
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral gem-difluoroalkyl reagents: gem-difluoroalkyl propargylic borons and gem-difluoroalkyl α-allenols†
Hui-Na
Zou
,
Meng-Lin
Huang
,
Ming-Yao
Huang
,
Yu-Xuan
Su
,
Jing-Wei
Zhang
,
Xin-Yu
Zhang
and
Shou-Fei
Zhu
 *
*
Frontiers Science Center for New Organic Matter, State Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: sfzhu@nankai.edu.cn
First published on 8th August 2023
Abstract
Chiral fluorinated reagents provide new opportunities for the discovery of drugs and functional materials because the introduction of a fluorinated group significantly alters a molecule's physicochemical properties. Chiral gem-difluoroalkyl fragments (R–CF2–C*) are key motifs in many drugs. However, the scarcity of synthetic methods and types of chiral gem-difluoroalkyl reagents limits the applications of these compounds. Herein, we report two types of chiral gem-difluoroalkyl reagents chiral gem-difluoroalkyl propargylic borons and gem-difluoroalkyl α-allenols and their synthesis by means of methods involving rhodium-catalyzed enantioselective B–H bond insertion reactions of carbenes and Lewis acid-promoted allenylation reactions. The mild, operationally simple method features a broad substrate scope and good functional group tolerance. These two types of reagents contain easily transformable boron and alkynyl or allenyl moieties and thus might facilitate rapid modular construction of chiral molecules containing chiral gem-difluoroalkyl fragments and might provide new opportunities for the discovery of chiral gem-difluoroalkyl drugs and other functional molecules.
Introduction
Fluorine-containing compounds have unusual physicochemical properties and have had a considerable impact on the discovery of new medicines, agrochemicals, catalysts, and functional materials.1 Thus, the development of fluorine-containing building blocks has recently been receiving increasing attention. The gem-difluoromethylene group is considered to be a bioisostere2 of carbonyl groups and oxygen atoms of ethers and can modulate the pKa of neighboring functional groups.3gem-Difluoroalkyl groups (–CF2–R) are key moieties in many fluorine-containing drugs, including lubiprostone,4 oteseconazole,5 vinflunine,6 and gemcitabine7 (Scheme 1a). The introduction of a gem-difluoroalkyl group into bioactive molecules is an effective strategy for studying structure–activity relationships and tuning the pharmacological activity of drugs and drug candidates.8 | ||
| Scheme 1 Background and strategy. | ||
The efficient construction of chiral gem-difluoroalkyl compounds has attracted substantial research interest over the past few decades.9 However, the types of chiral gem-difluoroalkyl compounds are still limited in number because of lack of efficient synthetic methods. Since organoboron compounds,10 alkynes, and allenes11 are common building blocks in organic synthesis, gem-difluoroalkyl-substituted chiral boron compounds and allenes are expected to become novel chiral gem-difluoroalkyl reagents. To our knowledge, there is only one catalytic method for the synthesis of boron-substituted chiral gem-difluoroalkyl compounds (Scheme 1b),12 while chiral gem-difluoroalkyl propargylic borons and chiral gem-difluoroalkyl-substituted allenes remain unknown. Therefore, the development of efficient, convenient methods for the synthesis of easily transformable chiral gem-difluoroalkyl-substituted boron compounds bearing alkyne and allene motifs would be highly desirable. Herein, we report a method for dirhodium-catalyzed B–H bond insertion reactions using gem-difluoroalkyl alkynyl N-triftosylhydrazones as carbene precursors for the preparation of a wide range of novel, stable chiral gem-difluoroalkyl propargylic borons in high yields with high enantioselectivities (Scheme 1c). We also developed a method for BF3·Et2O-promoted allenylation of aldehydes with a chiral gem-difluoroalkyl propargylic boron; this method offers rapid access to a wide range of chiral gem-difluoroalkyl α-allenols with adjacent axial and central chiralities. These two types of chiral gem-difluoroalkyl reagents, which contain easily transformable boron and alkynyl or allenyl moieties, have high value for facilitating the rapid, modular construction of chiral molecules containing gem-difluoroalkyl groups. We demonstrated the synthetic potential of the gem-difluoroalkyl α-allenols by transforming one of them into chiral gem-difluoroalkyl 2,5-dihydrofuran and tetrahydrofuran derivatives.
