
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulation of supramolecular chirality by stepwise axial coordination in a nano-size trizinc(II)porphyrin trimer†‡
Avinash
Dhamija§
,
Dolly
Chandel§
and
Sankar Prasad
Rath
 *
*
Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur-208016, India. E-mail: sprath@iitk.ac.in
First published on 10th May 2023
Abstract
Herein, we report a chiral guest's triggered spring-like contraction and extension motions coupled with unidirectional twisting in a novel flexible and ‘nano-size’ achiral trizinc(II)porphyrin trimer host upon step-wise formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:2, and 1:4 host–guest supramolecular complexes based on the stoichiometry of the diamine guests for the first time. During these processes, porphyrin CD responses have been induced, inverted, and amplified, and reduced, respectively, in a single molecular framework due to the change in the interporphyrin interactions and helicity. Also, the sign of the CD couplets is just the opposite between R and S substrates which suggests that the chirality is dictated solely by the stereographic projection of the chiral center. Interestingly, the long-range electronic communications between the three porphyrin rings generate trisignate CD signals that provide further information about molecular structures.
:1, 1:2, and 1:4 host–guest supramolecular complexes based on the stoichiometry of the diamine guests for the first time. During these processes, porphyrin CD responses have been induced, inverted, and amplified, and reduced, respectively, in a single molecular framework due to the change in the interporphyrin interactions and helicity. Also, the sign of the CD couplets is just the opposite between R and S substrates which suggests that the chirality is dictated solely by the stereographic projection of the chiral center. Interestingly, the long-range electronic communications between the three porphyrin rings generate trisignate CD signals that provide further information about molecular structures.
Introduction
Supramolecular chirality is an intriguing phenomenon widely observed in natural systems, including the DNA double helix, the secondary-α-helix structure of proteins and tropocollagen triple helix, and plays a vital role in the evolution of life, genetic information storage and replication, and transmembrane transport.1–3 In artificial systems, supramolecular chirality provides a tremendous platform to construct and explore highly complex architectures for various applications.4–14 Among numerous macromolecular or supramolecular systems, helical molecules are of significant interest not only because of their topological structures15–20 but also for their fascinating spring-like contraction–extension motions emulating muscle-like functions or mechanical motions.21–25 Several synthetic helical molecules such as oligomers and foldamers have been reported to exhibit such motions triggered by ion binding or by changing pH, solvent, or temperature.26–35 Some of them also undergo unidirectional twisting during contraction and extension, a feature commonly observed in biological systems.29,30 In contrast to intrinsically helical molecules, inducing helicity in an achiral molecule and achieving spring-like motions is much more challenging but vital for fabricating nanoscale mechanical machines.Porphyrin-based chromophores are highly attractive for investigating the induction and amplification of supramolecular chirality due to their unique spectral and physicochemical properties.36,37 In particular, dimeric and oligomeric porphyrin frameworks are of great interest due to their high sensitivity towards through-space interchromophoric electronic communication, even over relatively large distances.38–43 When the interacting porphyrin rings are associated with a chiral substrate, the coupling transitions can be detected by CD spectroscopy in the porphyrin spectral region.44–50 Guest stoichiometry-controlled helicity induction and amplification/inversion in a metalloporphyrin dimer with bifunctional guests are well established and known to occur via stepwise 1:1 and 1:2 host–guest complexation mechanisms.51–53 However, such a behaviour in a flexible porphyrin trimer rendering unidirectional spring-like motions has not been reported yet.
Herein, we report helicity induction, control, and spring-like contraction and extension motions in a novel ‘nano-size’ achiral trizinc(II) porphyrin trimer (1) host upon gradual addition of chiral diamines as substrates (Scheme 1). A clear rationalization of helicity induction and spring-like motions is demonstrated by stepwise induction, amplification, and reduction of the circular dichroism (CD) signal upon formation of 1:1, 1:2, and 1:4 host–guest complexes, respectively. X-ray structures of 1·(CHDA(R,R))2 and 1·(CHDA(R,R))4 exhibited the origin of chirality induction upon coordination of the CHDA(R,R) leading to the contraction and expansion of 1, respectively, with unidirectional twisting. The exciton coupling between the neighbouring and distant porphyrin rings in the host–guest complexes cumulatively originates from the trisignate CD couplet in the porphyrinic Soret band region. This work may pave the way for developing future nanoscale devices that can incorporate unidirectional twisting.
