariana W. Hobsteter,
Marcos J. Lo Fiego‡
 * and
Gustavo F. Silbestri‡*
* and
Gustavo F. Silbestri‡*
INQUISUR, Departamento de Química, Universidad Nacional del Sur (UNS)-CONICET, Av. Alem 1253, B8000CPB Bahía Blanca, Argentina. E-mail: ariana.hobsteter@uns.edu.ar; marcos.lofiego@uns.edu.ar; gsilbestri@uns.edu.ar
First published on 4th January 2024
Abstract
Following a rational design, three novel palladium(II) complexes bearing galactopyranoside-based N-heterocyclic carbene ligands have been synthesized via transmetalation of the corresponding Ag(I) complexes. Palladium(II) complexes have been characterized by NMR, FT-IR and elemental analysis. Catalytic studies, using the Stille and Suzuki–Miyaura cross-coupling reactions as model C–C coupling, reveal that the complexes are active and reusable. The best results in terms of TON values were achieved in aqueous medium using either the in situ deacetylation of the catalyst or the previously deacetylated catalyst. The catalytic condition using in situ deacetylation was more efficient because it avoids an additional deprotection step.
1. Introduction
Carbohydrates and their derivatives are important biomolecules with essential physiological functions, e.g., energy production and storage, structural integrity, and biological recognition processes.1 Incorporating glycan units into the N-heterocyclic carbene (NHC) backbone provides ligands with some unique properties including hydrophilicity, chirality, and biocompatibility.2 In addition, their metal complex derivatives are notably attractive as water-soluble catalysts.2b,d,3 However, a limited number of sugar-incorporated NHC complexes with different transition metals have been reported in recent years, holding a variety of bonds between the NHC core and the carbohydrate unit, using generally D-glucose as a representative sugar.4 Sugar-incorporated NHC complex catalysts are promising for their low cost and the possibility of replacing organic reaction media with aqueous media in catalysis. The aqueous medium allows easy separation of organic products and their reuse, as well as replacing volatile and flammable organic solvents. Enlargement of these catalysts has been an approach toward environmentally friendly chemical processes.5Palladium-catalyzed cross-coupling reactions are significant methods for carbon–carbon and carbon–heteroatom bond formation. For more than 40 years, the Stille reaction has proven to be an accessible and efficient method for organic synthesis.6 The main advantages include the stability and functional group tolerance of stannanes and the broad reaction scope of aryl halides. Therefore, this reaction has increased interest in pharmaceutical and fine chemical industries.7 Stille coupling reactions take place in organic solvents and are usually catalyzed homogeneously. Therefore, it is difficult to recover the expensive catalyst from the reaction mixture, leading to waste. Development of a recyclable and reusable catalyst system is valuable in green chemistry terms and practical application. Several strategies have developed involving the use of supported palladium complexes. Besides, implementing green solvents such as water simplifies catalyst separation by creating a biphasic reaction mixture.8 In 1990, Casalnuovo et al., reported the Suzuki reaction between aryl halides and boron reagents in water.9 This reaction has been crucial in the advancements of aqueous phase cross-coupling reactions.10 As a consequence, numerous catalysts capable of promoting Suzuki coupling under mild conditions in pure water or in combination with co-solvents are currently known.11
Regarding carbohydrate-based Pd–NHC, in 2007 Glorius et al.,4b reported the synthesis and characterization of the first Pd(II)–(NHC)2 complex starting from α-D-glucopyranosyl bromide and mesityl imidazole via Ag(I)–(NHC)2 complex. Later, Lin et al.4e prepared three different Pd–NHC complexes based on C6-substituted glucopyranose through a five-step sequential approach. The debenzoylated complexes were active and reusable for three cycles in a Suzuki–Miyaura type reaction, in water at 100 °C. On the other hand, Nishioka et al.12 reported the first carbohydrate-based C–C–N pincer-type Pd–NHC complexes via a Click type reaction. Moreover, the same group synthesized a series of similar complexes by changing the terminal alkyl groups (methyl, isopropyl, benzyl, and D-glucopyranosyl) in the NHC moiety.3b All complexes were investigated as catalysts in aqueous Suzuki–Miyaura reactions, using K2CO3 to remove the acetyl groups which improved both solubility and yield. Finally, the same group reported the synthesis of complexes Pd–[bis-NHC-(CH2)2]Cl2 and Pd–[bis-NHC-(CH2)3]Cl2 and their catalytic ability in water.13 These complexes exhibited a moderate catalytic activity, decomposing after three uses. The yields improved with the addition of tetrabutylammonium bromide, enhancing the solubility of the aryl halide. Recently, Zhou et al.3c synthesized and fully characterized glucopyranoside-functionalized N-heterocyclic carbenes based pyridine-enhanced precatalyst preparation, stabilization and initiation (PEPPSI) type palladium(II) complexes and their catalytic activity in Suzuki reaction. The synthesis involves three steps starting from D-glucose to obtain the NHC precursors. They found that a less flexible and bulkier substituent around the palladium metal probably contributed to the formation of isomers mixture. All complexes show good catalytic activity and recyclability in the Suzuki–Miyaura reaction carried out in a solvent mixture of EtOH/H2O. Furthermore, the same group reported the synthesis of four Pd–NHC PEPPSI complexes that contain glucose and different alkyl chains (Cn, with n = 1, 3, 8 and 16).3d The catalyst with the longest alkyl chain proved to be the most efficient in the Suzuki–Miyaura transformation.
Inspired by our previous work on the synthesis and catalytic evaluation of galactopyranoside-incorporated Au(I)–NHC complexes,14 we performed a rational design of analogous Pd(II)–PEPPSI type precatalyst complexes. The new targets were based on D-galactopyranose due to the commercial availability of its precursors and their almost unexplored application in this topic. On the other hand, the substituents on the imidazole nitrogen will allow us to understand correlations between complex structures and catalyst activities. The complexes were fully characterized and evaluated as catalysts in Stille and Suzuki–Miyaura cross-coupling reactions in aqueous medium with excellent results.