Results and discussion
Inspired by our earlier work on asymmetric B–H bond insertion,13 we hypothesized that gem-difluoroalkyl alkynyl N-triftosylhydrazones could serve as carbene precursors for the construction of chiral gem-difluoroalkyl reagents through asymmetric B–H bond insertion reactions. We began by using gem-difluoroalkyl alkynyl N-triftosylhydrazone 1a as a model substrate, trimethylamine–borane adduct 2a as a boron source, and NaH as a base (Table 1). First, we evaluated commercially available chiral dirhodium catalysts 4a–4i (0.5 mol%) in reactions at 0 °C in Et2O (entries 1–9). Of the tested catalysts, 4d gave the highest yield and enantioselectivity (85% yield, 89% ee, entry 4). We evaluated several alternative bases (entries 10–13) and found that they significantly decreased the yield but had little influence on the enantioselectivity. The solvent screening revealed that the weakly coordinating solvent methyl tert-butyl ether improved the yield to 99% (entry 14). In contrast, the chlorinated solvent dichloromethane substantially decreased both the yield and the enantioselectivity (entry 15). Lowering the reaction temperature to −10 °C had beneficial effects on the enantioselectivity: desired product 3aa was obtained in 99% yield with 93% ee (entry 16). However, the reaction at −20 °C gave a reduced yield and enantioselectivity (entry 17).|
|
|||||
|---|---|---|---|---|---|
| Entry | [Rh] | Solvent | Base | Yield (%) | ee (%) |
a Reaction conditions: 4/1a/2a = 0.0005![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.15:0.1 (mmol), 0.45 mmol base, 2.5 mL solvent; all the reactions were complete within 24 h. DCM, dichloromethane; MTBE, methyl tert-butyl ether. Isolated yields are given. The ee values were determined by HPLC.
b Performed at −10 °C.
c Performed at −20 °C. :0.15:0.1 (mmol), 0.45 mmol base, 2.5 mL solvent; all the reactions were complete within 24 h. DCM, dichloromethane; MTBE, methyl tert-butyl ether. Isolated yields are given. The ee values were determined by HPLC.
b Performed at −10 °C.
c Performed at −20 °C.
|
|||||
| 1 | 4a | Et2O | NaH | 41 | 31 |
| 2 | 4b | Et2O | NaH | 40 | 70 |
| 3 | 4c | Et2O | NaH | 71 | 85 |
| 4 | 4d | Et2O | NaH | 85 | 89 |
| 5 | 4e | Et2O | NaH | 52 | 11 |
| 6 | 4f | Et2O | NaH | 34 | 11 |
| 7 | 4g | Et2O | NaH | 67 | 63 |
| 8 | 4h | Et2O | NaH | 72 | 12 |
| 9 | 4i | Et2O | NaH | 15 | 26 |
| 10 | 4d | Et2O | NaOH | 64 | 90 |
| 11 | 4d | Et2O | K2CO3 | 60 | 89 |
| 12 | 4d | Et2O | K3PO4 | 56 | 90 |
| 13 | 4d | Et2O | LiOtBu | 54 | 86 |
| 14 | 4d | MTBE | NaH | 99 | 89 |
| 15 | 4d | DCM | NaH | 28 | 36 |
| 16b | 4d | MTBE | NaH | 99 | 93 |
| 17c | 4d | MTBE | NaH | 59 | 86 |
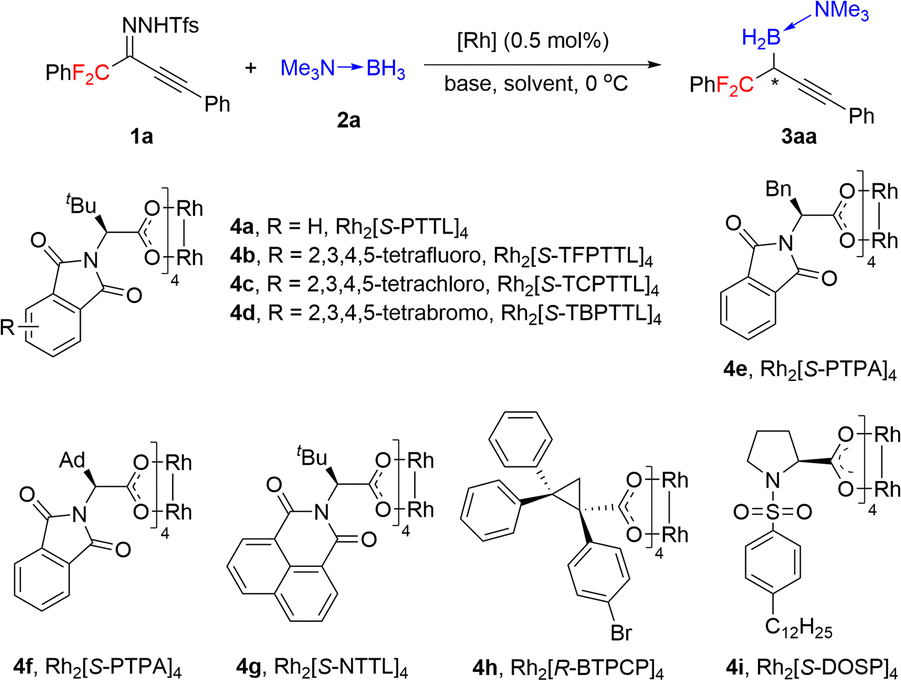
Under the optimal conditions (Table 1, entry 16), we evaluated B–H bond insertion reactions of various gem-difluoroalkyl alkynyl N-triftosylhydrazones 1 with trimethylamine–borane adduct 2a (Scheme 2). Reactions of N-triftosylhydrazones bearing an aryl group attached to the alkynyl moiety (1b–1t) gave the corresponding B–H bond insertion products (3ba–3ta) in 74–99% yields with 73–99% ee. The steric properties of the substituent on the aryl group clearly affected the enantioselectivity of the reaction. A substrate with an ortho-methyl group gave the expected product 3ba in high yield with high enantioselectivity, whereas the corresponding meta-methyl-substituted compound showed lower enantioselectivity (3ca). However, the position of a chlorine substituent had little effect on the enantioselectivity (3da–3fa). Furthermore, a substrate with an ortho-fluorine substituent gave the corresponding product (3ga) with good results. Transformation of a 1-naphthyl-substituted N-triftosylhydrazone afforded product 3ha with satisfactory results. Substrate 1i, which has a para-nitro group, gave access to the corresponding product (3ia) in 80% yield with 82% ee. We also evaluated substrates bearing an aryl or a heteroaryl group attached to the alkynyl moiety. Substrates with an electron-donating methyl group or an electron-withdrawing fluorine or chlorine atom or a trifluoromethyl group on the 1-phenyl ring were tolerated (3ja–3qa). Compounds with a 1-naphthyl or 1-thienyl group attached to the alkynyl moiety showed high yields and enantioselectivities (3ra, 3sa). When 4b was the catalyst, a substrate with a methyl group attached to the gem-difluoromethylene group afforded 3ta in good yield with moderate enantioselectivity. We also evaluated reactions of 1a with a series of borane adducts 2 and found that only trialkylamine–borane adducts afforded good results (see the ESI† for details). The structure and absolute configuration of (R)-3fa were determined by X-ray diffraction analysis of a single crystal.
