
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic tandem dehydrochlorination–hydrogenation of PVC towards valorisation of chlorinated plastic waste†
Galahad
O'Rourke
 a,
Tess
Hennebel
a,
Maxime
Stalpaert
a,
Alina
Skorynina
b,
Aram
Bugaev
c,
Kwinten
Janssens
a,
Lisa
Van Emelen
a,
Vincent
Lemmens
a,
Rodrigo
De Oliveira Silva
a,
Christel
Colemonts
d,
Philippe
Gabriels
d,
Dimitrios
Sakellariou
a and
Dirk
De Vos
*a
a,
Tess
Hennebel
a,
Maxime
Stalpaert
a,
Alina
Skorynina
b,
Aram
Bugaev
c,
Kwinten
Janssens
a,
Lisa
Van Emelen
a,
Vincent
Lemmens
a,
Rodrigo
De Oliveira Silva
a,
Christel
Colemonts
d,
Philippe
Gabriels
d,
Dimitrios
Sakellariou
a and
Dirk
De Vos
*a
aCentre for Membrane Separations, Adsorption, Catalysis and Spectroscopy for Sustainable Solutions (cMACS), KU Leuven, 3001 Leuven, Belgium. E-mail: dirk.devos@kuleuven.be
bALBA Synchrotron, Cerdanyola del Valles, E-08290 Barcelona, Spain
cPaul Scherrer Institute, Forschungsstrasse 111, 5232, Villigen, Switzerland
dVynova-group, 3980 Tessenderlo, Belgium
First published on 4th April 2023
Abstract
Chemical treatment of end-of-life PVC at high temperature often results in the formation of polyacetylene and eventually aromatic char. These insoluble conjugated polymers lead to industrial reactor blockages, and limit the efficiency in recycling chlorinated plastic waste. To address this challenge, a solvent-based tandem dehydrochlorination–hydrogenation process is proposed for the conversion of PVC to a saturated polymer backbone. When combining tetrabutylphosphonium ionic liquids and homogeneous Rh catalysts under H2 pressure, 81% dehydrochlorination is reached in 2 h, with the hydrogenation proceeding smoothly with minimal catalyst use of 0.5–2.0 mol% Rh. This process for PVC dechlorination yields soluble products that lack aromatics, have high degrees of dechlorination and possess a tunable content of double bonds. The chemical structures of the partially unsaturated polymer products and of the different structural motifs in the product are accurately monitored by a liquid 1H-NMR method. Finally, X-ray absorption spectroscopy (XAS) sheds light on the catalytic Rh species during the tandem process, which are stabilized by the ionic liquid. This tandem process enables rapid PVC conversion to a saturated organic product, with polyethylene segments giving the opportunity for ensuing recycling steps.
Introduction
Plastic waste management is a pressing matter in contemporary consumer society. The end-of-life processing of polyvinyl chloride (PVC), polyvinylidene chloride (PVDC) and other chlorinated plastics remains challenging due to their high chloride content.1 These chlorinated polymers, and especially PVC, which is the third most produced polymer worldwide, have many uses in construction, healthcare, electronics and the automotive sector with applications ranging from flooring to packaging material.2,3 However, various additives with potential toxicity (e.g. phthalate plasticizers), or mixed plastic contaminants can impede mechanical recycling of PVC and limit the use of recyclates to low-end applications.4 On the other hand, PVC valorization by thermal treatment is problematic, since beside potential dioxin formation, PVC has a flame retardant effect that limits net energy recovery by incineration.5Recent studies have considered ionic liquids as reaction media for thermal dehydrochlorination of PVC. Ionic liquids accelerate the dehydrochlorination by lowering the decomposition temperature, and act as solvents for PVC.6 The cation and especially the anion of these ionic salts have been found to play a role in the dechlorination efficiency.7 Additionally, phosphonium-based ionic liquids have shown promising results for the recycling of chlorinated multilayered blister packs.8 In an effort to avoid landfilling or incineration, such chemical recycling by controlled thermal decomposition of PVC can allow the recovery of HCl that is eliminated from the chains. However, open literature procedures barely focus on the dechlorination products, which, due to their nature, would be difficult to process by industry. The main dehydrochlorination product, polyacetylene, is an extensively conjugated polymer which tends to crosslink to polyaromatic tar.7,9 The insolubility of these products in practically all solvents leads to plugging of industrial reactors, thereby impeding further upcycling of the polymer carbon chain. If polyacetylene formation could be prevented, one could make PVC recycling more chemically feasible, with the potential of subsequent product transformations. Of the very few reports on such tandem transformations of PVC, a recent study demonstrated a tandem dechlorination–reduction–Friedel–Crafts alkylation.10 While this divergent silylium catalysed process effectively converts PVC to polyethylene–polystyrene-like copolymers, it requires stoichiometric amounts of Et3SiH, produces Et3SiCl as a by-product and consumes part of the aromatic solvent for functionalization of the polymer backbone. So far, no atom efficient methods for PVC valorisation exist, that avoid simultaneous incorporation of carbon groups on the polymer backbone. In order to reduce the masses of the reactants needed, the dehydrochlorination is yet to be combined with maximally atom-efficient consecutive reactions.
Therefore, this investigation on PVC conversion looks beyond the formation of polyacetylene by simple dehydrochlorination and explores the possibility of a tandem dehydrochlorination–hydrogenation of PVC by the usage of H2, as a potentially green reagent, and a homogeneous Rh hydrogenation catalyst. We hypothesize that in this combined catalytic reaction, the produced double bonds are hydrogenated, thereby ideally forming polyethylene type structures (Fig. 1). Minimal catalyst use, and avoiding stoichiometric reagents for dehydrochlorination such as silanes or metal hydrides, are important criteria on the road to a sustainable process that allows to tune the polymer product.10–12
 | ||
| Fig. 1 Recycling of PVC. (top) Polyacetylenes, formed via pyrolysis and thermal degradation tend to crosslink to polyaromatic tar. (bottom) Solvent based tandem dehydrochlorination–hydrogenation, using ionic liquids and Rh catalysts, can lead to saturated polymers. | ||
Results and discussion
Solvent-based tandem dehydrochlorination–hydrogenation