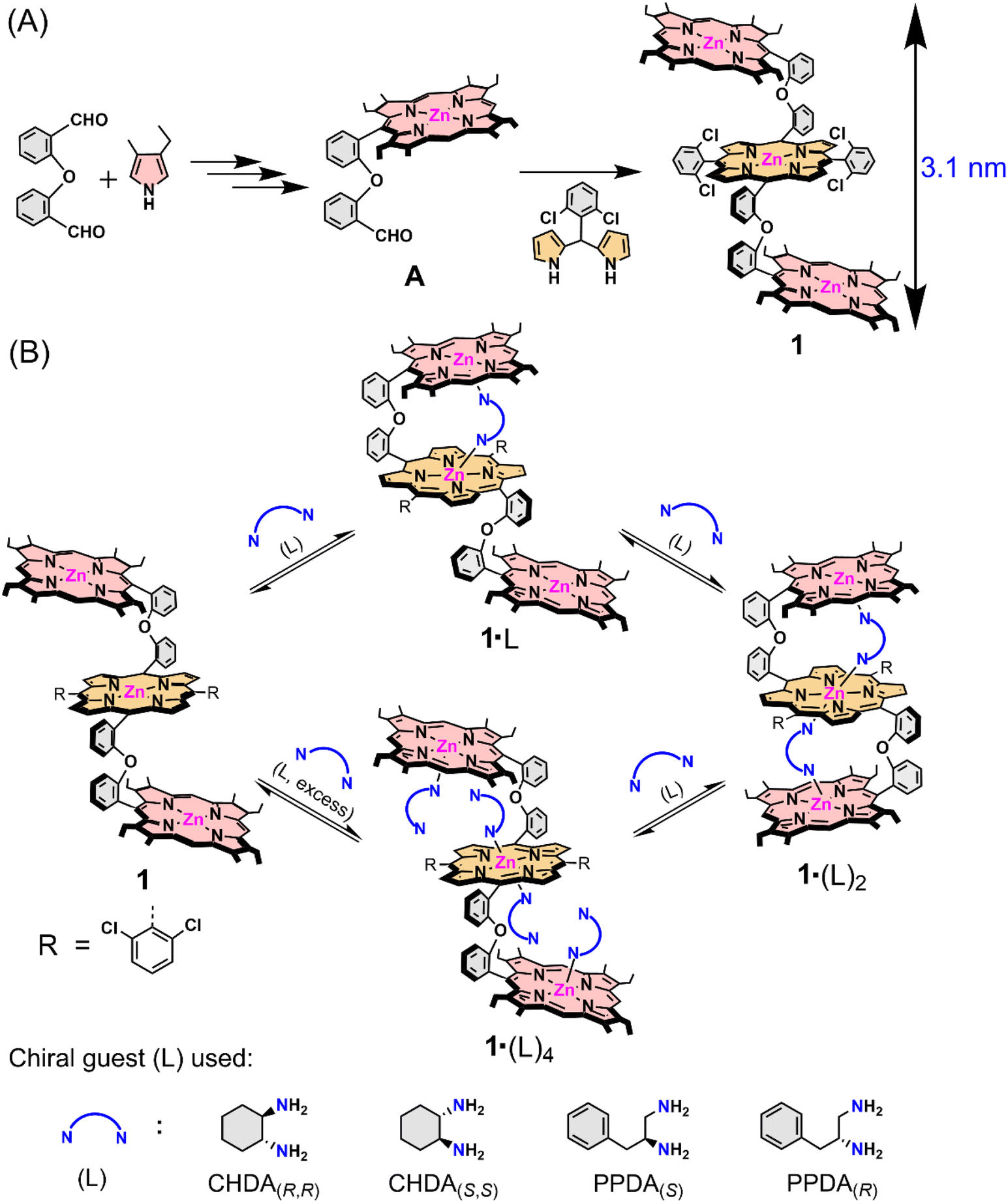 | ||
| Scheme 1 (A) Synthetic outline of the trizinc(II)porphyrin trimer host (1) and (B) host–guest complexation between 1 and chiral guests (L). | ||
Results and discussion
In order to develop a host for chiral guest encapsulation, we first prepared 2-(2-(porphyrin-5-yl)phenoxy)benzaldehyde (A) from 2,2′-oxydibenzaldehyde and 3-ethyl-4-methylpyrrole following the reported procedure.54 Afterward, the reaction of A and 2,2′-((2,6-difluorophenyl)methylene)bis(pyrrole) in the presence of a catalytic amount of TFA formed a freebase porphyrin trimer, which upon zinc metalation using Zn(OAc)2 converted to 1 (Scheme 1A; for a detailed synthesis procedure, see the ESI†). The ESI mass spectrum of 1 revealed a peak centred at m/z 2073.5229 corresponding to [1 + H]+; the isotopic distribution pattern of experimental mass is also nicely correlated with the theoretical pattern (Fig. S1‡). The formation of 1 was further characterized by NMR spectroscopy (Fig. S2–S4‡) and X-ray structural (for crystallization conditions, see the ESI‡) analysis (Fig. 1A and S5‡).55 Due to the flexible nature of diphenylether linkers, 1 is stabilized in a ladder-like conformation with a Zn1⋯Zn2/Zn2⋯Zn3 distance of 8.40 Å and a Zn1⋯Zn3 distance of 16.81 Å (Tables S1 and S2†).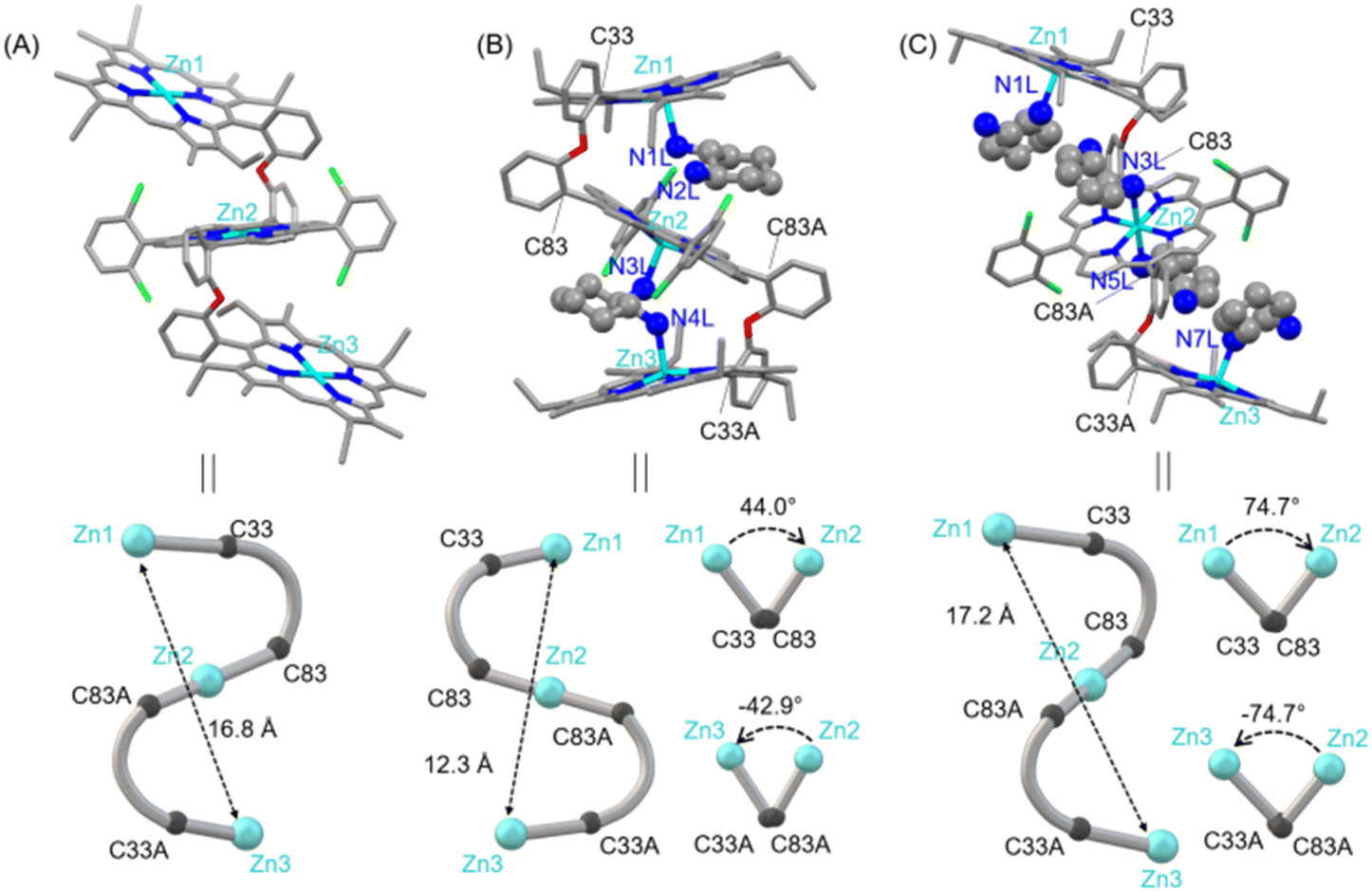 | ||
| Fig. 1 (A) Molecular structure of 1. Bond lengths (Å) Zn1⋯Zn2/Zn2⋯Zn3, 8.40 and Zn1⋯Zn3, 16.81. (B) Molecular structure of 1·(CHDA(R,R))2. Bond lengths (Å) and dihedral angles (deg) Zn1⋯Zn2, 6.46; Zn2⋯Zn3, 5.88; Zn1⋯Zn3, 12.34; Zn1–N1L, 2.156(4); Zn2–N2L, 3.521(4); Zn2–N3L, 2.193(4); Zn3–N4L, 2.191(4); Zn1–C33–C83–Zn2, 44.09; Zn2–C83A–C33A–Zn3, −42.91. (C) Molecular structure of 1·(CHDA(R,R))4. Bond lengths (Å) and dihedral angles (deg): Zn1⋯Zn2/Zn2⋯Zn3, 8.63; Zn1⋯Zn3, 17.25; Zn1–N1L/Zn3–N7L, 2.183(12); Zn2–N3L/Zn2–N5L, 2.397(14); Zn1–C33–C83–Zn2, 74.69; Zn2–C83A–C33A–Zn3, −74.69. | ||
The structure of 1·(CHDA(R,R))2 has been unveiled by single crystal X-ray crystallography56 (Fig. 1B and S6†) which showed that the complex crystallizes in a triclinic crystal system with P1 chiral space group. The molecular structure showed that all three porphyrins are oriented in a cofacial fashion and that the two molecules of CHDA(R,R) are encapsulated inside the cavities of 1via Zn–N coordination bonds. Overall, the complex attains a clockwise helical structure in which the dihedral angle between the first and second porphyrins (ϕ1, Zn1–C33–C83–Zn2) is 44.09°, and that between the second and third porphyrins (ϕ2, Zn2–C83A–C33A–Zn3) is −42.91°. The non-bonding distances of Zn1⋯Zn2 and Zn2⋯Zn3 are 6.46 and 5.88 Å, respectively, and the Zn1⋯Zn3 distance is 12.34 Å. The comparison of the non-bonding Zn⋯Zn distances of 1 and 1·(CHDA(R,R))2 clearly shows the contraction of the structure upon encapsulation of two molecules of CHDA(R,R).
