
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atypical hydrogenation selectivity of platinum by reactive environment modulation†
Jurjen
Cazemier‡
 a,
Moussa
Zaarour‡
a,
Sarah
Komaty
a,
Polina
Lavrik
a,
Antonio
Aguilar Tapia
b,
Sudheesh Kumar
Veeranmaril
a,
Jean-Louis
Hazemann
c and
Javier
Ruiz-Martinez
*ad
a,
Moussa
Zaarour‡
a,
Sarah
Komaty
a,
Polina
Lavrik
a,
Antonio
Aguilar Tapia
b,
Sudheesh Kumar
Veeranmaril
a,
Jean-Louis
Hazemann
c and
Javier
Ruiz-Martinez
*ad
aKAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: javier.ruizmartinez@kaust.edu.sa
bInstitut de Chimie Moléculaire de Grenoble, UAR2607 CNRS Université Grenoble Alpes, Grenoble F-38000, France
cInstitut Néel, UPR 2940 CNRS - Université Grenoble Alpes, Grenoble F-38000, France
dChemical Engineering Program, Physical Science and Engineering (PSE) Division, King Abdullah University of Science and Technology, Saudi Arabia
First published on 10th September 2024
Abstract
Controlling selectivity in the hydrogenation of compounds with several functional groups remains a challenging task in heterogeneous catalysis. We report herein the design and use of simple catalysts composed of Pt nanoparticles confined in Silicalite-1 (S-1, MFI type zeolite) for the inversion of the inherent C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O selective hydrogenation of cinnamaldehyde over Pt. The encapsulated catalyst achieved an unconventional 98% hydrocinnamaldehyde selectivity at high conversion levels (88%), which bypasses the inherent selectivity of Pt for CO versus CC hydrogenation. This high selectivity originates from the combination of the low polarity of the S-1 support and the toluene solvent that favor interaction of the catalyst with the CC over the CO group, and to the efficient encapsulation of the platinum in the zeolite channels which restricts the growth of the nanoparticles and consequently decreases the number of undesirable Pt(111) facets active for CO hydrogenation.
O selective hydrogenation of cinnamaldehyde over Pt. The encapsulated catalyst achieved an unconventional 98% hydrocinnamaldehyde selectivity at high conversion levels (88%), which bypasses the inherent selectivity of Pt for CO versus CC hydrogenation. This high selectivity originates from the combination of the low polarity of the S-1 support and the toluene solvent that favor interaction of the catalyst with the CC over the CO group, and to the efficient encapsulation of the platinum in the zeolite channels which restricts the growth of the nanoparticles and consequently decreases the number of undesirable Pt(111) facets active for CO hydrogenation.
1. Introduction
Selective hydrogenations are highly attractive reactions due to their widespread use, relatively low cost, and full atom efficiency.1 Hydrogenation steps are involved in roughly a quarter of all chemical transformations.2–4 However, selectively obtaining the targeted product in the hydrogenation of substrates with multiple reducible groups remains challenging. One example is the hydrogenation of α,β-unsaturated aldehydes, which can be converted to their corresponding aldehydes, unsaturated alcohols, or saturated alcohols by selectively hydrogenating the CC, CO, or both, respectively. At present, the catalytic hydrogenation of this class of compounds is not widely applied in industry, despite the excellent atom economy and the potential for decreased waste generation, due to an insufficient selectivity to the desired product.5 As such, much attention is paid to developing effective hydrogenation catalysts and to understanding the factors that influence selectivity.
Most research utilizes the inherent selectivity of the catalytically active metal i.e., Pd for CC hydrogenation and Pt, Au, and Ir for CO hydrogenation.6 This can be clearly observed in the hydrogenation of cinnamaldehyde, a commonly used model substrate where selectively hydrogenating either the CC or the CO is challenging as a result of its conjugated phenyl group.7 In this context, nickel-based heterogeneous catalysts were also employed for the selective CC hydrogenation and gave values ranging from 3% to 100% depending on the catalyst properties.5 One interesting example was reported by Zeng who demonstrated excellent CC selectivity at full conversion using the catalyst mSiO2@Ni/SiO2@mSiO2.8 This catalyst did not require a promoter and operated at relatively low temperature (80 °C). Despite the relatively lower cost of nickel, Ni-based heterogeneous catalysts often suffer from low activity and require the use of high metal loadings (sometimes exceed 30 wt%),9 high hydrogen pressure (above 30 bars),8 or the incorporation of promoters5 to enhance the activity. Noteworthy, employing promoters may involve a multi-step synthesis process that affect the Ni particle size, metal–support interactions, and consequently the catalyst's selectivity. Additionally, the properties of the promoter and the interactions between Ni and the promoter can also affect selectivity, potentially reducing the selectivity for hydrocinnamaldehyde.5 Therefore, developing facile approaches for preparing highly active noble-metal based heterogeneous catalyst with low metal loading, that do not require promoters or high hydrogen pressures is highly beneficial.
Recent reviews on the cinnamaldehyde hydrogenation show that only a few Pt-based catalysts were developed for high CC hydrogenation selectivity5,6 whereas the vast majority of Pt-based heterogeneous catalysts are used for the selective CO hydrogenation. The existence of these examples is very important to show that the selectivity of the catalyst can be modulated by controlling the catalyst properties and the reaction conditions.10–16 In more detail, alcohols, typically used as solvents for this reaction, give a higher selectivity to CO hydrogenation and can interact with the carbonyl group giving rise to acetal and ketal derivatives. This problem is avoided when using an apolar aromatic solvent such as toluene. However, in this case, the selectivity becomes higher for CC hydrogenation with a decrease in the catalyst activity due to blocking of active sites by the solvent molecules.12 In terms of the catalyst properties, using more polar supports enhances the selectivity to CO hydrogenation. In one example, Lee and coworkers demonstrated that replacing Pt/SiO2 catalyst by the more polar Pt/SBA-15 increases the cinnamyl alcohol (COL) selectivity from 56% to 90%. They attributed this enhancement to the more favored CO activation and suppressed CC reduction over the more polar catalyst.15 As such, we expect that using a support with a lower polarity than SiO2 should enhance the selectivity to CC versus CO hydrogenation. However, no detailed studies were made in this regard, to the best of our knowledge. Nevertheless, alternative methods to improve the CC selectivity such as adding Lewis acid functionality to the catalyst were used. Pt NPs supported on MIL-101 showed one the highest reported hydrocinnamaldehyde (HCAL) selectivities (>99.9%) due to the large amount of Lewis acid groups in MIL-101 that strongly interact with the CO group, making its hydrogenation highly unfavorable.17 Similarly, Huang et al. demonstrated that the addition of Lewis acidic Al additives could shift the selectivity from excellent CO hydrogenation to almost exclusively CC hydrogenation (>97.1%) when using PtFe nanospheres.7 Electronic effects from the supports were also shown to affect the selectivity. Huang et al. achieved 87.9% HCAL selectivity over Pt3Ni@Ni32Cu(OH)2-2 nanowires (NWs).1 This high selectivity to CC hydrogenation is attributed to the reduced electron density of platinum in the presence of nickel and copper in the support.1 Zhang et al. attained a similar selectivity of 87% over (Pt-enriched cage)@CeO2 consisting of hollow CeO2 shell and inner Pt–Ag coating.18 They suggested that Ag in the Pt wall, support effects from CeO2, and the very thin layer of Pt sites are the primary reasons behind the high selectivity.