Regarding the choice of the hydrogenation catalyst, we initially selected a homogeneous Rh(I) complex as the precatalyst, as prior investigations suggest activity of Rh for the hydrogenolysis of chlorobenzene derivatives and for hydrogenation of aromatics.13,14 Initial reactions of PVC (1 mmol based on the molar mass of a monomer unit) in tetrabutylphosphonium chloride (Bu4PCl, 3.5 eq.) with 2.0 mol% H(CO)Rh(PPh3)3 and H2-pressure (20 bar) at 180 °C for 40 min, yielded a black reaction mixture (Fig. 2). This mixture was washed with water–ethanol, to remove the excess of ionic liquid solvent, whereafter a black powder was obtained. It was found that this product was insoluble in known solvents for PVC (THF, butanone, cyclohexanone).15 The color of this product and the limited solubility are caused by extensive cross-linking reactions which can occur with double bonds formed during the dehydrochlorination. Hence, the hydrogenation does not seem to work adequately when performed in pure ionic liquid, suggesting that the H2 has a too limited solubility in Bu4PCl.16,17 Instead of using pure ionic liquid, the reactions were therefore performed with the same reagents, but with the addition of a good solvent for H2 and PVC. This gave a significant difference in product outcome. After 40 minutes reaction in 2-butanone, at 180 °C and with 3.5 eq. Bu4PCl, a precipitated yellow product was obtained (Fig. 2, 2A), while reactions in THF (Fig. 2, 2B) and cyclohexanone (Fig. 2, 2C) yielded again black products. The precipitation of the polymers occurred spontaneously during reaction, without the need for an antisolvent. Additionally, by 1H NMR no residual dissolved PVC was found in the solvent layer, indicating complete precipitation of the polymer product. Quantification by acid–base titration (see Experimental) showed that for each reaction after 40 min, 41 ± 5% of the chlorine content of the PVC was released as HCl.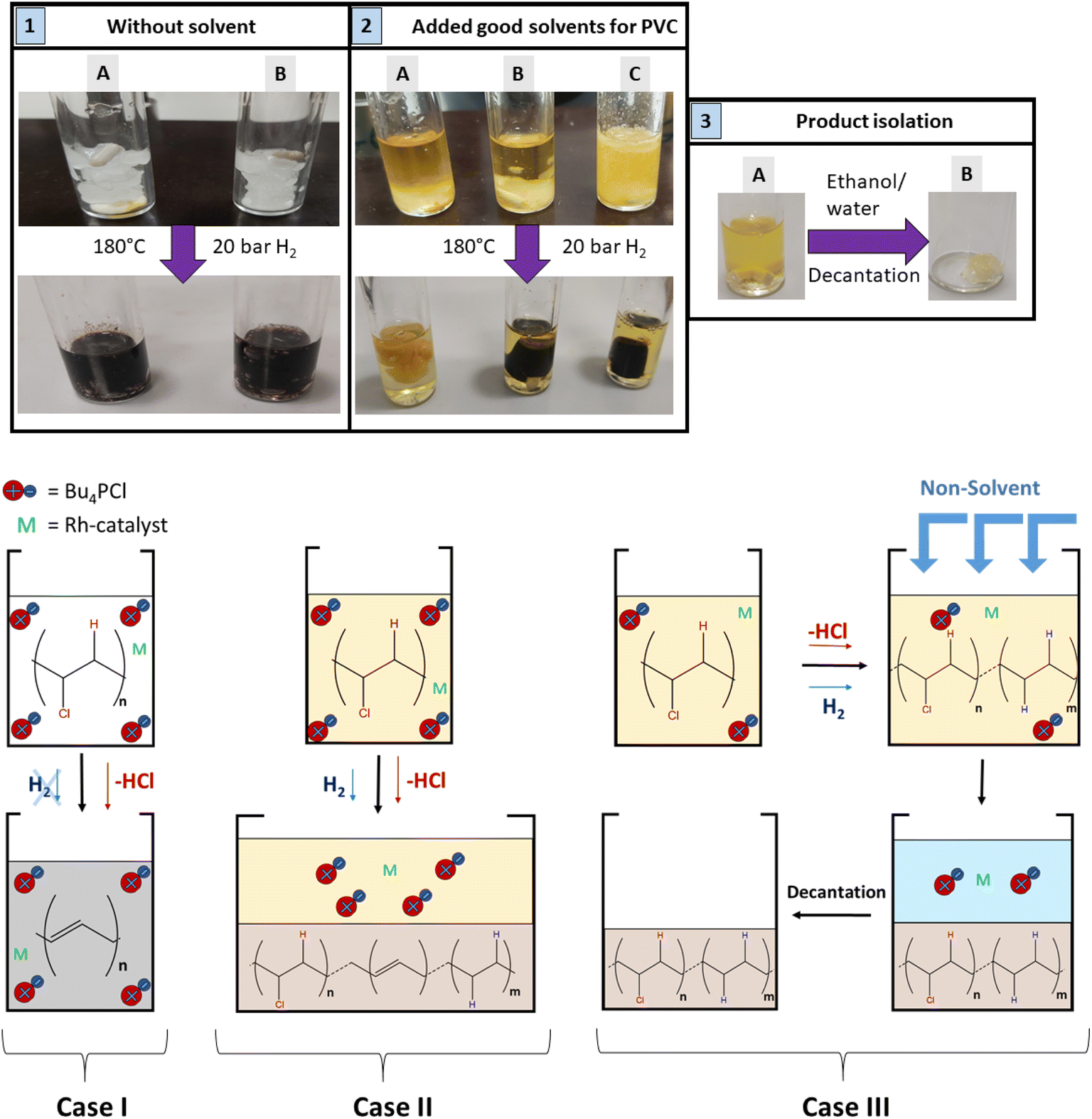 | ||
| Fig. 2 (top) Pictures of reaction mixtures before and after the reactions: (1) PVC with only 3.5 eq. Bu4PCl, without and with 2 mol% H(CO)Rh(PPh3)3-catalyst (1A and 1B, respectively); (2) PVC with 3.5 eq. Bu4PCl and 2 mol% H(CO)Rh(PPh3)3 in butanone (2A), THF (2B), cyclohexanone (2C); (3) PVC product with 0.85 eq. Bu4PCl and 2 mol% H(CO)Rh(PPh3)3 in butanone solvent (3A) and after product isolation (3B). (bottom) Three scenarios: case I: reaction in pure ionic liquid only leads to dehydrochlorination; case II: using an excess of ionic liquid in a solvent, the polymer precipitates spontaneously during reaction; case III: using a limited amount of ionic liquid in a solvent, spontaneous precipitation is avoided and largely saturated products are obtained. | ||
When applying the same reactions conditions, but limiting the amount of Bu4PCl to only 0.85 eq. and using butanone as the solvent, no spontaneous product precipitation was observed after 40 min of reaction (Fig. 2, 3A). Hence it was necessary to recover the product by non-solvent addition (ethanol–water or a mixture thereof), and centrifugation to obtain the solid product in the form of a gel (Fig. 2, 3B). The mass of the recovered product was 51% of the initial mass of PVC. Titration on the other hand, showed that 60% of HCl was released in this reaction using a limited amount of ionic liquid. This indicates that a higher degree of dechlorination can be obtained when the product remains dissolved during the entire tandem process, than when spontaneous precipitation occurs during the tandem reaction. ATR FTIR on the purified polymer products demonstrated a qualitative difference between PVC (Fig. 3A), the black products obtained after reaction in cyclohexanone with 3.5 eq. Bu4PCl (Fig. 3B), and the yellow products, as obtained in butanone (with 3.5 eq. or 0.85 eq. Bu4PCl; Fig. 3C). The black products had a non-zero baseline, indicating char formation due to the absorption across the entire spectrum by carbon black. Beside this, double bonds were identified based on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bending vibration (1077 cm−1). When exposed to air, the double bonds can easily get oxidized, leading to the detection of CO and C–O vibrational peaks in the insufficiently hydrogenated product; this is consistent with literature relating to thermal degradation of PVC.18 The FTIR spectrum of the yellow product (Fig. 3C) demonstrated effective tandem dehydrochlorination–hydrogenation, as evidenced by the intensity increase of the C–H vibrational peaks at 2913 cm−1 (s, νasCH2) and 2846 cm−1 (s, νsCH2), and a weakening of the C–Cl stretching (690 cm−1) and specific C–H bending vibration, which is associated to the presence of chloride (δ CHCl, 1250 cm−1).
C bending vibration (1077 cm−1). When exposed to air, the double bonds can easily get oxidized, leading to the detection of CO and C–O vibrational peaks in the insufficiently hydrogenated product; this is consistent with literature relating to thermal degradation of PVC.18 The FTIR spectrum of the yellow product (Fig. 3C) demonstrated effective tandem dehydrochlorination–hydrogenation, as evidenced by the intensity increase of the C–H vibrational peaks at 2913 cm−1 (s, νasCH2) and 2846 cm−1 (s, νsCH2), and a weakening of the C–Cl stretching (690 cm−1) and specific C–H bending vibration, which is associated to the presence of chloride (δ CHCl, 1250 cm−1).
 | ||
| Fig. 3 ATR FTIR spectra and assignments of vibrational peaks. (A) PVC before reaction; (B) black precipitate formed after reaction with insufficient hydrogenation: significant CO stretching and CC bending peaks and char formation, indicated by a non-zero baseline (product obtained using 3.5 eq. of Bu4PCl, 2 mol% H(CO)Rh(PPh3)3, in cyclohexanone at 180 °C for 2 h); (C) yellow product from effective tandem dehydrochlorination–hydrogenation, with intense C–H stretching vibrations and weakened C–Cl stretching vibrations (product obtained using 0.85 eq. of Bu4PCl, butanone and 2 mol% H(CO)Rh(PPh3)3 at 180 °C). | ||
Hence, while the IL is useful as a dehydrochlorination promoter, its use in excessive amounts seems unfavourable for the tandem dehydrochlorination–hydrogenation of PVC. Fig. 2 illustrates the three possible scenarios. Using pure ionic liquid (case I) leads to extensive dehydrochlorination but impedes hydrogenation, resulting in black char. When combining a large amount of IL with a solvent (case II), the hydrogenation is enhanced, but depending on the employed solvent, the product may still precipitate during the reaction and thus become inaccessible to the homogeneous Rh catalyst. In the ideal approach, the polymer remains entirely dissolved during dehydrochlorination thereby allowing an efficient consecutive hydrogenation in solution (case III). When using water as the non-solvent for product precipitation, one can easily determine the HCl yield in the water layer by titration, as a measure for the degree of dechlorination of PVC. When monitoring this reaction with 2 mol% H(CO)Rh(PPh3)3, 0.85 eq. Bu4PCl and 1 mmol of PVC over the course of 2 h (Fig. 4), it can be seen that the dehydrochlorination extent increases rapidly during the first hour when the ionic liquid is used. In opposition, a control reaction (without Bu4PCl, but with 2 mol% H(CO)Rh(PPh3)3 and heating of PVC at 180 °C in butanone), did not yield any HCl.