Dark red crystals of 1·(CHDA(R,R))4 are also grown for X-ray structural analysis.57 The complex crystallizes in a triclinic crystal system with P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Fig. 1C and S7†). From the crystal structure, it can be seen that one molecule of CHDA(R,R) coordinates to each zinc centre of the terminal porphyrins and two molecules of CHDA(R,R) bind to the zinc centre of the central porphyrin through the –NH2 binding site. Thus, the two terminal Zn(II) centres adopt five-coordinate square pyramidal geometry, and the central Zn(II) centre adopts six-coordinate octahedral geometry. Zn(II)porphyrins generally prefer to form five-coordinated states; however, the presence of four electron-withdrawing chloro groups in the central Zn(II)porphyrin enables it to attain the six-coordination state.58 Interestingly, 1·(CHDA(R,R))4 stabilizes in an extended clockwise helical structure in which porphyrin rings are away from each other because of the repulsion between four CHDA(R,R) molecules. The dihedral angles ϕ1 and ϕ2 are 74.69° and −74.69°, respectively. The Zn1⋯Zn2/Zn2⋯Zn3 non-bonding distance is 8.63 Å and the Zn1⋯Zn3 distance is 17.25 Å, indicating the extension of the structure compared to the 1:2 host–guest complex.
space group (Fig. 1C and S7†). From the crystal structure, it can be seen that one molecule of CHDA(R,R) coordinates to each zinc centre of the terminal porphyrins and two molecules of CHDA(R,R) bind to the zinc centre of the central porphyrin through the –NH2 binding site. Thus, the two terminal Zn(II) centres adopt five-coordinate square pyramidal geometry, and the central Zn(II) centre adopts six-coordinate octahedral geometry. Zn(II)porphyrins generally prefer to form five-coordinated states; however, the presence of four electron-withdrawing chloro groups in the central Zn(II)porphyrin enables it to attain the six-coordination state.58 Interestingly, 1·(CHDA(R,R))4 stabilizes in an extended clockwise helical structure in which porphyrin rings are away from each other because of the repulsion between four CHDA(R,R) molecules. The dihedral angles ϕ1 and ϕ2 are 74.69° and −74.69°, respectively. The Zn1⋯Zn2/Zn2⋯Zn3 non-bonding distance is 8.63 Å and the Zn1⋯Zn3 distance is 17.25 Å, indicating the extension of the structure compared to the 1:2 host–guest complex.