2. Results and discussion
According to our recent report,14 we carried out the synthesis of the imidazolium salt 1a from imidazole, in 51% yield as pure β anomer. Subsequently, we adapted the synthetic strategies reported by Lin et al., to synthesize palladium(II) complex 3a using 2a as NHC transfer agents.4e This method employed PdCl2 as the metal precursor and incorporated stoichiometric amounts of pyridine as the fourth coordinating ligand (Scheme 1; see Experimental section for details).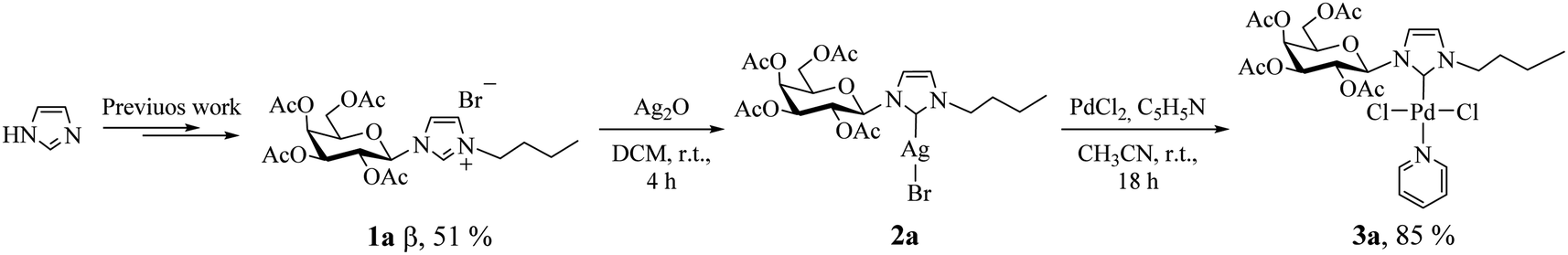 | ||
| Scheme 1 Procedure for synthesis of Pd(II)–NHC complex 3a. | ||
It is noteworthy to mention that, Lee et al.15 reported the exclusive formation of PdCl2Py2 using PdCl2 as palladium source by attempting to synthesize palladium(II) complexes using imidazolium salts as precursors and pyridine as both base and solvent. Nevertheless, in our optimized reaction conditions, we obtained PdCl2Py2 as a byproduct. All the efforts to prevent its formation by modifying reaction variables including temperature and stoichiometry did not produce favorable results. However, it was successfully separated by column chromatography (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate 1:1). Another aspect to highlight is that the reaction was carried out at room temperature, while the literature suggests 80 or 100 °C.3c,d Besides, under the employed reaction conditions formation of bis-carbene complex [Pd(II)–(NHC)2] was not detected. Palladium(II)–carbene complex 3a was fully characterized by 1H, 13C NMR, and 2D-HSQC, FT-IR spectroscopy and elemental analysis (see Experimental section for details). The 1H NMR data unambiguously confirmed the metal coordination by the disappearance of the proton signal of the imidazole ligand (singlet at δ 10.57 ppm). In addition, the 13C NMR spectra display the characteristic signal of the carbene carbon bound to palladium with value of 151.5 ppm,16 shifted to higher ppm, relative to the starting salts (136.8 ppm). Complex 3a was found to be stable at air and can be stored for prolonged periods.
:ethyl acetate 1:1). Another aspect to highlight is that the reaction was carried out at room temperature, while the literature suggests 80 or 100 °C.3c,d Besides, under the employed reaction conditions formation of bis-carbene complex [Pd(II)–(NHC)2] was not detected. Palladium(II)–carbene complex 3a was fully characterized by 1H, 13C NMR, and 2D-HSQC, FT-IR spectroscopy and elemental analysis (see Experimental section for details). The 1H NMR data unambiguously confirmed the metal coordination by the disappearance of the proton signal of the imidazole ligand (singlet at δ 10.57 ppm). In addition, the 13C NMR spectra display the characteristic signal of the carbene carbon bound to palladium with value of 151.5 ppm,16 shifted to higher ppm, relative to the starting salts (136.8 ppm). Complex 3a was found to be stable at air and can be stored for prolonged periods.
In order to study the catalytic activity of complex 3a, we assessed the Stille cross-coupling reaction in both organic solvents and aqueous medium. Table 1 summarizes the main results obtained.
|
|||||
|---|---|---|---|---|---|
| Entry | Complex | Solvent (1.5 mL) | Additive | Temp (°C) | Yieldb,c (%) |
| a Relation ArSn/PhBr 1:1.2.b Determinated by GC, with an external standard.c Homo-coupling product <10–15%.d 48 h.e DMF/H2O = 1:5.f TON = 43.g TON = 44. |
|||||
| 1 | 3a | Toluene | — | 80 | 27 |
| 2 | 3a | DMF | — | 80 | 63 |
| 3d | 3a | DMF | — | 80 | 69 |
| 4 | 3a | DMF | — | 100 | 60 |
| 5e | 3a | DMF/H2O | — | 80 | 36 |
| 6e | 3a | DMF/H2O | K2CO3 | 80 | 85f |
| 7 | 3a | H2O | K2CO3 | 80 | 38 |
| 8e | 4a | DMF/H2O | — | 80 | 87g |
| 9 | 4a | H2O | — | 80 | 30 |

We initiated the study by performing a cross-coupling reaction between (p-methylphenyl)tributylstannane and bromobenzene as a model system. The transformation was conducted in toluene at 80 °C with addition of 2.0 mol% of catalyst. Under these reaction conditions, the desired product was achieved after 18 h in 27% yield (Table 1, entry 1). A second experiment carried out in DMF, a polar solvent suitable for both reagents and catalyst, significantly increased the yield of the reaction to 63% (entry 2). However, a 48-hours reaction did not cause any change in performance (entry 3). Likewise, raising the temperature did not enhance the reaction; instead, it resulted in a decline in the catalytic efficacy (entry 4). Next, we continued the study using DMF/H2O (1:5) mixture as solvent, at 80 °C. Water was added to the reaction medium for its environmental benefits as well as for its role to facilitate the separation of compounds that are insoluble in water. In these conditions the reaction yield significantly reduced to 36%, probably due to the low catalyst solubility (entry 5). To improve the solubility of the complex in water, we conducted an experiment by adding K2CO3 to achieve in situ deacetylation of the carbohydrate unit. Interestingly, the conversion rate was quantitative (entry 6). Additionally, after a second catalytic cycle, we verified that the catalyst remained active, although its activity decreased significantly to 19% yield (see Experimental section for details of catalyst reuse process).