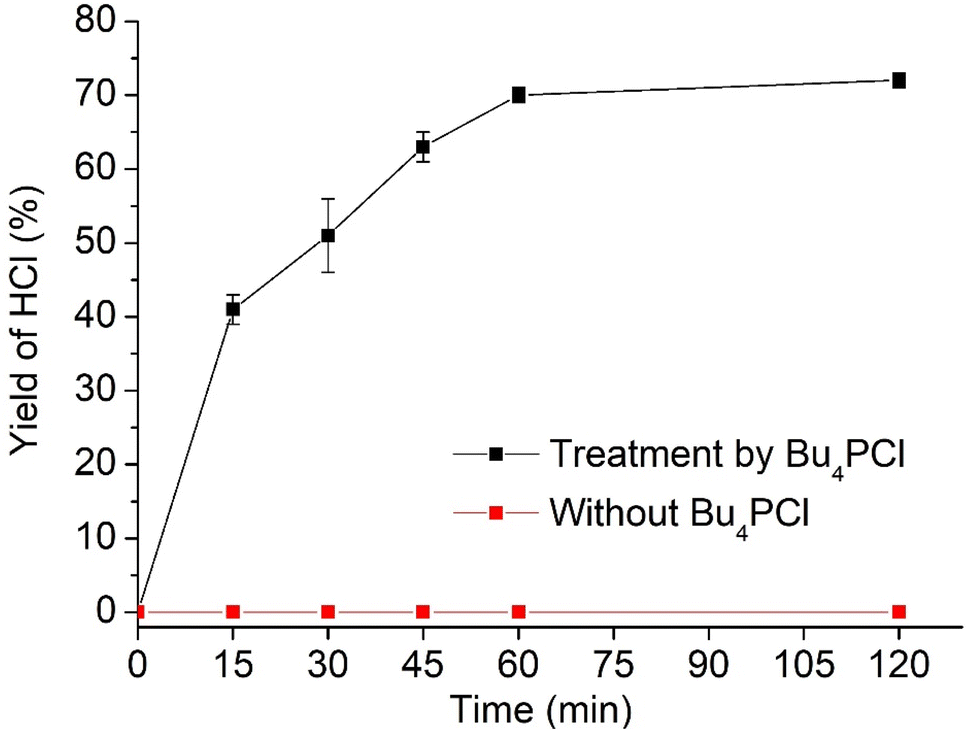 | ||
| Fig. 4 Yield of HCl, determined by acid–base titration, during reaction of PVC with 0.85 eq. Bu4PCl, 2 mol% Rh at 180 °C in butanone, with rapid dehydrochlorination during the initial 60 minutes. Error bars represent the standard deviation. Control reaction of PVC without Bu4PCl in butanone yielded no HCl (conditions: 1 mmol PVC, 180 °C, 20 bar H2, with 2 mol% H(CO)Rh(PPh3)3). | ||
Structure of polymer product
To give more solid proof for the reactions occurring during the proposed tandem dehydrochlorination–hydrogenation, solid state and liquid NMR were performed on the polymer products obtained using 0.85 eq. Bu4PCl and 20 bar H2 in butanone. While 2 mol% HCORh(PPh3)3 safely leads to a non-charred product, a Rh-catalyst loading of 0.5 mol% was found to be the minimum required to get a soluble product under these conditions. Lower catalyst loadings led to the black products as discussed before. A polymer product obtained after 2 h with this lowest catalyst loading was chosen for analysis by 13C-NMR, since it offers the best opportunity to observe intermediates. Firstly, the solid state spectrum of PVC (Fig. 5A) is marked by a loss of the tacticity information of the original polymer triad structure (VC–VC triads with VC representing vinyl chloride diads) due to peak broadening; the CHCl and CH2 groups have distinct broad peaks at 58 and 47 ppm, respectively. By liquid 13C NMR in d8-THF (Fig. 5B) it can be observed that these two broad peaks are in fact composed of several signals representing the atactic composition of PVC. The atactic triad structure of PVC, which contains racemo (r) and meso diads (m), is reflected in three distinct peak groups in liquid 13C-NMR, both for the –CHCl– signals (55–58 ppm) and the –CH2– signals (44–47 ppm). Secondly, the solid state NMR spectrum of the product (Fig. 5C) displayed a larger number of better resolved peaks than the PVC starting material using the same technique, which is indicative of a higher mobility of the polymer chains of the product.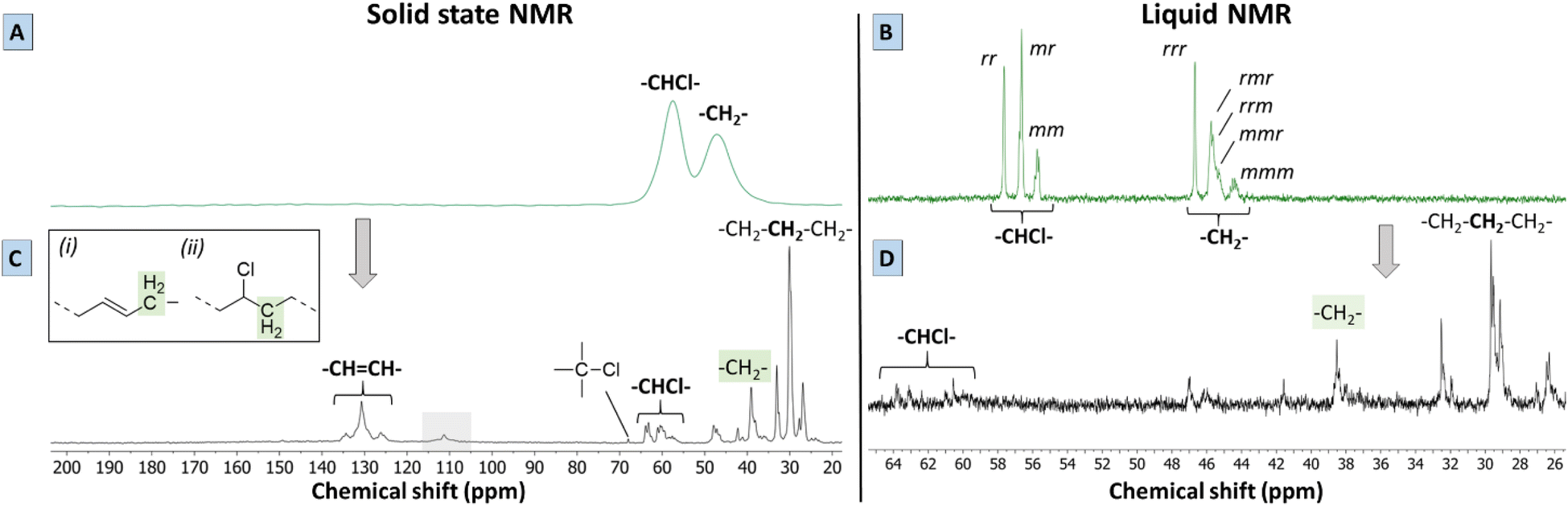 | ||
| Fig. 5 Solid state (MAS) NMR spectra of PVC (A) and of the product (C) containing double bonds (130 ppm). In C, the signal at 110 ppm (grey box) is background signal from probe. Marked ‘–CH2–’ peaks comprise (i) allylic CH2, (ii) α-CH2 next to –CHCl– groups. Liquid 13C NMR (d8-THF, 400 MHz) shows tacticity of PVC (B) and microstructure of polymer product (D). The product has 81% dechlorination and 7 double bonds per 100 C from tandem dehydrochlorination–hydrogenation (0.5 mol% H(CO)Rh(PPh3)3, 0.85 mmol Bu4PCl, 1 mL 2-butanone per 1 mmol PVC, 20 bar H2 at 180 °C). | ||
The solid state MAS 13C spectrum of the dehydrochlorination–hydrogenation product is drastically different from that of the starting PVC, with the peaks of aliphatic hydrocarbon moieties now dominating the spectrum. Some peaks of residual –CH(Cl)– appear in the δ = 57–64 ppm region, which is the expected shift range for secondary alkyl chlorides. The weak signals at 46–47 ppm are as well reminiscent of the –CH2– signals in the starting PVC. The very weak signal at 67 ppm may well arise from a small amount of tertiary chlorides. New signals appear in the δ = 125–135 ppm region. Based on liquid 13C NMR (Fig. 5D), a clear correlation was found with 1H signals between 5.39 and 5.50 ppm in the HSQC-DEPT135 spectrum of the dissolved product (Fig. S4†). Therefore, these signals at 125–135 ppm can be unambiguously assigned to olefinic double bonds that were formed by dehydrochlorination, and which were not hydrogenated by the Rh catalyst. Additionally, no correlations with aromatic protons were found by HSQC, indicating that the product did not contain aromatic groups. The intense 13C peak at 29 ppm indicates that sequences of –CH2– groups are now dominant in the polymer backbone, but the observation of a range of other signals (25–42 ppm) indicates that the structure does not resemble much that of pure polyethylene.19 The peaks around 39 ppm could be ascribed either to (i) –CH2– groups adjacent to isolated secondary –CHCl– units, or to (ii) allylic –CH2– groups, in α of newly formed CC bonds. Finally, while the strong –CH2– signals at ∼27 and ∼33 ppm are clearly of an aliphatic nature, they do not belong to a simple polyethylene chain. Candidate structures are (substituted) cyclohexyl rings, as well as –CH2– groups adjacent to a tertiary –CH– branching point. The latter are more difficult to observe due to their lower abundance, and especially on cyclohexyl rings, tertiary signals can have variable shifts. The mechanisms leading to such structures will be discussed below. It is however clear that these rings or branched structures occur much more frequently than any of the known defects in the native PVC, where defects, such as methyl branches, affect typically ≤1.5% of the carbon atoms in the chain.12,20,21 Thus, the branched or cyclic motifs observed in the reaction product must stem from the reaction itself, rather than from the original PVC backbone. Summarizing, the tandem PVC dehydrochlorination–hydrogenation in the ionic liquid leads to a highly saturated but non-linear polymer structure. Finally, it is to be remarked that the NMR spectra of the polymer product do not show any of the characteristic peaks of the Bu4PCl ionic liquid, proving that the IL is readily and fully removed.