The formation of the 1:1 host–guest complex in solution was confirmed from its 1H NMR spectrum which showed a large upfield shift of the chiral guest protons as the guest emerges in the porphyrin ring current (Fig. S8‡). For example, the CHDA(R,R) protons in 1·CHDA(R,R) resonated at −0.24 (H1), −1.78, −1.94 (H2), −2.05 (H3), −3.83, −3.94 (H4), −5.13 (H5) and −5.78, −7.49 (NH2) ppm. H2, H4 and NH2 protons were split in 1·CHDA(R,R) due to their being in close vicinity to the two different porphyrin rings. The identical 5, 15 meso protons of the porphyrin ring (once the substrate binds to the Zn centre) were split into two resonances and shifted to 10.16 and 9.89 ppm, due to the stereospecific twisting of the porphyrin rings in the host–guest complexes. In the 1:2 complex 1·(CHDA(R,R))2, a slight downfield shift of guest protons was observed as compared to 1·CHDA(R,R) (Fig. S8–S11‡). A similar shift of the guest protons was also observed with other substrates as well (Fig. S12‡). In contrast, the addition of 1.0 M equivalent of cyclohexylamine to 1 produced a 1:1 host–guest complex that results in smaller upfield shifts of guest protons due to the monotopic binding of the guest (Fig. S8‡). Moreover, ESI mass spectrometry of 1 with 100 eq. of CHDA(R,R) revealed peaks at m/z 2187.5136 and 2301.6658, respectively, which were assigned to [1·CHDA(R,R) + H]+ and [1·(CHDA(R,R))2 + H]+ (Fig. S13‡), confirming the formation of 1:1 and 1:2 host–guest complexes.
Host–guest complexation between 1 and chiral diamines (Scheme 1B and Fig. 2A) in solution state was also investigated by UV-vis spectroscopy. Upon addition of a chiral diamine guest to a dichloromethane solution of 1, three stepwise UV-visible spectral changes were observed depending on the concentration of the guest molecule. For instance, the addition of CHDA(R,R) from 0 to 50 equivalents to the dichloromethane solution of 1 resulted in, at first, a large red-shift of the Soret (from 407 to 419 nm), shoulder (from 423 to 436 nm) and Q bands (from 536 to 547 and 572 to 583 nm) along with a decrease in the Soret band intensity (Fig. 2B) due to the formation of a 1:1 sandwich complex, 1·CHDA(R,R). In the second step, upon further addition of CHDA(R,R) from 50 to 500 equivalents, more red-shift of the Soret (from 419 to 421 nm) and Q bands (from 547 to 549 and 583 to 584 nm) was observed accompanied by an increase in the Soret band intensity due to the conversion of 1·CHDA(R,R) to a 1:2 sandwich complex, 1·(CHDA(R,R))2 (Fig. 2C). In the third step, upon the addition of CHDA(R,R) from 500 to 4000 equivalents to 1·(CHDA(R,R))2, the UV-visible spectra changed again! The Soret (421 to 420 nm) and shoulder bands (436 to 435 nm) were shifted slightly to a lower wavelength (Fig. 2D). After that, no further shifting of the Soret band was observed upon the addition of excess CHDA(R,R), indicating the formation of a 1:4 open complex, 1·(CHDA(R,R))4 at 4000 eq. Similar UV-vis spectral changes of 1 were also observed with other substrates as well (Fig. S14‡). Moreover, the emission band of 1 decreases upon the addition of the CHDA(R,R) guest (Fig. S15‡).
