
Peptide–polyurea hybrids: a platform for tunable, thermally-stable, and injectable hydrogels†
Jessica A.
Thomas
a,
Zachary R.
Hinton
 ab and
LaShanda T. J.
Korley
*ab
ab and
LaShanda T. J.
Korley
*ab
aDepartment of Materials Science and Engineering, University of Delaware, Newark, DE 19716, USA. E-mail: lkorley@udel.edu
bDepartment of Chemical and Biomolecular Engineering, University of Delaware, Newark, DE 19716, USA
First published on 14th September 2023
Abstract
Drawing inspiration from natural systems, such as the highly segmented structures found in silk fibroin, is an important strategy when designing strong, yet dynamic biomaterials. Polymer–peptide hybrids aim to incorporate the benefits of hierarchical polypeptide structures into synthetic platforms that are promising materials for hydrogel systems due to aspects such as their biocompatibility and structural tunability. In this work, we demonstrated the utility of poly(ethylene glycol) (PEG) peptide–polyurea hybrids as self-assembled hydrogels. Specifically, poly(ε-carbobenzyloxy-L-lysine)-b-PEG-b-poly(ε-carbobenzyloxy-L-lysine) and poly(β-benzyl-L-aspartate)-b-PEG-b-poly(β-benzyl-L-aspartate) triblock copolymers were used as the soft segments in linear peptide–polyurea (PPU) hybrids. We systematically examined the effect of peptide secondary structure and peptide segment length on hydrogelation, microstructure, and rheological properties of our PPU hydrogels. Polymers containing α-helical secondary structures resulted in rapid gelation upon the addition of water, as driven by hierarchical assembly of the peptide segments. Peptide segment length dictated gel strength and resistance to deformation via complex relationships. Simulated injection experiments demonstrated that PPU hydrogels recover their original gel network within 10 s of cessation of high shear. Finally, we showed that PPU hydrogels remain solid-like within the range of 10 to 80 °C; however, a unique softening transition occurs at temperatures corresponding to slight melting of secondary structures. Overall, this bioinspired PPU hybrid platform provides opportunities to design synthetic, bioinspired polymers for hydrogels with tunable microstructure and mechanics for a wide range of thermal and injection-based applications.
Introduction
Dynamic polymer networks are of great importance in the development of advanced biomaterials. The ability of a hydrated polymer network to respond to stimuli and withstand mechanical stress is vital for hydrogel materials to succeed in desirable applications, such as in tissue scaffolds and injectable therapeutics.1,2 Additionally, networks that are stable under certain environmental conditions, such as temperature, but can dissociate when exposed to a trigger, such as pH, are sought after for emerging applications, including for enzyme immobilization or stabilization of biopharmaceuticals.3 However, it often is challenging to balance the requirement of material dynamics with the need for robust mechanical properties, as hydrated non-covalent networks are often weak and do not perform well under multiple deformation cycles.4 New understanding of design principles for improving the mechanics of dynamic hydrogels is needed to address these shortcomings.5 Towards this goal, significant effort in the field has been focused on the development and study of non-covalent hydrogel platforms, most notably, peptide-based systems. Examples from natural systems, such as spider silk, harness hierarchical assembly through polypeptide interactions to achieve strong, yet dynamic materials.6–8 Polymer–peptide hybrids are ideal candidates for the development of robust biomaterials, as they are biocompatible, adaptable, and can impart specific structural or mechanical properties to hydrogel systems through self-assembly.9–11The most common synthetic routes toward peptide–polymer hybrid materials include solid-phase peptide synthesis (SPPS) or recombinant synthesis methods that incorporate a polymer conjugation step.12 While these methods often allow for precise control over peptide sequence structure, yields mostly remain limited to the laboratory scale, thereby hindering options for bulk material analysis, mechanical characterization, and eventual scale-up for applications. For example, Sarker et al. achieved silk-like, PEG–polyalanine assemblies through a strategy of combining SPPS with click chemistry protocols, but only high molecular weight species displayed self-assembly of the peptide motifs, which challenges wider use as current synthetic methods inherently limit chain length.13 In contrast, ring opening polymerization of N-carboxy anhydride peptide derivatives initiated by functional synthetic polymers results in limited control over peptide sequences, but have the advantage of being fast, scalable, and accommodating to high molecular weight hybrids.14
Beyond synthetic challenges, understanding the role of supramolecular interactions in polymer–peptide hybrid systems under aqueous conditions is key to designing dynamic hydrogels while maintaining control over material performance. For example, Wang and coworkers established that the interplay between peptide secondary structures and directional hydrogen bonding pendant groups allowed for control over the extent of self-assembly in poly(lactic acid-co-glycolic acid-co-ethylene oxide) copolymers, and by extension, moduli, toughness, and swelling ratios of resultant hydrogels via unique annealing protocols.15 Polyurethane–peptide hybrids combine uniquely blocky structures and high molecular weights with peptide hydrogen bonding and assembly to form elegant material architectures with potential synergy from inter-block interactions.16–18 For instance, Zhang et al. leveraged these property advantages to synthesize self-healing hybrid polyurethane hydrogels that form via co-assembly of peptide secondary structures and small drug molecules.19 To this end, polymers with intrinsic molecular, structural diversity enhanced by the incorporation of structure-forming peptide segments are of particular interest.
An additional class of nature-inspired materials, peptide–polyurea (PPU) hybrids, harness interactions between peptides and synthetic polymer blocks resulting in tunable mechanics and stimuli responsiveness in the solid state.20–22 This PPU platform offers an opportunity to develop dynamic hydrated networks, as it is facile to achieve higher molecular weights while also incorporating peptide functionalities that result in solid-state, tunable hierarchical assembly. However, these systems are not typically utilized as hydrated biomaterials with only limited understanding of the structural factors that influence hydrogel mechanics and morphology. In this work, we report hydrogels formed from self-assembled, linear PPUs. Block copolymers containing poly(ethylene glycol) and peptidic segments were synthesized with varying peptide repeat length within the chain. Poly(ε-carbobenzyloxy-L-lysine) (PZLY) and poly(β-benzyl-L-aspartate) (PBLA) were employed as the peptide segments to observe the effects of secondary structure on hydrogel self-assembly. Mechanical and microstructural characterizations were performed to demonstrate the tunability of this hydrogel platform for a variety of applications, including as injectable, thermally stable soft materials that rapidly gelate in water. Overall, we illustrate the applicability of polymer–peptide hybrids in forming robust hydrogels via multi-scale structures dictated by molecular design.
Experimental
Materials
Tetrahydrofuran (THF, Optima™ LC/MS grade), anhydrous N,N-dimethylacetamide (DMAc), deuterated chloroform, and diethyl ether were purchased from Fisher Scientific. THF was dried using a solvent purification system (Vacuum Atmosphere Company). Other solvents were used as received. ε-Carbobenzyloxy-L-lysine (ZLY), β-benzyl-L-aspartate (BLA), triphosgene, 1,6-hexamethylene diisocyanate (HDI) (≥99%), and dibutyltin dilaurate (DBTDL), were purchased from Sigma-Aldrich and used as received. Carbobenzyloxy-L-lysine N-carboxyanhydride (ZLY-NCA) and β-Benzyl-L-aspartate N-carboxyanhydride (BLA-NCA) were prepared following established literature procedures.15 α,ω-Bis(amine)poly(ethylene glycol) (PEG diamine, 3400 g mol−1) was purchased from SINOPEG (Xiamen, P. R. China) and dried under vacuum at room temperature for 16 h and then at 60 °C for >4 h prior to use. All glassware was thoroughly dried before use.Triblock synthesis
Polyurea–peptides were constructed from precursor triblock copolymers PZLY-b-PEG-b-PZLY and PBLA-b-PEG-b-PBLA, which were synthesized via ring opening polymerization of ZLY-NCA or BLA-NCA, respectively. The peptide triblocks were initiated by PEG diamine under nitrogen atmosphere in a glovebox, as previously reported.15 The ratio of NCA monomer to PEG diamine was used to target specific peptide repeat lengths (e.g., 5, 20, 40) (peptide units per block). As an example: for the 20 peptide repeat length PZLY-b-PEG-b-PZLY, PEG diamine (0.277 g) was pre-dissolved in THF (7 mL) in a glass vial. In an oven-dried 50 mL single-neck round bottom flask equipped with a magnetic stirrer, ZLY-NCA (1 g, 3.3 mmol) was dissolved in 25 vol% DMAc in THF (7 mL). The PEG diamine solution was then added to the flask dropwise before capping with a Vigreux condenser. The reaction proceeded for 20 h at 22 °C under stirring. The product polymer was precipitated into diethyl ether, collected via vacuum filtration, then dried under vacuum at 22 °C overnight.Peptide–polyurea synthesis
The resulting PZLY-b-PEG-b-PZLY or PBLA-b-PEG-b-PBLA triblock copolymers were used as the soft segment in non-chain extended PPUs by reacting with PEG diamine and hexamethylene diisocyanate.15 Samples are labeled with the nomenclature ZN-X, or AN-X where Z or A refer to PZLY or PBLA, respectively, N denotes the peptide repeat length, and X represents peptide weight percent in the polyurea. All polymerizations were conducted under nitrogen atmosphere in a glovebox. The final weight percent of peptide present in the polyurea was calculated through eqn S1 (ESI†). As an example, the synthesis of Z20-10 is described. Hexamethylene diisocyanate (HDI) (1 mol) was added to a solution of 25 vol% DMAc in THF (10 mL) in a 50 mL single-neck round bottom flask equipped with a magnetic stirrer. Z20-b-PEG-b-Z20 (0.20 g, 0.04 mol) and PEG diamine (1.25 g, 0.96 mol) were dissolved in 10 mL of 25 vol% THF in DMAc in a glass vial, along with 5 drops of DBTDL as a catalyst. The soft segment solution was then added dropwise to the round bottom flask, before capping with a Vigreux condenser. The reaction was allowed to proceed at 60 °C for 18 hours. The polymer was precipitated into diethyl ether, collected via vacuum filtration, and dried under vacuum at 22 °C overnight. A non-peptidic control (PEG–PU) also was prepared as follows: PEG diamine (1 g) was dissolved in 25 vol% THF in DMAc (8 mL) in a glass vial, and 5 drops of DBTDL were added. Separately, a solution of HDI (46 μL) in of 25/75 THF/DMAc (8 mL) was prepared. The PEG diamine + DBTDL solution was added dropwise to the HDI solution under stirring and then capped with a Vigreux condenser. The reaction was allowed to proceed at 60 °C for 20 h, after which the final polymer was precipitated into diethyl ether and dried under vacuum at 22 °C overnight.Nuclear magnetic resonance spectroscopy (NMR)
The structures of ZLY-NCA, BLA-NCA, and triblock copolymers were confirmed via1H nuclear magnetic resonance spectroscopy (1H-NMR). Samples were prepared in CDCl3, and spectra were collected on a Bruker AVIII 400 MHz spectrometer at room temperature. Spectra (Fig. S1 and S2) and peak assignments are shown in the ESI.† Data was processed using MNOVA software.Gel permeation chromatography (GPC)
The average molecular weights and molecular weight distributions of PPUs were measured using a TOSOH EcoSEC Elite GPC system equipped with a refractive index detector and TSKgel columns (three SuperH and one SuperAW5000 column). 0.08 mL of sample was injected and eluted at 0.4 mL min−1 with 0.5 wt% LiBr in DMAc used at the mobile phase. Distributions were generated using the calibration curve constructed for six poly(methyl methacrylate) standards (Agilent) in the range of 4.76 to 675.5 kg mol−1.Gel fabrication
To form the hydrogels, PPUs were weighed into glass vials to which appropriate amounts of 0.01 M PBS buffer (pH ∼ 7.4) were added to fabricate 5, 10, or 25 wt% solutions/gels, and mixed using an orbital shaker at 22 °C until homogenous. The resulting gels were clear and passed the gel inversion test (i.e., did not flow under gravity).Circular dichroism (CD) spectroscopy
CD spectra were measured for the PPUs in dilute conditions (2 mg mL−1 in deionized water) using a Jasco 1500 Spectrophotometer. Samples were shaken overnight and sonicated for 1 minute in 10 second pulses to ensure full dissolution. Sample solutions were loaded into quartz cuvettes (Hellma Analytics) with a path length of 1 mm. Three accumulations were taken for each sample at a scanning speed of 50 nm min−1 and data pitch of 1 nm. Temperature ramps were performed from 25 °C to 80 °C and then cooled back down to 25 °C, at a heating/cooling rate of 1 °C min−1. Data was processed using OriginLab software.Scanning electron microscopy (SEM)
SEM micrographs were taken on an Aurgia 60 Crossbeam microscope with a 3 kV acceleration voltage. Hydrogel samples were frozen with liquid nitrogen, lyophilized for 72 hours, and sputter-coated with gold/palladium for one minute prior to imaging. Pore diameters were measured using ImageJ software.Cryogenic transmission electron microscopy (Cryo-TEM)
Hydrogels were lightly touched to the surface of a 400-mesh lacey carbon grid, and then smeared by hand using filter paper to generate a thin area for imaging and then immediately plunged into liquid ethane using a Leica EMCPC. Samples were maintained at 170 °C using a Gatan Model 626 cryo-holder and imaged on a Zeiss Libra 120 transmission electron microscope operating at 120 kV using low dose imaging methods to prevent sample damage from high electron beam sensitivity. Images were acquired with a Gatan Ultrascan 1000 CCD camera. Fiber diameters were measured using ImageJ software (≥100 measurements per sample).Rheological characterization
The mechanical responses of the fabricated gels were measured on a stress-controlled rheometer (DHR-3, TA Instruments) using a parallel plate geometry (20 mm diameter) with Peltier heating. Samples were loaded with a spatula by placing the desired quantity on the bottom plate, which was already equilibrated at the testing temperature. The sample was then slowly squeezed to fill the gap between the two plates (nominally between 0.5 and 0.8 mm) and trimmed around the edge of the plate. The exposed edge of the sample was covered with a thin film of silicone oil to prevent evaporation of water during the measurement. Axial force and temperature were allowed to equilibrate prior to starting the experiment.First, mechanical characterization was performed on all gel systems at 37 °C. Samples were pre-sheared for 300 s at 0.1% strain amplitude and 1 rad s−1 to further equilibrate potential stresses due to loading. A frequency sweep was then run between 0.1 rad s−1 and 100 rad s−1 at a strain amplitude of 0.5%. This strain was selected a priori to avoid breakup of the gels prior to characterization. A strain amplitude sweep was then performed to validate that the chosen amplitude was within the linear viscoelastic regime (LVR) and to observe critical phenomena (i.e., breakup). This amplitude sweep was performed at 1 rad s−1, between 0.01% and 1000% strain; however, in some cases, the test was halted after a few points were measured beyond the critical strain to prevent significant fracture or slip from occurring. For all gels, the strain amplitudes selected for the frequency sweeps were within the LVR.
Next, injectability studies were carried out on gels after strain sweeps were complete. Although injectability is more accurately determined using a steady shear mode, edge fracture and slip of the gels between parallel plates preclude this method from measuring microstructural changes during shear.23,24 Instead, oscillatory shear was used for this study, during which six segments were performed. A 30 s shear at 1 rad s−1 and 200% strain amplitude was applied, immediately followed by a testing period at 1 rad s−1 and 0.1% strain. Moduli were observed, and the next step was initiated when both moduli remained constant. These two steps were then repeated. For the last steps, deformation occurred for 60 s at the same conditions used for the previous stages, followed by the same procedure to monitor re-formation. In all cases, a ‘fast sampling’ procedure was used such that the stress and strain waveforms are continuously acquired. This method enables faster observation of microstructural changes to the gel; however, the applied strain amplitudes and the resulting moduli go through an induction period (i.e., are not at steady state) because of start-up inertia. We expect that the initial building and overshoot of applied strain does not affect the interpretation of the resulting moduli. The strains applied during simulated breakup were above the measured critical values in all cases; however, 25 wt% Z5-10, and 10 wt% and 25 wt% Z40-10 were not subjected to injectability studies because of the higher critical strains required to break up the gel. Specifically, upon applying this strain, significant slip was observed likely due to the overshoot in amplitude at the start of the breakup cycle.
Finally, thermorheological properties were measured by changing the temperature during oscillation. The samples were cooled from 37 °C to 5 °C, heated from 5 °C to 80 °C, and cooled from 80 °C to 5 °C, all at a rate of 2 °C min−1. During this thermal cycling, a discrete frequency sweep was performed at 0.1% strain and 1, 10, and 100 rad s−1, such that a continuous temperature ramp at each frequency could be constructed. Although this narrow range of frequencies would normally be insufficient for quantification of gelation phenomena, no phase transition to liquid-like behavior was observed, eliminating the need for such an analysis of frequency-independent gelation. Instead, transition temperatures were identified by determining the intersection of two lines fit to the measured storage moduli between 20–35 °C and 65–80 °C.
Results & discussion
Synthesis of hybrid PPUs
This work draws on previous synthetic strategies to formulate PPUs with tunable secondary structure composition, mechanical properties, and morphology in the solid state.21,25 To facilitate macromolecular design that dictates self-assembly, flow properties, and structure of hydrated hybrids, a series of PPUs were synthesized. NCA ring opening polymerization initiated by PEG diamine was used to form the triblock precursors (step 1, Fig. 1), as this method is scalable and produces high yields of protein–polymer conjugates in comparison to other pathways, such as solid-phase peptide synthesis or recombinant methods.12,26 Peptide repeat length of each triblock precursor was confirmed via nuclear magnetic resonance (NMR) spectroscopy end group analysis of the carbobenzoxy group (benzylic protons, peak at ∼5.0 ppm) (Fig. S2, ESI†). The resulting triblocks were then used as the soft segment in non-chain extended polyureas by reacting with PEG diamine and HDI (step 2, Fig. 1). The final PPUs had number average molecular weights ranging from 81–105 kg mol−1 (Table S1, ESI†), which are in the range sufficient to result in inter- and intra-chain interactions known to dictate the mechanical properties of silk fibers.24,25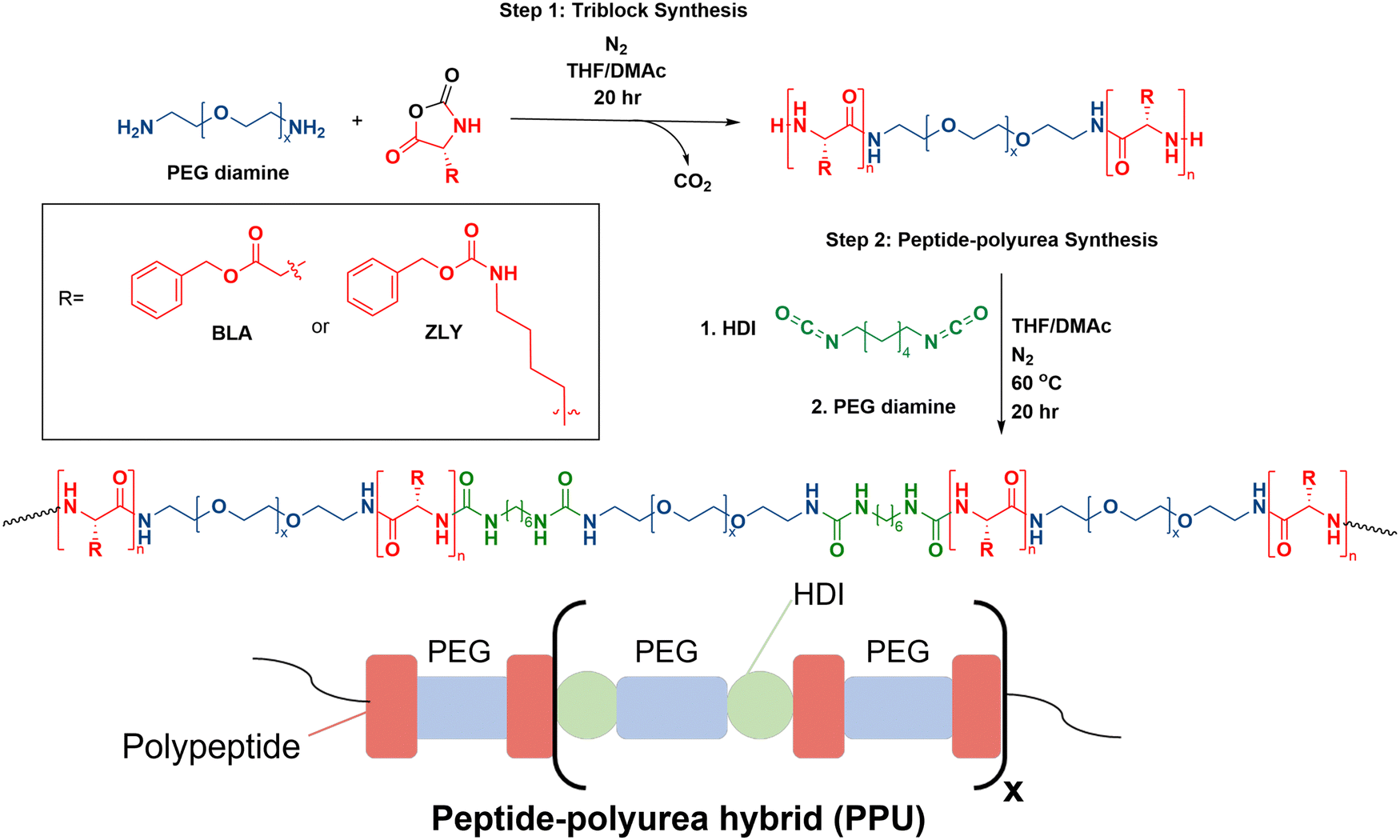 | ||
| Fig. 1 Overall reaction scheme of PPU hybrids and polymer structure. | ||
To examine the effects of secondary structure and hybrid chain architecture on hydrogelation, morphology, and mechanical properties, the synthesized series of PPUs were hydrated. A PBLA-b-PEG-b-PBLA triblock precursor with a peptide repeat length of 5, and three different PZLY-b-PEG-b-PZLY triblock precursors with peptide repeat lengths (5, 20, 40) were prepared to study the effect of peptide segment length on hydrogen bonding interactions between polymer chains and, by extension, hydrogel mechanics. Based on previous research, the PZLY segment length is expected to modulate the relative amount of α-helix/β-sheet structures present in the polyurea hybrids.21,25 The peptide content was maintained at 10 wt% in all PPU samples to directly target the impact of peptide secondary structure and chain length, as variations in peptide content also can influence PPU properties.20 Additionally, a control polyurea (PEG–PU) was prepared without peptide in the soft segment to examine the impact of PZLY and PBLA segments on gelation behavior. The average molecular weights and molecular weight distributions of the final PPUs were determined via gel permeation chromatography (GPC) to confirm all samples were of suitable molecular weight to result in hierarchical assembly (Fig. S3, ESI†).
Hydrogel formation and network morphology
To examine the gelation behavior of the PPU hybrids at various concentrations, solutions of Z5-10, Z20-10, Z40-10, and A5-10 polymers in PBS buffer were prepared at 5, 10, or 25 wt%. Solutions were then shaken at room temperature until homogenous and subjected to a vial inversion test, resulting in the Z series rapidly forming non-covalent hydrogels. In contrast, A5-10 did not form a hydrogel at these concentrations (Fig. S3, ESI†). A PEG–PU with a comparable molecular weight and hard segment content to the PPUs also was prepared as a control, but as shown in Fig. 2, did not form a gel at any of the prepared concentrations. These PEG–PU control results confirm that the mechanism of gelation involves peptide segments, and not simply hydrogen bonding between polymers, which is in agreement with Bilalis and co-workers.27 | ||
| Fig. 2 Vial inversion experiments of 10 wt% (a) Z5-10, (b) Z20-10, (c) Z40-10, and (d) PEG–PU polymers in 0.01 M PBS buffer. All PZLY hybrids form hydrogels, while the PEG–PU and PBLA (not shown) controls remain free-flowing solutions. SEM images of (e) Z5-10, (f) Z20-10, and (g) Z40-10 hydrogels reveal continuous porous networks. All micrographs were taken at 2k × magnification. | ||
The porosity of polymer hydrogels can have significant influence on the materials use in specific scenarios, such as serving as a scaffold for cell seeding or diffusion of therapeutic cargo.4 To examine the morphology of non-covalent PZLY-PEG PPU gels, scanning electron microscopy (SEM) was employed. In all samples, uniform porous structures were observed. Z5-10, Z20-10, and Z40-10 hydrogels displayed average pore sizes of approximately 30 μm, 7 μm, and 18 μm, respectively (Fig. 2(e)–(g)), which are in the range required for infiltration and proliferation of cell types, such as fibroblasts and skin cells.28 It has been frequently reported that the modulus of both covalent and non-covalent hydrogel networks is inversely proportional to pore size.29–31 However, as will be discussed in more detail in a later section, there is no significant correlation between the observed morphology or pore size with the modulus of these PPU dynamic hydrogels. This finding may indicate that the peptide secondary structure contributes more significantly to modulus than morphology. Thus, our results suggest that the mechanical properties of our system can be adjusted independently of pore size, which has important implications for the development of robust biomaterials without sacrificing diffusive properties.32,33