 | ||
| Scheme 2 Preparation of chiral gem-difluoroalkyl propargylic borons by rhodium-catalyzed B–H bond insertion reactions. a Reaction conditions: 4d/1/2 = 0.0005:0.15:0.1 (mmol), 0.45 mmol NaH, 2.5 mL MTBE, −10 °C. All reactions were complete within 24 h. Isolated yields are given. The ee values were determined by HPLC. b Performed at room temperature. c Catalyst 4b was used. | ||
Chiral α-allenols,14 which have both axial and central chiralities, not only are found in hundreds of natural products but also serve as valuable synthetic intermediates in a wide range of transformations. gem-Difluoroalkyl-substituted chiral α-allenols have great potential as novel chiral gem-difluoroalkyl reagents with possible applications for drug discovery. To the best of our knowledge, chiral gem-difluoroalkyl-substituted allenes have not been reported. Serendipitously, we found that BF3·Et2O-promoted addition reactions between (R)-3ba and aldehydes generated chiral gem-difluoroalkyl α-allenols, which have axial and central chiralities (Scheme 3; see the ESI† for optimization of the reaction conditions). Having discovered this, we evaluated a broad array of aldehydes, including formaldehyde and aromatic and aliphatic aldehydes in reactions with (R)-3ba. The addition reaction between formaldehyde and (R)-3ba gave chiral gem-difluoroalkyl α-allenol 5a in good yield with excellent regioselectivity and well-retained ee. Aromatic aldehydes with a 4-phenyl, 4-chloro, or 4-bromo substituent gave corresponding α-allenols 5b–5d in good yields with excellent regio-, diastereo-, and enantioselectivities. However, the yield of 5e from the reaction of 4-methoxy benzaldehyde was relatively low. 2-Thenaldehyde and 2-furfural gave good results (5f, 5g). Aliphatic aldehydes were also appropriate substrates, generating the desired products (5h–5j) in good yields with high regio- and diastereoselectivities. In addition, α-chiral amino aldehydes derived from natural amino acids reacted smoothly with (R)-3ba under the standard conditions, diastereoselectively providing gem-difluoroalkyl β-amino α-allenols 5k and 5l, which have three contiguous chiral centers. We propose that this reaction proceeds via transition state PT (Scheme 3). Coordination of BF3·Et2O to the carbonyl group of the aldehyde enhances the electrophilicity of the carbonyl carbon atom,15 and the concerted addition process ensures the high diastereoselectivity.
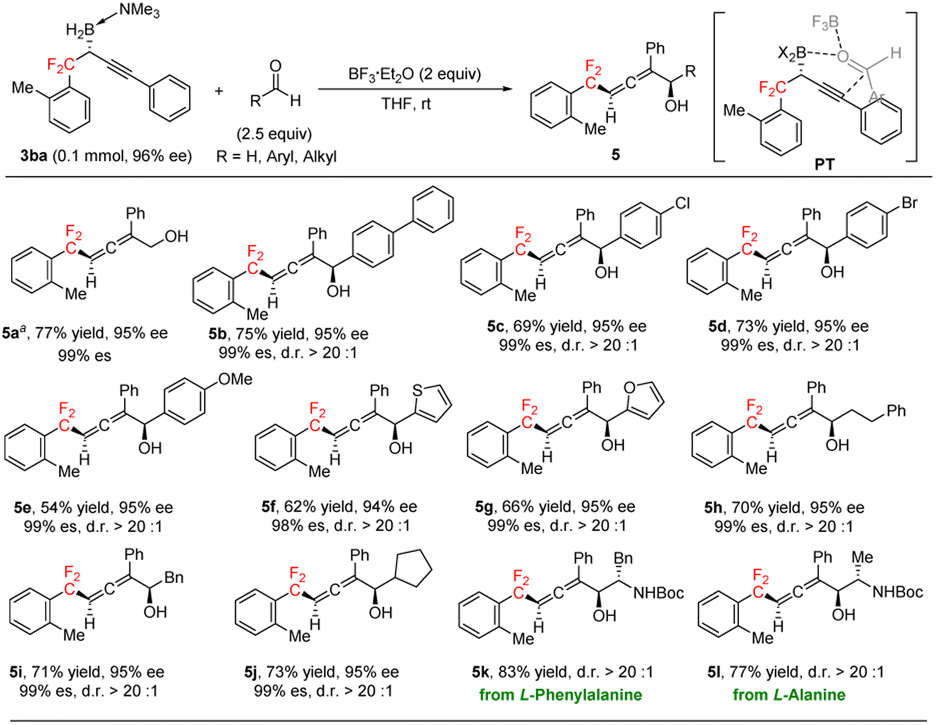 | ||
| Scheme 3 Preparation of chiral gem-difluoroalkyl α-allenols through addition reactions of gem-difluoroalkyl propargylic boron with aldehydes. a (CH2O)n (10 equiv.). | ||