A similar reaction as in entry 6 using pure water gave the same result as the base-free reaction, indicating that the reactants are insoluble in these conditions (entry 7).
Consequently, we decided to isolate the deacetylated complex (4a) and evaluate it as a catalyst. To perform the deacetylation step, the complex 3a was dissolved in DCM/MeOH and K2CO3 was added.17 The reaction mixture was stirred at room temperature for 24 h and after removing the solvent the deacetylated complex was obtained as a yellow solid (Scheme 2, see Experimental section for details). Complex 4a was characterized by NMR noting the disappearance of signals corresponding to acetyl groups.18
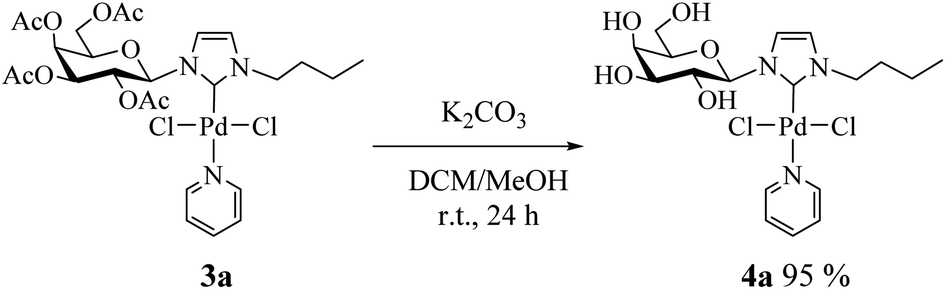 | ||
| Scheme 2 Procedure for preparation of deacetylated Pd(II)–NHC complex 4a. | ||
The catalytic evaluation of 4a, was carried out employing the conditions described in entry 5. This reaction using 4a yielded similar results to those obtained through the in situ deprotection conditions (Table 1, compare entries 6 and 8). However, as can be seen in entry 9, the yield decreased significantly when the reaction was performed in pure water, similar to the observations in entry 7.
Following a rational design, our main goal was to evaluate the impact of substituents on the imidazole nitrogen in Pd(II)–NHC complexes in terms of stability and catalytic activity. Based on the previously reported bulky imidazolium salts,14 we synthesized two novel compounds following a similar procedure employed for complex 3a (Scheme 3, see Experimental section).
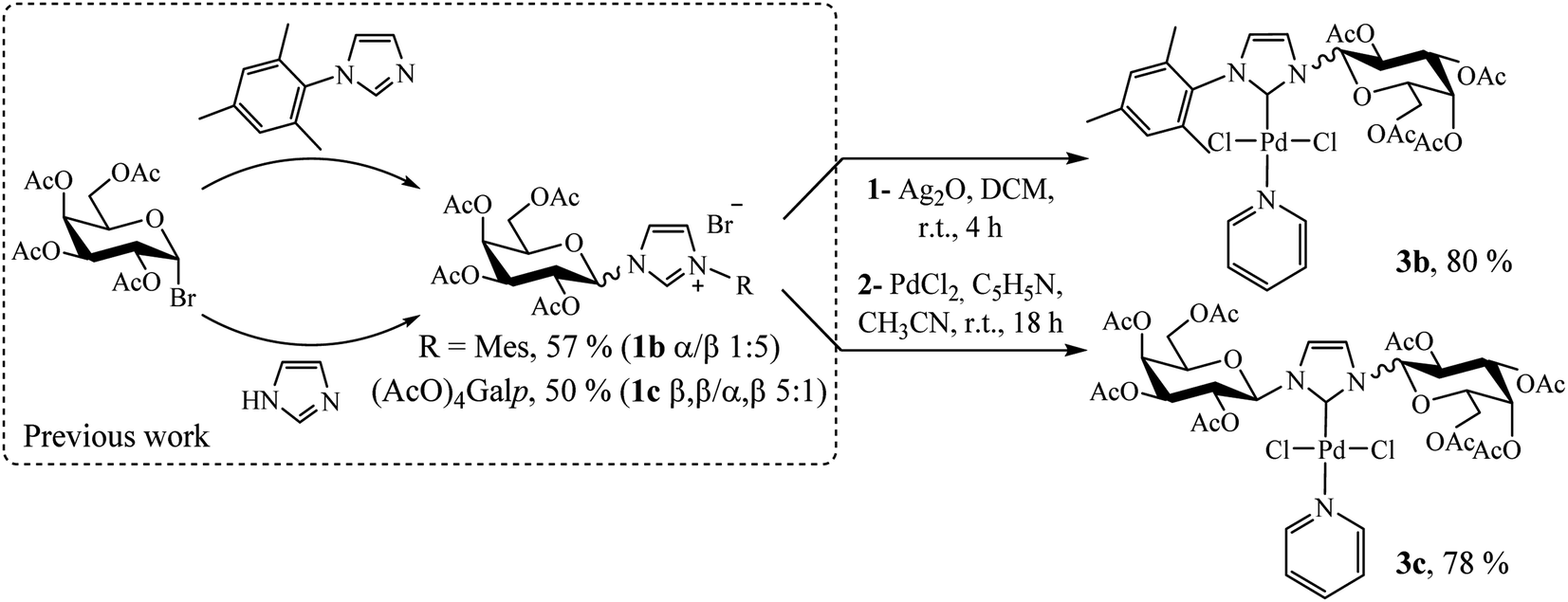 | ||
| Scheme 3 Procedure for preparation of Pd(II)–NHC complexes 3b and 3c. | ||
The relevant results for 3b and 3c complexes as catalysts in the Stille cross-coupling reaction with K2CO3, in DMF/H2O, at 80 °C are summarized in Table 2. As can be seen, the catalytic activity of both complexes is similar in terms of TON values or CG yield, with a slightly higher activity for complex 3c (entries 1 and 2). Even in an experiment with a reduced loading of 3c (1.0 mol%) no significant changes were observed (entry 3). In contrast, a decrease in catalytic activity was observed with the addition of 0.2 mol% of 3c. However, it is important to highlight that the turnover number increased more than 7 times compared with 2 mol% and more than 3 times compared with 1 mol% (entry 4). In comparison, commercial PdCl2 and mixed PdCl2/1c (1:1) catalyst with 1.0 mol% loading provided 2% and 6% yields, respectively (entries 5 and 6). Furthermore, when PdCl2/1c/C5H5N (1:1:1) was added, 13% was obtained (entry 7).
|
|||
|---|---|---|---|
| Entry | Catalyst (mol%) | TON | Yieldb,c (%) |
| a Relation ArSn/PhBr 1:1.2; DMF/H2O (1:5).b Determinated by GC, with an external standard.c Homo-coupling product <10–15%. |
|||
| 1 | 3b (2.0) | 43 | 86 |
| 2 | 3c (2.0) | 45 | 90 |
| 3 | 3c (1.0) | 87 | 87 |
| 4 | 3c (0.2) | 315 | 63 |
| 5 | PdCl2 (1.0) | 2 | |
| 6 | PdCl2/1c (1.0, 1:1) |
6 | |
| 7 | PdCl2/1c/C5H5N (1.0, 1:1:1) |
13 | |
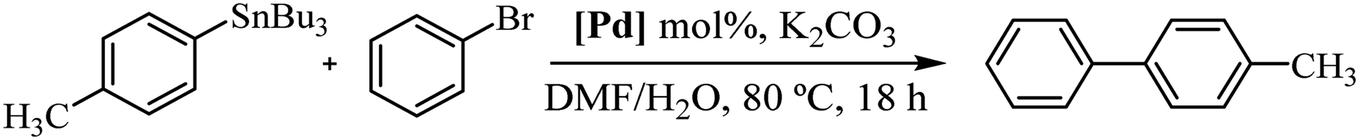
All complexes were stable, but since that 3c showed better performance, we decided to study the reactivity of a set of arylstannanes and aryl bromides with different steric and electronic demands (Table 3).

As shown in Table 3, the catalyst was found to be active and efficient with all selected reactant pairs. In general, no significant correlations were found between the electronic and steric effects of substituents on the arylstannane and the outcome of the reactions. Nevertheless, the highest yields are achieved when electron-donating groups are present in the haloarene. This study represents the first report of carbohydrate-containing Pd–NHC catalysts in the Stille reaction.
Recognizing the prominence of Suzuki–Miyaura type reactions in forming C–C bonds and their efficiency in an aqueous medium, we aimed to evaluated our catalyst in such transformations. To achieve this, we have replaced the arylstannane with boronic acid and conducted the Suzuki–Miyaura reaction study using complex 3c (Table 4).19
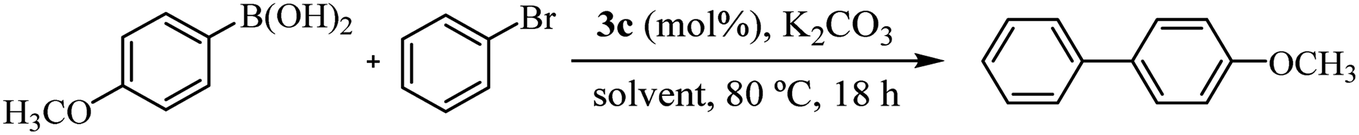
As shown in Table 4, the cross-coupling product was obtained in a 73% yield in pure water (TON = 73). A similar result was found with the tin derivative in DMF/H2O (compare with Table 3, entry 5, TON = 77). Notably, when the reaction was carried out in DMF/H2O, the yield increased to 87% (TON = 87), possibly due to the improved solubility of bromobenzene in the reaction mixture. Additionally, after a second catalytic cycle, we verified that the catalyst remained active without a significant loss of activity (72% yield). The most important advantage lies on the recyclability and reuse of 3c being reused up to two times in our reaction scale. However, on a larger scale probably it can be reused multiple times. Similar to the reaction carried out with an arylstannane, decreasing the catalyst loading to 0.2 mol% increased the turnover number by more than 3 times (compare entry 3 with entry 4 in Table 2). Complex 3c showed moderate catalytic activity in the Suzuki–Miyaura reaction compared to similar carbohydrate-containing Pd–NHC catalysts tested under similar conditions.3c,d
3. Experimental section
General procedures
The solvents were distilled, dried and stored according to standard procedures.20 Unless otherwise stated, reagents were obtained from commercial sources and were used as received. The imidazolium salts were synthesized according to our previously reported method.14 1H, 13C NMR, and 2D-HSQC spectra were recorded with a Bruker Advance 300 spectrometer. Chemical shifts (δ) are reported in ppm with the residual solvent resonance signal: δ H/C 7.27:77.2 for CDCl3; and δ H 4.79 for D2O. Melting points were determined on a Reichert–Kofler hot-stage microscope and were uncorrected. Microanalytical data were obtained using an Exeter Analytical Inc. CE-440 microanalyzer. Infrared spectra were collected on an FTIR spectrometer Nicolet Nexus-470. Stille coupling and Suzuki–Miyaura reactions mixture were analyzed by gas–liquid chromatography using a Shimadzu GC-14B instrument equipped with a flame-ionization detector and a HP-5MS column (30 m × 0.25 mm × 0.25 mm), using nitrogen as carrier gas. Mass spectra (EI) were obtained at 70 eV on a Hewlett Packard HP-5890 GC/MS instrument equipped with a HP-5972 selective mass detector.
