
Application of MOF-derived materials as electrocatalysts for CO2 conversion
Jiahe
Li
,
Haiqiang
Luo
,
Bo
Li
*,
Jian-Gong
Ma
 and
Peng
Cheng
*
and
Peng
Cheng
*
Department of Chemistry, Key Laboratory of Advanced Energy Material Chemistry, Renewable Energy Conversion and Storage Center, Frontiers, Science Center for New Organic Matter, Haihe Laboratory of Sustainable Chemical Transformations (Tianjin), College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: libo2015@nankai.edu.cn; pcheng@nankai.edu.cn
First published on 31st August 2023
Abstract
Electrochemical reduction of CO2 into fuel/high-value chemicals using electricity generated from renewable energy is one of the most promising ways to achieve carbon neutrality. Recently, metal–organic frameworks (MOFs)-derived materials, originating from the MOF architecture, show competitive performance as electrocatalysts in CO2 reduction. This review systematically summarizes several synthesis strategies to fabricate diverse and functional MOF-derived materials including pyrolysis, precursor regulation, post-modification and other synthesis. Additionally, the application of MOF-derived materials in the ECO2RR is classified in detail according to the reduction products. Eventually, some challenges and prospects of the synthesis of MOF-derived electrocatalysts and their application in ECO2RR are presented.
 Jiahe Li | Jiahe Li received her Bachelor's degree in Chemistry from Northeast Normal University (2018). She is currently pursuing her Master's degree under the supervision of Prof. Peng Cheng and Prof. Jian-Gong Ma in Nankai University. Her research focuses on the synthesis of metal–organic frameworks/composite and their application in catalytic CO2 conversion. |
 Haiqiang Luo | Haiqiang Luo received his BE degree in applied chemistry (2018) from Chongqing University. He is currently a PhD student at the College of Chemistry under the supervision of Prof. Peng Cheng and Prof. Jian-Gong Ma, Nankai University. His main research interest focuses on the synthesis of metal–organic frameworks/composite and their applications in electrochemistry CO2 reduction. |
 Bo Li | Bo Li received his BE degree in chemical engineering and technology (2011) and ME (2015) degree in chemical engineering from Lanzhou University. After receiving his PhD degree (2018) under the supervision of Prof. Peng Cheng and Prof. Jian-Gong Ma, he is currently a postdoctoral research fellow at the College of Chemistry, Nankai University. His main research interest focuses on the synthesis of metal–organic frameworks/composite and their applications in catalysis. |
 Jian-Gong Ma | Jian-Gong Ma received both Bachelor's and Master's degree in chemistry under the supervision of Prof. Peng Cheng at Nankai University in 2003 and 2006, respectively. After receiving PhD degree in 2011 under the supervision of Prof. Matthiass Driess, Technische Universität Berlin, he went back to China and joined Nankai University. Currently, Jian-Gong Ma is a professor and young academic leader at the Department of Chemistry, Nankai University. His scientific focus is the synthesis and application of metal–organic frameworks (MOFs) composites, especially in the catalytic conversion of CO2 and electrochemical sensing applications. |
 Peng Cheng | Peng Cheng is currently the Cheung Kong Professor at College of Chemistry, Nankai University, China. He received his BSc, MSc, and PhD from Nankai University in 1985, 1991, and 1994, respectively. Subsequently, he started his academic career at the same university and was appointed a full professor in 1996. From 1997 to 1999, he worked at Laboratoire de Chimie de Coordination du CNRS as a visiting professor and at Texas A&M University as a postdoctoral research fellow. His current research works focus on functional coordination polymers and molecule-based materials. |
1. Introduction