Secondary structure analysis and hydrogel microstructure
To gain insight on molecular interactions that drive the mechanical and morphological properties of the PPU dynamic hydrogel, we probed the secondary structure of the polyurea peptides (Z5-10, Z20-10, Z40-10, and A5-10) in solution via circular dichroism (CD) spectroscopy. All PZLY PPUs display negative peaks at 209 and 222 nm (Fig. 3), which are characteristic of α-helical conformations, whereas the non-gelating PBLA hybrid exhibits peaks indicative of β-turn conformations.34 Thus, it is clear that hydrogel formation is driven by specific interactions derived from the peptide secondary structure, not purely amphiphilic behavior that may arise from the segmented architectures or π–π stacking of the aromatic protecting groups.35,36 Based on prior understanding of non-hydrated PZLY-PEG PPUs, we would expect a mixture of α-helical and β-sheet secondary structures for Z5-10. A slight downturn in the CD spectrum can be seen at 215 nm, possibly indicating the presence of a small amount of β-sheets as a result of the short lysine segments; however, the dominating secondary structure is α-helical.37,38 We conclude that intramolecular α-helices are the driving force for gelation in PZLY-PEG PPU hydrogels via self-assembly of lysine segments, which has been observed in amphiphilic polylysine copolymer systems.39–41 Simultaneously, cooperative intramolecular bonding of α-helical domains can provide significant mechanical reinforcement, which has been shown to dictate properties, such as moduli and recovery of hybrid networks.27,42,43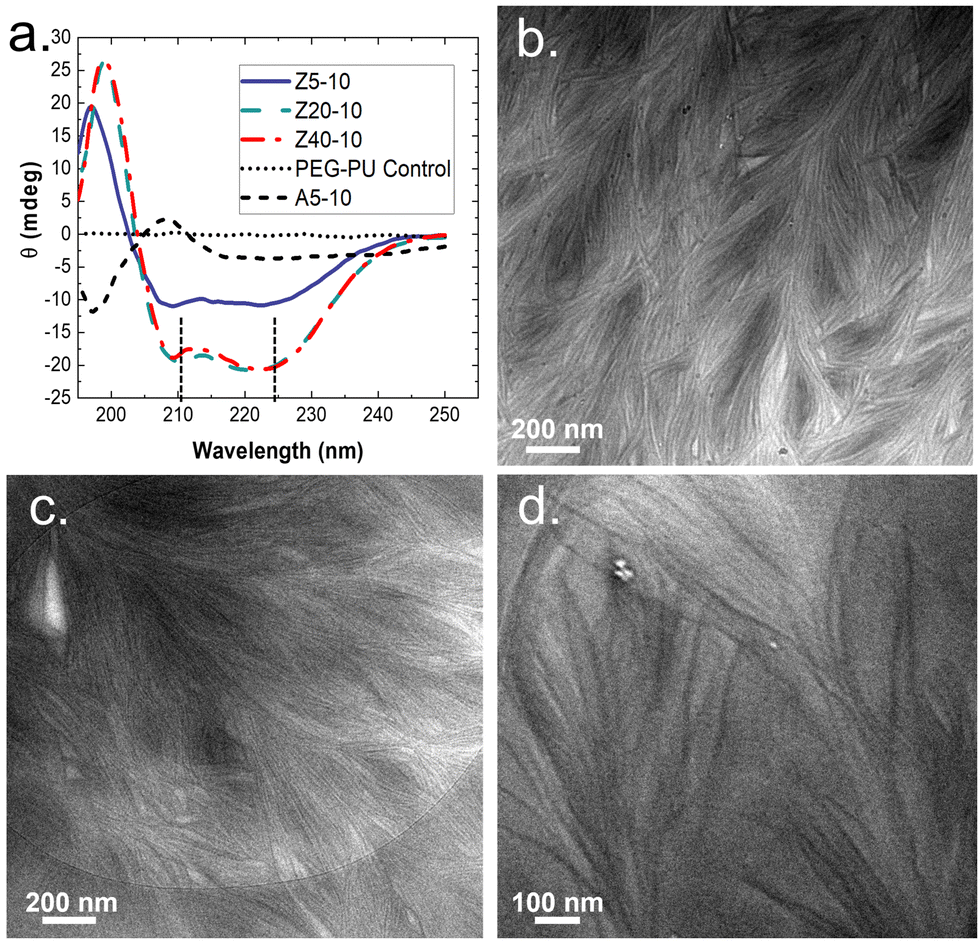 | ||
| Fig. 3 (a) CD Spectra of peptide-PEG polyureas. All measurements were taken at 37 °C at a concentration of 2 mg mL−1 (i.e., 0.2 wt%) of polymer in water. Z5-10, Z20-10, and Z40-10 all exhibit strong alpha helical patterns, with a positive cotton band present at 195 nm and characteristic negative peaks appearing at 208 and 222 nm, indicated with dashed lines. Cryo-TEM images of (b) Z5-10, (c) Z20-10 and (d) Z40-10 hydrogels. Dense networks of fibril structures can be observed in all samples. All samples were prepared at a concentration of 10 wt% polymer in water. | ||
Cryogenic transmission electron microscopy (cryo-TEM) was performed to further confirm the role of self-assembly in gelation. The micrographs, shown in Fig. 3(b)–(d), highlighted the presence of dense networks consisting of long fibrils in the PZLY-PEG PPU hydrogels. The individual fibrils display average diameters of 8–10 nm, which, considering that the diameter of a typical α-helix is about 1.2 nm, suggests multiple helices are associating to form the fibril networks driven by a non-covalent association between polymer chains.44 Homopolymers of PZLY have been shown to form α-helical fibrils resulting in aggregated networks, although this behavior is typically derived from significantly longer polypeptide chains than those present in our hybrid soft segments.45 In the case of the PZLY-PEG PPU hybrids, it is likely that the similarity of the α-helical pitch from both the PZLY segments and 72 PEG promote cooperative hierarchical assembly, which has been identified as the mechanism to induce ribbon-like assemblies in PEG-PBLA block copolymers.21 However, the fibrillar architectures observed in these PPU hydrogels are significantly longer and more ordered, which may be attributed to a combination of secondary structure hydrogen bonding of the peptide segment and the amphiphilic character of the PZLY and PEG blocks, resulting in packing of the nanofibers to form the hydrogel.42 The assembly of PPUs into long fiber bundles exhibits similarity to structures arising from the self-assembly of small peptide amphiphiles, where the mechanism of assembly relies first on the association of small peptide aggregates, followed by nanofiber growth and twisting into physically crosslinked hydrogels.46,47
Rheological behavior
Rheological measurements were used to assess the mechanical robustness of the non-covalent PZLY PPU gels and to investigate the effects of hybrid design and gel formulation on the mechanical response of the hydrogels at physiological temperatures. We note that salt concentration was kept at 0.01 M throughout all experiments in order to correlate hydrogel mechanics with observed circular dichroism results, which requires dilute solution for analysis. Increasing salt concentration could affect peptide segment assembly, however this is outside the scope of the current study. Gels were first subjected to strain sweeps to determine the linear viscoelastic regime and the critical strain amplitude (γc0), at which breakup of the hydrogel is initiated. As shown by Fig. 4(a), γc0 monotonically decreases with increasing peptide segment length, whereas the modulus (at fixed frequency) correlates directly with the peptide segment length. This trend is consistent with an increase in the apparent number of topological constraints (e.g., aggregate junctions, physical crosslinks), or a decrease in the length of strands between junction points, according to network theory.48 For the ZX series of PPU dynamic hydrogels, this relationship suggests that a significant portion of the PZLY segment is involved in the formation of supramolecular structure (e.g., aggregates, dynamically crosslinked network). In the absence of peptide segments, PEG–PUs of comparable molecular weights to the PPUs behave as liquids with relatively low viscosities (Fig. S5, ESI†). For all PZLY PPU hydrogels, the critical strain increased with increasing polymer concentration (Fig. 5(a)). For both Z5-10 and Z40-10, the critical strain steadily increases with concentration and exhibits an approximate order of magnitude increase from 10 wt% to 25 wt%; however, the critical strain plateaus at higher concentrations for Z20-10. This finding suggests that, for intermediate peptide segment lengths, increasing the peptide concentration of the solution does not increase the strain at which network breakup occurs (or the gel modulus). One explanation could be that additional peptide segments are able to aggregate such that the number of effective junctions remains constant upon increasing the polymer concentration.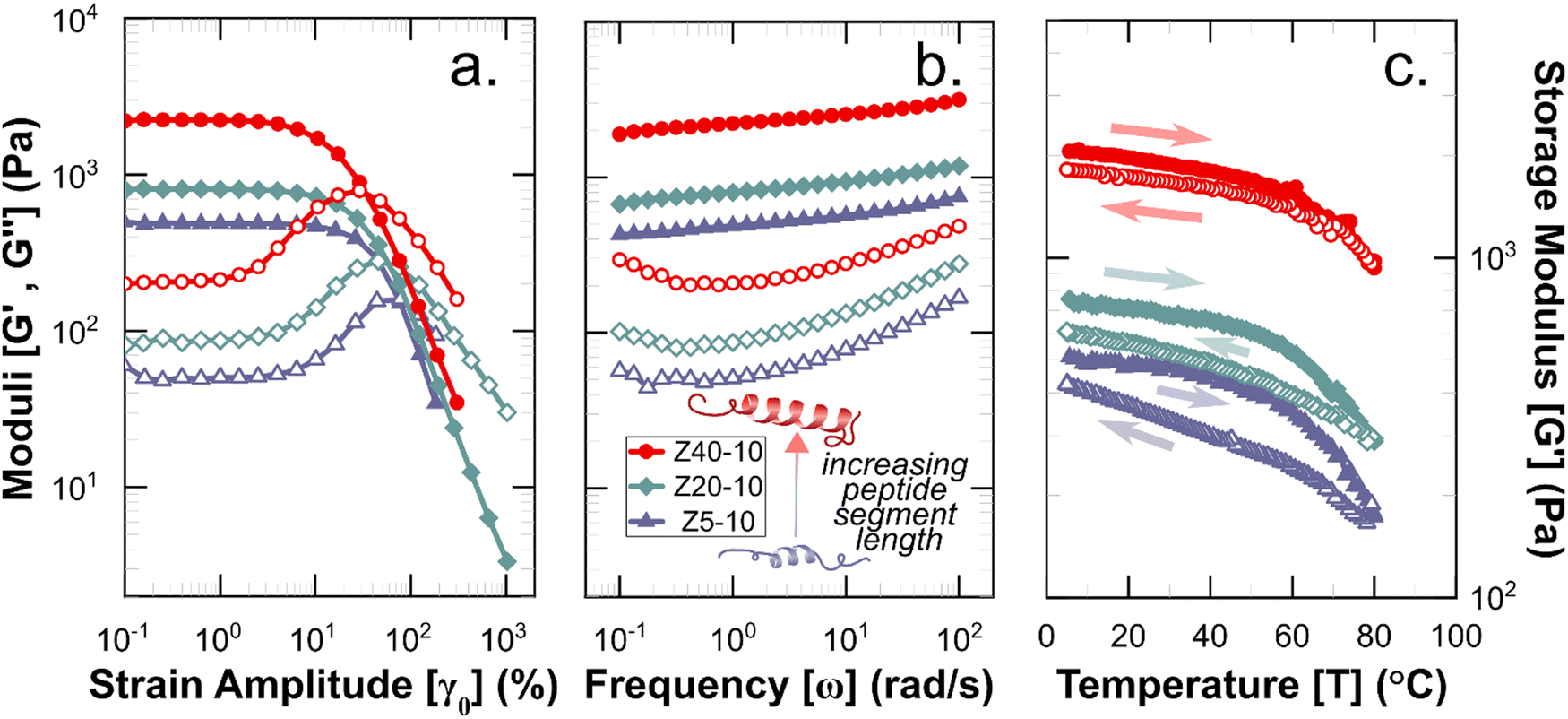 | ||
| Fig. 4 Rheological characterization of 10 wt% hydrogels with varying peptide segment length. (a) Strain amplitude and (b). Frequency responses, wherein filled symbols indicate storage moduli (G′) and open symbols indicate loss moduli (G′′). (c) Storage moduli during temperature ramps performed at ω = 1 rad s−1, with filled and open symbols corresponding to heating and cooling steps, respectively. Full datasets for amplitude and frequency sweeps are given in Fig. S6 (ESI†), and for temperature ramps in Fig. S11–S13 (ESI†). | ||
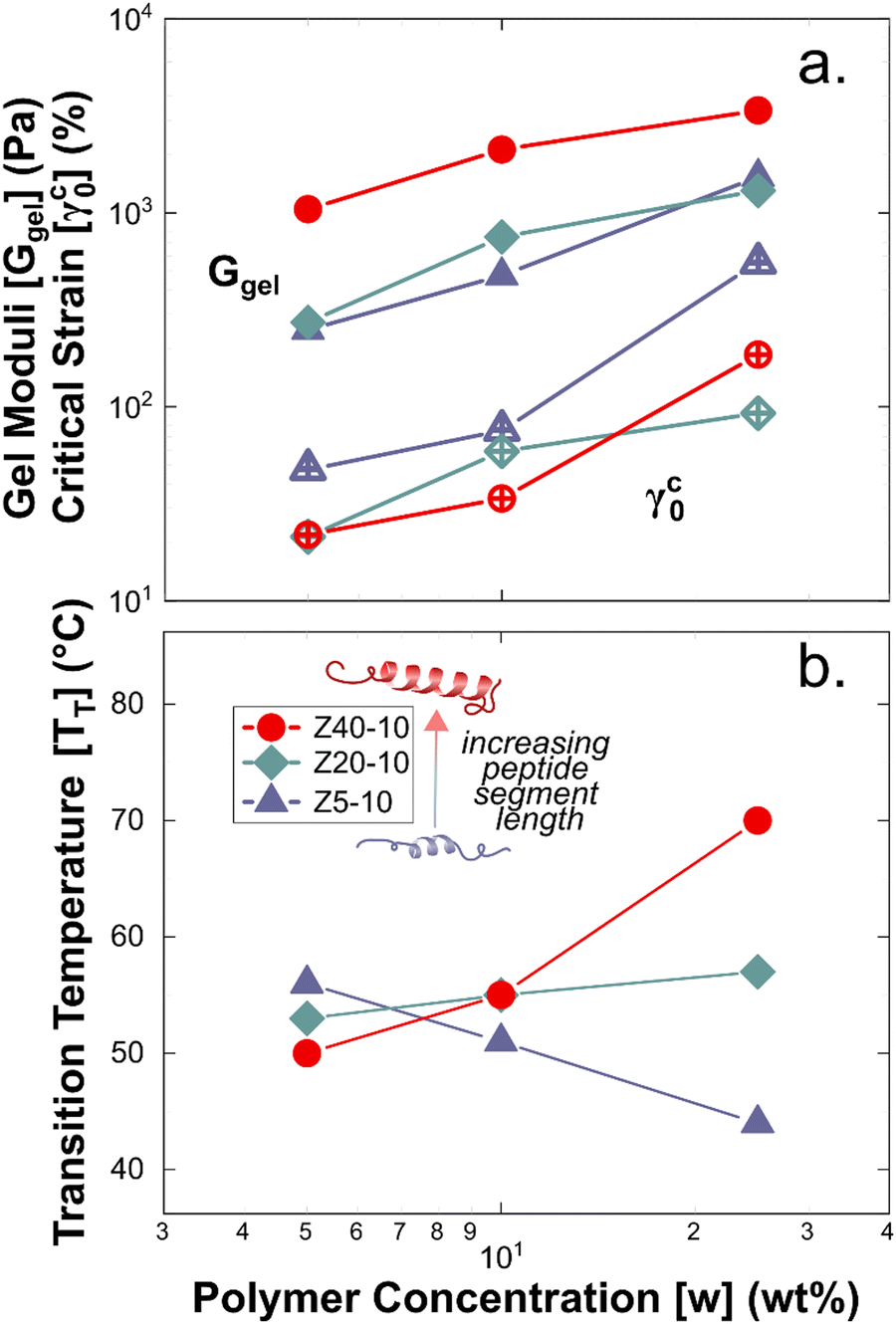 | ||
| Fig. 5 Summary of the rheology of PPU hydrogels as a function of concentration and peptide segment length. (a) Ggel (filled symbols) and γc0 (open symbols). (b) Transition temperature measured under increasing temperature at a frequency of 100 rad s−1. | ||
Frequency sweeps of the PPU ZX hydrogels confirm that in all cases a viscoelastic network is achieved, with a slight dependence of the storage modulus (G′) on frequency (ω). As shown in Fig. 4, for the 10 wt% hydrogels increasing PZLY length leads to increasing gel strength (i.e., the overall magnitude of G′ and the loss modulus (G′′)). Interestingly, the frequency dependence can be categorized by three different qualitative behaviors, depending on both PZLY length and concentration: (i) weak gel behavior where G′ approaches G′′ at low ω (i.e., transition to a fluid state is imminent); (ii) typical gel behavior where G′ is nearly independent of ω, and G′′ goes through a minimum, and (iii) transitional gel behavior where G′ approaches G′′ at high ω (i.e., a transitional regime is approached). Both Z5-10 and Z20-10 hydrogels exhibit moduli that follow cases i and ii with concentration dependence. For the Z40-10 gels, all three cases are observed, suggesting the formation of larger tertiary structures (i.e., fibrillar aggregates) at moderate concentrations that eventually become densely packed.49
The gel moduli [Ggel, defined as the complex modulus, G* = (G′2 + G′′2)1/2, at the minimum in G′′] of the hydrogels studied are summarized in Fig. 5(a). All hydrogels were of comparable stiffness to soft biological tissues, such as those found in the brain and liver.50 For Z5-10, Ggel follows a power-law in the polymer concentration, whereas with both Z20-10 and Z40-10, a slight plateau in Ggel is reached at the highest concentration. This plateau is strongest for Z40-10 (i.e., the smallest range of Ggel across concentrations is reached). The observed trend suggests that smaller peptide segments are able to form an aggregate structure that is less densely packed most likely because of the longer distance between peptide segments within the same chain and, thus, yield a stronger dependence of modulus on concentration.49 With a shorter spacing between the longer PZLY segments, there is a smaller contribution from additional PPU in solution to the overall hydrogel modulus, perhaps because these segments contribute to the same aggregates without significantly increasing their volume fraction or because larger aggregates are initiated at lower concentrations with sufficient size to generate a dense fibrillar hydrogel that is insensitive to packing fraction.49 This hypothesis is consistent with the cryo-TEM results in Fig. 3(d), which demonstrate a closely packed network of fibrils.
A key requirement for any material to be suitable for applications using injection processes is reversibility of the underlying structure (i.e., liquid-like flow followed by re-gelation), as clinical injections of such biomaterials are predicted to introduce significant shear, possibly up to 105 s−1.51,52 To quantify the potential of the PPU hydrogels to act as injectable biomaterials, a stepwise oscillatory experiment was performed to quantify the reformation rate of hydrogels after repeated application of non-linear strains. Fig. 6 illustrates how the PPU Z series hydrogels were deformed beyond γc0 for 30 s, during which there is a transition to liquid-like behavior (i.e., G′′ > G′). Under a smaller testing strain within the linear viscoelastic regime, solid-like behavior is regained in <10 s. Although injection applications are likely to impose steady shear flows, oscillation avoids unwanted edge fracture or slip at the plates while allowing for linear viscoelastic characterization. Importantly, 25 wt% Z5-10, and 10 and 25 wt% Z40-10 were not tested because overcoming γc0 required much higher strains, risking significant edge fracture. The 2nd cycle data shows the measured recovery is repeatable across multiple injections (i.e., the structure being reformed is an equilibrium state). The 3rd cycle, which shears the gel further from equilibrium using a longer deformation period, shows the order of magnitude difference in the moduli of the broken and reformed states. These results are consistent with similar experiments performed by Skoulas and coworkers, wherein amphiphilic PEG–peptide hybrid hydrogels with α-helical secondary structures recovered in the same timescale and by equivalent magnitudes when subjected to the same rheological protocol.53
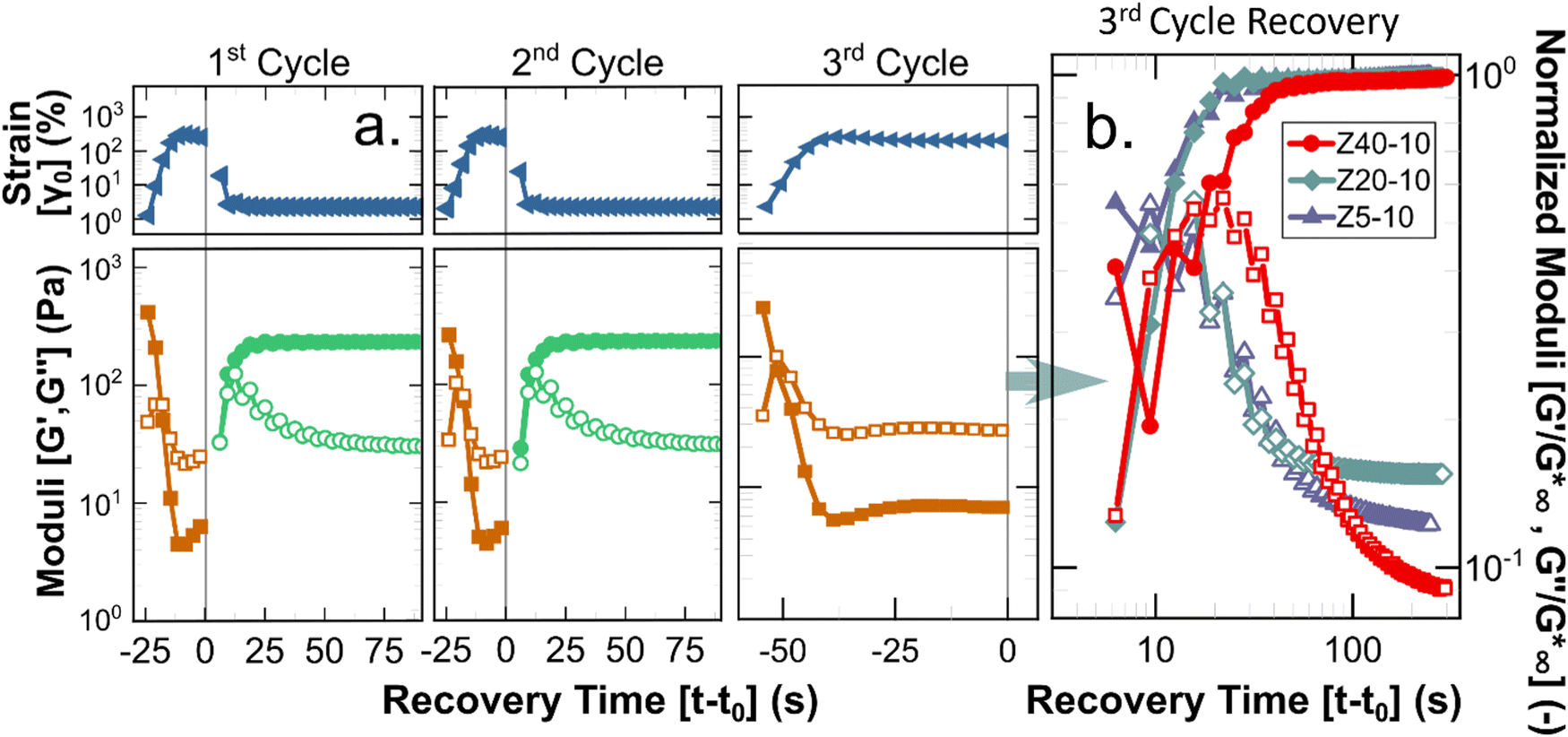 | ||
Fig. 6 Results of simulated injection experiments. (a) Three injection-recovery cycles performed for 5 wt% Z20-10 hydrogel. Recovery time is defined as the experimental time (t) relative to the cessation of a non-linear oscillatory strain (ending at t0). The recovery period from the 3rd cycle for all 5 wt% hydrogels is shown in (b), wherein filled and open symbols indicate G′ and G′′, respectively. Moduli have been normalized by the value of the complex modulus at full recovery ( ). ). | ||
Shear experiments after the 3rd breakup cycle provide additional insight into the dynamics of recovery for different PPU hydrogels. As highlighted in Fig. 6(b), the time required to achieve solid-like behavior is constant for Z5-10 and Z20-10 (∼10 s) and a factor of 2 larger for Z40-10. This extended recovery time is likely a function of the amount of strain applied relative to γc0, which is comparable for Z5-10 and Z20-10 and almost a factor of 2 smaller for Z40-10. The kinetics of recovery also are polymer concentration dependent, as shown by experimental data depicted in Fig. S7–S9 and summarized in Fig. S10 (ESI†). The trend observed with peptide segment length holds for different PPU concentrations, that is, recovery time is inversely related to γc0. These data highlight an important design criterion in the development of injectable hydrogels, whereby network reformation is dictated by the applied strain relative to a particular gel's breakup strain.
Thermal stability of hydrogels
A common feature of peptide-based hydrogels is thermal reversibility of structural features driven by the temperature dependence of peptidic assemblies, including gelation induced by both increasing and decreasing temperature.53,54 However, it is advantageous to retain a robust gel at a wide range of temperatures to enable stability under storage or incubation conditions outside of the body. As illustrated in Fig. 4(c), PZLY-PEG PPU hydrogels display remarkable resilience (i.e., minimal reduction in moduli) over a wide range of temperatures. Additionally, no transition to a liquid-like state (where G′′ > G′) is observed within the range of 10 to 80 °C (Fig. S11–S13, ESI†). A more rigorous analysis of frequency-independent gelation was attempted (Fig. S14, ESI†), but the convergence of the phase angles with temperature did not occur within the measured temperature range. This behavior suggests a potential disruption of the gel network is only theoretically possible outside of hydration. As shown in Fig. 4(c), hysteresis in G′ is measured between the heating and cooling cycles. This hysteretic behavior is most prominent at low frequency and is increasingly prominent with lower concentration and at a shorter peptide segment length. Temperature modulated CD measurements (Fig. 7) of the dilute Z20-10 PPU exhibit a similar hysteresis in the secondary structure content, suggesting that the observed thermorheological response is related to slight reorganization within self-assembled structures. Interestingly, no change to the secondary structure type (e.g., from α-helix to β-sheet) was observed, as may have been expected based on previous studies of polylysine in hydrated conditions.55 Thus, reorganization of inter-chain peptide assemblies reduces the density of effective network junctions and, therefore, modulus. Because the reorganization process presumably requires time for chains to diffuse, cooling cycles exhibit considerable hysteresis in moduli.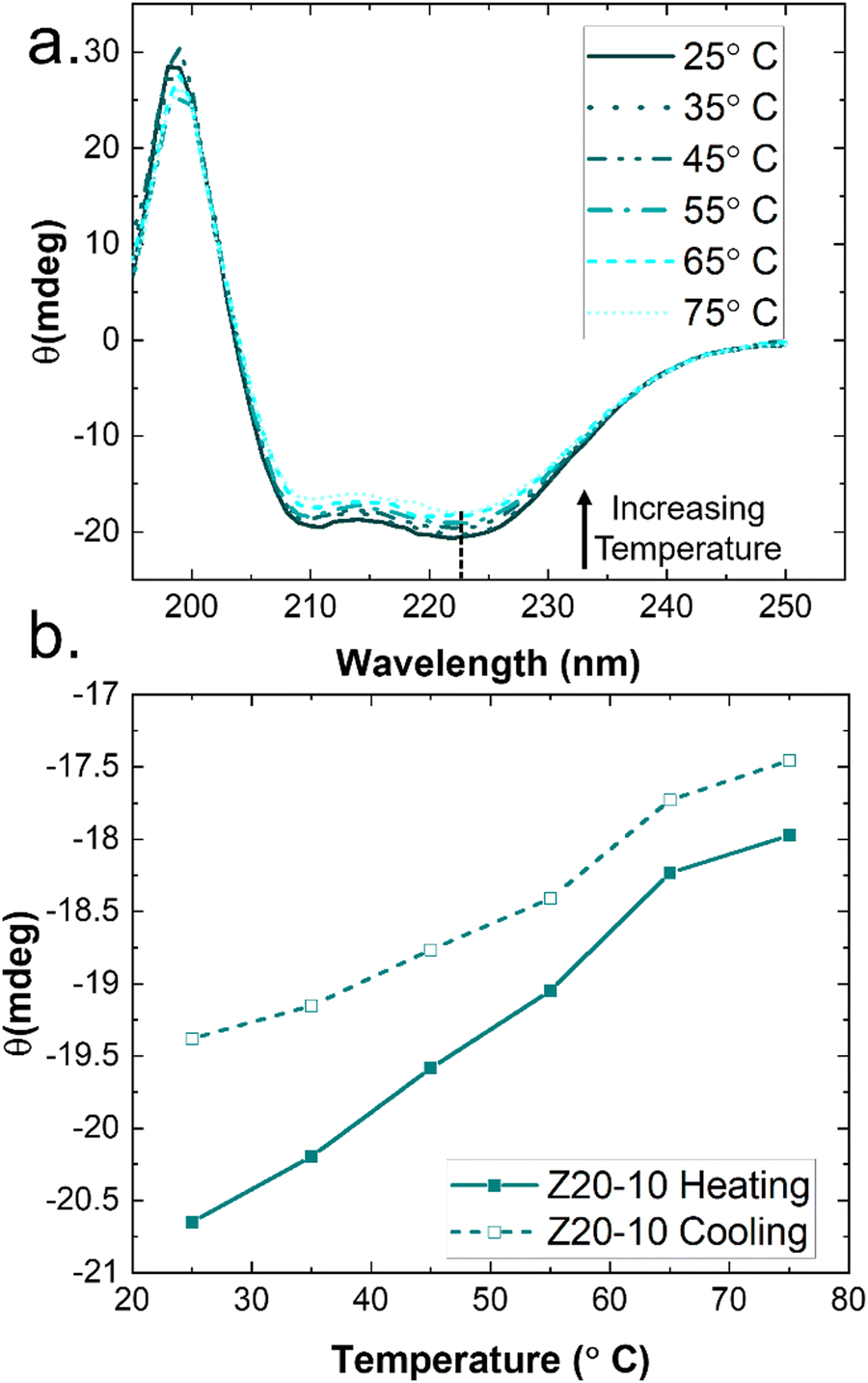 | ||
| Fig. 7 Temperature modulated CD spectra of Z20-10. (a) Full wavelength scan shows α-helical secondary structure is maintained over the entire temperature range; only very slight melting can be observed. (b) Monitoring the peak intensity at 222 nm reveals hysteresis in secondary structure recovery in a heating/cooling cycle. Points are taken from the temperature scans shown in (a). | ||
Interestingly, a weak thermal transition is observed in all of the measured PPU hydrogels, as most clearly indicated by an inflection in G′(T). The approximate transition temperatures (TT, at 100 rad s−1 under a heating ramp) of all hydrogels are shown in Fig. 5(b). It is important to note that the observed trends are not necessarily upheld at all frequencies, suggesting that the transition occurring most strongly affects fast modes in the network structure. For the shortest peptide segments (Z5-10), thermal stability is lost with increasing concentration. The temperature modulated CD experiments showed the least thermal dependence for Z5-10, which suggests that TT is likely related to both polymer chain hydration, conformation, and secondary structure formation, as has been seen in lysine-PEG diblocks.56 For Z20-10, negligible change to TT occurs as a function of concentration, and the value of TT corresponds to a sharp change to the α-helix peak intensity from CD. For long peptide segments (Z40-10), TT increases with increasing concentration, indicating that additional secondary structure forming segments contribute to thermal stability, perhaps through increasing aggregation of α-helices.
It is clear that the rheological behavior in the PPU hydrogels is a complex function of composition due to the hierarchical structures afforded by hybrid polymers. With the peptide secondary structure in mind, the observed mechanical properties of the networks can be attributed to a combination of polymer entanglement, inter-chain hydrogen bonding, and the α-helical conformations of PZLY, where the α-helices may act as entropic springs, contributing to hydrogel stiffness and recovery.40,53,57 However, additional polymer configurations (e.g., micelles) also may be possible at some compositions and conditions. Furthermore, ternary strucutures (i.e., fibrils) also are apparent from cryo-TEM and thermorheological studies. Overall, the the utility of our PPU hydrogels has been demonstrated as a platform to develop, control, and understand the effects of hybrid polyurea architecture on gel assembly and network dynamics.
Conclusions
In this work, we have shown that silk-inspired PZLY-PEG PPUs can be harnessed as a platform for dynamic, physically assembled hydrogels. These high molecular weight polymer–peptide hybrids facilitate hierarchical assembly, including the formation of α-helical secondary structures and higher order fibrillar networks. Varying peptide segment length in PZLY-PEG PPUs shows little impact on secondary structure conformation, but enables concentration-dependent tailoring of gel modulus and yield strain. Both the secondary structures and resultant mechanics of the hydrogels are stable up to temperatures of 80 °C. Additionally, the porosity of the PZLY-PEG PPU hydrogel networks is tunable independent of modulus, and the underlying hydrogel structure rapidly recovers after strong shear flows. Overall, these properties highlight the versatility of a PPU platform for non-covalent hydrogels with multi-scale structure, with potential applications as a stabilizing matrix for biomolecules prone to aggregation. Further studies will enable understanding of structural development of these materials, including small-angle X-ray scattering (SAXS) to probe multi-scale organization in these hydrogel systems. Possible opportunities to expand the usefulness of hybrid polyurea gels could include adding stimuli-responsive functionalities, bioconjugation with molecules of interest, and fitting to models of injectability to probe desired material specifications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Science Foundation (NSF) PIRE: bio-inspired Materials and Systems under grant number OISE 1844463. The authors acknowledge the use of facilities and instrumentation supported by NSF through the University of Delaware Materials Research Science and Engineering Center DMR 2011824. Cryo-TEM access was supported by grants from the NIH-NIGMS (P20 GM103446), the NIGMS (P20 GM139760), and the State of Delaware. We thank Shannon Modla in the DBI Bio-Imaging Center for assistance with obtaining cryo-TEM images. Access to the SEM was provided by the Keck Microscopy Center at the University of Delaware.References
- S. J. Buwalda, T. Vermonden and W. E. Hennink, Biomacromolecules, 2017, 18, 316–330 CrossRef CAS PubMed.
- O. Chaudhuri, L. Gu, D. Klumpers, M. Darnell, S. A. Bencherif, J. C. Weaver, N. Huebsch, H. P. Lee, E. Lippens, G. N. Duda and D. J. Mooney, Nat. Mater., 2016, 15, 326–334 CrossRef CAS PubMed.
- C. M. Meis, E. E. Salzman, C. L. Maikawa, A. A. A. Smith, J. L. Mann, A. K. Grosskopf and E. A. Appel, ACS Biomater. Sci. Eng., 2021, 7, 4221–4229 CrossRef CAS PubMed.
- S. Correa, A. K. Grosskopf, H. L. Hernandez, D. Chan, A. C. Yu, L. M. Stapleton and E. A. Appel, Chem. Rev., 2020, 121, 11385–11457 CrossRef PubMed.
- K. Zhang, Q. Feng, Z. Fang, L. Gu and L. Bian, Chem. Rev., 2021, 121, 11149–11193 CrossRef CAS PubMed.
- H. Fan and J. P. Gong, Macromolecules, 2020, 53, 2769–2782 CrossRef CAS.
- L. Montero De Espinosa, W. Meesorn, D. Moatsou and C. Weder, Chem. Rev., 2017, 117, 12851–12892 CrossRef CAS PubMed.
- K. Numata, Polym. J., 2020, 52, 1043–1056 CrossRef CAS.
- G. Ghosh, R. Barman, J. Sarkar and S. Ghosh, J. Phys. Chem. B, 2019, 123, 5909–5915 CrossRef CAS PubMed.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- R. Otter and P. Besenius, Org. Biomol. Chem., 2019, 17, 6719–6734 RSC.
- I. W. Hamley, Biomacromolecules, 2014, 15, 1543–1559 CrossRef CAS PubMed.
- A. Sarkar, C. Edson, D. Tian, T. D. Fink, K. Cianciotti, R. A. Gross, C. Bae and R. H. Zha, Biomacromolecules, 2021, 22, 95–105 CrossRef CAS PubMed.
- Ö. Ucak, G. Hause and W. H. Binder, Macromol. Chem. Phys., 2023, 224, 2200344 CrossRef.
- Q. Wang, Z. Shi, Y. Shou, K. Zhang, G. Li, P. Xia, S. Yan and J. Yin, ACS Biomater. Sci. Eng., 2020, 6, 1715–1726 CrossRef CAS PubMed.
- M. Kamaci, Eur. Polym. J., 2020, 123, 109444 CrossRef CAS.
- L. E. Matolyak, J. K. Keum, K. M. Van De Voorde and L. T. J. Korley, Org. Biomol. Chem., 2017, 15, 7607–7617 RSC.
- B. D. Ulery, L. S. Nair and C. T. Laurencin, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 832–864 CrossRef CAS PubMed.
- F. Zhang, C. Hu, Q. Kong, R. Luo and Y. Wang, ACS Appl. Mater. Interfaces, 2019, 37147–37155 CrossRef CAS PubMed.
- L. Matolyak, J. Keum and L. S. T. J. Korley, Biomacromolecules, 2016, 17, 3931–3939 CrossRef CAS PubMed.
- L. E. Matolyak, C. B. Thompson, B. Li, J. K. Keum, J. E. Cowen, R. S. Tomazin and L. S. T. J. Korley, Biomacromolecules, 2018, 19, 3445–3455 CrossRef CAS PubMed.
- D. Jang, C. B. Thompson, S. Chatterjee and L. T. J. Korley, Mol. Syst. Des. Eng., 2021, 6, 1003–1015 RSC.
- B. L. Walter, J. P. Pelteret, J. Kaschta, D. W. Schubert and P. Steinmann, Polym. Test., 2017, 61, 430–440 CrossRef CAS.
- E. J. Hemingway and S. M. Fielding, J. Rheol., 2019, 750, 735–750 CrossRef.
- J. C. Johnson, N. D. Wanasekara and L. T. J. Korley, J. Mater. Chem. B, 2014, 2, 2554–2561 RSC.
- A. Isidro-Llobet, M. N. Kenworthy, S. Mukherjee, M. E. Kopach, K. Wegner, F. Gallou, A. G. Smith and F. Roschangar, J. Org. Chem., 2019, 84, 4615–4628 CrossRef CAS PubMed.
- P. Bilalis, D. Skoulas, A. Karatzas, J. Marakis, A. Stamogiannos, C. Tsimblouli, E. Sereti, E. Stratikos, K. Dimas, D. Vlassopoulos and H. Iatrou, Biomacromolecules, 2018, 19, 3840–3852 CrossRef CAS PubMed.
- K. Whang, D. Ph, K. E. Healy, D. Ph, D. R. Elenz and E. K. Nam, Tissue Eng., 1999, 5, 35–51 CrossRef CAS PubMed.
- A. Panitch, J. Chmielewski and C. M. R. Pe, Macromol. Biosci., 2011, 11, 1426–1431 CrossRef PubMed.
- M. K. Lee, M. H. Rich, K. Baek, J. Lee and H. Kong, Sci. Rep., 2015, 5, 1–7 Search PubMed.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS PubMed.
- I. L. Chin, Z. Wei, H. Li, R. Atkin and S. Choi, Chem. Commun., 2021, 57, 773–776 RSC.
- A. E. Rowan and P. H. J. Kouwer, Nat. Commun., 2018, 9, 1–8 CrossRef PubMed.
- N. J. Greenfield, Nat. Protoc., 2007, 1, 2876–2890 CrossRef PubMed.
- V. Castelletto and I. W. Hamley, Biophys. Chem., 2009, 141, 169–174 CrossRef CAS PubMed.
- S. Santra, S. Kolay, S. Sk, D. Ghosh, A. Mishra, L. Roy, K. Sarkar and M. R. Molla, Polym. Chem., 2022, 13, 3294–3303 RSC.
- C. Bonduelle, Polym. Chem., 2018, 9, 1517–1529 RSC.
- J. U. Izunobi and C. L. Higginbotham, Polym. Int., 2013, 62, 1169–1178 CrossRef CAS.
- J. S. Lee, M. J. Kang, J. H. Lee and D. W. Lim, Biomacromolecules, 2022, 23, 2051–2063 CrossRef CAS PubMed.
- S. C. O. Neill, Z. H. Bhuiyan and R. S. Tu, Soft Matter, 2017, 13, 7521–7528 RSC.
- C. Cai, J. Lin, T. Chen, X. S. Wang and S. Lin, Chem. Commun., 2009, 2709–2711 RSC.
- C. Chen, J. Lan, Y. Li, D. Liang, X. Ni and Q. Liu, Chem. Mater., 2020, 32, 1153–1161 CrossRef CAS.
- C. Lu, L. Jiang, W. Xu, F. Yu, W. Xia, M. Pan, W. Zhou, X. Pan, C. Wu and D. Liu, Colloids Surf., B, 2019, 182, 110384 CrossRef CAS PubMed.
- R. R. Sinden, DNA Structure and Function, 1994, pp. 287–335 Search PubMed.
- K. Cie, Biochime, 2017, 137, 106–114 CrossRef PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5133–5138 CrossRef CAS PubMed.
- H. S. Liao, J. Lin, Y. Liu, P. Huang, A. Jin and X. Chen, Nanoscale, 2016, 8, 14814–14820 RSC.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, 2003 Search PubMed.
- G. M. Conley, C. Zhang, P. Aebischer, J. L. Harden and F. Scheffold, Nat. Commun., 2019, 10, 1–8 CrossRef CAS PubMed.
- I. Levental, C. Georges and P. A. Janmey, Soft Matter, 2007, 3, 299–306 RSC.
- M. H. Chen, L. L. Wang, J. J. Chung, Y. H. Kim, P. Atluri and J. A. Burdick, ACS Biomater. Sci. Eng., 2017, 3, 3146–3160 CrossRef CAS PubMed.
- H. Lopez, J. W. Souza and E. A. Appel, Macromol. Biosci., 2021, 21, 2000295 CrossRef PubMed.
- D. Skoulas, E. Stratikos, D. Vlassopoulos, H. Frielinghaus and H. Iatrou, Macromolecules, 2021, 54, 10786–10800 CrossRef CAS.
- E. S. Gil, R. J. Spontak and S. M. Hudson, Macromol. Biosci., 2005, 5, 702–709 CrossRef CAS PubMed.
- W. Dzwolak, R. Ravindra, C. Nicolini and R. Jansen, J. Am. Chem. Soc., 2004, 126, 3762–3768 CrossRef CAS PubMed.
- A. Harada, S. Cammas and K. Kataoka, Macromolecules, 1996, 9297, 6183–6188 CrossRef.
- M. Kageshima, M. A. Lantz and S. P. Jarvis, Chem. Phys. Lett., 2001, 343, 77–82 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sm00780d |
| This journal is © The Royal Society of Chemistry 2023 |