Next, we explored the synthetic potential of our method (Scheme 4). We found that the B–H bond insertion reaction of gem-difluoroalkyl alkynyl sulfonylhydrazone 1b and trimethylamine–borane adduct 2a could be conducted at a gram scale with 0.2 mol% 4d as the catalyst to afford (R)-3ba in good yield with excellent enantioselectivity (Scheme 4a). Gold-catalyzed cyclization of 5b stereoselectively furnished gem-difluoroalkyl 2,5-dihydrofuran 6, which has two chiral centers (Scheme 4b). Hydrogenation of the trisubstituted olefin moiety of 6 over Pd/C afforded chiral gem-difluoroalkyl-substituted tetrahydrofuran 7, which has three chiral centers (Scheme 4b). Recently, various compounds containing tetrahydrofuran units bearing chiral gem-difluoroalkyl substituents have been proposed for the treatment of cancers and other diseases.16
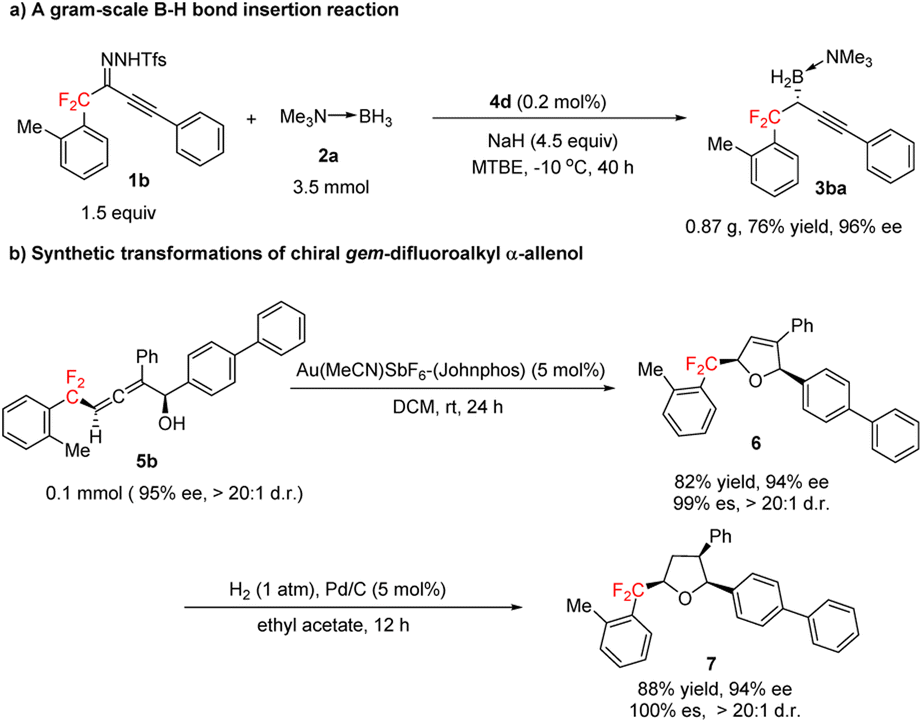 | ||
| Scheme 4 Gram-scale B–H bond insertion reaction and transformations of gem-difluoroalkyl α-allenol 5b. | ||
Conclusions
In conclusion, we have developed two types of chiral gem-difluoroalkyl reagents: gem-difluoroalkyl propargylic borons and gem-difluoroalkyl α-allenols. First, a wide range of novel, stable chiral gem-difluoroalkyl propargylic borons were synthesized in high yields with high enantioselectivities by means of dirhodium-catalyzed B–H bond insertion reactions. Then, aldehydes (formaldehyde and aromatic and aliphatic aldehydes) were allenylated with chiral gem-difluoroalkyl propargylic boron in the presence of BF3·Et2O for rapid access to a wide range of chiral gem-difluoroalkyl α-allenols with two or three contiguous chiral centers, including adjacent axial and central chiralities. Moreover, a gem-difluoroalkyl α-allenol was readily derivatized to afford chiral gem-difluoroalkylated 2,5-dihydrofuran and tetrahydrofuran derivatives, demonstrating the considerable potential utility of chiral gem-difluoroalkyl reagents for organic synthesis.Data availability
All experimental data were provided in ESI.†Author contributions
S. F. Z. and H. N. Z. conceived and guided the study; H. N. Z., M. L. H., and M. Y. H. designed the experiments and analysed the data; H. N. Z. performed the reactions; Y. X. S. performed the DFT calculations; M. L. H., J. W. Z. and X. Y. Z. made some of the substrates; S. F. Z. and H. N. Z. wrote the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the National Key R&D Program of China (2021YFA1500200), National Natural Science Foundation of China (92256301, 92156006, and 22221002), “111” project (B06005) of the Ministry of Education of China, Haihe Laboratory of Sustainable Chemical Transformations, Fundamental Research Funds for the Central Universities, and New Cornerstone Science Foundation through the XPLORER PRIZE for financial support.Notes and references
- (a) T. Hiyama, Organofluorine Compounds: Chemistry and Applications, Springer, New York, 2000 CrossRef; (b) P. Kirsch, Modern Fluoroorganic Chemistry, Wiley-VCH, Weinheim, 2013 CrossRef; (c) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- G. M. Dubowchik, V. M. Vrudhula, B. Dasgupta, J. Ditta, T. Chen, S. Sheriff, K. Sipman, M. Witmer, J. Tredup, D. M. Vyas, T. A. Verdoorn, S. Bollini and A. Vinitsky, Org. Lett., 2001, 3, 3987–3990 CrossRef CAS PubMed.
- (a) G. L. Grunewald, M. R. Seim, J. Lu, M. Makboul and K. R. Criscione, J. Med. Chem., 2006, 49, 2939–2952 CrossRef CAS PubMed; (b) M. S. Smyth, H. Ford and T. R. Burke, Tetrahedron Lett., 1992, 33, 4137–4140 CrossRef CAS.
- K. McKeage, G. L. Plosker and M. A. Siddiqui, Drugs, 2006, 66, 873–879 CrossRef CAS PubMed.
- S. M. Hoy, Drugs, 2022, 82, 1017–1023 CrossRef CAS PubMed.
- J. E. Frampton and M. D. Moen, Drugs, 2010, 70, 1283–1293 CrossRef CAS PubMed.
- S. Noble and K. L. Goa, Drugs, 1997, 54, 447–472 CrossRef CAS PubMed.
- (a) X. Yue, X.-L. Qiu and F.-L. Qing, J. Fluorine Chem., 2008, 129, 866–874 CrossRef CAS; (b) R.-W. Wang and F.-L. Qing, Org. Lett., 2005, 7, 2189–2192 CrossRef CAS PubMed; (c) Y.-Y. Wu, X. Zhang, W.-D. Meng and F.-L. Qing, Org. Lett., 2004, 6, 3941–3944 CrossRef CAS PubMed; (d) K. Nakayama, H. C. Kawato, H. Inagaki, R. Nakajima, A. Kitamura, K. Someya and T. Ohta, Org. Lett., 2000, 2, 977–980 CrossRef CAS PubMed; (e) Z. Zhang, Y.-L. Xiao and X. Zhang, Acc. Chem. Res., 2018, 51, 2264–2278 CrossRef PubMed.
- (a) C. Ni, F. Wang and J. Hu, Beilstein J. Org. Chem., 2008, 4, 21–27 Search PubMed; (b) P. Zhang and C. Wolf, Angew. Chem., Int. Ed., 2013, 52, 7869–7873 CrossRef CAS PubMed; (c) Y.-L. Liu, J.-S. Yu and J. Zhou, Asian J. Org. Chem., 2013, 2, 194–206 CrossRef CAS; (d) X. Gao, Y.-L. Xiao, X. Wan and X. Zhang, Angew. Chem., Int. Ed., 2018, 57, 3187–3191 CrossRef CAS PubMed; (e) X. Gao, R. Cheng, Y.-L. Xiao, X.-L. Wan and X. Zhang, Chem, 2019, 5, 2987–2999 CrossRef CAS; (f) L. An, F.-F. Tong, S. Zhang and X. Zhang, J. Am. Chem. Soc., 2020, 142, 11884–11892 CrossRef CAS PubMed; (g) J. Liu, L. Yu, C. Zheng and G. Zhao, Angew. Chem., Int. Ed., 2021, 60, 23641–23645 CrossRef CAS PubMed; (h) H. Uno, K. Kawai, T. Araki, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2022, 61, e202117635 CrossRef CAS PubMed.
- (a) D. G. Hall, Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, Wiley-VCH, Weinheim, 2nd edn, 2011 CrossRef; (b) E. Fernández, Advances in Organoboron Chemistry towards Organic Synthesis, Georg Thieme Verlag, Stuttgart, 2020 Search PubMed.
- (a) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954–1993 CrossRef CAS PubMed; (b) J. Ye and S. Ma, Acc. Chem. Res., 2014, 47, 989–1000 CrossRef CAS PubMed; (c) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074–3112 CrossRef CAS PubMed.
- B. Liu, H.-H. Wu and J. Zhang, ACS Catal., 2018, 8, 8318–8323 CrossRef CAS.
- For reviews, see: (a) S.-F. Zhu, Chin. J. Chem., 2021, 39, 3211–3218 CrossRef CAS; (b) M.-Y. Huang and S.-F. Zhu, Chem. Sci., 2021, 12, 15790–15801 RSC; (c) M.-Y. Huang and S.-F. Zhu, Chem. J. Chin. Univ., 2020, 41, 1426–1448 CAS; For examples, see: (d) Q.-Q. Cheng, S.-F. Zhu, Y.-Z. Zhang, X.-L. Xie and Q.-L. Zhou, J. Am. Chem. Soc., 2013, 135, 14094–14097 CrossRef CAS PubMed; (e) Q.-Q. Cheng, H. Xu, S.-F. Zhu and Q.-L. Zhou, Acta Chim. Sin., 2015, 73, 326–329 CrossRef CAS; (f) J.-M. Yang, Z.-Q. Li, M.-L. Li, Q. He, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2017, 139, 3784–3789 CrossRef CAS PubMed; (g) Y. Pang, Q. He, Z.-Q. Li, J.-M. Yang, J.-H. Yu, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2018, 140, 10663–10668 CrossRef CAS PubMed; (h) Y.-T. Zhao, Y.-X. Su, X.-Y. Li, L.-L. Yang, M.-Y. Huang and S.-F. Zhu, Angew. Chem., Int. Ed., 2021, 60, 24214–24219 CrossRef CAS PubMed; (i) M.-Y. Huang, Y.-T. Zhao, C.-D. Zhang and S.-F. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202203343 CAS; (j) H.-N. Zou, Y.-T. Zhao, L.-L. Yang, M.-Y. Huang, J.-W. Zhang, M.-L. Huang and S.-F. Zhu, ACS Catal., 2022, 12, 10654–10660 CrossRef CAS.
- (a) J. M. Alonso and P. Almendros, Chem. Rev., 2021, 121, 4193–4252 CrossRef CAS PubMed; (b) X. Fan, Y. He and X. Zhang, Chem. Rec., 2016, 16, 1635–1646 CrossRef CAS PubMed; (c) M. Deliaval, R. Jayarajan, L. Eriksson and K. J. Szabó, J. Am. Chem. Soc., 2023, 145, 10001–10006 CrossRef CAS PubMed.
- N. Okamoto, T. Sueda, H. Minami and R. Yanada, Org. Lett., 2019, 21, 8847–8851 CrossRef CAS PubMed.
- (a) G. Wang, D. B. Smith, L. Beigelman and C. A. Jekle, US Pat., 0176910A1, 2016 Search PubMed; (b) M. Gmachl and M. H. Hofmann, US Pat., 0249492A1, 2022 Search PubMed; (c) M. Smith and K. G. Klumpp,US Pat., 10092649B2, 2018 Search PubMed; (d) D. J. Mergott and B. A. Willis, US Pat., 0231791A1, 2019 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2246187. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03266c |
| This journal is © The Royal Society of Chemistry 2023 |