CO2 is one of the greenhouse gases from fossil fuel combustion, which causes global warming and destroys the ecosystem, even endangering human health. Therefore, the utilization of CO2 is a hotspot that people are currently concerned about. However, CO2 conversion requires high energy due to the thermodynamic and kinetic stability of CO2; thus, the development of efficient catalysts is a promising strategy to harness CO2 as a resource.1–3 Electrocatalytic CO2 reduction is a promising reduction strategy that uses renewable electricity to convert CO2 into high value-added products.4–9 At the same time, electrocatalysis has attracted extensive attention from researchers due to its unique advantages among many transformation pathways.Metal–organic frameworks (MOFs) are a type of porous materials composed of metal ions or clusters as nodes and multidentate organic compounds as linkers. Due to their special structure and composition, MOFs and their composite have advantages including highly crystallinity, high porosity and specific surface area, mechanically tunable pore surface, designable framework, and structure diversity, which have been widely used in luminescence, host–guest recognition, adsorption/separation, magnetic materials, energy conversion/storage, catalysis, and other fields.10–15 Among them, the application of MOFs as catalysts has attracted increasing attention. The high specific surface area of MOFs exposes more catalytic active sites, while the ultra-high pore and framework configuration promote the transport efficiency of reactants to the active sites, which is an advantage that many traditional catalysts do not have.16–18 Benefiting from these unique physical and chemical properties, MOFs are used as electrocatalysts or catalyst supports for the electroreduction of CO2. For example, in 2012, a copper rubeanate metal organic framework was first reported for use as an electrocatalyst in CO2 reduction, which showed a more positive onset potential than metal Cu in CO2 saturated solution, and produced almost pure HCOOH (faradaic efficiency higher than 98%).19
However, the poor electronic conductivity and structural stability of MOFs hinder their industrial application in CO2 electrocatalysis to some extent. To solve this problem, MOFs have been used as precursors or templates for the preparation of MOF-derived electrocatalysts. In 2008, Xu et al. obtained nanoporous carbon materials firstly by calcining MOF-5. Compared with pristine MOF, the new carbon materials has higher specific surface area, large pore volume, and good capacitance characteristics, which are used as electrode materials for electrochemical double-layer capacitors.20 MOF-derived materials maintain the advantages of MOFs including high surface area and porosity, meanwhile modifying the chemical and thermal stability of original MOFs. In addition, more diverse and functional materials are obtained by changing the conditions of pyrolysis or post-modification, which is helpful to expand the diversity and application of MOF materials.21–23 Some representative MOFs such as ZIF-8, ZIF-67, HKUST-1, MOF-5, Bi-BTB (CAU-7), MIL-101, and some composite materials24–26 are listed in Fig. 1, which are often used to synthesize MOF-derived materials. In this review, the main synthesis strategies for MOF-derived materials as well as their application in CO2RR are systematically summarized. We divide the synthesis methods of MOF-derived materials into four parts, including direct pyrolysis of pristine MOFs, pyrolysis of MOF composite, post-modification of MOF-derived materials, and other synthesis methods. Representative examples are given to illustrate the purpose and advantages of each synthesis method. In addition, the application of MOF-derived materials as catalysts for the electrochemical reduction of CO2 according to the classification of reduction products are discussed and summarized, such as C1, C2, and C2+ products. It is hoped that this review will provide a valuable reference for researchers concerned in this field and accelerate the innovative and industrial application of MOF-derived materials in CO2RR.
