
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible alkaline hydrogen evolution and oxidation reactions using Ni–Mo catalysts supported on carbon†
Rituja B.
Patil
 ag,
Manjodh
Kaur
a,
Stephen D.
House
ab,
Lance
Kavalsky
d,
Keda
Hu
e,
Shirley
Zhong
f,
Dilip
Krishnamurthy
d,
Venkatasubramanian
Viswanathan
d,
Judith
Yang
abc,
Yushan
Yan
e,
Judith
Lattimer
f and
James R.
McKone
*a
ag,
Manjodh
Kaur
a,
Stephen D.
House
ab,
Lance
Kavalsky
d,
Keda
Hu
e,
Shirley
Zhong
f,
Dilip
Krishnamurthy
d,
Venkatasubramanian
Viswanathan
d,
Judith
Yang
abc,
Yushan
Yan
e,
Judith
Lattimer
f and
James R.
McKone
*a
aDepartment of Chemical and Petroleum Engineering, University of Pittsburgh, Pittsburgh, PA 15260, USA. E-mail: jmckone@pitt.edu; Tel: +1 (412)-384-740
bEnvironmental TEM Catalysis Consortium (ECC), University of Pittsburgh, Pittsburgh, PA 15260, USA
cDepartment of Physics and Astronomy, Pittsburgh, USA
dDepartment of Mechanical Engineering, Carnegie Mellon University, Pittsburgh, PA 15213, USA
eVersogen, Wilmington, DE 19803, USA
fGiner Labs, Newton, MA 02466, USA
gCurrently: Intel Corp., Hillsboro, OR 97124, USA
First published on 21st August 2023
Abstract
Coupling water electrolysis and fuel cell energy conversion is an attractive strategy for long-duration energy storage. This mode of operation depends on the ability to reversibly catalyze hydrogen evolution and oxidation, and ideally using nonprecious catalyst materials. Here we report the synthesis of Ni–Mo catalyst composites supported on oxidized Vulcan carbon (Ni–Mo/oC) and demonstrate the ability to catalyze reversible hydrogen evolution and oxidation. For the hydrogen evolution reaction, we observed mass-specific activities exceeding 80 mA mg−1 at 100 mV overpotential, and measurements using hydroxide exchange membrane electrode assemblies yielded full cell voltages that were only ∼100 mV larger for Ni–Mo/oC cathodes than for Pt–Ru/C cathodes at current densities exceeding 1 A cm−2. For hydrogen oxidation, Ni–Mo/oC films required <50 mV overpotential to achieve half the maximum anodic current density, but activity at larger overpotentials was limited by internal mass transfer and oxidative instability. Nonetheless, estimates of the mass-specific exchange current for Ni–Mo/oC from micropolarization measurements showed its hydrogen evolution/oxidation activity is within 1 order of magnitude of commercial Pt/C. Density functional theory calculations helped shed light on the high activity of Ni–Mo composites, where the addition of Mo leads to surface sites with weaker H-binding energies than pure Ni. These calculations further suggest that increasing the Mo content in the subsurface of the catalyst would result in still higher activity, but oxidative instability remains a significant impediment to high performance for hydrogen oxidation.
Introduction
Hydrogen is an important chemical feedstock, used extensively for syntheses of ammonia, methanol, and in petroleum refining.1 Powering water electrolysis with renewable and nuclear electricity is among the most straightforward ways to decarbonize hydrogen production.2,3 Interest has grown recently in radically reducing the cost of hydrogen generated from low-carbon power sources.4 Reaching these goals would open up opportunities to use electrolytic hydrogen not only as a chemical feedstock, but also as a fuel and for long-term storage of renewable energy.5One way to use hydrogen for energy storage is by deploying unitized regenerative fuel cells (URFCs), which combine the functions of water electrolysis and fuel cell electricity generation into a single device.6 Due to the reversible nature of URFCs, these devices demand the use of bifunctional catalysts or stable mixtures of two different catalysts on each electrode. Moreover, the well-established issues of scarcity and cost for precious metal catalysts hinder the practical deployment of proton-exchange membrane (PEM) electrolyzers and fuel cells on the scale of the global electric grid.7 In this context, hydroxide exchange membrane (HEM) electrolyzers and fuel cells are promising because the non-corrosive nature of HEM electrolytes enables the use of non-precious catalysts and ancillary materials with high terrestrial abundance.8–10
Ni-based catalysts have shown particular promise for alkaline hydrogen evolution and oxidation.11–16 Members of our research team have extensively investigated Ni–Mo bimetallic catalysts for the hydrogen evolution reaction (HER).17–22 Table S1 in the ESI† compiles results of a structured literature review covering prior reports of catalysts containing Ni and Mo with the highest activity toward the alkaline HER. Building on this prior work, we have improved on a straightforward synthetic strategy to create Ni–Mo composite catalysts supported on oxidized carbon supports (Ni–Mo/oC) whose mass activity toward the alkaline HER and the hydrogen oxidation reaction (HOR) is within one order of magnitude of commercial Pt nanoparticles. We have also constructed HEM electrolyzers using Ni–Mo/oC composite cathodes and IrOx anodes that require <100 mV of additional overvoltage relative to those using Pt–Ru/C cathodes when operating at current densities of 1 A cm−2 and above.
We also observed that Ni–Mo/oC is an active alkaline HOR catalyst, but the maximum obtainable current density for hydrogen oxidation was ∼30% lower than the external mass transfer limit predicted from the Levich Equation and observed for Pt/C catalyst. We attribute this behavior to the combined effects of internal mass transfer limitations and oxidative catalyst deactivation. Finally, first-principles calculations showed Mo substitution in the subsurface of the Ni crystal lattice has a favorable impact on hydrogen binding energies, implying increased Mo loading in the alloy phase could produce even higher HER/HOR activity.
Experimental
Syntheses of Ni-Mo/oC nanocomposites
The synthesis procedure was adopted from a prior report that we modified to incorporate oxidized carbon black as the catalyst support.20 In a representative procedure, 1 g carbon black (Vulcan XC-72, Fuel Cell Earth) was added to 50 mL of 2 M aqueous nitric acid solution (Certified ACS Plus, Fisher Scientific) in a 100 mL beaker. The beaker was capped with a Petri dish and the mixture was magnetically stirred at room temperature at 550 rpm for 2 days. The contents were then collected by centrifugation (Sorvall ST8, Fisher Scientific) at 3000 rpm for 5 min and washed with water 2–3 times. The product was dried in a drying oven (Isotemp, 1660 W, Fisher Scientific) at 60 °C.In a typical Ni–Mo/oC synthesis targeting 25 wt% metal content, 22.5 mL of diethylene glycol (99%, Alfa Aesar) was added to a 100 mL beaker; 0.5 g of the dry oxidized carbon was then added to the diethylene glycol. The mixture was further agitated in an ultrasonic bath (Branson Ultrasonics Series M) to generate a homogeneous slurry. A precursor solution was then prepared in a separate beaker by mixing 0.75 g nickel nitrate hexahydrate (98%, Alfa Aesar), 0.3 g ammonium molybdate dihydrate (ACS reagent grade, ACROS Organics), 2.5 mL deionized water (purified to ≥18.2 MΩ cm resistivity, ≤3 ppb total organic content using a Millipore Milli-Q Advantage A10) and 1 mL ammonium hydroxide solution (28–30 w/w%, ACS reagent grade). This blue precursor solution was added to the carbon/glycol slurry all at once and stirred for 1 min at room temperature.
The beaker was then transferred to a preheated hotplate set at 400 °C, and the reaction temperature was monitored using a thermometer. A mixed-phase Ni–Mo oxide was precipitated by heating the mixture to 110 °C; the beaker was then removed from the hotplate and allowed to cool for a few seconds. The still-hot solution was centrifuged at 3000 rpm for 8 min and re-suspended 2–3 times in water and then acetone. Finally, the carbon-supported Ni–Mo oxide catalyst precursor was washed with methanol (99.5%, Fisher Scientific) and dried in an oven at 60 °C. Finally, the catalyst precursor was reduced in a single-zone tube furnace (Lindberg Mini-Mite, Fisher Scientific) at 200 °C for 30 min and 450 °C for 1 h under forming gas (5% H2 and 95%N2) (Matheson gas) atmosphere to generate the final Ni–Mo/oC catalyst.
Safety note
The final catalyst comprises oxidatively unstable metal nanoparticles, which are pyrophoric and may ignite upon exposure to atmosphere. We observed that catalysts synthesized on carbon supports were less likely to ignite (perhaps due to the relatively high thermal conductivity of graphitic carbon), but carbon also constitutes a fuel source for aerobic combustion. Care must be taken to ensure the catalyst composite is cool before removing to atmosphere. We have had no difficulty handling batch sizes up to 1 g, but we recommend the adoption of additional safety protocols if the synthesis is to be scaled further.Characterization tools
X-ray diffraction (XRD) measurements were made using a Bruker D8 system with Cu Kα radiation (λ = 1.54 Å). Measurements were collected over the 2θ angle from 10 to 90° with a step size of 0.02°. The data collection times were tuned empirically to produce XRD patterns with adequate signal-to-noise ratios.Scanning electron microscopy (SEM) analysis was performed using Zeiss Sigma 500 VP microscope at an accelerating voltage of 10 kV. A small amount of catalyst was placed on carbon tape for analysis. Imaging was performed using secondary electron and backscatter detectors.
Transmission electron microscopy (TEM) analysis was performed using a Titan Themis G2 200 Probe Cs Corrected STEM (equipped with a windowless Super-X energy-dispersive X-ray spectrometer) and a Hitachi H-9500 environmental TEM. High-angle annular dark-field scanning TEM (HAADF-STEM) and STEM energy-dispersive x-ray spectroscopy (EDS) were performed on the Titan Themis. Specimens for TEM examination were prepared by drop-casting catalyst composite diluted in isopropanol (without the addition of polymer binders) onto 400 mesh ultrathin carbon-coated Cu TEM grids (Ted Pella Inc.). Both instruments were predominately operated at an accelerating voltage of 200 kV for these studies, though some of the H-9500 imaging was performed at 300 kV.
For STEM-EDS mapping, a probe current of 330 pA and pixel dwell time of 5 μs was used, with a total mapping duration of 15 min per area. No appreciable changes in the specimens were observed during TEM analysis, even during the extended EDS mapping, indicating that the beam interaction did not result in discernible sample modifications under these conditions. The observed morphologies were consistent across all microscopes and imaging modes.
X-ray photoelectron spectroscopy (XPS) was performed on a Thermo Scientific ESCALAB 250Xi instrument. An initial survey scan identified the major constituents of the sample and subsequent high-resolution scans were completed for the Ni 2p region and Mo 3d region in the range of 845–880 eV and 225–240 eV binding energies, respectively.
Thermogravimetric analysis (TGA) was used to determine the metal/carbon ratio in supported catalysts. TGA measurements employed a TA Instruments SDTQ600 thermo-gravimetric analyzer. Argon was flowed continuously at a rate of 100 sccm as purge gas. Samples were loaded into an alumina pan, introduced into the sample chamber, and equilibrated under an additional 20 sccm of N2 gas. Two sequential heating–cooling steps were then completed first under oxidizing (20 sccm air) and then reducing (10 sccm H2) conditions to remove all nonmetallic elements. The Electronic Supplementary Information includes a representative TGA dataset.
Electrochemical measurements
Electrochemical measurements were carried out using rotating disk electrode (RDE) voltammetry by depositing catalyst films on glassy carbon substrates. Colloidal catalyst inks were made by mixing 20 mg reduced Ni–Mo/oC catalyst and 20 mg of Nafion solution (20 wt% dispersed in water/alcohol mixture, Ion Power) with 1 mL isopropanol (99.5%, Fisher Scientific) and sonicated for at least 20 min. For catalyst deposition, 10 μL of this solution was drop cast on a 5 mm glassy carbon electrode (Pine Research) via two serial depositions of 5 μL each and dried under an infrared lamp (BR-40 type bulb, 125 watts, McMaster Carr). The glassy carbon substrate was then immersed in ethanol for 5 seconds followed by immersion in 0.1 M KOH for another 5 seconds to wet the catalyst film. Note, however, that we found the ethanol treatment step was not always required to ensure complete wetting of the catalyst film by KOH (aq) electrolyte.Electrochemical data were collected using a digital potentiostat/galvanostat (Gamry Reference 600+) and a commercial rotator (Pine MSR) for the working electrode. We used a single-chamber polytetrafluoroethylene cell (Pine Research) with a large-area (several cm2) Ni foil counter electrode and an Ag/AgCl reference electrode (1 M KCl, Thermo Scientific). The 0.1 M KOH (ACS reagent grade, Sigma Aldrich) electrolyte was freshly prepared prior to each measurement session comprising no more than a few hours. The cell was purged with H2 gas (99.999%, Matheson gas) for at least 2–3 min before beginning measurements to maintain electrolyte saturation. The Ag/AgCl reference electrode was calibrated to the reversible hydrogen electrode (RHE) potential using commercial Pt/C (10 wt% or 60 wt%, Alfa Aesar) composite deposited onto glassy carbon in the same way as for Ni–Mo/oC composites. To minimize the possibility of contamination, measurements of Pt-containing films were always carried out after experimentation with nonprecious catalysts was complete, and the cell was cleaned thoroughly between uses.
Cyclic voltammograms were collected over a range of overpotentials bracketing the equilibrium potential for the hydrogen evolution/oxidation reaction at a scan rate of 10 mV s−1 unless otherwise noted. Chronoamperometry measurements were also collected over a range of potentials with sampling intervals of 20 mV. Solution resistance was measured using a protocol built in to the Gamry potentiostat hardware, which comprises high-frequency impedance analysis at a DC bias of 0 V vs. RHE. These measurements generally fell in the range from 30 to 50 Ω, in good agreement with estimates based on an analytical expression developed by Myland and Oldham.23 Resistance was compensated by subtracting the IR drop from the empirical overpotential data after experiments were complete.
We report catalytic activity normalized to superficial electrode area or catalyst mass. For mass normalization, mass loadings were taken on a metals basis from TGA measurements and the known loading of total solids in the colloidal catalyst ink. For example, a film reported as 0.25 mgcat cm−2 comprised 25 wt% Ni–Mo/oC deposited at a total loading of 1 mg cm−2 of metal + carbon composite on the glassy carbon substrate.
Membrane electrode assembly fabrication and testing
We measured potential vs current data for full water electrolysis using a single-cell hydroxide exchange membrane (HEM) electrolyzer. The membrane was 80 μm thick PiperION A (Versogen) and the catalyst binder was a hydroxide exchange ionomer (HEI) PiperION A (5 wt% in ethanol; Versogen). The cathode catalyst comprised 25 wt% Ni–Mo/oC (0.5–1.0 mgcat cm−2) or 75 wt% Pt–Ru/C (0.15 mgcat cm−2, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of Pt to Ru) and the anode was unsupported nanoparticulate IrO2 (3 mgcat cm−2). The films were prepared by manually air-spraying a catalyst ink suspension in 20 vol% water, 80 vol% isopropanol onto the anion exchange membranes in a clean room. This was followed by a solvent annealing step in which the coated membrane was incubated in a sealed container next to a small vessel filled with 20 vol% ethanol in water. Finally, coated membranes were compressed at 140 psi to form stable and tightly adhering membrane electrode assemblies (MEAs).
:1 ratio of Pt to Ru) and the anode was unsupported nanoparticulate IrO2 (3 mgcat cm−2). The films were prepared by manually air-spraying a catalyst ink suspension in 20 vol% water, 80 vol% isopropanol onto the anion exchange membranes in a clean room. This was followed by a solvent annealing step in which the coated membrane was incubated in a sealed container next to a small vessel filled with 20 vol% ethanol in water. Finally, coated membranes were compressed at 140 psi to form stable and tightly adhering membrane electrode assemblies (MEAs).
MEAs were pre-conditioned by soaking in 3 M KOH for 1 h prior to assembly into a 5 cm2 electrolyzer using H23C6 carbon paper (Freudenberg) as the cathode gas-diffusion layer (GDL) and platinized Titanium sinter as the anode GDL. The cell was further conditioned by flowing ultrapure water at 0.35 mL min−1 through the anode while the cell was heated to 80 °C with the current held at 0.1 A cm−2. This flow rate was maintained throughout the experiment and found empirically to support current densities in excess of 2 A cm−2 without evidence of transport limitations. The conditioning was continued until a steady state voltage was obtained; this was found to require 12–18 hours.
Polarization curves were recorded after the conditioning process was complete. Chronopotentiometric polarization curves were collected over the current range from 0.02 A cm−2 to 2.0 A cm−2 at 80 °C under anode feed conditions, with either 0.1M KOH or ultra-pure water flowed at 0.35 mL min−1. The curves were recorded at fixed current for 1–2 min during to obtain a stable cell voltage. A full polarization curve was therefore completed in ∼2 h. Between the collection of polarization curves, the cell was held at 0.1 A cm−2.
Computational studies
Density functional theory (DFT) calculations were conducted using the GPAW package via the atomic simulation environment.24,25 A target grid spacing of 0.16 Å and a Monkhorst–Pack k-mesh of 4 × 4 × 1 were used.26 To improve the convergence of the self-consistent field algorithm, a Fermi smearing of 0.05 eV was applied. Ion-electron interactions were considered through the projector-augmented wave approach.27 Geometries were optimized until a maximum force of 0.05 eV Å−1 was met. Zero-point energy and entropic effects were assumed to be uniformly 0.24 eV for all systems, as done previously.28To identify all symmetrically distinct hollow sites, Delaunay triangulation was used. This approach forms a 2D Voronoi tessellation of the surface to recognize these sites and has been successfully demonstrated on multiple types of systems with varying geometries and compositions.29 Through the implementation of this method within the pymatgen package, this approach can be systematically applied for every surface.30
The Ni–Mo catalyst was represented with a face-centered cubic (fcc) slab model oriented with the (111) plane exposed to investigate the trends in adsorption of hydrogen at 1/4 ML coverage of adsorbed hydrogen. Use of this hydrogen coverage for investigating trends is in line with previous work.28
The energy associated with swapping a Mo atom from the bulk to the surface was calculated to be 0.75 eV, so we considered the surface layer to consist exclusively of Ni. The binding energy of hydrogen was also found to vary by no more than ∼0.005 eV when Mo was substituted for Ni in the second sublayer. Therefore, to obtain a full range of potential adsorption behavior, we enumerated all possible concentrations of Mo in the first sublayer of our 2 × 2 × 1 supercell via the ICET python package.31 For each of these generated structures, varying degrees of the surface symmetry were broken, and we calculated the hydrogen adsorption energy on each of the unique surface sites.
Moreover, for each calculated hydrogen adsorption energies, there exists an inherent uncertainty stemming from an approximation of the electron–electron exchange–correlation (XC), as a closed-form of this functional is currently unknown. To capture the sensitivity of this system towards the choice of exchange correlation functional and quantify the associated uncertainties, we used a Bayesian approach via the Bayesian error estimation functional (BEEF-vdW).32
Results and discussion
Fig. 1 compiles representative SEM images and XRD patterns of the carbon-supported Ni–Mo oxide precursor and the final catalyst. The images in Fig. 1a and b depict nanoparticulate composites with no clear demarcation between the carbon support and the transition metals. Therefore, backscatter SEM analysis was performed, as shown in Fig. 1c. The bright white spots in the backscatter SEM image correspond to Ni–Mo (denser and higher atomic mass) while the grey regions belong to the carbon support. These data show that the catalyst contains nanoparticles and/or agglomerates that are uniformly dispersed on the carbon support and tens of nm or smaller in size. EDS analysis using a spot size on the micron scale gave a 7/3 ratio of Ni to Mo as an aggregate composition in the final catalyst (see ESI,† Fig. S1).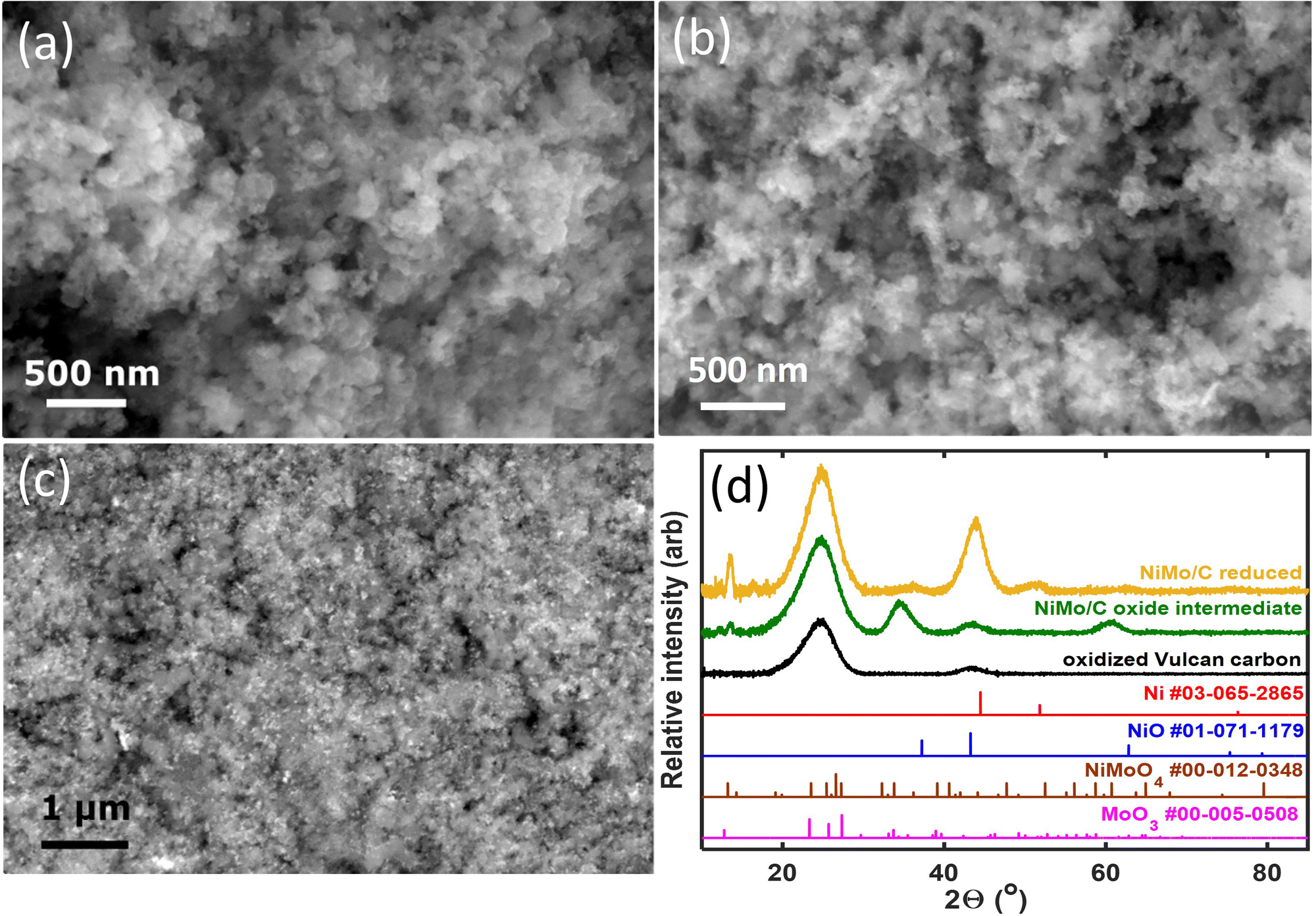 | ||
| Fig. 1 Representative materials characterization data for Ni–Mo/o-C: (a) SEM image of the carbon-supported Ni–Mo oxide precursor after precipitation from diethylene glycol; (b) SEM image of the final catalyst composite collected with a secondary electron detector; (c) SEM image of the final catalyst composite collected using a backscatter detector to increase elemental contrast; (d) compiled XRD data and standard patterns (annotated with numerical indexes from the ICDD database). | ||
XRD measurements (Fig. 1d) of the catalyst precursor show two broad peaks at 2θ = 25 and 43°, consistent with the diffraction of Vulcan carbon alone; this suggests the precursor is an amorphous or poorly crystallized mixture of Ni and Mo oxides. The final reduced Ni–Mo/oC catalyst shows two more features at 44 and 52° corresponding to diffraction for face-centered cubic Ni. Additional weak diffraction features at 2θ = 37, 50, 60, and 76° correspond to oxides of Ni and Mo. The low intensity of these peaks indicates the residual oxides are less prevalent or poorly crystalline. The Mo content in the Ni–Mo alloy was calculated from the shift in fcc Ni peaks and found to be 10 ± 1 mol%, although we cannot rule out the possibility that diffraction from residual NiO or the Vulcan carbon causes the maximum intensity of the Ni diffraction peak to shift to lower 2θ values.33 Thus, 10 mol% is likely an upper bound for Mo content in the Ni–Mo alloy component of the catalyst. The metal crystallite size, calculated from Scherrer analysis, was 3.6 ± 0.2 nm. The total metal (Ni + Mo) content in the reduced catalyst, obtained from thermogravimetric analysis (TGA; Fig. S2, ESI†) was 25 wt% relative to the total quantity of solid in the catalyst powder (this does not include polymer binder). Note that this mass ratio was obtained empirically by varying the initial ratio of metal precursors to carbon in the synthesis mixture. XPS measurements further confirm the prevalence of Ni and Mo oxides on the catalyst surface, as discussed in the ESI† (Fig. S3).
Fig. 2 shows representative TEM micrographs and EDS mapping of the 25 wt% Ni–Mo/oC catalyst. The dispersion of Ni–Mo on oxidized Vulcan carbon is evident in the bright field image: the darker particles, with an average diameter of 7.7 ± 0.5 nm, correspond to Ni–Mo while the lighter particles on the order of 50 nm are the carbon support. The HAADF-STEM image shows Ni–Mo catalyst (brighter regions) dispersed as irregularly shaped agglomerates on carbon (darker regions). The EDS maps confirm that each Ni-rich particle is surrounded by Mo and O.
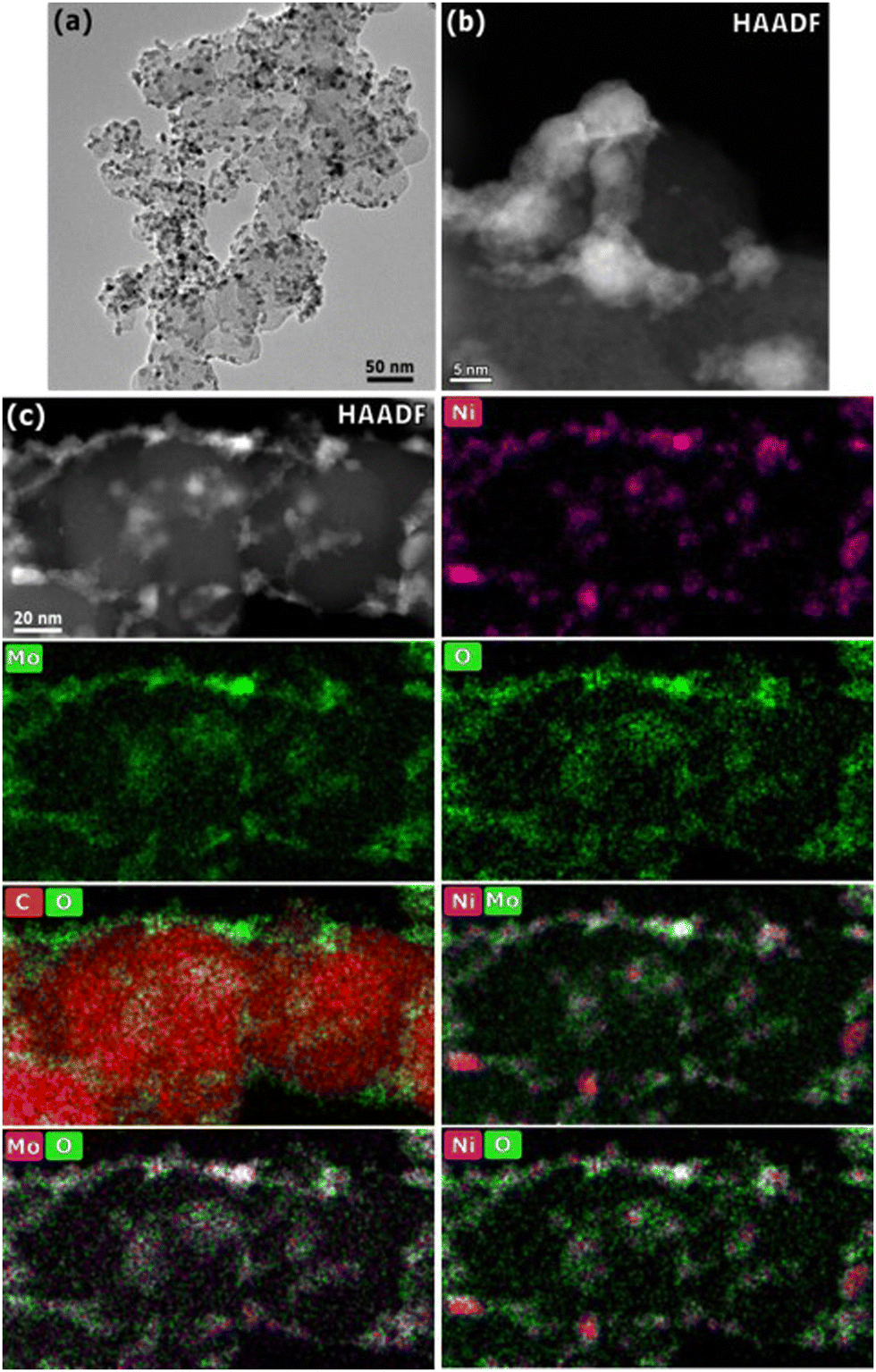 | ||
| Fig. 2 TEM and HAADF-STEM image of 25 wt% Ni–Mo/o-C. (a) Bright field image depicting a representative nanoparticle/carbon composite; (b) HAADF dark field image depicting a nanoparticle agglomerate on the carbon support; (c) multipanel overlay of HAADF image and EDS maps demonstrating the spatial distribution of Ni, Mo, and O. | ||
These characterization data together suggest that Ni–Mo/oC catalyst comprises Ni-rich nanoparticles that are coated and interconnected by Mo-rich oxides. This morphology is similar to what we observed in prior studies.20,21 We attribute the high prevalence of Mo-rich oxides to incomplete reduction of the mixed Ni–Mo oxide precursor along with the limited solubility of metallic Mo in fcc Ni. Notably, the EDS maps in Fig. 2c show that the Mo oxides are distributed more broadly than the Ni-rich particles. This is consistent with favorable surface wetting that results from pre-oxidizing the carbon support.
Representative voltammetry and chronopotentiometry data for Ni–Mo/oC composites catalyzing the alkaline HER are shown in Fig. 3. Polarization data in Fig. 3a clearly demonstrate the higher activity of NiMo/oC compared to unsupported Ni and Ni–Mo nanoparticles. The activity of Ni–Mo/oC normalized to the total mass of metal at 100 mV overpotential and 0.25 mgcat cm−2 loading was 82 ± 5 mA mgcat−1, where the error bound represents a 95% confidence interval from 50 catalyst films (ESI,† Fig. S4). This value was approximately 10 times higher than unsupported Ni–Mo nanoparticles, 100 times higher than unsupported Ni nanoparticles, and 5 times lower than commercial 10 wt% Pt/C under similar experimental conditions (Fig. S5 in the ESI† depicts comparisons to Pt/C over a wider range of overpotentials).
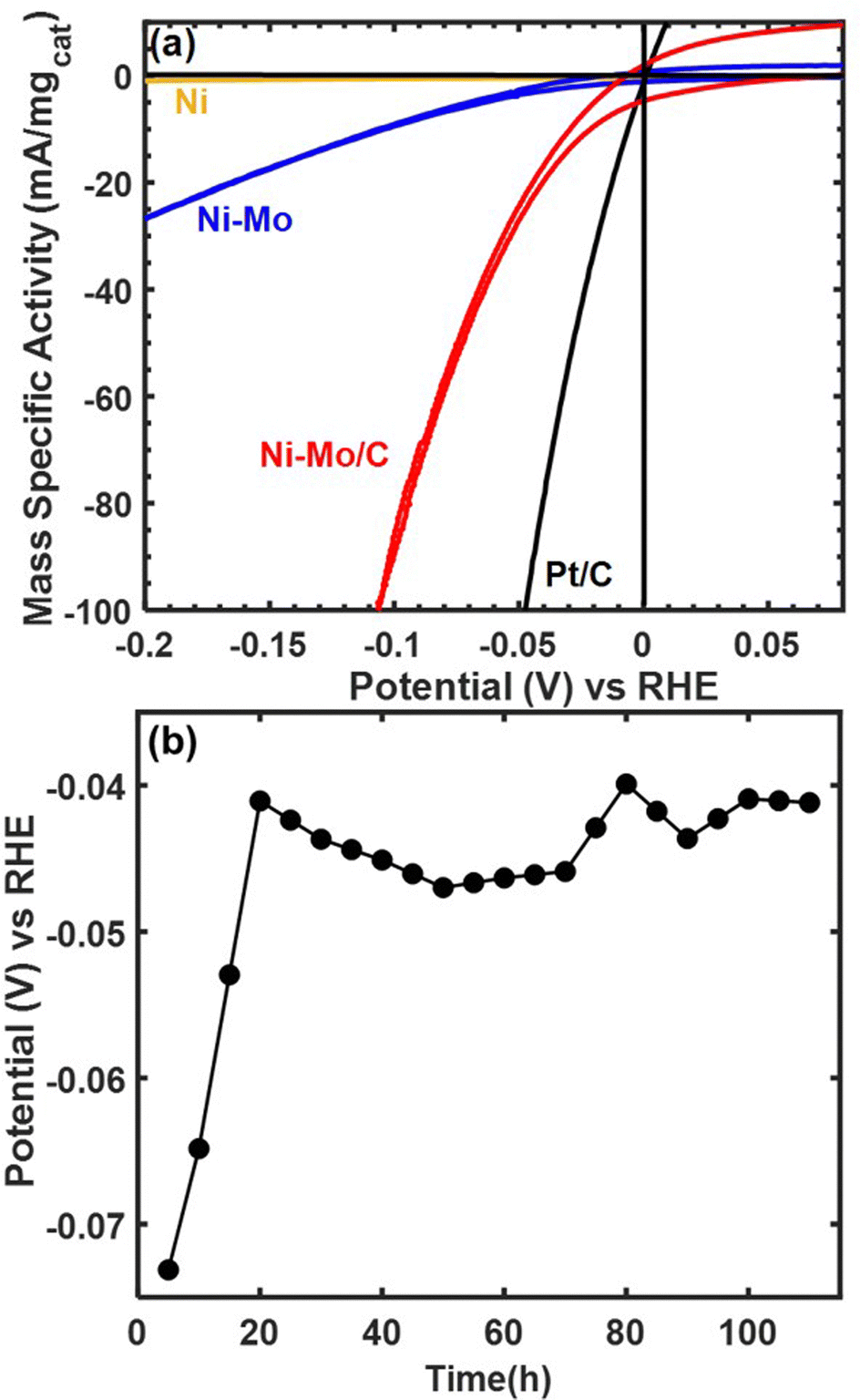 | ||
| Fig. 3 HER activity and stability data collected via RDE at 1600 rpm in 0.1 M KOH. (a) Representative polarization data for 25 wt% Ni–Mo/o-C (0.25 mgcat cm−2), unsupported Ni–Mo (1 mgcat cm−2), unsupported Ni nanoparticles (1 mgcat cm−2) and commercial 10 wt% Pt/C (0.025 mgcat cm−2). (b) Chronopotentiometry data for 0.375 mgcat cm−2 Ni–Mo/oC at −10 mA cm−2. The data points in (b) represent trailing average values of the cathode potential over 5 hour intervals. To improve adhesion at 0.375 mgcat cm−2 (1.5 mg cm−2 total loading), the glassy carbon substrate was first roughened by brief abrasion with 240 grit SiC grinding paper. | ||
We also varied mass loading on the electrode to measure the resulting impact on HER activity (ESI,† Fig. S6). Despite considerable sample-to-sample variability, we observed a monotonic increase in the apparent HER mass activity with decreasing mass loading, which is broadly indicative of mass transfer limitations.34,35 This behavior extends at least as low as 10 μgcat cm−2, which was the lowest we measured.
We also measured the stability of Ni–Mo/oC by performing chronopotentiometry experiments at a constant current density of −10 mA cm−2 (−27 mA mgcat−1) for 110 h, as shown in Fig. 3b. The overpotential required to maintain this current density began at nearly 80 mV and then decreased to 40–50 mV, where it remained for the duration of the stability experiment. This type of improved activity over time has been observed previously and attributed to dissolution of Mo-oxides.36 Note that this measurement used the maximum catalyst loading that would reliably adhere to a roughened glassy carbon electrode to match conditions under which a nonprecious catalyst is likely to be used in a device setting. The cumulative charge passed corresponds to >4 mol H2 per mol of total metal content in the catalyst.
The ESI† includes a tabulated comparison of performance metrics for alkaline HER catalysts containing Ni and Mo. References were downselected from a keyword search returning ∼2000 prior publications to capture a subset with the highest reported geometric current densities at 100 mV overpotential while also explicitly reporting catalyst mass loading on the electrode substrate. Our Ni–Mo/oC catalyst compares favorably with these in terms of geometric and mass-normalized activity, and it also benefits from the ability to be processed and coated as a catalyst “ink” as is common practice in proton- and hydroxide-exchange membrane fuel cells and electrolyzers. Hence, we undertook further experiments to measure the practical performance of Ni–Mo/oC in full HEM electrolyzer cathodes. Representative polarization data comparing Ni–Mo/oC with Pt–Ru/C cathode catalysts are shown in Fig. 4.
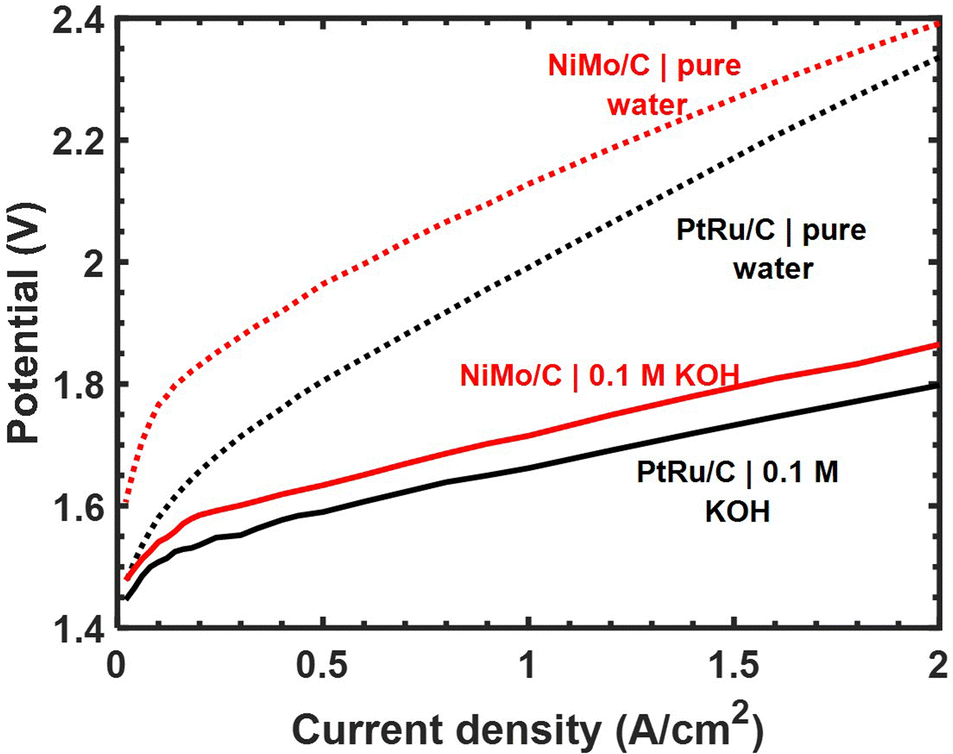 | ||
| Fig. 4 Polarization data for full HEM electrolysis using 25 wt% Ni–Mo/o-C and Pt–Ru/C (50 wt% Pt, 25 wt% Ru) cathodes using aryl-ammonium based membrane/binder polymers and IrOx anode catalysts. The total catalyst loading of Ni–Mo/oC was 1 mgcat cm−2 and for Pt–Ru/C it was 0.15 mgcat cm−2. Unsupported IrOx nanoparticle anodes were loaded at 3 mgcat cm−2. | ||
When feeding aqueous alkaline electrolyte, the total cell voltage required to electrolyze water was only 50–70 mV higher with a Ni–Mo/oC cathode catalyst compared to Pt–Ru/C over the current densities ranging from 0.2 to 2 A cm−2. By contrast, with a feed of pure water, the potential difference between the same two MEAs was nearly 200 mV at 0.2 A cm−2 and diminished to <100 mV at 2 A cm−2. The generally larger cell voltages required in the latter case are attributable to additional mass transfer limitations in pure water associated with hydroxide ion conduction through the alkaline ionomer binder. This mass transfer limit may be further exacerbated by the use of Ni–Mo/oC at higher mass loading than Pt–Ru/C. Nonetheless, the total cell potential required to generate 1 A cm−2 in a pure water feed was ∼2.1 V when using a Ni–Mo/oC cathode, and this compares favorably to several of the highest performing HEM electrolyzers reported to date.37–39
Considering the promising results with respect to HER activity, we continued to explore applications of Ni–Mo/oC composites as catalysts for the hydrogen oxidation reaction. Representative polarization data are compiled in Fig. 5. The capacitive response of Ni–Mo/oC dominated the electrochemical behaviour under potential cycling, which made it difficult to quantify HOR activity. We instead performed a series of chronopotentiometric experiments by progressively stepping the potential over 20 mV increments every 120 seconds to extract the steady-state current response in the potential region bracketing 0 V vs. RHE (Fig. 5a).
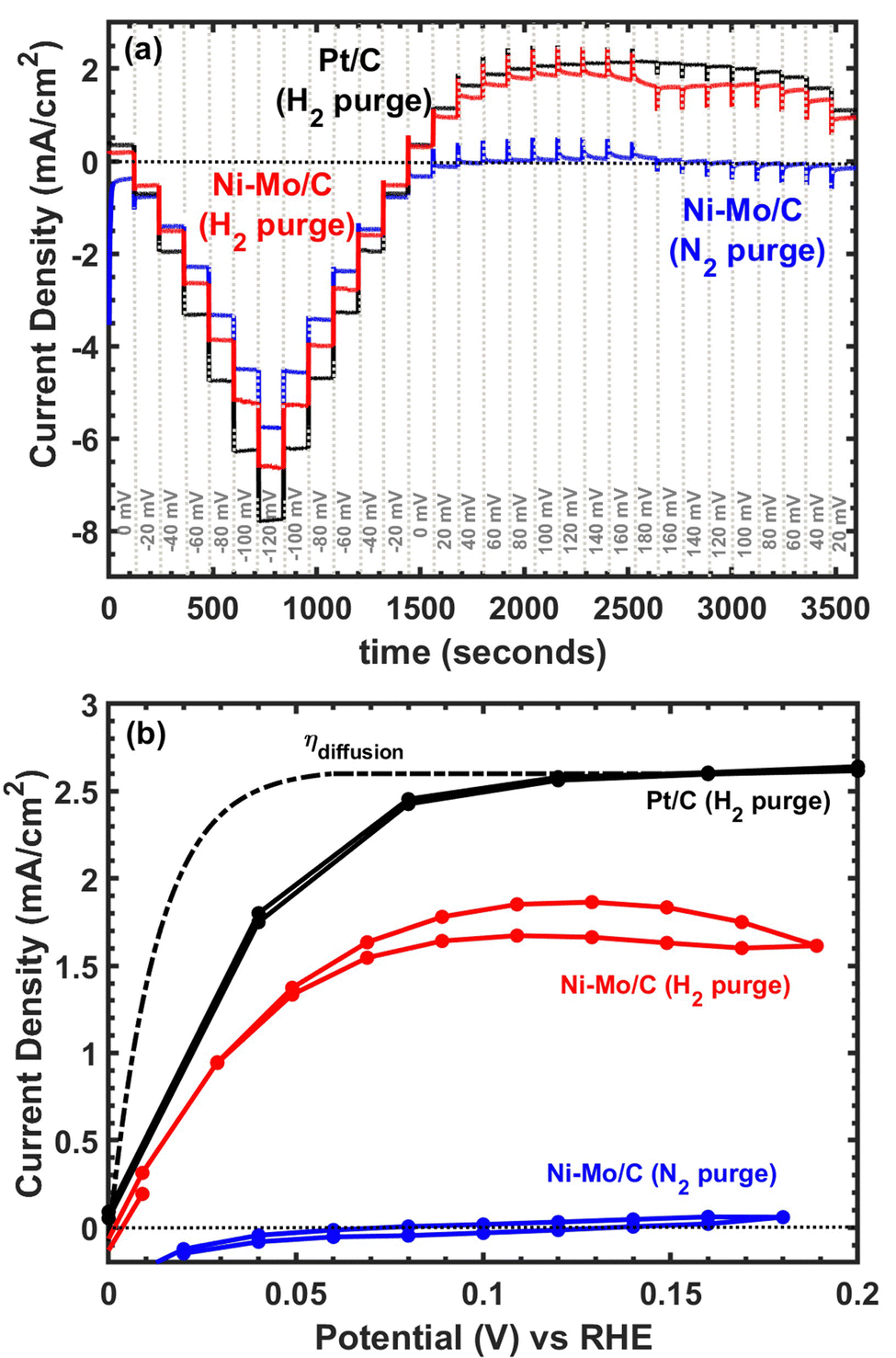 | ||
| Fig. 5 Representative data for alkaline hydrogen oxidation. (a) Chronoamperometry data depicting sequential potential steps with 120 second holds used to minimize the impact of interfacial capacitance. (b) Steady-state voltammograms for Ni–Mo/oC catalyst under hydrogen and nitrogen purge compared to 60 wt% Pt/C and a simulated external transport limited overpotential curve. | ||
Under N2 purge, chronopotentiometry results showed negligible anodic current flow, whereas under H2 Ni–Mo/oC showed a clear onset of oxidation at 0 V vs. RHE. Only ∼30 mV overpotential was required to reach half the anodic limiting current, and this half-wave potential was indistinguishable from that of the 60 wt% Pt/C control. However, the maximum observed anodic current density was ∼1.75 mA cm−2 for Ni–Mo/oC, considerably lower than that of Pt/C at ∼2.5 mA cm−2. The dotted line in Fig. 5b depicts a simulated concentration overpotential curve corresponding to the expected HOR behaviour of a catalyst whose reaction rate constant is much larger than the mass transfer coefficient (i.e., external mass transfer controlled).40,41
The predicted external mass transfer limited current density—which is described by the Levich equation and does not depend on the catalyst composition—was 2.6 mA cm−2 at 1600 rpm in 0.1 M KOH, in close agreement with the data collected for a Pt/C catalyst. The fact that Ni–Mo/oC catalysts did not reach 2.6 mA cm−2 suggests that a different type of mass transfer limitation may negatively impact the observed activity. Others have made similar observations of low anodic limiting currents in Ni-based alkaline HOR catalysts, albeit without additional discussion.42,43
As an additional screen for mass transfer limitations, we varied catalyst loading on the electrode. Fig. 6 collects representative results showing a non-monotonic relationship between loading and HOR activity. We therefore collected replicate measurements to improve statistical confidence, and the relationship between mass loading and current density at a fixed HOR overpotential of 50 mV (measured directly or by linear interpolation from steady-state polarization data at 40 and 60 mV) is compiled in Fig. 6b. The slope of the relationship between geometric current density and mass loading becomes shallower as mass loading increases, and the highest mass loading we tested was insufficient to achieve the expected external mass transfer limited current of 2.6 mA cm−2—even at more positive applied potentials. Based on these results (along with additional data compiled in Fig. S7 in the ESI†), we conclude that Ni–Mo/oC exhibits internal mass transfer limited behavior at HOR overpotentials below 100 mV and mass loadings above 0.1 mgcat cm−2. However, this does not explain the inability to achieve an external mass transfer limit at larger HOR overpotentials.
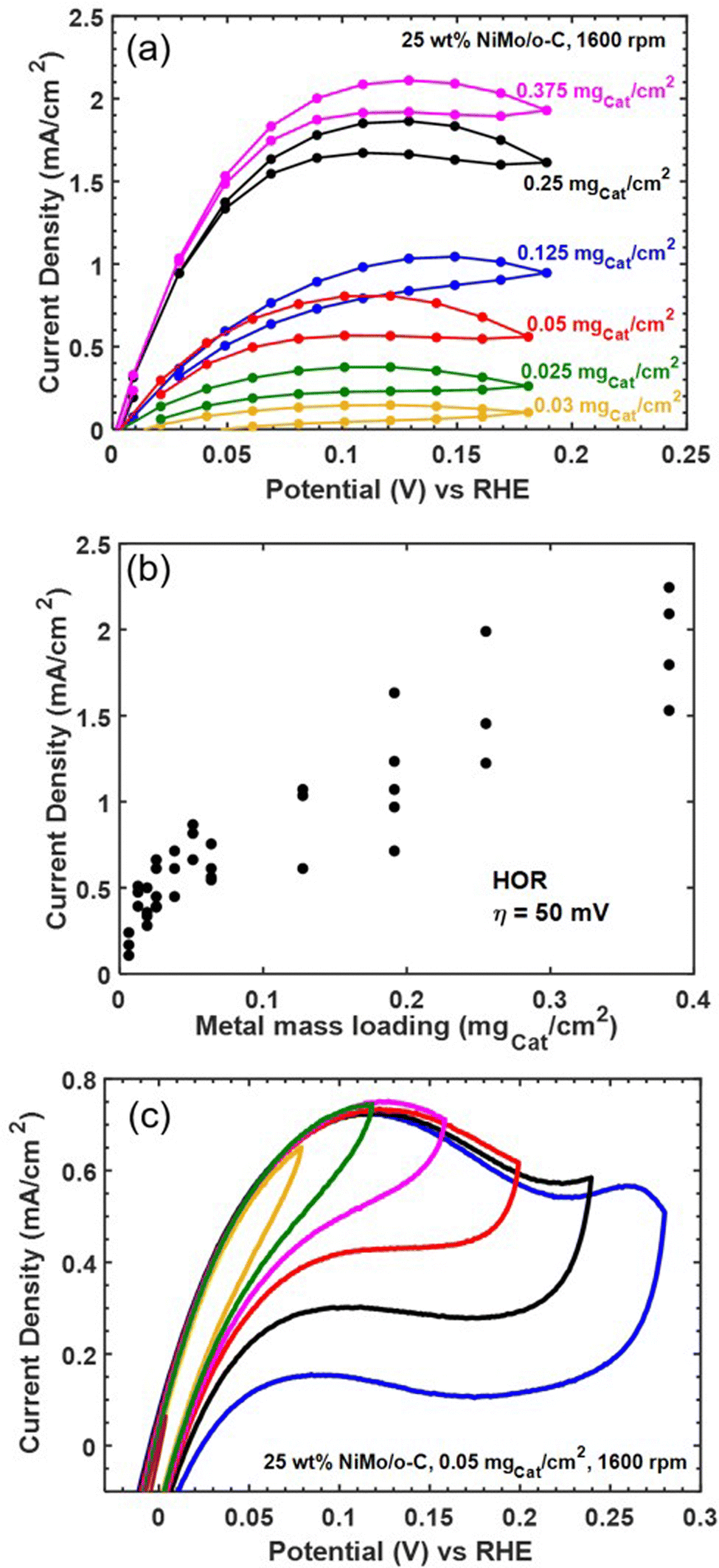 | ||
| Fig. 6 Evidence for mass transfer limitations and oxidative catalyst deactivation. (a) Representative steady-state voltammograms for the HOR on Ni–Mo/oC over a range of catalyst mass loadings. (b) Full dataset compiling HOR current density as a function of mass loading. (c) Representative cyclic voltammetry data collected at a scan rate of 0.5 mV s−1 over a range of positive potential limits. | ||
We hypothesized that Ni–Mo/oC is further limited by instability toward oxidative deactivation, since the most stable forms of Ni and Mo are oxides (or oxo anions in the case of Mo) in alkaline pH at potentials positive of 0 V vs. RHE.44 To address this hypothesis, we ran cyclic voltammetry experiments on Ni–Mo/oC at 0.05 mgcat cm−2 loading while progressively increasing the positive scan limit from 80 to 280 mV vs. RHE in 40 mV increments (Fig. 6c). Increasing the limit from 80 mV to 120 mV vs. RHE increased the anodic current density, as expected for an active HOR catalyst. However, further increases in the positive potential limit yielded a distinct peak in the anodic current density at ∼120 mV vs. RHE, and reversing the sweep direction at potentials positive of this peak gave markedly reduced anodic current densities in the reverse sweep. We take this as strong evidence for the onset of catalyst poisoning or deactivation at ∼120 mV vs. RHE. At least some of the deactivation appears to be reversible, as evidenced by the slight recovery in anodic current density over the potential range from 0.2 to 0.05 V vs. RHE in the negative-going return sweeps. Finally, CV data obtained by scanning to 280 mV vs. RHE show the onset of an additional anodic feature, which we attribute to irreversible catalyst oxidation as evidence by the near-complete loss of anodic current in the reverse sweep.20
These results enable us to construct a more complete picture of the HOR at Ni–Mo/oC catalyst composites. Reaction kinetics dominate the current-overpotential behavior at modest overpotentials and low catalyst loadings (up to 0.05 mgcat cm−2). Attempting to increase geometric HOR activity by increasing catalyst loading then leads to internal mass transfer limitations at overpotentials over which the catalyst remains stable (< 150 mV vs. RHE). Finally, oxidative catalyst deactivation, which appears to be only partially reversible, takes over at HOR overpotentials exceeding 150 mV.
In view of the ability of Ni–Mo/oC to catalyze the alkaline HER and HOR (and despite its stability limitations in the latter case), we sought to make direct comparisons between this catalyst and commercial Pt/C for reversible hydrogen electrocatalysis. We therefore performed sequential catalyst cycling wherein the potential was stepped between −100 mV and +100 mV vs. RHE on a 10 min duty cycle (Fig. 7a). In both cases, mass loading was optimized to maximize the superficial current density for the HOR with a minimum quantity of active catalyst. This resulted in 60 wt% Pt/C composites at 0.12 mgcat cm−2 loading and Ni–Mo/oC at 0.375 mgcat cm−2. In the latter case, this was the highest loading we could achieve without delaminating the catalyst layer.
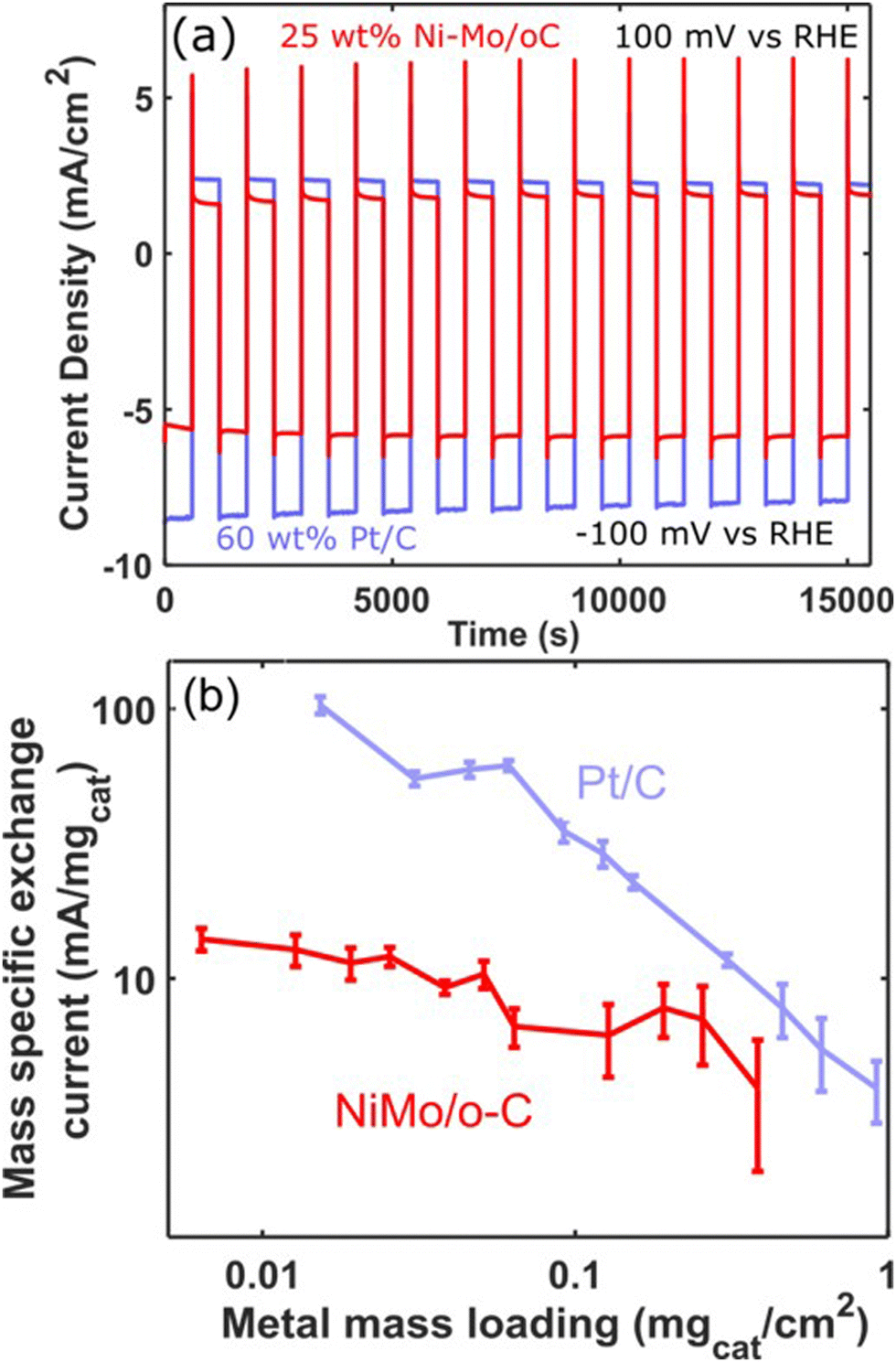 | ||
| Fig. 7 Direct comparisons of HER/HOR reversibility between Ni–Mo/oC and Pt/C catalysts. (a) Chronoamperometry measurements comprising a series of 10 minute potential steps between +100 and −100 mV vs. RHE. The mass loading of 25 wt% Ni–Mo/oC was 0.375 mgcat cm−2 and 60 wt% Pt/C was 0.12 mgcat cm−2 (b) Mass-specific exchange currents as a function of mass loading, collected from micropolarization measurements. | ||
Generally, the Pt/C catalyst produced modestly larger geometric HER and HOR current densities than Ni–Mo/oC. However, the HER and HOR activity of Pt/C also marginally decreased over 15 cycles (5 h). We speculate this is predominantly due to catalyst poisoning that results from the use of 0.1 M KOH electrolyte without extensive pre-purification.45 By contrast, the HER and HOR activity of Ni–Mo/oC increased slightly over the course of the experiment, which is again consistent with prior results attributed to dissolution of inactive oxides.36
As a final comparison between Ni–Mo/oC and Pt/C catalysts, we took advantage of their reversible HER/HOR activity to extract mass-specific exchange currents in the micropolarization region. We applied the linearized form of the Butler Volmer equation to relate the slope of the current–voltage relationship about 0 V vs. RHE to the exchange current density via:
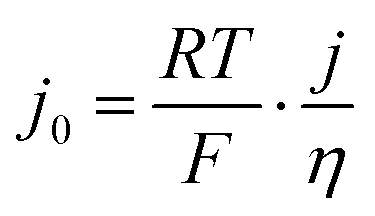
It should be possible to further close the gap in mass activity between Ni- and Pt-based catalysts through several means. One straightforward modification would involve increasing the mass fraction of metal in a carbon-supported composite, which would enable the use of higher mass loadings of active catalyst without incurring large losses due to mass transfer. Another way to increase activity is to decrease particle size, but this strategy may ultimately be limited by the tendency for Ni-based catalysts to spontaneously oxidize in aerobic environments. A third way to increase HOR activity in an oxidatively unstable catalyst might be to increase the temperature while limiting the applied potential to the range in which the catalyst remains stable, as has been shown for Co–Ni–Mo composites.50
In another recent report, an unsupported Ni–Mo composite containing 20 mol% Mo reached the expected external mass transfer limited current density at HOR overpotentials up to at least 200 mV even at room temperature.51 This result suggests that Ni–Mo alloys with higher Mo content are more catalytically active and may also be more stable toward oxidation. To this effect, prior reports have also suggested that Mo atoms vicinal to surface Ni atoms have a favorable impact on hydrogen binding.52
To further probe the influence of Mo content on Ni–Mo alloy HER/HOR activity, hydrogen binding energies on Ni–Mo composites were obtained from DFT calculations (Fig. 8). We first found that it is thermodynamically more favorable for Mo atoms to be present in the sub-surface of the catalyst, as opposed to on the surface, in agreement with previous findings.53,54 The adsorption energies of hydrogen as a function of Mo content in the first sublayer is shown in Fig. 8a. In all cases, the presence of Mo near the surface weakens the hydrogen bond at the ideal site relative to pure Ni, which is advantageous for HER activity because it shifts the H-binding energy nearer to thermoneutral (horizontal dashed line in Fig. 8a). The error-bars are ±1 standard deviation, σ, of the BEEF ensemble for the ideal sites. Interestingly, a non-monotonic trend emerges with a maximum at 75 mol% Mo. This implies that there is a limit to the weakening of the hydrogen bond through the presence of Mo near the surface. Moreover, the H-binding energetics of all surface sites exhibit a similar overall trend, highlighting the general synergy between Ni and Mo.
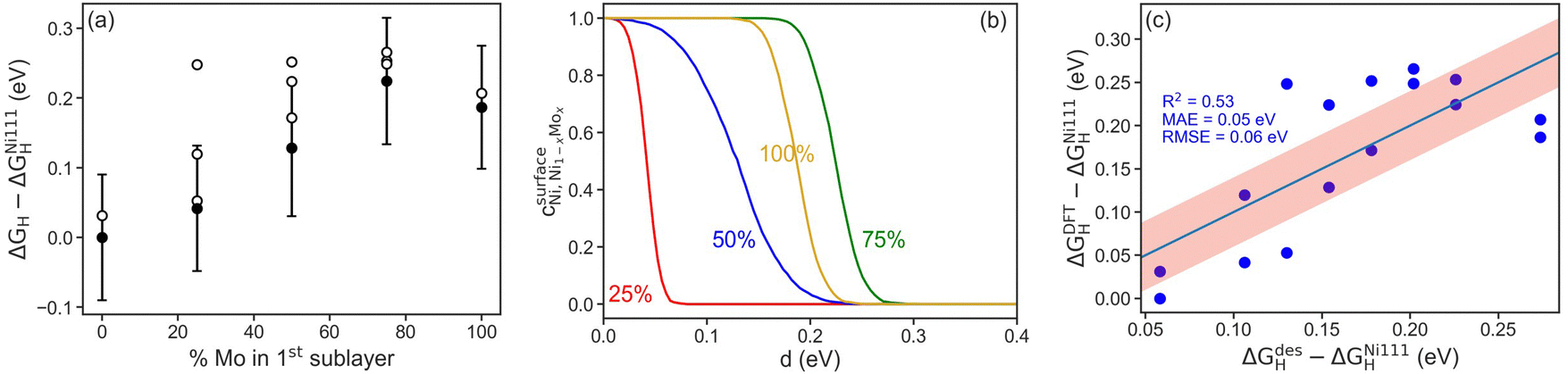 | ||
| Fig. 8 Compiled results of density functional theory (DFT) calculations to elucidate the impact of Mo alloying on the binding of hydrogen intermediates in Ni–Mo. (a) Hydrogen binding energy on Ni–Mo alloys with different Mo content. Filled circles are ideal sites and open circles are non-ideal sites. Error bars are 1σ of the BEEF ensemble average. (b) Distinguishability metric for Ni–Mo alloys compared to pure Ni. (c) Parity plot correlating a geometric model based on nearest neighbors with DFT-calculated H-binding energies. | ||
To further investigate the distinguishability between H-adsorption energies in Ni–Mo alloys relative to pure Ni, we leveraged BEEF ensemble distributions. Prior work has shown that through the use of this Bayesian analysis, additional insights into the uncertainty associated with DFT predictions can be obtained.55,56 For this application, every functional within the ensemble will yield varying differences between the adsorption energies relative to pure Ni. Here we used the following distinguishability metric:57
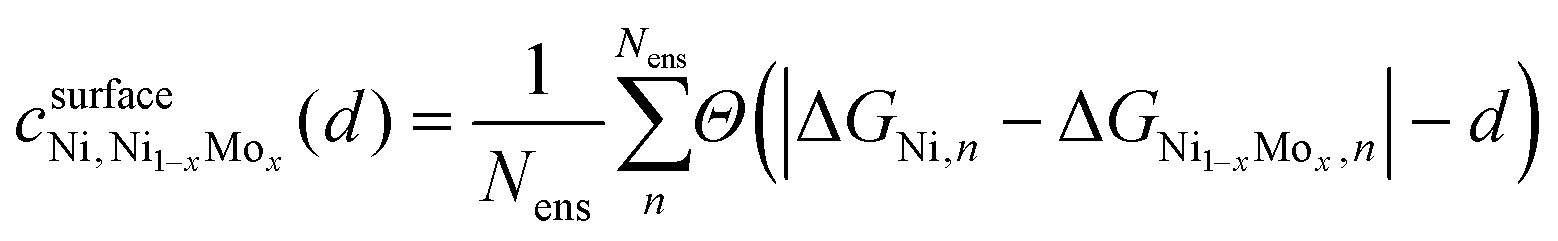
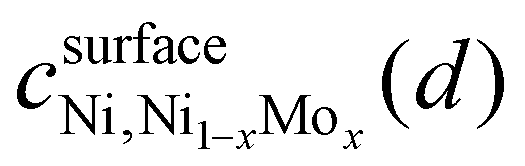 is the confidence that the adsorption energy difference between Ni and the Ni–Mo alloy is at least d eV. Applying this expression, confidence curves comparing the most thermodynamically favorable site of each alloy (i.e., the site with the most negative H-adsorption energy) to that of pure Ni are visualized in Fig. 8b, where a steeper decline is indicative of greater distinguishability between the adsorption energy distributions. At all considered Mo concentrations, a steep decline in the curve is observed over a range of ≤0.1 eV, implying a high degree of distinguishability. This further reinforces the proposed weakening of H-binding to surface Ni sites due to the presence of subsurface Mo atoms.
is the confidence that the adsorption energy difference between Ni and the Ni–Mo alloy is at least d eV. Applying this expression, confidence curves comparing the most thermodynamically favorable site of each alloy (i.e., the site with the most negative H-adsorption energy) to that of pure Ni are visualized in Fig. 8b, where a steeper decline is indicative of greater distinguishability between the adsorption energy distributions. At all considered Mo concentrations, a steep decline in the curve is observed over a range of ≤0.1 eV, implying a high degree of distinguishability. This further reinforces the proposed weakening of H-binding to surface Ni sites due to the presence of subsurface Mo atoms.
Motivated by previous successful applications of local geometric descriptors to predict adsorption energies, we also adopted an approach to further probe the influence of subsurface Mo.58–61 Each active hollow site consists of three surface Ni atoms with 9 nearest neighbors. The identity of each of these nearest neighbors, either Ni or Mo, modulates the character of the active site Ni by varying their local environments. Moreover, since the surface facet considered is fcc (111) across all slabs studied, there will always be a consistent number of nearest neighbors for the hollow active sites, with only the local composition changing. This constraint simplifies the development of a linear geometric model based on quantifying nearest-neighbors.
In consideration of these factors, we propose the following geometric model:
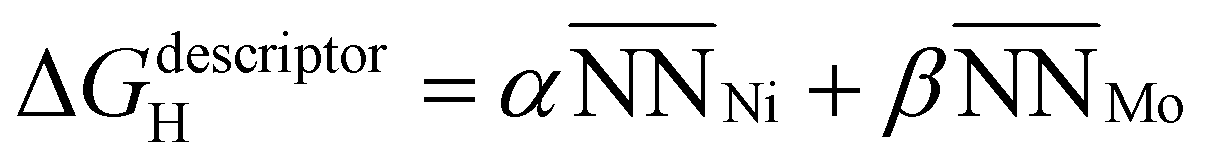
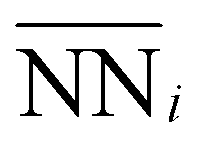 is the average number of species i neighboring each of the three Ni that constitute the active site. Fitting this to all obtained adsorption energies via a least squares regression, we found an α value of −0.015 eV and a β value of 0.057 eV. A negative value of α means that increasing the prevalence of Ni around each of the active site atoms approaches the pure Ni configuration, with a concomitant strengthening in H-binding. On the other hand, a positive value of β means increasing Mo occupancy around the active site in the sublayer weakens the H-adsorption, in agreement with the analysis above. Moreover, as the magnitudes of α and β are unequal, there is an imbalance on the effect of introducing Mo versus removing Ni, leading to a net decrease in H-adsorption energy with Mo subsurface doping. To gauge the accuracy of this geometric model, Fig. 8c depicts the results as a parity plot against the corresponding DFT-based free energies of H-binding. The relationship gives an R2 value of 0.53, indicating that the geometric approach indeed captures the trend. Hence, the results of these first-principles calculations clearly support the potential benefit of increasing the Mo content in Ni–Mo alloys, provided Mo remains stable in a subsurface configuration under reaction conditions. It also remains to be seen whether these general trends hold in the presence of surface oxide/hydroxide phases, which may enhance or inhibit HER/HOR activity.
is the average number of species i neighboring each of the three Ni that constitute the active site. Fitting this to all obtained adsorption energies via a least squares regression, we found an α value of −0.015 eV and a β value of 0.057 eV. A negative value of α means that increasing the prevalence of Ni around each of the active site atoms approaches the pure Ni configuration, with a concomitant strengthening in H-binding. On the other hand, a positive value of β means increasing Mo occupancy around the active site in the sublayer weakens the H-adsorption, in agreement with the analysis above. Moreover, as the magnitudes of α and β are unequal, there is an imbalance on the effect of introducing Mo versus removing Ni, leading to a net decrease in H-adsorption energy with Mo subsurface doping. To gauge the accuracy of this geometric model, Fig. 8c depicts the results as a parity plot against the corresponding DFT-based free energies of H-binding. The relationship gives an R2 value of 0.53, indicating that the geometric approach indeed captures the trend. Hence, the results of these first-principles calculations clearly support the potential benefit of increasing the Mo content in Ni–Mo alloys, provided Mo remains stable in a subsurface configuration under reaction conditions. It also remains to be seen whether these general trends hold in the presence of surface oxide/hydroxide phases, which may enhance or inhibit HER/HOR activity.
Conclusions
We synthesized catalytically active Ni–Mo composites supported on oxidized Vulcan carbon for reversible hydrogen evolution and oxidation under alkaline conditions. The mass specific HER activity of this catalyst, as measured via exchange current or a cathodic current at 100 mV overpotential, was within one order of magnitude of a commercial Pt/C catalyst composite. Similar results were obtained when comparing Ni–Mo/oC to Pt–Ru/C in HEM electrolyzer cathodes. Hydrogen oxidation activity was also comparable to Pt/C at overpotentials below 100 mV, but catalyst oxidation inhibits the HOR at larger overpotentials.We also observed persistent evidence for internal mass transfer limitations at catalyst loadings even as low at tens of μgcat cm−2, implying that our observations constitute a lower-bound estimate of the intrinsic HER/HOR activity of Ni–Mo composites. Further insights about intermediate energetics, obtained from density functional theory calculations, showed that increasing the Mo content in the Ni–Mo alloys weakens hydrogen adsorption and increases HER/HOR activity. This work points to several strategies to further close the gap in activity between precious and nonprecious HER catalysts, but considerable work remains in learning how to stabilize these catalysts under HOR conditions if they are to be used for unitized regenerative fuel cells. Further efforts to address durability and failure modes under HER conditions are also warranted in future studies on this catalyst system.62
Author contributions
Experimental design was conceptualized by R. B. P., J. R. M., J. L., Y. Y., and V. V. R. B. P. and M. K. performed catalyst synthesis, electroanalytical studies, and the associated data analysis. S. H. and J. Y. performed the TEM measurements and analysis. L. K., V. V., and D. K. performed the computational calculations and associated analysis. Y. Y. and K. H. developed and synthesized the HEM membranes and ionomers. S. Z. and J. L. performed the electrolyzer experiments and associated analysis. R. B. P., M. K., and J. R. M. compiled the data and prepared the manuscript. All the authors contributed to manuscript revisions.Conflicts of interest
K. H., Y. Y., S. Z., and J. L. are employed by companies that are commercializing hydroxide exchange membrane electrolyzers and/or components thereof.Acknowledgements
R. B. P., M. K., K. H., Y. Y., J. L., and J. R. M. acknowledge the U.S. Department of Energy for financial support of this work under award number DE-SC0020576. J. R. M. acknowledges additional support from the Arnold and Mabel Beckman Foundation. The electron microscopy work was performed at the Nanoscale Fabrication and Characterization Facility at the University of Pittsburgh. This research used resources of the Environmental TEM Catalysis Consortium (ECC), which is supported by the University of Pittsburgh and Hitachi High Technologies and S.D.H and J.Y are thankful for this support. D. K. and V.V. gratefully acknowledge funding support from the National Science Foundation under Award No. CBET-1554273. L. K. acknowledges the support of the Natural Sciences and Engineering Research Council of Canada (NSERC).References
- The Future of Hydrogen – Analysis, International Energy Agency, Paris, 2019.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- J. Bartlett and A. Krupnick, Decarbonized Hydrogen in the US Power and Industrial Sectors: Identifying and Incentivizing Opportunities to Lower Emissions, Resources for the Future, 2020.
- U.S. National Clean Hydrogen Strategy and Roadmap, U.S. Dept. of Energy, Washington, DC, 2023.
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y.-M. Chiang, C. T. M. Clack, A. Cohen, S. Doig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B.-M. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. E. Trancik, C.-J. Yang and K. Caldeira, Science, 2018, 360, eaas9793 CrossRef PubMed.
- Z. Pu, G. Zhang, A. Hassanpour, D. Zheng, S. Wang, S. Liao, Z. Chen and S. Sun, Appl. Energy, 2021, 283, 116376 CrossRef CAS.
- M. A. Hubert, L. A. King and T. F. Jaramillo, ACS Energy Lett., 2022, 7(1), 17–23 CrossRef CAS.
- S. Marini, P. Salvi, P. Nelli, R. Pesenti, M. Villa, M. Berrettoni, G. Zangari and Y. Kiros, Electrochim. Acta, 2012, 82, 384–391 CrossRef CAS.
- M. David, C. Ocampo-Martínez and R. Sánchez-Peña, J. Energy Storage, 2019, 23, 392–403 CrossRef.
- H. Ito, N. Miyazaki, S. Sugiyama, M. Ishida, Y. Nakamura, S. Iwasaki, Y. Hasegawa and A. Nakano, J. Appl. Electrochem., 2018, 48, 305–316 CrossRef CAS.
- G. Wang, W. Li, B. Huang, L. Xiao, J. Lu and L. Zhuang, ACS Appl. Energy Mater., 2019, 2, 3160–3165 CrossRef CAS.
- J. Zhang, T. Wang, P. Liu, Z. Liao, S. Liu, X. Zhuang, M. Chen, E. Zschech and X. Feng, Nat. Commun., 2017, 8, 15437 CrossRef CAS PubMed.
- M. Y. Gao, C. Yang, Q. B. Zhang, J. R. Zeng, X. T. Li, Y. X. Hua, C. Y. Xu and P. Dong, J. Mater. Chem. A, 2017, 5, 5797–5805 RSC.
- I. A. Raj, J. Mater. Sci., 1993, 28, 4375–4382 CrossRef CAS.
- P. Kuang, T. Tong, K. Fan and J. Yu, ACS Catal., 2017, 7, 6179–6187 CrossRef CAS.
- V. Pundir, A. Gaur, R. Kaur, A. Bagchi and V. Bagchi, Energy Adv., 2023, 2, 321–327 RSC.
- J. R. McKone, B. F. Sadtler, C. A. Werlang, N. S. Lewis and H. B. Gray, ACS Catal., 2013, 3, 166–169 CrossRef CAS.
- M. R. Shaner, J. R. McKone, H. B. Gray and N. S. Lewis, Energy Environ. Sci., 2015, 8, 2977–2984 RSC.
- E. L. Warren, J. R. McKone, H. A. Atwater, H. B. Gray and N. S. Lewis, Energy Environ. Sci., 2012, 5, 9653–9661 RSC.
- R. B. Patil, A. Mantri, S. D. House, J. C. Yang and J. R. McKone, ACS Appl. Energy Mater., 2019, 2, 2524–2533 CrossRef CAS.
- R. B. Patil, S. D. House, A. Mantri, J. C. Yang and J. R. McKone, ACS Catal., 2020, 10, 10390–10398 CrossRef CAS.
- P. M. Csernica, J. R. McKone, C. R. Mulzer, W. R. Dichtel, H. D. Abruña and F. J. DiSalvo, ACS Catal., 2017, 3375–3383 CrossRef CAS.
- J. C. Myland and K. B. Oldham, Anal. Chem., 2000, 72, 3972–3980 CrossRef CAS PubMed.
- J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dułak, L. Ferrighi, J. Gavnholt, C. Glinsvad, V. Haikola, H. A. Hansen, H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M. Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen, V. Petzold, N. A. Romero, J. Stausholm-Møller, M. Strange, G. A. Tritsaris, M. Vanin, M. Walter, B. Hammer, H. Häkkinen, G. K. H. Madsen, R. M. Nieminen, J. K. Nørskov, M. Puska, T. T. Rantala, J. Schiøtz, K. S. Thygesen and K. W. Jacobsen, J. Phys.: Condens. Matter, 2010, 22, 253202 CrossRef CAS PubMed.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef.
- J. H. Montoya and K. A. Persson, npj Comput. Mater., 2017, 3, 1–4 CrossRef CAS.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef CAS.
- M. Ångqvist, W. A. Muñoz, J. M. Rahm, E. Fransson, C. Durniak, P. Rozyczko, T. H. Rod and P. Erhart, Adv. Theory Simul., 2019, 2, 1900015 CrossRef.
- J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D. Landis, J. K. Nørskov, T. Bligaard and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235149 CrossRef.
- V. Petkov, Y. Maswadeh, J. A. Vargas, S. Shan, H. Kareem, Z.-P. Wu, J. Luo, C.-J. Zhong, S. Shastri and P. Kenesei, Nanoscale, 2019, 11, 5512–5525 RSC.
- E. Kemppainen, A. Bodin, B. Sebok, T. Pedersen, B. Seger, B. Mei, D. Bae, P. C. K. Vesborg, J. Halme, O. Hansen, P. D. Lund and I. Chorkendorff, Energy Environ. Sci., 2015, 8, 2991–2999 RSC.
- J. N. Hansen, H. Prats, K. K. Toudahl, N. Mørch Secher, K. Chan, J. Kibsgaard and I. Chorkendorff, ACS Energy Lett., 2021, 6, 1175–1180 CrossRef CAS.
- D. E. Brown, M. N. Mahmood, M. C. M. Man and A. K. Turner, Electrochim. Acta, 1984, 29, 1551–1556 CrossRef CAS.
- D. Li, E. J. Park, W. Zhu, Q. Shi, Y. Zhou, H. Tian, Y. Lin, A. Serov, B. Zulevi, E. D. Baca, C. Fujimoto, H. T. Chung and Y. S. Kim, Nat. Energy, 2020, 5, 378–385 CrossRef CAS.
- B. Motealleh, Z. Liu, R. I. Masel, J. P. Sculley, Z. Richard Ni and L. Meroueh, Int. J. Hydrogen Energy, 2021, 46, 3379–3386 CrossRef CAS.
- G. A. Lindquist, S. Z. Oener, R. Krivina, A. R. Motz, A. Keane, C. Capuano, K. E. Ayers and S. W. Boettcher, ACS Appl. Mater. Interfaces, 2021, 13, 51917–51924 CrossRef CAS PubMed.
- M. Sulaiman, E. Climent, A. Hammouti and A. Wachs, Chem. Eng. Sci., 2019, 199, 496–507 CrossRef CAS.
- Q. He, N. S. Suraweera, D. C. Joy and D. J. Keffer, J. Phys. Chem. C, 2013, 117, 25305–25316 CrossRef CAS.
- Y. Yang, X. Sun, G. Han, X. Liu, X. Zhang, Y. Sun, M. Zhang, Z. Cao and Y. Sun, Angew. Chem., Int. Ed., 2019, 58, 10644–10649 CrossRef CAS PubMed.
- E. S. Davydova, F. D. Speck, M. T. Y. Paul, D. R. Dekel and S. Cherevko, ACS Catal., 2019, 9, 6837–6845 CrossRef CAS.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, National Association of Corrosion, Houston, 2nd edn, 1974 Search PubMed.
- R. Patil, Q. Meng and J. McKone, ChemRxiv, 2021, preprint DOI:10.26434/chemrxiv-2021-t2qzn.
- B. L. Abrams, P. C. K. Vesborg, J. L. Bonde, T. F. Jaramillo and I. Chorkendorff, J. Electrochem. Soc., 2008, 156, B273 CrossRef.
- S. Kobayashi, D. A. Tryk and H. Uchida, Electrochem. Commun., 2020, 110, 106615 CrossRef CAS.
- W. Sheng, M. Myint, J. G. Chen and Y. Yan, Energy Environ. Sci., 2013, 6, 1509–1512 RSC.
- S. M. Alia and B. S. Pivovar, J. Electrochem. Soc., 2018, 165, F441–F455 CrossRef CAS.
- W. Sheng, A. P. Bivens, M. Myint, Z. Zhuang, R. V. Forest, Q. Fang, J. G. Chen and Y. Yan, Energy Environ. Sci., 2014, 7, 1719–1724 RSC.
- Y. Duan, Z.-Y. Yu, L. Yang, L.-R. Zheng, C.-T. Zhang, X.-T. Yang, F.-Y. Gao, X.-L. Zhang, X. Yu, R. Liu, H.-H. Ding, C. Gu, X.-S. Zheng, L. Shi, J. Jiang, J.-F. Zhu, M.-R. Gao and S.-H. Yu, Nat. Commun., 2020, 11, 4789 CrossRef CAS.
- N. N. T. Pham, S. G. Kang, H.-J. Kim, C. Pak, B. Han and S. G. Lee, Appl. Surf. Sci., 2021, 537, 147894 CrossRef CAS.
- K. K. Rao, Q. K. Do, K. Pham, D. Maiti and L. C. Grabow, Top. Catal., 2020, 63, 728–741 CrossRef CAS.
- A. V. Ruban, H. L. Skriver and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 15990–16000 CrossRef.
- A. J. Medford, J. Wellendorff, A. Vojvodic, F. Studt, F. Abild-Pedersen, K. W. Jacobsen, T. Bligaard and J. K. Nørskov, Science, 2014, 345, 197–200 CrossRef CAS PubMed.
- G. Houchins, D. Krishnamurthy and V. Viswanathan, MRS Bull., 2019, 44, 204–212 CrossRef.
- O. Vinogradova and V. Viswanathan, J. Phys. Chem. C, 2021, 125, 4468–4476 CrossRef CAS.
- Y. Nanba and M. Koyama, ACS Omega, 2021, 6, 3218–3226 CrossRef CAS PubMed.
- D. Friebel, V. Viswanathan, D. J. Miller, T. Anniyev, H. Ogasawara, A. H. Larsen, C. P. O’Grady, J. K. Nørskov and A. Nilsson, J. Am. Chem. Soc., 2012, 134, 9664–9671 CrossRef CAS PubMed.
- F. Calle-Vallejo, J. Tymoczko, V. Colic, Q. H. Vu, M. D. Pohl, K. Morgenstern, D. Loffreda, P. Sautet, W. Schuhmann and A. S. Bandarenka, Science, 2015, 350, 185–189 CrossRef CAS PubMed.
- B. Yan, D. Krishnamurthy, C. H. Hendon, S. Deshpande, Y. Surendranath and V. Viswanathan, Joule, 2017, 1, 600–612 CrossRef CAS.
- D. Li, A. R. Motz, C. Bae, C. Fujimoto, G. Yang, F.-Y. Zhang, K. E. Ayers and Y. Seung Kim, Energy Environ. Sci., 2021, 14, 3393–3419 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization data; compiled performance metrics for prior reports of Ni-based alkaline HER catalysts. See DOI: https://doi.org/10.1039/d3ya00140g |
| This journal is © The Royal Society of Chemistry 2023 |