Besides the detailed study of the 13C spectrum of the polymer product, its liquid 1H-NMR spectrum was used to quantify the degree of dechlorination of the product. The analysis, as elaborated in Section 4 of the ESI,† is based on integration of the signals of various CHCl groups and CH2 groups, in order to determine their relative abundance. In general, the products contain vinylic groups (–CH; δ 5.3–5.6 ppm), representing the formed double bonds; –CHCl– groups, adjacent to –CH2–CHCl– segments, as subsisting from the initial VC–VC structure (δ 4.10–4.65 ppm); and isolated –CHCl– that is adjacent to ethylene units (E–VC–E, shift at 3.9 ppm); the latter are formed after partial dehydrochlorination with subsequent hydrogenation. Peaks of CHCl (VC–VC type), and CHCl (E–VC–E) contribute to the total fraction of chloride in the products and these peaks were found to correlate perfectly in HSQC with the 13C peaks at 58–64 ppm (Fig. S4†). Additionally, one observes CH2– groups either in the α-position to CHCl groups (VC units at δ 2.4–2.0 ppm), as in the original PVC structure, allylic protons (δ 2.0 ppm) and CH2-groups (δ 1.28 ppm) in completely hydrogenated segments of the polymer product backbone, which are in so-called ethylene–ethylene (E–E) structures. This analysis allows quantification of all types of groups on the product and thus allows to determine the degree of dechlorination. By this method, the same polymer product that was investigated by 13C-NMR (Fig. 5) specifically showed a degree of dechlorination of 81%, and 7 double bonds per 100 carbon atoms. The 1H-spectra of products at various stages of dehydrochlorination–hydrogenation (obtained after variable reaction times with 1 mol% Rh) are shown in Fig. 6. More extensive reaction leads to a larger CH2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CHCl ratio and thus higher degrees of dechlorination (Experimental, ESI Section 4†).
:CHCl ratio and thus higher degrees of dechlorination (Experimental, ESI Section 4†).
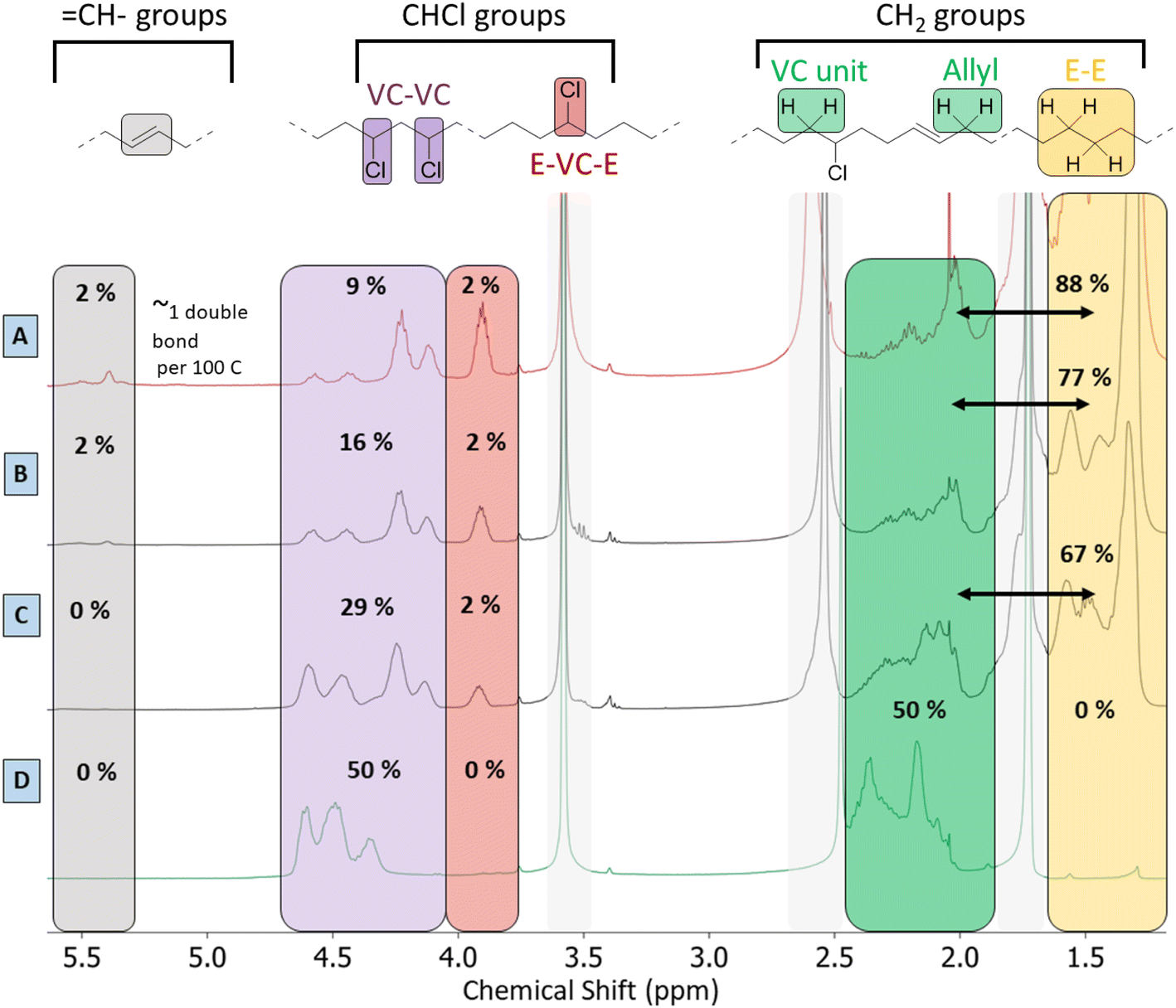 | ||
| Fig. 6 1H-NMR spectra (d8-THF, 400 MHz) of the tandem dehydrochlorination–hydrogenation products in various stages of dechlorination. Grey boxes indicate residual THF (δ 3.58, 1.71 ppm) and water (δ 2.51 ppm) peaks. The indicated percentages represent the determined carbon type fractions in each spectrum. PVC (62 mg, 1 mmol) was reacted with 0.85 mmol Bu4PCl, 1.0 mol% H(CO)Rh(PPh3)3 in 2-butanone at 180 °C, 20 bar H2. With products obtained after (A) 2 h (with 78% dechlorination), (B) 45 min (with 64% dechlorination), (C) 15 min (with 38% dechlorination) and (D) virgin PVC (with 0% dechlorination). | ||
Optimisation of reaction conditions
A search for the optimal conditions for the tandem reaction of PVC was performed with 0.85 mmol of Bu4PCl as the reaction promotor and H(CO)Rh(PPh3)3 as the hydrogenation catalyst, focusing on the degree of dechlorination of the polymer backbone as determined by 1H-NMR as the relevant optimization parameter. Firstly, variation of the temperature (Fig. 7A) showed that the reaction is fastest at 180 °C, with the dechlorination degree gently increasing up to 75% after 2 h of reaction. When comparing this curve with the HCl yield, as determined by titration (Fig. 4), the trends are very similar. Such data indicate that large amounts of HCl are available during the later phase of the reaction, and this will influence the structural changes of the eventual polymer product. Additionally, the NMR method allows quantification of each type of group on the polymer backbone. When following the tandem reaction with 2 mol% of H(CO)Rh(PPh3)3 catalyst at 180 °C (Fig. 7B), it is seen that after a 2 h reaction, only one double bond remains in the product; parallelly, the composition of the polymer backbone evolves from 50 to 88% CH2 groups. Additionally, in the first 15 minutes of the reaction, isolated CHCl– groups, as part of ethylene–vinyl chloride–ethylene sequences (E–VC–E; –(CH2CH2)–(CH2CHCl)–(CH2CH2)–) appear in the 1H NMR spectrum at 3.9 ppm; these –CHCl– groups represent a small fraction of 2% of the total number of carbon atoms. This fraction remained constant during the entire reaction, while the fraction of other CHCl groups, typically in VC–VC arrangements (4.1–4.6 ppm), decreases from 50 to 13%. This asserts that –CHCl– groups next to –(CH2CHCl)– units are dechlorinated faster, while the isolated –CHCl– groups (e.g. in E–VC–E arrangements) react much more slowly. Varying the Rh-catalyst loading between 2 and 0.5 mol% strongly influences the number of double bonds in the polymer backbone. When reducing the catalyst loading down to 0.5 mol% H(CO)Rh(PPh3)3, up to 7 double bonds remain per 100 C atoms. Additionally, this reduced catalyst loading results in an enhanced dechlorination degree of the product of 81% (Fig. 7C and D). Further lowering of the catalyst loading resulted in product insolubility, suggesting that a too large number of (conjugated) double bonds increases the likelihood of cross-linking in the presence of released HCl.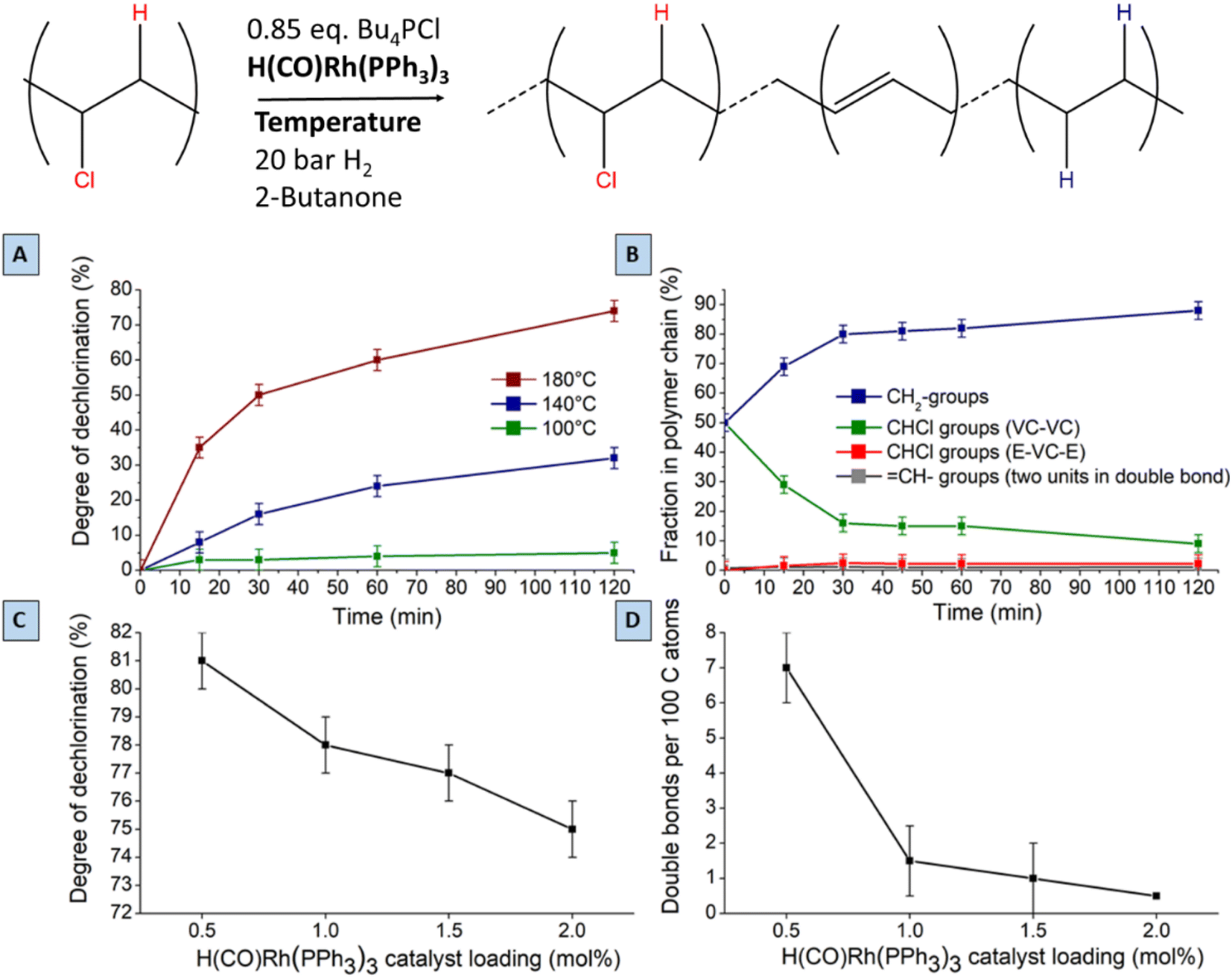 | ||
| Fig. 7 Dechlorination of PVC by tandem dehydrochlorination–hydrogenation (1 mmol PVC based on monomer, 0.85 mmol Bu4PCl). (A) Temperature influence on the extent of dechlorination using 2 mol% H(CO)Rh(PPh3)3. (B) Fractions of groups in backbone of polymer product during reaction at 180 °C using 2 mol% H(CO)Rh(PPh3)3. (C and D) Influence of H(CO)Rh(PPh3)3 loading on the degree of dechlorination and on the number of residual double bonds per 100 carbon atoms in the polymer product, at 180 °C. Quantification of products by liquid 1H-NMR (d8-THF, 400 MHz). | ||