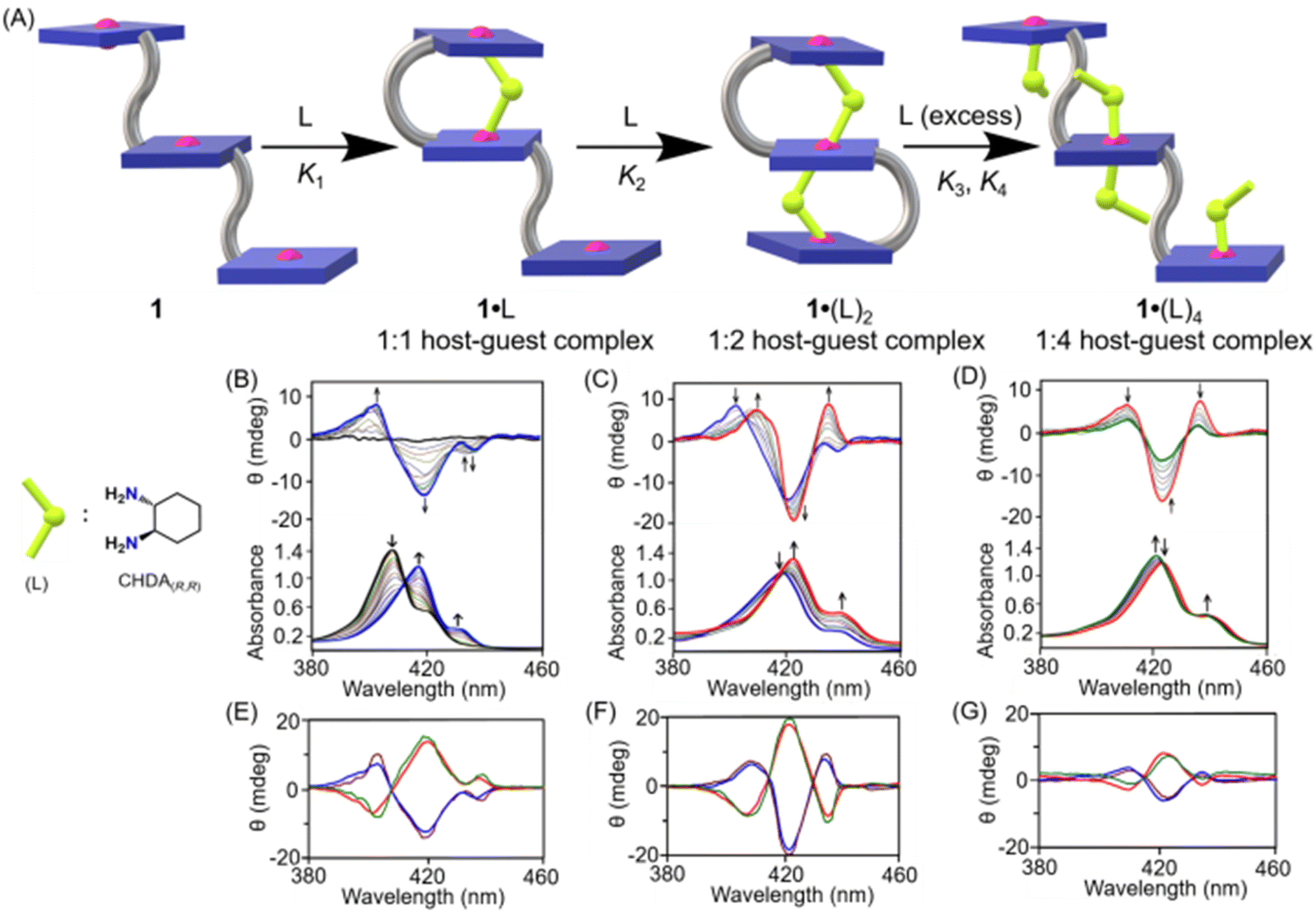 | ||
| Fig. 2 (A) Schematic representation of the formation of 1:1, 1:2 and 1:4 host–guest complexes between 1 and CHDA(R,R). CD and UV spectral changes (in CH2Cl2 at 295 K) of 1 (at 3 × 10−6 M) upon addition of CHDA(R,R) as the host–guest molar ratio changes from (B) 1:0 to 1:50, (C) 1:50 to 1:500 and (D) 1:500 to 1:4000. Calculated (cal) and observed (obs) CD spectra (E) of 1·CHDA(R,R) (brown, cal), 1·CHDA(S,S) (green, cal), 1·CHDA(R,R) (blue, obs) and 1·CHDA(S,S) (red, obs); (F) 1·(CHDA(R,R))2 (brown, cal), 1·(CHDA(S,S))2 (green, cal), 1·(CHDA(R,R))2 (blue, obs) and 1·(CHDA(S,S))2 (red, obs); and (G) 1·(CHDA(R,R))4 (brown, cal), 1·(CHDA(S,S))4 (green, cal), 1·(CHDA(R,R))4 (blue, obs) and 1·(CHDA(S,S))4 (red, obs). | ||
CD spectroscopy also exhibited three distinct spectral patterns at different concentration regions associated with 1·L, 1·(L)2 and 1·(L)4 complexes. Upon gradual addition of CHDA(R,R) from 0 to 50 equivalents to the dichloromethane solution of 1, a negative CD couplet (Acal = −220 M−1 cm−1) appeared due to the formation of 1·CHDA(R,R) (Fig. 2B). Interestingly, a similar sign of the CD couplet was also observed with the related diphenylether-bridged dizinc(II)porphyrin dimer upon formation of a 1:1 sandwich complex with CHDA(R,R), in which two porphyrin rings were oriented in an anticlockwise direction to have a minimum host–guest steric clash.47 Upon further addition of the substrate from 50 to 500 equivalents, 1·CHDA(R,R) gets converted to 1·(CHDA(R,R))2, which displayed an enhanced trisignate CD signal (Acal1 = +276 M−1 cm−1; Acal1 is the amplitude between the first and second CD bands) with ‘positive–negative–positive’ signs (Fig. 2C). The trisignate CD signal of 1·(CHDA(R,R))2 and its larger CD amplitude compared to 1·CHDA(R,R) indicate that the three porphyrin rings came closer to each other with a positive helical twist, causing a strong intramolecular exciton coupling between them.59,60 On the other hand, the addition of a chiral monoamine, (R)-phenylethylamine (up to 500 equiv.), to 1·CHDA(R,R) resulted in no change in the sign and intensity of the CD couplet, which confirmed that the CD inversion and amplification only take place when the chiral substrate binds in a bidentate fashion (Fig. S16‡). Upon further addition of the CHDA(R,R) guest from 500 to 4000 equivalents to 1·(CHDA(R,R))2, the CD signal gradually decreased to give a low intensity trisignate CD signal (Acal1 = +75 M−1 cm−1) due to the formation of 1·(CHDA(R,R))4 (Fig. 2D). Similar observations but with opposite CD responses were also obtained with the opposite enantiomer of the substrate which demonstrates that the chirality is dictated solely by the stereographic projection of the chiral centre of the substrates (Fig. 2E–G).
Similar spring-like motions were also observed for 1 upon interaction with the PPDA(S) and PPDA(R) guest by CD spectroscopy (Fig. S17 and S18‡). Table S3‡ summarizes the experimental spectral parameters for all the complexes reported here. To understand the helicity induction process, we also performed CD spectroscopic studies with a dizinc(II)nickel(II)porphyrin trimer (2), in which nickel was present in the central porphyrin ring (Schemes S2 and S3‡). The addition of CHDA(R,R) or PPDA(S) from 0 to 1000 equivalents to the dichloromethane solution of 2 resulted in only low-intensity CD signals due to monotopic binding of the guests with the host in both 1:1 and 1:2 complexes (Fig. S19‡). Thus, the binding of the chiral diamine to 1 in a bidentate fashion induces helicity in both 1·L and 1·(L)2 complexes and is responsible for intense bisignate and trisignate CD signals. The binding constant between 1 and the chiral diamines (L) was determined by a CD and UV-visible spectroscopic titration method. For complexation between 1 and CHDA(R,R), K1, K2, K3 and K4 are found to be 1.8 ± 0.2 × 105 M−1, 9.5 ± 0.2 × 103 M−1, 2.1 ± 0.1 × 103 M−1 and 1.5 ± 0.2 × 103 M−1 (Fig. S20‡), respectively. Similarly, binding constants were also calculated for other complexes (Fig. S21–S23‡).
Geometry optimization of both clockwise and anticlockwise conformers of 1·CHDA(R,R) was performed with the help of density functional theory (DFT), in which the anticlockwise conformation of 1·CHDA(R,R) was found to be more stable than the clockwise conformer by 10.8 kcal mol−1 (Fig. S24A‡) due to the preorganized projection of the NH2 group in CHDA(R,R). TDDFT calculated CD spectra of the clockwise and anticlockwise conformers of 1·CHDA(R,R) were also calculated separately (Fig. S24B‡); the experimental CD spectrum matched well with the calculated one using the anticlockwise conformer. The DFT optimized structure of 1·(CHDA(R,R))2 also matches well with the X-ray structure with a much longer Zn2–N2 distance (Fig. 3A and S25‡). The trisignate nature of the CD spectrum of 1·(CHDA(R,R))2 was reproduced nicely in the TDDFT calculation (Fig. 3B). Molecular orbital calculations showed that not only the +ve and −ve coupling interaction between distal porphyrins (first–second and second–third) but also the +ve interaction between proximal porphyrins (first–third) is involved in the trisignate CD signal (Fig. S26 and S27, and Table S5‡). Although multisignate CD signals have been observed earlier for porphyrin aggregates,15,16 the origin of these bands has seldom been reported.
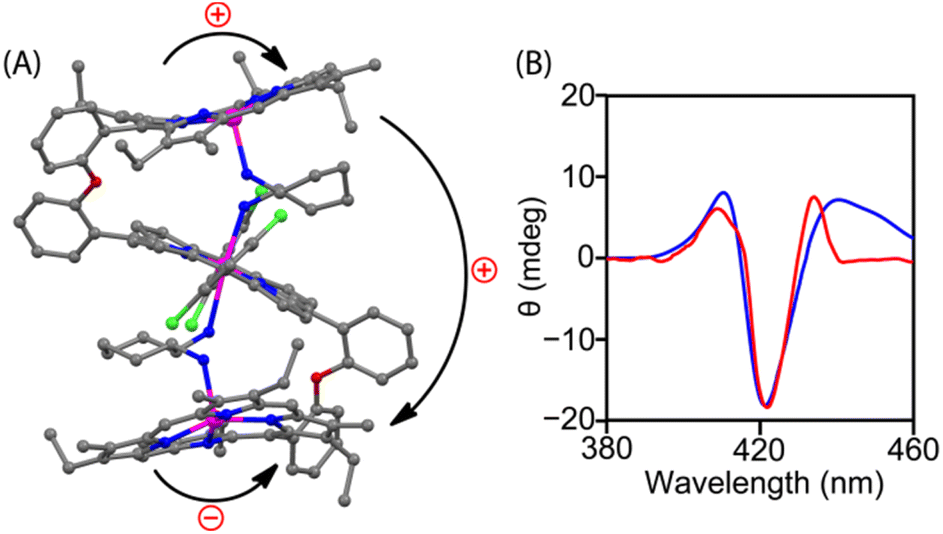 | ||
| Fig. 3 (A) DFT-optimized structure of 1·(CHDA(R,R))2; bent arrows represent the direction of interporphyrin twisting. (B) TDDFT-calculated (blue line) and experimental (red line) CD spectra of 1·(CHDA(R,R))2. | ||
Conclusions
In summary, the present work demonstrates unique chiral-guest-regulated spring-like contraction and extension motions coupled with unidirectional twisting in a flexible and achiral ‘nano-size’ trizinc(II)porphyrin trimer host (1). The work also rationalized the origin of chirality transfer from an optically active diamine guest (L) to an achiral host 1 upon step-wise formation of 1:1, 1:2 and 1:4 host–guest supramolecular complexes during which porphyrin CD responses have been induced, inverted and amplified and also reduced in a single molecular framework for the first time. Moreover, the strong intramolecular exciton coupling between the three porphyrin rings generates trisignate CD signals, which also provide information about the distances and relative orientations of the porphyrin chromophores. These results provide a design strategy for the development of novel ‘nano-size’ molecular machines with elastic motions for various applications.
Data availability
A detailed description of the synthesis and experimental procedures along with complete characterization data, instrumentation and computational details can be found in the ESI.† Data related to the X-ray crystal structures reported in this study have been deposited in the Cambridge Crystallographic Data Center (CCDC) under accession codes 2214044 (for 1), 2214045 (for 1·(CHDA(R,R))2), and 1849072 (for 1·(CHDA(R,R))4).Author contributions
A. D. and S. P. R. conceptualized and S. P. R. supervised the overall work. A. D. and D. C. performed the experiments. A. D., D. C. and S. P. R. analysed the results and wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank the Council of Scientific and Industrial Research (CSIR), New Delhi , the Science and Engineering Research Board (SERB), New Delhi, India and SERB-STAR for financial support. SPR thanks SERB for giving him the Science and Technology Award for Research (SERB-STAR). The CARE scheme of IIT Kanpur is gratefully acknowledged for the CD facility.Notes and references
- T. Taniguchi and T. Usuki, Supramolecular Chemistry: From Molecules to Nanomaterials, John Wiley & Sons Ltd, New York, 2012 Search PubMed.
- J. Gal, Top. Curr. Chem., 2013, 340, 1 CrossRef CAS.
- R. A. Hegstrom and D. K. Kondepudi, Sci. Am., 1990, 262, 108 CrossRef.
- E. Schwartz, S. L. Gac, J. L. M. J. Cornelissen, R. J. M. Nolte and A. E. Rowan, Chem. Soc. Rev., 2010, 39, 1576 RSC.
- D. M. Bassani, J.-M. Lehn, G. Baum and D. Fenske, Angew. Chem., Int. Ed., 1997, 36, 1845 CrossRef CAS.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304 CrossRef CAS PubMed.
- G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1 CrossRef CAS.
- A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948 CrossRef CAS.
- D. Pijper and B. L. Feringa, Soft Matter, 2008, 4, 1349 RSC.
- K. J. Hartlieb, A. K. Blackburn, S. T. Schneebeli, R. S. Forgan, A. A. Sarjeant, C. L. Stern, D. Cao and J. F. Stoddart, Chem. Sci., 2014, 5, 90 RSC.
- X. Wang, S. Xiang, C. Qi, M. Chen, X. Su, J.-C. Yang, J. Tian, H.-T. Feng and B. Z. Tang, ACS Nano, 2022, 16, 8223 CrossRef CAS PubMed.
- X. Li, S. Wan, Y. Xu, J. Tian, L.-C. Shen, H. Zuilhof, M. Zhang and A. C.-H. Sue, Chem, 2022, 8, 2136 Search PubMed.
- B. A. F. Le Bailly and J. Clayden, Chem. Commun., 2016, 52, 4852 RSC.
- Y. Wang, D. Niu, G. Ouyang and M. Liu, Nat. Commun., 2022, 13, 1710 CrossRef CAS PubMed.
- Q.-P. Hu, H. Zhou, T.-Y. Huang, Y.-F. Ao, D.-X. Wang and Q.-Q. Wang, J. Am. Chem. Soc., 2022, 144, 6180 CrossRef CAS PubMed.
- S. Akine and H. Miyake, Coord. Chem. Rev., 2022, 468, 214582 CrossRef CAS.
- A. Scarso and J. Rebek Jr, Top. Curr. Chem., 2006, 265, 1 CrossRef CAS.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752 CrossRef CAS PubMed.
- T. Leigh and P. Fernandez-Trillo, Nat. Rev. Chem., 2020, 4, 291 CrossRef CAS PubMed.
- Z. Ashbridge, S. D. P. Fielden, D. A. Leigh, L. Pirvu, F. Schaufelberger and L. Zhang, Chem. Soc. Rev., 2022, 51, 7779 RSC.
- K. Kinbara and T. Aida, Chem. Rev., 2005, 105, 1377 CrossRef CAS PubMed.
- D. J. V. Dijken, J. M. Beierle, M. C. A. Stuart, W. Szymański, W. R. Browne and B. L. Feringa, Angew. Chem., Int. Ed., 2014, 53, 5073 CrossRef PubMed.
- Y. Yoshida, Y. Mawatari, A. Motoshige, R. Motoshige, T. Hiraoki, M. Wagner, K. Müllen and M. Tabata, J. Am. Chem. Soc., 2013, 135, 4110 CrossRef CAS PubMed.
- O.-S. Jung, Y. J. Kim, Y.-A. Lee, J. K. Park and H. K. Chae, J. Am. Chem. Soc., 2000, 122, 9921 CrossRef CAS.
- F. Li, X. Devaux, D. Sluysmans, F. Zerbetto, I. Huc and A.-S. Duwez, Chem, 2021, 7, 1333 Search PubMed.
- H.-J. Kim, E. Lee, H.-S. Park and M. Lee, J. Am. Chem. Soc., 2007, 129, 10994 CrossRef CAS PubMed.
- T. Nabeshima, Y. Yoshihira, T. Saiki, S. Akine and E. Horn, J. Am. Chem. Soc., 2003, 125, 28 CrossRef CAS PubMed.
- P. Pengo, L. Pasquato, S. Moro, A. Brigo, F. Fogolari, Q. B. Broxterman, B. Kaptein and P. Scrimin, Angew. Chem., Int. Ed., 2003, 42, 3388 CrossRef CAS.
- S. Yamamoto, H. Iida and E. Yashima, Angew. Chem., Int. Ed., 2013, 52, 6849 CrossRef CAS PubMed.
- Y. Suzuki, T. Nakamura, H. Iida, N. Ousaka and E. Yashima, J. Am. Chem. Soc., 2016, 138, 4852 CrossRef CAS PubMed.
- D. Mondal, M. Ahmad, B. Roy, A. Mondal and P. Takuldar, Nat. Commun., 2022, 13, 6507 CrossRef CAS.
- E. Ohta, H. Sato, S. Ando, A. Kosaka, T. Fukushima, D. Hashizume, M. Yamasaki, K. Hasegawa, A. Muraoka, H. Ushiyama, K. Yamashita and T. Aida, Nat. Chem., 2011, 3, 68 CrossRef CAS PubMed.
- N. Ousaka, K. Shimizu, Y. Suzuki, T. Iwata, M. Itakura, D. Taura, H. Iida, Y. Furusho, T. Mori and E. Yashima, J. Am. Chem. Soc., 2018, 140, 17027 CrossRef CAS PubMed.
- M. Barboiu, G. Vaughan, N. Kyritsakas and J. M. Lehn, Chem. – Eur. J., 2003, 9, 763 CrossRef CAS PubMed.
- T. Hashimoto, T. Nishimura, J. M. Lim, D. Kim and H. Maeda, Chem. – Eur. J., 2010, 16, 11653 CrossRef CAS.
- H. Lu and N. Kobayashi, Chem. Rev., 2016, 116, 6184 CrossRef CAS PubMed.
- V. Valderrey, G. Aragay and P. Ballester, Coord. Chem. Rev., 2014, 258, 137 CrossRef.
- N. Berova, G. Pescitelli, A. G. Petrovic and G. Proni, Chem. Commun., 2009, 5958 RSC.
- M. Harmata, Acc. Chem. Res., 2004, 37, 862 CrossRef CAS PubMed.
- J. W. Canary, Chem. Soc. Rev., 2009, 38, 747 RSC.
- G. A. Hembury, V. V. Borovko and Y. Inoue, Chem. Rev., 2008, 108, 1 CrossRef CAS PubMed.
- G. Pescitelli, L. D. Bari and N. Berova, Chem. Soc. Rev., 2014, 43, 5211 RSC.
- A. Dhamija, P. Mondal, B. Saha and S. P. Rath, Dalton Trans., 2020, 49, 10679 RSC.
- I. C. Pintre, S. Pierrefixe, A. Hamilton, V. Valderrey, C. Bo and P. Ballester, Inorg. Chem., 2012, 51, 4620 CrossRef CAS.
- H. Gholami, D. Chakraborty, J. Zhang and B. Borhan, Acc. Chem. Res., 2021, 54, 654 CrossRef CAS PubMed.
- (a) S. Brahma, S. A. Ikbal, A. Dhamija and S. P. Rath, Inorg. Chem., 2014, 53, 2381 CrossRef CAS PubMed; (b) A. Dhamija, S. A. Ikbal and S. P. Rath, Inorg. Chem., 2016, 55, 13014 CrossRef CAS PubMed.
- S. Brahma, S. A. Ikbal, S. Dey and S. P. Rath, Chem. Commun., 2012, 48, 4070 RSC.
- (a) A. Dhamija, B. Saha, D. Chandel, H. Malik and S. P. Rath, Inorg. Chem., 2020, 59, 801 CrossRef CAS; (b) B. Saha, D. Chandel and S. P. Rath, Inorg. Chem., 2022, 61, 2154 CrossRef CAS PubMed.
- B. Saha, A. G. Petrovic, A. Dhamija, N. Berova and S. P. Rath, Inorg. Chem., 2019, 58, 11420 CrossRef CAS PubMed.
- A. Dhamija, B. Saha and S. P. Rath, Inorg. Chem., 2017, 56, 15203 CrossRef CAS PubMed.
- S. A. Ikbal, A. Dhamija, S. Brahma and S. P. Rath, J. Org. Chem., 2016, 81, 5440 CrossRef CAS PubMed.
- S. A. Ikbal, S. Brahma and S. P. Rath, Chem. Commun., 2014, 50, 14037 RSC.
- J. Etxebarria, A. V. Ferran and P. Ballester, Chem. Commun., 2008, 5939 RSC.
- M. Tanaka, K. Ohkubo, C. P. Gros, R. Guilard and S. Fukuzumi, J. Am. Chem. Soc., 2006, 128, 14625 CrossRef CAS PubMed.
- Crystal data for 1, CCDC-2214044, triclinic, space group P, a = 11.2120(9) Å, b = 13.3082(11) Å, c = 19.7177(15) Å, α = 73.666(2)°, β = 84.633(2)°, γ = 68.645(2)°, V = 2629.4(4) Å3, Z = 1, Dc = 1.289 Mg per mm3, T = 100(2) K, no. of reflections used = 9766, θmax = 25.498°, R1 = 0.0712 (for I > 2σ(I)), wR2 (all data) = 0.1071, goodness of fit on F2 = 1.057.
- Crystal data for 1·(CHDA(R,R))2, CCDC-2214045, triclinic, space group P1, a = 12.967(2) Å, b = 14.683(2) Å, c = 17.866(3) Å, α = 82.663(4)°, β = 72.777(4)°, γ = 72.012(4)°, V = 3087.6(8) Å3, Z = 1, Dc = 1.241 Mg per mm3, T = 100(2) K, no. of reflections used = 22655, θmax = 25.50°, R1 = 0.0452 (for I > 2σ(I)), wR2 (all data) = 0.1086, goodness of fit on F2 = 1.007.
- Crystal data for 1·(CHDA(R,R))4, CCDC-1849072, triclinic, space group P, a = 12.199(5) Å, b = 14.356(5) Å, c = 21.640(5) Å, α = 83.953(5)°, β = 86.895(5)°, γ =66.880(5)°, V = 3466(2) Å3, Z = 2, Dc = 1.210 Mg per mm3, T = 100(2) K, no. of reflections used = 12311, θmax = 25.10°, R1 = 0.1591 (for I > 2σ(I)), wR2 (all data) = 0.4507, goodness of fit on F2 =1.013.
- L. Favereau, A. Cnossen, J. B. Kelber, J. Q. Gong, R. M. Oetterli, J. Cremers, L. M. Herz and H. L. Anderson, J. Am. Chem. Soc., 2015, 137, 14256 CrossRef CAS.
- G. Sargsyan, B. M. Leonard, J. Kubelka and M. Balaz, Chem. – Eur. J., 2014, 20, 1878 CrossRef CAS.
- A. Mammana, G. Pescitelli, T. Asakawa, S. Jockusch, A. G. Petrovic, R. R. Mona-co, R. Purrello, N. J. Turro, K. Nakanishi, G. A. Ellestad, M. Balaz and N. Berova, Chem. – Eur. J., 2009, 15, 11853 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Vadapalli Chandrasekhar on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Text, figures, and tables depicting detailed experimental procedures and characterisation data including UV-vis, X-ray crystallography and DFT. CCDC 2214044, 2214045, and 1849072. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00858d |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |