
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulation of IL-17 backbone dynamics reduces receptor affinity and reveals a new inhibitory mechanism†
Daniel J.
Shaw
 a,
Lorna C.
Waters
b,
Sarah L.
Strong
b,
Monika-Sarah E. D.
Schulze
c,
Gregory M.
Greetham
d,
Mike
Towrie
d,
Anthony W.
Parker
d,
Christine E.
Prosser
c,
Alistair J.
Henry
c,
Alastair D. G.
Lawson
c,
Mark. D.
Carr
b,
Richard J.
Taylor
c,
Neil T.
Hunt
*a and
Frederick W.
Muskett
*b
a,
Lorna C.
Waters
b,
Sarah L.
Strong
b,
Monika-Sarah E. D.
Schulze
c,
Gregory M.
Greetham
d,
Mike
Towrie
d,
Anthony W.
Parker
d,
Christine E.
Prosser
c,
Alistair J.
Henry
c,
Alastair D. G.
Lawson
c,
Mark. D.
Carr
b,
Richard J.
Taylor
c,
Neil T.
Hunt
*a and
Frederick W.
Muskett
*b
aDepartment of Chemistry and York Biomedical Research Institute, University of York, Heslington, York, YO19 5DD, UK. E-mail: neil.hunt@york.ac.uk
bDepartment of Molecular and Cell Biology/Leicester Institute of Structural and Chemical Biology, University of Leicester, University Road, Leicester, LE1 7RH, UK. E-mail: fwm1@leicester.ac.uk
cUCB Pharma, UCB Biopharma UK, 216 Bath Road, Slough, SL1 3WE, UK
dCentral Laser Facility, Research Complex at Harwell, STFC Rutherford Appleton Laboratory, Harwell Oxford, Didcot, Oxon OX11 0QX, UK
First published on 16th June 2023
Abstract
Knowledge of protein dynamics is fundamental to the understanding of biological processes, with NMR and 2D-IR spectroscopy being two of the principal methods for studying protein dynamics. Here, we combine these two methods to gain a new understanding of the complex mechanism of a cytokine:receptor interaction. The dynamic nature of many cytokines is now being recognised as a key property in the signalling mechanism. Interleukin-17s (IL-17) are proinflammatory cytokines which, if unregulated, are associated with serious autoimmune diseases such as psoriasis, and although there are several therapeutics on the market for these conditions, small molecule therapeutics remain elusive. Previous studies, exploiting crystallographic methods alone, have been unable to explain the dramatic differences in affinity observed between IL-17 dimers and their receptors, suggesting there are factors that cannot be fully explained by the analysis of static structures alone. Here, we show that the IL-17 family of cytokines have varying degrees of flexibility which directly correlates to their receptor affinities. Small molecule inhibitors of the cytokine:receptor interaction are usually thought to function by either causing steric clashes or structural changes. However, our results, supported by other biophysical methods, provide evidence for an alternate mechanism of inhibition, in which the small molecule rigidifies the protein, causing a reduction in receptor affinity. The results presented here indicate an induced fit model of cytokine:receptor binding, with the more flexible cytokines having a higher affinity. Our approach could be applied to other systems where the inhibition of a protein–protein interaction has proved intractable, for example due to the flat, featureless nature of the interface. Targeting allosteric sites which modulate protein dynamics, opens up new avenues for novel therapeutic development.
Introduction
The interleukin-17 (IL-17) family of proteins are proinflammatory cytokines that play important roles in both the innate and adaptive human immune response. The family consists of six members (IL-17A through to IL-17F), which function as disulphide linked homodimers (e.g., IL-17AA). In addition, IL-17A and IL-17F also form a mixed protomer hetero-dimer (IL-17AF). The physiological roles of IL-17AA and IL-17FF are in protection against bacterial and fungal infection, primarily at epithelial and dermatological barriers; however unregulated accumulation in these tissues is also associated with several serious autoimmune diseases including psoriasis, ankylosing spondylitis and psoriatic arthritis. As such, IL-17AA in particular, has been the target of successful therapeutic intervention by two licensed biological entities, secukinumab1,2 and ixekizumab.3,4 In addition, next generation therapies, such as bimekizumab,5,6 target complexes containing both IL-17A and IL-17F. There is growing evidence that targeting both the IL-17AA and FF isoforms may bring added patient benefit over IL-17AA alone, stimulating a detailed study into the origins of their functional differences.We focus upon the role of IL-17 structural dynamics in the formation of complexes between the cytokines and cell-surface receptors, which is part of the signalling process. The IL-17 receptors have five family members (IL-17RA through to IL-17RE) each of which contain subunits comprising an extracellular cytokine binding domain, a transmembrane region, and an intracellular signalling domain. The long-accepted model of IL-17AA, IL-17FF and IL-17AF signalling is based upon the formation of a hetero-trimeric complex in which the cytokine binds two different receptor molecules, one RA and one RC, leading to receptor complex dimerization.7,8 Recent studies have suggested that IL-17FF may also be able to signal via a complex formed with only IL-17RC.9
Crystal structures of the three IL-17 dimers (AA, AF and FF) in complex with the extracellular domains of receptor IL-17RA (D1 and D2) have been reported.10–14 Whilst the symmetry of the IL-17 homodimers presents the opportunity for binding two molecules of IL-17RA, in practice only a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex has been observed, and it has been proposed that conformational changes induced in the cytokine upon binding preclude recruitment of a second IL-17RA molecule. To date, only the structure of IL-17FF in complex with receptor IL-17RC D1–D4 has been reported.9 In contrast to IL-17RA, IL-17RC binding induces very few structural changes in IL-17FF, thus permitting the recruitment of two IL-17RC molecules. This structural data points to the possibility of the existence of different binding mechanisms between the receptor classes. Recently, the crystal structure of the heterotrimeric complex of IL-17FF in complex with IL-17RA and IL-17RC has been reported.15
:1 complex has been observed, and it has been proposed that conformational changes induced in the cytokine upon binding preclude recruitment of a second IL-17RA molecule. To date, only the structure of IL-17FF in complex with receptor IL-17RC D1–D4 has been reported.9 In contrast to IL-17RA, IL-17RC binding induces very few structural changes in IL-17FF, thus permitting the recruitment of two IL-17RC molecules. This structural data points to the possibility of the existence of different binding mechanisms between the receptor classes. Recently, the crystal structure of the heterotrimeric complex of IL-17FF in complex with IL-17RA and IL-17RC has been reported.15
The essence of therapeutic intervention strategies has been the inhibition of signalling complex formation through ligand binding to IL-17 dimers, which then prevents interaction with the receptors. Several studies have shown that small molecule inhibitors of IL17AA are able to bind in the central hydrophobic pocket, located between the two interior hairpins at the interface between the two halves of the homodimer16–19 (see Fig. 1), while a recent study has identified a new binding site at the C-terminus.20 These ligands have been shown to be capable of either directly inhibiting IL-17AA binding to IL-17RA and/or inhibiting IL-17AA signalling.
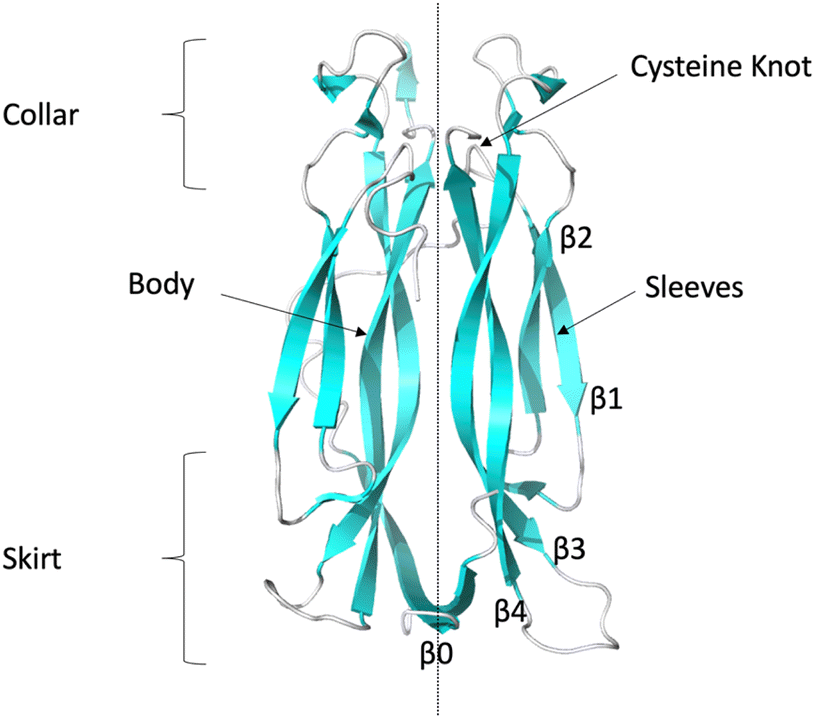 | ||
| Fig. 1 Ribbon representation of IL-17AA illustrating major structural elements (generated using PDB code: 7UWM). The illustration shows one protomer to the left and the other to the right of the dotted line resulting, in the case of the homo-dimers, a symmetric dimer. | ||
Structural data available for a limited number of the IL-17AA/inhibitor complexes that target the central hydrophobic pocket suggest that there is only very limited overlap between one of the binding sites of IL-17RA and the inhibitors, while only moderate structural changes occur to the cytokine. Recent NMR studies21 have however highlighted the markedly different levels of conformational heterogeneity observed in the IL-17AA, IL-17FF and IL-17AF dimers, with the higher amount of flexibility exhibited by the IL-17AA dimer being linked to the significantly tighter affinity observed for IL-17RA binding to IL-17AA relative to IL-17FF and IL-17AF. This leads to the possibility that these small molecule inhibitors may also function by altering the flexibility of particular regions of IL-17AA and this is the hypothesis that we test here.