Lowering the H2 pressure (from 15 to 5 bar H2) gave a similar effect as applying a low Rh catalyst loading. These findings suggest that the initial dehydrochlorination is sufficiently fast, and that the ensuing hydrogenation proceeds readily if enough catalyst and hydrogen are available. The fact that the eventual degree of dechlorination is higher at lower Rh loading indicates that internal allylic chlorides on the polymer backbone play a significant role in the dehydrochlorination. Previous reports suggest a zipper-like mechanism for dehydrochlorination, in which initially formed allylic chloride moieties act as preferred sites for a quickly propagating dehydrochlorination.7,22 When the rate of hydrogenation is too high, these allylic chlorides are hydrogenated to a less reactive –CHCl– in a saturated environment. Therefore, the rates of dehydrochlorination and hydrogenation must be matched to each other to achieve sufficient dechlorination degrees while avoiding char formation. Under the optimised conditions, with 0.5 mol% of Rh-catalyst, 81% of dechlorination was realized. Further increasing the reaction time under identical conditions to 26 h, gave only a 5% increase, to 86% dechlorination. In contrast to the non-hydrogenated dehydrochlorination products, which appear as black and brittle materials, the products of tandem dehydrochlorination–hydrogenation were yellow-whitish with an elastomeric appearance as a gel. Other catalyst precursors than H(CO)RhPPh3 were also considered; it was found that 0.5–2 mol% of RhCl3·H2O displays the same hydrogenation activity in the tandem reaction of PVC with 0.85 mmol of Bu4PCl as H(CO)Rh(PPh3)3 (ESI, Fig. S7†).
Mechanism of tandem dehydrochlorination–hydrogenation
Based on the 13C-NMR results, the 1H-NMR observations regarding the degree of dechlorination, and on the amounts of HCl released, as determined by acid–base titrations, a vision on the mechanism of the tandem dehydrochlorination–hydrogenation of PVC can be proposed. In the presence of an ionic liquid, PVC dehydrochlorination is generally considered to start by an ionic mechanism, rather than via radicals.7,23 By analogy with the Bu4PCl-mediated dehydrochlorination of 1,2-dichloroethane, an E2 mechanism is likely, with the chloride in non-aqueous media acting as a base accepting the proton. The acidity of the hydrogen atoms in PVC is also enhanced by the electron-withdrawing properties of the Cl– substituents on the rest of the chain, which raise the susceptibility of –(CH2–CHCl)– groups to E2 reactions. The E2 reaction initially leads to allylic chlorides, which react even faster since the formation of increasingly conjugated double bonds is a strong driving force for the elimination. Conversely, isolated –CHCl– groups in saturated parts of the chain are significantly less reactive. Thus, the overall dehydrochlorination is favored by maintaining a sufficient degree of unsaturation on the chains, e.g. by working with relatively low catalyst concentrations or hydrogen pressures. However, the availability of increasing amounts of HCl and (conjugated) double bonds also raises the likelihood of carbocation formation on the chain (Fig. 8). An isolated secondary carbocation in a hydrocarbon chain could be expected to undergo isomerization, e.g. to form a methyl branch via a protonated cyclopropane intermediate. However, the NMR data (13C, 1H) do not evidence any significant presence of methyl groups in the product. Therefore, reaction of the carbocation with a CC bond, either in the same chain or on a different chain is much more likely. Intramolecular reaction could lead to cyclic structures, which after hydrogenation appear as saturated cycloalkyl groups. Intermolecular reaction leads to two chains being linked via a bond between two tertiary centers (HR2C–CHR2). The 13C NMR data are consistent with both intra- and intermolecular routes.
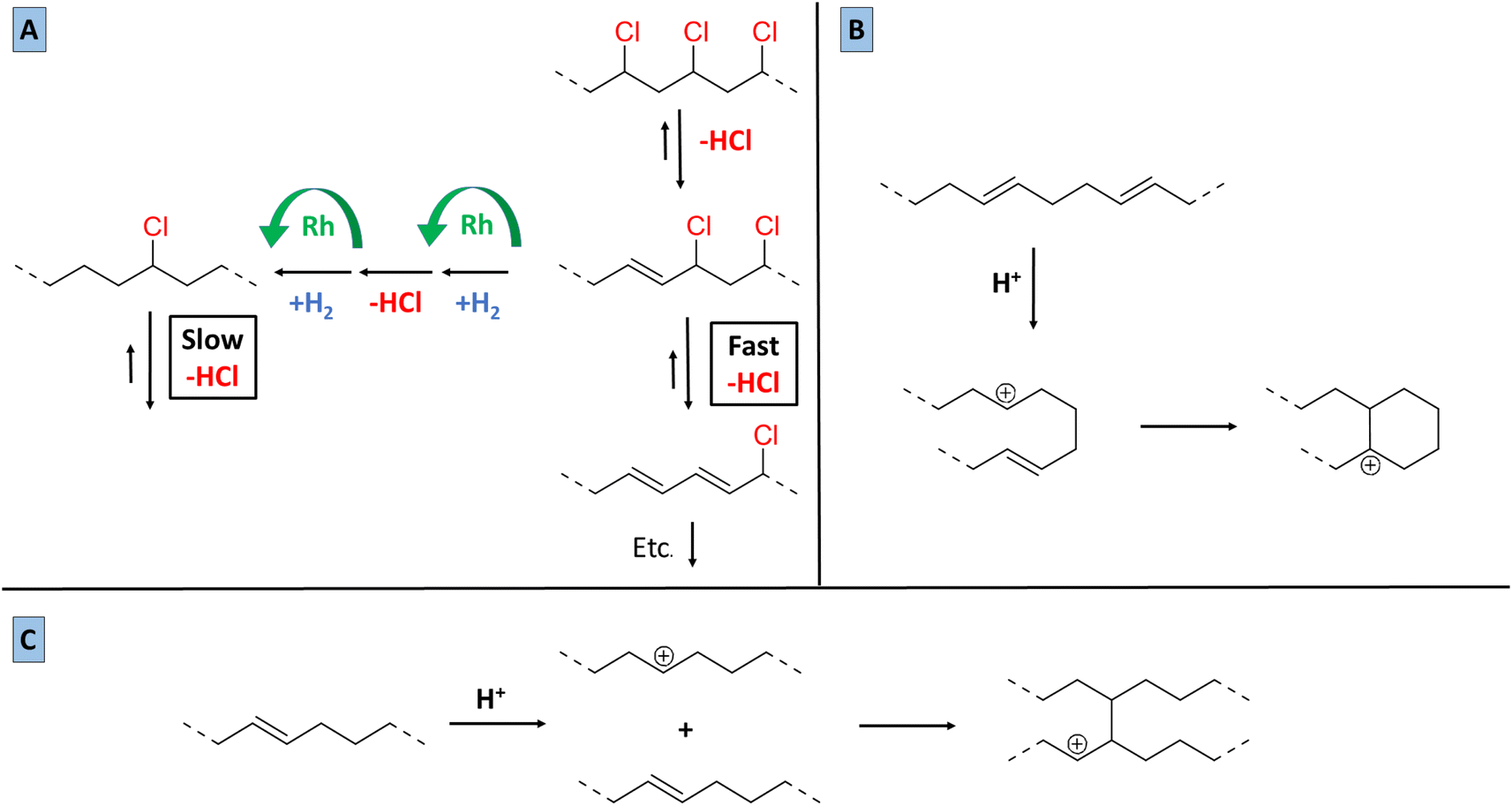 | ||
| Fig. 8 Mechanism of dehydrochlorination–hydrogenation of PVC: (A) dehydrohalogenation is faster for allylic chlorides than for isolated chlorides in a saturated chain; (B) intramolecular cyclization in a chain with several unsaturated bonds; (C) intermolecular reaction leading to cross-linking. | ||
To give a qualitative proof for the role of HCl in promoting intermolecular reactions between polymer chains, a tandem dehydrochlorination–hydrogenation reaction was performed with 0.85 eq. Bu4PCl, 2 mol% H(CO)Rh(PPh3)3 in butanone and with 2 mmol HCl added in a concentrated dioxane solution from the start on. By 1H-NMR, the product after 30 minutes of reaction showed only 17% dechlorination, versus 50% in the absence of added HCl. This suggests that the dehydrochlorination could be to some extent reversible, especially since in our conditions, the HCl is not quenched, e.g. by addition of a strong base. More importantly, when HCl was added initially, a black insoluble product was obtained after 1 h of reaction, while in the reference reaction without added HCl, the product remained soluble even after 2 h of reaction, and reached a high degree of saturation. Thus, the accumulation of HCl clearly plays a role in the formation of insoluble products when the hydrogenation activity is not sufficiently high.
Role of the ionic liquid
Since the best performance in the tandem process was observed with only 0.85 equivalents Bu4PCl in butanone (or 25 mol% with respect to PVC), the effects of the chemical structure and concentration of the ionic liquid were investigated in more detail. In previous work using ILs for PVC dehydrochlorination, large excesses of IL with respect to PVC were used. For instance, Zhao used 13 equivalents of 1-butyl-3-methylimidazolium chloride with respect to the PVC monomer;24 in earlier work on the non-reductive dehalogenation of PVC, we used 4.3 equivalents of Bu4PCl.7 From previous reports, clear influence of the halide of the ionic liquid on the decomposition temperature of PVC was observed. When using various halide anions in the ionic liquids, soluble products were only obtained with Bu4PCl and Bu4PBr, while Bu4PI led to high product insolubility even with the lowest equivalents. Hence, in order to better understand these halide effects, model reactions were performed with 2-chloropentane as a reactant, in the same ionic liquids, at 140 °C for 2 h (Fig. 9A and C). When changing the anion of the IL, the chloropentane conversion increases in the order Cl− < Br− < I−. While elimination of HCl to form pentenes is the only reaction observed in Bu4PCl, substitution reactions, with formation of 2-bromopentane or 2-iodopentane become increasingly dominant in the same order Br− < I−. This obviously reflects an increasing nucleophilicity as the size and polarizability of the halide anion increase (Fig. 9C). As is apparent from the chloropentane conversion, the change of the halide from Cl− to Br− enhances the dechlorination degree of the product, with the Br-phosphonium salt performing better than its Cl-based counterpart. Changing the cation of the ionic liquid from Bu4PCl to Ph4PCl gave almost no reaction of the model compound; this again underscores the unique effect that tetraalkylphosphonium cations have on the properties of the halide ions, especially on their basicity and nucleophilicity.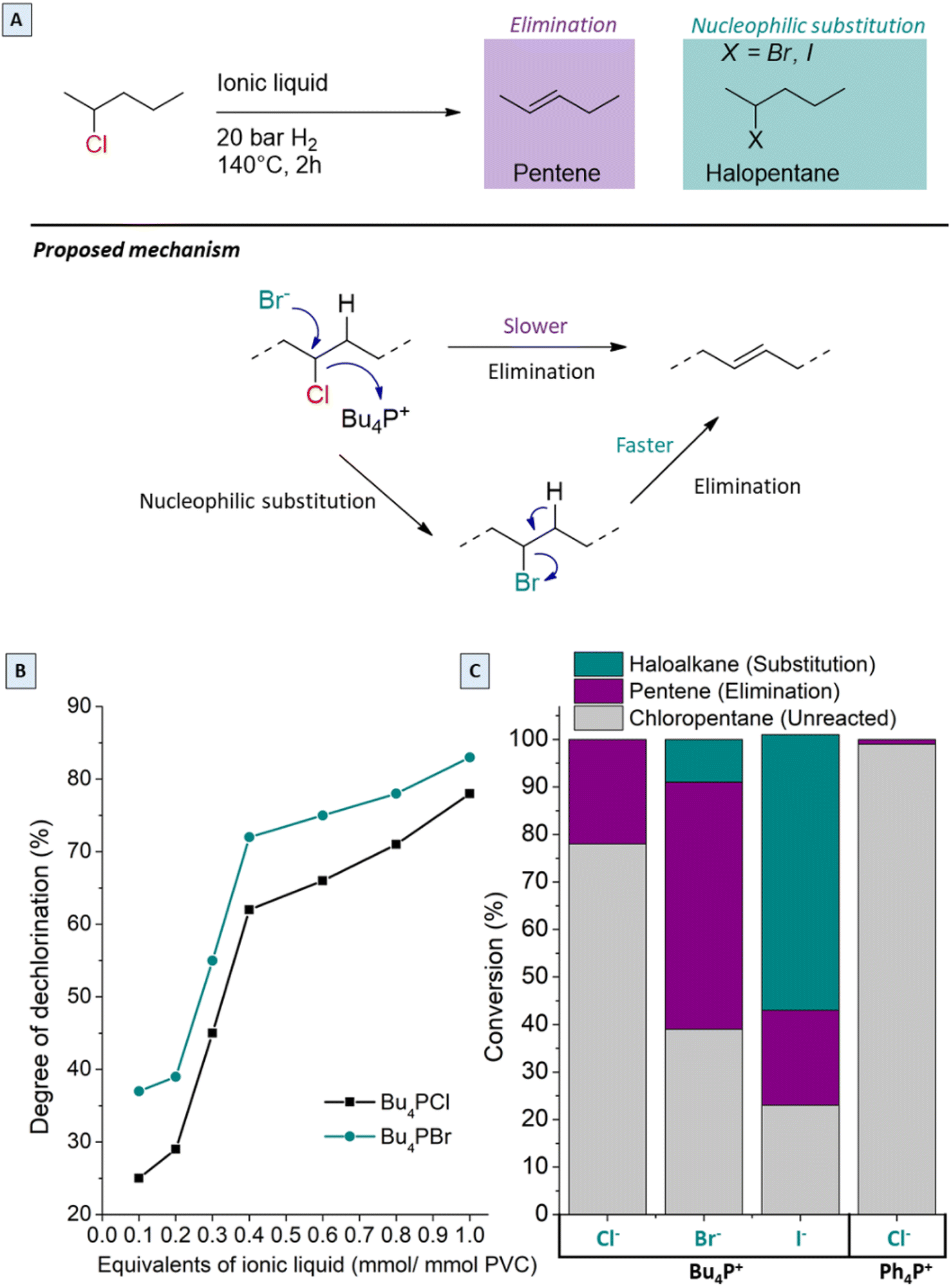 | ||
| Fig. 9 Role of the ionic liquid counter ion. (A) Conversion of 2-chloropentane in various ionic liquids at 140 °C (0.5 mmol chloropentane with 0.85 mmol ionic liquid); proposed reactions of PVC in Bu4PBr. (B) Variation of ionic liquid loading and type for tandem reaction of PVC (2.0 mol% H(CO)Rh(PPh3)3, variable equivalents of Br or Cl-based ionic liquid, 1 mL 2-butanone per 1 mmol PVC, 20 bar H2 at 180 °C, 2 h). (C) Conversion of 2-chloropentane using various ionic liquids (determined by gas chromatography) by nucleophilic substitution and elimination, as promoted by ionic liquids. | ||
In view of these results, the higher PVC dechlorination rates in Bu4PBr and Bu4PI ionic liquids can be ascribed to nucleophilic substitutions on the PVC backbone, creating C–Br or C–I bonds. Because of the superior leaving group properties of bromide in comparison to chloride, the subsequent dehydrohalogenation proceeds more readily, causing acceleration of the entire dehalogenation process of PVC (Fig. 9A). In the Bu4PI ionic liquid, the PVC dehydrochlorination is so rapid that an insoluble product is formed even before sufficient saturation of the chains can occur. Hence, the analysis of the influence of different substoichiometric amounts of ionic liquids in the tandem dehydrochlorination–hydrogenation of PVC shows that no soluble products for quantification were obtained using Bu4PI, but using Bu4PBr as the ionic liquid systematically resulted in higher dehydrochlorination rates than with Bu4PCl, in all applied molar ratios (Fig. 9B) because of the additional occurrence of nucleophilic substitution reactions.
