
From animal testing to in vitro systems: advancing standardization in microphysiological systems
Darwin R.
Reyes
 *a,
Mandy B.
Esch
a,
Lorna
Ewart
b,
Rohollah
Nasiri
c,
Anna
Herland
c,
Kyung
Sung
d,
Monica
Piergiovanni
e,
Carolina
Lucchesi
f,
James T.
Shoemaker
g,
Jelena
Vukasinovic
g,
Hiroki
Nakae
h,
James
Hickman
i,
Kapil
Pant
j,
Anne
Taylor
k,
Niki
Heinz
l and
Nureddin
Ashammakhi
*m
*a,
Mandy B.
Esch
a,
Lorna
Ewart
b,
Rohollah
Nasiri
c,
Anna
Herland
c,
Kyung
Sung
d,
Monica
Piergiovanni
e,
Carolina
Lucchesi
f,
James T.
Shoemaker
g,
Jelena
Vukasinovic
g,
Hiroki
Nakae
h,
James
Hickman
i,
Kapil
Pant
j,
Anne
Taylor
k,
Niki
Heinz
l and
Nureddin
Ashammakhi
*m
aNational Institute of Standards and Technology (NIST), Gaithersburg, MD, USA. E-mail: darwin.reyes@nist.gov
bEmulate, Inc., Boston, Massachusetts, USA
cRoyal Institute of Technology, Stockholm, Sweden
dFood and Drug Administration (FDA), Silver Spring, Maryland, USA
eEuropean Commission, Joint Research Centre (JRC), Ispra, Italy
fBioneXus Foundation, ATCC, Manassas, VA, USA
gLena Biosciences, Inc., Atlanta, Georgia, USA
hJMAC Japan bio Measurement & Analysis Consortium, Tokyo, Japan
iHesperos, Inc., Orlando, Florida, USA
jSynVivo, Inc., Huntsville, Alabama, USA
kXona Microfluidics, Inc., Research Triangle Park, North Carolina, USA
lAltis Biosystems, Inc., Durham, North Carolina, USA
mInstitute for Quantitative Health Science and Engineering, Department of Biomedical Engineering, College of Engineering, and College of Human Medicine, Michigan State University, East Lansing, MI, USA. E-mail: ashammak@msu.edu
First published on 15th February 2024
Abstract
Limitations with cell cultures and experimental animal-based studies have had the scientific and industrial communities searching for new approaches that can provide reliable human models for applications such as drug development, toxicological assessment, and in vitro pre-clinical evaluation. This has resulted in the development of microfluidic-based cultures that may better represent organs and organ systems in vivo than conventional monolayer cell cultures. Although there is considerable interest from industry and regulatory bodies in this technology, several challenges need to be addressed for it to reach its full potential. Among those is a lack of guidelines and standards. Therefore, a multidisciplinary team of stakeholders was formed, with members from the US Food and Drug Administration (FDA), the National Institute of Standards and Technology (NIST), European Union, academia, and industry, to provide a framework for future development of guidelines/standards governing engineering concepts of organ-on-a-chip models. The result of this work is presented here for interested parties, stakeholders, and other standards development organizations (SDOs) to foster further discussion and enhance the impact and benefits of these efforts.
1. Introduction
Therapeutics have been continuously developed to treat various diseases and they have classically been tested using two-dimensional cell cultures1 and pre-clinical experimental animals.2 The former does not represent in vivo events and therefore cannot completely predict what would happen in the body.3 Experimental animals provide a full in vivo environment and have been extensively used before clinical trials. However, animals are different species and have a physiology different from that of humans.3 Often, drugs that have proved safe and efficient in animals may have side effects which can, in extreme cases, be fatal when the drugs are used in humans. This has led to withdrawal of those drugs even in the post-marketing phase.4 Given the fact that the cost of developing a single drug is about one billion dollars, withdrawal of a drug after all research is done (pre-clinical and clinical) and its marketing has been engaged represents a big loss to developers.5The search for alternative testing approaches resulted in the recognition of opportunities that became available because of the developments made in tissue engineering, organoid biology, and microfluidic devices.6 These paved the way to the introduction of so-called microphysiological systems (MPS). These MPS are engineered microdevices (containing human cells and tissues) that are designed to mimic certain organ structure(s) and function(s) in vitro.7 Thus, they can be used to study function and disease or reproduce and monitor organ reactions after exposure to compounds.8,9 Although each type of MPS can be used for these purposes and applications, each one of them has its own advantages and limitations, and thus combinations of these have also emerged as in the case of the integration of organoids into these systems.
MPS have the advantage of mimicking organs at a small scale while also representing the circulatory flow system of the human body and can also be used to study compounds in very minute volumes.10 In addition to their use for assessing primary toxicity, the integration of multiple MPS units as multi-organ-MPS (MoMPS) or body-on-a-chip systems can be used to study secondary and systemic toxicity. Furthermore, induced pluripotent stem cells (iPSC) can be used to devise personalized MPS and MoMPS. In the future, body- or human-on-a-chip systems can provide an approach to assess the progress of the disease, design individualized therapeutic regimens,11 and monitor response to treatment to provide appropriate adjustments when needed, thus enabling better and more accurate counseling of patients. We expect that MPS will enable reductions in the use of experimental animals, and will greatly reduce cost2 and losses in the drug development process. MPS can be used to develop clinical trials-on-a-chip and help with selecting patients for clinical trials. It is expected that these systems will complement and replace some of the tools currently used for drug development.
To expedite the translation of MPS technology into industrial use and clinical applications, several issues need to be addressed, among which standardization represents an important aspect. However, for standardization to succeed, stakeholders should provide input to metrology labs and regulatory agencies regarding specific recommendations for standardizing this technology and qualifying the existing models.12–15 Standardization should encompass terminology, measurement protocols, and external components that control internal conditions within the system (Fig. 1). This approach will enable the development and utilization of a common language among the scientific community and stakeholders working in the field. A common language would include the definition of various components, processes, and systems involved in the technology, such as materials used, units of measurement, and protocols for building platforms and/or biological models. Ideally, standards should be robust, reliable, and affordable, incorporating the perspectives of end-users and stakeholders. To be adopted, they require the consensus of all parties involved, including developers, regulatory agencies, metrology institutions, and stakeholders. Users are most likely to adopt those standards when they recognize the value and benefits they offer.
 | ||
| Fig. 1 Standards are developed as a result of the work of different collaborating stakeholders. Standards will help with characterizing and comparing different microphysiological systems, and with communication between stakeholders. | ||
There have been numerous activities in the standardization of MPS worldwide.16 These efforts by various working groups aim to address different aspects of this rapidly advancing field. Currently, efforts towards defining terminology in both the microfluidics and the MPS fields have already provided three standards, two under ISO and one under ASTM International.17–19 The standard terminology specifically related to MPS describes these systems as devices that either contain one or more engineered organs, or organ substructures, or a functional organ unit (or units) in a controlled microenvironment. Thus, representing one or more aspects of a specific organ, for example, its functionality, dynamic processes, and/or physiology/pathology. All those aspects are studied under a number of stimuli, such as exposure to biologics (e.g., monoclonal antibodies, vaccines), mechanical changes, electromagnetic light or radiation, and pharmaceuticals (e.g., small molecules). Also, an MPS should be able to monitor cells (i.e., mono-cultures, co-cultures, explants from tissues or organoids) in real-time. On the other hand, OoCs are described in the existing ASTM International standard as a subset of MPS that can replicate one or more features of organ(s) functionality, dynamic processes, or physiological/pathophysiological behavior. No mention of other aspects like real-time monitoring or external stimuli are included in this definition. Since we have these definitions at hand, and in the spirit of promoting the use of standards more broadly, we will refer to the systems described in this article mainly as MPS or OoC, based on the definitions mentioned above. In addition, we use the terms system and platform interchangeably throughout the article when referring to a microfluidic network or chip along with other components such as pumps and sensors.
The working groups leading the efforts towards the development of MPS standards consist of stakeholders from academia, funding agencies, regulators, and industry. At the “Workshop on Standards for Microphysiological Systems” held at Michigan State University (USA) April 2023, the participants emphasized the importance of sharing results and ideas generated by different working groups. This workshop was organized by the OoC/ToC Engineering Standards Working Group (USA) and included members from the working group and other representatives from academia, industry, Food and Drug Administration (FDA), National Institute of Standards and Technology (NIST), National Institutes of Health (NIH), and the European Commission. Such collaborations will enhance the understanding and direction of each group, helping to identify gaps and define future efforts. Consequently, this Perspective article is being written to provide a summary and insights into crucial aspects of standardization, including available technology (section 2. MPS design and engineering), advances in different regions of the world (section 3. Availability of standards and guidelines), their impact (section 4. Impact of standards), existing challenges, and future prospects (section 5. Current challenges and future outlook), and conclusions (section 6).
2. MPS design and engineering
2.1. Flow systems
More than two decades ago, microfluidic technologies began to emerge with the potential to revolutionize modern biology. Microfluidic-based systems can process small fluid volumes (ranging from 10−9 L to 10−15 L) by utilizing microscale channels with typical dimensions of tens to hundreds of micrometers.6 Indisputably, this technology has brought new capabilities and made substantial contributions to the field of biology and medical research,20 serving as a valuable tool for developing innovative biological models. Among these advancements, the introduction of MPS technology garnered significant interest within the scientific community as a promising model. Researchers have demonstrated that such systems can more accurately represent the in vivo physiological functions of tissues and organs in both normal and disease states.21MPS technology has witnessed remarkable advancements in recent years that resulted in a wide array of microfluidic network designs tailored to support specific tissue and organ microenvironments for various applications. As a result, numerous companies have emerged, offering plug-and-play and user-friendly systems to cater to end-users' needs, which include systems that only require the addition of cells to the cell culture chambers, similar to what is done with the multi-well plates (or microtiter plates) to fully connected systems to external components such as pumps. However, due to the unique structures and functions of different organs, these systems exhibit significant variations from one another. Consequently, end-users need to consider several factors before adopting commercially available technologies. Among these considerations, end-users must verify specific system characteristics, such as ensuring that shear stress levels fall within the expected physiological range. They should also assess the flow direction, as some platforms feature unidirectional flow with or without recirculation, while others have bidirectional flow. Additionally, the desired throughput, mechanical stimulation options, such as stretching or compression, or electrical stimulation, and other factors like air exposure, e.g., air–liquid-interface (ALI), co-culture capabilities, cell–cell interactions, and single versus multi-organ requirements need to be taken into account. Moreover, end-users should evaluate gas permeability, optical clearance, compound absorption and adsorption, pore size, porosity, membrane thickness, and chemical surface properties of the materials used. Considering these diverse issues can make the decision-making process challenging. Ultimately, end-users may require different systems for different organs, thereby introducing the challenge of training on multiple systems that utilize distinct upstream and downstream protocols, including cell seeding, system operation, sample collection, and processing. Currently, researchers from various sectors are pooling their expertise to overcome these challenges and establish cross-platform standards, aiming to provide guidelines that facilitate the interpretation of results obtained through experimenting with these systems.
2.2. Actuation and sensing
Actuation and sensing are two critical aspects of MPS. Actuation systems encompass liquid handling, perfusion, operational systems, and external stimulations (mechanical and electrical). On the other hand, sensing plays a crucial role in real-time monitoring of cell and tissue functions within the system.15 In the human body, cells are subjected to various biomechanical stimuli that are tissue-specific and may change in response to diseases or injuries. Therefore, it is crucial to generate an appropriate physiological or pathological biomechanical environment to successfully replicate in vivo conditions and behavior in MPS models.22 Extensive efforts have been devoted to constructing diverse materials and devices that enable the delivery of mechanical cues to cells and tissues, thereby exploring the impact of such signals on cell and tissue function.23Standardized testing protocols for experimental setups and actuation are also necessary. These protocols should include comprehensive instructions on the selection and calibration of actuation parameters, as well as the configuration and utilization of actuation systems. Clear guidelines on the preparation of culture media, handling of cells and tissues, and placement of sensors with relation to the position of the cells in the platform (critical when combining multiple organs in one system) should be provided to ensure consistent and repeatable actuation experiments. Standards for actuation in MPS are essential to guarantee the reproducibility, comparability, and reliability of experimental results. The standardization of actuation modalities, systems, procedures, and documentation will contribute to the further advancement of MPS technology.
2.2.1.1 Mechanical actuation. Creating dynamic microenvironments around cells and tissues within MPS is crucial for influencing cellular responses and functions, particularly in relation to physiologically relevant mechanical stimuli. External syringe pumps have emerged as the preferred method for delivering mechanical stimuli to these systems due to their high precision and programmability. Additionally, microfluidic pumps have been integrated within the chips themselves to reduce their size. Alternative delivery methods, such as rocking, passive delivery, or hydrostatic pressure, have also been demonstrated. Pistons and pressure controllers on a diaphragm have been employed, in the case of compression stress, to apply the necessary forces to the MPS. Furthermore, the utilization of multiple mechanical stimuli has been proposed and demonstrated to enhance the replication of physiologically relevant microenvironments for tissues and organs, such as in lung-on-a-chip22 and kidney-on-a-chip, which are subjected to both shear flow and cyclic strain. Other studies have also reported the application of mechanical stimulation in gut-on-a-chip24 models to create a microenvironment that closely resembles in vivo conditions for cells. The incorporation of mechanical stimuli using pressure controllers provides an affordable and easily manipulatable platform for conducting conclusive testing of biological hypotheses.25
Integrating multiple stimuli presents technical challenges due to interactive effects and increased biological complexity. The mechanical features of MPS contribute to stimulating realistic tissue formation and function as well as capturing integrative elements of tissue function in response to external insults and injuries and have emerged as a crucial consideration in the design of these systems.26
Mechanical actuation using fluid flow and shear stress, is one of the most common forms of actuation and stimulation in MPS. The pumping and control system should be standardized to ensure the delivery of the appropriate flow rate for specific operations. To ensure the reliability, comparability, and robustness of the MPS across different research groups and laboratories, standardization of these actuation procedures and parameters is essential, and having standards related to specifications about pumps and other flow control components like connectors and tubing will greatly improve the reproducibility of the actuation methods in these systems.