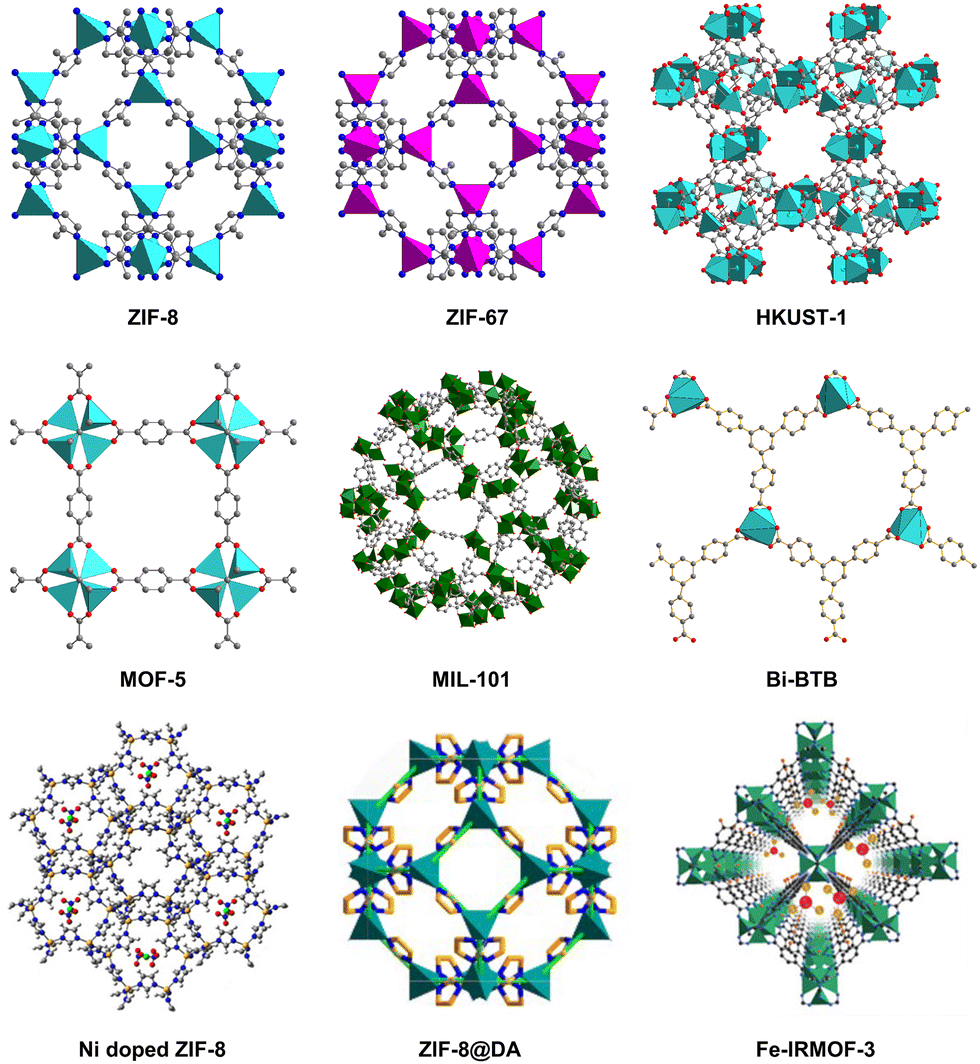 | ||
| Fig. 1 Some representative MOFs and MOF-related materials are used to synthesize MOF-derived materials. | ||
2. Synthesis strategies for MOF-derived materials
2.1 Direct pyrolysis of pristine MOFs
In general, MOFs are directly calcined to obtain functional MOF-derived materials without templates and complicated synthesis steps. Due to its simplicity and rapidity, direct pyrolysis method is widely used for the synthesis of various MOF-derived materials, such as porous carbon materials, mono/multi-metallic compounds, single-site catalysts, and their composites.27–29MOFs have diverse framework structures, high surface area, controllable pore structure, as well as morphology. MOF-derived carbon materials always retain the original structure and morphology of pristine MOFs after calcination, which can broaden the structure and properties of carbon materials. At the same time, the direct calcination of MOFs overcomes the disadvantage that porous carbon materials usually require a large number of complex synthesis steps.30–32 Therefore, MOFs are often used as ideal precursors for the preparation of porous carbon materials. The typical synthesis method is carbonization under proper atmosphere (mainly N2 and Ar) at a specific temperature, followed by simple acid treatment or evaporation of metal atoms at high temperature to obtain the MOF-derived materials. It is well known that Zn has a low boiling point (907 °C); when Zn-based MOF is calcined at high temperature, ZnO is completely removed by the reduction of organic ligands to metal during carbonization. Thus, MOF-5,20 ZIF-8,33 ZIF-68, ZIF-69,34 IRMOF-3, IRMOF-8,35 and other Zn-based MOFs are used as precursors for the synthesis of porous carbon materials. On the other hand, MOFs composed of other metal ions (non-Zn transition metal elements such as Co, Cr, and Fe and some main group metal elements such as Al) and organic ligands are also used as precursors to produce metal-free porous carbon materials after carbonization and acid treatment. For example, Yusuke Yamauchi et al. successfully obtained nanoporous carbon materials with high surface area and large pore volume by calcining and acid treatment using typical Al-based MOF (Al-PCP).36 By the treatment of HF, Al2O3 generated at high temperature was removed. In particular, the material obtained by calcination at 800 °C has a surface area of up to 5500 m2 g−1 and an average pore volume of 4.3 cm3 g−1, which can be applied to catalysis and adsorption.
In general, MOFs contain metal nodes and organic ligands with specific functional groups (–NH2, –SO3H, –CN, etc.); therefore, heteroatom-doped carbon materials are obtained by controlling the temperature and reaction time during pyrolysis. In particular, ZIF is a class of zeolite-like MOF material formed by the coordination of Zn2+ or Co2+ metal ions and organic imidazole ligands, in which N-containing imidazole ligands are used as the precursor of N-doped carbon materials. For example, ZIF-8 was prepared by 2-methylimidazole containing N as an organic ligand and Zn2+, and ZIF-8 derived N-doped C/ZIF composites was obtained by the pyrolysis of ZIF-8 from 600 °C to 1000 °C for 3 h under Ar atmosphere.37 When the temperature was close to the boiling point of Zn, Zn was evaporated and the content was reduced. Pure N-doped porous carbon materials were obtained after HCl cleaning. The N species were pyrrole N and pyridine N atoms. The use of ZIF-8 as a template and precursor contributed to the uniform distribution of N species throughout the material.
In addition to pyrolysis in an inert atmosphere, MOFs are also used for pyrolysis in oxygen or air, where non-metallic atoms such as C, N, and O in the organic ligands are oxidized to gases, ultimately yielding MOF-derived metal oxides. In contrast to metal oxides prepared by conventional methods, the framework structure of the MOF avoids aggregation/degradation and enhances the catalytic activity of the catalyst.38,39 Salunkhe et al. found that the high temperature pyrolysis of ZIF-67 in an inert atmosphere followed by acid treatment yielded nanoporous carbon materials, while pyrolysis at 500 °C under N2 atmosphere for 30 min followed by thermal treatment at a lower temperature of 350 °C in air atmosphere for 2 h yielded nanoporous Co3O4.40 The morphology of MOF-derived materials is usually affected by the original MOF; therefore, the pyrolysis of Co-MOF with different morphologies can yield Co3O4 with different morphologies. The product obtained from the pyrolysis of ZIF-67 at lower temperatures is a spherical dodecahedron (Fig. 2a), while the Co3O4 sample prepared using Co3(NDC)3(DMF)4 as a template and pyrolyzing at 600 °C for 1 h in air formed an agglomerated nanostructure (Fig. 2b), in which large particles with an average size of about 250 nm are composed of small particles with a