Characterization of the homogeneous Rhodium catalyst
The solidification of the IL at room temperature (Tmelting = 64 °C) was used as a unique feature to trap the active catalytic species during the tandem dehydrochlorination–hydrogenation.25 X-ray absorption spectroscopy (XAS) was performed at the European Synchrotron Radiation facility (ESRF, Grenoble) for the characterization of the Rh species at high energies (23.2 keV). To unravel the speciation of the catalyst, ex situ samples of Rh catalysts immobilized in IL were measured before and after reaction under optimal conditions (vide supra) and compared to reference spectra (ESI, Table S1†). The obtained X-ray absorption near edge structure (XANES) and the extended X-ray absorption fine structure (EXAFS) spectra are reported in Fig. 10.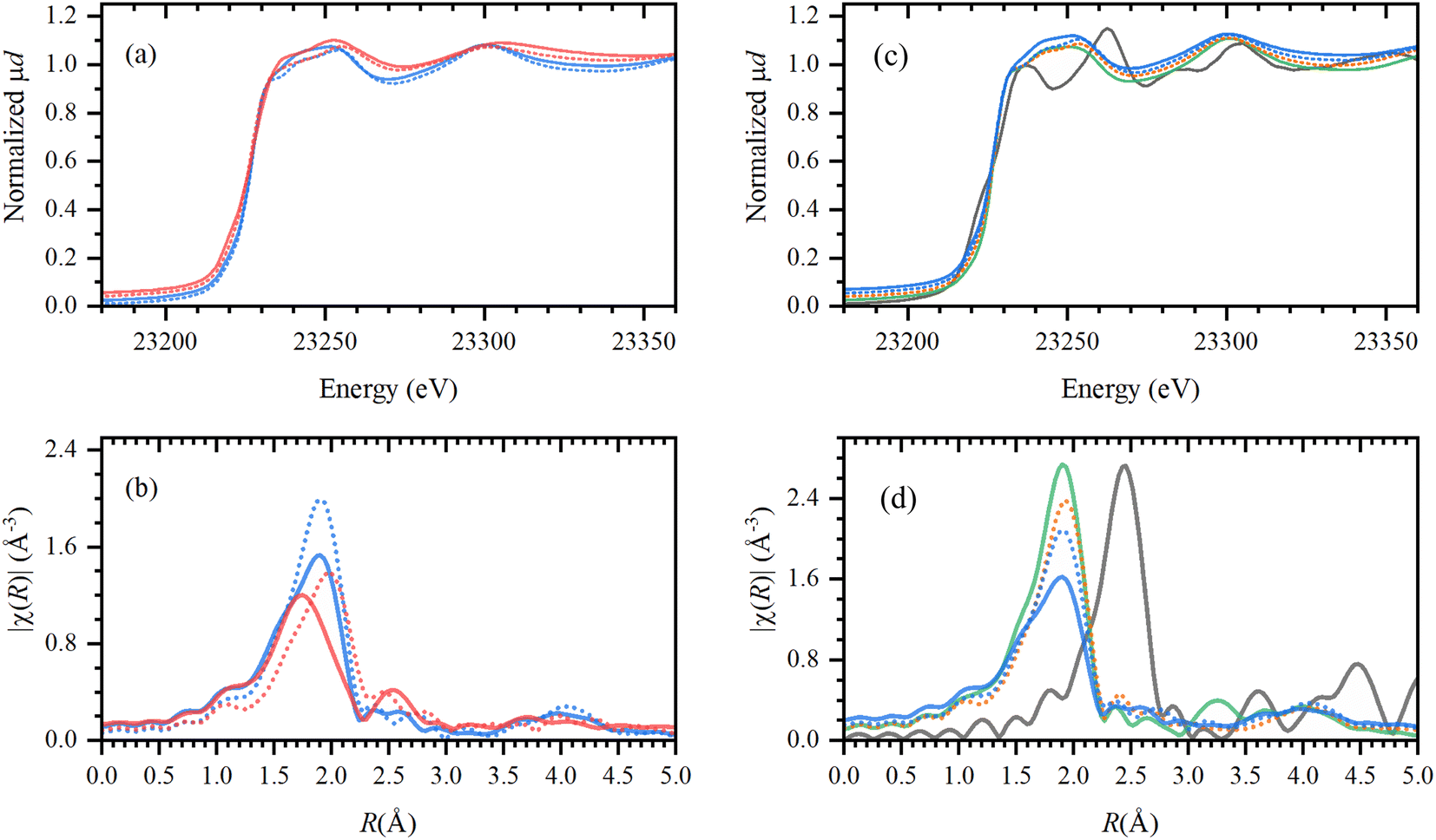 | ||
| Fig. 10 Left: XANES (a) and phase-uncorrected FT-EXAFS (b) data for the active Rh catalyst (H(CO)Rh(PPh3)3) trapped in Bu4PCl (blue) and Bu4PBr (red); characterized before (solid line) and after reaction (dashed line) with PVC. Right: XANES (c) and phase-uncorrected FT-EXAFS (d) data for the Rh catalyst in Bu4PCl (blue, solid and dashed, before and after reaction), and comparison to RhCl3 in Bu4PCl after reaction with PVC (orange); reference compounds RhCl3 (green) and Rh-foil (gray). Detailed descriptions of the samples can be found in the ESI, Table S1.† | ||
First, the H(CO)Rh(PPh3)3 catalyst was characterized in both Bu4PCl and Bu4PBr, before and after reaction, to investigate structural changes in the Rh coordination sphere during reaction, with respect to the applied ionic liquid. Overall, the catalyst maintains a similar oxidation state before and after reaction (solid and dashed line), as no shift in the edge jump was observed in the XANES. However, in EXAFS there is a clear shift towards larger bond distances in the Rh coordination sphere. Presumably, this results from a loss of the lighter elements (H, CO, PPh3) in favor of significantly heavier halides (Cl−, Br−), as a consequence of the high halide content in the reaction medium.25 The observed shift is more pronounced in the brominated Bu4PBr IL, where after reaction one finds a high concentration of Cl− originating from the high degree of PVC dechlorination, and of Br− from the ionic liquid itself (cf. supra, Role of the ionic liquid). After reaction in Bu4PCl, the final Rh catalyst shows similar features as obtained with RhCl3. This provides additional proof of the ligand substitution with halides under the applied conditions. In order to confirm the true homogeneous nature of the Rh catalyst, a comparison was made with Rh foil. This indicated that the catalyst is ionic and monometallic, as no Rh0, nor Rh–Rh contributions were observed in the experimental samples. Relating to catalyst stability, it could also be verified experimentally that the formed Rh–Cl species is stable in the ionic liquid during at least three reaction cycles (ESI, Section 7, Fig. S9 and S10†).
Conclusion
Conversion of PVC by tandem catalytic dehydrochlorination–hydrogenation was proposed as a highly effective method to achieve high dechlorination degrees of PVC. As additional advantages, soluble products are obtained, and H2 is an efficient, waste-free reagent for the reduction. In contrast to other methods for PVC recycling which use reducing agents like silanes, or metal hydrides it was found the limited quantities of ionic liquid and Rh-catalyst can reach high conversion to PE-like structures. Additionally, the PVC does not have to be pretreated by drying before reaction because dehydrochlorination–hydrogenation promoters are not as sensitive to moisture.10 The double bond content of the product can be tuned by varying the concentration of the Rh hydrogenation catalyst. The active Rh species was found to be of ionic nature, and to occur predominantly as homogeneous Rh chloride complexes, which are formed in the ionic liquid medium during the PVC dehydrochlorination reaction. Mechanistically, the HCl elicits rearrangements on residual double bonds of the product, leading to saturated tertiary branching points or cycloaliphatic structures. The findings indicate that the ionic liquids play a critical role in facilitating the HCl elimination reactions.8 Initial recyclability tests indicate that the applied Rh-catalysts are stable under the reaction conditions for three consecutive reaction cycles.Further work will be directed towards functionalizing or selectively splitting the double bonds for enhanced PVC valorization after the tandem conversion process.26,27 Additionally, the influence of different types of additives on the tandem process will be investigated for typical PVC waste applications.22,28–31
Experimental
PVC powder (Mw ∼ 43000, Mn ∼ 22000), Bu4PCl (96%), Bu4PBr (98%) and Bu4PI (98%) were supplied as solid powders by Sigma-Aldrich. 2-Butanone (technical grade, 99%), THF (>99.9%), and cyclohexanone (99%) solvents were acquired from the same supplier. RhCl3·H2O and H(CO)Rh(PPh3)3 were supplied by TCI. The reagents nor the PVC were additionally purified or dried before use.
Tandem dehydrochlorination–hydrogenation reactions
The reactions were performed in homemade metal alloy autoclave reactors which can withstand pressures up to 60 bar. PVC (1 mmol based on the monomer unit with molar mass of 62.6 g mol−1, 62.5 mg), Bu4PCl (0.85 mmol, 250 mg) and H(CO)Rh(PPh3)3 (2 mol%, 18.4 mg) or varying quantities as described otherwise, were brought into a glass liner. Thereafter, the solvent (1 mL) was added to the glass liner, which was then placed inside the reactor. Subsequently, the reactor was sealed and purged twice with N2-gas before filling it with 20 bar of H2-gas. The reaction mixture was then stirred (750 rpm) at elevated temperature (100–180 °C). After heating for the designated time, the reaction was quenched by cooling the reactor in an ice bath. The solvent layer was decanted and the precipitated polymer was dissolved in THF for purification. Thereafter, the polymer was precipitated again by the dropwise addition of water to the solvent layer. After isolation of the precipitated polymer product, it was washed three times with water (2 mL) and ethanol (2 mL). Finally, the product was subjected to an air stream to remove excess solvent and dried at 60 °C under reduced pressure overnight. Optimisation reactions were done in triplicate. Curves show mean values ± standard deviation as error margin.HCl yield determination
The HCl yields in tandem PVC dehydrochlorination reactions were quantified by acid–base titration of the recovered solvent layer (1 mL). This layer was diluted with water (6 mL) and titrated with a 0.05 M KOH solution. A correction was taken into account for the acidity of the ionic liquid by titrating Bu4PCl (0.85 mmol, 250 mg) diluted in water (6 mL). Experiments were done in triplicate.Determination of dechlorination degree by NMR
The dechlorinated polymer products (samples of ±5 mg) were dissolved at 50 °C in d8-THF. The obtained polymer solutions were measured by liquid 1H-NMR at room temperature on a 400 MHz Bruker Avance III HD spectrometer. The dechlorination degree was determined by integrating the 1H-NMR spectrum and calculating the relative fractions of each type of carbon in the polymer chain. Each integral area is divided by the number of protons per corresponding carbon group. The sum of all types of carbon groups is then equalled to 100%, and the fraction of each carbon type in the polymer backbone can be calculated. The degree of dechlorination is defined as: 100% − (number of residual CHCl in product)/(number of CHCl groups initially in PVC). An example is described in the ESI.†Influence of ionic liquids
2-Chloropentane conversion in the presence of ionic liquids was determined by gas chromatography (GC), using a Shimadzu GC-2010 device, with CP Sil-5 column. Quantification and calibration of the flame ionisation detector (FID) response were done by the effective carbon number (ECN) method (ECN (2-halopentane): 4.88, ECN (pentene): 4.9, ECN (pentane): 5). Reactions were performed in autoclaves with a 2-chloropentane stock solution (0.5 mmol, 1.0 M, 1 mL) and the ionic liquid (0.85 mmol of Bu4PCl, Bu4PBr, Bu4PI or Ph4PCl) added to the glass liner. After stirring for 2 h at 140 °C under 20 bar H2, the autoclaves with reaction mixtures were cooled to 0 °C in an ice bath before GC analysis.XAS data collection and analysis
XAS measurements were performed in transmission mode by continuous scanning of a Si(311) double-crystal for Rh K-edge at the BM23 beamline of the ESRF (Grenoble, France). The RhCl3 reference was pressed into a pellet to optimize the absorption jump at the Rh K-edge. The samples were sealed inside 5 mm Teflon rings of optimal thickness and covered with Kapton tape. For each sample, 10 spectra were collected and then averaged to improve signal-to-noise ratio. The energy sampling steps were 0.5 eV. The Rh foil was used as a reference sample and was installed between 2nd and 3rd ionization chambers. Experimental XAS data processing, including normalization, energy calibration, extraction of χ(k) signals and further Fourier analysis of EXAFS, were performed in the Demeter package.32Data availability
Original data are available within KULeuven’s OneDrive system. The ESI† contains additional characterisation data and conditions of experiments.Author contributions
O'Rourke G. wrote the original draft, did editing, conceptualization and conducted experimental work. Hennebel T., Stalpaert M., Van Emelen L., Lemmens L. measured samples related to XAS or conducted experiments. De Oliveira R. and Sakellariou D. enabled the solid state NMR methodology and performed measurements. Bugaev A. and Skorynina A. conducted XAS modelling. Janssens K. was involved in the communication and reporting of the XAS data. Colemonts C. and Gabriels P. gave advice in the investigation. De Vos D. supervised the investigation, edited the manuscript with conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Vynova and the authors thank Peter Van der Veken and Viviane Vandenbergh for their industrial expertise on the project. The authors are also grateful to Jannick Vercammen, Sam Van Minnebruggen and Simon Smolders for measuring the XAS samples. G. O., K. J., V. L., L. V. E. and D. D. V. thank FWO (G0F2320N) for financial support. We acknowledge the European Synchrotron Radiation Facility for providing the beamtime (proposal MA-4443 and MA4926) at BM23 beamline and Dr Kirill Lomachenko for the professional support.References
- T. Okada, S. Sutoh, K. Sejima, H. Tomohara and S. Mishima, Polym. Degrad. Stab., 2020, 171, 109040 CrossRef CAS
.
- PlasticsEurope, Plast. – Facts 2018, 2018, p. 38 Search PubMed.
- J. Yu, L. Sun, C. Ma, Y. Qiao and H. Yao, Waste Manag., 2016, 48, 300–314 CrossRef CAS PubMed
- S. Windels, T. Diefenhardt, N. Jain, C. Marquez, S. Bals, M. Schlummer and D. E. De Vos, Green Chem., 2022, 24, 754–766 RSC
-
J. Hiltz, R. S. Underhill, H. Moyst, Technical Report, Defence R&D Canada – Atlantic, 2007 Search PubMed
- T. Zhao, Q. Zhou, X. L. He, S. Di Wei, L. Wang, J. M. N. van Kasteren and Y. Z. Wang, Green Chem., 2010, 12, 1062–1065 RSC
- D. Glas, J. Hulsbosch, P. Dubois, K. Binnemans and D. E. Devos, ChemSusChem, 2014, 7, 610–617 CrossRef CAS PubMed
- K. Oster, A. Tedstone, A. J. Greer, N. Budgen, A. Garforth and C. Hardacre, Green Chem., 2020, 22, 5132–5142 RSC
- Y. J. Choi, J. H. Kim and J. S. Im, J. Mater. Res. Technol., 2021, 14, 2845–2852 CrossRef CAS
- M. K. Assefa and M. E. Fieser, J. Mater. Chem. A, 2023, 11, 2128–2132 RSC
- L. Monsigny, J. C. Berthet and T. Cantat, ACS Sustain. Chem. Eng., 2018, 6, 10481–10488 CrossRef CAS
- W. H. Starnes, J. Polym. Sci. Part A Polym. Chem., 2005, 43, 2451–2467 CrossRef CAS
- F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2002, 102, 4009–4091 CrossRef CAS PubMed
- C. Vangelis, A. Bouriazos, S. Sotiriou, M. Samorski, B. Gutsche and G. Papadogianakis, J. Catal., 2010, 274, 21–28 CrossRef CAS
- G. Grause, S. Hirahashi, H. Toyoda, T. Kameda and T. Yoshioka, J. Mater. Cycles Waste Manag., 2017, 19, 612–622 CrossRef CAS
- A. Kordi and F. Sabzi, Int. J. Hydrogen Energy, 2018, 43, 18296–18305 CrossRef CAS
- B. R. Mellein, A. M. Scurto and M. B. Shiflett, Curr. Opin. Green Sustain. Chem., 2021, 28, 100425 CrossRef CAS
- R. R. Stromberg, S. Straus and B. G. Achhammer, J. Res. Natl. Bur. Stand., 1958, 60, 147 CrossRef CAS
- Z. Zhou, C. Anklin, R. Kuemmerle, R. Cong, X. Qiu, J. Decesare, M. B. Kapur and R. Patel, Macromolecules, 2021, 54, 5985–5990 CrossRef CAS
- D. Braun, G. Holzer and T. Hjertberg, Polym. Bull., 1981, 5, 367–372 CrossRef CAS
- W. H. Stames, F. C. Schilling, I. M. Plitz, R. E. Cais and F. A. Bovey, Polym. Bull., 1981, 4, 555–562 CrossRef
- P. Zhao, T. Li, W. Yan and L. Yuan, Environ. Technol., 2018, 39, 977–985 CrossRef PubMed
- V. H. Tran, A. Guyot, T. P. Nguyen and P. Molinié, Polym. Degrad. Stab., 1992, 37, 209–216 CrossRef CAS
-
T. Zhao, Q. Zhou, X.-H. Jiang, A.-K. Du and Y.-Z. Wang, 5th ISFR, 2009, pp. 260–262 Search PubMed
- K. Janssens, A. L. Bugaev, E. G. Kozyr, V. Lemmens, A. A. Guda, O. A. Usoltsev, S. Smolders, A. V. Soldatov and D. E. De Vos, Chem. Sci., 2022, 13, 10251–10259 RSC
- P. Liu and C. Ai, Ind. Eng. Chem. Res., 2018, 57, 3807–3820 CrossRef CAS
- A. Dewaele, T. Renders, B. Yu, F. Verpoort and B. F. Sels, Catal. Sci. Technol., 2016, 6, 7708–7717 RSC
- S. S. Suresh, S. Mohanty and S. K. Nayak, J. Clean. Prod., 2017, 149, 863–873 CrossRef CAS
- M. Chantreux, D. Ricard, L. Asia, S. Rossignol and P. Wong-Wah-Chung, Radiat. Phys. Chem., 2021, 188, 109671 CrossRef CAS
- J. Miliute-Plepiene, A. Fråne and A. M. Almasi, Clean. Eng. Technol., 2021, 4, 100246 CrossRef
- S. Kim, Waste Manag., 2001, 21, 609–616 CrossRef CAS PubMed
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00945a |
| This journal is © The Royal Society of Chemistry 2023 |