General procedure for the synthesis of galactopyranoside-substituted-NHC palladium(II) complexes (3a–c)
In a 25 mL round-bottom flask, equipped with a nitrogen inlet, was prepared a solution of [Ag(I)–NHC–Br], from the imidazolium salt (0.4 mmol) and silver oxide (0.2 mmol), in dry DCM (4 mL). The reaction mixture was stirred at room temperature for 4 h in dark. The solution was filtered through a pad of Celite and the solvent was removed in vacuum. Then, a [Ag(I)–NHC–Br] solution in 1 mL acetonitrile under N2, PdCl2 (0.4 mmol) and pyridine (0.4 mmol) was added and the mixture was stirred at room temperature. After 18 h, the solution was filtered through a pad of Celite and the solvent was removed in vacuum. The palladium complex was purified by column chromatography (hexane/ethyl acetate 1:1).
1-Butyl-3-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)imidazol-2-ylidene palladium(II) pyridine dichloro (3a)
Pale yellow solid, 85% yield; mp 82–84 °C; Rf = 0.50 (hexane/ethyl acetate 4:6). 1H NMR (300 MHz, CDCl3) δ 8.98 (d, J = 4.9 Hz, 2H, HPy); 7.78 (t, J = 8.4 Hz, 1H, HPy); 7.36 (t, 2H, HPy); 7.23 (d, J = 2.2 Hz, 1H, H4Imi); 6.96 (d, J = 2.1 Hz, 1H, H5Imi); 6.77 (d, J = 9.2 Hz, 1H, H1β); 5.63–5.48 (m, 2H, H4,2β); 5.41 (dd, J = 10.3, 3.3 Hz, 1H, H3β); 4.87–4.64 (m, 1H, NCH2); 4.38–4.16 (m, 3H, H5,6β, NCH2); 4.09 (dd, J = 10.6, 6.3 Hz, 1H, H6β); 2.19 (s, 3H, CH3CO); 2.05 (s, 3H, CH3CO); 2.02 (s, 3H, CH3CO); 2.00 (s, 5H, CH3CO, NCH2CH2); 1.48 (q, J = 7.4 Hz, 2H, CH2CH3); 1.01 (t, J = 7.3 Hz, 3H, CH3). 13C NMR (75 MHz, CDCl3) δ 170.6 (CO); 170.4 (CO); 170.1 (CO); 169.9 (CO); 152.6 (C2Imi); 151.5 (CHPy); 138.3 (CHPy); 124.6 (CHPy); 122.5 (C5Imi); 119.5 (C4Imi); 86.9 (C1); 73.8 (C5); 71.3 (C3); 67.8 (C2); 67.3 (C4); 61.1 (C6); 51.2 (NCH2); 32.5 (NCH2CH2); 21.2 (CH3CO); 20.8 (CH3CO); 20.8 (CH3CO); 20.6 (CH3CO); 20.0 (CH2CH3); 13.8 (CH3). FTIR (neat): 3053.8; 2985.6; 2962.1; 2929.4; 1751.4; 1605.6; 1444.8; 1429.6; 1370.3; 1260.6; 1227.6; 1090.2; 1065.2; 921.7; 896.4; 749.9; 700.7. Elemental analysis calcd for C26H35Cl2N3O9Pd: C 43.93, H 4.96, N 5.91; found C 43.79, H 4.87, N 5.57.
1-Mesityl-3-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)imidazol-2-ylidene palladium(II) pyridine dichloro (3b)
Yellow solid, 80% yield; mp 122–124 °C; Rf = 0.62 (hexane/ethyl acetate 4:6). 1H NMR (300 MHz, CDCl3) δ 8.77 (d, J = 5.0 Hz, 2H, HPy); 7.68 (t, J = 7.6 Hz, 1H, HPy), 7.44 (d, J = 1.9 Hz, 1H, H4Imi); 7.28–7.21 (m, 2H, HPy); 7.03 (s, 1H, HAr); 6.98 (d, J = 9.2 Hz, 1H, H1β); 6.95 (s, 1H, HAr); 6.90 (d, J = 2.0 Hz, 1H, H5Imi); 5.67–5.53 (m, 2H, H4,2β); 5.47 (dd, J = 10.3, 3.2 Hz, 1H, H3β); 4.43–4.08 (m, 3H, H5,6β); 2.38 (s, 3H, CH3Ar), 2.34 (s, 3H, CH3CO), 2.22 (s, 3H, CH3CO), 2.12 (s, 3H, CH3CO), 2.07 (s, 3H, CH3CO), 2.04 (s, 3H, CH3Ar), 2.03 (s, 3H, CH3Ar). 13C NMR (75 MHz, CDCl3) δ 170.6 (CO); 170.5 (CO); 170.2 (CO); 169.9 (CO); 154.3 (C2Imi); 151.6 (CHPy); 139.4 (CAr); 138.3 (CHPy); 136.3 (CAr); 135.9 (CAr); 134.8 (CHAr); 129.3 (CHAr); 125.2 (C5Imi); 124.4 (CHPy); 119.8 (C4Imi); 87.2 (C1); 73.9 (C5); 71.2 (C3); 68.2 (C2); 67.3 (C4); 61.0 (C6); 21.3 (CH3Ar); 21.2 (CH3CO); 20.9 (CH3CO); 20.8 (CH3CO); 20.7 (CH3CO); 19.3 (CH3Ar); 18.6 (CH3Ar). FTIR (neat): 3054.0; 2986.8; 1752.6; 1422.3; 1370.6; 1262.7; 1224.4; 896.2; 749.7; 703.5. Elemental analysis calcd for C31H37Cl2N3O9Pd: C 48.17, H 4.83, N 5.44; found C 47.95, H 4.91, N 4.99.
1-(2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl)-3-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)imidazol-2-ylidene palladium(II) pyridine dichloro (3c)
Pale yellow solid, 78% yield; mp 102–124 °C; Rf = 0.40 (hexane/ethyl acetate 1:1). 1H NMR (300 MHz, CDCl3) δ 8.98 (d, J = 5.1 Hz, 2H, HPy); 7.80 (t, J = 7.6 Hz, 1H, HPy); 7.39 (t, J = 6.9 Hz, 2H, HPy); 7.26 (s, 2H, H4,5Imi); 6.76 (d, J = 9.2 Hz, 2H, H1β); 5.59–5.46 (m, 4H, H4,2β); 5.40 (dd, J = 10.2, 3.2 Hz, 2H, H3β); 4.33–4.02 (m, 6H, H5,6β); 2.20 (s, 6H, CH3CO); 2.04 (s, 6H, CH3CO); 1.99 (s, 12H, 2 × CH3CO). 13C NMR (75 MHz, CDCl3) δ 170.5 (CO); 170.3 (CO); 170.2 (CO); 169.9 (CO); 156.5 (C2Imi); 151.7 (CHPy); 138.4 (CHPy); 124.7 (CHPy); 120.3 (C4,5Imi); 87.2 (C1); 74.0 (C5); 71.2 (C3); 67.9 (C2); 67.3 (C4); 61.1 (C6); 21.0 (CH3CO); 20.8 (CH3CO); 20.7 (CH3CO); 20.6 (CH3CO). FTIR (neat): 3054.5; 2985.0; 1746.2; 1437.5; 1368.0; 1263.8; 1221.3; 1094.0; 1063.1; 742.8; 704.2. Elemental analysis calcd for C36H45Cl2N3O18Pd: C 43.89, H 4.60, N 4.27; found C 43.40, H 4.61, N 4.12.
Procedure for the synthesis of deacetylated complex (4a)
In a 10 mL round bottom flask, 0.1 mmol of 3a was dissolved in 3 mL of DCM/MeOH (1:1) and 0.03 mmol of K2CO3 was added. The reaction mixture was stirred at room temperature for 24 h. The mixture was filtered through a pad of Celite and concentrated in vacuum to give the desired deprotected complex.
1-Butyl-3-(β-D-galactopyranosyl)imidazol-2-ylidene palladium(II) pyridine dichloro (4a)
Yellow solid, 95% yield. 1H NMR (300 MHz, D2O) δ 8.87 (d, J = 5.4 Hz, 2H, HPy); 7.81 (t, J = 7.6 Hz, 1H, HPy); 7.34 (s, 1H, H4Imi); 7.27 (s, 1H, H5Imi); 7.22 (t, 2H, HPy); 5.10 (d, J = 9.4 Hz, 1H, H1β); 4.79 (s, 2H, NCH2); 4.15–4.08 (m, 2H, H3,2β); 4.09–4.00 (m, 1H, H4β); 3.87–3.76 (m, 2H, H6β); 3.48 (dd, J = 8.7, 4.0 Hz, 1H, H5β); 1.99–1.86 (m, 2H, NCH2CH2); 1.63–1.52 (m, 2H, CH2CH3); 1.04 (t, J = 7.3 Hz, 3H, CH3). 13C NMR (75 MHz, D2O) δ 153.2 (C2Imi); 139.0 (CHPy); 126.0 (C5Imi); 125.1 (CHPy); 121.4 (C4Imi); 89.4 (C1); 86.0 (C5); 78.0 (C3); 77.2 (C2); 72.3 (C4); 62.3 (C6); 52.0 (NCH2); 32.2 (NCH2CH2); 19.5 (CH2CH3), 13.1 (CH3).General procedure for the Stille cross-coupling reaction catalyzed by palladium(II) complexes (3a–c)
In a Schlenk tube loaded with 1.5 mL of DMF/H2O (1:5), 2 mol% of palladium(II) complex and 0.007 mmol K2CO3 were added. After stirring for 5 min, 0.48 mmol of bromobenzene and 0.4 mmol of arylstannane were added consequently. The mixture was stirred for 18 h at 80 °C and then ethyl ether (10 mL × 2) was added and extracted. The organic layer was dried over MgSO4 and injected on GC-MS to determine to reaction conversion. The product was purified by flash chromatography on silica gel doped with 10% of KF (hexane:EtOAc, 9:1–7:3). To the aqueous phase containing hydrophilic catalyst, bromobenzene and arylstannane were added. The mixture was stirred at room temperature for 18 h. This process was repeated several times until the transformation was no longer efficient.
C–C coupling product characterization
4. Conclusions
Galactopyranosyl imidazolium salts from aryl- or β-D-galactosyl imidazole as precursors were used to synthesize three new stable Pd(II)–PEPPSI type precatalyst complexes, in excellent yield under mild conditions, employing PdCl2 as metal precursor and stoichiometric amounts of pyridine.Catalytic studies indicate that the complexes are active and reusable, in the C–C cross-coupling reactions in aqueous medium. It is important to note that the Stille reaction necessarily requires the use of organic solvents to dissolve the reactants. Excellent yields have been obtained using a mixture of DMF/H2O, reducing the use of organic solvent by up to 80%. On the other hand, in the Suzuki–Miyaura reaction, excellent yields were observed using pure water or DMF/H2O due to the solubility of boronic acid. In both C–C cross-coupling type reactions, a decrease in catalytic activity and an increase of more than 3 times in the turnover number were observed with 0.2 mol% of catalyst. These methodologies allow for excellent conversion percentages and provide a more ecological perspective to these types of transformations.
Further work is under development in our laboratory focused on the effectiveness of these Pd(II)–NHC complexes in other aqueous phase reactions.
Author contributions
Ariana W. Hobsteter: methodology, validation, formal analysis, investigation, writing original draft. Marcos J. Lo Fiego: conceptualization, methodology, validation, supervision, formal analysis, writing original draft, writing – review & editing. Gustavo F. Silbestri: conceptualization, funding acquisition, investigation, methodology, project administration, resources, visualization, writing original draft, writing – review & editing. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by Agencia Nacional de Promoción Científica y Tecnológica (PICT 2019-2522), and Universidad Nacional del Sur (PGI 24/Q108), Bahía Blanca, Argentina. CONICET is thanked for a research fellowship to A. W. H.Notes and references
- Essentials of glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, A. G. Darvill, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 4th edn, 2022 Search PubMed.
- (a) P. de Frémont, E. D. Stevens, M. D. Eelman, D. E. Fogg and S. P. Nolan, Synthesis and characterization of gold(I) N-heterocyclic carbene complexes bearing biologically compatible moieties, Organometallics, 2006, 25, 5824–5828 CrossRef; (b) W. Zhao, V. Ferro and M. V. Baker, Carbohydrate–N-heterocyclic carbene metal complexes: synthesis, catalysis and biological studies, Coord. Chem. Rev., 2017, 339, 1–16 CrossRef CAS; (c) M. E. Cucciolito, M. Trinchillo, R. Iannitti, R. Palumbo, D. Tesauro, A. Tuzi, F. Ruffo and A. D'Amora, Sugar-incorporated N-heterocyclic-carbene-containing gold(I) complexes: synthesis, characterization, and cytotoxic evaluation, Eur. J. Inorg. Chem., 2017, 42, 4955–4961 CrossRef; (d) J. P. Byrne, P. Musembi and M. Albrecht, Carbohydrate-functionalized N-heterocyclic carbene Ru(II) complexes: synthesis, characterization and catalytic transfer hydrogenation activity, Dalton Trans., 2019, 48, 11838–11847 RSC; (e) A. Annunziata, A. Amoresano, M. E. Cucciolito, R. Esposito, G. Ferraro, I. Iacobucci, P. Imbimbo, R. Lucignano, M. Melchiorre, M. Monti, C. Scognamiglio, A. Tuzi, D. M. Monti, A. Merlino and F. Ruffo, Pt(II) versus Pt(IV) in carbene glycoconjugate antitumor agents: minimal structural variations and great performance changes, Inorg. Chem., 2020, 59, 4002–4014 CrossRef CAS PubMed.
- (a) M. Guitet, F. Marcelo, S. A. de Beaumais, Y. Zhang, J. Jiménez-Barbero, S. Tilloy, E. Monflier, M. Ménand and M. Sollogoub, Diametrically Opposed Carbenes on an α-Cyclodextrin: Synthesis, Characterization of Organometallic Complexes and Suzuki–Miyaura Coupling in Ethanol and in Water, Eur. J. Org Chem., 2013, 18, 3691–3699 CrossRef; (b) Y. Imanaka, H. Hashimoto and T. Nishioka, Syntheses and catalytic ability of sugar-incorporated N-heterocyclic carbene pincer Pd complexes possessing various N-substituents, Bull. Chem. Soc. Jpn., 2015, 88, 1135–1143 CrossRef CAS; (c) Z. Zhou, Q. Xie, J. Li, Y. Yuan, Y. Liu, Y. Liu, D. Lu and Y. Xie, Glucopyranoside-functionalized NHCs-Pd(II)-PEPPSI complexes: anomeric isomerism controlled and catalytic activity in aqueous Suzuki reaction, Catal. Lett., 2022, 152, 838–847 CrossRef CAS; (d) Q. Xie, J. Li, X. Wen, Y. Huang, Y. Hu, Q. Huang, G. Xu, Y. Xie and Z. Zhou, Carbohydrate-substituted N-heterocyclic carbenes palladium complexes: high efficiency catalysts for aqueous Suzuki–Miyaura reaction, Carbohydr. Res., 2022, 512, 108516 CrossRef CAS PubMed.
- (a) T. Nishioka, T. Shibata and I. Kinoshita, Sugar-incorporated N-heterocyclic carbene complexes, Organometallics, 2007, 26, 1126–1128 CrossRef CAS; (b) F. Tewes, A. Schlecker, K. Harms and F. Glorius, Carbohydrate-containing N-heterocyclic carbene complexes, J. Organomet. Chem., 2007, 692, 4593–4602 CrossRef CAS; (c) J. C. Shi, N. Lei, Q. Tong, Y. Peng, J. Wei and L. Jia, Synthesis of chiral imidazolinium carbene from a carbohydrate and its rhodium(I) complex, Eur. J. Inorg. Chem., 2007, 15, 2221–2224 CrossRef; (d) B. K. Keitz and R. F. Grubbs, Ruthenium olefin metathesis catalysts bearing carbohydrate-based N-heterocyclic carbenes, Organometallics, 2010, 29, 403–408 CrossRef CAS PubMed; (e) C. C. Yang, P. S. Lin, F. C. Liu, I. J. B. Lin, G. H. Lee and S. M. Peng, Glucopyranoside-incorporated N-heterocyclic carbene complexes of silver(I) and palladium(II): efficient water-soluble Suzuki–Miyaura coupling palladium(II) catalysts, Organometallics, 2010, 29, 5959–5971 CrossRef CAS; (f) T. Shibata, S. Ito, M. Doe, R. Tanaka, H. Hashimoto, I. Kinoshita, S. Yanod and T. Nishioka, Dynamic behaviour attributed to chiral carbohydrate substituents of N-heterocyclic carbene ligands in square planar nickel complexes, Dalton Trans., 2011, 40, 6778–6784 RSC; (g) T. Shibata, H. Hashimoto, I. Kinoshita, S. Yanod and T. Nishioka, Unprecedented diastereoselective generation of chiral-at-metal, half sandwich Ir(III) and Rh(III) complexes via anomeric isomerism on “sugar-coated” N-heterocyclic carbene ligands, Dalton Trans., 2011, 40, 4826–4829 RSC; (h) Z. Zhou, J. Qiu, L. Xie, F. Du, G. Xu, Y. Xie and Q. Ling, Synthesis of chiral imidazolium salts from a carbohydrate and their application in Pd-catalyzed Suzuki–Miyaura reaction, Catal. Lett., 2014, 144, 1911–1918 CrossRef CAS; (i) A. S. Henderson, J. F. Bower and M. C. Galan, Pseudo-enantiomeric carbohydrate-based N-heterocyclic carbenes as promising chiral ligands for enantiotopic discrimination, Org. Biomol. Chem., 2020, 18, 3012–3016 RSC; (j) Z. Zhou, Y. Zhao, H. Zhen, Z. Lin and Q. Ling, Poly(ethylene glycol)- and glucopyranoside-substituted N-heterocyclic carbene precursors for the synthesis of arylfluorene derivatives using efficient palladium-catalyzed aqueous Suzuki reaction, Appl. Organomet. Chem., 2016, 30, 924–931 CrossRef CAS; (k) S. Denizalti, F. Çetin telli, S. Yildiran, A. Y. Salman and B. Cetinkaya, Newly synthesized furanoside-based NHC ligands for the arylation of aldehydes, Turk. J. Chem., 2016, 40, 689–697 CrossRef CAS.
- (a) C. J. Li and T. H. Chan, Organic reactions in aqueous media, Wiley, New York, 1997 Search PubMed; (b) P. A. Grieco, Organic synthesis in water, Blackie Academic & Professional, London, 1998 CrossRef.
- (a) M. Kosugi and K. Fugami, A historical note of the Stille reaction, J. Organomet. Chem., 2002, 653, 50–53 CrossRef CAS; (b) C. Cordovilla, C. Bartolomé, J. M. Martinez-Ilarduya and P. Espinet, The Stille reaction, 38 years later, ACS Catal., 2015, 5, 3040–3053 CrossRef CAS.
- K. Shaughnessy and R. DeVasher, Palladium-Catalyzed Cross-Coupling in Aqueous Media: Recent Progress and Current Applications, Curr. Org. Chem., 2005, 9, 585–604 CrossRef CAS.
- J. C. Flores, G. F. Silbestri and E. de Jesús, Water-soluble transition-metal complexes with hydrophilic N-heterocyclic carbene ligands for aqueous-phase applications, Adv. Organomet. Chem., 2022, 77, 169–242 CrossRef CAS.
- A. L. Casalnuovo and J. C. Calabrese, Palladium-catalyzed alkylations in aqueous media, J. Am. Chem. Soc., 1990, 112, 4324–4330 CrossRef CAS.
- (a) N. E. Leadbeater, Fast, easy, clean chemistry by using water as a solvent and microwave heating: the Suzuki coupling as an illustration, Chem. Commun., 2005, 2881–2902 RSC; (b) C. J. Li, Organic reactions in aqueous media with a focus on carbon–carbon bond formations: a decade update, Chem. Rev., 2005, 105, 3095–3166 CrossRef CAS PubMed; (c) V. Polshettiwar, A. Decottignies, C. Len and A. Fihri, Suzuki–Miyaura Cross-Coupling Reactions in Aqueous Media: Green and Sustainable Syntheses of Biaryls, ChemSusChem, 2010, 3, 502–522 CrossRef CAS PubMed; (d) K. H. Shaughnessy, Chapter 1: Metal-Catalyzed Cross-Couplings of Aryl Halides to Form C–C Bonds in Aqueous Media, in Metal-Catalyzed Reactions in Water, ed. P. H. Dixneuf and V. Cadierno, Wiley-VCH, Weinheim, Germany, 2013, pp. 1–46 Search PubMed.
- (a) A. Chatterjee and T. R. Ward, Recent advances in the palladium catalyzed Suzuki–Miyaura cross-coupling reaction in water, Catal. Lett., 2016, 146, 820–840 CrossRef CAS; (b) D. Q. Dong, H. Yang, M. Y. Zhou, Z. H. Wei, P. Wu and Z. L. Wang, Recent Advances in Palladium-Catalyzed Reactions in Water, Curr. Opin. Green Sustainable Chem., 2023, 40, 100778 CrossRef CAS.
- Y. Imanaka, H. Hashimoto, I. Kinoshita and T. Nishioka, Incorporation of a sugar unit into a C–C–N pincer Pd complex using click chemistry and its dynamic behavior in solution and catalytic ability toward the Suzuki–Miyaura coupling in water, Chem. Lett., 2014, 43, 687–689 CrossRef CAS.
- Y. Imanaka, K. Nakao, Y. Maeda and T. Nishioka, Sugar-incorporated chelating bis-N-heterocyclic carbene palladium complexes. Synthesis, structures, and catalytic ability for Suzuki-Miyaura cross-coupling reactions in water, Bull. Chem. Soc. Jpn., 2017, 90, 1050–1057 CrossRef CAS.
- A. W. Hobsteter, M. A. Badajoz, M. J. Lo Fiego and G. F. Silbestri, Galactopyranoside-substituted N-heterocyclic carbene gold(I) complexes: synthesis, stability, and catalytic applications to alkyne hydration, ACS Omega, 2022, 7, 21788–21799 CrossRef CAS PubMed.
- C. Y. Liao and H. M. Lee, Trans-dichlorodipyridinepalladium(II), Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m680–m681 CrossRef CAS.
- Similar displacement for analogous species. Ref. 3d.
- B. Wang, D. D. McClosky, C. T. Anderson and G. Chen, Synthesis of a suite of click-compatible sugar analogs for probing carbohydrate metabolism, Carbohydr. Res., 2016, 433, 54–62 CrossRef CAS PubMed.
- 4a is stable at room temperature, but not in D2O solution, having a spectrum that is not superimposable after 3 days. Furthermore, black palladium was not observed.
- In a Schlenk tube loaded with 1.5 mL of solvent, 1 mol% of palladium(II) complex (3c) and 0.8 mmol K2CO3 were added. After stirring for 5 min, 0.4 mmol of bromobenzene and 0.6 mmol of (p-methoxyphenyl) boronic acid were added consequently. The mixture was stirred for 18 h at 80 °C and then ethyl ether (10 mL × 2) was added and extracted. The organic layer was dried over MgSO4 and injected on GC-MS to determine to reaction conversion. To the aqueous phase containing hydrophilic catalyst, bromobenzene and (p-methoxyphenyl) boronic acid were added. The mixture was stirred at room temperature for 18 h. This process was repeated several times until the transformation was no longer efficient.
- D. D. Perrin, W. L. F. Armarego and D. R. Perrin, Purification of laboratory chemicals, 2nd edn, Pergamon Press, 1980 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR, 2D-HSQC and FTIR spectra for compounds 3a–c and 4a. See DOI: https://doi.org/10.1039/d3ra08031e |
| ‡ Member of CONICET. |
| This journal is © The Royal Society of Chemistry 2024 |