2.2.2.1 Electrical sensors. Sensing is an essential aspect MPS as it involves the continuous monitoring and measurement of cellular behavior, tissue function, and environmental parameters. The establishment of standards for sensors is crucial to ensure the reliability, reproducibility, and comparability of sensing data across different MPS. Overall, reliability standard testing protocols provide a way to measure a system's performance under specific conditions for a period of time, whereas standard protocols for reproducibility provide a systematic way to constantly and reliably obtain results that could be compared between measurements of different batches (e.g., cells in culture). Thus, to be able to know up to what point a sensor will be within specifications and to be able to confidently compare results taken at different times and with different sets of cells, we need standard protocols for reliable and reproducible measurements. This is critical since decisions regarding, for example, the efficacy and toxicity of a drug will depend on the readout of those sensors, thus making those readouts crucial. These standard protocols encompass multiple elements, including sensor types, fabrication techniques, measurement protocols, and data analysis methodologies. One significant aspect of sensor standardization relates to the careful selection and characterization of sensor types. A diverse range of sensors have been developed for MPS including electrochemical, optical, and impedance sensors. They have been employed to effectively monitor cellular responses, biomarkers, and environmental conditions.27 To facilitate standardization efforts, it is important to focus on identifying the most appropriate sensor types for specific applications and defining their characteristics, such as sensitivity, selectivity, dynamic range, and response time. Furthermore, standardized sensor fabrication techniques and materials need to be developed to ensure consistent performance and compatibility across different MPS.
The electrochemical sensors developed for MPS applications typically implement a three-electrode setup. By modifying the working electrode with a biorecognition element such as an enzyme, antibody, aptamer, or nanoparticles, the analyte of interest can be detected through a redox reaction on the working electrode. The generated electrical signal, i.e., current/voltage, corresponds to the concentration of the analyte of interest.28 Various electrochemical biosensors have been proposed by different research groups to monitor tissue function in terms of metabolic parameters and biomarker secretion.29,30
When considering standards for electrochemical (EC) biosensors used in MPS, several parameters need to be taken into account, including electrode materials, functionalization protocols, experimental setup, and electrolyte type. Technical and cell-based standards can enhance instrument compatibility, ensure reliable operation, and improve supply chains. Standardized quality criteria, minimum viability and lifetime requirements, and other standards based on organ types can improve the selection of providers and compatibility, while also better serving the intended use.31
Trans-epithelial/endothelial electrical resistance (TEER) sensors have been developed as a useful tool to evaluate barrier integrity in tissue barrier platforms like blood-brain barrier-on-a-chip, gut-on-a-chip, and other tissue barrier platforms.32 However, when comparing different systems, caution must be exercised as the absolute TEER values are influenced by various factors, including medium formulation, temperature, electrode geometry, measurement technique, and specific cell properties of interest. Therefore, accurate comparisons require considering the configuration and environment, highlighting the need for developing standards for such sensors.27
2.2.2.2 Optical sensors. Optical sensors have been employed in MPS to monitor cells and tissues. Unlike electrode integration methods that require direct contact with living cells or cell effluents, optical sensors utilize electromagnetic radiation and do not need to physically touch the living system. Optical sensors offer several advantages, including durability, low noise, and high temporal resolution, making them well-suited for use in MPS, though so far, they are mainly used as an endpoint measurement. Methods such as surface plasmon resonance (SPR), optical waveguide light mode spectroscopy (OWLS), photonic crystals (PC), and resonant waveguide grating (RWG) utilize surface-bound evanescent electromagnetic waves to detect changes in refractive index resulting from interactions such as cellular responses or analyte secretion near the sensor surface. These methods are particularly valuable for integration with MPS as they minimize electromagnetic radiation exposure to the sample.27
The establishment of standardized measurement protocols is a crucial component of sensor standardization in any system. These protocols delineate the procedures for sensor calibration, sensor integration into the system, consistency checks between batches, evaluation of sensor performance under varying conditions, and data acquisition. Furthermore, standardized data acquisition protocols should consider establishing parameters such as sampling rates, temporal resolution, and data storage formats. Validation protocols should also be considered to compare sensor measurements with established standards or reference methods to estimate the accuracy and reliability of sensors in MPS.
In summary, the establishment of sensor standards in MPS is crucial to guarantee the replicability and comparability of sensor data. These standards include, but are not limited to, the selection of sensors, techniques for fabrication, procedures for measurement, methodologies for data analysis, and quality assurance measures. By implementing robust sensing and actuation standards, the progress of the field toward more precise and effective models can be accelerated.
2.3. Control and automation
MPS are benefiting from advances in automation, control systems, and robotics. While these advancements bring the potential for increased speed and application in various industries, they also introduce complexity. The role, processes, and use of control and automation aspects will require standardization. Given the nature of this field, in the future, teams working on MPS technology should involve engineers, information technology (IT) specialists, and experts in artificial intelligence (AI) systems,11 as these areas are already making an impact. The integration of AI and its potential for use in MPS have been discussed by scientists working in the field.33,34 It is an emerging field that is expanding exponentially and will raise significant ethical and regulatory concerns. However, due to the lack of standards, addressing these concerns will be more difficult and challenging within the current state of affairs. However, as US Congress and stakeholders get involved in regulating the use of AI and other automated processes requiring the use of computerized systems in daily aspects of our lives, the possibilities for an earlier-than-expected standardization of some of these processes integrated into MPS will likely be possible at a pace faster than other aspects that are not as controversial.2.4. Multi-organ-MPS (MoMPS)
MoMPS systems combine multiple MPS within a single system. The OoC chambers are interconnected through a fluidic circuit, allowing for the recirculation of a common cell culture medium. The advantage of linking multiple MPS is that soluble components can travel from one OoC compartment to another, thus resulting in effects that cannot be captured by a single MPS alone. For instance, a drug metabolite generated in the liver OoC compartment can move through the fluidic stream and impact the function of heart cells in the heart OoC compartment.35,36 Similarly, secondary effects can be observed with environmental chemicals, such as naphthalene.37,38 MoMPS systems are well-suited for detecting both the primary effects of a drug and its cytotoxicity as well as possible secondary toxic effects from drug metabolites. However, the inherent complexity of these devices can present barriers to their widespread adoption.Several authors have reviewed how MoMPS systems can contribute to early-stage drug development.39–41 Some chemicals are highly toxic and cannot be directly tested on humans, making MPS a valuable tool for chemical risk assessment. When designing MoMPS systems, two broad criteria must be considered. The primary objective is to create systems that closely mimic the human body or specific parts of it, effectively simulating a patient's response to a drug. This requires ensuring that the tissues within the system function well and closely resemble their in vivo counterparts. For instance, drug-metabolizing cells should exhibit sufficiently high metabolic rates. Second, the fluidic platform used in the system must be designed with human physiology in mind, particularly in relation to ADME (absorption, distribution, metabolism, and excretion) and PBPK (physiologically based pharmacokinetic) assessments. The goal is to demonstrate that, based on scaling, the system can replicate pharmacokinetic parameters observed in humans, such as peak concentration (Cmax), half-life (T1/2), clearance, and excretion. Additionally, it is essential to validate whether parent compound elimination and metabolite formation occur as expected. Merely combining relevant cells in appropriate ratios within a model does not guarantee superior performance compared to a monolayer culture system. Evaluating the agreement between in vitro results with preclinical animal testing and, where available, human clinical results can demonstrate the accuracy of the MoMPS model.
Applying a chemical engineering approach, tissues involved in drug metabolism or in altering drug concentrations by way of storing or filtering them (as opposed to tissues monitoring drug effects) should be represented in physiological volume ratios as well.42,43 Similarly, while not mandatory for every system, it can be advantageous for the medium flow through tissue chambers to mimic physiological flow rates. In a related approach, a MoMPS system can be mathematically simulated using physiologically based pharmacokinetic models (PBPK).44–46 However, it is not possible to completely mimic the complexity of the human body, and developers of MoMPS systems must make compromises and prioritize features that are essential to create practical and functional systems (domains of validity or context of use). The proper representation of the human body is still a subject of debate. Therefore, we propose that each system is evaluated based on a predefined set of experiments with expected outcomes, or where results are correlated to physiological outcomes.
3. Availability of standards and guidelines
The process of developing standards will go through different phases as this field is still growing and, in many aspects, not yet mature. To overcome technical and biomedical challenges, reach a consensus on terminology, establish experimental and reporting methods, and enable interoperability and benchmarking, standards are essential tools that can ensure a solid, widely adopted, and consensus-based approach.47,48 For this purpose, it is necessary to actively involve standard development organizations (SDOs) as the main actors in the formal standardization process. Thus, reports and guidelines will be created by working groups and SDOs, which will eventually be transformed into standards (Fig. 2).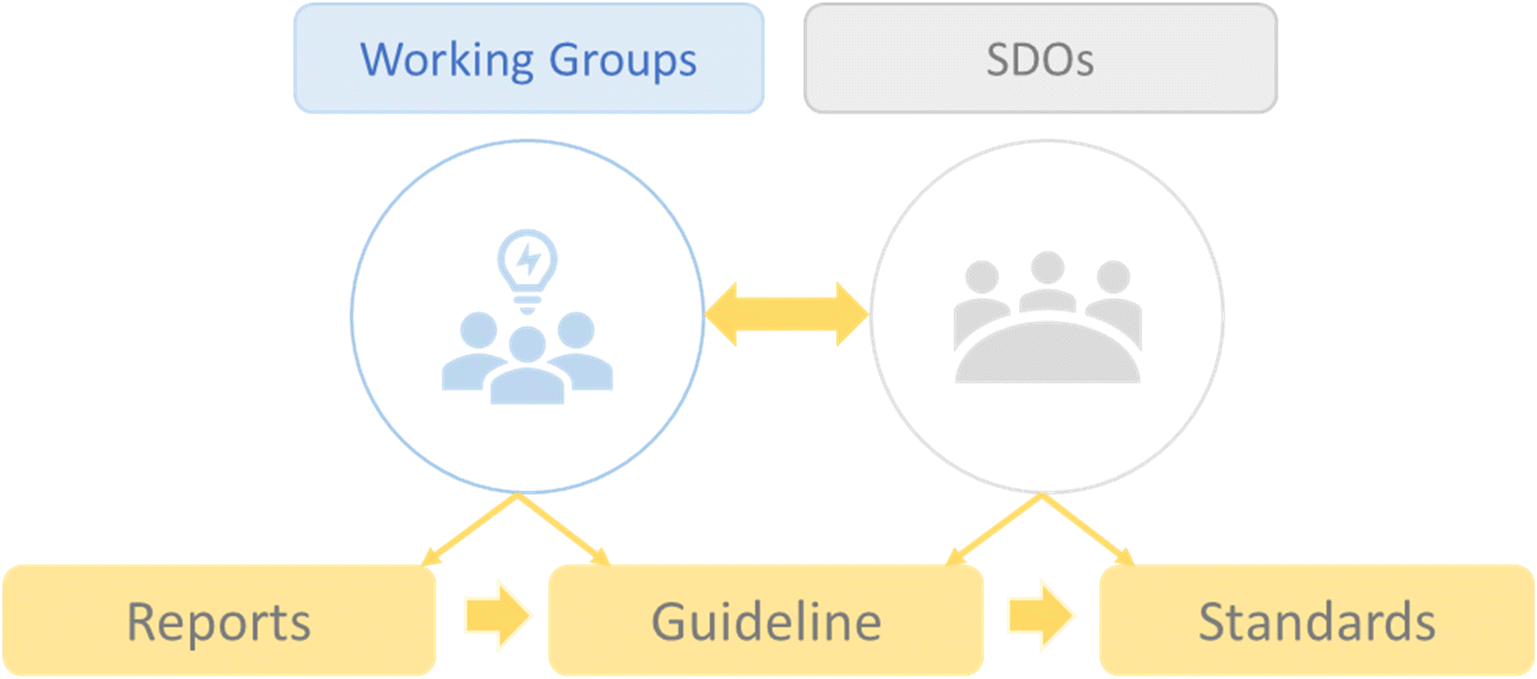 | ||
| Fig. 2 Illustration of the process of standards development and the role of working groups and standard development organizations (SDOs). | ||
There is a strong justification for translating scientific evidence into standards, supporting the advancement of the MPS field towards wide acceptance by stakeholders and creating a robust marketplace for human-relevant alternatives to animal testing. End users are asking for simple-to-use, cost-effective MPS that can be purchased off-the-shelf and then adapted to their specific applications. To fully trust these products, the characterization of technological components such as materials or biomechanical properties is necessary to facilitate industry uptake. Standards could play an important role in this regard by describing specific requirements and performance of the components in an open, clear, and structured manner. Although progress along this path currently varies in different parts of the world, updates and global-level collaboration will be necessary. Below, we discuss the status of microfluidics/MPS standardization in the USA, Europe, and Asia.
3.1. USA
Early efforts in the USA to develop standards for microfluidic systems began to yield results in 2007 when SEMI (Semiconductor Equipment and Materials International) published their first standard on microfluidics: Guide for Design and Materials for Interfacing Microfluidic Systems (SEMI MS6). Subsequent efforts led to the publication of five more standards in the following years, with three directly related to microfluidic systems and the other two applicable to microfluidic systems. These standards include: 1) Specification for Microfluidic Interfaces to Electronic Device Packages (SEMI MS7); 2) Specification for High-Density Permanent Connections Between Microfluidic Devices (SEMI MS9); 3) Specification for Microfluidic Port and Pitch Dimensions (SEMI MS11); 4) Guide to Evaluating Hermeticity of Microelectromechanical Systems (SEMI MS8); and 5) Test Method to Measure Fluid Permeation Through MEMS Packaging Materials (SEMI MS10).49 While these standards were the first ones published in the USA, their reception was not widespread, and many individuals in the microfluidics field are unaware of their existence. However, parallel efforts continued in Europe, beginning with the establishment of a common vocabulary for microfluidic terms. These international efforts within the microfluidics community led by the Microfluidics Association (MFA)50 with support from their members and CEA-Leti, have produced the first ISO (International Organization for Standardization) standards in microfluidics, the first one published in January 2022 and the second one in September 2023.49Directly related to MPS, the Standards Coordinating Body (SCB), a US-based SDO, published first standard for microphysiological systems titled “Standard Terminology Relating to Microphysiological Systems” (Designation: F3570 – 22). This ASTM (American Society for Testing and Materials) International standard was published in July 2022.19 The SCB is also actively developing other standards and is working towards the publication of a standard on Cardiac MPS.51
3.2. European Union (EU)
In 2021, the European Commission's Joint Research Centre, along with the European Standardization Organizations Comité Européen de Normalisation (CEN) and Comité Européen de Normalisation Électrotechnique (CENELEC), decided to set the MPS standardization process in motion by the “Putting Science into Standards” workshop which brought together developers, end-users, and standardization experts. To encourage the development of OoC/MPS-specific standards, CEN-CENELEC has initiated concrete actions by establishing the OoC Focus Group (OoC-FG), a European coordination platform that aims to stimulate and coordinate interaction among all relevant European stakeholders interested in potential standardization in the field of OoC/MPS. The secretariat of the OoC-FG is held by NEN, the Royal Netherlands Standardization Institute, and activities started in March 2022 with expected outcomes after two years. The OoC-FG is comprised of five working groups that cover a wide range of topics, ranging from research and development (R&D) to manufacturing, and from terminology to regulatory applications:- WG1 – Terminology, ecosystem, interdependencies
- WG2 – Biosciences
- WG3 – Engineering
- WG4 – Experimental design and data management
- WG5 – User perspective and regulatory, legal and ethical aspects
The first objective of the OoC-FG is to build a roadmap by identifying standardization gaps and setting priorities, providing concrete suggestions on how new standards could look. Based on the roadmap developed by the OoC-FG, the CEN and CENELEC Technical Boards can initiate further standardization actions. One of the objectives of the OoC-FG is to establish liaisons with technical bodies that address areas related to OoC, such as ISO/TC 276 Biotechnology, ISO/TC (ISO/Technical Committee) 215 Health Informatics, CEN/TC 140 in vitro diagnostic medical devices, CEN/TC 251 Health Informatics, and other key stakeholders in the OoC ecosystem. To achieve this, the OoC-FG has established a strong synergy with the European OoC Society (EUROoCS).
3.3. Asia: Japan
Standardization activities for OoC/MPS are currently underway in Asia, particularly in Japan. Japan has been actively involved in cell-related standardization through ISO/TC 276 Biotechnology. One of the standards developed by Japan in collaboration with the U.S. is ISO 23033:2021, titled “Biotechnology – Analytical methods – General requirements and considerations for the testing and characterization of cellular therapeutic products,” which is applicable to cell characterization in OoC.Japanese convenorship manages TC 276/WG 4 working group (Bioprocessing), specifically focusing on cell processing. So far, WG 4 has issued standards for ancillary materials used in cell production (ISO 20399:2022), cell transportation (ISO 21973:2020), equipment related to cell production (ISO/TS 23565:2021), and packaging (ISO 20404:2023).
Japan recognizes that these cell-related standards can be applied to OoC/MPS and is currently harmonizing them with device standards specific to OoC. The Japan Bio Measurement & Analysis Consortium (JMAC), an industry group that promotes standardization on the device side, is in the process of launching a new project called MF4MPS (Microfluidics for MPS) to bring together relevant companies. Key members of this project have already initiated discussions with MFA and other European-based organizations. Currently, within ISO, a Japanese member serves as a liaison representative between TC 48 and TC 276, working towards harmonization at the international standard level.
4. Impact of standards
4.1. Impact on translational applications
The availability of standards will have an impact on translating the technology to the clinic and related processes. The effects of these standards will affect several aspects within the MPS arena. First, it will be easier to adapt these standardized testing methods alongside the current ones that are exclusively dependent on 2D cell culture and experimental animals. Second, in addition to demonstrating their impact on cost, MPS will require standards to be used more widely for pre-clinical applications. Third, standards will facilitate communication and comparison of results, which can be effectively communicated to users such as clinicians and those working in clinics or providing patient care. Fourth, the availability of standardized terminology and technology will aid the integration of the system into current lab and clinical setups. Fifth, having standardized technology will pave the way for developing more advanced healthcare systems and create new opportunities for innovation. And sixth, the impact on patients for making more efficient diagnoses, providing appropriate and less toxic therapeutic regimens (e.g., in cancer patients) will make this technology even more attractive when standards are in place. There are still other untapped benefits of the technology that will significantly impact patient care, and we have no doubt that standards will make this possible.4.2. Impact on regulatory process
Standardization is an important factor for the successful commercialization of cell culture products. This provides the end user with assurance that the products meet certain criteria in terms of material properties, dimensions, tolerances, sterilization, and quality control, facilitating their integration into standard operating procedures (SOPs). Cell culture microplates or multi-well plates have been used by generations of researchers because they are standardized and fit into routine laboratory workflows and SOPs. The engineering characteristics of multi-well plates, such as the number of wells, well dimensions, and well spacing, are established by the Society for Laboratory Automation and Screening (SLAS) and the American National Standards Institute (ANSI) standards.52 These standards enable interoperability between the microplates, readout instruments, and laboratory automation equipment, thereby increasing productivity. Interoperability facilitates usage and saves capital resources because switching to another multi-well plate supplier does not require acquiring new equipment. Similarly, standardizing organ-on-a-chip devices is expected to have a positive impact on their commercialization and adoption.52,53In addition to engineering parameters, establishing specific performance standards is also important for cell culture disposables. There is a wealth of information that prior standards and the prior use of well-established products can provide in this context. While the multi-well plate cell culture market is considered somewhat of a commodity, it represents 40% of the global microplate market, which was estimated at $892 M in 2021 based on three reports.53–55 Multi-well plate manufacturers generally recommend a cell plating density and volume of culture medium for standard use, thus enabling end users to easily compare the results and troubleshoot. Therefore, it is important to establish performance standards for specific applications or context of use (CoU) to promote technology adoption and provide benchmarking and troubleshooting capabilities.
Finally, reliable operation and consistent performance from device to device and batch to batch are required for commercial products. Outgassing, evaporative losses, and non-specific protein adsorption are likely to require standardization, and additional standards may be needed for port-to-tube connections. While the use of standards in regulatory processes is generally voluntary, they play a crucial role in the regulatory process of the US Food and Drug Administration (FDA) where they significantly impact the safety and quality of products released to the public. Meeting regulatory requirements and consistently manufacturing high-quality products can pose unique challenges for novel medical products. Increasing the development and utilization of consensus standards will undoubtedly aid in product development, characterization, and regulatory predictability. Therefore, the FDA encourages sponsors of regulatory submissions and manufacturers to appropriately utilize voluntary consensus standards.56 Several consensus standards provide a framework for the development, manufacturing, and testing of various medical products to ensure compliance with necessary safety and quality requirements. The regulatory process can be streamlined by using relevant consensus standards, which ensure data consistency, predictability, and credibility while reducing uncertainty. It is important to note that when incorporating consensus standards into product development and testing for pre-marketing applications, rigorous conformity assessment, as described in the FDA standards and conformity assessment program,57 is an integral part of a robust regulatory framework that incorporates the appropriate use of consensus standards.
The FDA, specifically the Center for Devices and Radiological Health (CDRH), has decided to recognize standards to streamline the regulatory review process. The FDA Standards Recognition Program evaluates consensus standards for their applicability to the evaluation of the safety and performance of medical devices. Standards recognition is the procedure through which the FDA identifies standards to which producers of medical devices may submit a declaration of conformity, demonstrating compliance with appropriate requirements of the Federal Food, Drug, and Cosmetic Act (FD&C Act). Similarly, the Center for Biologics Evaluation and Research (CBER) launched a program to identify and recognize consensus standards to facilitate the development and assessment of regenerative medicine therapies.58 The FDA may recognize all, part of, or none of a consensus standard. The Federal Register Documents page contains the historical record of all FDA recognition determinations, including whether a standard is recognized in full, in part, or not recognized at all.59 Once the FDA decides to recognize a standard, the information is updated in the FDA online database even before the standard is formally recognized and published in the Federal Register.
5. Current challenges and future outlook
Within the microfluidics community, there has been some resistance to the idea of developing standards for microfluidics.60 This resistance has made it somewhat challenging to engage a larger number of stakeholders in the standardization process for microfluidic systems. However, as mentioned earlier, efforts have persisted, and progress has been made not only within the microfluidics community but also in the field of MPS. Therefore, to make significant advancements, the initial hurdle is to convince a greater number of stakeholders in both the microfluidic and MPS communities about the importance of developing standards. As the number of submissions for microfluidics-based systems to FDA continues to increase61 and with the signing into law of the FDA Modernization Act 2.0,62 it is expected a growing interest in the development of guidelines and standards. The expectation, with Modernization Act 2.0, is that systems like the MPS will become practical alternatives to animal testing by demonstrating their capacity to provide reliable and more translatable data, leading to better predictions and far lower costs compared to clinical trials. As MPS applications evolve from basic and academic research to alternatives for pre-clinical studies and, as envisioned in the future of the MPS field, for clinical trials, the interests of many industry stakeholders will shift towards the utilization of these systems in applications that require approval from regulatory agencies.The adoption of this technology will be facilitated by the industry's need to minimize the effort required to demonstrate the efficient and reliable performance of their systems. Initially, the small number of companies submitting data to regulatory agencies will have to demonstrate the viability of their systems using their own protocols. Therefore, this first wave of submissions will result in a reduced number of companies showcasing the utility of this technology for generating high-quality regulatory data due to the associated costs and efforts. However, having standards will offer both small and large companies a set of validated protocols to showcase the efficacy and safety of their technologies. This will lower the barrier to providing acceptable data to regulatory agencies, ensuring that all companies have equal opportunity to demonstrate the efficiency of their technologies.
Microfluidic-based cultures are poised to have a considerable impact across various industries, including pharmaceuticals, cosmetics, chemical engineering, and agriculture. Despite a significant increase in the number of peer-reviewed publications in the last five years that describe the advantages of microfluidic cell culture, the widespread adoption of this technology remains limited in the different industries that could benefit from it.61,63–65 Therefore, there is a growing call to establish standards for model developers in order to accelerate the realization of the anticipated benefits.
The development of standards for microfluidic cell culture does not have to start with a blank piece of paper. Indeed, some existing frameworks, which draw from best practices in other areas such as medical devices, have been referenced above. To make progress, it is recommended to establish an association or consortium comprising engineers, biologists, regulators, and quality assurance professionals. Ideally, the group should also include international representation, considering that different countries may have unique requirements. While it may not be possible to satisfy all requirements in the final outcome, they should be discussed and debated before reaching a consensus position. Establishing a consortium would also prevent a situation where a leading manufacturer of a current platform gains significant market share and ultimately dictates the standard for other developers to follow. Precedent exists in the laboratory analytical devices field, and history teaches us that these instruments were not always the best choice for setting the standard.
An early task of such a consortium will be to strike a balance between setting restrictive standards and fostering ongoing innovation. The MPS field has gained prominence only in the last decade, and there is still significant potential for further development. It is also advisable for the consortium to initially focus on the engineering aspects of these systems. This approach would simplify end-user training requirements, allowing them to dedicate more time to the biological aspects. A standardized platform is likely to be more readily integrated into laboratory workflows and may enhance efficiencies when combined with automation. Consequently, usage rates would increase significantly, promoting large-scale manufacturing and eventually driving down the platform cost.
Another compelling reason to prioritize standardization of the microfluidic platform over biological models is the inherent complexity of biology. Apart from models described within the International Congress on Harmonisation (ICH) guidelines for safety pharmacology, genetic toxicology, and reproductive toxicology, standards are not widely established in biology. However, well-trained researchers are familiar with extensive guidelines, including those for good cell culture practice, aimed at improving the quality of their work. Since the development of standards is a time-consuming process, it is crucial for consortium members to stay informed about technological advancements that could lead to engineering and/or biological improvements. This can be achieved by sharing early drafts of proposed standards with expert stakeholder groups, ensuring their input and keeping them up to date.
6. Conclusions
Microphysiological systems are expected to have a significant impact on disease research, drug development, and future healthcare. For this technology to be effectively utilized and to benefit the industry, regulators, users, and other stakeholders, it is crucial to establish clear definitions for its terminology, processes, and systems. To achieve this, the development of standards is necessary. Currently, there is a small number of such standards in this area, but various groups have been collaborating to address this issue. These collective efforts and discussions emphasize the importance of having standards to promote the adoption of the technology by industry, regulators, and clinicians. Support from societies and focused projects is required to bring together and integrate fragmented initiatives. Furthermore, funding from government, industry, and foundations is essential to support the development of standards in this rapidly evolving field, which is expected to have a profound impact on industry and patients' health.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DRR acknowledges NIST-on-a-Chip Initiative funding. NA acknowledges funds received from the National Institutes of Health (1UG3TR003148-01), and support from the office of Michigan State University Vice President for Research and the Institute of Institute for Quantitative Health Science and Engineering, Michigan State University.References
- S. E. Jenkinson, G. W. Chung, E. Van Loon, N. S. Bakar, A. M. Dalzell and C. D. A. Brown, Pfluegers Arch., 2012, 464, 601–611 CrossRef CAS PubMed.
- T. Hartung, Nature, 2009, 460, 208–212 CrossRef CAS PubMed.
- T. Hartung, ALTEX, 2008, 25, 3–16 CrossRef.
- R. K. Harrison, Nat. Rev. Drug Discovery, 2016, 15, 817–818 CrossRef CAS PubMed.
- O. J. Wouters, M. McKee and J. Luyten, JAMA, 2020, 323, 844–853 CrossRef PubMed.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS PubMed.
- B. Zhang, A. Korolj, B. F. L. Lai and M. Radisic, Nat. Rev. Mater., 2018, 3, 257–278 CrossRef.
- L. Ewart, K. Fabre, A. Chakilam, Y. Dragan, D. B. Duignan, J. Eswaraka, J. Gan, P. Guzzie-Peck, M. Otieno, C. G. Jeong, D. A. Keller, S. M. De Morais, J. A. Phillips, W. Proctor, R. Sura, T. Van Vleet, D. Watson, Y. Will, D. Tagle and B. Berridge, Exp. Biol. Med., 2017, 242, 1579–1585 CrossRef CAS.
- L. A. Low, C. Mummery, B. R. Berridge, C. P. Austin and D. A. Tagle, Nat. Rev. Drug Discovery, 2021, 20, 345–361 CrossRef CAS PubMed.
- N. Ashammakhi, E. Elkhammas and A. Hasan, J. Biomed. Mater. Res., 2019, 107, 2006–2018 CrossRef CAS PubMed.
- K. L. Fetah, B. J. DiPardo, E. Kongadzem, J. S. Tomlinson, A. Elzagheid, M. Elmusrati, A. Khademhosseini and N. Ashammakhi, Small, 2019, 15, 1901985 CrossRef CAS PubMed.
- U. Marx, T. Akabane, T. B. Andersson, E. Baker, M. Beilmann, S. Beken, S. Brendler-Schwaab, M. Cirit, R. David, E.-M. Dehne, I. Durieux, L. Ewart, S. C. Fitzpatrick, O. Frey, F. Fuchs, L. G. Griffith, G. A. Hamilton, T. Hartung, J. Hoeng, H. Hogberg, D. J. Hughes, D. E. Ingber, A. Iskandar, T. Kanamori, H. Kojima, J. Kuehnl, M. Leist, B. Li, P. Loskill, D. L. Mendrick, T. Neumann, G. Pallocca, I. Rusyn, L. Smirnova, T. Steger-Hartmann, D. A. Tagle, A. Tonevitsky, S. Tsyb, M. Trapecar, B. Van de Water, J. Van den Eijnden-van Raaij, P. Vulto, K. Watanabe, A. Wolf, X. Zhou and A. Roth, ALTEX, 2020, 37, 365–394 Search PubMed.
- M. Mastrangeli, S. Millet, The Orchid Partners and J. Van Den Eijnden-van Raaij, ATLEX, 2019, 650–668 CrossRef.
- K. Fabre, B. Berridge, W. R. Proctor, S. Ralston, Y. Will, S. W. Baran, G. Yoder and T. R. Van Vleet, Lab Chip, 2020, 20, 1049–1057 RSC.
- M. Piergiovanni, S. B. Leite, R. Corvi and M. Whelan, Lab Chip, 2021, 21, 2857–2868 RSC.
- M. Piergiovanni, O. Cangar, S. B. Leite, L. Mian, A. Jenet, R. Corvi, M. Whelan, F. Taucer and A. Ganesh, Stem Cell Rep., 2021, 16, 2076–2077 CrossRef PubMed.
- ISO 22916:2022 Microfluidic devices - Interoperability requirements for dimensions, connections and initial device classification, https://www.iso.org/standard/74157.html, Accessed August 2, 2023.
- ISO 10991:2023 Microfluidics - Vocabulary, https://www.iso.org/standard/82146.html, Accessed October 15, 2023.
- ASTM International F3570–22 - Standard Terminology Relating to Microphysiological Systems, https://www.astm.org/f3570-22.html, Accessed June 22, 2023.
- E. K. Sackmann, A. L. Fulton and D. J. Beebe, Nature, 2014, 507, 181–189 CrossRef CAS PubMed.
- S. N. Bhatia and D. E. Ingber, Nat. Biotechnol., 2014, 32, 760–772 CrossRef CAS PubMed.
- C. L. Thompson, S. Fu, H. K. Heywood, M. M. Knight and S. D. Thorpe, Front. Bioeng. Biotechnol., 2020, 8, 602646 CrossRef PubMed.
- Y. Zhang and P. Habibovic, Adv. Mater., 2022, 34, 2110267 CrossRef CAS PubMed.
- H. J. Kim, H. Li, J. J. Collins and D. E. Ingber, Proc. Natl. Acad. Sci. U. S. A., 2015, 113, E7–E15 Search PubMed.
- K. Kaarj and J.-Y. Yoon, Micromachines, 2019, 10, 700 CrossRef.
- I. A. Morales, C.-M. Boghdady, B. E. Campbell and C. Moraes, Front. Bioeng. Biotechnol., 2022, 10, 1060895 CrossRef.
- H. Kavand, R. Nasiri and A. Herland, Adv. Mater., 2022, 34, 2107876 CrossRef CAS PubMed.
- Y. S. Zhang, J. Aleman, S. R. Shin, T. Kilic, D. Kim, S. A. Mousavi Shaegh, S. Massa, R. Riahi, S. Chae, N. Hu, H. Avci, W. Zhang, A. Silvestri, A. Sanati Nezhad, A. Manbohi, F. De Ferrari, A. Polini, G. Calzone, N. Shaikh, P. Alerasool, E. Budina, J. Kang, N. Bhise, J. Ribas, A. Pourmand, A. Skardal, T. Shupe, C. E. Bishop, M. R. Dokmeci, A. Atala and A. Khademhosseini, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E2293–E2302 CAS.
- J. Aleman, T. Kilic, L. S. Mille, S. R. Shin and Y. S. Zhang, Nat. Protoc., 2021, 16, 2564–2593 CrossRef CAS PubMed.
- Y. Zhu, K. Mandal, A. L. Hernandez, S. Kawakita, W. Huang, P. Bandaru, S. Ahadian, H.-J. Kim, V. Jucaud, M. R. Dokmeci and A. Khademhosseini, Curr. Opin. Biomed. Eng., 2021, 19, 100309 CrossRef PubMed.
- A. Shinde, K. Illath, U. Kasiviswanathan, S. Nagabooshanam, P. Gupta, K. Dey, P. Chakrabarty, M. Nagai, S. Rao, S. Kar and T. S. Santra, Anal. Chem., 2023, 95, 3121–3146 CrossRef CAS PubMed.
- C. M. Leung, P. De Haan, K. Ronaldson-Bouchard, G.-A. Kim, J. Ko, H. S. Rho, Z. Chen, P. Habibovic, N. L. Jeon, S. Takayama, M. L. Shuler, G. Vunjak-Novakovic, O. Frey, E. Verpoorte and Y.-C. Toh, Nat. Rev. Methods Primers, 2022, 2, 33 CrossRef CAS.
- B. M. Maoz, A. Herland, O. Y. F. Henry, W. D. Leineweber, M. Yadid, J. Doyle, R. Mannix, V. J. Kujala, E. A. FitzGerald, K. K. Parker and D. E. Ingber, Lab Chip, 2017, 17, 2294–2302 RSC.
- A. Polini and L. Moroni, Biomater. Biosyst., 2021, 1, 100012 CAS.
- W. Gao, C. Wang, Q. Li, X. Zhang, J. Yuan, D. Li, Y. Sun, Z. Chen and Z. Gu, Front. Bioeng. Biotechnol., 2022, 10, 985692 CrossRef.
- C. Oleaga, C. Bernabini, A. S. T. Smith, B. Srinivasan, M. Jackson, W. McLamb, V. Platt, R. Bridges, Y. Cai, N. Santhanam, B. Berry, S. Najjar, N. Akanda, X. Guo, C. Martin, G. Ekman, M. B. Esch, J. Langer, G. Ouedraogo, J. Cotovio, L. Breton, M. L. Shuler and J. J. Hickman, Sci. Rep., 2016, 6, 20030 CrossRef CAS.
- C. W. McAleer, A. Pointon, C. J. Long, R. L. Brighton, B. D. Wilkin, L. R. Bridges, N. Narasimhan Sriram, K. Fabre, R. McDougall, V. P. Muse, J. T. Mettetal, A. Srivastava, D. Williams, M. T. Schnepper, J. L. Roles, M. L. Shuler, J. J. Hickman and L. Ewart, Sci. Rep., 2019, 9, 9619 CrossRef PubMed.
- K. Viravaidya, A. Sin and M. L. Shuler, Biotechnol. Prog., 2004, 20, 316–323 CrossRef CAS PubMed.
- K. Viravaidya and M. L. Shuler, Biotechnol. Prog., 2002, 18, 174–181 CrossRef CAS PubMed.
- M. E. Andersen, ALTEX, 2014, 31, 364–367 CrossRef.
- C. D. Edington, W. L. K. Chen, E. Geishecker, T. Kassis, L. R. Soenksen, B. M. Bhushan, D. Freake, J. Kirschner, C. Maass, N. Tsamandouras, J. Valdez, C. D. Cook, T. Parent, S. Snyder, J. Yu, E. Suter, M. Shockley, J. Velazquez, J. J. Velazquez, L. Stockdale, J. P. Papps, I. Lee, N. Vann, M. Gamboa, M. E. LaBarge, Z. Zhong, X. Wang, L. A. Boyer, D. A. Lauffenburger, R. L. Carrier, C. Communal, S. R. Tannenbaum, C. L. Stokes, D. J. Hughes, G. Rohatgi, D. L. Trumper, M. Cirit and L. G. Griffith, Sci. Rep., 2018, 8, 4530 CrossRef PubMed.
- A. S. Smith, C. J. Long, B. J. Berry, C. McAleer, M. Stancescu, P. Molnar, P. G. Miller, M. B. Esch, J.-M. Prot, J. J. Hickman and M. L. Shuler, Stem Cell Res. Ther., 2013, 4, S9 CrossRef PubMed.
- M. B. Esch, A. S. T. Smith, J.-M. Prot, C. Oleaga, J. J. Hickman and M. L. Shuler, Adv. Drug Delivery Rev., 2014, 69–70, 158–169 CrossRef CAS PubMed.
- M. Malik, Y. Yang, P. Fathi, G. J. Mahler and M. B. Esch, Front. Cell Dev. Biol., 2021, 9, 721338 CrossRef PubMed.
- H. E. Abaci and M. L. Shuler, Integr. Biol., 2015, 7, 383–391 CrossRef PubMed.
- J. H. Sung, C. Kam and M. L. Shuler, Lab Chip, 2010, 10, 446–455 RSC.
- J. H. Sung, Y. Wang and M. L. Shuler, APL Bioeng., 2019, 3, 021501 CrossRef PubMed.
- A. Lorenz, M. Raven and K. Blind, J. Technol. Transf., 2019, 44, 1097–1133 CrossRef.
- Anonymous, Microfluidic, https://store-us.semi.org/search?=microfluidic, Accesses January 24, 2023.
- D. R. Reyes and H. Van Heeren, J. Res. Natl. Inst. Stand. Technol., 2019, 124, 124001 CrossRef CAS PubMed.
- Project: Microphysiological Systems, https://www.standardscoordinatingbody.org/project-organonachip-standards-landscape-assessment, Accessed June 20, 2023.
- https://www.slas.org/education/ansi-slas-microplate-standards, Accessed June 20, 2023.
- Microplates Market, Microplates Market, https://www.credenceresearch.com/report/microplates-market, Accessed June 20, 2023.
- V. Allwardt, A. J. Ainscough, P. Viswanathan, S. D. Sherrod, J. A. McLean, M. Haddrick and V. Pensabene, Bioengineering, 2020, 7, 112 CrossRef CAS PubMed.
- Microplate Systems Market, By Type (Multi-mode Microplate Readers, Single-mode Microplate Readers, and Others), By Application (Drug discovery, Clinical diagnostics, Efficacy studies, and Others), By End-Use, and By Region Forecast To 2030, 2023.
- Microplates Market Size, Market Share, Application Analysis, Regional Outlook, Growth Trends, Key Players, Competitive Strategies and Forecasts - 2023 to 2031, https://www.researchandmarkets.com/reports/5393576/microplates-market-share, Accessed June 20, 2023.
- Katzen, Sally, Circular No. A-119 -- Federal Register (Federal Participation in the Development and Use of Voluntary Consensus Standards and in Conformity Assessment Activities), https://obamawhitehouse.archives.gov/omb/circulars_a119_a119fr, Accessed June 20, 2023.
- Standards and Conformity Assessment Program, https://www.fda.gov/medical-devices/premarket-submissions-selecting-and-preparing-correct-submission/standards-and-conformity-assessment-program, Accessed June 20, 2023.
- Voluntary Consensus Standards Recognition Program for Regenerative Medicine Therapies, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/voluntary-consensus-standards-recognition-program-regenerative-medicine-therapies, Accessed June 20, 2023.
- Federal Register Documents, https://www.fda.gov/medical-devices/standards-and-conformity-assessment-program/federal-register-documents, Accessed June 20, 2023.
- D. R. Reyes, H. Van Heeren, S. Guha, L. Herbertson, A. P. Tzannis, J. Ducrée, H. Bissig and H. Becker, Lab Chip, 2021, 21, 9–21 RSC.
- S.5002 - FDA Modernization Act 2.0, https://www.congress.gov/bill/117th-congress/senate-bill/5002/text, Accessed January 5, 2024.
- L. Ewart and A. Roth, Nat. Rev. Drug Discovery, 2021, 20, 327–328 CrossRef CAS PubMed.
- S. W. Baran, P. C. Brown, A. R. Baudy, S. C. Fitzpatrick, C. Frantz, A. Fullerton, J. Gan, R. N. Hardwick, K. M. Hillgren, A. K. Kopec, J. L. Liras, D. L. Mendrick, R. Nagao, W. R. Proctor, D. Ramsden, A. J. S. Ribeiro, D. Stresser, K. E. Sung, R. Sura, K. Tetsuka, L. Tomlinson, T. Van Vleet, M. P. Wagoner, Q. Wang, S. Y. Arslan, G. Yoder and J. E. Ekert, ALTEX, 2022, 39, 297–314 Search PubMed.
- P. Hargrove-Grimes, L. A. Low and D. A. Tagle, Cells Tissues Organs, 2022, 211, 269–281 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |