
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent progress in understanding the catalyst layer in anion exchange membrane electrolyzers – durability, utilization, and integration
Emily K.
Volk
 a,
Melissa E.
Kreider
b,
Stephanie
Kwon
*c and
Shaun M.
Alia
*b
a,
Melissa E.
Kreider
b,
Stephanie
Kwon
*c and
Shaun M.
Alia
*b
aAdvanced Energy Systems Graduate Program, Colorado School of Mines, Golden, Colorado 80401, USA
bChemistry and Nanoscience Center, National Renewable Energy Laboratory, Golden, Colorado 80401, USA. E-mail: salia@nrel.gov
cDepartment of Chemical and Biological Engineering, Colorado School of Mines, Golden, Colorado 80401, USA. E-mail: kwon@mines.edu
First published on 7th November 2023
Abstract
Anion exchange membrane water electrolyzers (AEMWEs) are poised to play a key role in reducing capital cost and materials criticality concerns associated with traditional low-temperature electrolysis technologies. To accelerate the development and deployment of this technology, an in-depth understanding of cell materials integration is essential. Notably, the complex chemistries and interactions within the catalyst layer (consisting of the anode/cathode catalyst, anion exchange ionomer, and their interfaces with the transport layers and membrane) collectively influence overall cell performances, lifetimes, and costs. This review outlines recent advances in understanding the catalyst layer in AEMWEs. Specifically, electrode development strategies (including catalyst deposition techniques and configurations as well as transport layer design strategies) and our current understanding of catalyst–ionomer interactions are discussed. Effects of cell assembly and operational variables (including compression, temperature, pressure, and electrolyte conditions) on cell performance are also discussed. Lastly, we consider cutting-edge in situ and ex situ diagnostic techniques to study the complex chemistries within the catalyst layer as well as discuss degradation mechanisms that arise due to the integration of cell components. Simultaneously, comparisons are made to proton exchange membrane water electrolyzers (PEMWEs) and liquid alkaline water electrolyzers (LAWE) throughout the review to provide context to researchers transitioning into the AEMWE space. We also include recommendations for standard operating procedures, configurations, and metrics for comparing activity and stability.
 Emily K. Volk | Emily Volk is currently a PhD candidate at the Colorado School of Mines in the Advanced Energy Systems Graduate Program. She is co-advised by Dr Stephanie Kwon and Dr Shaun Alia. Her thesis work centers on non-platinum group metal oxide catalysts for the oxygen evolution reaction in alkaline media and their integration into catalyst layers for electrochemical devices. Specifically, she is interested in the electrochemical and physical characterization of activation/deactivation mechanisms relevant to the catalyst layer. Before coming to Mines, Emily worked as a Scientist for Pall Corporation and obtained her BS in Chemical Engineering from the University of Rochester. |
 Melissa E. Kreider | Melissa Kreider is a postdoctoral researcher in the Electrochemical Engineering and Materials Chemistry Group at NREL. Her current work focuses on materials integration and catalyst layer engineering to improve the performance and durability of anion exchange membrane water electrolyzers. She is interested in using electrochemical and materials characterization to understand electrocatalyst activation and degradation processes. Prior to joining NREL, she completed her PhD in Chemical Engineering at Stanford University with Prof. Thomas Jaramillo, where she developed electrocatalysts for fuel cell applications and utilized in situ X-ray spectroscopic, diffraction, and mass spectrometric techniques to characterize catalyst evolution during reaction. |
 Stephanie Kwon | Stephanie Kwon is an assistant professor in the Chemical and Biological Engineering department at Colorado School of Mines. Her research interest centers on heterogeneous catalysis and sustainability. She brings together cutting-edge research tools, including in situ spectroscopy, microkinetic modeling, and density functional theory calculations, to understand surface reactions at a molecular-level. In doing so, she aims to tackle our current energy and environmental problems by providing novel catalytic systems with improved energy and atomic efficiency. Her current research projects include CO2 conversion, low temperature H2O electrolysis for H2 production, biomass conversion (via aldol condensation catalysis), benzene alkylation, and alkene oxidation catalysis. |
 Shaun M. Alia | Shaun Alia is a senior scientist in the Electrochemical Engineering and Materials Chemistry Group at NREL. His research interests involve understanding electrochemical and degradation processes, materials integration, and component development. Within the HydroGEN Energy Materials Network, Dr Alia is a Technology Lead in Low Temperature Electrolysis and a capability owner in materials development and testing. He is further active within in situ durability, diagnostics, and accelerated stress test development as a Durability Liaison for the H2NEW consortium. Previously, he worked in several areas related to electrochemical energy conversion and storage, including hydrogen and direct methanol fuel cells, capacitors, and batteries. |
Broader contextHydrogen (H2) production from water electrolysis allows for the efficient conversion of low-cost renewable electrons to stored chemical energy, thereby limiting the curtailment of variable renewable energies (e.g., wind and solar) and creating a promising long-term energy storage medium. This ultimately provides green pathways for carbon-intensive and hard-to-decarbonize sectors including transportation; steel, cement, and ammonia production; and chemical synthesis, and facilitates the energy transition from fossil fuels to variable renewable energy sources. Current barriers to water electrolysis penetration in the market originate from the costly and rare materials required by current state-of-the-art PEMWEs and the low efficiency and incompatibility with intermittent operation of commercial LAWEs. AEMWEs have the potential to offset these concerns by providing green H2 production at high operating current densities and with the potential for intermittent operation, while utilizing earth-abundant, non-platinum group metal (PGM) materials. Understanding the materials, interfaces, and interactions within the catalyst layer of these devices is a critical next step to facilitate the development and deployment of AEMWE technology. |
1. Introduction
Green H2 produced via electrochemical water splitting has emerged as a key energy carrier to facilitate the integration and storage of low-cost, variable renewable electrons and, ultimately, decarbonization across sectors.1–3 Current industrial methods, however, face cost and scalability challenges.2,4 Liquid alkaline water electrolyzers (LAWEs) traditionally suffer from low operating current densities (on the order of 100 s of mA cm−2),5 highly caustic electrolyte (>7 M KOH) with increased maintenance and operational costs, and incompatibility with H2 backpressure, thus requiring costly H2 gas separation and compression upstream for industrial implementation.2 State-of-the-art proton exchange membrane water electrolyzers (PEMWEs) allow for both high operating current densities (2–5 A cm−2 at 1.8–2.0 V)6–8 and significantly decreased H2 separation costs with the ability to apply H2 backpressure.2,4 The harsh, acidic near-surface environment in PEMWE systems, however, requires the use of expensive and rare platinum group metal (PGM) materials for catalysts and component coatings (transport layers and separators),2 imposing capital cost4 and materials criticality9,10 concerns. Further, there is interest to move away from the fluoropolymer materials often used for the membrane and ionomer in PEMWE systems due to high costs and growing concerns over the lasting effects of fluoropolymers on human health and the environment.11Anion exchange membrane water electrolyzers (AEMWE) are an emerging technology that encompass the benefits of both LAWE and PEMWE technologies. Like PEMWEs, AEMWE systems utilize polymer-based electrolytes and a zero-gap approach to achieve high operating current densities (>1 A cm−2 at 1.8 V).1,12–14 AEMWE systems can also allow for the application of H2 backpressure to minimize H2 separation costs.2,15,16 More importantly, these systems operate with a near-surface alkaline environment, which allows for the use of non-PGM materials for catalysts and other components,2,15,17 thus significantly reducing capital cost and materials criticality concerns. Furthermore, AEMWE systems typically operate with dilute supporting electrolytes (i.e., 0.1–1 M KOH or K2CO3), or, possibly, with pure water feeds,18 reducing operating cost and materials durability concerns related to the high-concentration electrolytes typically used in LAWE systems.
Over the last decade, interest in low temperature electrolysis technologies has risen rapidly. Fig. 1 shows Web of Science search results for the keywords “alkaline electrolysis” (grey diamonds), “liquid alkaline electrolysis” (green squares), “proton exchange membrane electrolysis” (red triangles) and “anion exchange membrane electrolysis” (blue circles) by year. Alkaline electrolysis is by far the largest field, with renewed interest developing in the late 20th century. AEMWE and PEMWE have comparatively skyrocketed in relevance in the last 5 years; search results for “anion exchange membrane electrolysis” yielded 761 publications, 485 of which were published in the last 5 years (i.e., 2019–present). Because “alkaline electrolysis” may have inadvertently included results from AEMWE, we also looked at the trends for “liquid alkaline electrolysis”; interestingly, this term emerges in 1991 and doesn’t increase above 5 publications per year until 2015, at the same time as AEMWE interest beings to rapidly increase, suggesting its emergence was to help distinguish research in traditional LAWE vs. novel AEMWE. As the AEMWE system is an emerging technology within the field, there is a need to translate the lessons learned from PEMWE and LAWE to AEMWE and to elucidate the differences among these technologies.
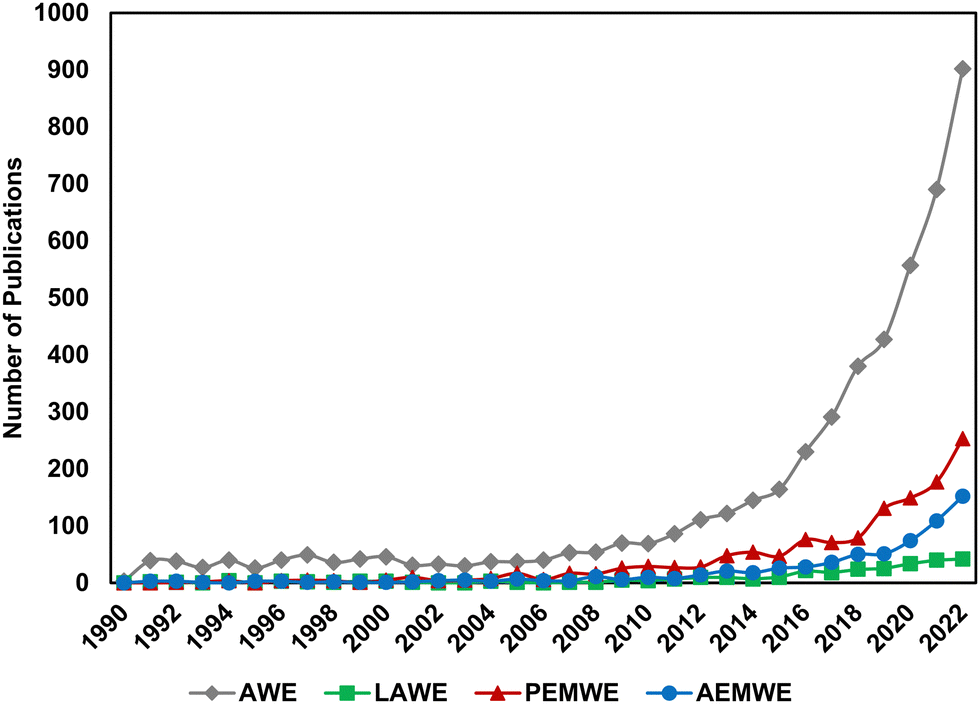 | ||
| Fig. 1 Number of publications mentioning “alkaline electrolysis (grey, AWE)”, “liquid alkaline electrolysis” (green, LAWE), “proton exchange membrane electrolysis” (red, PEMWE), and “anion exchange membrane electrolysis” (blue, AEMWE) over the last 30 years. Data obtained from Web of Science on 08/01/2023. | ||
Several reviews have covered recent component-level advances in AEMWE technology,19–23 including for membranes/ionomers24–29 and catalysts.30–34 A significant improvement has been made over the course of the last 5–10 years in enhancing the ionic conductivity of anion-exchange polymers (>100 mS cm−1)35 and prolonging the stability of AEMWE cells (stability tests of up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h have been reported).24–27,29,35–42 More recently, several studies have reported novel strategies for transport layer design by modifying the porosity and morphology of materials to enhance AEMWE performance.43,44 These improvements, alongside advances in catalyst development, have enabled AEMWE cell performances exceeding 3 A cm−2 at reasonable voltages (1.8–2.2 V).13,14
000 h have been reported).24–27,29,35–42 More recently, several studies have reported novel strategies for transport layer design by modifying the porosity and morphology of materials to enhance AEMWE performance.43,44 These improvements, alongside advances in catalyst development, have enabled AEMWE cell performances exceeding 3 A cm−2 at reasonable voltages (1.8–2.2 V).13,14
The reported performances of AEMWE, however, still fall short of PEMWE counterparts. Both the oxygen (OER) and hydrogen evolution (HER) reactions typically require higher overpotentials in AEMWE relative to their PEMWE analogues, with the OER as the more-limiting reaction in both conditions.8 HER kinetics drop by several orders of magnitude when moving from acid to base,45,46 and although intrinsic OER kinetics have been shown to be similar or slightly improved in alkaline vs. acidic electrolytes, OER kinetics are more limited in AEMWE than PEMWE at the device level. This discrepancy is likely due to differences in catalyst–ionomer interactions and less-than-optimal understanding of materials integration, ink rheology, and electrode properties that can limit OER kinetics at the device level. Furthermore, transport layer development in AEMWE is significantly lacking compared to the PEMWE space, and advances are needed to improve catalyst-transport layer interfaces and the transport of liquid reactants and gaseous products. Many questions remain regarding key device-level variables, including ideal operating conditions and the role of the ionomer within the catalyst layer. Few reviews have discussed such questions and research related to the integration of the catalyst layer with other device components.
This review aims to cover recent advances in understanding the catalyst layer in AEMWEs, with a focus on the chemistry and physics behind the integration of materials at the device level. To provide context for this review, the current state-of-the-art in AEMWE cell components, including catalyst, membrane, and transport layer design, will be briefly summarized in Section 2. Next, recent advances in electrode development, including catalyst deposition techniques and configurations, catalyst-transport layer design strategies, and catalyst–ionomer integration will be discussed in Section 3. The effects of cell assembly and operational variables (Section 4) as well as in situ and ex situ diagnostic techniques that can be used to understand processes within the catalyst layer will be reviewed (Section 5). Lastly, single-cell stability, with a focus on degradation mechanisms that arise due to the integration of components, will be discussed in Section 6. Throughout this review, key comparisons between AEMWE and LAWE and PEMWE will be made to facilitate a knowledge transfer for researchers transitioning into the emerging AEMWE space.
2. Overview of AEMWE cell components and recent component-level advances
AEMWEs commonly use a cell hardware configuration analogous to that widely used in PEMWE and PEM fuel cell (PEMFC) technologies (Fig. 2a). However, there is yet no consensus on a standard configuration for AEMWE. This is in sharp contrast to the PEMWE components and assembly protocols that have been standardized in several benchmarking efforts across the US, EU, and internationally,47–49 including in the Future Generation Membrane Electrode Assembly (FuGeMEA) configuration outlined by the U.S. Department of Energy (DOE) H2NEW Consortium.48,50 This current lack of effective benchmarking and baselining within AEMWE reflects the current status of the field and demonstrates the potential for developing and optimizing components across AEMWEs. Common cell components used in AEMWEs, as well as areas for future research, are outlined below. Note, the cell components discussed here specifically relate to research and development efforts at the single-cell level; commercial stack components and configurations likely have different design considerations, which will be addressed but are not the focus of this review.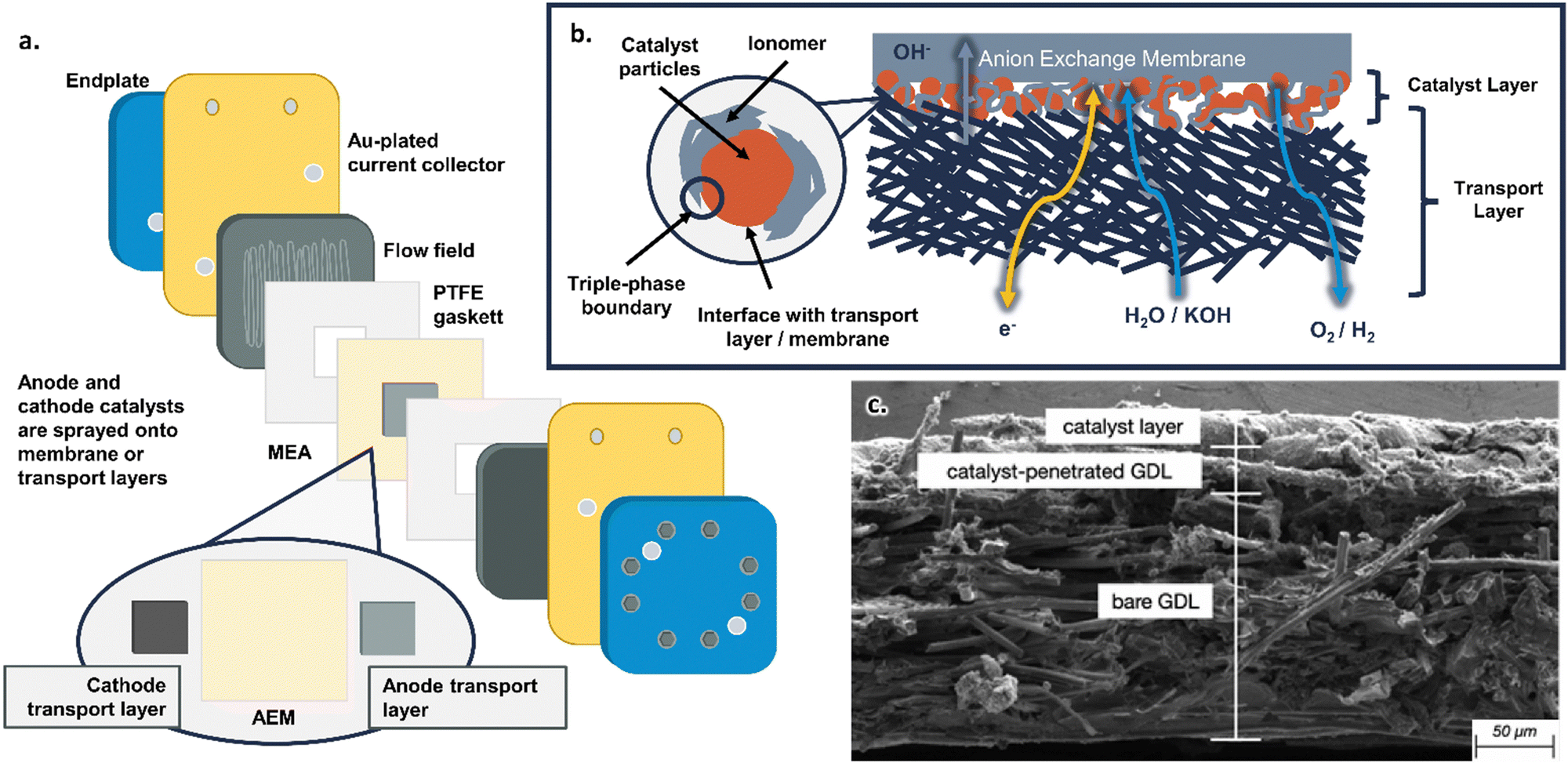 | ||
| Fig. 2 (a) Schematic of the components in an AEMWE. (b) Schematic of the catalyst layer in an AEMWE. (c) SEM image of an anode catalyst layer in an AEM fuel cell showing the transport layer fibers, catalyst layer, and interfacial region. Reprinted from J. Marie Mora et al., Analytical-based simulation approach for an anion exchange membrane fuel cell, Energy Conversion and Management, 273, 116382, Copyright (2022), with permission from Elsevier.65 | ||
At either end of an AEMWE are the cell end plates (shown in blue, Fig. 2a). These end plates have threading for bolts that provide compression to the cell. The end plates are often made of aluminium (Al) or stainless steel, which are chosen to apply necessary compression and to withstand the mild temperatures of AEMWE (60–80 °C). Beyond performing the desired function, these materials choices should not have a significant impact on electrolyzer performance or other materials integration. It is worth noting that stainless steel exhibits a higher alkaline stability than Al,17 which may be beneficial in case of a cell leakage. In PEMWE systems, the end plates are often made of anodized Al.51
The next components in the cell are the current collectors (shown in gold, Fig. 2a). The current (or voltage) is supplied through the current collectors by leads connected to a power supply or a potentiostat. They thus must be made of materials with high electronic conductivity to minimize their resistance contributions to the overall cell configuration. Most commonly, Au-plated Cu is used for the current collectors for both AEMWE and PEMWE.47
The current collectors are then in contact with the flow fields or bipolar plates (shown in grey, Fig. 2a) through which the liquid reactant (i.e., water or supporting electrolyte) flows into the cell. Flow fields for AEMWEs are most often made of stainless steel or Ni due to their alkaline compatibility, analogous to the materials choices in LAWE, without the need for the PGM coatings used in PEMWEs. Yet, one must be cautious in using stainless steel because it is prone to dissolution and can introduce Fe contaminants into the electrolyte, which have been shown to significantly impact the performance of Ni and Co materials; specifically, Fe contaminants have been shown to boost the OER activity of these catalytic systems.52,53 Fe contamination in the electrolyte has also been shown to negatively impact long-term durability in pure-water feeds due to interactions with the ionomer (details in Section 6).54 The use of Ni for flow field materials also has some concerns. Although Ni is stable against dissolution in alkaline and oxidizing environments, it is likely to passivate (i.e., form a less-conductive oxide layer) and deform over time, decreasing cell performance. It is therefore necessary to routinely polish Ni flow fields to avoid complications such as increased contact and high frequency resistances.
In comparison, PEMWE systems utilize flow fields made of Ti due to its acidic stability. Pt or Au protective coatings are often used to help with longevity and minimize contact resistances,55 although the use of PGM or noble-metal-based materials can contribute to cost concerns. Ti oxidation (i.e., to TiO2) can lead to increased contact resistances, which can result in a decrease in overall cell performance.56 Learning from these experiences in the PEMWE field, the use of such conductive coatings may be a good strategy to pursue for AEMWE flow fields to prevent increased contact resistances caused by Ni oxidation and thus to suppress cell degradation.
At the center of the cell is the membrane electrode assembly (MEA). The MEA is composed of the anion exchange membrane, anode/cathode catalyst layers, and transport layers, all framed by polytetrafluoroethylene (PTFE) gasketing (for research cells with no H2 backpressure) with a typical total thickness of <1000 μm. Each individual component within the MEA will be discussed in the following paragraphs.