The importance of dynamics has also been alluded to previously by groups who have determined cytokine structures either free or bound to receptors, ligands or antibodies using X-ray crystallography.9,12 Indeed, as observed “a potential shortcoming of such structural comparisons is that the crystal structures of neither the free- nor of the liganded-state may be fully representative of the solution state(s)”.9
Taken together this leads to the conclusion that a detailed study of the structural dynamics of IL-17 and its ligand complexes is needed to establish the presence of a link between IL-17 function and the degree of flexibility exhibited by the IL-17 dimers. To achieve this we combine an extensive set of NMR relaxation parameters with 2D-IR spectroscopy of the IL-17 amide I vibrational mode, which is sensitive to secondary structure and structural dynamics.
Protein dynamics refers to the time dependant fluctuations of the proteins structure and 15N NMR provides a residue-by-residue readout of backbone motions occurring on the picosecond to nanosecond (ps–ns; 10−12–10−9 s) timescale as well as probing motions on the microsecond to millisecond (μs–ms; 10−6–10−3 s) timescales. Motions in the ps-ns regime include ligand binding events and the kinetics of reactions, whereas the μs–ms regime encompasses many enzyme reactions and allosteric effects. Therefore, measuring these timescales by 15N NMR can potentially link protein dynamic nature to these functional processes. Comparison of these backbone timescales between closely related proteins or free and ligand/receptor bound proteins, as we do here, allows an insight into the roles of dynamic differences in these processes.
To complement NMR data we also apply 2D-IR spectroscopy. 2D-IR uses a sequence of ultrashort laser pulses to generate a 2D-spectroscopic map which arises directly from the protein secondary structure and the spatial arrangement and interactions of peptide units. Of particular relevance here, the 2D-IR spectrum of the amide I vibrational mode (essentially the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching motion of the peptide link) is highly sensitive to the vibrational coupling that occurs when peptide carbonyl groups form extended 3D structures such as alpha helixes via H-bonding.22 In combination with the ultrafast femtosecond to picosecond (fs–ps, 10−15–10−12 s) time resolution of 2D-IR, the 2D map of the amide I band allows us to evaluate the overall relative rigidity of the backbone of each protein on ultrafast timescales.
O stretching motion of the peptide link) is highly sensitive to the vibrational coupling that occurs when peptide carbonyl groups form extended 3D structures such as alpha helixes via H-bonding.22 In combination with the ultrafast femtosecond to picosecond (fs–ps, 10−15–10−12 s) time resolution of 2D-IR, the 2D map of the amide I band allows us to evaluate the overall relative rigidity of the backbone of each protein on ultrafast timescales.
In combination, NMR and 2D-IR approaches allow us to probe dynamics across a broad time scale from femtoseconds to milliseconds and length scales ranging from single residue to the whole molecule. By comparing results for IL-17AA, IL-17AF and IL-17FF individually and in complex with a tool compound that is known to affect receptor affinity (the inhibitory macrocyclic compound MC – ensemble 369 (ref. 23)) clear differences between the isoforms have been established alongside distinctive impacts of the MC on their respective dynamics, which correlate with changes in receptor binding affinities. The new understanding of the function of these important proteins and the inhibitory mechanism of the MC compounds17 provides new insights into opportunities for modulation of the conformational flexibility of IL-17 isoforms.
As this flexibility is directly related to the assembly of the signalling complex, this information will serve to guide future campaigns seeking to target multiple family members, whilst avoiding others, with either small molecules or biologics. Clearly, such an approach can be applied to other complexes so aiding the understanding of the mechanism of ligand–receptor formation and advancing therapeutic development.
Experimental
Detailed methods are provided in the ESI.†Protein sample preparation
Unlabelled and uniformly 15N, 15N/13C, 15N/2H and 15N/13C/2H labelled samples of mature human IL-17AA and IL-17FF homodimers (residues 24–155 and 31–163 respectively) and IL-17AF hetero-dimers were prepared as described previously.212D-IR spectroscopy
For 2D-IR measurements, a 20 μL aliquot of a solution of IL-17 (∼100 μM, in a deuterated 50 mM sodium phosphate buffer at pH 7.4 also containing 100 mM NaCl) was placed between two CaF2 windows with a path-length of 25 μm. All 2D-IR spectra were acquired using the ULTRA B laser system at the STFC Rutherford Appleton Laboratory, which has been described elsewhere.24 Each 2D-IR spectrum was the average of three scans. Full details are given in the ESI.†NMR spectroscopy
Detailed methods for the backbone assignment of IL-17AF in complex with MC, ligand mapping and backbone dynamics experiments are provided in the ESI.†NMR spectra of the IL-17 dimers were acquired from 350 μL samples of approximately 200 μM monomer IL-17AA, and IL-17FF, and 75–100 μM protomer IL-17AF (only one protomer labelled). NMR spectra of the IL-17 hetero-dimers with macrocycle (MC – ensemble 369, purchased from WuXi AppTec) were acquired from 350 μL samples of 200 μM 15N/13C/2H labelled IL-17A in complex with unlabelled IL-17F and 110–200 μM unlabelled IL-17A in complex with 15N/13C/2H labelled IL-17F and at a saturating concentration of MC (1.5 molar excess). Control experiments (data not presented) were collected to assess the effects of DMSO on the spectra of the various protomers at the maximum amount required to achieve saturation with MC (5% v/v). Only minor effects on the spectra were observed (i.e., chemical shift changes at a maximum of less than one half line width) consistent with many protein systems and interpreted as changes in solvation of the protein and/or very low affinity (KD » 100 mM) DMSO interactions.
NMR data were acquired at 308 K on either a 600 MHz Bruker Avance HD III or 800 MHz Bruker AVII spectrometer equipped with 5 mm, z-gradient TCI cryoprobes. Spectra assignments for the apo-IL-17 homo- and hetero-dimers have been reported previously.2115N relaxation data were acquired on a Bruker AV III HD 600 MHz spectrometer using in-house written, standard trosy based T1, T2 and NOE experiments.25,26 All NMR data were processed using NMRPipe27 with linear prediction used to extend the effective acquisition times by up to 2-fold in nitrogen in regular sampled data. The non-uniform sampled data was reconstructed using the IST algorithm implemented in NMRPipe. Spectra were analysed, and relaxation parameters calculated, using the NMRFAM Sparky package.28 Values for the reduced spectral density function were calculated using the software Relax version 4.1.3.29,30
X-ray crystallography
Human IL-17AA protein (amino acids 34–155, N68D, C129S) was concentrated to 8 mg mL−1 and crystals were grown by sitting drop vapour diffusion in 100 mM sodium citrate buffer, pH 5.5, 8% v/v 2-propanol, and 10% w/v PEG10k. Crystals were then soaked with the macrocycle (10 mM in DMSO) for 2 hours at 18 °C. Crystals were then cryoprotected with 25% ethylene glycol and flash-cooled at 100 K. Data was collected on beamline I04-1 at the synchrotron Diamond Light Source, UK. The diffraction data was indexed, integrated, and scaled with FastDP.31–34 The crystal structure was determined by molecular replacement method using Phaser35 and the published human IL-17AA structure (PDB code 4HR9)13 as the search model. The model was refined using the CCP4 suite36 and Phenix37 with alternating cycles of manual model building with Coot.38 Data collection and refinement statistics are listed in the ESI, Table S1.† The coordinates and structure factors have been deposited in the Protein Data Bank (PDB code 8CDG). All structures were analysed, and figures prepared using Pymol.39 In addition, all structures used in this work were compared using the software DSSP40 with the results summarised in the ESI (Fig. S9†).Microfluidic diffusional sizing (MDS)
MDS was performed by Fluid Analytics (https://www.fluidic.com/), detailed description of the assay method and results are provided in the ESI.†Biolayer interferometry binding assays (BLI)
The dissociation constant (KD), Kon and Koff for IL-17AA and IL-17AF binding to IL-17RA were determined by biolayer interferometry on an Octet Qke system (ForteBio). Equivalent experiments were also recorded in the presence of a saturating amount of MC and experiments were carried out at 25 °C and 1000 rpm orbital shaking. DMSO was included in all experiments at the concentration used with a saturating amount of MC (5% v/v).Raw data was corrected by double referencing and analysed using the Octet data analysis software (ForteBio) and exported into Prism (version 9.3.1) for analysis. The KD values were derived from the steady state equilibrium analysis by fitting the steady state maximum response binding levels. Association and dissociation rate constants were calculated based on fitting to the heterogeneous ligand model in the Octet Data Analysis software (version 12.0.2.11).
Equivalent experiments were also acquired to characterise the interaction of IL-17AA and IL-17AF with IL-17RC. For these experiments, the Protein G biosensors were coated with C-terminal Fc tagged IL-17RC isoform 1 (residues 21–465) (R&D Systems).
Results and discussion
IL-17 structure and dynamics
To investigate our hypothesis that the dynamics of IL17 are important for cytokine-receptor binding we chose to combine two techniques that have been used extensively in the study of protein dynamics: 2D-IR22,41 and NMR spectroscopy. Combining these methods allow us to probe dynamics not only across a broad time scale (femtoseconds to milliseconds) but also from individual residues to entire secondary structural elements.We begin with a discussion of the secondary structure of the IL-17 homodimers before progressing to measurements of their dynamics via 2D-IR and NMR spectroscopy. Thereafter we introduce the effect of binding the tool compound MC to assess the impact of ligand binding on IL-17 structural dynamics.
The collar region contains four conserved (two per protomer) intra-chain disulphide bonds which link the internal β-hairpin of each protomer to both the external β-hairpin and the loop which links the two hairpins. These disulphide bonds bestow a “cysteine knot like” fold, although the central disulphide bond, which should be threaded through the eye of the knot, is missing in this instance. A long, poorly conserved coil region links the N-terminal region to the external β-hairpin.
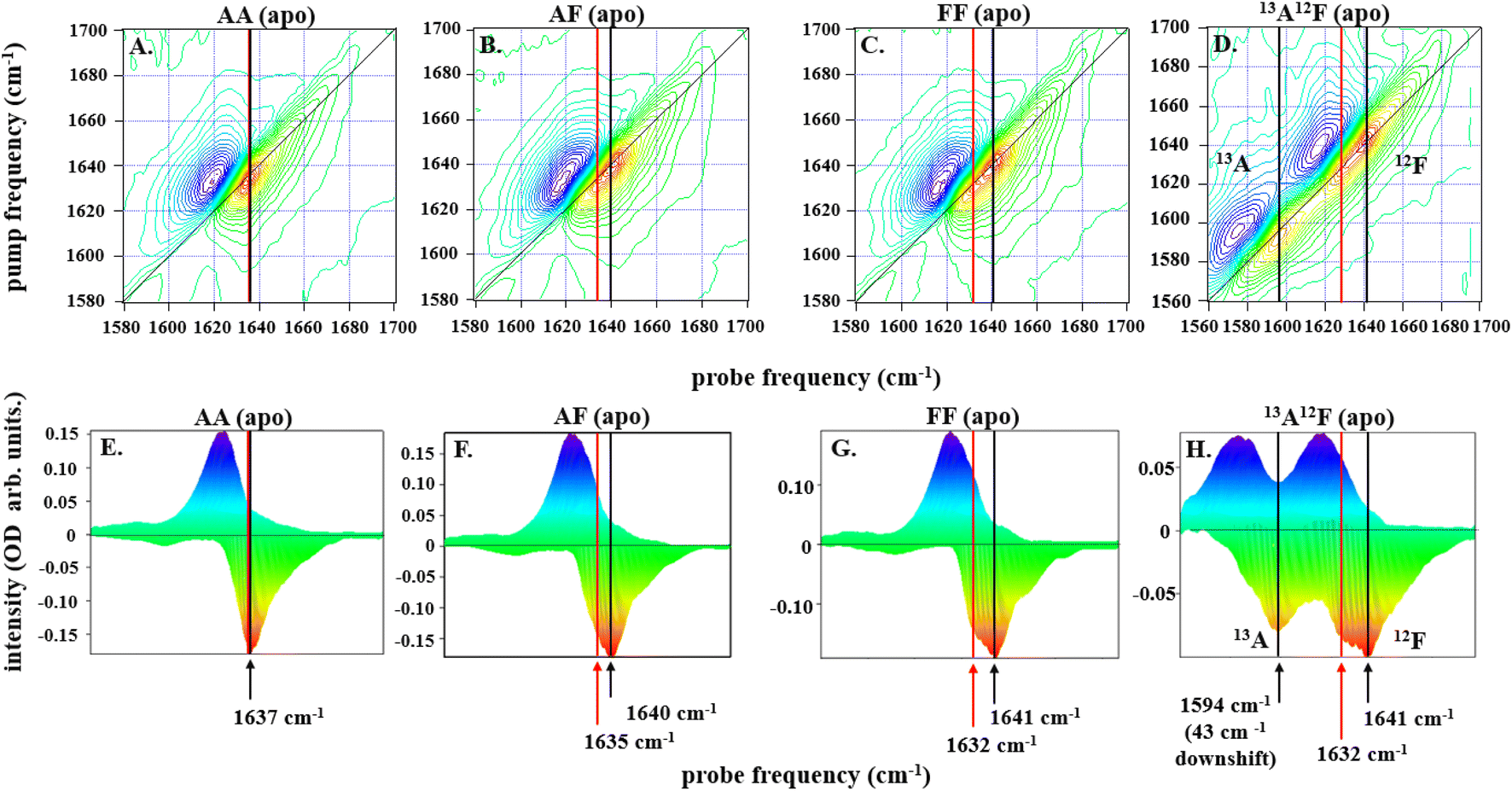 | ||
| Fig. 2 2D-IR spectra for all dimers, IL-17AA in panels A and E, IL-17AF in panels B and F, IL-17FF in panels C and G, and IL-17AF13A12F in panels D and H, as labelled at a pump–probe delay time (Tw) of 250 fs are depicted along the top row of (A–D). The color scale runs from red (negative) to blue (positive). Diagonal projections of the spectra in A–D are shown in E–H. The frequency positions of the main band and shoulder described in the text are marked with arrows (black: main band; red, shoulder). The 13A12F sample contains IL17AF in which the A protomer has been isotopically enriched with 13C to shift the amide I band to lower frequency and remove overlap between the amide I bands of the A and F protomers. | ||
Subtracting the 2D-IR spectrum of IL-17AA from that of IL-17FF reveals the spectral signature of the changes occurring between the two homodimers (Fig. 3B). Specifically, gains in amplitude at 1620 and 1640 cm−1 on the spectrum diagonal reflect the change from a single peak (IL-17AA) to a double-peaked structure (IL-17FF) shown by comparing Fig. 2(E and G). A similar peak pattern is obtained in the IL-17AF-IL-17AA difference 2D-IR spectrum. This difference spectral signature will be referred to again in discussions of MC binding (below).
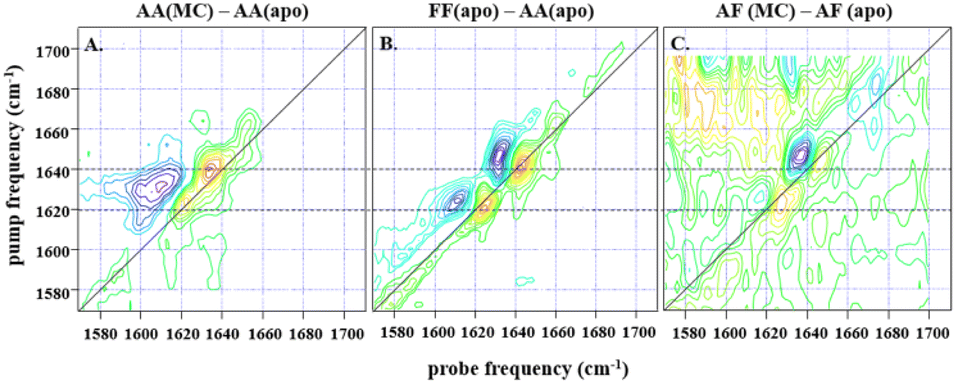 | ||
| Fig. 3 (A) shows the difference spectrum obtained by subtraction of IL-17AA apo-2D-IR data from a 1:2 macrocycle (MC) bound sample at waiting time (Tw) of 250 fs and at parallel polarisation, whilst (B) shows the subsequent difference obtained from subtraction of the same IL-17AA apo-data from that for an IL17-FF apo-sample at the same waiting time. A similar difference is shown in (C), obtained for an AF macrocycle minus AF apo-subtraction, again at a Tw of 250 fs, but with a much lower signal-to-noise level that is indicative of the low binding affinity of the AF isoform. The dashed horizontal lines highlight the similarity in peak positions for both subtractions, indicating that the binding of the macrocycle to both IL-17AA and IL-17AF induces changes that are much more comparable both spectroscopically and structurally to the IL-17FF apo-form. | ||
To shed light on the molecular origins of the shoulder feature, which represents the key difference between the 2D-IR spectra of IL-17 AA and IL-17 FF (cf.Fig. 2E and G), the 2D-IR spectrum of a sample of IL-17AF in which the A protomer contained 13C isotope substitution was obtained (Fig. 2D and H). The spectroscopic impact of this isotopic labelling is to shift the amide I band of the A protomer down in frequency by ∼40 cm−1 enabling it to be separated from that of the 12C-containing F protomer. The diagonal projection of the resulting spectrum (Fig. 2H) clearly shows that the shoulder is present only in the amide I band of the F protomer (red arrow, Fig. 2H), while the A protomer contains a single peak as observed for IL-17AA (Fig. 2A).
The form of the 2D-IR spectrum of the amide I band of IL-17AA is consistent with a structure containing both β-sheet and random coil elements, as indicated by the crystal structure. The main peak at 1636 cm−1 is assigned to the ν⊥ mode of a β-sheet.41 The broad ridge is assigned to a combination of random coil elements, unstructured contributions to the β-sheet and β-turns.41 In the spectra of IL-17FF and IL-17AF the appearance of the shoulder feature (red arrows Fig. 2F–H) is consistent with part of the ν⊥ mode shifting to lower frequency, leading to it manifesting as a shoulder and creating the double-peaked structure that isn't present in IL-17AA (Fig. 2E). Such a down-shift of the ν⊥ mode is observed for more extensive β-sheet structures or ones in which the amide oscillators (CO bonds) are more strongly coupled. The latter would be observed if structural dynamics or conformational fluctuations are reduced.41,44 The data on the partially isotopically labelled IL-17AF sample shows clearly that the differences are specific to the β-sheet structures of the IL-17F protomer.
Overall, the evidence from the 2D-IR data is that the β-sheet structures of the F protomer include a component that differs in terms of its structural dynamics from that of the A protomer, with the results consistent with IL-17AA being more flexible than IL-17FF. Measurements of the vibrational relaxation time of the v = 0–1 (blue) of the amide I bands of IL-17AA, IL-17AF and IL-17FF using IR pump-probe spectroscopy (Fig. S1A–C†) provide further support for this hypothesis. This experiment measures the rate at which the amide I band of the protein dissipates excess vibrational energy following excitation. Vibrational relaxation times of 0.87 ± 0.08, 0.81 ± 0.04 ps and 0.74 ± 0.05 ps for the β-sheet portion of the IL-17AA, AF, and FF amide I bands respectively show that the shift to lower frequency of the ν⊥ mode observed via 2D-IR also correlates with faster vibrational relaxation. It has been shown that, in the absence of significant changes in solvent access to a particular residue, which is not the case here, vibrational relaxation of peptides is dominated by intramolecular vibrational relaxation.45,46 Such a process would be expected to be facilitated by stronger intra-sheet interactions arising from reduced structural dynamics. Leading to the observed shorter vibrational lifetime and consistent with the assignment of the differences in IL-17 AA and FF to structural dynamics.
The line-shape of resonances in 2D-trosy spectra varies between isoforms (as discussed previously21), with the spectra obtained for the ‘A’ homodimer generally showing a greater range of line widths and signal intensities than those of the ‘F’ homodimer. In addition, spectra acquired at 800 MHz were significantly broadened compared to those at 600 MHz, indicating the system is approaching the intermediate exchange regime at this magnetic field. High quality spectra that yielded reliable relaxation parameters for the majority of backbone amides were obtained at 600 MHz. The 15N relaxation rate constants R1, R2 and the 1H–15N steady-state NOE, R2/R1vs. R1R2 plots and reduced spectral density parameters47–50 calculated for the IL-17 isoforms are given in the ESI.†
Analysis of R1 and R2 plots show significant variation throughout the sequence for all protomers, particularly in the first 50 residues and the last 5 C-terminal residues (see ESI, Fig. S3†). The N- terminal region has significantly elevated R1 and reduced R2 values (indicating ps-ns flexibility and/or anisotropic rotation) compared to the majority of residues of the core β-hairpins. These regions also have reduced hetNOE, again indicating internal flexibility (τe). The C-terminal residues 148 to 153 (158 to 163 for the F protomer) have more core like values but with the last few residues being significantly smaller, again suggesting considerable backbone flexibility in these regions. Both the N- and C- termini are effectively tethered to the core of the protein via the disulphide bonds in the cysteine knot and the interchain disulphide bond. These bonds stabilize the backbone motions in the adjacent residues, but the affect is limited, particularly in the N-terminal which shows internal flexibility up to about residue 63 (70 in the F protomer).
Further analysis using R2/R1vs. R1R2 plots51 indicate that all systems investigated show anisotropic rotational diffusion, indicative of deviation from a rotationally averaged spherically globular structure. Also, a significant number of residues are in either chemical exchange (Rex) or experience fast internal motions (τe). Accordingly, we used reduced spectral density mapping47–50 to analyse the complex dynamics observed in these systems. Combining both these qualitative methods enabled a more detailed analysis of the backbone dynamics, where we have chosen to describe the results in terms of the protomer contribution in the AA, Af, aF (where the upper-case letter refers to the 15N labelled protomer) and FF isoforms, and to subsequently show the effect of binding of an inhibitory macrocycle to the hetero-dimer.
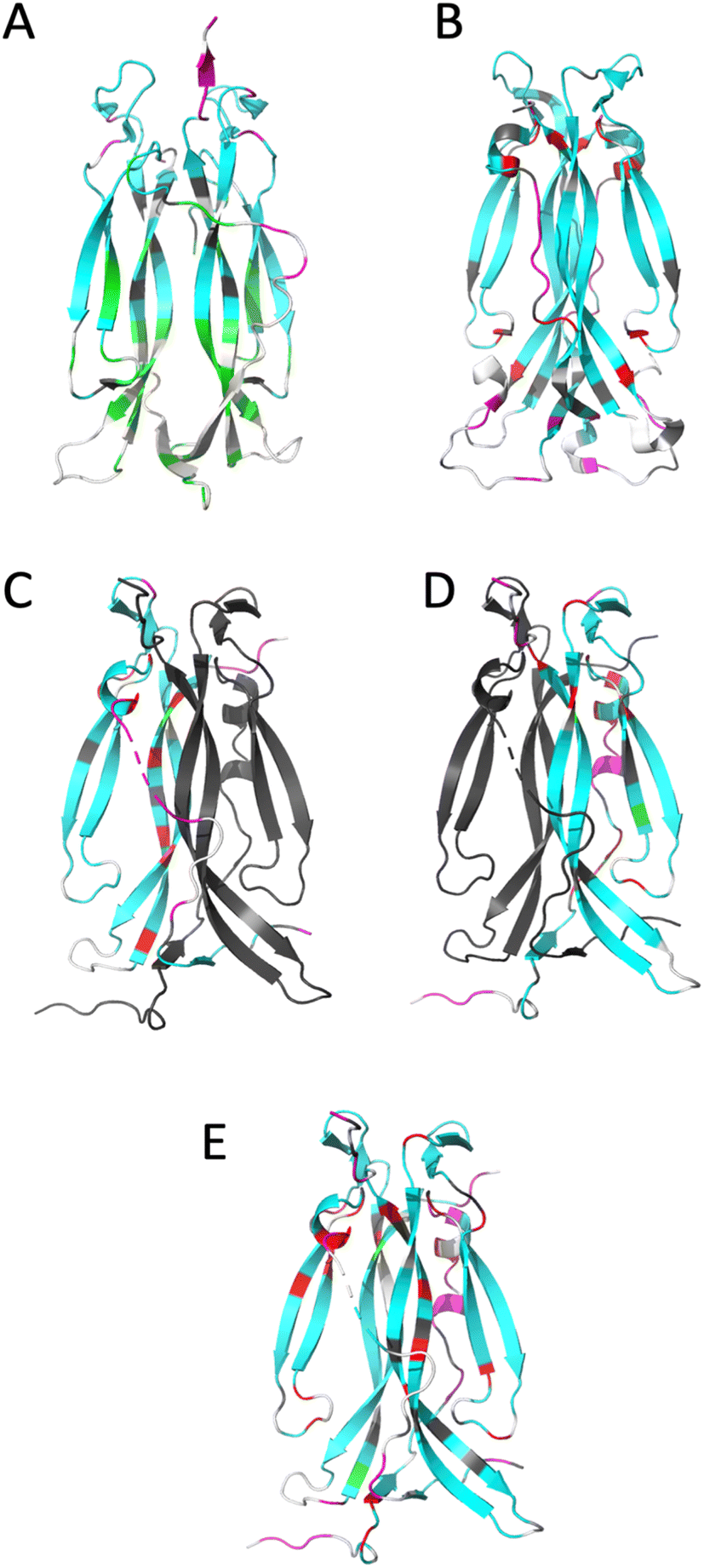 | ||
| Fig. 4 Ribbon representation of IL-17 isoforms. (A) IL-17AA (PDB code 4HSA), (B) IL-17FF (PDB code 1JPY), (C) IL-17Af (PDB code 5N92), (D) IL-17Fa and (E) IL-17AF:MC complex. The backbone is colour coded by motion type. Colours are, white – not assigned, grey – overlapped resonance therefore no reliable data, cyan – relaxation dominated by overall correlation time (τc), magenta – motions faster (τe) than the overall correlation time (τc) dominate relaxation, green – resonance decayed too quickly to determine R2 and therefore are interpreted as undergoing conformational exchange (Rex), red – exchange contribution (Rex) to relaxation of resonance. Due to the absence of a significant portion of the long interface coil region in the crystal structure of free IL-17AA, the more complete structure obtained in complex with IL-17RA was used. | ||
For IL-17AA, a significant number of amides (26 in total) decayed very rapidly during the course of the R2 NMR experiment with no visible signal after the third or fourth time-point (48 ms), hence it was not possible to determine reliable relaxation parameters for these residues. However, a conservative lower limit could be estimated of about 0.5 s−1 of R2 for these residues. Such a rapid decay in the signal intensity could be interpreted as residues that are undergoing significant chemical exchange i.e., existing in two or more conformational states with different magnetic environments.
Conversely, the IL-17FF homodimer has relatively few residues that show a significant right shift in the J (0.87ωH) vs. J (0) plot (i.e., greater than the median average deviation) that would indicate chemical exchange in the μs–ms timescale (summarised in ESI† and see Fig. 4). The exchange observed for residues 74–75, 79–80, 92 and 154 are at the cystine knot and is possibly due to disulphide bond isomerization.
However, most significant is the loss of conformational exchange in the bottom part of the second β-hairpin and the ‘skirt’ region. The few residues (95, 97, 118, 131, 136, 140 and 143) that are in conformational exchange are mostly located in the loop between the two β-hairpins and in strand 4 of the second β-hairpin (the other residues are in the ‘skirt’ region of the molecule). Those at the top of strand 4 are in the ‘collar’ region of the molecular near the cysteine knot and the observed exchange could, as with the homodimers, be a consequence of disulphide bond isomerization.
The dynamics of the IL-17Fa protomer of the heterodimer show a very similar pattern to the IL-17FF homodimer but with the N-terminal residues 60–64 and 74–80 showing conformational exchange and conversely residues 132–139 becoming more stable. The increased conformational exchange may be due to the loss of polar contacts these residues make with residues 143–145 with the second ‘F’ protomer in the homodimer. The stabilization of residues 132–139 may be as a consequence of this region now making polar contacts with N91 in the ‘F’ protomer and K39 of the ‘A’ protomer. In the ‘FF’ homo-dimer polar contacts are still made with N91 but K39 is replaced with a serine residue, with F41 and Q42 forming a short helical turn so re-orienting that segment away from the ‘A’ protomer. This re-orientation of the N-terminal residues changes the sidechain and backbone contacts dramatically between the two protomers.
In general terms, the results from 2D-IR and NMR experiments complement each other very well. The observation that the IL-17AA isoform has a hydrophobic core that is considerably more dynamic is supported by the 2D-IR data, where the β-sheet signal is not clearly separated from the main amide I band leading to a single ‘averaged’ frequency distribution (see Fig. 2). The ‘F’ homodimer is a much less dynamic protein with the relaxation of the majority (70%) of the residues being dominated by molecular tumbling. The heterodimer is more F- like, with the A-protomer being stabilized at the expense of the F-protomer.
Impact of ligand binding on IL-17 structure and dynamics
Co-crystallisation of IL-17AA:MC revealed a binding site located at the dimer interface. As with other reported cytokine–ligand complexes16,52 the MC binds in a central pocket formed at the dimer interface. Although covalently linked by disulphide bridges, the monomers move apart slightly so widening the pocket to accommodate the MC. This movement is not as dramatic as reported for other related ligands16 as, relative to the apo structure (PDB accession code 4HR9), the distance between Cα atoms for L120 increases by only 1.5 Å. Indeed, the Cα RMSD between the apo IL-17AA and our structure is calculated at 0.74 Å, again indicating only moderate structural changes upon ligand binding. However, as with all reported ligand complexes L120 forms two hydrogen bonds with the amide groups of the MC and a fifth hydrogen bond is formed by the amide group of W90 of one protomer with a carboxylic acid moiety of the MC (see ESI, Fig. S10†). The calculated dimer interface energy (calculated using PDBePISA) remains unchanged (ΔG ∼ −23 kcal mol−1) as does the dimer interface area (∼2100 Å2). Therefore, it would appear that this macrocycle causes only moderate structural changes, and although the macrocycle would undoubtedly block receptor binding at one site the other site would still be available for receptor binding.
Comparison of the apo-IL-17AA13,53 crystal structures available with those bound to a receptor13–15,54 show that the IL-17AA does undergo conformational change upon binding. This indicates that, for the IL-17AA:MC complex to bind to a receptor, it too would need to undergo a similar conformational change. However, there are no major changes to the secondary structural elements (as indicated by DSSP analysis, see ESI, Fig. S9†) or the tertiary fold.
A number of previous studies have shown that quantitative receptor binding affinities show some variation however, trends in affinities between all studies are consistent.11–14,16 In order to study the effect of the MC on receptor binding, biolayer interferometry, using tethered proteins and microfluidic diffusional sizing (MDS), which uses fluorescently labelled, untethered protein, were used to provide quantitative data to compare with spectroscopy results.
:1 stoichiometry.
| Microfluidic diffusional sizing | ||
|---|---|---|
| K D (nM) | K D with MC (nM) | |
| IL-17AA:RA | 2.85 (0.49–8.72) | 45.7 (16.52–110.8) |
| IL-17AF:RA | 65.3 (34.98–120.10) | 92.8 (47.04–185.20) |
| IL-17FF:RA | 221 (93.83–533.8) | 668 (379.2–1198) |
| IL-17AA:RC | 2.95 (1.02–6.19) | 11.8 (5.23–24.13) |
| IL-17AF:RC | 1.10 (0.12–2.86) | 2.19 (0.21–6.59) |
| IL-17FF:RC | 5.10 (3.02–8.06) | 7.02 (4.45–10.59) |
|
||
| Biolayer interferometry | ||
| IL-17AA:RA | 5.5 ± 1.7 | 16.5 ± 6.5 |
| IL-17AF:RA | 14.6 ± 3.0 | 23.0 ± 7.5 |
:1 model. Due to a limiting signal-to-noise ratio we were unable to optimise conditions to simplify the binding event so accurate determination of kon and koff was not possible. However, from steady state kinetics (see ESI, Fig. S12†) an estimation of KD was possible, and a qualitative evaluation of the binding isotherms revealed significant differences between IL-17AA and AF both in the absence and presence of MC. The results are summarized in Table 1.
Qualitative assessment of the binding isotherms of the IL-17AA and IL-17AF isoforms to IL-17RA suggest similar kon values, however notably the IL-17AA isoform shows no dissociation during the course of the experiment, whereas IL-17AF has a measurable koff. Previous studies16,17,19 have suggested that small molecule inhibitors function by either causing structural changes that prevent receptor binding or by causing steric clashes. Our experiments, run in the presence of MC, suggests that IL-17AA binds both MC and IL-17RA simultaneously as only significant effects on koff are observed resulting in approximately a 3–4-fold weaker KD.
Binding of MC to IL-17AF also reduces the affinity for IL-17RA, although to a lesser extent (∼2 fold). However, in contrast to IL-17AA, the MC has little effect on the off-rate. This perhaps suggests a different mechanism of AF inhibition by the MC.
The MC has a clear impact on KD for IL-17AA binding to receptor RA and less so for the IL-17AF and IL-17FF. Here, we recall that NMR and 2D-IR both indicated that IL-17AA was the most structurally dynamic of the three isoforms, while those containing F were more rigid in nature. Interestingly, for the IL-17AA isoform, kon is relatively unaffected but koff is significantly increased, indicating that the ternary complex (of IL-17AA:MC:RA) forms but is unstable.
Clearly, this is not the case for receptor C, where affinities are similar for all three isoforms, and with only modest affects observed in the presence of MC. However, this difference may reflect the nature of receptor C's ability to accommodate the binding of a more rigid molecule (e.g., receptor C itself being more dynamic than receptor A), it may also reflect structural differences between the receptors.
As can be seen from published crystal structures of IL-17 isoforms bound to either RA or RC,9,10,12–15 RA binding induces significant structural changes in the cytokine whereas when binding to RC far more modest structural changes occur.
Effects of an inhibitory macrocycle on IL-17 dynamics
Having established the varying dynamic nature of the IL-17 isoforms, and that the impact of the MC upon receptor binding is isoform dependent, it is clear that the possibility exists for MC binding to alter isoform dynamics in a manner that inhibits the interaction of IL-17AA with IL-17RA.16 We now test this hypothesis by considering the results of NMR and 2D-IR spectroscopy of the MC-bound IL-17 isoforms in a similar manner to that reported above for the MC-free IL-17 dimers.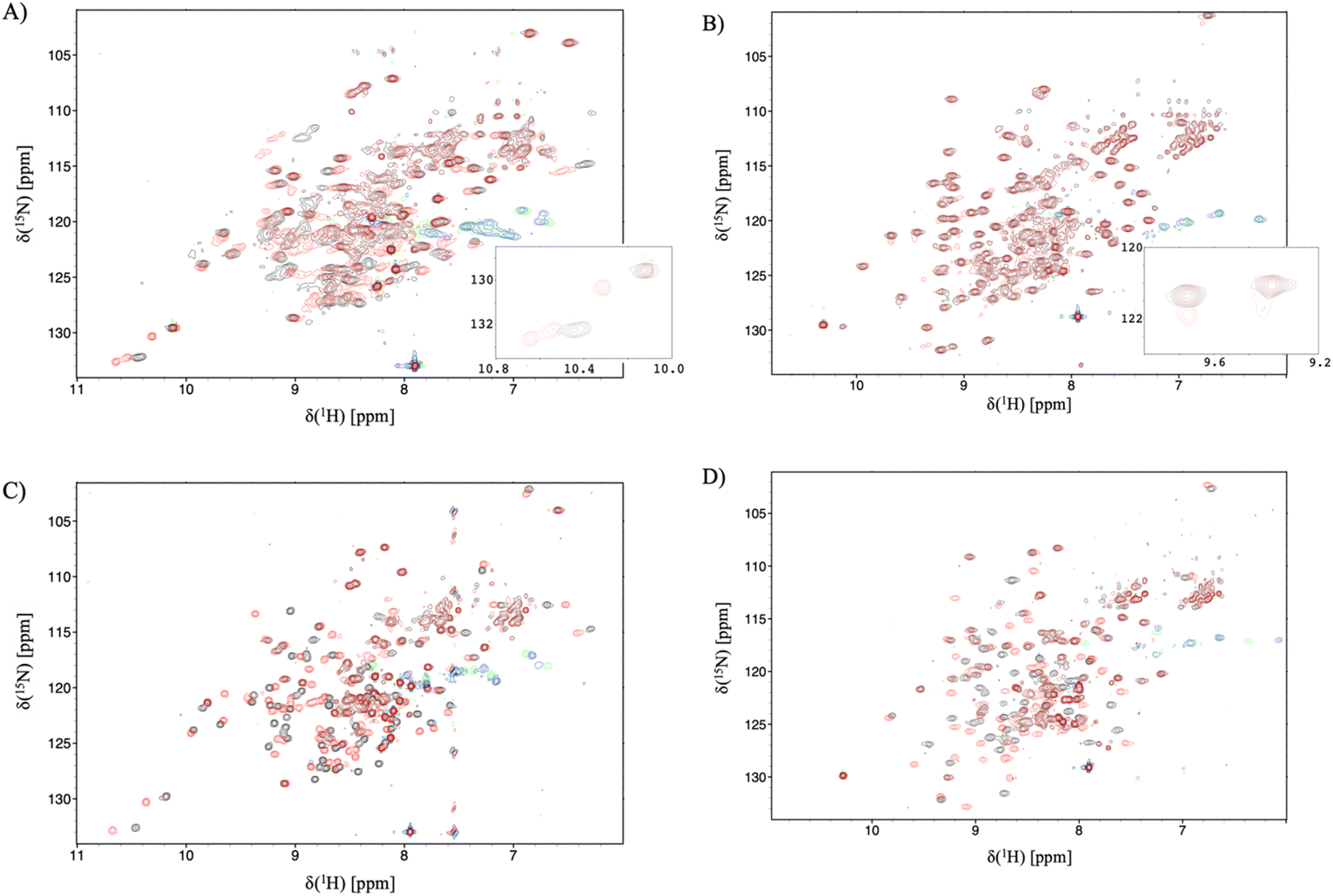 | ||
| Fig. 5 15N-trosy spectra of the IL-17 isoforms in the presence and absence of MC. Black contours are apo protein and red contours are in the presence of MC, green/blue contours are aliased arginine sidechain resonances. (A) IL-17AA (B) IL-17FF (C) 15N labelled A protomer of IL-17AF (D) 15N labelled F protomer of IL-17AF. Insert panel A shows additional signals due to de-symmetrisation of the Il-17AA dimer. and the insert to panel B shows additional signals resulting from incomplete saturation of the IL-17FF homodimer. | ||
For the IL-17FF isoform additional, but weak, signals were observed (Fig. 5B). This was interpreted as incomplete saturation of the binding site (estimated as about 10% based on peak height ratios) due to a combination of very low ligand affinity, limiting solubility of the ligand and a slow kon.
At such a low occupancy, it was not possible to obtain reliable NMR dynamics data. Consequently, only the IL-17AF:MC complex afforded spectra suitable for an NMR dynamics analysis.
The dynamics of the F protomer show a very similar pattern to both the IL-17FF homo- and IL-17Fa hetero-dimer. However, unlike the ‘A’ protomer in the complex residues (E96 and L147) near the MC binding site now appear to go into conformational exchange.
:2 molar ratio of IL-17 to MC (see Fig. 3). Binding of MC to IL-17AA was found to induce clear changes in the 2D-IR spectrum of the homodimer, characterised by gains in amplitude at 1620 and 1640 cm−1 on the spectrum diagonal (Fig. 3A). Indeed, it is striking that the form of the difference spectrum is almost identical to that obtained between spectra of the apo IL-17FF and AA homodimers (Fig. 3B). It is important to stress that, at the concentrations studied, the MC does not contribute to the IR spectrum in this region. The similarity of the IL-17AA:MC – apo IL-17AA and the apo IL-17FF- apo IL-17AA – difference spectra (see Fig. 3A & B) clearly shows that MC binding to IL-17AA induces changes in the 2D-IR spectrum of the IL-17AA molecule that make it spectroscopically more similar to the IL-17FF homodimer. Given the attribution of the spectral differences between IL-17AA and IL-17FF to reduced structural dynamics affecting the portion of the amide I band originating from β-sheet structures in IL-17FF, above, this implies that a reduction in flexibility occurs upon binding MC to IL-17AA, such that its spectrum closely resembles that of the FF isoform.
A similar result is obtained for the AF:MC complex though the signal-to-noise ratio obtained for the IL-17AF:MC – apo IL-17AF difference spectrum is lower than the equivalent one for IL-17AA (Fig. 3). This observation correlates with a smaller overall change in the flexibility upon MC binding, as observed in the NMR dynamics analysis. In the case of the IL-17FF:MC complex, the 2D-IR spectral distribution is essentially the same in the presence and absence of the MC, indicating that no appreciable change in dynamics could be detected, which is consistent with a very low binding affinity for IL-17FF compared to the other isoforms.
:2 excess of the MC ligand compound, the vibrational relaxation time for the IL-17AA:MC complex was significantly shorter than observed for the apo protein, with an 18% decrease to 0.70 ± 0.04 ps. This value in the IL-17AA:MC complex is remarkably close to that observed for IL-17FF in the absence of the MC molecule (0.74 ± 0.05 ps) as shown in Fig. S1.† In marked contrast, macrocyclic addition to IL-17FF resulted in no change in its relaxation dynamics, consistent with the observed low binding affinity for MC.
In contrast, macrocyclic addition to IL-17 FF resulted in no change in the relaxation dynamics, consistent with the observed low binding affinity.
Conclusions
Previous studies on a number of cytokines have highlighted their dynamic nature, which is believed to play an important role in allowing them a range of diverse binding partners.55 Indeed, structural studies of the IL-17 dimers have often failed to fully explain important features of the IL-17 receptor complexes, leading to the suggestion that the crystal structures might not be fully representative of the solution state.12 We have previously reported21 a phenomenological analysis of solution NMR data for IL-17A, AF and F, which established a link between the varying dynamic nature of the IL-17 dimers and their affinity for IL-17RA. The quantitative dynamics data reported in this paper, using a combination of NMR and 2D-IR spectroscopy, sheds considerably more light on the molecular mechanism of IL-17AA:RA binding and that by which a series of small molecule inhibitors modulate such interactions.Based on measurements of structural dynamics spanning a range of time and length scales alongside structural and biophysical studies, we confirm that differences in the structural dynamics of the IL-17 isoforms play an important role in defining their relative affinities for receptor A. The IL-17AA isoform is the most structurally dynamic and has the highest affinity for the receptor, while the more rigid IL-17FF isoform has the lowest receptor affinity. This implies the presence of an induced fit model of receptor ligand binding: IL-17FF being the most rigid of the three isoforms studied would have a higher energy barrier to overcome before it can bind to receptor RA.
Whilst a number of therapeutic antibodies targeting IL17A have been approved, to date only one small molecule antagonist has progressed to clinical trials.56 However, a number of small molecule inhibitors that target IL-17A have been reported.16–19 Of these, perhaps the best studied have been based on the series of macrocycles patented by Ensemble,23 which have been shown to bind in the central binding pocket. Previous crystallographic studies of IL-17:peptide:ligand complexes have suggested that these inhibitors of IL-17 function either by sterically blocking and/or inducing structural changes that preclude receptor binding. We report here the first structure of a IL17AA:MC complex in the absence of any additional binding partners and this structure has confirmed that there are only moderate structural changes to IL-17AA induced upon ligand binding. Comparison of the IL-17AA:MC and published IL-17:receptor structures show that structural changes induced by either ligand or receptor would sterically block binding of the second partner, but this does not preclude the possibility that the IL-17 dimer could undergo structural rearrangements to accommodate the simultaneous binding of both partners. In fact, the BLI data presented here shows that the MC has a dramatic effect on the dissociation of IL-17RA from IL-17AA providing clear evidence for the ability of IL-17AA to form a ternary complex with both MC and RA, hinting that there could be a further previously unidentified inhibitory mechanism.
In order to more fully elucidate the molecular mechanisms by which MC-369 inhibits the interaction of IL-17AA with IL-17RA, and based on the dynamic analysis of the apo-IL-17 dimers presented in this paper, we obtained dynamics data for IL-17:MC complexes. Comparison of the apo-IL-17 and IL-17:MC data provides compelling evidence that these ligands function, at least in part, by modulating the dynamic nature of the IL-17AA dimer. In fact, the MC ligand reduces the dynamic nature of IL-17AA to a level comparable to the IL-17FF isoform. Moreover, the impact of MC binding on receptor affinity is greatest for IL-17AA. We propose therefore, that MC binding dramatically reduces the dynamics of IL-17AA, so inhibiting IL-17AA from adopting the conformation required for a high affinity interaction rather than just causing structural changes or steric clashes. Such a conclusion is consistent with the limited structural changes observed upon MC binding to IL-17AA.
Our findings identify an important new mechanism by which one could target IL17:RA binding in the search for new therapeutics. On a more global scale the studies presented here highlight the importance of studying protein dynamics in solution, using complimentary techniques such as NMR and 2D-IR spectroscopy. Understanding the dynamic nature of a protein not only greatly adds to our understanding of their function, including their interaction with binding partners, but in the case of relatively flexible molecules, including many cytokines, could also open up new avenues in the design of inhibitors, potentially resulting in the development of new unexpected therapeutic options. Indeed, our approach paves the way for the development of allosteric inhibitors that modulate the interaction of proteins which bind via flat, featureless surfaces which to date have proved intractable to small molecule inhibition.
Data availability
The atomic coordinates of the presented structure were deposited in the RCSB PDB with accession code 8CDG. Experimental data and its characterisation are available in the ESI† and upon request from the authors.Author contributions
D. J. S. & L. C. W.: conceptualization, formal analysis, investigation, methodology, validation, visualization, data curation, funding acquisition, writing – review & editing. S. L. S., M-S. E. D. S., G. M. G, M. T., A. W. P: investigation, resources, methodology. C. E. P.: conceptualization, resources, project administration, supervision, funding acquisition. A. J. H.: conceptualization, project administration, supervision, funding acquisition. A. D. G. L.: conceptualization, resources, project administration, supervision, funding acquisition, writing – review & editing. M. D. C.: conceptualization, resources, project administration, supervision, funding acquisition, R. J. T.: conceptualization, resources, project administration, supervision, funding acquisition, writing – original draft, writing – review & editing. N. T. H.: conceptualization, investigation, methodology, resources, validation, writing – original draft, writing – review & editing. F. W. M.: conceptualization, investigation, methodology, resources, formal analysis, validation, visualisation, data curation, software, writing – original draft, writing – review & editing.Conflicts of interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Daniel J. Shaw, Monika-Sarah E. D. Schulze, Christine E. Prosser, Alastair D. G. Lawson, Alistair J. Henry and Richard J. Taylor are or have been employees of UCB and may hold UCB shares and/or stock options.Acknowledgements
The work reported here was supported by a long-term research partnership between UCB Biopharma and Prof. Carr’s group at the University of Leicester, and between UCB Biopharma and Prof. Hunt’s group at the University of York. Structural biology applications used in this project were compiled and configured by SBGrid https://elifesciences.org/articles/01456.Notes and references
- J. G. Krueger, K. A. Wharton, Jr, T. Schlitt, M. Suprun, R. I. Torene, X. Jiang, C. Q. Wang, J. Fuentes-Duculan, N. Hartmann, T. Peters, I. Koroleva, R. Hillenbrand, M. Letzkus, X. Yu, Y. Li, A. Glueck, A. Hasselberg, B. Flannery, M. Suarez-Farinas and W. Hueber, J. Allergy Clin. Immunol., 2019, 144, 750–763 CrossRef CAS PubMed.
- K. Reich, K. A. Papp, R. T. Matheson, J. H. Tu, R. Bissonnette, M. Bourcier, D. Gratton, R. A. Kunynetz, Y. Poulin, L. A. Rosoph, G. Stingl, W. M. Bauer, J. M. Salter, T. M. Falk, N. A. Blodorn-Schlicht, W. Hueber, U. Sommer, M. M. Schumacher, T. Peters, E. Kriehuber, D. M. Lee, G. A. Wieczorek, F. Kolbinger and C. C. Bleul, Exp. Dermatol., 2015, 24, 529–535 CrossRef CAS PubMed.
- J. G. Krueger, S. Fretzin, M. Suarez-Farinas, P. A. Haslett, K. M. Phipps, G. S. Cameron, J. McColm, A. Katcherian, I. Cueto, T. White, S. Banerjee and R. W. Hoffman, J. Allergy Clin. Immunol., 2012, 130, 145–154 CrossRef CAS PubMed.
- L. Liu, J. Lu, B. W. Allan, Y. Tang, J. Tetreault, C. K. Chow, B. Barmettler, J. Nelson, H. Bina, L. Huang, V. J. Wroblewski and K. Kikly, J. Inflammation Res., 2016, 9, 39–50 CrossRef CAS PubMed.
- K. Reich, R. B. Warren, M. Lebwohl, M. Gooderham, B. Strober, R. G. Langley, C. Paul, D. De Cuyper, V. Vanvoorden, C. Madden, C. Cioffi, L. Peterson and A. Blauvelt, N. Engl. J. Med., 2021, 385, 142–152 CrossRef CAS PubMed.
- R. B. Warren, A. Blauvelt, J. Bagel, K. A. Papp, P. Yamauchi, A. Armstrong, R. G. Langley, V. Vanvoorden, D. De Cuyper, C. Cioffi, L. Peterson, N. Cross and K. Reich, N. Engl. J. Med., 2021, 385, 130–141 CrossRef CAS PubMed.
- S. L. Gaffen, Nat. Rev. Immunol., 2009, 9, 556–567 CrossRef CAS PubMed.
- L. Monin and S. L. Gaffen, Cold Spring Harbor Perspect. Biol., 2018, 10, a028522 CrossRef PubMed.
- A. Goepfert, S. Lehmann, J. Blank, F. Kolbinger and J. M. Rondeau, Immunity, 2020, 52, 499–512 CrossRef CAS PubMed.
- S. C. Wilson, N. A. Caveney, M. Yen, C. Pollmann, X. Xiang, K. M. Jude, M. Hafer, N. Tsutsumi, J. Piehler and K. C. Garcia, Nature, 2022, 609, 622–629 CrossRef CAS PubMed.
- L. K. Ely, S. Fischer and K. C. Garcia, Nat. Immunol., 2009, 10, 1245–1251 CrossRef CAS PubMed.
- A. Goepfert, S. Lehmann, E. Wirth and J. M. Rondeau, Sci. Rep., 2017, 7, 8906 CrossRef PubMed.
- S. Liu, X. Song, B. A. Chrunyk, S. Shanker, L. R. Hoth, E. S. Marr and M. C. Griffor, Nat. Commun., 2013, 4, 1888 CrossRef PubMed.
- S. Liu, J. Desharnais, P. V. Sahasrabudhe, P. Jin, W. Li, B. D. Oates, S. Shanker, M. E. Banker, B. A. Chrunyk, X. Song, X. Feng, M. Griffor, J. Jimenez, G. Chen, D. Tumelty, A. Bhat, C. W. Bradshaw, G. Woodnutt, R. W. Lappe, A. Thorarensen, X. Qiu, J. M. Withka and L. D. Wood, Sci. Rep., 2016, 6, 26071 CrossRef CAS PubMed.
- A. Goepfert, C. Barske, S. Lehmann, E. Wirth, J. Willemsen, J. E. Gudjonsson, N. L. Ward, M. K. Sarkar, R. Hemmig, F. Kolbinger and J. M. Rondeau, Cell Rep., 2022, 41, 111489 CrossRef CAS PubMed.
- S. Liu, L. A. Dakin, L. Xing, J. M. Withka, P. V. Sahasrabudhe, W. Li, M. E. Banker, P. Balbo, S. Shanker, B. A. Chrunyk, Z. Guo, J. M. Chen, J. A. Young, G. Bai, J. T. Starr, S. W. Wright, J. Bussenius, S. Tan, A. Gopalsamy, B. A. Lefker, F. Vincent, L. H. Jones, H. Xu, L. R. Hoth, K. F. Geoghegan, X. Qiu, M. E. Bunnage and A. Thorarensen, Sci. Rep., 2016, 6, 30859 CrossRef CAS PubMed.
- A. Espada, H. Broughton, S. Jones, M. J. Chalmers and J. A. Dodge, J. Med. Chem., 2016, 59, 2255–2260 CrossRef CAS PubMed.
- C. Q. F. Wang, Y. T. Akalu, M. Suarez-Farinas, J. Gonzalez, H. Mitsui, M. A. Lowes, S. J. Orlow, P. Manga and J. G. Krueger, J. Invest. Dermatol., 2013, 133, 2741–2752 CrossRef CAS PubMed.
- S. Kostrun, A. Fajdetic, D. Pesic, K. Brajsa, V. Bencetic Mihaljevic, D. Jelic, A. Petrinic Grba, I. Elenkov, R. Rupcic, S. Kapic, I. Ozimec Landek, K. Butkovic, A. Grgicevic, D. Ziher, A. Cikos, J. Padovan, G. Saxty, K. Dack, H. Bladh, T. Skak-Nielsen, S. Feldbaek Nielsen, M. Lambert and M. Stahlhut, J. Med. Chem., 2021, 64, 8354–8383 CrossRef CAS PubMed.
- E. R. Goedken, M. A. Argiriadi, J. D. Dietrich, A. M. Petros, N. Krishnan, S. C. Panchal, W. Qiu, H. Wu, H. Zhu, A. M. Adams, P. M. Bodelle, L. Goguen, P. L. Richardson, P. F. Slivka, M. Srikumaran, A. K. Upadhyay, B. Wu, R. A. Judge, A. Vasudevan, S. M. Gopalakrishnan, P. B. Cox, V. S. Stoll and C. Sun, Sci. Rep., 2022, 12, 14561 CrossRef CAS PubMed.
- L. C. Waters, V. Veverka, S. L. Strong, F. W. Muskett, N. Dedi, A. D. G. Lawson, C. E. Prosser, R. J. Taylor, A. J. Henry and M. D. Carr, Cytokine, 2021, 142, 155476 CrossRef CAS PubMed.
- N. T. Hunt, Chem. Soc. Rev., 2009, 38, 1837–1848 RSC.
- WO2013116682A1, 2013.
- L. Minnes, D. J. Shaw, B. P. Cossins, P. M. Donaldson, G. M. Greetham, M. Towrie, A. W. Parker, M. J. Baker, A. J. Henry, R. J. Taylor and N. T. Hunt, Anal. Chem., 2017, 89, 10898–10906 CrossRef CAS PubMed.
- G. Zhu, Y. Xia, L. K. Nicholson and K. H. Sze, J. Magn. Reson., 2000, 143, 423–426 CrossRef CAS PubMed.
- N. A. Lakomek, J. F. Ying and A. Bax, J. Biomol. NMR, 2012, 53, 209–221 CrossRef CAS PubMed.
- F. Delaglio, S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer and A. Bax, J. Biomol. NMR, 1995, 6, 277–293 CrossRef CAS PubMed.
- W. Lee, M. Tonelli and J. L. Markley, Bioinformatics, 2015, 31, 1325–1327 CrossRef PubMed.
- E. J. d'Auvergne and P. R. Gooley, J. Biomol. NMR, 2008, 40, 107–119 CrossRef PubMed.
- E. J. d'Auvergne and P. R. Gooley, J. Biomol. NMR, 2008, 40, 121–133 CrossRef PubMed.
- G. Winter and K. E. McAuley, Methods, 2011, 55, 81–93 CrossRef CAS PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 133–144 CrossRef CAS PubMed.
- R. W. Grosse-Kunstleve, N. K. Sauter, N. W. Moriarty and P. D. Adams, J. Appl. Crystallogr., 2002, 35, 126–136 CrossRef CAS.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Struct. Biol., 2011, 67, 235–242 CrossRef CAS PubMed.
- A. J. Mccoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- N. Collaborative Computational Project, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1994, 50, 760–763 CrossRef PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkoczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L. W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213–221 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC, https://pymol.org/2/ Search PubMed.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed.
- P. Hamm and M. Zanni, Concepts and Methods of 2D Infrared Spectroscopy, Cambridge University Press, Cambridge, 2011 Search PubMed.
- S. G. Hymowitz, E. H. Filvaroff, J. P. Yin, J. Lee, L. Cai, P. Risser, M. Maruoka, W. Mao, J. Foster, R. F. Kelley, G. Pan, A. L. Gurney, A. M. de Vos and M. A. Starovasnik, EMBO J., 2001, 20, 5332–5341 CrossRef CAS PubMed.
- S. Gerhardt, W. M. Abbott, D. Hargreaves, R. A. Pauptit, R. A. Davies, M. R. Needham, C. Langham, W. Barker, A. Aziz, M. J. Snow, S. Dawson, F. Welsh, T. Wilkinson, T. Vaugan, G. Beste, S. Bishop, B. Popovic, G. Rees, M. Sleeman, S. J. Tuske, S. J. Coales, Y. Hamuro and C. Russell, J. Mol. Biol., 2009, 394, 905–921 CrossRef CAS PubMed.
- D. J. Shaw, R. E. Hill, N. Simpson, F. S. Husseini, K. Robb, G. M. Greetham, M. Towrie, A. W. Parker, D. Robinson, J. D. Hirst, P. A. Hoskisson and N. T. Hunt, Chem. Sci., 2017, 8, 8384–8399 RSC.
- P. Hamm, M. H. Lim and R. M. Hochstrasser, J. Phys. Chem. B, 1998, 102, 6123–6138 CrossRef CAS.
- C. T. Middleton, L. E. Buchanan, E. B. Dunkelberger and M. T. Zanni, J. Phys. Chem. Lett., 2011, 2, 2357–2361 CrossRef CAS PubMed.
- N. A. Farrow, O. W. Zhang, A. Szabo, D. A. Torchia and L. E. Kay, J. Biomol. NMR, 1995, 6, 153–162 CrossRef CAS PubMed.
- J. W. Peng and G. Wagner, Biochemistry, 1995, 34, 16733–16752 CrossRef CAS PubMed.
- R. Ishima and K. Nagayama, Biochemistry, 1995, 34, 3162–3171 CrossRef CAS PubMed.
- R. Ishima, K. Yamasaki and K. Nagayama, J. Biomol. NMR, 1995, 6, 423–426 CAS.
- J. M. Kneller, M. Lu and C. Bracken, J. Am. Chem. Soc., 2002, 124, 1852–1853 CrossRef CAS PubMed.
- M. D. Andrews, K. N. Dack, M. J. de Groot, M. Lambert, C. J. Sennbro, M. Larsen and M. Stahlhut, J. Med. Chem., 2022, 65, 8828–8842 CrossRef CAS PubMed.
- J. P. Ting, F. Tung, S. Antonysamy, S. Wasserman, S. B. Jones, F. F. Zhang, A. Espada, H. Broughton, M. J. Chalmers, M. E. Woodman, H. A. Bina, J. A. Dodge, J. Benach, A. Zhang, C. Groshong, D. Manglicmot, M. Russell and S. Afshar, PLoS One, 2018, 13, e0190850 CrossRef PubMed.
- S. C. Wilson, N. A. Caveney, M. Yen, C. Pollmann, X. Xiang, K. M. Jude, M. Hafer, N. Tsutsumi, J. Piehler and K. C. Garcia, Nature, 2022, 609, 622–629 CrossRef CAS PubMed.
- J. Y. Cui and G. P. Lisi, Front. Mol. Biosci., 2021, 8, 773252 CrossRef CAS PubMed.
- DICE Therapeutics Announces Positive Topline Data from Phase 1 Clinical Trial of Lead Oral IL-17 Antagonist, DC-806, for Psoriasis, https://www.globenewswire.com/news-release/2022/10/11/2531642/0/en/DICE-Therapeutics-Announces-Positive-Topline-Data-from-Phase-1-Clinical-Trial-of-Lead-Oral-IL-17-Antagonist-DC-806-for-Psoriasis.html) Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00728f |
| This journal is © The Royal Society of Chemistry 2023 |