Besides the role of the support, the Pt NPs size and geometry also have a distinctive role in the CAL hydrogenation. The Pt(111) facets, typically found in larger NPs (>1.2 nm), preferentially adsorb the CAL molecule through the CO bond thus producing more unsaturated alcohol, on the other hand, Pt(100) facets adsorb CAL through both the CC and CO and thus yielding more saturated alcohol, meanwhile the low coordination sites found in small particles and Pt(110) sites preferentially adsorb CAL through the CC bond and yield more aldehyde.19 Finally, decreasing the hydrogen pressure increases HCAL selectivity by lowering the hydrogen density on the platinum surface. This lower hydrogen density allows cinnamaldehyde to adsorb through the more sterically demanding modes, thus permitting the CC to bind to Pt to a greater extent.15
Finally, a relatively new approach used to influence selectivity is the encapsulation of metal nanoparticles in porous supports, such as zeolites.20–24 Following this approach, the narrow pore cavities of the supports may restrict certain adsorption conformations onto the metal, thereby endowing catalysts with chemoselectivity.25,26 Additionally, encapsulation restricts the growth of metal particles during catalyst activation due to internal size constraints leading to small and sintering resistant nanoparticles.26–28 This approach allowed for the formation of COL with almost 99% selectivity due to the restricted adsorption conformation of CAL when hydrogenated over Pt@S-1 using methanol as a solvent.
While most literature focuses on engineering the catalyst and optimizing reaction conditions to enhance CO selectivity during CAL hydrogenation over Pt-based catalysts, taking advantage of the inherent selectivity of Pt for CO hydrogenation, inverting selectivity to obtain HCAL is less considered as it requires the incorporation of acid functionalities or the introduction of additional metals. Herein we demonstrate the selective CC hydrogenation over Pt-based heterogeneous catalyst prepared in a simple approach, without the need for an acid promoter or other metal functionalities. An excellent 98% selectivity to CC hydrogenation was achieved at 88% conversion using Pt NPs confined in Silicalite-1 (S-1), an MFI type zeolite, and toluene solvent. This exceptional selectivity originates from the nonpolar reactive environment composed of the S-1 pore walls and toluene solvent, combined with ultrafine Pt nanoparticles free from Pt(111) facets, formed in the restricted spaces within the zeolite channels. This environment reduces the hydrogenation of the CO bond and thus favors the formation of HCAL.
2. Experimental
2.1. Chemicals and materials
All the commercial products were used as received. Tetrapropylammonium hydroxide (TPAOH, 1 M in water, Sigma Aldrich), tetraethyl orthosilicate (TEOS, ≥99%, Sigma Aldrich), ethylene diamine (99%, Sigma Aldrich), ethylene glycol (99.8%, Sigma Aldrich), sodium hydroxide (≥98%, NaOH, Sigma Aldrich), platinum on silica (1 wt%, Sigma Aldrich), fumed silica (Sigma Aldrich, 0.007 μm, 390 ± 40 m2 g−1), dodecane (≥99%, Sigma Aldrich), 1-hexene (97%, Sigma Aldrich), trans-cinnamaldehyde (99%, Sigma Aldrich), sodium hexachloroplatinate hexahydrate (Na2PtCl6·6H2O, 34.17 wt% Pt by ICP, Alfa Aesar), tetraamineplatinum nitrate ([Pt(NH3)4](NO3)2, 99.99%, Alfa Aesar), Reichardt's dye (90%, Sigma Aldrich), absolute ethanol (99.95%, AnalaR NORMAPUR® ACS, VWR), methanol (≥99.9%, HiPerSolv CHROMANORM, VWR), toluene (≥99.8%, HiPerSolv CHROMANORM, VWR), dichloromethane (DCM, ≥99.8%, VWR), cyclododecene (cis- and trans-mixture, >95.0%, TCI), deionized water (MilliQ Direct-Q® 5, 18 MΩ cm).2.2. Material synthesis
Silicalite-1 (S-1) was synthesized based on a procedure reported by Yu et al. using a gel composition of 1.0 SiO2: 0.4 TPAOH: 36.1 H2O: 4.0 EtOH.29 Typically, TEOS (4.0 g) was added dropwise to a solution of H2O (5.9 g) and TPAOH (1 M in water, 7.8 g) under vigorous stirring. TEOS was hydrolyzed by stirring for 4 h and a clear suspension was obtained. The suspension was transferred to a PTFE-lined stainless-steel 50 mL autoclave and crystallization was conducted in a static oven at 170 °C for 72 h. The as-synthesized solid product was centrifuged and washed with water until the pH reached 7. The product was freeze-dried and calcined in static air at 550 °C for 6 h (1.5 °C min−1 ramp).Pt@S-1-in was synthesized based on an in situ procedure reported by Yu et al. using a gel composition of 1.0 SiO2: 0.4 TPAOH: 34.9 H2O: 4.0 EtOH: 0.001 Na2PtCl6·6H2O: 0.074 ethylene diamine.29 Typically, TEOS (4.1 g) was added to a solution of H2O (5.8 g) and TPAOH (1 M in water, 7.8 g) under vigorous stirring, and hydrolyzed completely over 4 h. [Pt(NH2CH2CH2NH2)2]Cl2 was prepared by mixing ethylene diamine (95 μL) with an aqueous Na2PtCl6·6H2O solution (0.1 M, 192 μL). The Pt precursor was added in two portions into the synthesis gel with 0.5 h of stirring after each addition. The resulting clear colorless suspension was transferred to a PTFE bottle and crystallized at 100 °C for 65 h (Pt@S-1-in) or transferred to a PTFE-lined stainless-steel 50 mL autoclave and crystallized at 170 °C for 72 h (Pt@S-1-in-l). The solid products were centrifuged and washed with water until pH 7. The product was freeze-dried and calcined in static air at 550 °C for 6 h (1.5 °C min−1 ramp).
Pt@S-1-im was prepared using an incipient-wetness impregnation. Briefly, calcined S-1 (1.0 g) is added to a Schlenk flask and dried overnight at 120 °C under vacuum. [Pt(NH3)4](NO3)2 (9.0 mg, 0.02 mmol) is dissolved in deionized water (190 μL). The platinum precursor solution is added dropwise to the S-1 under stirring. After the addition is complete, the powder is stirred for another 4 h before drying under vacuum at 120 °C overnight. The final Pt@S-1-im is obtained by calcination in static air at 300 °C for 3 h (1.5 °C min−1 ramp).
Pt nanoparticles stabilized in ethylene glycol were synthesized using a reported procedure.30 Briefly, a glycol solution of NaOH (1.5 mL, 0.5 M) was added into a glycol solution of Na2PtCl6·6H2O (41 mg in 1.5 mL). The transparent yellow solution is heated at 160 °C for 3 h under an Ar flow to remove water and organic byproducts. A black homogeneous colloidal suspension of Pt metal nanoclusters was obtained.
Pt/S-1-ex was obtained using an excess solution impregnation. Pt nanoparticles in ethylene glycol (1.2 mL) are dispersed in methanol (10.8 mL) by sonicating for 15 min. To a vial equipped with magnetic stirring is added calcined S-1 (1.0 g) and the Pt solution. The suspension is homogenized by vigorous stirring followed by sonication for 15 min. The methanol is removed by heating at 50 °C while stirring until a thick paste remained. This paste was dried and calcined in static air (200 °C for 1 h (1.5 °C min−1) to remove ethylene glycol, followed by 300 °C for 2 h (1.5 °C min−1) for calcination) to obtain the final product.
To obtain Pt/SiO2-ex, the S-1 is replaced by silica (1.0 g) and additional methanol (11 mL) is added before homogenizing the suspension.
2.3. Characterization
High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) micrographs were obtained with a Cs-probe corrected Titan microscope (Thermo Fischer Scientific) at the acceleration voltage of 300 kV. Catalysts were reduced at 300 °C (10 °C min−1) in pure hydrogen (8 mL min−1) for 1 h followed by cooling under inert atmosphere (10 mL min−1) prior to analysis. For the establishment of the particle size distribution of platinum, over 200 particles from different micrographs were analysed using the Fiji image processing package. Integrated differential phase contrast (iDPC) STEM imaging was performed on a double aberration-corrected Themis Z microscope (Thermo Fischer Scientific) operated at 300 kV and acquired with a 4-quadrant DF4 detector. Prior to each measurement the microscopes were aligned with a cross-grating standard sample. The dispersions were estimated from average particle sizes by assuming a cuboctahedral geometry.31Silanol density was determined by FTIR using self-supporting pellets in a Nicolet 6700 equipped with a MCT detector and a ZnSe window. Samples are heated to 200 °C for 2 h at 10 °C min−1 prior to in-vacuo spectral acquisition. Spectra were collected at the range of 4000–400 cm−1 by accumulating 64 scans at a 4 cm−1 resolution using Nicolet Omnic Software in Absorbance mode. The aperture was set to 80 and the auto gain option was used. Absorbance and integrals were normalized by pellet mass post treatment.
Polarity measurements were performed using Reichardt's dye. A solution of Reichardt's dye in DCM (0.2 mg ml−1) was added to approximately 100 mg support until a change in color is observed. The solvent was removed, and the sample was dried under vacuum. The samples were analyzed by UV-Vis Jasco V-670 spectrophotometer equipped with a PSH-001 Powder Sample Holder, using a diffuse reflectance mode. BaSO4 was used as a reference. The surface polarity parameter ET was calculated from eqn (1) where λmax is the longest wavelength of the adsorbed Reichardt's dye. The unitless normalized polarity parameter ETN is calculated from eqn (2).32
ET = 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 591/λmax 591/λmax | (1) |
| ETN = (ET − 30.7)/32.4 | (2) |
The contact angle (θ) measurements were carried out on a Drop Shape Analyzer DSA100E instrument from Krüss scientific equipped by a camera placed on the goniometer. Approximately 3 μL liquid droplets were placed onto the surface of self-supporting pellets through a micro syringe with a 2.67 μl s−1 dosing speed and images were record when the liquid droplets achieved stable steady state. The contact angle is determined by the tangential angle measurement obtained from the equilibrium of surface tensions at the interface between the solid and liquid using the Ellipse (Tangent-1) fitting method.
The CO chemisorption experiments were carried out using a Thermo Scientific iS50 FTIR spectrometer equipped with a customized heatable stainless steel SPECAC transmission cell with CaF2 windows and a MCT detector. Spectra were collected at the range of 4000–400 cm−1 by accumulating 64 scans at a 4 cm−1 resolution using Nicolet Omnic Software in Absorbance mode. The aperture was set to 80 and the auto gain option was used. The self-supporting pellets were placed in the cell and pretreated prior to each experiment under hydrogen (6%, gas balance Ar) with a 30 mL min−1 flow at 300 °C (ramp rate 10 °C min−1) for 1 h then purged with He with 30 mL min−1 flow for 1 h and cooled down to room temperature (RT) under the inert atmosphere (He flow). After these conditions, the background was collected. The sample was treated under a CO flow of 20 mL min−1 (3% in He) until the sample is completely saturated with CO (t = 30 min). After this step the sample was purged with 30 mL min−1 of He to remove the excess and the physisorbed CO species then the spectrum was collected.
2.4. Catalyst evaluation
To obtain spent catalysts, the reaction mixture is centrifuged at 10000 rpm for 5 m and washed twice with abs. ethanol.
:763 Pt:CAL) are added to a stainless-steel reactor equipped with mechanical stirring (Mettler Toledo MultiMax). The reactor is stirred at 800 rpm and purged three times with nitrogen followed by three times with hydrogen before pressurizing to 10 bar with hydrogen. The reactor is heated at 60 °C for the desired duration and samples are taken at regular intervals using a dip tube. Samples were filtered over a 0.45 μm filter and analyzed using a GC-FID equipped with HP-5 column.
3. Results and discussion
Silicalite-1 (S-1), a pure silica MFI zeolite, was chosen as support for the confined catalysts. The absence of strong acidic sites is beneficial for reducing side reactions such as aldol condensation and etherification that can occur when using aldehydes as reactants. We aimed to encapsulate Pt nanoparticles in the S-1 micropores in order to restrict the size of the Pt NPs, thereby reducing or preventing the formation of undesired Pt(111) and Pt(100) facets. For that purpose, the sample (Pt@S-1-in) was prepared by an in situ synthesis procedure where the platinum precursor was added into the synthesis gel in the form of a platinum-ethylene diamine complex to avoid its precipitation as hydroxides.29 This facilitates the incorporation of platinum into the micropores of the growing zeolite.27,33 The results were compared with other catalysts bearing Pt NPs and Silicalite-1 or SiO2 supports.The MFI structure of Pt@S-1-in and the other MFI-based catalyst employed in this work (vide infra) were confirmed using powder X-ray diffraction (Fig. S1†). In addition, no reflections from Pt crystalline phases were observed for the Pt@S-1-in sample, indicating the absence of large Pt crystals. Nitrogen physisorption experiments of the MFI-based catalysts revealed a surface area (368–403 m2 g−1) and micropore volumes (0.11–0.14 cm3 g−1, Table S1 and Fig. S2†) in the range of literature values.34 The Pt loading of Pt@S-1-in was determined to be 0.40 wt% through ICP-OES elemental analysis (Table S2†).
HAADF-STEM analysis revealed Pt@S-1-in as small ellipsoidal zeolite crystals (Fig. S3a†) with Pt NPs homogeneously distributed and a narrow particle size distribution (PSD) of 1.2 ± 0.3 nm (Fig. 1a), indicating that the zeolite channels are indeed restricting the growth of Pt NPs. The location of Pt NPs in the zeolite crystals was further studied by integrated differential phase contrast-scanning transmission electron microscopy (iDPC-STEM) (Fig. S5†).33,35 The Pt NPs appear to be larger than the zeolite channels, which is especially noticeable in Fig. S5a† indicating that the NPs are growing also through defects.20 However, the center of the majority of Pt NPs is adjacent to the straight channels (dark lines and holes), as such the Pt NPs seem to be preferentially formed near the sinusoidal channels. Such preference for the sinusoidal channels has been previously observed and attributed to the occupation of the intersectional channels by the template molecule, leaving the straight and sinusoidal channels available for Pt species, with a higher preference for the sinusoidal ones due to their larger size. This was also supported by calculations showing that it is favorable for the ethylene diamine complex to be located in the sinusoidal channels.29,36In situ X-ray absorption spectroscopy (Fig. S6-S8†) suggests that the Pt NPs of the employed catalysts are fully reduced under the reduction and the hydrogenation reaction conditions used. On the other hand, HAADF-STEM analysis of Pt/SiO2 catalyst revealed a fully amorphous support morphology (Fig. S3f†) and Pt NPs on the surface with a wide PSD of 3.0 ± 1.2 nm (Fig. 1b).
 | ||
| Fig. 1 Comparative study between the structure and performance of Pt@S-1-in and the commercial Pt/SiO2 catalyst. HAADF-STEM images and particle size distributions of (a) Pt@S-1-in and (b) Pt/SiO2. (c) Activity comparison in the hydrogenation of 1-hexene. (d) Evaluation of the confinement through the cyclododecene hydrogenation, lower conversion indicates better confinement. (e) Main structural differences between Pt@S-1-in and Pt/SiO2 catalysts. (f) HCAL selectivity plotted against CAL conversion. | ||
2D electron microscopy and XRD only provide an indication of encapsulation. To conclusively demonstrate the degree of encapsulation of Pt@S-1-in, size-exclusion hydrogenation experiments with cyclododecene were performed. This molecule is too large to be accommodated in the S-1 pores and therefore can only be reduced over platinum on the external surface. We also performed 1-hexene hydrogenation experiments to verify the activity of this catalyst as 1-hexene is small enough to fit inside the pores of S-1 and should be readily hydrogenated in case the catalyst is active.
Apparent TOFs determined from 1-hexene hydrogenations (Fig. 1c) at initial conversion rates over Pt@S-1-in and Pt/SiO2 showed that, while both catalysts are capable of hydrogenating 1-hexene, Pt/SiO2 is significantly more active. We initially suspected that the lower activity of Pt@S-1-in originated from diffusion limitations induced by encapsulation. However, further investigations with Pt NPs supported on S-1 (Pt/S-1-ex) revealed only a slightly higher apparent TOF compared to in the confined catalyst (Fig. S9 and Table S3†), indicating that the difference in apparent TOF mainly results from the type of the support and not the presence of diffusion limitations. The larger substrate, cyclododecene, on the other hand, was fully converted over the commercial Pt/SiO2 within 24 h of reaction at 80 °C (Fig. 1d). Meanwhile, no conversion is observed over Pt@S-1-in in the investigated timespan (24 h). All the characterization results strongly confirm that our synthesis strategy renders ultrafine Pt NPs encapsulated in the zigzag pores of S-1. The structure of the confined sample and commercial reference are illustrated in Fig. 1e.
After evaluating the encapsulation of Pt NPs within Pt@S-1-in, the performance in the selective hydrogenation of CAL was assessed and compared with the Pt/SiO2 catalyst (Fig. 1f). The hydrogenation of CAL can give multiple products (Scheme 1). The reaction conditions were chosen to maximize selectivity to HCAL and minimize the formation of COL. Specifically, a mild temperature (100 °C) was used to reduce the overhydrogenation to COL,37 and a moderate hydrogen pressure (10 bar) was employed to avoid limitations arising from low hydrogen solubility while still preventing the excess formation of COL associated with high hydrogen pressures.15,16,37 Finally, the toluene solvent was selected due to its ability to suppress the formation COL.37,38
 | ||
| Scheme 1 Hydrogenation products of cinnamaldehyde. | ||
CAL hydrogenation in the presence of bare S-1 showed negligible conversion after 49 h time on stream, confirming that S-1 is not catalytically active. Under the selected reaction conditions, selectivity to HCAL over commercial Pt/SiO2 reached a maximum of 85% at 75% conversion, such observation, despite the inherent selectivity of Pt to CO hydrogenation, is a consequence of the optimized reaction conditions. Nonetheless, the primary side products remain COL, formed by undesired CO hydrogenation of CAL, and hydrocinnamyl alcohol (HCOL) originated primarily from further hydrogenation of COL (Fig. S11a†), as evidenced by the increase of HCOL in parallel to the decrease of COL. The HCAL selectivity decreases at higher conversion levels to 75% due to the overhydrogenation of the HCAL to HCOL.
The confinement of Pt NPs in the pores of S-1 greatly enhances HCAL selectivity. Under the same reaction conditions as Pt/SiO2, Pt@S-1-in achieves 99% selectivity to HCAL at 78% conversion. No cinnamyl alcohol (COL) could be detected over the confined catalyst even at high conversions (Fig. S12a†). Nonetheless, selectivity to CAL slightly decreased to 98% at 88% conversion due to the limited transformation of HCAL into HCOL. Given that the reactions were performed under similar conditions, the difference in selectivity between Pt/SiO2 and Pt@S-1-in is attributed to the catalysts structure. This excellent selectivity is accompanied, nevertheless, with a decrease in the apparent initial TOF (58 h−1) compared to Pt/SiO2 (141 h−1), this observation, similar to the lower apparent TOF of Pt@S-1-in in the hydrogenation of 1-hexene can be attributed to the type of support used.
To understand the origin of the high selectivity of Pt@S-1-in compared to the commercial catalyst, several parameters such as the support type, Pt particle size, and confinement were investigated. Lee et al. observed an increase in HCAL selectivity when using the less polar SiO2 support instead of the more polar SBA-15.15 To evaluate the importance of the support polarity in our case, catalysts with Pt NPs supported on silica (Pt/SiO2-ex, 0.45 wt% Pt, Table S2†) and on Silicalite-1 (Pt/S-1-ex, 0.49 wt% Pt, Table S2†) were prepared by impregnation with a colloidal Pt NPs solution. STEM images and PSDs, shown in Fig. 2a and b, confirm that the Pt particles in both catalysts have a similar size distribution of 2.3 ± 1.1 nm (Pt/S-1-ex) and 2.0 ± 0.8 nm (Pt/SiO2-ex). These particles are substantially larger than the S-1 pore entrance (0.55 nm) and ensures that Pt is only present on the external surface.30,39,40
 | ||
| Fig. 2 Investigation of the effect of support polarity on HCAL selectivity by comparing catalysts of similar Pt NP size and different support polarity. HAADF-STEM images and particle size distributions of (a) a nonpolar Pt/S-1-ex and (b) a polar Pt/SiO2-ex catalysts. (c) FTIR spectra normalized to mass showing the silanol bands. (d) Diffuse reflectance (DR) UV-vis spectra of Reichardt's dye adsorbed onto SiO2 and S-1 supports. Water contact angle measurements on self-supported pellet of (e) SiO2 and (f) S-1. (g) HCAL selectivity plotted against CAL conversion. | ||
To confirm if replacing the SiO2 supports S-1 results in a decreased polarity, we performed a series of experiments including silanol concentration determination, adsorption of Reichardt's dye, and water contact angle determination and the results are summarized in Fig. 2c–f, Fig. S15–S18, and Table S4.† FTIR spectrum of the Pt/S-1-ex catalyst shows a small band at 3742 cm−1 ascribed to OH stretching vibrations of isolated silanols (Fig. 2c).15 For the Pt/SiO2-ex sample, the same band is observed but with much higher intensity. Additionally, the Pt/SiO2-ex catalyst shows two overlapping broad contributions at lower wavenumbers centered around 3665 cm−1 and 3550 cm−1, attributed to geminal and vicinal silanol groups.15 The relative ratios of silanol density between catalysts, determined by comparing the total band area normalized by pellet mass, revealed that Pt/SiO2-ex possesses over 40 times more silanol groups. To better understand how this difference in silanol concentration affects polarity, we adsorbed Reichardt's dye on the corresponding supports and monitored the resulting absorption profile by diffuse reflectance (DR) UV-vis absorption spectroscopy. Reichardt's dye is used an indicator for comparing the polarity of surfaces or solvents due to the solvatochromism of its longest absorption wavelength (λmax), which is redshifted when the dye is adsorbed on a lower polarity surface.41–44 In our case, adsorbing the dye on S-1 gave an absorption band centered at round 315 nm and a shoulder at 375 nm corresponding to the dye charge transitions, independent of the support (Fig. S15†). A third, less-intense broad band centered at 495 nm in the visible region (λmax) which is used as an indicator of relative support polarity (Fig. 2d). This band was blueshifted to 480 nm in the case of SiO2. Additionally, the surface polarity parameter ET and normalized ETN values increased from 57.8 kcal mol−1 and 0.84, respectively, for S-1 to 59.6 kcal mol−1 and 0.89 in the case of SiO2 indicating a higher polarity of the SiO2 support. Finally, water contact angle measurements showed approximately 22° for S-1, which is larger than the 10° observed for SiO2 (Fig. 2e, f, Fig. S17, S18, and Table S4†) indicating a more hydrophobic nature, in line with its lower silanol concentration and polarity. In conclusion, the lower silanol concentration, higher λmax of adsorbed Reichardt's dye, and larger water contact angle observed for S-1 confirm its lower polarity compared to SiO2.
Catalytic performance tests of Pt/S-1-ex and Pt/SiO2-ex, shown in Fig. 2g, show similar trends in selectivity as function of conversion with a maximum and stable selectivity until around 65% conversion, then selectivity decreases with the increase in conversion. Throughout the reaction, the less polar Pt/S-1-ex catalyst demonstrated around 8% higher selectivity. Notably, the product distributions also differ over these catalysts. A lower HCAL selectivity was observed over Pt/SiO2-ex, due to the formation of COL and overhydrogenation of HCAL and COL into HCOL (Fig. S11b†). In contrast, the formation of COL is hindered in the Pt/S-1-ex catalyst (Fig. S11c†), where the decrease in HCAL selectivity is related to its overhydrogenation to HCOL. Based on these observations, we claim that the nonpolar S-1 support reduces the activity of the catalyst towards CO hydrogenation resulting in a higher HCAL selectivity than the homologous catalysts with the more polar SiO2 support.45 On the other hand, in the case of CAL, the Pt NPs can interact with the olefin group only when CAL is adsorbed in a flat conformation (Fig S18†). Considering that the carbonyl and olefin are conjugated vicinal groups, their simultaneous interactions with the catalyst becomes easier, thus increasing the chances of CO hydrogenation which is readily an inherent property of Pt.15,46
The effect of polarity on the selective hydrogenation reaction was further investigated by using methanol, a polar solvent, instead of toluene. Zhang et al. have recently reported high selectivity to COL by using methanol as solvent in a Pt catalyst encapsulated in Silicalite-1.20 In order to make a proper comparison with those results, a Pt-confined catalyst was prepared at similar hydrothermal synthesis conditions as described by Zhang et al. The sample, named Pt@S-1-in-l, has Pt NPs encapsulated in S-1 with similar particle size as Pt@S-1-in, but with larger zeolite particle sizes (Fig. 3a). The catalytic performance results, in Fig. 3b, show a significant change in product selectivity when changing the solvent. Under a polar solvent, HCAL selectivity drastically drops to 49% at 65% conversion, which is similar to the results obtained by Zhang et al. with a catalyst prepared in a similar procedure as reported here.20 Ma et al., who used a Pd0.6Ni@S-1 catalyst, explained these solvent effects by the more favorable activation of the carbonyl double bond by more polar solvents, thus leading to a decreased HCAL selectivity.16 In our case, we observed a higher initial CO hydrogenation rate over Pt@S-1-in-l in methanol (0.51 × 10−2 mol l−1 h−1, Table S14†) compared to toluene (0.02 × 10−2 mol l−1 h−1), in agreement with the role of the polar methanol in the activation of the carbonyl. These results highlight the role of polarity on the reactive environment composed of both the solvent and the catalyst for controlling product selectivity.
 | ||
| Fig. 3 (a) HAADF-STEM image and particle size distribution of Pt@-S-in-l catalyst used to study the effect of solvent polarity on Hydrocinnamaldehyde (HCAL) selectivity. (b) HCAL selectivity plotted as a function of conversion on Pt@S-1-in-l with toluene as a solvent at 100 °C or methanol as a solvent at 60 °C using similar conditions to those reported by Zhang et al.20 | ||
It was noticeable from Fig. 2g that selectivity of the Pt@S-1-ex catalyst at low conversion levels is very similar to the Pt@S-1-in (Fig. 1f), which confirms that the type of support is also critical for product selectivity, however, the decrease in selectivity in the case of Pt/S-1-ex at higher conversion indicates that other factors in the catalyst structure, such as Pt NP size and confinement, may have also contributed to the obtained results. It is noteworthy that the FTIR spectra of the Silicalite-1-based catalysts show similar profiles in the silanol region, with similar band positions and comparable intensities, which are significantly lower than that of Pt/SiO2-ex (Fig. 2c and Fig. S16†). Therefore, we confirm that the differences in selectivity observed over the Silicalite-1-based catalysts are not related to differences in polarity.
To investigate the effect of Pt particle-size on product selectivity, we compared the catalyst Pt@S-1-in bearing 1.1 nm Pt NPs with Pt@S-1-im and Pt/S-1-ex, comprising 1.7 ± 0.5 nm (Fig. S4†) and 2.3 ± 0.8 Pt NPs confined in or supported on S-1 respectively. According to experimental and theoretical studies,10,15 high coordination Pt(111) facets favor the adsorption and hydrogenation of CO group leading to a higher COL selectivity. The number of these sites is known to decrease with the decreasing particle size and they become completely absent in particles smaller than 1.7 nm.47 These calculations assume perfect cuboctahedral Pt NP geometry. Since the zeolite pores can induce a strong geometrical effect, these calculations might be less accurate for the investigated systems. In order to experimentally evaluate the structure of the Pt NPs, CO chemisorption followed by transmission FTIR was used.48–51 The spectra are presented in Fig. 4a and the reference Pt/SiO2 sample is shown in Fig. S21.† The reference Pt/SiO2 sample shows absorption bands at 2096 and 2054 cm−1, which can be attributed to the stretching vibration of CO linearly adsorbed on high and low Pt0 coordination sites, respectively.50,52–54 In the case of the Pt/S-1-ex, Pt@S-1-im and Pt@S-1-in catalysts, the component at high frequency is completely absent, suggesting that the Pt NPs only expose low coordination atoms, which is in line with the small particle size observed by electron microscopy. The remaining bands appearing in the range of 1900–1780 cm−1 in these catalysts are associated with CO adsorbed in a bridged configuration between two Pt atoms.53
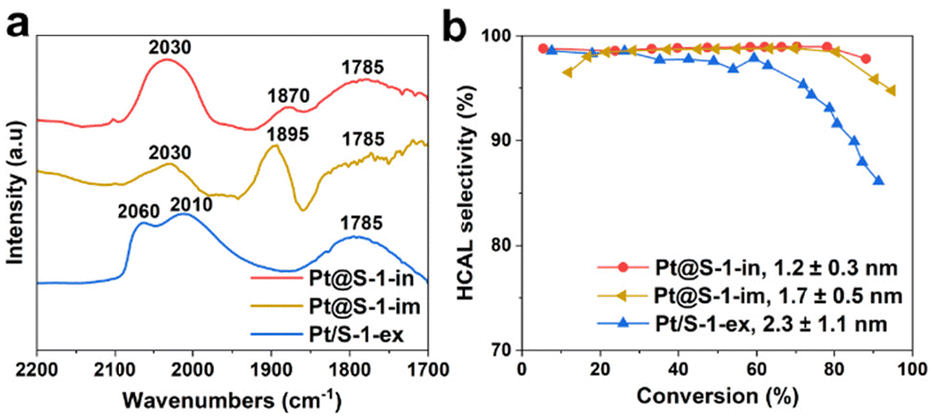 | ||
| Fig. 4 Investigation of the effect of Pt particle size on the HCAL selectivity. (a) FTIR spectra of CO chemisorbed onto the Pt/S-1-ex, Pt@S-1-im, and Pt@S-1-in catalysts at room temperature. Spectra are offset for clarity. (b) HCAL selectivity plotted against CAL conversion on catalysts with small (Pt@S-1-in), medium (Pt@S-1-im), and large (Pt/S-1-ex) Pt particles. | ||
The selectivity to HCAL versus conversion plots for the samples with different particle sizes is presented in Fig. 4b. All the samples show a high selectivity at low conversion levels.
However, a less selective behavior is observed for the samples with the largest Pt NPs (Pt/S-1-ex) at conversions higher than 30%. When comparing catalysts supported on silica, the same behavior is observed, the samples with the largest Pt NPs (Pt/SiO2, see Fig. 1b) has lower HCAL selectivity compared to Pt/SiO2-ex (see Fig. 2g).
Next, the origin of the loss of selectivity at high conversion levels was explored. This was investigated by monitoring the concentration of the undesired HCOL produced versus CAL conversion on Pt@S-1-in and Pt/S-1-ex (Fig. 5), which are the catalysts exhibiting the maximum and minimum HCAL selectivity among the S-1 based catalysts (Fig. S13a†), respectively. Fig. 5a shows that no HCOL was initially produced from the hydrogenation of 83 mM of CAL over both catalysts. Over Pt@S-1-in, a low HCOL concentration of 0.8 mM was only detected at ∼90% conversion (48 h TOS). In contrast, in the case of Pt/S-1-ex the HCOL concentration increased after 30% CAL conversion and reached 6.4 mM of HCOL at 85% conversion, 8 times higher than that found over the confined Pt@S-1-in at a similar conversion level.
 | ||
| Fig. 5 Evolution of particle size with reaction progression and the resulting effect on selectivity. (a) Concentration of hydrocinnamyl alcohol (HCOL) produced versus conversion of 83 mM cinnamaldehyde over Pt@S-1-in and Pt/S-1-ex catalysts. HAADF-STEM images and particle size distributions prior to the reaction and after ∼85% cinnamaldehyde (CAL) conversion of (b and c) Pt@S-1-in at 0 and 48 h TOS, and (d and e) of Pt/S-1-ex at 0 and 72 h TOS. | ||
An inspection of the structure of both catalysts was done after 85% conversion by HAADF-STEM. The Pt@S-1-in shows an overall increase in the average Pt particle size of 0.4 nm (Fig. 5b and c). However, most of the particles are less than 2 nm. In the case of the Pt/S-1-ex, the average NP size have increased more than in the Pt@S-1 counterpart and the sizes are more heterogeneous (Fig. 5d and e). The results suggest that the formation of larger NPs is the main origin of the loss of selectivity in the reaction, highlighting the importance to encapsulate the Pt NP to avoid the formation of large NPs. Finally, we investigated the reusability of the catalyst to evaluate how does the change in Pt particle size during the reaction affect the performance in the next hydrogenation cycle. For this reason, we compensated the CAL converted during the first 24 h and then run the reaction for another cycle. Interestingly, the obtained conversion (47%) and selectivity (94%) from the second cycle were close to those obtained from the first cycle with fresh catalyst (53% and 99%, respectively, Fig. S22†). The small decrease in selectivity, nevertheless, is in line with the observed increase in Pt particle size during the reaction which became more prone to CO hydrogenation than the fresh catalyst.
4. Conclusions
Pt NPs in Silicalite-1 shows an exceptional 98% selectivity to CC hydrogenation of cinnamaldehyde, a model substrate for α,β-unsaturated aldehydes which is atypical selectivity for Pt-based heterogeneous catalysts. Confinement enhances the selectivity to hydrocinnamaldehyde through two main factors. The first and most important is through creating a nonpolar reactive environment that promotes selectivity to CC bond hydrogenation. The second is the Silicalite-1 micropores that impose constraints to the size of Pt NPs, which reduces the number of unselective Pt (111) facets. Reducing the polarity of the reaction medium by replacing the toluene solvent by the more polar methanol decreased HCAL selectivity, which confirms that polarity is the main factor controlling selectivity. These results also suggest that confinement in the zeolite pores is not promoting the reaction by a geometrical effect of the reactant or by promoting a new transition state. Consequently, by precisely engineering the catalyst structure and controlling the reaction conditions, we can guide the selectivity of α,β-unsaturated aldehydes to produce almost only the saturated aldehyde derivative over a Pt metal-based catalysts inherently selective for CO hydrogenation. We have shown the importance of catalyst design and reaction conditions in guiding the selectivity for α,β-unsaturated aldehydes.
Author contributions
Jurjen Cazemier: writing – original draft, writing – review & editing, methodology, investigation, formal analysis, data curation. Moussa Zaarour: writing – original draft, writing – review & editing, methodology, investigation, formal analysis, data curation, supervision. Sarah Komaty: writing – review & editing, methodology, investigation, formal analysis. Polina Lavrik: writing – review & editing, methodology, investigation, formal analysis. Antonio Aguilar Tapia: methodology, investigation, formal analysis. Sudheesh Kumar Veeranmaril: methodology, investigation, formal analysis. Jean-Louis Hazemann: methodology, investigation. Javier Ruiz-Martinez: writing – original draft, writing – review & editing, conceptualization, methodology, funding acquisition, data curation, supervision.The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Funding for this work was provided by baseline grant (BAS/1/1402-01-01) from King Abdullah University of Science and Technology (KAUST). The authors acknowledge Samy Ould-Chikh & Mustafa Caglayan for assisting in the Synchrotron experiments, Mohamad Abou Daher for water contact angle measurements, and Eganathan Kaliyamoorthy for the elemental analysis. We acknowledge the European Synchrotron Radiation Facility for provision of synchrotron radiation facilities, and we would like to thank Eric Lahera for assistance in using beamline BM30.References
- P. Wang, Q. Shao, X. Cui, X. Zhu and X. Huang, Hydroxide-Membrane-Coated Pt3Ni Nanowires as Highly Efficient Catalysts for Selective Hydrogenation Reaction, Adv. Funct. Mater., 2018, 28, 1705918 CrossRef.
- R. A. Sheldon and H. Van Bekkum, Fine Chemicals through Heterogeneous Catalysis, Wiley-VCH, 2008 Search PubMed.
- S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, Wiley, New York, 2001 Search PubMed.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Selective Hydrogenation over Supported Metal Catalysts: From Nanoparticles to Single Atoms, Chem. Rev., 2020, 120, 683–733 CrossRef CAS PubMed.
- X. Wang, X. Liang, P. Geng and Q. Li, Recent Advances in Selective Hydrogenation of Cinnamaldehyde over Supported Metal-Based Catalysts, ACS Catal., 2020, 10, 2395–2412 CrossRef CAS.
- K. N. Patil, P. Manikanta, P. M. Srinivasappa, A. H. Jadhav and B. M. Nagaraja, State-of-the-art and perspectives in transition metal-based heterogeneous catalysis for selective hydrogenation of cinnamaldehyde, J. Environ. Chem. Eng., 2023, 11, 109168 CrossRef CAS.
- C. Yang, S. Bai, Y. Feng and X. Huang, An On–Demand, Selective Hydrogenation Catalysis over Pt−Fe Nanocatalysts under Ambient Condition, ChemCatChem, 2019, 11, 2265–2269 CrossRef CAS.
- B. Li and H. C. Zeng, Formation Combined with Intercalation of Ni and Its Alloy Nanoparticles within Mesoporous Silica for Robust Catalytic Reactions, ACS Appl. Mater. Interfaces, 2018, 10, 29435–29447 CrossRef CAS PubMed.
- R. Xie, G. Fan, Q. Ma, L. Yang and F. Li, Facile synthesis and enhanced catalytic performance of graphene-supported Ni nanocatalyst from a layered double hydroxide-based composite precursor, J. Mater. Chem. A, 2014, 2, 7880–7889 RSC.
- F. Delbecq and P. Sautet, Competitive C=C and C=O Adsorption of α-β unsaturated aldehydes on Pt and Pd surfaces in relation with the selectivity of hydrogenation reactions: A theoretical approach, J. Catal., 1995, 152, 217–236 CrossRef CAS.
- A. S. Nagpure, L. Gurrala, P. Gogoi and S. V. Chilukuri, Hydrogenation of cinnamaldehyde to hydrocinnamaldehyde over Pd nanoparticles deposited on nitrogen-doped mesoporous carbon, RSC Adv., 2016, 6, 44333–44340 RSC.
- Y. Li, H. Cheng, W. Lin, C. Zhang, Q. Wu, F. Zhao and M. Arai, Solvent effects on heterogeneous catalysis in the selective hydrogenation of cinnamaldehyde over a conventional Pd/C catalyst, Catal. Sci. Technol., 2018, 8, 3580–3589 RSC.
- E. V. Ramos-Fernández, J. M. Ramos-Fernández, M. Martínez-Escandell, A. Sepúlveda-Escribano and F. Rodríguez-Reinoso, Selective hydrogenation of cinnamaldehyde over (111) preferentially oriented Pt particles supported on expanded graphite, Catal. Lett., 2009, 133, 267–272 CrossRef.
- M. L. Toebes, Y. Zhang, J. Hájek, T. Alexander Nijhuis, J. H. Bitter, A. Jos Van Dillen, D. Y. Murzin, D. C. Koningsberger and K. P. De Jong, Support effects in the hydrogenation of cinnamaldehyde over carbon nanofiber-supported platinum catalysts: Characterization and catalysis, J. Catal., 2004, 226, 215–225 CrossRef CAS.
- L. J. Durndell, C. M. A. Parlett, N. S. Hondow, M. A. Isaacs, K. Wilson and A. F. Lee, Selectivity control in Pt-catalyzed cinnamaldehyde hydrogenation, Sci. Rep., 2015, 5, 9425 CrossRef CAS PubMed.
- Q. Lu, H. Wang, J. Sun, X.-Z. Wei, Q. Zhang, X. Zhang, L. Chen, J. Liu, Y. Chen and L. Ma, Solvent effect in selective hydrogenation of cinnamaldehyde over Pd–Ni nanoclusters encapsulated within siliceous zeolite, Microporous Mesoporous Mater., 2024, 367, 112979 CrossRef CAS.
- H. Liu, Z. Li and Y. Li, Chemoselective hydrogenation of cinnamaldehyde over a Pt-Lewis acid collaborative catalyst under ambient conditions, Ind. Eng. Chem. Res., 2015, 54, 1487–1497 CrossRef CAS.
- S. Song, X. Liu, J. Li, J. Pan, F. Wang, Y. Xing, X. Wang, X. Liu and H. Zhang, Confining the Nucleation of Pt to In Situ Form (Pt-Enriched Cage)@CeO2 Core@Shell Nanostructure as Excellent Catalysts for Hydrogenation Reactions, Adv. Mater., 2017, 29, 1700495 CrossRef PubMed.
- M. Deng, D. Wang and Y. Li, Rational design of catalysts for heterogeneous selective hydrogenation of unsaturated aldehydes/ketones: From nanoparticles to single atoms, Appl. Catal., A, 2023, 666, 119423 CrossRef CAS.
- C. Liu, P. Zhu, J. Wang, H. Liu and X. Zhang, Geometrically embedding dispersive Pt nanoparticles within silicalite-1 framework for highly selective α, β-unsaturated aldehydes hydrogenation via oriented C = O adsorption configuration, Chem. Eng. J., 2022, 446, 137064 CrossRef CAS.
- L. Alfilfil, J. Ran, C. Chen, X. Dong, J. Wang and Y. Han, Highly dispersed Pd nanoparticles confined in ZSM-5 zeolite crystals for selective hydrogenation of cinnamaldehyde, Microporous Mesoporous Mater., 2022, 330, 111566 CrossRef CAS.
- P. Gallezot, B. Blanc, D. Barthomeuf and M. I. Païs da Silva, Selective hydrogenation of cinnamaldehyde controlled by host/guest interactions in beta zeolite, Stud. Surf. Sci. Catal., 1994, 84, 1433–1439 CrossRef CAS.
- H. Liu, L. Chang, L. Chen and Y. Li, Nanocomposites of Platinum/Metal-Organic Frameworks Coated with Metal-Organic Frameworks with Remarkably Enhanced Chemoselectivity for Cinnamaldehyde Hydrogenation, ChemCatChem, 2016, 8, 946–951 CrossRef CAS.
- Z. Guo, C. Xiao, R. V. Maligal-Ganesh, L. Zhou, T. W. Goh, X. Li, D. Tesfagaber, A. Thiel and W. Huang, Pt nanoclusters confined within metal-organic framework cavities for chemoselective cinnamaldehyde hydrogenation, ACS Catal., 2014, 4, 1340–1348 CrossRef CAS.
- Q. Chen, M. Wang, C. Zhang, K. Ren, Y. Xin, M. Zhao and E. Xing, Selectivity Control on Hydrogenation of Substituted Nitroarenes through End-On Adsorption of Reactants in Zeolite-Encapsulated Platinum Nanoparticles, Chem. – Asian J., 2018, 13, 2077–2084 CrossRef CAS PubMed.
- M. Zaarour, J. Cazemier and J. Ruiz-Martínez, Recent developments in the control of selectivity in hydrogenation reactions by confined metal functionalities, Catal. Sci. Technol., 2020, 10, 8140–8172 RSC.
- S. Goel, Z. Wu, S. I. Zones and E. Iglesia, Synthesis and catalytic properties of metal clusters encapsulated within small-pore (SOD, GIS, ANA) zeolites, J. Am. Chem. Soc., 2012, 134, 17688–17695 CrossRef CAS PubMed.
- Z. Wu, S. Goel, M. Choi and E. Iglesia, Hydrothermal synthesis of LTA-encapsulated metal clusters and consequences for catalyst stability, reactivity, and selectivity, J. Catal., 2014, 311, 458–468 CrossRef CAS.
- N. Wang, Q. Sun, R. Bai, X. Li, G. Guo and J. Yu, In Situ Confinement of Ultrasmall Pd Clusters within Nanosized Silicalite-1 Zeolite for Highly Efficient Catalysis of Hydrogen Generation, J. Am. Chem. Soc., 2016, 138, 7484–7487 CrossRef CAS PubMed.
- P. Wang, J. Zhao, X. Li, Y. Yang, Q. Yang and C. Li, Assembly of ZIF nanostructures around free Pt nanoparticles: Efficient size-selective catalysts for hydrogenation of alkenes under mild conditions, Chem. Commun., 2013, 49, 3330–3332 RSC.
- A. Borodziński and M. Bonarowska, Relation between crystallite size and dispersion on supported metal catalysts, Langmuir, 1997, 13, 5613–5620 CrossRef.
- C. Reichardt, Pyridinium-N-phenolate betaine dyes as empirical indicators of solvent polarity: Some new findings, Pure Appl. Chem., 2008, 80, 1415–1432 CrossRef CAS.
- L. Liu, M. Lopez-Haro, C. W. Lopes, C. Li, P. Concepcion, L. Simonelli, J. J. Calvino and A. Corma, Regioselective generation and reactivity control of subnanometric platinum clusters in zeolites for high-temperature catalysis, Nat. Mater., 2019, 18, 866–873 CrossRef CAS PubMed.
- P. A. Zielinski, A. Van Neste, D. B. Akolekar and S. Kaliaguine, Effect of high-energy ball milling on the structural stability, surface and catalytic properties of small-, medium- and large-pore zeolites, Microporous Mater., 1995, 5, 123–133 CrossRef CAS.
- L. Liu, M. Lopez-Haro, C. W. Lopes, S. Rojas-Buzo, P. Concepcion, R. Manzorro, L. Simonelli, A. Sattler, P. Serna, J. J. Calvino and A. Corma, Structural modulation and direct measurement of subnanometric bimetallic PtSn clusters confined in zeolites, Nat. Catal., 2020, 3, 628–638 CrossRef CAS.
- Z. Wei, X. Zhu, X. Liu, H. Xu, X. Li, Y. Hou and Y. Liu, Pt-Re/rGO bimetallic catalyst for highly selective hydrogenation of cinnamaldehyde to cinnamylalcohol, Chin. J. Chem. Eng., 2019, 27, 369–378 CrossRef CAS.
- A. Al-Shathr, Z. M. Shakor, B. Y. Al-Zaidi, H. Sh. Majdi, A. A. AbdulRazak, S. Aal-Kaeb, A. A. Shohib and J. McGregor, Reaction Kinetics of Cinnamaldehyde Hydrogenation over Pt/SiO2: Comparison between Bulk and Intraparticle Diffusion Models, Int. J. Chem. Eng., 2022, 2022, 303874 Search PubMed.
- Z. M. Shakor, A. A. AbdulRazak and A. A. Shuhaib, Optimization of process variables for hydrogenation of cinnamaldehyde to cinnamyl alcohol over a Pt/SiO2 catalyst using response surface methodology, Chem. Eng. Commun., 2022, 209, 827–843 CrossRef CAS.
- C. Liu, J. Liu, S. Yang, C. Cao and W. Song, Palladium Nanoparticles Encapsulated in a Silicalite-1 Zeolite Shell for Size-Selective Catalysis in Liquid-Phase Solution, ChemCatChem, 2016, 8, 1279–1282 CrossRef CAS.
- T. Humplik, R. Raj, S. C. Maroo, T. Laoui and E. N. Wang, Framework water capacity and infiltration pressure of MFI zeolites, Microporous Mesoporous Mater., 2014, 190, 84–91 CrossRef CAS.
- S. J. Tavener, J. H. Clark, G. W. Gray, P. A. Heath and D. J. Macquarrie, Reichardt's dye as a probe for surface polarity of chemically and thermally treated silicas, Chem. Commun., 1997, 1147–1148 RSC.
- D. J. Macquarrie, S. J. Tavener, G. W. Gray, P. A. Heath, J. S. Rafelt, S. I. Saulzet, J. J. E. Hardy, J. H. Clark, P. Sutra, D. Brunel, F. Di Renzo and F. Fajula, The use of Reichardt's dye as an indicator of surface polarity, New J. Chem., 1999, 23, 725–731 RSC.
- F. Cataldo, Application of Reichardt's Solvent Polarity Scale (ET(30)) in the Selection of Bonding Agents for Composite Solid Rocket Propellants, Liquids, 2022, 2, 289–302 CrossRef CAS.
- S. Spange, A. Reuter and D. Lubda, ET(30) surface polarity parameters of alkyl- and aryl-group-functionalized silica particles: Differentiating the surface environments by means of the application of differently substituted Reichardt's dyes, Langmuir, 1999, 15, 2103–2111 CrossRef CAS.
- Z. J. Berkson, M. Bernhardt, S. L. Schlapansky, M. J. Benedikter, M. R. Buchmeiser, G. A. Price, G. J. Sunley and C. Copéret, Olefin-Surface Interactions: A Key Activity Parameter in Silica-Supported Olefin Metathesis Catalysts, JACS Au, 2022, 2, 777–786 CrossRef CAS PubMed.
- A. J. Plomp, H. Vuori, A. O. I. Krause, K. P. de Jong and J. H. Bitter, Particle size effects for carbon nanofiber supported platinum and ruthenium catalysts for the selective hydrogenation of cinnamaldehyde, Appl. Catal., A, 2008, 351, 9–15 CrossRef CAS.
- J. Aarons, M. Sarwar, D. Thompsett and C.-K. Skylaris, Perspective: Methods for large-scale density functional calculations on metallic systems, J. Chem. Phys., 2016, 145, 220901 CrossRef PubMed.
- R. K. Brandt, M. R. Hughes, L. P. Bourget, K. Truszkowska and R. G. Greenler, The interpretation of CO adsorbed on Pt/SiO2 of two different particle-size distributions, Surf. Sci., 1993, 286, 15–25 CrossRef CAS.
- D. C. Koningsberger, D. E. Ramaker, J. T. Miller, J. De Graaf and B. L. Mojet, The direct influence of the support on the electronic structure of the active sites in supported metal catalysts: Evidence from Pt-H anti-bonding shape resonance and Pt-CO FTIR data, Top. Catal., 2001, 15, 35–42 CrossRef CAS.
- C. Lentz, S. P. Jand, J. Melke, C. Roth and P. Kaghazchi, DRIFTS study of CO adsorption on Pt nanoparticles supported by DFT calculations, J. Mol. Catal. A: Chem., 2017, 426, 1–9 CrossRef CAS.
- J. Ruiz-Martínez, A. Sepúlveda-Escribano, J. A. Anderson and F. Rodríguez-Reinoso, Influence of the preparation method on the catalytic behaviour of PtSn/TiO2 catalysts, Catal. Today, 2007, 123, 235–244 CrossRef.
- A. Corma, P. Serna, P. Concepción and J. J. Calvino, Transforming nonselective into chemoselective metal catalysts for the hydrogenation of substituted nitroaromatics, J. Am. Chem. Soc., 2008, 130, 8748–8753 CrossRef CAS PubMed.
- P. Bazin, O. Saur, J. C. Lavalley, M. Daturi and G. Blanchard, FT-IR study of CO adsorption on Pt/CeO2: Characterisation and structural rearrangement of small Pt particles, Phys. Chem. Chem. Phys., 2005, 7, 187–194 RSC.
- V. Perrichon, L. Retailleau, P. Bazin, M. Daturi and J. C. Lavalley, Metal dispersion of CeO2-ZrO2 supported platinum catalysts measured by H2 or CO chemisorption, Appl. Catal., A, 2004, 260, 1–8 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures and tables on the catalysts' characterization and activity. See DOI: https://doi.org/10.1039/d4qi01820f |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2024 |