The anion exchange membrane functions as the electrolyte material in AEMWEs, analogous to the proton-conducting membranes used in PEMWEs. The membrane is typically made of a conductive polymer containing positively charged functional groups (often N-based) to allow for the transport of OH−, although there is yet no standardization for the membrane choice. This contrasts with PEMWEs, where Nafion is often used as the standard proton-conducting material. Several reviews have highlighted recent advances in polymer chemistry for anion exchange membranes and ionomers,24–29 including those that are commercially available.26 Recent developments in cation functional groups, side chain engineering, cross-linking strategies, and backbone development have resulted in anion exchange polymers that have high ionic conductivity and alkaline stability. Such developments allow for extended durability testing for AEMWE cells and bring the technology closer to commercialization.
On either side of the anion exchange membrane are the transport layers, which are also called gas diffusion layers (GDLs) or porous transport layers (PTLs). The transport layer facilitates the transport of liquid reactants (i.e., water or supporting electrolyte) and gaseous products (i.e., H2 at the cathode, O2 at the anode; Fig. 2b) and must be electronically conductive to allow electrons to move between the catalyst particles and the current collectors. It is also important to note that, on the anode side, the transport layer must be able to withstand high potentials (i.e., often up to and exceeding 2 V) and an aqueous, alkaline environment (i.e., pH of 12–14). Common materials for transport layers in AEMWEs include C paper and Ni, Ni alloy, or stainless steel meshes and foams,21,57,58 although C is not a viable choice for the anode transport layer because it oxidizes and degrades at high potentials and pH.59,60
Comparatively, transport layers in PEMWEs often consist of C paper at the cathode and Pt-coated Ti at the anode.61 Ti is chosen due to its acid compatibility (i.e., it passivates but does not corrode) and manufacturability, while Pt provides necessary electronic conductivity.61 Transport layer properties are critical for facilitating interfacial contact with the catalyst layer and can heavily affect site accessibility. For PEMWEs, research has suggested that the use of a microporous layer (MPL) at the catalyst-layer interface can help facilitate interfacial properties, including improved ionic and electronic contacts and increased active-site accessibility.62,63 The MPL is usually a few microns thick and has a smaller mesh size than the bulk of the transport layer to facilitate these improved interfacial contacts. Such use of an MPL is currently less studied in the AEMWE community, although a recent study by Razmjooei et al. showed an improvement in performance by utilizing a transport layer topped with a Ni MPL in an AEMWE.43 This work suggests that AEMWE performance could be improved by engineering the interface between the catalyst and transport layer material; this will be discussed further in Section 3.2. Lastly, transport layer design also impacts membrane durability; transport layers can provide structural support to the membrane and prevent excessive creep (caused by swelling) into the catalyst/transport layers. Such behaviors can occur when surface porosity is too high, especially under H2 backpressure operation, as has been shown in PEMWEs.64
PTFE gaskets frame the transport layers and facilitate desired compression to the cell. Note that gasketing can be used for ambient pressure cells, but other methods are likely required for high-pressure operation; pressurized operation and effects of cell compression are discussed in Section 4.1. The thickness of the gaskets can be adjusted individually for the cathode and anode, allowing for different compressions as well as the use of different transport layer thickness on either side.
In between the transport layers and anion exchange membrane are the anode and cathode catalyst layers. In this review, the catalyst layer specifically refers to (1) the catalyst particles themselves, (2) the anion-conducting polymer (referred to as the ionomer) dispersed with the catalyst particles, (3) their interface with the porous transport layer, and (4) their interface with the anion exchange membrane (shown schematically in Fig. 2b and as a cross-sectional SEM image from JM Mora et al.65 in Fig. 2c). Anode and cathode catalysts can be deposited either on the membrane (catalyst coated membrane, or CCM, approach) or on the transport layer (catalyst coated substrate, or CCS, approach); the pros and cons of these approaches are discussed in Section 3.2. The most common deposition methods for anode and cathode catalyst layers involve spraying from an ink (which can ultimately translate to manufacturing techniques, such as a roll-to-roll coating), though other deposition methods will be discussed in detail in Section 3.1. The catalysts are often deposited as a mixture with the ionomer (Fig. 2b); the role of ionomers and details of catalyst–ionomer integrations are discussed in Section 3.3.
Both anode and cathode catalyst layer designs involve many of the same variables and considerations, including catalyst loading, ionomer content, and electrolyte conditions, among others. Anode catalyst layers, however, unlike cathode catalyst layers, must be able to withstand high voltages and thus highly oxidative conditions. Further, cathode catalyst layers have different hydration considerations, especially when AEMWEs are operated cathode dry (discussed further in Section 4.2). Similarly, the electrolyte flow configuration (i.e., DIW vs. supporting electrolyte, cathode wet vs. dry) will affect necessary ionomer contents and these needs may vary between anode and cathode catalyst layers. These topics are discussed further in Section 3.3.
The most commonly used catalysts in PEMWE are Pt/C and IrO2 for HER and OER, respectively.2 The acidic near-surface environment of PEMWEs necessitates the use of these expensive and rare PGM materials, as non-PGMs oxidize into soluble forms at such conditions.17 In fact, even Ir-based materials become thermodynamically unstable at the harsh OER reaction conditions in both acid and base;17,66,67 Ir has been shown to dissolve via multiple routes at high anodic potentials, including via the formation of IrO3 (g) and, subsequently, to soluble IrO42−,67 raising concerns over the long-term durability of Ir in electrolyzers.
Like in PEMWE, Pt/C and IrO2 have historically been common choices for AEMWE catalysts. However, the alkaline environment of AEMWEs also allows for the use of non-PGM materials. First-row transition metals (e.g., Ni, Fe, Co, and Mn) have been studied in many forms, including metallic nanoparticles, polycrystalline metals, sulfides, nitrides, and oxides,68–71 which are shown to be active for OER in alkaline media. Metallic species often exhibit higher OER reactivities than the respective oxides, but oxidative conditions (i.e., high potential and pH) at the anode likely mean that any starting material eventually converts to an oxidized (i.e., oxide or (oxy)hydroxide) form in situ.72 Recent works have explored the effects of varying composition (e.g., mixed metals and/or heteroatom X-ides, including nitrides, sulfides, phosphides, etc.),73–75 of the use of different oxide forms,76–78 and the use of supports79–81 to enhance catalytic activity, active surface area, and electronic conductivity; such advances in catalyst development have been reviewed previously.30–34
For the HER in AEMWE systems, many studies still use PGM-based catalysts, primarily Pt or PtRu on high surface area C, analogous to those used in PEMWE.82–84 More recent works have focused on developing alternative non-PGM materials, including CoNiO2,85 NiCu oxide,86 and Ni/CeO2–La2O3/C.87 Performance enhancements have been observed for transition metal phosphides and sulfides, such as VCo-P88 and Fe0.2Ni0.8–P0.5S0.5,89 and there has been significant interest in NiMo-based alloys.12,90–92 It is instructive to note that the optimal loadings for non-PGM cathode catalysts in an MEA are typically several times greater than those for PGM catalysts; for example Tricker et al. found optimized cathode loadings of 0.3 mg cm−2 and 1.6 mg cm−2 for Pt/C and CoNiO2/C, respectively.85 The necessity of higher loading may be due to lower accessible surface areas and/or lower per site activity compared to PGM catalysts supported on high surface area carbons. These materials are often unstable and induce performance drops on the order of 100–200 mV at 10 mA cm−2 in three-electrode, rotating disc electrode (RDE) studies vs. PGM cathode catalysts;93–95 more work is therefore required to develop non-PGM catalyst options for HER. Promising catalysts must also be tested in AEMWE devices, since RDE activity is not necessarily predictive of performance in a device due to significant differences in the catalyst layer environment and transport processes, as has been demonstrated for PEMFCs.96
For both anode (OER) and cathode (HER) catalysts, considerations must also be made to material cost, availability, and criticality. As discussed, PGMs, such as Ir, have been identified as having high material criticality,9,10 motivating the development of non-PGM materials for OER and HER catalysts as well as, more largely, AEMWE cell components. However, not all non-PGMs have equal material costs or criticalities. For example, Co has appeared alongside PGMs on critical materials lists globally;9,10,97 considerations to the long-term viability and costs of sourcing different catalyst materials must be weighed alongside measures of activity and stability.
The following sections will dive deeper into recent advances in understanding how these individual components integrate in AEMWEs, with specific focus on the catalyst layer and how key interfaces impact the overall cell performance (e.g., overpotentials, accessibility of active sites, stability, etc.).
3. Recent advances in electrode development and configurations
Electrode manufacturing, including catalyst deposition methods, configurations, and composition, have been shown to impact the overall performance of PEMFCs, PEMWEs, and, more recently, AEMWEs. The following sections will explore recent advances in electrode development, including catalyst deposition techniques (Section 3.1), strategies for integrating catalysts with membranes and transport layers (Section 3.2), and catalyst–ionomer interactions and integration strategies (Section 3.3).3.1 Catalyst deposition techniques
There are numerous strategies for depositing catalytic materials onto the membrane or transport layer. These deposition strategies can affect the type and nature of active sites (i.e., chemical structures of the catalytic materials), the degree to which active sites are exposed, and the network of electronic and ionic conductivities between catalysts particles as well as between the catalyst and the transport layer or membrane.The most commonly used technique is spraying a catalyst ink, composed of catalyst particles, ionomer, water, and alcohol (e.g., n-propyl or isopropyl alcohol, ethanol, etc.), onto a substrate (e.g., membrane or transport layer).13,98–100 This method has also been widely implemented in PEMWE research.101 Typically, spraying can be performed by hand using an airbrush (Fig. 3a) or by using an automated ultrasonic spray system (Fig. 3b), both of which provide varying degrees of control regarding deposition homogeneity. Typical, the tuning variables for these spraying techniques include ink composition (including solvent choice and concentration as well as catalyst/ionomer ratios) and preparation (including sonication and icing procedures), deposition temperature, and deposition time. Ink compositions need to be optimized for each catalyst–ionomer pair, which can otherwise lead to significant discrepancy between research groups and research publications, as shown previously in the PEMWE and PEMFC spaces.102–107 For example, the water and alcohol content in the ink solution affects the surface tension of the ink, which, in turn, impacts the dispersion of the catalyst in the ink and the ink drying rates, all of which can significantly impact catalyst layer microstructure (Fig. 3c), as shown previously for PEMFCs.105,107 The ionomer content can similarly impact ink rheology as well as catalyst dispersion, which has been shown to have significant impacts on catalyst layer microstructure in PEMFCs103,105 and PEMWEs.106 Catalyst agglomeration in inks can lead to less-than-ideal surface areas of nanoparticle catalysts, impacting activity. Different sonication procedures can help mitigate catalyst and ionomer agglomeration, as has been shown in PEMFCs (Fig. 3d),102 and such procedures must be developed on a case-by-case basis depending on the catalyst–ionomer pair used. The results depicted in Fig. 3c and d show that ink design choices can have substantial effects on catalyst layer structure; such lessons from these results are importantly transferable to AEMWEs. Although optimizing each variable can be challenging and time consuming, the abundance of tuning variables can provide a high degree of control in terms of catalyst layer thickness and microstructure. These spraying techniques also have the advantage of facile scalability, adaptation to existing commercial manufacturing methods (i.e., roll-to-roll coating),108 and the ability to further optimize for cost and processing needs.
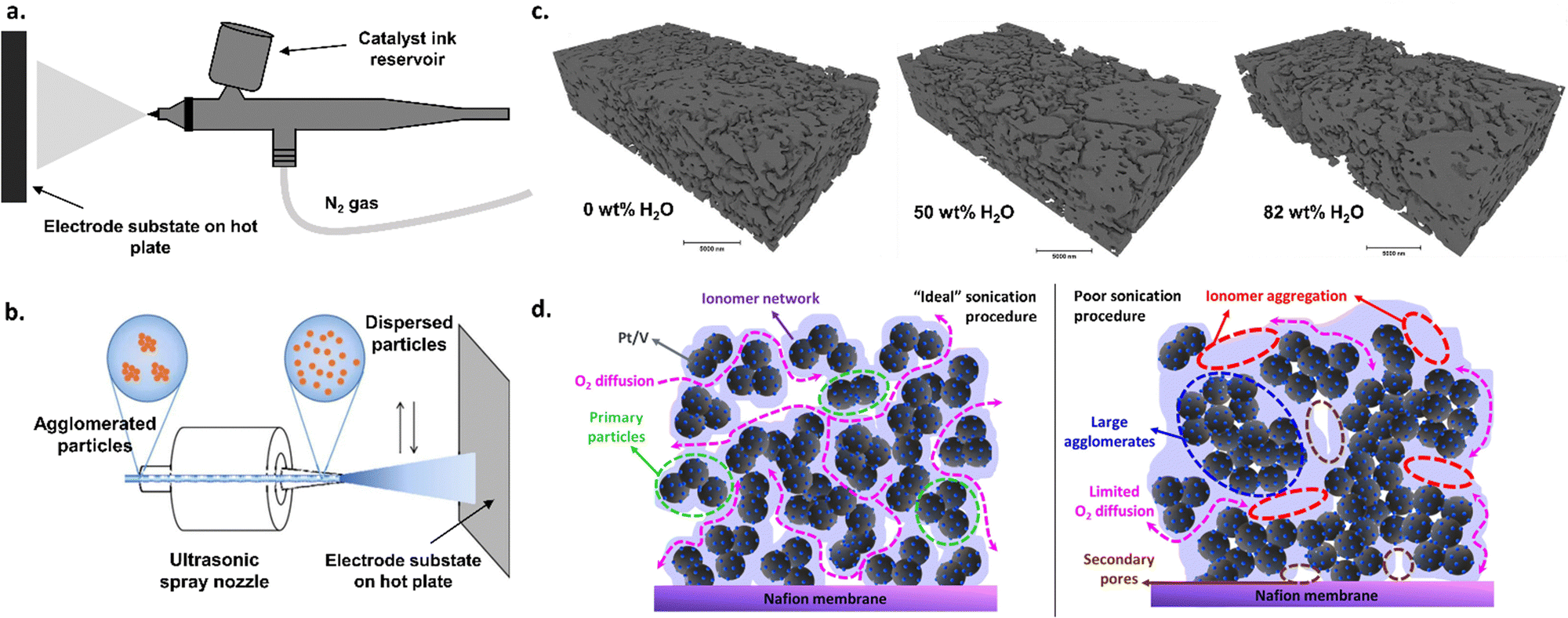 | ||
| Fig. 3 (a) Schematic of the airbrush spraying method for catalyst deposition. (b) Schematic of an ultrasonic spray system. Adapted from Yarlagadda et al., J. Electrochem. Soc., 2017 with permission.204 (c) Nano-CT segmented phase contract images for catalyst layers prepared with different water contents in the ink. Reprinted from L. Osmieri et al., Utilizing ink composition to tune bulk-electrode gas transport, performance, and operational robustness for a Fe–N–C catalyst in polymer electrolyte fuel cell, Nano Energy, 75, 104943, Copyright (2020), with permission from Elsevier.102 (d) proposed catalyst layer structure for a PEMFC cathode with an “ideal” sonication procedure vs. a “poor” sonication procedure. Reprinted with permission from Wang et al., ACS Appl. Energy Mater. Copyright (2019) American Chemical Society.104 | ||
Ink-spraying methods generally require the use of an ionomer or a polymeric binder to adhere the catalyst particles to the substrate (i.e., membrane or transport layer). The ionomer may also serve as an ion conductor that helps to shuttle OH− ions through the catalyst layer to/from catalyst active sites. This is especially critical in pure water operation, where the ionomer phase is the sole ion conductor. Yet, such ionomers or polymers can inhibit electronic conductivity pathways between catalyst particles and between the catalyst and transport layers, as well as block catalyst active sites, leading to decreased cell performance. The catalyst can also facilitate oxidation and loss of the ionomer, leading to instability within the catalyst layer and to long-term durability concerns,54,109,110 which will be discussed in more detail in Sections 3.3 and 6.
In supporting electrolyte, anions in the bulk-liquid phase provide additional OH− conductivity and an ionomer may not be required as an ion conductor. As a result, catalyst deposition techniques that do not require the use of ionomer or polymeric binder can be used, reducing concerns of site-blocking by the ionomer and of inhibited electronic conductivity. If desired, an ionomer can be integrated after electrode coating to facilitate operation in pure water feeds. Such “direct” deposition methods that bypass the use of ionomers include electrodeposition, chemical vapor deposition, physical vapor deposition (e.g., sputtering),111 and plasma deposition,91,112,113 which differ in the ways to deposit active catalysts on to the cell components.
In the electrodeposition method, the substrate material (often the transport layer) is submerged in an electrolyte containing metal ions of the desired catalyst type, and potential is applied to deposit the metals on the substrate.114,115 These materials can be grown as oxides or oxidized ex situ to the desired form. Unlike airbrush spraying, electrodeposition methods avoid ink rheology concerns and discrepancies; instead, catalyst layer thickness and structure can be precisely controlled via changes to the concentration and identity of the electrolyte solution and to the electrodeposition time and potential.41,116–118 This then allows for the formation of catalyst layers on the nanoscale, which can be advantageous in terms of maximizing active surface areas and minimizing mass transport losses related to evolved gasses navigating the catalyst and transport layers. For example, Sanchez et al. utilized an electrodeposition technique on oxidized C-based transport layers to achieve low loadings of Co-based HER catalysts, decreasing mass transport resistances and increasing overall cell performance.119 Relatedly, several other recent studies have employed this technique to create three-dimensional structures and enhance both OER and HER performance by increasing active surface areas and tuning morphological properties.41,116–118,120
Other strategies include thermochemical deposition, where the catalyst materials are grown directly onto the transport layer or membrane without the use of applied potential.121 Vapor deposition techniques typically involve vaporizing metal precursors into inert carrier gases (e.g., Ar, N2, or He), which then deposit on a substrate material to form thin catalyst layers. These vapor deposition techniques include chemical vapor deposition,122 physical vapor deposition (e.g., sputtering),111 and plasma deposition.91,112,113
Lastly, considerations of the compatibility of different electrode deposition methods with established manufacturing methods (e.g., doctor blade, slot die, gravure, roll-to-roll, etc.) are also important. For example, spray techniques, and the associated ink developments required, lend themselves readily to roll-to roll coating techniques, as has been shown for PEMFCs.108
3.2 Catalyst-membrane and catalyst-transport layer integration strategies
As mentioned above, catalysts can be deposited on the membrane or on the transport layer (CCM or CCS). For PEMWE, the CCM approach is most popular. In the AEMWE space, however, the preferred configuration (CCM vs. CCS) is still being determined.Several studies have utilized the CCM deposition technique for AEMWEs.14,98,123–127 Hnát et al. investigated the CCM approach with NiCo2O4 and NiFe2O4 catalysts airbrush-sprayed directly onto a polystyrene-block-poly(ethylene-ranbutylene)-block-polystyrene membrane. The authors found that there was good contact between catalyst and membrane in cross-sectional SEM images and saw no catalyst delamination after testing (20 h at 250 mA cm−2).123 More recently, Koch et al. utilized a direct bar coating method to create CCMs; their AEMWE cell exhibited a high current density of 2 A cm−2 at 1.8 V and low degradation rates of <1 mV h−1, showing the promise of such a method.
Despite these successes, concerns remain over the CCM approach for AEMWE, and several studies have reported durability concerns related to the CCM approach. For example, spraying an ink, typically composed of water and alcohols, directly on the membrane may causing swelling, mechanical deformation, and pinholes. In a study by Ito et al., the authors tested a NiFe catalyst and the same ionomer/membrane (i.e., AS-4 and A201 from Tokuyama), and saw rapid MEA deactivation, attributed to delamination of the catalyst layer caused by poor adhesion of the catalyst particles to the membrane.124 They hypothesized that this was due to (1) mechanical failure of the membrane, (2) sheer force imposed by the recirculating electrolyte, or (3) chemical degradation of the ionomer. They suggested that the ionomer was not robust enough to serve as a binder in these systems, and suggested that the CCS approach may be the preferred choice in AEMWE due to higher stability.124 These results emphasize the importance of understanding the role of the ionomer in AEMWE catalyst layers; if it can be identified that ionomers are not required for ion-transport through the catalyst layer in supporting-electrolyte-fed cases, such concerns over poor binding characteristics for anion exchange ionomers may be mitigated. Ito et al. further explored the use of PTFE as a binder;124 this is discussed in more detail in Section 3.3.
Electrode deposition with the CCS approach has been proposed to enhance catalyst layer stability by reducing losses due to catalyst layer delamination. The CCS approach introduces other considerations and concerns, however, such as increased ionic resistance at the membrane–catalyst layer interface and poor control of catalyst distribution onto/within the transport layer. Catalyst layer design strategies, including in how the catalyst particles are integrated with transport layer and novel transport layer designs, can help overcome these concerns.
As shown in the schematic in Fig. 4a, transport layer morphology and porosity can be modified to increase interfacial contact with the membrane and improve catalyst layer utilization, as well to improve liquid and gas transport. Several approaches have been employed for engineering transport layers, including utilizing different pore sizes, pore size gradients, microporous layers, and 3D printed transport layers.
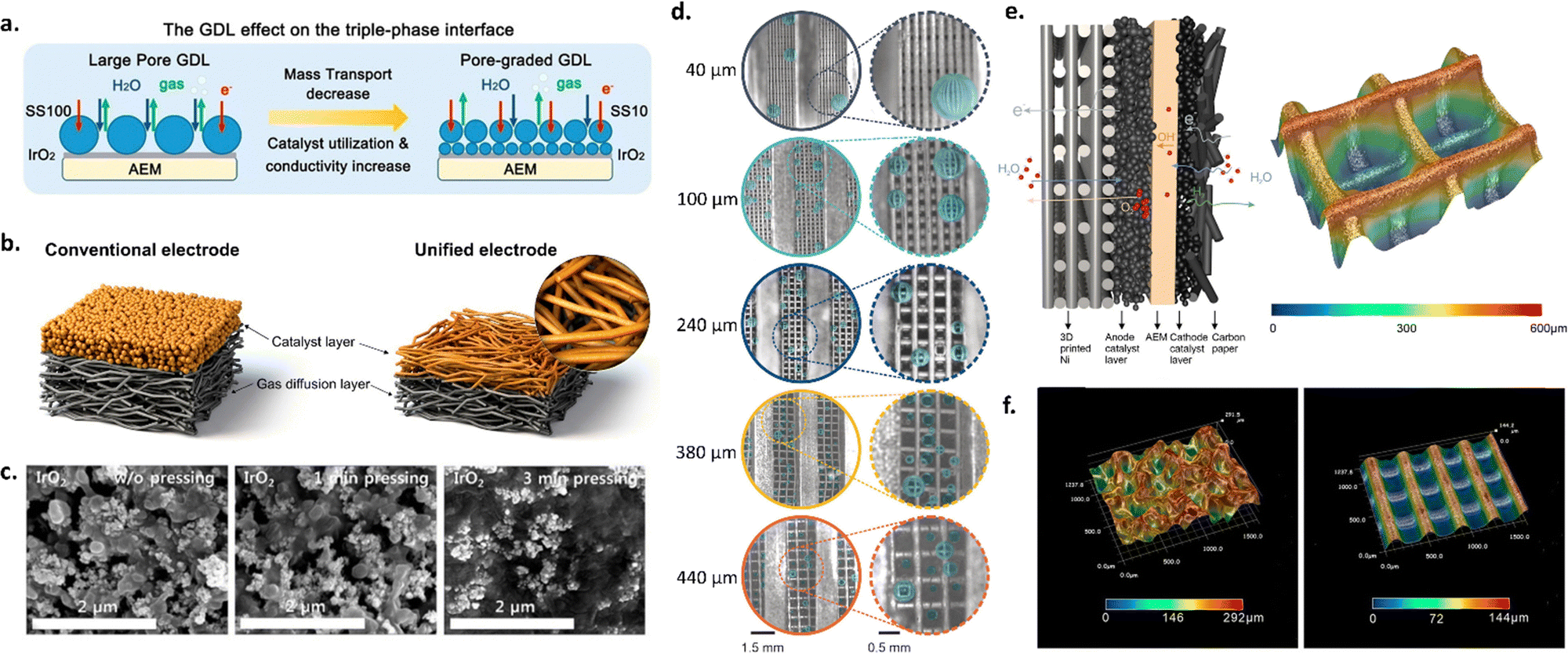 | ||
| Fig. 4 (a) Schematic of different transport layer strategies and the impact on the catalyst layer-AEM interface. Reprinted with permission from Xu et al., ACS Energy Letters. Copyright (2021) American Chemical Society.126 (b) Unified electrode strategy. Reprinted with permission from Park et al., ACS Catal. Copyright (2022) American Chemical Society.123 (c) SEM images of catalyst layer surfaces with different hot-pressing times. Reprinted from A. Lim et al., A study on electrode fabrication and operation variables affecting the performance of anion exchange membrane water electrolysis, Journal of Industrial and Engineering Chemistry, 76, 410–418, Copyright (2019), with permission from Elsevier.133 (d) Photos of O2 gas bubbles evolved at 1 A cm−2 in AEM electrolyzers with different 3D-printed Ni transport layer pore sizes (40 μm, 100 μm, 240 μm, 380 μm, 440 μm). (e) Schematic of the anode and cathode catalyst layers in an AEM electrolyzer (left) and optical microscope image of the 3D-printed Ni transport layer with a pore size of 240 μm (right). (f) Optical microscope images of a commercial Ni foam transport layer vs. a 3D-printed Ni transport layer with a pore size of 240 μm. (d)–(f) Were reprinted with permission from Huang et al., Angewandte Chemie International Edition, Copyright (2023) John Wiley and Sons.44 | ||
Park et al. investigated the use of an MPL with two different anode catalysts (i.e., IrO2 and NiFe alloy) to improve interfacial contact between the catalyst and transport layer. This resulted in improved catalyst utilization, lower ohmic and charge transfer resistances, and improved cell durability. The formation of a microporous layer was facilitated by deposition via the CCS approach due to the pores in the transport layer.99 In another study, Razmjooei et al. utilized a Ni MPL on top of a Ni mesh transport layer to facilitate improved catalyst layer contact with both the transport layer and membrane, leading to improved mass transport properties. The authors found that the MPL provided reduced capillary pressure, allowing more-rapid transport of liquid reactants through the catalyst layer to catalyst active sites. They also observed decreased ohmic resistances due to increased catalyst-transport layer contact.43 Similarly, Park et al. used a pore-graded transport layer and unified electrode design to improve catalyst layer contact, utilization and conductivity,128 as shown in Fig. 4b.
Hot pressing is another technique to help improve interfacial properties for CCS-deposited electrodes; this technique has been studied extensively for PEMFCs, and, more recently, for PEMWEs.129–131 Hot pressing utilizes a heated hydraulic pump to compress the sprayed catalyst layer at different pressures and temperatures. Hot pressing has several advantages, including decreasing ohmic resistances. Several studies have utilized this technique for AEMWE systems. For example, Cho et al. studied the effects of changing the temperature of hot pressing (i.e., room temperature, 50, and 80 °C) for AEMWEs consisting of an IrO2 anode and Pt/C cathode both with PTFE as the binder material. The authors found that increasing the hot-pressing temperature decreased polarization resistances and led to increased activity; for a cell hot-pressed at 80 °C, the activity was 1.9× higher than for a sample with no pressing.132 Later, Lim et al. investigated differences in pressing time and found that cell performance decreased with increased pressing time (i.e., from 161 to 118 mA cm−2 at 1.6 V as the pressing time increased from 0 to 3 min), which they attributed to structural deformation of the catalyst layer and increased blocking of catalyst active sites by the PTFE binder; this deformation with increased hot-pressing time is shown in Fig. 4c.133 The results of these two studies suggest that hot pressing can be advantageous, but that considerations must be made to treatment times to avoid deformation and performance losses.
Focusing on the challenges of transporting electrons, gases, and water through the anode transport layer, Huang et al. fabricated 3D Ni transport layers with straight-through pores and variable grid sizes, which were then coated with a NiFe catalyst. They found that there was a trade-off with grid (pore) size between mass transport and catalyst utilization, and thus the best performance was achieved with a gradient in grid size. Fig. 4d shows the results of their study and the differences in bubble formation for transport layers with different pore sizes. Compared with a transport layer with a pore size of 240 μm throughout, using a top layer with a pore size of 240 μm over a bulk pore size of 550 μm led to a decrease in overpotential of 38 and 100 mV at 1 and 2 A cm−2, respectively.44 The surface morphology and porosities of these 3D Ni transport layers led to improved mass transport vs. a commercial Ni foam transport layer; the differences in these transport layers are shown in optical images from the authors in Fig. 4e and f. Other works have also used transport layer materials directly as electrodes, similar to the approach used in liquid alkaline electrolysis; such works include the study of Ni foam,134–137 Ni felt,57,138 and SS felt57 directly as catalysts in AEMWEs.
3.3 Ionomer content and catalyst/ionomer interactions
The ionomer is often used in the catalyst layer for multiple purposes: (a) to adhere catalyst materials to the membrane or transport layer, (b) to provide ionic conductivity for OH− species (product in HER and reactant in OER) through the catalyst layers, and (c) to facilitate ideal rheological characteristics in catalyst inks for spraying deposition methods. In most studies, the same polymer is used for the ionomer and membrane. Many ionomers/membranes have been developed by research groups139–141 and commercially; typical commercial ionomers include Aemion, Sustainion, Fumasep FAA-3, and PiperION.26 Important properties for ionomers include their ion exchange capacity (IEC), water uptake, and swelling, which can deviate from the properties of the bulk membrane depending on the catalyst layer morphology. These properties are often correlated; high IEC is desirable for good ionic conductivity but can also lead to high water uptake and swelling, which can then lead to poor mechanical integrity and delamination of the catalyst layer. These effects have been studied in part by the Kohl group.142,143 There are therefore benefits to matching ionomer properties to the needs of the catalyst layer.The specific role of the ionomer in AEMWE can vary significantly depending on the materials and operating conditions used. An ionomer is required for pure-water AEMWE operation to provide transport pathways for reacting OH− species to/from catalyst sites. In supporting electrolyte, however, it has been proposed that the supporting electrolyte (e.g., 0.1–1 M KOH) provides ample ionic conductivity through the catalyst layer, rendering the ionomer unnecessary for such a role.28 Lei et al. demonstrated this in their study of a NiFeCo OER catalyst; they showed that discontinuities in ionomer-catalyst contact (caused by pH-driven structural changes) had a less significant impact on OER reactivity in supporting electrolyte vs. operation in pure water.82 This work is discussed in more detail in Section 4.2.
Several studies have identified “optimal” values for ionomer content, with the majority of studies suggesting 5–20 wt% ionomer in the catalyst layer.83,98,124,125,132,144–148 This optimization is based on a trade-off between having enough ionomer to provide adhesion and ionic conductivity, and not having so much that catalyst active sites are blocked or catalyst particles are electronically isolated.28 Ideal ionomer loading is system-dependent, however, and will likely be affected by the ionomer properties, the electrode, and the operating conditions, as will be discussed in more depth in Section 4.2.
Relatedly, the role of the ionomer likely varies between anode and cathode catalyst layers. At the anode, the ionomer may be more susceptible to oxidative degradation pathways due to the local high potential environments; this is expected to be less of a concern at the cathode. At the cathode, the ionomer may play a critical role in providing site accessibility and reactant/product transport through the cathode catalyst layer, especially when operated under cathode dry conditions, even in supporting electrolyte. Understanding specifically how cathode ionomer content affects performance under different flow configurations and operating conditions is therefore necessary.
Preferred ionomer contents may also vary between anode and cathode catalyst layers. For example, Faid et al. showed that changing the Fumion ionomer content in the cathode catalyst layer (Ni/C), for a cell fed with 1 M KOH at both electrodes, had a more significant impact on cell overpotentials compared to changing the ionomer content in the anode catalyst layer (NiO). The authors hypothesized that increased ionomer content negatively affected the morphology of the catalyst layer, reducing exposed active sites; this hypothesis was supported by SEM imaging.147 In contrast, using Pt/C and IrO2 catalysts with Aemion ionomer, Koch et al. found anode ionomer loading to be the more significant variable.145 More work is required to understand how the membrane, ionomer, catalysts, catalyst deposition method, and electrolyte used affect optimal ionomer loading.
Ionomer degradation is a significant problem with currently available materials, especially in pure-water operation. As previously discussed, one such degradation mechanism is mechanical instability due to swelling. This can be addressed through tuning of polymer properties to decrease water uptake and/or increase the elastic modulus.28 Other degradation mechanisms are related to reactions between the ionomer and the electrolyte1,148,149 and catalyst materials.1,54,110,150 In a study on degradation in AEMFC devices, Diesendruck and Dekel found that the low-hydration environment present at the cathode led to non-solvated OH− with increased nucleophilicity and ability to degrade the ionomer.151 Similar degradation pathways may exist in AEMWE cathodes under dry cathode operation, motivating the use of ionomers with high water uptake in the cathode catalyst layer. Understanding these system-specific interactions will be critical to preventing catalyst-layer reconstruction or delamination, as well as ionic conductivity losses.
Catalyst–ionomer interactions are particularly likely to lead to ionomer degradation at the oxidative conditions at the anode (i.e., high pH, high potential). For example, phenyl groups in the ionomer backbone have been shown to adsorb to catalytic active sites, which then facilitate oxidation of these groups leading to ionomer degradation. Particularly in pure water feeds, if electronically connected catalyst sites have insufficient OH− available to perform OER, these sites may preferentially oxidize the ionomer instead. Perspectives on phenyl oxidation have been summarized in a recent review by Matanovik and Kim.109 Catalysts can also interact strongly with ionomer functional groups, with negative impacts on both activity and stability. For example, Ghoshal et al. showed that the pore-filled (PF)AEM Gen2 ionomer developed at the National Renewable Energy Laboratory reacted with a Co3O4 catalyst, suppressing the Co(III/IV) transition and leading to decreased kinetic reactivity.110 Krivina et al. have further shown that different non-PGM catalysts can react differently with ionomers, leading to variable levels of ionomer degradation and ionic-conductivity losses; their work is discussed in more detail in Section 6.54 It is therefore critical to study possible reactions between relevant catalyst–ionomer pairs to improve stabilities.
In supporting electrolyte feeds, where the ionomer's primary role is likely catalyst layer adhesion, alternative polymers with improved operational stability can be used. For example, PTFE has been used in electrolyte-fed AEMWEs with promising results.100,124,132,146 Cho et al. studied the use of PTFE as a binder in an AEMWE single cell consisting of a Pt/C cathode catalyst, IrO2 anode catalyst, and an A201 anion exchange membrane from Tokuyama. They found that 5–9 wt% of the PTFE binder showed superior initial performance than higher PTFE contents, but that there was rapid degradation caused by catalyst delamination. When they increased the PTFE content to 20 wt%, they saw stable AEMWE operation for 1600 cycles of cyclic voltammetry from 1.5 to 2.2 V, suggesting that PTFE (or another polymeric binder) is viable as an alternative binding agent to ionomer.132,146
Careful consideration of catalyst–ionomer pairs and investigation into any possible reactions between such materials are critical to gain deeper understanding of possible AEMWE degradation mechanisms. Such knowledge has direct implications for catalyst layer design, where electronic and ionic conductivity pathways must be created and maintained. Ultimately, understanding ionomer degradation mechanisms and implementing this knowledge into catalyst layer design can help facilitate the development of AEMWEs with long operational lifetimes and assist their wide-scale deployment.
4. Effects of cell configuration and operating conditions
While many cell assembly variables and cell operating conditions have been standardized for PEMWE systems,48,50 such variables remain undecided in the AEMWE space. As discussed in Section 2, discrepancies remain over material choices for cell hardware, including end plates, flow fields, and transport layers. Such material choices alongside changes to assembly and operational variables can directly affect reaction kinetics and mass transport rates, and thereby lead to differences in cell overpotentials and stabilities. This emphasizes the need for understanding and optimizing for these choices, as well as for standardization across laboratories to ensure comparable results. Changes to assembly and operational variables, including cell compression, temperature, and pressure will be discussed in Section 4.1, electrolyte variables and pre-treatment procedures will be discussed in Section 4.2, and the need for and recommendations for establishing baseline operation and standardized protocols will be discussed in Section 4.3.4.1 Effects of cell assembly and operational variables
Cell assembly variables related to material choices and electrode configurations have been discussed in Sections 2 and 3, respectively. Cell compression is another variable that has been studied for AEMWEs and PEMWEs, which can be changed by altering transport layer or PTFE gasket thicknesses. Gasket stiffness can also play an important role in cell compression; more-stiff gaskets will prevent additional compression occurring when the cell is closed, whereas less-stiff gaskets can lead to additional compression. Higher cell compression can lead to better contact between the catalyst layer and the transport layers/membrane, which, in turn, can decrease Ohmic and kinetic losses and improve cell performance. However, overly high cell compressions can cause membrane tearing or deformation, decreasing cell longevity. Therefore, understanding the implications of cell compression on performance and durability as well as determining preferred compression ranges are important for advancing AEMWE design.In the PEMWE space, increasing cell compression has been shown to reduce interfacial contact resistances and decrease cell overpotentials, although it may have detrimental effects on the mass transport of gaseous products by constricting pores in the transport layer and increasing hydrogen crossover through the membrane.152 For AEMWE systems, recent works provide insights into the effects of varying cell compression on overall performance. For example, Xu et al. systematically evaluated several AEMWE single cells with different levels of cell compression. They found that increasing cell compression led to higher cell activities, which they hypothesized was related to increased interfacial contact.126
Temperature is an important operational variable, as changes to temperature directly affect kinetic rate constants, thermodynamic equilibrium potentials, and ionic conductivities. The temperature range for low temperature electrolysis technologies (including PEMWE and AEMWE) is generally limited to below 100 °C (i.e., the boiling point for water), with operation commonly between 50 and 90 °C.2 Lim et al. evaluated the effects of changing cell operating temperatures in the range of 50 to 90 °C and found that at a constant overpotential of 591 mV, there was a +50 mA cm−2 increase in current density for every 10 °C increase in temperature. They attributed this higher activity to increased ionic conductivity in the membrane, ionomer, and electrolyte and to increased electrochemical kinetics.133 Ionic conductivity is sensitive to temperature in the range of 0 to 100 °C, which is known to increase with temperature. For example, OH− transport via the Grotthuss ion hopping mechanism is sensitive to temperature in this range.153 The authors further saw a 22.6% decrease in Ohmic resistance and a 21.9% decrease in kinetic resistances measured by in situ electrochemical impedance spectroscopy (EIS) as the temperature increased from 50 to 90 °C, demonstrating that higher temperatures are desirable for high cell activities.
Similarly, in a recent study by Caprì et al., the authors studied an AEMWE containing Fumasep FAA3-50 membrane and ionomer, NiFe2O4 anode catalyst, and Pt/C cathode catalyst at different temperatures (30–60 °C) and saw a similar increase in performance with increasing temperature. At 2.2 V, they reported an increase of + 500 mA cm−2 with every 10 °C increase in temperature; their cell produced 1.35 A cm−2 of current at 30 °C and 3 A cm−2 at 60 °C, one of the highest reported current densities for AEMWE. The authors similarly attribute this performance gain to an increase in both ionic conductivity and electrochemical kinetics, supported by decreasing Ohmic and kinetic resistances observed in in situ EIS.13
These works have clearly shown that operating at higher temperatures has positive effects on cell activities. Increased temperatures, however, can also accelerate component degradation rates, leading to longevity concerns. Furthering understanding of how temperature impacts component-level and cell durability will be critical, including membrane/ionomer degradation pathways, catalyst–ionomer interactions, and transport layer and flow field oxidation.
Application of H2 backpressure is another cell operational variable of interest. H2 backpressure applied at the cathode can reduce H2 separation and compression costs by mitigating downstream processing costs.2,4 In fact, commercial PEMWEs often operate with 15–30 bar of backpressure at the cathode.154,155 Fewer studies have investigated the effects of H2 backpressure on performance in AEMWEs than in PEMWEs, likely due to difficulties in designing and obtaining the specialized cell hardware and test stand configurations required to perform this work.
Recent works studying H2 backpressure in AEMWE suggest a rising interest in this area. Ito et al. evaluated an AEMWE composed of a CuCoOx anode catalyst, Pt/C cathode catalyst, and A201 membrane (Tokuyama) at 1, 5, and 8.5 bar of cathode backpressure. They found that there were negligible increases in cell overpotential and high-frequency resistances with increasing cell pressure, and suggested that the pressure range tested was not sufficient to determine a trend for these parameters.156 More interestingly, they found that H2 gas humidity decreased with increasing pressure; specifically, relative humidity decreased by one order of magnitude from 1 to 8.5 bar.156 Low relative humidity can decrease the need to dry evolved H2 gas downstream, reducing post-processing and overall operating costs for H2 production from AEMWEs. Such decreased relative humidity in the product stream, however, may be indicative of membrane dehydration, and future works to investigate the implications of H2 backpressure on water management throughout the cell will be important to understand and mitigate water transport limitations to the cathode. Such water-management concerns are discussed further in Section 4.2.
Applying H2 backpressure also has important efficiency and safety considerations; recent reports from PEMWE have shown that increasing cathode backpressure can increase rates of H2 gas crossover from cathode to anode, leading to H2 loss and creating unstable H2/O2 mixtures and introducing safety concerns.152,157,158 Like for PEMWE, anion exchange membranes have some permeability to H2(g), and H2 crossover may also be a concern. Nafion, the standard membrane used in PEMWE, has been reported to have a H2 gas permeability of approximately 1.73 × 10−11 mol cm−1 s−1 bar−1 at 30 °C when fully hydrated.158 Comparatively, Ito et al. found that the A201 membrane (Tokuyama) had a H2(g) permeability of 5.6 × 10−12 mol cm−1 s−1 bar−1 at 50 °C.159 Other anion exchange membrane chemistries will have different permeabilities to H2, and these values should be studied and reported. Ito et al. further studied the possibility of H2 crossover at 1, 5, and 8.5 bar of backpressure, and concluded that H2(g) permeability of an AEM was 0.16× that of a PEMWE operating at the same conditions, indicating the strong possibility for H2 backpressure operation and the need to continue to study high-pressure AEMWE systems.156 Another important consideration is that H2 backpressure operation is only feasible when the cell is operated at cathode-dry conditions (i.e., no water or supporting electrolyte feed to the cathode), which can lead to problems related to water management in the membrane, which will be discussed in more detail in Section 4.2.
Cell pre-treatment and conditioning procedures are often employed to break-in electrolyzer cells and yield improved performance. Such protocols are standardized for PEMWE,47,48 but remain undetermined for AEMWE. This is, in part, due to a lack of knowledge about optimal operating procedures and practices, as well as the wide range of materials employed in the field; Section 4.3 discusses the need for benchmarking, baselining, and standardized protocols for AEMWE.
Several recent works have reported that conditioning the membrane with electrolyte, either ex situ or in situ, before cell operation is necessary to achieve high performance.133,146 For example, external membrane soaking before cell assembly is a common pretreatment procedure, with methods ranging from soaking in 0.5 to 3 M KOH for several hours up to several days. Oftentimes, such procedures are utilized to ion-exchange membranes and ionomers to OH− form, thereby improving ionic conductivity. These procedures also help to hydrate the membrane before operation. Other approaches utilize in situ membrane pretreatment procedures. For example, Cho et al. found that pre-feeding AEMWEs with 0.5 M KOH decreased Ohmic losses, likely by increasing ionic conductivity over time. They showed that pre-treating the cell with 0.5 M KOH before testing led to increased performance in the order of 0 h < 8 h < 24 h of pretreatment. They also suggested that flowing electrolyte through the cell was more effective than soaking the membrane in 1 M KOH for 24 h before assembly.146
Relatedly, differences in cell conditioning procedures may dramatically impact AEMWE performance. Conditioning with potentiostatic vs. galvanostatic control, holds vs. cycling protocols, and different timescales for these procedures undoubtedly impact cell activity and durability. Such variables need to be investigated moving forward, to aid both in advancing AEMWE performance and in understanding and developing preferred protocols that can be utilized across laboratories.
4.2 Electrolyte variables: including composition, concentration, and flow configuration
Electrolyte variables – including the choice and concentration of supporting electrolyte (or lack thereof) and the flow configuration to the cell – can further significantly impact cell performance by directly altering OH− concentrations (reactant for OER, product for HER) and ionic conductivities, with significant impacts on cell reaction kinetics and mass transport rates.Numerous studies have examined the effects of different electrolyte choices and concentrations, including the type of cations (Li+, Na+, K+) and anions (OH−, HCO3−, CO32−, PO43−) in concentrations ranging from pure water (no salt) to 5 M.82,110,146,159–161 Generally, studies agree that operation in supporting electrolyte yields higher performance and better cell stability than pure-water operation. This has been attributed to better ionic conductivity maintained throughout the cell,54,82,162 better stability of the anion conductive polymers used for cell membranes and ionomer,163 improved site accessibility,164 and near-surface pH effects that cause structural changes.82 Most commonly, supporting electrolytes containing OH− species provide higher cell reactivities than those with carbonates or phosphates, due to both higher ionic conductivities and reduced carbonation/phosphorylation of the membrane/ionomer. However, the exact role of a supporting electrolyte and how it can affect catalyst layer structure and overall cell activity and stability are still debated.
Near surface pH changes have been suggested to play a role in catalytic activities and stabilities. Lei et al. compared AEMWE performance in pure water vs. two different supporting electrolytes, 10 mM KOH and a phosphate buffer. These two supporting electrolytes had a similar pH of 12 but were expected to experience different near-surface pH swings during water splitting reactions. The authors showed that near-surface pH changes led to catalyst morphological changes for both pure water and 10 mM KOH operation; such changes are evident in SEM images of the anode catalysts before and after the reactions, as shown in Fig. 5a–d. They further demonstrated that using a phosphate buffer precluded this reconstruction, as shown in Fig. 5e and f. In the pure water and KOH-fed cases, the authors hypothesized that there was a loss in ionic contact related to this reconstruction (Fig. 5g and h). Such reconstruction led to cell deactivation in pure water operation, although this effect was less prominent in KOH-fed cells because the additional ionic conductivity provided by the supporting electrolyte was sufficient to overcome losses in the ionomer network.82
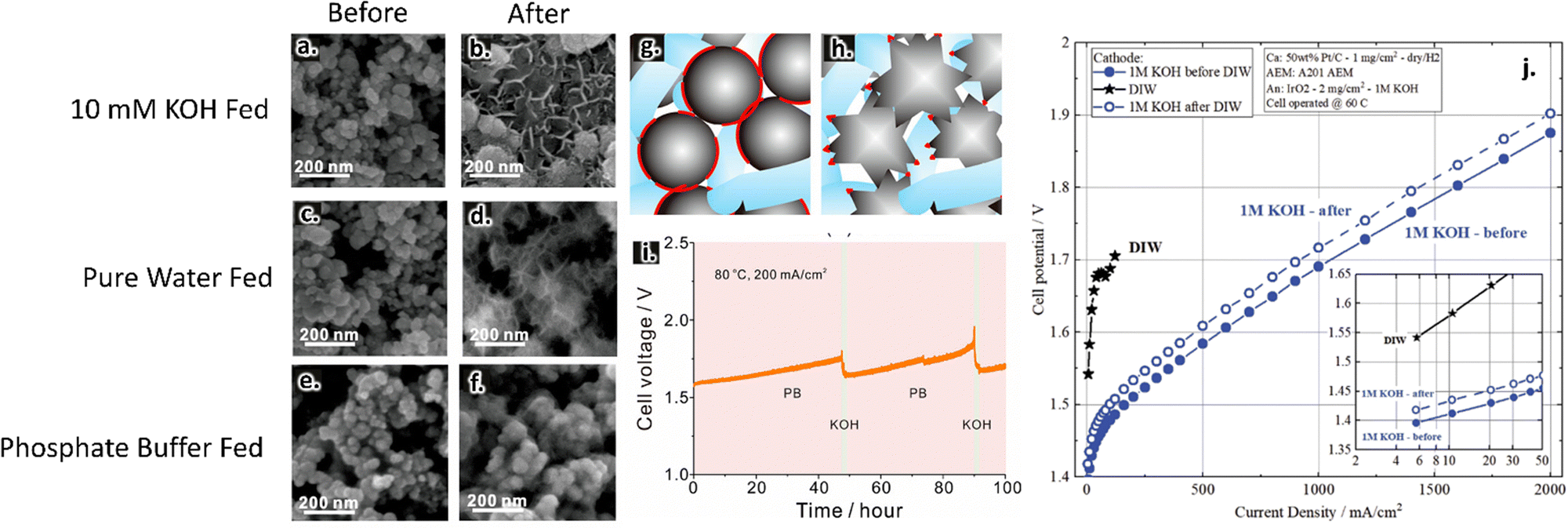 | ||
| Fig. 5 Anode catalyst layers before testing (a), (c) and (e) and after testing (b), (d) and (f) in 1 mM KOH (a) and (b), pure water (c) and (d), and phosphate buffer (e) and (f) at 200 mA cm−2. Images collected using SEM imaging. Proposed mechanism of catalyst deactivation in pure water feed; blue phase represents the ionomer, grey phase represents the catalyst, and the red phase indicates regions of catalyst–ionomer contact (g) and (h). Prolonged stability test in phosphate buffer where the solution was periodically refreshed with KOH (i). Reprinted with permission from Lei et al., ACS Sustainable Chemistry & Engineering, Copyright (2022) American Chemical Society.82 Effect of pre-feeding DIW on AEM activity (j). Reproduced from Kiessling et al., Journal of the Electrochemical Society, Copyright (2021).161 | ||
Zhang et al. similarly noted surface reconstruction effects for Ni, Fe, and Co-containing oxide catalysts at different pH conditions (i.e., pH = 7, 12, and 14). Specifically, the authors showed a decreased crystallinity for all samples in X-ray diffraction (XRD) measurements after testing (i.e., 6 h at 0.5 mA cm−2) in pure water conditions. They also suggested that high pH (i.e., 14) was necessary to enhance stability of these non-PGM catalysts, especially those containing Fe.165 Such results indicate the importance of understanding both near-surface pH effects and the role of the ionomer in AEMWEs with supporting electrolyte.
The identity of cations has also been reported to impact cell performance. For example, some works have suggested that cations can interact with surface-adsorbed *OH and affect OER activities.161,166 Kiessling et al. demonstrated that cations with larger charge densities have more significant stabilization of adsorbed *OH, resulting in cell activities in the order of K+ > Na+ > Li+, where K+ yielded the highest cell performance.161 Possible cation effects on HER have also been proposed. For example, Chen et al. proposed that, with increasing pH, positively charged cations co-adsorb with OH, shifting the H adsorption peak potential. They showed that this shift was also impacted by cation strength, with further delays in the H adsorption potential in the order of Li+ < K+ < Na+ < Cs+, where Cs+ resulted in the largest delay.167 These results further provide insights into the disputed non-Nernstian pH dependency of HER. Additional discussion on the role of cations in HER can be found in the review by Jia et al.168
Changes to electrolyte composition during testing have been shown to impact cell performance. For example, during operation in pure-water, the activity and durability can be affected if there is residual KOH (or other supporting electrolyte) in the reactor lines.146,160 Hassan et al. demonstrated this in their study of an AEMWE with an IrO2 anode catalyst, PtNi cathode catalyst, and XION membrane (from Xergy Inc.). They showed that when there was residual KOH in the lines, cell activity was initially higher but degraded faster.160 Similarly, Kiessling et al. reported significant activity loss after switching from 1 M KOH to pure water and back to 1 M KOH (Fig. 5j).161 Relatedly, Lei et al. showed that extended AEMWE operation using a phosphate buffer as the supporting electrolyte led to phosphorylation of the membrane/ionomer and subsequent performance losses. They demonstrated that these losses could be reversed with brief exposure to 0.01 M KOH feeds (Fig. 5i).82 These results suggest that electrolyte identity and flow protocols can significantly change performance results.
Electrolyte flow configuration can also play a role in overall cell performance. Cathode dry operation is desirable due to the ability to apply H2 backpressure and decrease H2 separation costs. Cathode dry operation has also been found to improve H2 gas diffusion through the transport layer, leading to more-uniform cathode utilization vs. a catholyte-fed case.146
Cathode dry operation, however, introduces water transport and membrane dehydration concerns, especially at high-current operation. This concern arises, in part, due to the differences in mass transport requirements for AEMWEs vs. PEMWEs. In PEMWE systems, water is both provided to and consumed at the anode. However, in AEMWEs, water is produced at the anode and consumed at the cathode, requiring water transport through the membrane to supply the reactant for HER; this is shown schematically in Fig. 6a.169,170 As a result, several studies have investigated the role of water content and water management in AEMWEs.142,143,169–171
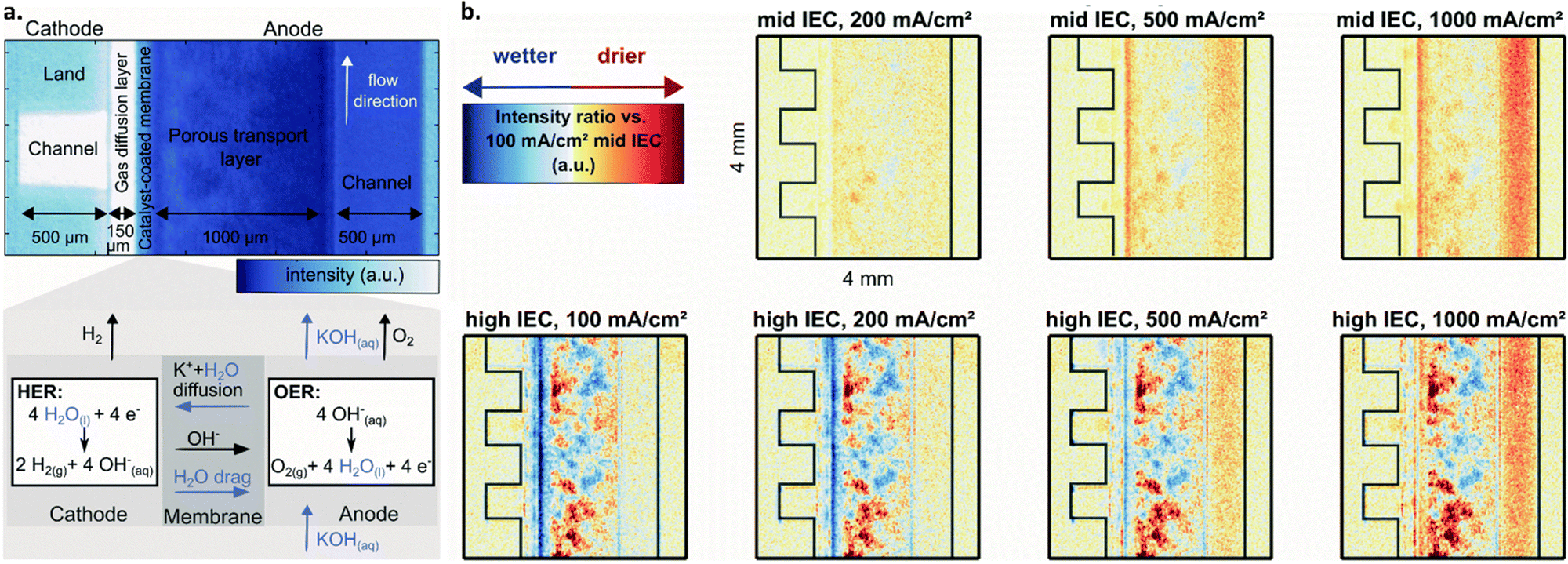 | ||
| Fig. 6 (a) Neutron image of an AEMWE single cell operated cathode-dry with, from left to right, cathode flow channel, gas diffusion layer, membrane, porous transport layer, anode flow channel. Darker blue color indicates higher water content. Below, a zoomed-in schematic of the membrane from above depicting the OER and HER at the anode and cathode as well as mechanisms of water and KOH transport within the cell. (b) Drying in an AEM cell with varied current densities and ion exchange capacities for the binder used in the cathode catalyst layer, shown as relative to the high IEC, 100 mA cm−2 case (bottom left). Blue corresponds to wetter conditions whereas red corresponds to dryer (see legend in top left). Top row is for cells with a mid-ion exchange capacity binder at three current densities, bottom for a high ion exchange capacity binder at three current densities. From left to right, cathode flow channel, gas diffusion layer, and catalyst coated membrane. Reproduced from Koch et al. with permission from The Royal Society of Chemistry.171 | ||
For example, Koch et al. utilized neutron imaging to study water content at different locations within an AEMWE with membranes having different IEC. The authors concluded that membrane dehydration does occur under cathode-dry operation, and that this effect becomes more pronounced at higher operating current densities; this is shown in Fig. 6b, where the membrane showed drier (redder) response from neutron imaging at 1000 mA cm−2vs. lower current densities. They further suggest that choosing a membrane/ionomer with a higher IEC can reduce cathode-drying complications by facilitating improved water retention at high current operation; Fig. 6b shows that wet (blue) regions are more evident for the high IEC vs. mid IEC case. Overall, they show that when the cathode is more dry (i.e., with cathode dry operation and with an anion exchange polymer with low IEC), cell performance is decreased.171
As discussed in Section 3.3, high IEC can also lead to increased ionomer swelling and cell deactivation, suggesting that there is a necessary balance. This phenomenon was investigated by Huang et al. in a two-part publication on ionomer optimization for water uptake in AEMWEs, covering both the anode and cathode. The authors suggest that by cross-linking the polymers in the ion exchange membrane and/or ionomer with high IEC, such water uptake issues can be mitigated.142,143
Pure-water and cathode dry operation, considered to be advantages of PEMWE technology, are likely also desirable in instances for commercialization of AEMWE technology. However, performance and durability are significantly improved in supporting electrolyte for traditional catalyst layers, and some works have suggested that the performance benefits of supporting electrolytes outweigh the arguments for pure-water operation.172,173 Furthermore, some authors have argued that dilute electrolytes may be more cost-effective than ultra-pure deionized water, which has significant production costs.164 Still, others have proposed routes for the implementation of water only operation.18 Therefore, future works are necessary to evaluate, simultaneously, (1) the necessity of operating under these proposed “ideal” conditions and (2) the types of materials that would enable pure-water, cathode-dry operation. In the meantime, operating in supporting electrolyte allows for significant developments in the field and improved understanding.
4.3 Baseline operation and standardized protocols
As discussed in the previous sections, AEMWE is at an earlier stage of development than PEMWE and LAWE technologies, and materials for catalysts, transport layers, and anion exchange polymers are still under active development. Furthermore, these materials behave differently from each other and have different optimal operating conditions. There are also well-known differences in achievable performance based on operating temperature, PGM vs. non-PGM catalysts, and pure water vs. supporting electrolyte operation, but little consensus has been reached in the field about what conditions should be considered standard. Consequently, there is a lack of baseline protocols and metrics for cell testing that would allow for direct comparison across research groups.As a start, the AEMWE field can borrow some best practices from PEMWE. There have been efforts within the EU49 and the U.S. DOE's H2NEW consortium48,50 to develop standard testing protocols with set MEA materials, loadings, and processing steps to serve as a benchmark for comparison. There have also been efforts to eliminate intra-group performance variation due to differences in test stations and protocols through round robin testing of identical MEAs.47 Importantly, these benchmarks do not need to have the highest performance or most advanced materials; instead, the emphasis is on using commercial materials that are widely available to all research groups. Initial efforts to standardize protocols for AEMWE have been conceptualized and started through the U.S. DOE's HydroGEN consortium174 and within the IEA,175 but, as of yet, decisions regarding appropriate baselining and benchmarking remain undecided.
Because there are several concurrent lines of research in AEMWE, there could be several benchmarks, for example covering PGM and non-PGM catalysts, pure water and supporting electrolyte operation, and different classes of membrane chemistries. Along with having defined materials, the benchmarks would also need a standard testing protocol and performance and/or durability metrics for comparison. Methods for taking polarization curves, impedance spectroscopy, and durability tests should be standardized. Standard metrics for comparing performance (e.g., voltage at 1 A cm−2, current density at 1.8 V or at a voltage efficiency of 68%176) and durability (e.g., degradation rate in mV h−1 at 1 A cm−2) are also necessary.
Based on the current state of the field, a reasonable benchmark cell, particularly to serve as a comparison for novel membranes and anode catalysts (i.e., PGM cathode, supporting electrolyte), would be a commercial NiFeOx anode catalyst and Pt/C cathode catalyst deposited onto Ni and C transport layers, respectively. While many forms of NiFeOx have been studied, one of the most common, commercially available forms is NiFe2O4. This material has been discussed in the literature as a possible non-PGM benchmark catalyst for alkaline OER.70 Although NiFe2O4 lacks desirable materials properties (its composition contains more Fe than has been reported preferable,52,177 it has relatively poor electronic conductivity, and available particle sizes are relatively large (∼30 nm)), it is commercially available in relatively large quantities. Furthermore, for catalysts with similar particle sizes, differences between three different commercial suppliers of NiFe2O4 have been shown to be low in RDE testing;70 NiFe2O4 may therefore be a reasonable candidate for a benchmark OER catalyst. The best choices for the anion exchange membrane and ionomer are yet to be known, which has implications for the choice of benchmarking temperature and supporting electrolyte. As a baseline to maximize performance, we recommend 1 M KOH and 80 °C, although these operating conditions may not be accessible to all membrane chemistries and can hasten degradation. While activity metrics at either a particular current density or voltage are relevant for demonstrating progress towards performance targets, such as the 3 A cm−2 at 1.8 V (2026 target for PEMWE from the U.S. DOE),8 a current density-based metric is preferred for comparison due to the widely varying high frequency resistances (HFR) in AEMWEs that significantly affect measured cell voltages. Specifically, we recommend reporting the HFR-free voltage (to account for differences in cell hardware and membrane resistances) at 1 A cm−2, an industrially relevant current density. For durability assessments, the U.S. DOE PEMWE and LAWE targets are stated in terms of a mV h−1 increase at a constant current density;8 we therefore recommend that durability measurements be conducted at a moderate, constant current density of 1 A cm−2.
In addition to establishing common performance metrics, it will be important to standardize best practices for materials characterization before and after testing. Component-level characterization of catalysts is common, but additional characterization of fully assembled cells is necessary to understand changes within the catalyst layer. The following section will discuss recent advances in in situ and ex situ techniques to understand such changes.
5. Ex situ and in situ diagnostic techniques to understand changes in the catalyst layer
Characterization of the used electrode and membrane materials is necessary to understand processes within the catalyst layer for AEMWEs. Changes to the chemical states of the catalysts, to the distribution of catalyst and ionomer in the catalyst layer, and to the morphology and porosity of the catalyst layer are all of interest to study. Relevant characterization techniques include microscopy, tomography, spectroscopy, X-ray techniques, inductively coupled plasma-mass spectrometry (ICP-MS), and gas chromatography-mass spectrometry (GC-MS) analysis, among others.Ex situ characterization, which often involves comparing materials before and after testing, provides valuable insights into changes that occurred during testing. It is also typically easier to implement than in situ/operando measurements since no specialized cell hardware is required. In RDE studies, it has become the standard to provide pre- and post-test characterizations of catalysts to show what changes have or have not occurred, such as the conversion of “X-ide” (i.e., sulfides, phosphides, etc.) OER catalysts to the (oxy)hydroxide form.72 There are many differences between the operating environments of RDE and MEAs, however, meaning that there is a need to also implement this type of ex situ, post-test characterization to study catalyst, ionomer, and membrane changes and/or degradation after AEMWE testing.38,41,54,82,178,179
Changes to catalyst layer morphology are most readily studied by ex situ microscopy techniques, including scanning electron microscopy (SEM) and transmission electron microscopy (TEM). In these studies, either top-down or cross-sectional images of the catalyst layer either on the membrane or transport layer are taken. For TEM imaging, the catalyst layer is embedded in an epoxy or resin and microtomed into a thin, transparent film. For example, cross-sectional scanning TEM has been used to understand changes to catalyst layer morphology and the membrane–catalyst interface after different accelerated stress tests in PEMWE.180 Several recent studies have employed SEM techniques to show changes to the catalyst layer structure in AEMWEs before and after testing.82,113,144,178,181 Pushkareva et al. used post-test, top-down SEM to understand the adhesion of the anode NiFe2O4 and cathode NiFeCo CCM catalyst layers on different membranes (i.e., Sustanion, Aemion, and Tokuyama A201 shown in Fig. 7a–c, respectively).181 Although the same Nafion binder was used for all of the MEAs in this study, catalyst layer homogeneity and bonding to the membrane were much better for Sustainion than for Aemion and A-201 membranes, in agreement with the performance and HFR trends. This is evident in the differences in lost catalyst observable in Fig. 7avs.Fig. 7b and c. Focusing on catalyst layer composition changes, Razmjooei et al. studied NiAl and NiAlMo anode catalyst layers on stainless steel anode transport layers using cross-section SEM and energy dispersive X-ray spectroscopy (EDX) elemental mapping.113 They observed increased porosity and increased O content after activation in KOH and durability testing, corresponding to Al and Mo leaching and conversion to oxidized Ni species. Simple optical images can also be used to show changes in color and macroscale morphology or coverage.98
 | ||
| Fig. 7 Top-down SEM images of NiFe2O4 anode catalyst layers on (a) Sustainion, (b) Aemion, and (c) Tokuyama A201 membranes at (top) 100× and (bottom) 400× magnification. Reprinted from Pushkareva et al., Comparative Study of Anion Exchange Membranes for Low-Cost Water Electrolysis, Int. J. Hydrog. Energy, 45(49), 26070–26079, Copyright (2020), with permission from Elsevier.179 (d) Schematic of PEMWE cell developed for in situ XAS measurements. Reprinted from Ampurdanés et al., Cobalt-Based Oxide Materials as Non-PGM Catalyst for HER in PEM Electrolysis and In situ XAS Characterization of Its Functional State, Catal. Today, 336, 161–168, Copyright (2019), with Permission from Elsevier.193 | ||
Structural changes to the catalyst and transport layers can be assessed using XRD measurements.38,43 For example, Chen et al. used post-test XRD to show that there were no bulk structural changes to the Ni transport layer, which they used as support for the argument that resistance changes were instead due to changes within the catalyst layer.98 It is important to note that surface structural changes or amorphization can be difficult to detect via bulk XRD, so complementary techniques are also needed.
X-ray photoelectron (XPS) and Raman spectroscopies are powerful tools to assess changes to catalyst and ionomer composition and oxidation state in the catalyst layer.82 Furthermore, ICP-MS and inductively coupled plasma-optical emission spectrometry (ICP-OES) can also be used to measure metal dissolution from the catalyst layer and transport layers after testing.82,90,182 Using a stainless steel felt as the anode catalyst, Sampathkumar et al. found that while there were minimal changes to the XRD pattern after cycling in 1 M KOH, XPS and Raman spectroscopy showed significant changes in surface composition and oxidation of the Ni, Fe, and Cr species.183 These surface changes, including leaching of Fe and the formation of an (oxy)hydroxide layer, resulted in a decrease in charge transfer resistance and an improvement in AEMWE performance. Using XPS, Campagna-Zignani et al. found that a NiMo cathode became less oxidized and its C overlayer was removed after testing, which may relate to the observed decrease in charge transfer resistance during the stability test.38 Furthermore, XPS studies by Krivina et al. on NiCoO2, NiFe2O4, and Ni0.5Co0.5Fe2O4 anode catalysts identified distinct changes in surface composition for the different structures after durability testing (20 h at 500 mA cm−2).54 These authors further utilized XPS to study the degradation behavior of the ionomer for different catalytic systems, revealing important details about catalyst–ionomer interactions and the implications on durability. This study is discussed in more detail in Section 6. These works demonstrate the importance of characterizing and considering changes to all components of the catalyst layer.
In situ (operando) characterization is commonly defined as measurement of materials under realistic operating conditions (during the reaction). Though logistically more challenging, in situ characterization can provide valuable insights into the nature of the catalyst layer and membrane in the high pH, high potential conditions of an AEMWE. As has been discussed in several recent reviews,184–186 these techniques have been used to study bulk and surface chemistries, as well as catalyst layer morphology. This is vital to understanding catalyst activity and degradation mechanisms involving catalysts, ionomers, and membranes.
In half-cell, three-electrode measurements, in situ X-ray absorption (XAS); XPS, Raman, and UV-visible spectroscopies; and XRD have been used to characterize compositional, phase, and structural changes of HER and OER catalysts.186–190 On-line ICP-MS has further been used to probe the pH- and potential-dependent dissolution of metals from catalysts. While significant insights have been gained from this work that have helped to shape catalyst development efforts, it is important to recognize that materials changes and rates of degradation can be very different in an MEA environment vs. in a liquid electrolyte. For example, an on-line ICP-MS study of a PEMFC gas diffusion electrode half-cell found an order of magnitude lower rate of Pt dissolution compared to an analogous study in a three-electrode flow cell.191 They attributed this change to differences in mass transport that shifted the equilibrium potential for dissolution and favored redeposition of dissolved species.
To specifically understand the interactions between the different components in the catalyst layer, in situ MEA characterization is needed. Peng et al. used operando X-ray computed tomography to understand catalyst layer utilization and mass transport in an ultra-low Ir loaded PEMWE using various Ti transport layers with uniform porosity.192 They found a significant inverse relationship between porosity and contact area, with an intermediate contact area percentage found to be optimal for catalyst utilization. Although mass transport resistance is minimized for a transport layer with the highest porosity, kinetic and ohmic overpotentials are minimized at intermediate porosities and contact areas. Ampurdanés et al. utilized a similar PEMWE cell with X-ray transparent Kapton windows in both the end plates and holes in the current collectors to conduct in situ Co K-edge and Ir L3-edge XAS measurements in transmission mode (Fig. 7d).193 Further development of these in situ cells, particularly for AEMWE devices, is needed.
Other characterization methods provide insights into system-level operation and the origin of performance losses. On-line GC-MS is often used for Faradaic efficiency and H2 crossover measurements. These measurements can be used to assess the effects of operating conditions on H2 crossover. In situ EIS is often used to study changes in ohmic, kinetic, catalyst layer, and mass transport resistances to better understand the origin of overpotentials within the MEA. The development of methods to quantify the electrochemical surface area (ECSA) of non-PGM oxide catalysts is also necessary. Currently, ECSA determination is largely limited to estimations of the double layer capacitance, either from cyclic voltammetry68,194 or EIS195,196 measurements. These methods have inherent errors and are reported to provide surface areas accurate to within an order of magnitude.68,194
Recently, there have been efforts to develop reference electrodes that can be integrated into the single cell to separate the contributions of the anode and the cathode.85,147 Xu et al. demonstrated the use of a Ag/AgCl reference electrode, connected to the cell via a hydrated strip of membrane, to measure polarization curves and impedance spectroscopy. They found that changes to the morphology of the anode transport layer led to decreased anode overpotential and charge transfer resistance with no impact on cathode performance, whereas an increase in cathode transport layer thickness affected both the anode and cathode.126 While there are challenges to successful implementation of reference electrodes, such as maintaining hydration and good connection with the MEA, reference electrodes will be a powerful tool for understanding the impacts of materials integration and operational choices on individual components of the MEA.
To understand and improve AEMWE performance and durability, it is necessary to understand the origin of losses, the condition of materials within the device, and degradation mechanisms. Ex situ characterization of cell components and the catalyst layer, such as through microscopy and spectroscopy, allows for post-mortem analysis of material changes and effects on interfaces and integration. In situ techniques have been employed extensively in half-cell, three-electrode measurements, but it should not be assumed that these insights will correlate directly with material behavior in single cells due to differences in the reaction environment and the increased importance of interactions between components in the single cell. Further efforts to design cells and techniques for in situ characterization in AEMWEs are needed, and ex situ materials characterization should be employed more frequently for improved understanding of the catalyst layer.
6. Catalyst layer stability and stability testing protocols
Currently, AEMWE cells have demonstrated lifetimes on the order of 10000 h, but this likely needs to increase by at least an order of magnitude to be competitive with other technologies.4
Individual, component-level stability of catalysts21,31,197 and membranes21,40,172,198 has been reviewed previously. For catalysts, it is generally understood that oxidation and dissolution are the biggest contributors to degradation. For membranes, swelling and oxidation of the backbone are key concerns. These effects can be compounded and made more complicated at the device level, where the integration of components and cell operating variables can have dramatic effects on stabilities. This section will discuss durability considerations for AEMWE operation, with a focus on degradation mechanisms that arise due to the integration of components and cell operational variables.
Membrane hydration is a concern for long-term AEMWE operation, especially in cathode dry operation (discussed in Section 4.2 based on the results of Koch et al.171). Wang et al. further assessed water transport within AEMWEs by evaluating the effect of changing catalyst layer properties on the mass-transport resistance of water in AEMWEs. In their first study, they modified catalyst distribution and thickness on a series of different transport layers, finding that denser catalyst layers promoted more efficient water transport.169 In a later study, these authors investigated the effects of membrane thickness and catalyst layer porosity to further elucidate the origins of concentration (i.e., transport) overpotentials and to understand water transport in AEMWEs. They concluded that water transport through the membrane is limiting. They further monitored the relative humidity of evolved H2, finding that when this value decreased, cell performance decreased concurrent with an increase in overall cell resistances.170 Long periods of rest (i.e., 12+ h) have also been shown to lead to membrane dehydration,199 which is important to consider especially for dynamic operation of AEMWEs.
Membranes, ionomers, and supporting electrolytes are also subjected to carbonation when the cell or electrolyte reservoirs are exposed to the atmosphere.198,200–202 Such carbonation can decrease the ionic conductivity of the ionomers, leading to decreased performance. Carbonation can also change the near-surface pH, affecting activities and stabilities. Parrondo et al. initially observed this degradation route for a water-fed AEMWE single cell with a polysulfone-based membrane; they found that short-term degradation was attributable to CO2 intrusion and subsequent carbonation.198 This degradation route has also been noted in anion exchange membrane fuel cells (AEMFCs)200,201 and CO2 electrolyzers,202 both of which often employ the same types of anion exchange membranes utilized in AEMWEs. For example, Zheng et al. concluded that CO2 intrusion in an AEMFC resulted in a Nernstian thermodynamic shift in anode potential caused by a pH change associated with increased carbonation as well as decreased kinetics resulting from a lack of OH− reactants.200 Such phenomena may translate to AEMWEs.
Other pH effects are critical for AEMWE stability. For example, Mayerhöfer et al. studied a CuCoOx catalyst with an Aemion membrane and ionomer from Ionomr in a single cell AEMWE in both pure water and 0.1 M KOH. They concluded that without supporting electrolyte, the near-surface pH was not basic enough to keep CuCoOx in a thermodynamically stable form and led to the dissolution of both Cu and Co; these materials are predisposed to dissolve at low pH conditions.17 They conclude that supporting electrolyte is therefore required to maintain the thermodynamic stability of non-PGM catalysts and that anion exchange polymers are insufficient to provide such a basic environment.148 Relatedly, Zhang et al. evaluated the pH-stability relationship for Ni, Fe, and Co-containing oxides and found that all materials tested had improved stability at high pH (i.e., pH = 14) vs. weakly alkaline (i.e., pH = 12) or pure-water (i.e., pH = 7) conditions. The stability of Fe-containing materials suffered the most with decreasing alkalinity (i.e., at pH 7 and 12 vs. 14) due to increased Fe dissolution measured with ICP-OES.165
Krivina et al. also observed Fe dissolution in pure-water operation. They found that, among a series of 5 non-PGM catalysts (Co3O4, NiO, NiCoO2, Ni0.5Co0.5Fe2O4, and NiFe2O4), a Ni-rich surface was formed after durability testing in pure water (20 h, 500 mA cm−2) for Co-containing catalysts, whereas for NiFe2O4 an Fe-rich surface was formed, as evidenced by XPS results (Fig. 8a). The authors attributed this change to preferential leaching of Fe and redeposition onto the catalyst surface. The authors then intentionally introduced Fe3+ species to the electrolyte, resulting in an increased degradation rate that they attributed to a disruption in the catalyst–ionomer network by the dissolved Fe species (Fig. 8b).54 Future works that investigate whether this effect persists in supporting electrolyte operation, as well as those focused on the development of methods to stabilize Fe in OER catalysts, are critical.
 | ||
| Fig. 8 (a) Metal ratios before and after testing in pure water for bimetallic oxide catalysts measured with XPS. (b) The role of solvated iron species in single-cell degradation, where Fe-spiked water showed increased oxidation of the ionomer vs. pure water evidenced with XPS (inserts). (c) Proposed mechanism of single-cell degradation for six oxide catalysts. Reprinted with permission from Krivina et al., Advanced Materials, Copyright (2022) John Wiley and Sons.54 | ||
Additionally, membrane and ionomer swelling at AEMWE operating conditions, especially in pure water feeds, can lead to cell degradation98,142,143 and catalyst detachment and delamination1,148 Chen et al. studied the effects of polymer swelling in the membrane vs. the ionomer in the catalyst layer separately, to probe if there was a difference in how performance was affected. This study used Ir as the anode catalyst, Pt/C as the cathode catalyst, and HMT-PMBI as the membrane and ionomer. In doing so, they demonstrated that their crosslinking strategy resulted in a 4× decrease in volumetric ionomer swelling. With different degrees of crosslinking in either the membrane or ionomer in the catalyst layer, the authors were able to selectively change the water uptake capacity and probe ionomer swelling for each location individually. They concluded that membrane swelling has a minimal impact on overall performance in pure-water feeds, but found that preventing ionomer swelling in the catalyst layer lead to a 4-fold increase in the lifetime of the cell.98 These results suggest that limiting ionomer swelling in the catalyst layer is essential to achieving high cell stabilities.
To isolate the electrochemical stability of ionomers (i.e., without interference from the catalyst) in different supporting electrolytes and oxidizing conditions, Krivina et al. used a quartz crystal microbalance to measure mass loss as a function of applied potential for films of ionomer. They found that the ionomers gained mass at open circuit voltage due to hydration, particularly in pH 10 and 14 electrolyte. Aemion fully delaminated at 700 mV overpotential in pH 10, whereas Sustainion and PiperION had 5–10% mass loss in pH 14 at the 700 mV overpotential.203 These results show that ionomer can be oxidized and degraded due to exposure to oxidizing potentials and alkaline electrolyte, even without the presence of a catalyst.
Catalyst–ionomer interactions can also lead to degradation of the ionomer and cell deactivation. This is especially critical in pure water feeds, where the ionomer is essential to provide ionic conductivity through the catalyst layer. Krivina et al. recently conducted a comprehensive assessment of degradation mechanisms for AEMWEs operated in pure water with different PGM (IrO2) and non-PGM (NiO, Co3O4, NiCoO2, Ni0.5Co0.5Fe2O4, and NiFe2O4) anode catalysts and with PiperION membranes and Versogen ionomers.54 Using post-test XPS (C, N, and F 1s) spectra, they found that IrO2 facilitated significant ionomer degradation, which they attributed to high electronic conductivity and more active sites for ionomer oxidation reactions. They found that non-PGM catalysts also facilitated ionomer degradation, but to a lesser extent due to the decreased electronic conductivity of these systems compared to IrO2. Fig. 8c shows the proposed effects of these losses on cell performance; IrO2 and Co3O4 are expected to have sufficient electronic conductivity to maintain accessibility to active sites that are co-located with remaining ionomer at the membrane interface. Conversely, NiCoO2, Ni0.5Co0.5Fe2O4, and NiFe2O4 are predicted to have insufficient electronic conductivity through the catalyst layer, creating a gap between regions accessible to electrons and to ions. Understanding and controlling these catalyst–ionomer interactions will be essential for improving durability in pure water operation.
AEMWE currently lacks standard operating and reporting protocols, including for durability testing. Many studies evaluate cells using potentiostatic or galvanostatic holds at suggested operating levels (e.g., around 2.0 V or 1 A cm−2).38,178,204 While these tests provide insights into degradation mechanisms that arise due to the harsh, oxidative operating conditions, the practical operation of AEMWE electrolyzers coupled with variable renewable energy sources will likely operate at on/off operation. To assess the effects of such operation on durability, other studies have investigated the use of cycling tests, where the voltage or current is cycled from high to low,40,179 and periodic voltage bias.205 Campagna-Zignani et al. evaluated cells with a 1 A cm−2 hold and with 0.2 to 1 A cm−2 cycles. They found that for the current hold case, the performance leveled off after initial deactivation. For the cycling test, they observed similar behavior, but the steady-state activity was less than that for the hold test. They attributed this to semi-reversible losses observed during the cycling tests.38 Further investigations into the effects of cycling on the chemistry of the catalysts (e.g., change to phase, crystal structure), the chemistry of the ionomer (e.g., if there are accelerated degradation/oxidation pathways) and to the catalyst layer structure are necessary to understand how these cells will behave in practice.
The timescales for comparing stability are also critical. Many cells show dramatic changes in the first 10s of hours of operation, and then level off to a steady state degradation rate. For example, Campagna-Zignani et al. tested a cell composed of NiFe anode, NiMo/KB cathode, and Fumatech FAA-3–50 membrane at 1 A cm−2 and observed initial rapid degradation for 100, which leveled off at 80% of the initial efficiency over the remaining 2000 h operation.38 Appropriate timescales for reporting and comparing stability must therefore be established. Further, the development and validation of accelerated stress tests that can be completed on shorter time scales, and which are employable by many researchers, are necessary.
7. Conclusions and future outlooks
Interest in AEMWE is rising rapidly as the need for lower-cost, green H2 production technologies compatible with intermittent variable renewable energy sources rise. Recently, component-level advances have dramatically enhanced AEMWE activities and stabilities, and cells with high operating current densities (e.g., >3 A cm−2) and up to 10000 h operation have been reported. While significant, these achievements fall short of commercialization goals, and a critical next step in AEMWE research will be to expand on these component-level advances and move towards a more-rigorous understanding of device integration, especially on understanding, designing, and developing the catalyst layer.
While AEMWE can adopt some catalyst-layer design strategies from the PEMWE and PEMFC spaces, fundamental differences in the relevant chemistries for water splitting reactions, ionomer and catalyst materials, and degradation mechanisms necessitate AEMWE-specific research efforts. In terms of materials, questions surrounding the relevance of the electronic conductivity of non-PGM oxide catalysts, which have much lower conductivities than the PGMs used in PEM systems, remain. Furthermore, while many PEMWE materials choices have been standardized, there is no such consensus in the AEMWE community regarding the preferred choices for catalysts, membranes and ionomers, transport layers, and/or electrolyte composition.
For AEMWE electrode design, decisions must be made about preferred deposition methods and configurations. Spray techniques lend themselves most readily to developed manufacturing techniques, such as roll-to-roll coating, used in the PEMFC space. Modification and optimization of ink formulations, rheological parameters, and spray temperature and times are avenues for further exploration and advancement in this space. In terms of configurations, there is not yet a consensus on whether CCM vs. CCS offers superior AEMWE performance. Here, the balance between interfacial contact resistances, mass transport effects, and catalyst layer stability must be studied and optimized.
Studies on transport layer design and integration have shown that C paper transport layers work well for cathode catalyst layers. On the anode side, transport layer properties are critical for electron and liquid/gas mass transport, and further developments focused on fiber dimensions, pore size, porosity, and type (e.g., foam vs. mesh vs. sinter) are necessary to improve performance. Oxidation and dissolution of transport layer materials may also be important considerations for cell durability. Transport layer design strategies that are often used in PEMWE, including the use of microporous layers and conductive, protective coatings, will likely be key areas of growth for AEMWE development.
The role of the ionomer in AEMWE remains ambiguous; in PEMWE, the ionomer serves as the sole proton conductor through the catalyst layer, and its integration is critical to achieve high catalyst utilization and performance. For AEMWE, operation in dilute supporting electrolyte offers significant performance gains over pure-water operation and may obviate the need for ionomer-facilitated ion conduction through the catalyst layer. Understanding the specific role of the ionomer – whether it serves as an ionic conductor, binder, and/or ink-design agent – is a critical next step in AEMWE development. Moreover, the chemical interactions between anion exchange ionomers, catalysts, and electrolyte need to be studied; such interactions have significant impacts on cell durability that are specific to AEMWEs.
Furthermore, there is a critical need to establish baseline testing procedures, benchmarks, and standardized performance metrics so that reasonable comparisons can be made between research groups. The optimization and standardization of cell compression, backpressure, temperature, pre-treatment procedures, and cell conditioning procedures are necessary to support the advancement of this technology. Also, diagnostic techniques, especially those that are in situ or operando, must be advanced alongside materials development and integration studies. Key parameters related to device integration, including how the catalyst layer structure, porosity, composition, and chemistries change during AEMWE operation, necessitate techniques that can probe these changes at high pH and high voltage conditions in real time.
AEMWE durability remains a significant challenge, as it involves both degradation mechanisms associated with individual components and the interactions between materials that can lead to additional degradation pathways. Furthermore, strategies to respond to degradation pathways that may emerge during different operation modes, including the impact of intermittent loads, high temperatures, and H2 backpressure, are needed. Additionally, water transport and hydration of the membrane and ionomer have been shown to play a role in cell longevity and should continue to be studied in the future. Interactions between the catalyst and the ionomer have been shown to vary between catalyst types, and future works that explore different degradation pathways for different catalyst–ionomer pairs are also critical. Cell losses have also been shown to dramatically improve with dilute supporting electrolyte, and therefore different stability targets should be considered for pure water- vs. supporting electrolyte-fed AEMWEs.
Research dedicated to understanding and designing catalyst layers for AEMWEs will be a critical next step in accelerating the commercialization of this technology. Such advances will, in turn, help enable the large-scale deployment of green H2 production by providing a cost-effective and performance-competitive complementary technology to commercial PEMWE and LAWE systems.
Author contributions
E. K. V.: conceptualization, writing – original draft. M. E. K.: writing – original draft, writing – review & editing. S. K. and S. M. A.: writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Noor Ul Hassan (NREL), Jesse Dugan (Mines), Manasi Vyas (Mines) and Kemakorn Ithisuphalap (Mines) for providing a careful proofreading of this manuscript. We acknowledge financial support from the HydroGEN 2.0 LTE consortium. SK acknowledges startup funding from Colorado School of Mines. This work was authored by the National Renewable Energy Laboratory, operated by Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains, and the publisher, by accepting the article for publication, acknowledges, that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.References
- H. N. Dinh, S. M. Alia, B. S. Pivovar, F. M. Toma, A. Z. Weber, D. Ding, G. Groenewold, A. McDaniel, A. Ambrosini, T. Ogitsu and B. Wood, HydroGEN Overview: A Consortium on Advanced Water Splitting Materials, Annual Merit Review, 2022 Search PubMed.
- S. Alia, D. Ding, A. McDaniel, F. M. Toma and H. N. Dinh, Chalkboard 2 - How to Make Clean Hydrogen, Electrochem. Soc. Interface, 2021, 30(4), 49, DOI:10.1149/2.F13214IF.
- B. Pivovar, N. Rustagi and S. Satyapal, Hydrogen at Scale (H2@Scale): Key to a Clean, Economic, and Sustainable Energy System, Electrochem. Soc. Interface, 2018, 27(1), 47, DOI:10.1149/2.F04181if.
- IRENA, Green Hydrogen Cost Reduction – Scaling up Electrolyzers to Meet the 1.5C Climate Goal, ISBN: 978-92-9260-295-6, International Renewable Energy Agency, 2020, https://www.irena.org/publications/2020/Dec/Green-hydrogen-cost-reduction, accessed 2022-08-18.
- U.S. Department of Energy, Technical Targets for Liquid Alkaline Electrolysis, https://www.energy.gov/eere/fuelcells/technical-targets-liquid-alkaline-electrolysis, accessed 2023-08-04.
- B. Pivovar, M. Ruth and R. Ahluwalia, H2NEW: Hydrogen (H2) from Next-Generation Electrolyzers of Water LTE Task 3c: System and Techno-Economic Analysis.
- Y. Wang, Y. Pang, H. Xu, A. Martinez and K. S. Chen, PEM Fuel Cell and Electrolysis Cell Technologies and Hydrogen Infrastructure Development – a Review, Energy Environ. Sci., 2022, 15(6), 2288–2328, 10.1039/D2EE00790H.
- U.S. Department of Energy Hydrogen and Fuel Cells Technology Office, Technical Targets for Proton Exchange Membrane Electrolysis, Energy.gov. https://www.energy.gov/eere/fuelcells/technical-targets-proton-exchange-membrane-electrolysis, accessed 2023-08-01.
- S. M. Fortier, N. T. Nassar, G. W. Lederer, J. Brainard, J. Gambogi and E. A. McCullough, Draft Critical Mineral List—Summary of Methodology and Background Information—U.S. Geological Survey Technical Input Document in Response to Secretarial Order No. 3359, 2018–1021, U.S. Geological Survey, 2018 DOI:10.3133/ofr20181021.
- European Commission, Report on Critical Raw Materials in the Circular Economy, 2018 DOI:10.2873/167813.
- R. Lohmann, I. T. Cousins, J. C. DeWitt, J. Glüge, G. Goldenman, D. Herzke, A. B. Lindstrom, M. F. Miller, C. A. Ng, S. Patton, M. Scheringer, X. Trier and Z. Wang, Are Fluoropolymers Really of Low Concern for Human and Environmental Health and Separate from Other PFAS?, Environ. Sci. Technol., 2020, 54(20), 12820–12828, DOI:10.1021/acs.est.0c03244.
- D. Li, E. J. Park, W. Zhu, Q. Shi, Y. Zhou, H. Tian, Y. Lin, A. Serov, B. Zulevi, E. D. Baca, C. Fujimoto, H. T. Chung and Y. S. Kim, Highly Quaternized Polystyrene Ionomers for High Performance Anion Exchange Membrane Water Electrolysers, Nat. Energy, 2020, 5(5), 378–385, DOI:10.1038/s41560-020-0577-x.
- A. Caprì, I. Gatto, C. Lo Vecchio, S. Trocino, A. Carbone and V. Baglio, Anion Exchange Membrane Water Electrolysis Based on Nickel Ferrite Catalysts, ChemElectroChem, 2023, 10(1), e202201056, DOI:10.1002/celc.202201056.
- S. Koch, L. Metzler, S. K. Kilian, P. A. Heizmann, F. Lombeck, M. Breitwieser and S. Vierrath, Toward Scalable Production: Catalyst-Coated Membranes (CCMs) for Anion-Exchange Membrane Water Electrolysis via Direct Bar Coating, Adv. Sustainable Syst., 2022, 2200332, DOI:10.1002/adsu.202200332.
- R. Abbasi, B. P. Setzler, S. Lin, J. Wang, Y. Zhao, H. Xu, B. Pivovar, B. Tian, X. Chen, G. Wu and Y. Yan, A Roadmap to Low-Cost Hydrogen with Hydroxide Exchange Membrane Electrolyzers, Adv. Mater., 2019, 31(31), 1805876, DOI:10.1002/adma.201805876.
- Ionomr, Hydrogen Production Cost by AEM Water Electrolysis, Document ID: FM-7024-B, 2020, https://ionomr.com/wp-content/uploads/2021/02/FM-7024-A-Hydrogen-Production-Cost-by-AEM-White-Paper-copy.pdf, accessed 2023-02-01.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, NACE, 1974, 13(4), 471 Search PubMed.
- G. Lindquist and S. Boettcher, Reports From The Frontier: Overcoming Limitations for Pure-Water Anion-Exchange-Membrane Electrolysis, Electrochem. Soc. Interface, 2023, 32(2), 32, DOI:10.1149/2.F05232IF.
- C. Santoro, A. Lavacchi, P. Mustarelli, V. Di Noto, L. Elbaz, D. R. Dekel and F. Jaouen, What Is Next in Anion-Exchange Membrane Water Electrolyzers? Bottlenecks, Benefits, and Future, ChemSusChem, 2022, 15(8), e202200027, DOI:10.1002/cssc.202200027.
- S. A. Lee, J. Kim, K. C. Kwon, S. H. Park and H. W. Jang, Anion Exchange Membrane Water Electrolysis for Sustainable Large-Scale Hydrogen Production, Carbon Neutralization, 2022, 1(1), 26–48, DOI:10.1002/cnl2.9.
- N. Du, C. Roy, R. Peach, M. Turnbull, S. Thiele and C. Bock, Anion-Exchange Membrane Water Electrolyzers, Chem. Rev., 2022, 122(13), 11830–11895, DOI:10.1021/acs.chemrev.1c00854.
- E. López-Fernández, C. G. Sacedón, J. Gil-Rostra, F. Yubero, A. R. González-Elipe and A. de Lucas-Consuegra, Recent Advances in Alkaline Exchange Membrane Water Electrolysis and Electrode Manufacturing, Molecules, 2021, 26(21), 6326, DOI:10.3390/molecules26216326.
- C. Li and J.-B. Baek, The Promise of Hydrogen Production from Alkaline Anion Exchange Membrane Electrolyzers, Nano Energy, 2021, 87, 106162, DOI:10.1016/j.nanoen.2021.106162.
- N. Chen and Y. M. Lee, Anion-Conducting Polyelectrolytes for Energy Devices, Trends Chem., 2022, 4(3), 236–249, DOI:10.1016/j.trechm.2021.12.009.
- W. You, K. J. T. Noonan and G. W. Coates, Alkaline-Stable Anion Exchange Membranes: A Review of Synthetic Approaches, Prog. Polym. Sci., 2020, 100, 101177, DOI:10.1016/j.progpolymsci.2019.101177.
- D. Henkensmeier, M. Najibah, C. Harms, J. Žitka, J. Hnát and K. Bouzek, Overview: State-of-the Art Commercial Membranes for Anion Exchange Membrane Water Electrolysis, J. Electrochem. Energy Convers. Storage, 2020, 18(2), 024001, DOI:10.1115/1.4047963.
- E. J. Park, C. G. Arges, H. Xu and Y. S. Kim, Membrane Strategies for Water Electrolysis, ACS Energy Lett., 2022, 7(10), 3447–3457, DOI:10.1021/acsenergylett.2c01609.
- P. Mardle, B. Chen and S. Holdcroft, Opportunities of Ionomer Development for Anion-Exchange Membrane Water Electrolysis, ACS Energy Lett., 2023, 3330–3342, DOI:10.1021/acsenergylett.3c01040.
- J. Xue, J. Zhang, X. Liu, T. Huang, H. Jiang, Y. Yin, Y. Qin and M. D. Guiver, Toward Alkaline-Stable Anion Exchange Membranes in Fuel Cells: Cycloaliphatic Quaternary Ammonium-Based Anion Conductors, Electrochem. Energy Rev., 2022, 5(2), 348–400, DOI:10.1007/s41918-021-00105-7.
- K. Chand and O. Paladino, Recent Developments of Membranes and Electrocatalysts for the Hydrogen Production by Anion Exchange Membrane Water Electrolysers: A Review, Arabian J. Chem., 2023, 16(2), 104451, DOI:10.1016/j.arabjc.2022.104451.
- F. Zeng, C. Mebrahtu, L. Liao, A. K. Beine and R. Palkovits, Stability and Deactivation of OER Electrocatalysts: A Review, J. Energy Chem., 2022, 69, 301–329, DOI:10.1016/j.jechem.2022.01.025.
- Z. Qin, W. Liu, W. Que, J. Feng, W. Shi, F. Wu and X. Cao, Non-Noble-Metal Electrocatalysts for Oxygen Evolution Reaction toward Seawater Splitting: A Review, ChemPhysMater, 2022, 2(3), 185–196, DOI:10.1016/j.chphma.2022.11.001.
- M. Plevová, J. Hnát and K. Bouzek, Electrocatalysts for the Oxygen Evolution Reaction in Alkaline and Neutral Media. A Comparative Review, J. Power Sources, 2021, 507, 230072, DOI:10.1016/j.jpowsour.2021.230072.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Electrocatalysis for the Oxygen Evolution Reaction: Recent Development and Future Perspectives, Chem. Soc. Rev., 2017, 46(2), 337–365, 10.1039/C6CS00328A.
- B. Yang and Z. Cunman, Progress in Constructing High-Performance Anion Exchange Membrane: Molecular Design, Microphase Controllability and In-Device Property, Chem. Eng. J., 2023, 457, 141094, DOI:10.1016/j.cej.2022.141094.
- Z. Zakaria and S. K. Kamarudin, A Review of Alkaline Solid Polymer Membrane in the Application of AEM Electrolyzer: Materials and Characterization, Int. J. Energy Res., 2021, 45(13), 18337–18354, DOI:10.1002/er.6983.
- M. Mandal, Recent Advancement on Anion Exchange Membranes for Fuel Cell and Water Electrolysis, ChemElectroChem, 2021, 8(1), 36–45, DOI:10.1002/celc.202001329.
- S. Campagna Zignani, M. L. Faro, A. Carbone, C. Italiano, S. Trocino, G. Monforte and A. S. Aricò, Performance and Stability of a Critical Raw Materials-Free Anion Exchange Membrane Electrolysis Cell, Electrochim. Acta, 2022, 413, 140078, DOI:10.1016/j.electacta.2022.140078.
- B. Motealleh, Z. Liu, R. I. Masel, J. P. Sculley, Z. Richard Ni and L. Meroueh, Next-Generation Anion Exchange Membrane Water Electrolyzers Operating for Commercially Relevant Lifetimes, Int. J. Hydrogen Energy, 2021, 46(5), 3379–3386, DOI:10.1016/j.ijhydene.2020.10.244.
- A. Carbone, S. C. Zignani, I. Gatto, S. Trocino and A. S. Aricò, Assessment of the FAA3-50 Polymer Electrolyte in Combination with a NiMn2O4 Anode Catalyst for Anion Exchange Membrane Water Electrolysis, Int. J. Hydrogen Energy, 2020, 45(16), 9285–9292, DOI:10.1016/j.ijhydene.2020.01.150.
- Y. S. Park, J. Yang, J. Lee, M. J. Jang, J. Jeong, W.-S. Choi, Y. Kim, Y. Yin, M. H. Seo, Z. Chen and S. M. Choi, Superior Performance of Anion Exchange Membrane Water Electrolyzer: Ensemble of Producing Oxygen Vacancies and Controlling Mass Transfer Resistance, Appl. Catal., B, 2020, 278, 119276, DOI:10.1016/j.apcatb.2020.119276.
- Z. Liu, S. D. Sajjad, Y. Gao, H. Yang, J. J. Kaczur and R. I. Masel, The Effect of Membrane on an Alkaline Water Electrolyzer, Int. J. Hydrogen Energy, 2017, 42(50), 29661–29665, DOI:10.1016/j.ijhydene.2017.10.050.
- F. Razmjooei, T. Morawietz, E. Taghizadeh, E. Hadjixenophontos, L. Mues, M. Gerle, B. D. Wood, C. Harms, A. S. Gago, S. A. Ansar and K. A. Friedrich, Increasing the Performance of an Anion-Exchange Membrane Electrolyzer Operating in Pure Water with a Nickel-Based Microporous Layer, Joule, 2021, 5(7), 1776–1799, DOI:10.1016/j.joule.2021.05.006.
- B. Huang, X. Wang, W. Li, W. Tian, L. Luo, X. Sun, G. Wang, L. Zhuang and L. Xiao, Accelerating Gas Escape in Anion Exchange Membrane Water Electrolysis by Gas Diffusion Layers with Hierarchical Grid Gradients, Angew. Chem., Int. Ed., 2023, 62, e202304230, DOI:10.1002/anie.202304230.
- W. Sheng, H. A. Gasteiger and Y. Shao-Horn, Hydrogen Oxidation and Evolution Reaction Kinetics on Platinum: Acid vs Alkaline Electrolytes, J. Electrochem. Soc., 2010, 157(11), B1529, DOI:10.1149/1.3483106.
- S. Ernst and C. H. Hamann, The PH-Dependence of the Hydrogen Exchange Current Density at Smooth Platinum in Alkaline Solution (KOH), J. Electroanal. Chem. Interfacial Electrochem., 1975, 60(1), 97–100, DOI:10.1016/S0022-0728(75)80206-3.
- G. Bender, M. Carmo, T. Smolinka, A. Gago, N. Danilovic, M. Mueller, F. Ganci, A. Fallisch, P. Lettenmeier, K. A. Friedrich, K. Ayers, B. Pivovar, J. Mergel and D. Stolten, Initial Approaches in Benchmarking and Round Robin Testing for Proton Exchange Membrane Water Electrolyzers, Int. J. Hydrogen Energy, 2019, 44(18), 9174–9187, DOI:10.1016/j.ijhydene.2019.02.074.
- N. Danilovic, G. Bender and A. Weber, H2NEW LTE: Performance and Benchmarking, 2021 Search PubMed.
- Joint Research Centre (European Commission), G. Tsotridis and A. Pilenga, EU Harmonized Protocols for Testing of Low Temperature Water Electrolysis, Publications Office of the European Union, LU, 2021 Search PubMed.
- J. A. Wrubel, C. Milleville, E. Klein, J. Zack, A. M. Park and G. Bender, Estimating the Energy Requirement for Hydrogen Production in Proton Exchange Membrane Electrolysis Cells Using Rapid Operando Hydrogen Crossover Analysis, Int. J. Hydrogen Energy, 2022, 47(66), 28244–28253, DOI:10.1016/j.ijhydene.2022.06.155.
- Y. Fu, M. Hou, X. Yan, J. Hou, X. Luo, Z. Shao and B. Yi, Research Progress of Aluminium Alloy Endplates for PEMFCs, J. Power Sources, 2007, 166(2), 435–440, DOI:10.1016/j.jpowsour.2007.01.018.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, Nickel–Iron Oxyhydroxide Oxygen-Evolution Electrocatalysts: The Role of Intentional and Incidental Iron Incorporation, J. Am. Chem. Soc., 2014, 136(18), 6744–6753, DOI:10.1021/ja502379c.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, Cobalt–Iron (Oxy)Hydroxide Oxygen Evolution Electrocatalysts: The Role of Structure and Composition on Activity, Stability, and Mechanism, J. Am. Chem. Soc., 2015, 137(10), 3638–3648, DOI:10.1021/jacs.5b00281.
- R. A. Krivina, G. A. Lindquist, S. R. Beaudoin, T. N. Stovall, W. L. Thompson, L. P. Twight, D. Marsh, J. Grzyb, K. Fabrizio, J. E. Hutchison and S. W. Boettcher, Anode Catalysts in Anion-Exchange-Membrane Electrolysis without Supporting Electrolyte: Conductivity, Dynamics, and Ionomer Degradation, Adv. Mater., 2022, 2203033, DOI:10.1002/adma.202203033.
- K. Zhang, X. Liang, L. Wang, K. Sun, Y. Wang, Z. Xie, Q. Wu, X. Bai, M. S. Hamdy, H. Chen and X. Zou, Status and Perspectives of Key Materials for PEM Electrolyzer, Nano Res. Energy, 2022, 1(3), e9120032, DOI:10.26599/NRE.2022.9120032.
- S.-H. Wang, J. Peng and W.-B. Lui, Surface Modification and Development of Titanium Bipolar Plates for PEM Fuel Cells, J. Power Sources, 2006, 160(1), 485–489, DOI:10.1016/j.jpowsour.2006.01.020.
- B. Chen, A. L. G. Biancolli, C. L. Radford and S. Holdcroft, Stainless Steel Felt as a Combined OER Electrocatalyst/Porous Transport Layer for Investigating Anion-Exchange Membranes in Water Electrolysis, ACS Energy Lett., 2023, 8(6), 2661–2667, DOI:10.1021/acsenergylett.3c00878.
- N. U. Hassan, E. Motyka, J. Kweder, P. Ganesan, B. Brechin, B. Zulevi, H. R. Colón-Mercado, P. A. Kohl and W. E. Mustain, Effect of Porous Transport Layer Properties on the Anode Electrode in Anion Exchange Membrane Electrolyzers, J. Power Sources, 2023, 555, 232371, DOI:10.1016/j.jpowsour.2022.232371.
- I. S. Filimonenkov, C. Bouillet, G. Kéranguéven, P. A. Simonov, G. A. Tsirlina and E. R. Savinova, Carbon Materials as Additives to the OER Catalysts: RRDE Study of Carbon Corrosion at High Anodic Potentials, Electrochim. Acta, 2019, 321, 134657, DOI:10.1016/j.electacta.2019.134657.
- S. G. Ji, H. Kim, W. H. Lee, H.-S. Oh and C. H. Choi, Real-Time Monitoring of Electrochemical Carbon Corrosion in Alkaline Media, J. Mater. Chem. A, 2021, 9(35), 19834–19839, 10.1039/D1TA01748A.
- C. Rakousky, U. Reimer, K. Wippermann, M. Carmo, W. Lueke and D. Stolten, An Analysis of Degradation Phenomena in Polymer Electrolyte Membrane Water Electrolysis, J. Power Sources, 2016, 326, 120–128, DOI:10.1016/j.jpowsour.2016.06.082.
- J. Polonský, R. Kodým, P. Vágner, M. Paidar, B. Bensmann and K. Bouzek, Anodic Microporous Layer for Polymer Electrolyte Membrane Water Electrolysers, J. Appl. Electrochem., 2017, 47(10), 1137–1146, DOI:10.1007/s10800-017-1110-1.
- T. Schuler, J. M. Ciccone, B. Krentscher, F. Marone, C. Peter, T. J. Schmidt and F. N. Büchi, Hierarchically Structured Porous Transport Layers for Polymer Electrolyte Water Electrolysis, Adv. Energy Mater., 2020, 10(2), 1903216, DOI:10.1002/aenm.201903216.
- C. Arthurs and A. Kusoglu, Compressive Creep of Polymer Electrolyte Membranes: A Case Study for Electrolyzers, ACS Appl. Energy Mater., 2021, 4(4), 3249–3254, DOI:10.1021/acsaem.0c03024.
- J. M. Mora, M. Sarker, Z. Najafianashrafi, Md. A. Rahman, A. C. Yang-Neyerlin, B. Pivovar and P.-Y. A. Chuang, Analytical-Based Simulation Approach for an Anion Exchange Membrane Fuel Cell, Energy Convers. Manage., 2022, 273, 116382, DOI:10.1016/j.enconman.2022.116382.
- S. M. Alia, B. Rasimick, C. Ngo, K. C. Neyerlin, S. S. Kocha, S. Pylypenko, H. Xu and B. S. Pivovar, Activity and Durability of Iridium Nanoparticles in the Oxygen Evolution Reaction, J. Electrochem. Soc., 2016, 163(11), F3105, DOI:10.1149/2.0151611jes.
- O. Kasian, J.-P. Grote, S. Geiger, S. Cherevko and K. J. J. Mayrhofer, The Common Intermediates of Oxygen Evolution and Dissolution Reactions during Water Electrolysis on Iridium, Angew. Chem., Int. Ed., 2018, 57(9), 2488–2491, DOI:10.1002/anie.201709652.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, Benchmarking Hydrogen Evolving Reaction and Oxygen Evolving Reaction Electrocatalysts for Solar Water Splitting Devices, J. Am. Chem. Soc., 2015, 137(13), 4347–4357, DOI:10.1021/ja510442p.
- G. C. Anderson, B. S. Pivovar and S. M. Alia, Establishing Performance Baselines for the Oxygen Evolution Reaction in Alkaline Electrolytes, J. Electrochem. Soc., 2020, 167(4), 044503, DOI:10.1149/1945-7111/ab7090.
- E. Volk, S. Kwon and S. M. Alia, Catalytic Activity and Stability of Non-Platinum Group Metal Oxides for the Oxygen Evolution Reaction in Anion Exchange Membrane Electrolyzers, J. Electrochem. Soc., 2023, 170(6), 064506, DOI:10.1149/1945-7111/acd605.
- H. Osgood, S. V. Devaguptapu, H. Xu, J. Cho and G. Wu, Transition Metal (Fe, Co, Ni, and Mn) Oxides for Oxygen Reduction and Evolution Bifunctional Catalysts in Alkaline Media, Nano Today, 2016, 11(5), 601–625, DOI:10.1016/j.nantod.2016.09.001.
- B. R. Wygant, K. Kawashima and C. B. Mullins, Catalyst or Precatalyst? The Effect of Oxidation on Transition Metal Carbide, Pnictide, and Chalcogenide Oxygen Evolution Catalysts, ACS Energy Lett., 2018, 3(12), 2956–2966, DOI:10.1021/acsenergylett.8b01774.
- P. Acharya, R. H. Manso, A. S. Hoffman, S. I. P. Bakovic, L. Kékedy-Nagy, S. R. Bare, J. Chen and L. F. Greenlee, Fe Coordination Environment, Fe-Incorporated Ni(OH)2 Phase, and Metallic Core Are Key Structural Components to Active and Stable Nanoparticle Catalysts for the Oxygen Evolution Reaction, ACS Catal., 2022, 12(3), 1992–2008, DOI:10.1021/acscatal.1c04881.
- M. Etesami, A. A. Mohamad, M. T. Nguyen, T. Yonezawa, R. Pornprasertsuk, A. Somwangthanaroj and S. Kheawhom, Benchmarking Superfast Electrodeposited Bimetallic (Ni, Fe, Co, and Cu) Hydroxides for Oxygen Evolution Reaction, J. Alloys Compd., 2021, 889, 161738, DOI:10.1016/j.jallcom.2021.161738.
- G. Borisov, V. Bachvarov, H. Penchev, R. Rashkov and E. Slavcheva, Multi-Metallic Electrodeposited Catalysts Applicable for Oxygen Evolution Reaction in AEM Water Electrolysis, Mater. Lett., 2021, 286, 129248, DOI:10.1016/j.matlet.2020.129248.
- Z. Cai, X. Bu, P. Wang, J. C. Ho, J. Yang and X. Wang, Recent Advances in Layered Double Hydroxide Electrocatalysts for the Oxygen Evolution Reaction, J. Mater. Chem. A, 2019, 7(10), 5069–5089, 10.1039/C8TA11273H.
- Y. Yuan, Y. Guo, W. Wu, Z. Mao, H. Xu and Y. Ma, Kinetics of Active Oxide Species Derived from a Metallic Nickel Surface for Efficient Electrocatalytic Water Oxidation, ACS Energy Lett., 2022, 7(10), 3276–3285, DOI:10.1021/acsenergylett.2c01493.
- W. Moschkowitsch, N. Zion, H. C. Honig, N. Levy, D. A. Cullen and L. Elbaz, Mixed-Metal Nickel–Iron Oxide Aerogels for Oxygen Evolution Reaction, ACS Catal., 2022, 12(19), 12162–12169, DOI:10.1021/acscatal.2c03351.
- E. Cossar, A. Oyarce Barnett, F. Seland and E. A. Baranova, The Performance of Nickel and Nickel-Iron Catalysts Evaluated As Anodes in Anion Exchange Membrane Water Electrolysis, Catalysts, 2019, 9(10), 814, DOI:10.3390/catal9100814.
- M. Zhiani, F. Jalili and S. Kamali, In Situ Cathode Polarization Measurement in Alkaline Anion Exchange Membrane Water Electrolyzer Equipped with a PdNiFeCo/C-Ceria Hydrogen Evolution Electrocatalyst, Int. J. Hydrogen Energy, 2017, 42(43), 26563–26574, DOI:10.1016/j.ijhydene.2017.09.038.
- P. Fortin, T. Khoza, X. Cao, S. Y. Martinsen, A. Oyarce Barnett and S. Holdcroft, High-Performance Alkaline Water Electrolysis Using AemionTM Anion Exchange Membranes, J. Power Sources, 2020, 451, 227814, DOI:10.1016/j.jpowsour.2020.227814.
- C. Lei, K. Yang, G. Wang, G. Wang, J. Lu, L. Xiao and L. Zhuang, Impact of Catalyst Reconstruction on the Durability of Anion Exchange Membrane Water Electrolysis, ACS Sustainable Chem. Eng., 2022, 10(50), 16725–16733, DOI:10.1021/acssuschemeng.2c04855.
- L. Osmieri, Y. He, H. T. Chung, G. McCool, B. Zulevi, D. A. Cullen and P. Zelenay, La–Sr–Co Oxide Catalysts for Oxygen Evolution Reaction in Anion Exchange Membrane Water Electrolyzer: The Role of Electrode Fabrication on Performance and Durability, J. Power Sources, 2023, 556, 232484, DOI:10.1016/j.jpowsour.2022.232484.
- S. S. Jeon, J. Lim, P. W. Kang, J. W. Lee, G. Kang and H. Lee, Design Principles of NiFe-Layered Double Hydroxide Anode Catalysts for Anion Exchange Membrane Water Electrolyzers, ACS Appl. Mater. Interfaces, 2021, 13(31), 37179–37186, DOI:10.1021/acsami.1c09606.
- A. W. Tricker, J. K. Lee, J. R. Shin, N. Danilovic, A. Z. Weber and X. Peng, Design and Operating Principles for High-Performing Anion Exchange Membrane Water Electrolyzers, J. Power Sources, 2023, 567, 232967, DOI:10.1016/j.jpowsour.2023.232967.
- A. Y. Faid, A. O. Barnett, F. Seland and S. Sunde, NiCu Mixed Metal Oxide Catalyst for Alkaline Hydrogen Evolution in Anion Exchange Membrane Water Electrolysis, Electrochim. Acta, 2021, 371, 137837, DOI:10.1016/j.electacta.2021.137837.
- C. C. Pavel, F. Cecconi, C. Emiliani, S. Santiccioli, A. Scaffidi, S. Catanorchi and M. Comotti, Highly Efficient Platinum Group Metal Free Based Membrane-Electrode Assembly for Anion Exchange Membrane Water Electrolysis, Angew. Chem., Int. Ed., 2014, 53(5), 1378–1381, DOI:10.1002/anie.201308099.
- L. Wan, Z. Xu, Q. Xu, P. Wang and B. Wang, Overall Design of Novel 3D-Ordered MEA with Drastically Enhanced Mass Transport for Alkaline Electrolyzers, Energy Environ. Sci., 2022, 15(5), 1882–1892, 10.1039/D2EE00273F.
- L. Wan, Z. Xu, P. Wang, P.-F. Liu, Q. Xu and B. Wang, Dual Regulation Both Intrinsic Activity and Mass Transport for Self-Supported Electrodes Using in Anion Exchange Membrane Water Electrolysis, Chem. Eng. J., 2022, 431, 133942, DOI:10.1016/j.cej.2021.133942.
- P. Chen and X. Hu, High-Efficiency Anion Exchange Membrane Water Electrolysis Employing Non-Noble Metal Catalysts, Adv. Energy Mater., 2020, 10(39), 2002285, DOI:10.1002/aenm.202002285.
- L. Wang, T. Weissbach, R. Reissner, A. Ansar, A. S. Gago, S. Holdcroft and K. A. Friedrich, High Performance Anion Exchange Membrane Electrolysis Using Plasma-Sprayed, Non-Precious-Metal Electrodes, ACS Appl. Energy Mater., 2019, 2(11), 7903–7912, DOI:10.1021/acsaem.9b01392.
- P. Thangavel, M. Ha, S. Kumaraguru, A. Meena, A. N. Singh, A. M. Harzandi and K. S. Kim, Graphene-Nanoplatelets-Supported NiFe-MOF: High-Efficiency and Ultra-Stable Oxygen Electrodes for Sustained Alkaline Anion Exchange Membrane Water Electrolysis, Energy Environ. Sci., 2020, 13(10), 3447–3458, 10.1039/D0EE00877J.
- M. Ďurovič, J. Hnát and K. Bouzek, Electrocatalysts for the Hydrogen Evolution Reaction in Alkaline and Neutral Media. A Comparative Review, J. Power Sources, 2021, 493, 229708, DOI:10.1016/j.jpowsour.2021.229708.
- W. Moschkowitsch, O. Lori and L. Elbaz, Recent Progress and Viability of PGM-Free Catalysts for Hydrogen Evolution Reaction and Hydrogen Oxidation Reaction, ACS Catal., 2022, 12(2), 1082–1089, DOI:10.1021/acscatal.1c04948.
- S. Wang, A. Lu and C.-J. Zhong, Hydrogen Production from Water Electrolysis: Role of Catalysts, Nano Convergence, 2021, 8(1), 4, DOI:10.1186/s40580-021-00254-x.
- L. Pan, S. Ott, F. Dionigi and P. Strasser, Current Challenges Related to the Deployment of Shape-Controlled Pt Alloy Oxygen Reduction Reaction Nanocatalysts into Low Pt-Loaded Cathode Layers of Proton Exchange Membrane Fuel Cells, Curr. Opin. Electrochem., 2019, 18, 61–71, DOI:10.1016/j.coelec.2019.10.011.
- S. Kiemel, T. Smolinka, F. Lehner, J. Full, A. Sauer and R. Miehe, Critical Materials for Water Electrolysers at the Example of the Energy Transition in Germany, Int. J. Energy Res., 2021, 45(7), 9914–9935, DOI:10.1002/er.6487.
- B. Chen, P. Mardle and S. Holdcroft, Probing the Effect of Ionomer Swelling on the Stability of Anion Exchange Membrane Water Electrolyzers, J. Power Sources, 2022, 550, 232134, DOI:10.1016/j.jpowsour.2022.232134.
- J. E. Park, H. E. Bae, M. Karuppannan, K. M. Oh, O. J. Kwon, Y.-H. Cho and Y.-E. Sung, Effect of Catalyst Layer Designs for High-Performance and Durable Anion-Exchange Membrane Water Electrolysis, J. Ind. Eng. Chem., 2022, 109, 453–460, DOI:10.1016/j.jiec.2022.02.033.
- M. Plevová, J. Hnát, J. Žitka, L. Pavlovec, M. Otmar and K. Bouzek, Optimization of the Membrane Electrode Assembly for an Alkaline Water Electrolyser Based on the Catalyst-Coated Membrane, J. Power Sources, 2022, 539, 231476, DOI:10.1016/j.jpowsour.2022.231476.
- Z. Turtayeva, F. Xu, J. Dillet, K. Mozet, R. Peignier, A. Celzard and G. Maranzana, Manufacturing Catalyst-Coated Membranes by Ultrasonic Spray Deposition for PEMFC: Identification of Key Parameters and Their Impact on PEMFC Performance, Int. J. Hydrogen Energy, 2022, 47(36), 16165–16178, DOI:10.1016/j.ijhydene.2022.03.043.
- L. Osmieri, G. Wang, F. C. Cetinbas, S. Khandavalli, J. Park, S. Medina, S. A. Mauger, M. Ulsh, S. Pylypenko, D. J. Myers and K. C. Neyerlin, Utilizing Ink Composition to Tune Bulk-Electrode Gas Transport, Performance, and Operational Robustness for a Fe–N–C Catalyst in Polymer Electrolyte Fuel Cell, Nano Energy, 2020, 75, 104943, DOI:10.1016/j.nanoen.2020.104943.
- S. Khandavalli, J. H. Park, N. N. Kariuki, D. J. Myers, J. J. Stickel, K. Hurst, K. C. Neyerlin, M. Ulsh and S. A. Mauger, Rheological Investigation on the Microstructure of Fuel Cell Catalyst Inks, ACS Appl. Mater. Interfaces, 2018, 10(50), 43610–43622, DOI:10.1021/acsami.8b15039.
- M. Wang, J. H. Park, S. Kabir, K. C. Neyerlin, N. N. Kariuki, H. Lv, V. R. Stamenkovic, D. J. Myers, M. Ulsh and S. A. Mauger, Impact of Catalyst Ink Dispersing Methodology on Fuel Cell Performance Using in situ X-Ray Scattering, ACS Appl. Energy Mater., 2019, 2(9), 6417–6427, DOI:10.1021/acsaem.9b01037.
- S. S. Kocha, J. W. Zack, S. M. Alia, K. C. Neyerlin and B. S. Pivovar, Influence of Ink Composition on the Electrochemical Properties of Pt/C Electrocatalysts, ECS Trans., 2013, 50(2), 1475, DOI:10.1149/05002.1475ecst.
- S. Khandavalli, J. H. Park, N. N. Kariuki, S. F. Zaccarine, S. Pylypenko, D. J. Myers, M. Ulsh and S. A. Mauger, Investigation of the Microstructure and Rheology of Iridium Oxide Catalyst Inks for Low-Temperature Polymer Electrolyte Membrane Water Electrolyzers, ACS Appl. Mater. Interfaces, 2019, 11(48), 45068–45079, DOI:10.1021/acsami.9b14415.
- T. Van Cleve, S. Khandavalli, A. Chowdhury, S. Medina, S. Pylypenko, M. Wang, K. L. More, N. Kariuki, D. J. Myers, A. Z. Weber, S. A. Mauger, M. Ulsh and K. C. Neyerlin, Dictating Pt-Based Electrocatalyst Performance in Polymer Electrolyte Fuel Cells, from Formulation to Application, ACS Appl. Mater. Interfaces, 2019, 11(50), 46953–46964, DOI:10.1021/acsami.9b17614.
- S. A. Mauger, M. Wang, F. C. Cetinbas, M. J. Dzara, J. Park, D. J. Myers, R. K. Ahluwalia, S. Pylypenko, L. Hu, S. Litster, K. C. Neyerlin and M. Ulsh, Development of High-Performance Roll-to-Roll-Coated Gas-Diffusion-Electrode-Based Fuel Cells, J. Power Sources, 2021, 506, 230039, DOI:10.1016/j.jpowsour.2021.230039.
- I. Matanovic and Y. S. Kim, Electrochemical Phenyl Oxidation: A Limiting Factor of Oxygen Evolution Reaction in Water Electrolysis, Curr. Opin. Electrochem., 2023, 101218, DOI:10.1016/j.coelec.2023.101218.
- S. Ghoshal, B. S. Pivovar and S. M. Alia, Evaluating the Effect of Membrane-Ionomer Combinations and Supporting Electrolytes on the Performance of Cobalt Nanoparticle Anodes in Anion Exchange Membrane Electrolyzers, J. Power Sources, 2021, 488, 229433, DOI:10.1016/j.jpowsour.2020.229433.
- D. M. Mattox, Handbook of Physical Vapor Deposition (PVD) Processing, William Andrew, 2010 Search PubMed.
- G. Schiller, R. Henne and V. Borck, Vacuum Plasma Spraying of High-Performance Electrodes for Alkaline Water Electrolysis, J. Therm. Spray Technol., 1995, 4(2), 185–194, DOI:10.1007/BF02646111.
- F. Razmjooei, A. Farooqui, R. Reissner, A. S. Gago, S. A. Ansar and K. A. Friedrich, Elucidating the Performance Limitations of Alkaline Electrolyte Membrane Electrolysis: Dominance of Anion Concentration in Membrane Electrode Assembly, ChemElectroChem, 2020, 7(19), 3951–3960, DOI:10.1002/celc.202000605.
- G. H. A. Therese and P. V. Kamath, Electrochemical Synthesis of Metal Oxides and Hydroxides, Chem. Mater., 2000, 12(5), 1195–1204, DOI:10.1021/cm990447a.
- M. W. Losey, J. J. Kelly, N. D. Badgayan, S. K. Sahu and P. S. Rama Sreekanth, Electrodeposition, Reference Module in Materials Science and Materials Engineering, Elsevier, 2017 DOI:10.1016/B978-0-12-803581-8.10137-7.
- X. Lu and C. Zhao, Electrodeposition of Hierarchically Structured Three-Dimensional Nickel-Iron Electrodes for Efficient Oxygen Evolution at High Current Densities, Nat. Commun., 2015, 6, 6616, DOI:10.1038/ncomms7616.
- W.-S. Choi, M. J. Jang, Y. S. Park, K. H. Lee, J. Y. Lee, M.-H. Seo and S. M. Choi, Three-Dimensional Honeycomb-Like Cu0.81Co2.19O4 Nanosheet Arrays Supported by Ni Foam and Their High Efficiency as Oxygen Evolution Electrodes, ACS Appl. Mater. Interfaces, 2018, 10(45), 38663–38668, DOI:10.1021/acsami.8b12478.
- Y. S. Park, W.-S. Choi, M. J. Jang, J. H. Lee, S. Park, H. Jin, M. H. Seo, K.-H. Lee, Y. Yin, Y. Kim, J. Yang and S. M. Choi, Three-Dimensional Dendritic Cu–Co–P Electrode by One-Step Electrodeposition on a Hydrogen Bubble Template for Hydrogen Evolution Reaction, ACS Sustainable Chem. Eng., 2019, 7(12), 10734–10741, DOI:10.1021/acssuschemeng.9b01426.
- J. Sanchez, T. R. Hellstern, L. A. King and T. F. Jaramillo, Surface Engineering of 3D Gas Diffusion Electrodes for High-Performance H2 Production with Nonprecious Metal Catalysts, Adv. Energy Mater., 2019, 9(40), 1901824, DOI:10.1002/aenm.201901824.
- Y. S. Park, J. H. Lee, M. J. Jang, J. Jeong, S. M. Park, W.-S. Choi, Y. Kim, J. Yang and S. M. Choi, Co3S4 Nanosheets on Ni Foam via Electrodeposition with Sulfurization as Highly Active Electrocatalysts for Anion Exchange Membrane Electrolyzer, Int. J. Hydrogen Energy, 2020, 45(1), 36–45, DOI:10.1016/j.ijhydene.2019.10.169.
- Y. S. Park, M. J. Jang, J. Jeong, S. M. Park, X. Wang, M. H. Seo, S. M. Choi and J. Yang, Hierarchical Chestnut-Burr Like Structure of Copper Cobalt Oxide Electrocatalyst Directly Grown on Ni Foam for Anion Exchange Membrane Water Electrolysis, ACS Sustainable Chem. Eng., 2020, 8(6), 2344–2349, DOI:10.1021/acssuschemeng.9b06767.
- J.-O. Carlsson and P. M. Martin, Chemical Vapor Deposition, in Handbook of Deposition Technologies for Films and Coatings, ed. P. M. Martin, William Andrew Publishing, Boston, 3rd edn, 2010, ch. 7, pp. 314–363 DOI:10.1016/B978-0-8155-2031-3.00007-7.
- J. Hnát, M. Plevova, R. A. Tufa, J. Zitka, M. Paidar and K. Bouzek, Development and Testing of a Novel Catalyst-Coated Membrane with Platinum-Free Catalysts for Alkaline Water Electrolysis, Int. J. Hydrogen Energy, 2019, 44(33), 17493–17504, DOI:10.1016/j.ijhydene.2019.05.054.
- H. Ito, N. Miyazaki, S. Sugiyama, M. Ishida, Y. Nakamura, S. Iwasaki, Y. Hasegawa and A. Nakano, Investigations on Electrode Configurations for Anion Exchange Membrane Electrolysis, J. Appl. Electrochem., 2018, 48(3), 305–316, DOI:10.1007/s10800-018-1159-5.
- J. E. Park, S. Y. Kang, S.-H. Oh, J. K. Kim, M. S. Lim, C.-Y. Ahn, Y.-H. Cho and Y.-E. Sung, High-Performance Anion-Exchange Membrane Water Electrolysis, Electrochim. Acta, 2019, 295, 99–106, DOI:10.1016/j.electacta.2018.10.143.
- Q. Xu, S. Z. Oener, G. Lindquist, H. Jiang, C. Li and S. W. Boettcher, Integrated Reference Electrodes in Anion-Exchange-Membrane Electrolyzers: Impact of Stainless-Steel Gas-Diffusion Layers and Internal Mechanical Pressure, ACS Energy Lett., 2021, 6(2), 305–312, DOI:10.1021/acsenergylett.0c02338.
- Y. Leng, G. Chen, A. J. Mendoza, T. B. Tighe, M. A. Hickner and C.-Y. Wang, Solid-State Water Electrolysis with an Alkaline Membrane, J. Am. Chem. Soc., 2012, 134(22), 9054–9057, DOI:10.1021/ja302439z.
- J. E. Park, S. Park, M.-J. Kim, H. Shin, S. Y. Kang, Y.-H. Cho and Y.-E. Sung, Three-Dimensional Unified Electrode Design Using a NiFeOOH Catalyst for Superior Performance and Durable Anion-Exchange Membrane Water Electrolyzers, ACS Catal., 2022, 12(1), 135–145, DOI:10.1021/acscatal.1c04117.
- A. Therdthianwong, P. Manomayidthikarn and S. Therdthianwong, Investigation of Membrane Electrode Assembly (MEA) Hot-Pressing Parameters for Proton Exchange Membrane Fuel Cell, Energy, 2007, 32(12), 2401–2411, DOI:10.1016/j.energy.2007.07.005.
- S. M. Andersen, R. Dhiman, M. J. Larsen and E. Skou, Importance of Electrode Hot-Pressing Conditions for the Catalyst Performance of Proton Exchange Membrane Fuel Cells, Appl. Catal., B, 2015, 172–173, 82–90, DOI:10.1016/j.apcatb.2015.02.023.
- Z. Kang, Z. Fan, F. Zhang, Z. Zhang, C. Tian, W. Wang, J. Li, Y. Shen and X. Tian, Studying Performance and Kinetic Differences between Various Anode Electrodes in Proton Exchange Membrane Water Electrolysis Cell, Materials, 2022, 15(20), 7209, DOI:10.3390/ma15207209.
- M. K. Cho, H.-Y. Park, S. Choe, S. J. Yoo, J. Y. Kim, H.-J. Kim, D. Henkensmeier, S. Y. Lee, Y.-E. Sung, H. S. Park and J. H. Jang, Factors in Electrode Fabrication for Performance Enhancement of Anion Exchange Membrane Water Electrolysis, J. Power Sources, 2017, 347, 283–290, DOI:10.1016/j.jpowsour.2017.02.058.
- A. Lim, H. Kim, D. Henkensmeier, S. Jong Yoo, J. Young Kim, S. Young Lee, Y.-E. Sung, J. H. Jang and H. S. Park, A Study on Electrode Fabrication and Operation Variables Affecting the Performance of Anion Exchange Membrane Water Electrolysis, J. Ind. Eng. Chem., 2019, 76, 410–418, DOI:10.1016/j.jiec.2019.04.007.
- C. Simari, A. Caprì, M. H. Ur Rehman, A. Enotiadis, I. Gatto, V. Baglio and I. Nicotera, Composite Anion Exchange Membranes Based on Polysulfone and Silica Nanoscale Ionic Materials for Water Electrolyzers, Electrochim. Acta, 2023, 462, 142788, DOI:10.1016/j.electacta.2023.142788.
- D. Chanda, J. Hnát, M. Paidar, J. Schauer and K. Bouzek, Synthesis and Characterization of NiFe2O4 Electrocatalyst for the Hydrogen Evolution Reaction in Alkaline Water Electrolysis Using Different Polymer Binders, J. Power Sources, 2015, 285, 217–226, DOI:10.1016/j.jpowsour.2015.03.067.
- J. Hnát, M. Paidar, J. Schauer, J. Žitka and K. Bouzek, Polymer Anion Selective Membranes for Electrolytic Splitting of Water. Part I: Stability of Ion-Exchange Groups and Impact of the Polymer Binder, J. Appl. Electrochem., 2011, 41(9), 1043–1052, DOI:10.1007/s10800-011-0309-9.
- J. Hnát, M. Plevová, J. Žitka, M. Paidar and K. Bouzek, Anion-Selective Materials with 1,4-Diazabicyclo[2.2.2]Octane Functional Groups for Advanced Alkaline Water Electrolysis, Electrochim. Acta, 2017, 248, 547–555, DOI:10.1016/j.electacta.2017.07.165.
- A. Khataee, A. Shirole, P. Jannasch, A. Krüger and A. Cornell, Anion Exchange Membrane Water Electrolysis Using AemionTM Membranes and Nickel Electrodes, J. Mater. Chem. A, 2022, 10(30), 16061–16070, 10.1039/D2TA03291K.
- M. Mandal, G. Huang and P. A. Kohl, Anionic Multiblock Copolymer Membrane Based on Vinyl Addition Polymerization of Norbornenes: Applications in Anion-Exchange Membrane Fuel Cells, J. Membr. Sci., 2019, 570–571, 394–402, DOI:10.1016/j.memsci.2018.10.041.
- Y. S. Kim and C. Bae, Poly(Arylene)-Based Anion Exchange Polymer Electrolytes, US9051431B2, June 9, 2015, https://patents.google.com/patent/US9051431B2/en?oq=9051431, accessed 2023-08-01.
- D. P. Leonard, M. Lehmann, J. M. Klein, I. Matanovic, C. Fujimoto, T. Saito and Y. S. Kim, Phenyl-Free Polynorbornenes for Potential Anion Exchange Ionomers for Fuel Cells and Electrolyzers, Adv. Energy Mater., 2023, 13(3), 2203488, DOI:10.1002/aenm.202203488.
- G. Huang, M. Mandal, N. U. Hassan, K. Groenhout, A. Dobbs, W. E. Mustain and P. A. Kohl, Ionomer Optimization for Water Uptake and Swelling in Anion Exchange Membrane Electrolyzer: Hydrogen Evolution Electrode, J. Electrochem. Soc., 2021, 168(2), 024503, DOI:10.1149/1945-7111/abde7b.
- G. Huang, M. Mandal, N. U. Hassan, K. Groenhout, A. Dobbs, W. E. Mustain and P. A. Kohl, Ionomer Optimization for Water Uptake and Swelling in Anion Exchange Membrane Electrolyzer: Oxygen Evolution Electrode, J. Electrochem. Soc., 2020, 167(16), 164514, DOI:10.1149/1945-7111/abcde3.
- E. Cossar, A. O. Barnett, F. Seland, R. Safari, G. A. Botton and E. A. Baranova, Ionomer Content Optimization in Nickel-Iron-Based Anodes with and without Ceria for Anion Exchange Membrane Water Electrolysis, J. Power Sources, 2021, 514, 230563, DOI:10.1016/j.jpowsour.2021.230563.
- S. Koch, P. A. Heizmann, S. K. Kilian, B. Britton, S. Holdcroft, M. Breitwieser and S. Vierrath, The Effect of Ionomer Content in Catalyst Layers in Anion-Exchange Membrane Water Electrolyzers Prepared with Reinforced Membranes (Aemion + TM), J. Mater. Chem. A, 2021, 9(28), 15744–15754, 10.1039/D1TA01861B.
- M. K. Cho, H.-Y. Park, H. J. Lee, H.-J. Kim, A. Lim, D. Henkensmeier, S. J. Yoo, J. Y. Kim, S. Y. Lee, H. S. Park and J. H. Jang, Alkaline Anion Exchange Membrane Water Electrolysis: Effects of Electrolyte Feed Method and Electrode Binder Content, J. Power Sources, 2018, 382, 22–29, DOI:10.1016/j.jpowsour.2018.02.025.
- A. Y. Faid, L. Xie, A. O. Barnett, F. Seland, D. Kirk and S. Sunde, Effect of Anion Exchange Ionomer Content on Electrode Performance in AEM Water Electrolysis, Int. J. Hydrogen Energy, 2020, 45(53), 28272–28284, DOI:10.1016/j.ijhydene.2020.07.202.
- B. Mayerhöfer, F. D. Speck, M. Hegelheimer, M. Bierling, D. Abbas, D. McLaughlin, S. Cherevko, S. Thiele and R. Peach, Electrochemical- and Mechanical Stability of Catalyst Layers in Anion Exchange Membrane Water Electrolysis, Int. J. Hydrogen Energy, 2022, 47(7), 4304–4314, DOI:10.1016/j.ijhydene.2021.11.083.
- J. Liu, Z. Kang, D. Li, M. Pak, S. M. Alia, C. Fujimoto, G. Bender, Y. S. Kim and A. Z. Weber, Elucidating the Role of Hydroxide Electrolyte on Anion-Exchange-Membrane Water Electrolyzer Performance, J. Electrochem. Soc., 2021, 168(5), 054522, DOI:10.1149/1945-7111/ac0019.
- D. Xu, M. B. Stevens, M. R. Cosby, S. Z. Oener, A. M. Smith, L. J. Enman, K. E. Ayers, C. B. Capuano, J. N. Renner, N. Danilovic, Y. Li, H. Wang, Q. Zhang and S. W. Boettcher, Earth-Abundant Oxygen Electrocatalysts for Alkaline Anion-Exchange-Membrane Water Electrolysis: Effects of Catalyst Conductivity and Comparison with Performance in Three-Electrode Cells, ACS Catal., 2019, 9(1), 7–15, DOI:10.1021/acscatal.8b04001.
- C. E. Diesendruck and D. R. Dekel, Water – A Key Parameter in the Stability of Anion Exchange Membrane Fuel Cells, Curr. Opin. Electrochem., 2018, 9, 173–178, DOI:10.1016/j.coelec.2018.03.019.
- A. Martin, P. Trinke, M. Stähler, A. Stähler, F. Scheepers, B. Bensmann, M. Carmo, W. Lehnert and R. Hanke-Rauschenbach, The Effect of Cell Compression and Cathode Pressure on Hydrogen Crossover in PEM Water Electrolysis, J. Electrochem. Soc., 2022, 169(1), 014502, DOI:10.1149/1945-7111/ac4459.
- E. Gileadi and E. Kirowa-Eisner, Electrolytic Conductivity—the Hopping Mechanism of the Proton and Beyond, Electrochim. Acta, 2006, 51(27), 6003–6011, DOI:10.1016/j.electacta.2006.03.084.
- PEM Electrolysis for Hydrogen Production: Principles and Applications, ed. N. Zhao, D. Bessarabov, H. Wang and H. Li, CRC Press, Boca Raton, 2015 DOI:10.1201/b19096.
- P. Millet and S. Grigoriev, Water Electrolysis Technologies, in Renewable Hydrogen Technologies, ed. L. M. Gandía, G. Arzamendi, P. M. Diéguez, Elsevier, Amsterdam, 2013, pp. 19–41 DOI:10.1016/B978-0-444-56352-1.00002-7.
- H. Ito, N. Kawaguchi, S. Someya and T. Munakata, Pressurized Operation of Anion Exchange Membrane Water Electrolysis, Electrochim. Acta, 2019, 297, 188–196, DOI:10.1016/j.electacta.2018.11.077.
- H. Ito, N. Miyazaki, M. Ishida and A. Nakano, Cross-Permeation and Consumption of Hydrogen during Proton Exchange Membrane Electrolysis, Int. J. Hydrogen Energy, 2016, 41(45), 20439–20446, DOI:10.1016/j.ijhydene.2016.08.119.
- M. Schalenbach, T. Hoefner, P. Paciok, M. Carmo, W. Lueke and D. Stolten, Gas Permeation through Nafion. Part 1: Measurements, J. Phys. Chem. C, 2015, 119(45), 25145–25155, DOI:10.1021/acs.jpcc.5b04155.
- H. Ito, N. Kawaguchi, S. Someya, T. Munakata, N. Miyazaki, M. Ishida and A. Nakano, Experimental Investigation of Electrolytic Solution for Anion Exchange Membrane Water Electrolysis, Int. J. Hydrogen Energy, 2018, 43(36), 17030–17039, DOI:10.1016/j.ijhydene.2018.07.143.
- N. U. Hassan, Y. Zheng, P. A. Kohl and W. E. Mustain, KOH vs. Deionized Water Operation in Anion Exchange Membrane Electrolyzers, J. Electrochem. Soc., 2022, 169(4), 044526, DOI:10.1149/1945-7111/ac5f1d.
- A. Kiessling, J. C. Fornaciari, G. Anderson, X. Peng, A. Gerstmayr, M. R. Gerhardt, S. McKinney, A. Serov, Y. S. Kim, B. Zulevi, A. Z. Weber and N. Danilovic, Influence of Supporting Electrolyte on Hydroxide Exchange Membrane Water Electrolysis Performance: Anolyte, J. Electrochem. Soc., 2021, 168(8), 084512, DOI:10.1149/1945-7111/ac1dcd.
- G. A. Lindquist, S. Z. Oener, R. Krivina, A. R. Motz, A. Keane, C. Capuano, K. E. Ayers and S. W. Boettcher, Performance and Durability of Pure-Water-Fed Anion Exchange Membrane Electrolyzers Using Baseline Materials and Operation, ACS Appl. Mater. Interfaces, 2021, 13(44), 51917–51924, DOI:10.1021/acsami.1c06053.
- G. A. Lindquist, J. C. Gaitor, W. L. Thompson, V. Brogden, K. J. T. Noonan and S. W. Boettcher, Oxidative Instability of Ionomers in Hydroxide-Exchange-Membrane Electrolyzers, Energy Environ. Sci., 2023, 16, 4373–4387, 10.1039/D3EE01293J.
- R. Rossi, R. Taylor and B. E. Logan, Increasing the Electrolyte Salinity to Improve the Performance of Anion Exchange Membrane Water Electrolyzers, ACS Sustainable Chem. Eng., 2023, 11(23), 8573–8579, DOI:10.1021/acssuschemeng.3c01245.
- L. Zhang, Q. Xu, Y. Hu, L. Chen and H. Jiang, Benchmarking the PH–Stability Relationship of Metal Oxide Anodes in Anion Exchange Membrane Water Electrolysis, ACS Sustainable Chem. Eng., 2023, 11(36), 13251–13259, DOI:10.1021/acssuschemeng.3c01619.
- G.-F. Li, M. Divinagracia, M. F. Labata, J. D. Ocon and P.-Y. Abel Chuang, Electrolyte-Dependent Oxygen Evolution Reactions in Alkaline Media: Electrical Double Layer and Interfacial Interactions, ACS Appl. Mater. Interfaces, 2019, 11(37), 33748–33758, DOI:10.1021/acsami.9b06889.
- X. Chen, I. T. McCrum, K. A. Schwarz, M. J. Janik and M. T. M. Koper, Co-Adsorption of Cations as the Cause of the Apparent PH Dependence of Hydrogen Adsorption on a Stepped Platinum Single-Crystal Electrode, Angew. Chem., Int. Ed., 2017, 56(47), 15025–15029, DOI:10.1002/anie.201709455.
- Q. Jia, E. Liu, L. Jiao, J. Li and S. Mukerjee, Current Understandings of the Sluggish Kinetics of the Hydrogen Evolution and Oxidation Reactions in Base, Curr. Opin. Electrochem., 2018, 12, 209–217, DOI:10.1016/j.coelec.2018.11.017.
- R. Wang, K. Inoguchi, M. Ohashi, S. Someya, T. Munakata, M. Ishida and H. Ito, Effect of Catalyst Distribution and Structural Properties of Anode Porous Transport Electrodes on the Performance of Anion Exchange Membrane Water Electrolysis, Int. J. Hydrogen Energy, 2021, 46(76), 37757–37767, DOI:10.1016/j.ijhydene.2021.09.078.
- R. Wang, M. Ohashi, M. Ishida and H. Ito, Water Transport Analysis during Cathode Dry Operation of Anion Exchange Membrane Water Electrolysis, Int. J. Hydrogen Energy, 2022, 47(97), 40835–40848, DOI:10.1016/j.ijhydene.2022.09.181.
- S. Koch, J. Disch, S. K. Kilian, Y. Han, L. Metzler, A. Tengattini, L. Helfen, M. Schulz, M. Breitwieser and S. Vierrath, Water Management in Anion-Exchange Membrane Water Electrolyzers under Dry Cathode Operation, RSC Adv., 2022, 12(32), 20778–20784, 10.1039/D2RA03846C.
- D. Li, A. R. Motz, C. Bae, C. Fujimoto, G. Yang, F.-Y. Zhang, K. E. Ayers and Y. S. Kim, Durability of Anion Exchange Membrane Water Electrolyzers, Energy Environ. Sci., 2021, 14(6), 3393–3419, 10.1039/D0EE04086J.
- C. B. Capuano, K. Ayers and A. Keane, AEM Electrolysis Testing Update and Industrial Device Requirements, ECS Meet. Abstr., 2019,(37), 1730, DOI:10.1149/MA2019-02/37/1730.
- S. Alia, HydroGEN: Low Temperature Electrolysis; DOE Hydrogen Program Annual Merit Reivew; HydroGEN Advanced Water Splitting Materials, 2023, https://www.hydrogen.energy.gov/pdfs/review23/p148a_alia_2023_p.pdf.
- International Energy Agency Technology Collaboration Programme, Advanced Fuel Cells – Introduction to Annex 30 Electrolysis, https://www.ieafuelcell.com/fileadmin/webfiles/Introduction_to_Annex_30.pdf, accessed 2023-08-04.
- Q. Li, A. Molina Villarino, C. R. Peltier, A. J. Macbeth, Y. Yang, M.-J. Kim, Z. Shi, M. R. Krumov, C. Lei, G. G. Rodríguez-Calero, J. Soto, S.-H. Yu, P. F. Mutolo, L. Xiao, L. Zhuang, D. A. Muller, G. W. Coates, P. Zelenay and H. D. Abruña, Anion Exchange Membrane Water Electrolysis: The Future of Green Hydrogen, J. Phys. Chem. C, 2023, 127(17), 7901–7912, DOI:10.1021/acs.jpcc.3c00319.
- M. W. Louie and A. T. Bell, An Investigation of Thin-Film Ni–Fe Oxide Catalysts for the Electrochemical Evolution of Oxygen, J. Am. Chem. Soc., 2013, 135(33), 12329–12337, DOI:10.1021/ja405351s.
- M. J. Jang, J. Yang, J. Lee, Y. S. Park, J. Jeong, S. M. Park, J.-Y. Jeong, Y. Yin, M.-H. Seo, S. M. Choi and K. H. Lee, Superior Performance and Stability of Anion Exchange Membrane Water Electrolysis: PH-Controlled Copper Cobalt Oxide Nanoparticles for the Oxygen Evolution Reaction, J. Mater. Chem. A, 2020, 8(8), 4290–4299, 10.1039/C9TA13137J.
- S. Campagna Zignani, M. Lo Faro, S. Trocino and A. S. Aricò, Investigation of NiFe-Based Catalysts for Oxygen Evolution in Anion-Exchange Membrane Electrolysis, Energies, 2020, 13(7), 1720, DOI:10.3390/en13071720.
- S. M. Alia, K. S. Reeves, H. Yu, J. Park, N. Kariuki, A. J. Kropf, D. J. Myers and D. A. Cullen, Electrolyzer Performance Loss from Accelerated Stress Tests and Corresponding Changes to Catalyst Layers and Interfaces, J. Electrochem. Soc., 2022, 169(5), 054517, DOI:10.1149/1945-7111/ac697e.
- I. V. Pushkareva, A. S. Pushkarev, S. A. Grigoriev, P. Modisha and D. G. Bessarabov, Comparative Study of Anion Exchange Membranes for Low-Cost Water Electrolysis, Int. J. Hydrogen Energy, 2020, 45(49), 26070–26079, DOI:10.1016/j.ijhydene.2019.11.011.
- D. H. Marin, J. T. Perryman, M. A. Hubert, G. A. Lindquist, L. Chen, A. M. Aleman, G. A. Kamat, V. A. Niemann, M. B. Stevens, Y. N. Regmi, S. W. Boettcher, A. C. Nielander and T. F. Jaramillo, Hydrogen Production with Seawater-Resilient Bipolar Membrane Electrolyzers, Joule, 2023, 7(4), 765–781, DOI:10.1016/j.joule.2023.03.005.
- S. Nuggehalli Sampathkumar, T. B. Ferriday, P. H. Middleton and J. Van Herle, Activation of Stainless Steel 316L Anode for Anion Exchange Membrane Water Electrolysis, Electrochem. Commun., 2023, 146, 107418, DOI:10.1016/j.elecom.2022.107418.
- S. Zuo, Z.-P. Wu, H. Zhang and X. W. Lou, Operando Monitoring and Deciphering the Structural Evolution in Oxygen Evolution Electrocatalysis, Adv. Energy Mater., 2022, 12(8), 2103383, DOI:10.1002/aenm.202103383.
- K. Zhu, X. Zhu and W. Yang, Application of In Situ Techniques for the Characterization of NiFe-Based Oxygen Evolution Reaction (OER) Electrocatalysts, Angew. Chem., Int. Ed., 2019, 58(5), 1252–1265, DOI:10.1002/anie.201802923.
- J. Timoshenko and B. Roldan Cuenya, In Situ/Operando Electrocatalyst Characterization by X-Ray Absorption Spectroscopy, Chem. Rev., 2021, 121(2), 882–961, DOI:10.1021/acs.chemrev.0c00396.
- M. E. Kreider and M. Burke Stevens, Material Changes in Electrocatalysis: An In Situ/Operando Focus on the Dynamics of Cobalt-Based Oxygen Reduction and Evolution Catalysts, ChemElectroChem, 2023, 10(3), e202200958, DOI:10.1002/celc.202200958.
- Y. Deng and B. S. Yeo, Characterization of Electrocatalytic Water Splitting and CO2 Reduction Reactions Using In Situ/Operando Raman Spectroscopy, ACS Catal., 2017, 7(11), 7873–7889, DOI:10.1021/acscatal.7b02561.
- Y. Han, H. Zhang, Y. Yu and Z. Liu, In Situ Characterization of Catalysis and Electrocatalysis Using APXPS, ACS Catal., 2021, 11(3), 1464–1484, DOI:10.1021/acscatal.0c04251.
- O. Kasian, S. Geiger, K. J. J. Mayrhofer and S. Cherevko, Electrochemical On-Line ICP-MS in Electrocatalysis Research, Chem. Rec., 2019, 19(10), 2130–2142, DOI:10.1002/tcr.201800162.
- K. Ehelebe, J. Knöppel, M. Bierling, B. Mayerhöfer, T. Böhm, N. Kulyk, S. Thiele, K. J. J. Mayrhofer and S. Cherevko, Platinum Dissolution in Realistic Fuel Cell Catalyst Layers, Angew. Chem., 2021, 133(16), 8964–8970, DOI:10.1002/ange.202014711.
- X. Peng, P. Satjaritanun, Z. Taie, L. Wiles, A. Keane, C. Capuano, I. V. Zenyuk and N. Danilovic, Insights into Interfacial and Bulk Transport Phenomena Affecting Proton Exchange Membrane Water Electrolyzer Performance at Ultra-Low Iridium Loadings, Adv. Sci., 2021, 8(21), 2102950, DOI:10.1002/advs.202102950.
- J. Ampurdanés, M. Chourashiya and A. Urakawa, Cobalt Oxide-Based Materials as Non-PGM Catalyst for HER in PEM Electrolysis and in Situ XAS Characterization of Its Functional State, Catal. Today, 2019, 336, 161–168, DOI:10.1016/j.cattod.2018.12.033.
- S. Trasatti and O. A. Petrii, Real Surface Area Measurements in Electrochemistry, J. Electroanal. Chem., 1992, 327(1), 353–376, DOI:10.1016/0022-0728(92)80162-W.
- S. Watzele, P. Hauenstein, Y. Liang, S. Xue, J. Fichtner, B. Garlyyev, D. Scieszka, F. Claudel, F. Maillard and A. S. Bandarenka, Determination of Electroactive Surface Area of Ni-, Co-, Fe-, and Ir-Based Oxide Electrocatalysts, ACS Catal., 2019, 9(10), 9222–9230, DOI:10.1021/acscatal.9b02006.
- S. S. Jeon, P. W. Kang, M. Klingenhof, H. Lee, F. Dionigi and P. Strasser, Active Surface Area and Intrinsic Catalytic Oxygen Evolution Reactivity of NiFe LDH at Reactive Electrode Potentials Using Capacitances, ACS Catal., 2023, 13(2), 1186–1196, DOI:10.1021/acscatal.2c04452.
- V. Charles, A. O. Anumah, K. A. Adegoke, M. O. Adesina, I. P. Ebuka, N. A. Gaya, S. Ogwuche and M. O. Yakubu, Progress and Challenges Pertaining to the Earthly-Abundant Electrocatalytic Materials for Oxygen Evolution Reaction, Sustainable Mater. Technol., 2021, 28, e00252, DOI:10.1016/j.susmat.2021.e00252.
- J. Parrondo, C. G. Arges, M. Niedzwiecki, B. E. Anderson, K. E. Ayers and V. Ramani, Degradation of Anion Exchange Membranes Used for Hydrogen Production by Ultrapure Water Electrolysis, RSC Adv., 2014, 4(19), 9875–9879, 10.1039/C3RA46630B.
- A. K. Niaz, J.-Y. Park and H.-T. Lim, Operational Parameters Correlated with the Long-Term Stability of Anion Exchange Membrane Water Electrolyzers, Int. J. Hydrogen Energy, 2021, 46(62), 31550–31562, DOI:10.1016/j.ijhydene.2021.07.078.
- Y. Zheng, T. J. Omasta, X. Peng, L. Wang, J. R. Varcoe, B. S. Pivovar and W. E. Mustain, Quantifying and Elucidating the Effect of CO2 on the Thermodynamics, Kinetics and Charge Transport of AEMFCs, Energy Environ. Sci., 2019, 12(9), 2806–2819, 10.1039/C9EE01334B.
- N. Ziv, A. N. Mondal, T. Weissbach, S. Holdcroft and D. R. Dekel, Effect of CO2 on the Properties of Anion Exchange Membranes for Fuel Cell Applications, J. Membr. Sci., 2019, 586, 140–150, DOI:10.1016/j.memsci.2019.05.053.
- P. Mardle, S. Cassegrain, F. Habibzadeh, Z. Shi and S. Holdcroft, Carbonate Ion Crossover in Zero-Gap, KOH Anolyte CO2 Electrolysis, J. Phys. Chem. C, 2021, 125(46), 25446–25454, DOI:10.1021/acs.jpcc.1c08430.
- R. A. Krivina, G. A. Lindquist, M. C. Yang, A. K. Cook, C. H. Hendon, A. R. Motz, C. Capuano, K. E. Ayers, J. E. Hutchison and S. W. Boettcher, Three-Electrode Study of Electrochemical Ionomer Degradation Relevant to Anion-Exchange-Membrane Water Electrolyzers, ACS Appl. Mater. Interfaces, 2022, 14(16), 18261–18274, DOI:10.1021/acsami.1c22472.
- V. Yarlagadda, S. E. McKinney, C. L. Keary, L. Thompson, B. Zulevi and A. Kongkanand, Preparation of PEMFC Electrodes from Milligram-Amounts of Catalyst Powder, J. Electrochem. Soc., 2017, 164, F845, DOI:10.1149/2.1461707jes.
- A. K. Niaz, A. Akhtar, J.-Y. Park and H.-T. Lim, Effects of the Operation Mode on the Degradation Behavior of Anion Exchange Membrane Water Electrolyzers, J. Power Sources, 2021, 481, 229093, DOI:10.1016/j.jpowsour.2020.229093.
| This journal is © The Royal Society of Chemistry 2024 |