size of about 25 nm in a densely stacked way.41 Li et al. pyrolyzed MOF-71 at a relatively low temperature to obtain mesoporous nanostructured Co3O442 (Fig. 2c), which had a high specific surface area and small size, exhibited significantly improved electrochemical properties, and also had excellent cycling stability. Interestingly, porous shell cage Co3O4 could be obtained by the pyrolysis of Prussian blue analogue (PBA) Co3[Co(CN)6]2 at 400 °C (Fig. 2d) due to the generation and release of internal CO2 and NxOy.43 The flexible and variable synthesis conditions endow MOF-derived metal oxides with more diverse morphology and crystal structure, which also regulates the mass transfer in the catalytic process and the chemical environment around the active site, thus influencing the selectivity and activity of the catalysts.44,45
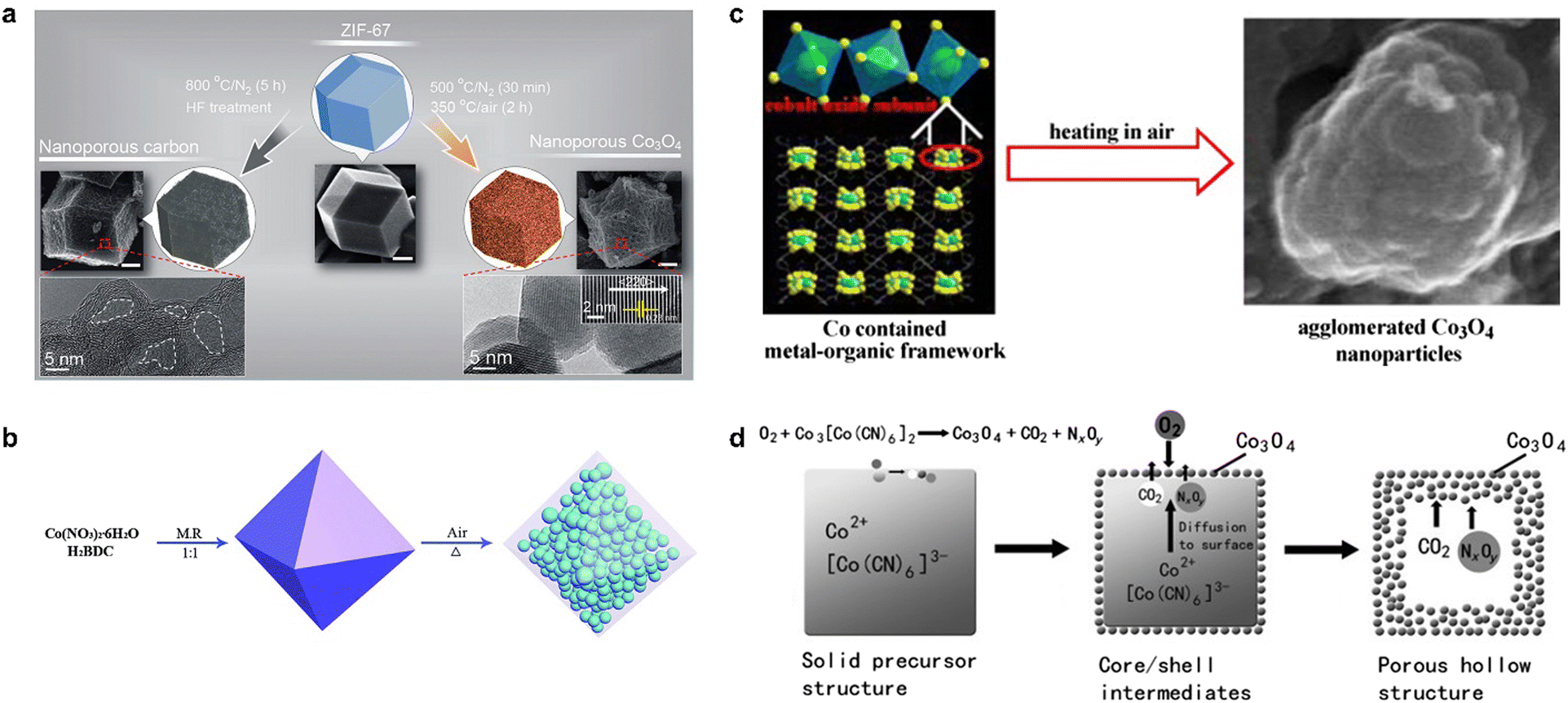 | ||
| Fig. 2 The synthetic routes of Co3O4 synthesized by (a) ZIF-67. Reproduced from ref. 40 with permission from American Chemical Society, copyright 2015. (b) Co3(NDC)3(DMF)4. Reproduced from ref. 41 with permission from Elsevier, copyright 2010. (c) MOF-71. Reproduced from ref. 42 with permission from Royal Society of Chemistry, copyright 2015. (d) (PBA) Co3[Co(CN)6]2 with different morphologies. Reproduced from ref. 43 with permission from John Wiley and Sons, copyright 2012. | ||
It was widely known that Cu0 and Cu+ were generally considered as the active sites for the formation of CH4 and CO, respectively, and obtained higher content of C2 or C2+ products in a certain proportion. Also, the change of oxidation state of other elements effected the reaction mechanism and thus changed the product distribution.46–48 For example, the oxidation number of the obtained metal oxide also could be adjusted by controlling the reaction conditions during the pyrolysis of MOFs. For example, MIL-88-Fe was used as a template to prepare spindle-type mesoporous α-Fe2O3 and magnetite Fe3O4.49 If there is organic residue on the surface of α-Fe2O3, hematite formed after two steps of calcination, including calcination in air for 50 min and calcination in N2 at a higher temperature in turn,50 in which, the organic residue obtained by the first incomplete calcination was used as a reducing agent for the reduction of Fe3+ to Fe2+ in the second step of calcination under an inert atmosphere.
Compared with single metal oxide catalysts, multi-metal oxide catalysts exhibit superior catalytic activity due to the synergistic effect of multiple components. Multi-metallic oxides and their composites are easily prepared by the direct pyrolysis of polymetallic MOFs, in which the metal nodes can be converted into metal oxides and organic ligands into carbon materials during carbonization, such as graphene and CNT (carbon nano tube). This makes the MOF-derived materials more functional.51–53 For example, Wu et al. transformed the bimetallic zeolite imidazole skeletons Zn–Co-ZIFs, which were converted by 2-methylimidazole and Co/Zn ions to spinel ZnxCo3−xO4 hollow dodecahedra.54 Porous carbon-coated CuCo2O4 concave polyhedral composites were obtained by the pyrolysis of ZIF-8 analogue Cu–Co-ZIF at 500 °C for 2 h under Ar atmosphere.55 Jia et al. obtained NiCo2O4/CoO/CNTs composites with excellent performance in the field of biosensing by the pyrolysis of NiCo-MOF.56
Recently, single-site catalysts have shown great application prospects because of their unique catalytic properties and maximum metal utilization efficiency. MOF with well-defined skeletons was prepared by uniformly dispersed metal nodes and heteroatoms such as N, S, and O as anchor sites, which were ideal precursors for single-site catalysts.57–60 Ma et al. prepared a Co single-site catalysts using Co ions in which uniformly distributed CoN4/C catalytic centers were constructed by the pyrolysis of the cobalt imidazole skeleton and acid treatment.61 Zn-containing bimetallic MOF is often used as a precursor for the preparation of M–N–C single-site catalyst. Zn acts as a sacrificial metal to increase the spacing between adjacent metal atoms and reduce agglomeration. For example, Zn ions were used as a “fence” to control the space distance between Co ions during the pyrolysis of Zn/Co bimetallic MOF. Zn ions were reduced to Zn atoms with low boiling point and evaporated, free N sites were generated during pyrolysis, and organic ligands were also reduced to Co ions.62 At this time, the stable Co atoms of N-doped porous carbon were confirmed to be atomically dispersed, without Co clusters and small crystals, and the Co loading was more than 4 wt%. The addition of Zn controlled the spatial separation between Co atoms and stabilized the separated Co single-site after evaporation at high temperature. Compared to ZIF-67 without Zn addition, Co NPs were obtained after pyrolysis.
2.2 Pyrolysis of MOF composite
Although MOF-derived materials show great performance in various applications, it is easy for the framework of MOF to collapse and lose the original crystal topology during direct calcination. In addition, the materials derived from pyrolysis largely depend on the type and structure of the MOF precursors, which limits the performance of the resulting catalysts. Considering the need for different compositions and properties of catalysts in actual catalytic reactions, MOFs are modified by adding other substances prior to pyrolysis. MOF composites combine MOF with one or more different functional materials to obtain new physical and chemical properties and enhanced properties by manipulating the electronic and chemical structure, which are not obtained by MOF alone.63–67In general, MOFs contain a large amount of carbon and do not require the introduction of an additional carbon source, but in most cases, the performance of the sample is constrained. Previous studies have shown that the addition of additional carbon sources as additives can improve the porous structure of carbon materials and sometimes provided higher surface area than the porous carbon generated without an additional carbon source.33,68 Organics such as glucose, furfuryl alcohol (FA), xylitol, glycerol, ethylene glycol, ethylenediamine, melamine, and phenolic resin have been used as carbon precursors.69,70 For example, Al-PCP was used as a carbon source and FA as a secondary carbon source for carbonization by Yusuke Yamauchi's team; it was found that higher the furfuryl alcohol loading, larger the BET surface area of the porous carbon materials, and the Al-PCP/FA composites with the highest furfuryl alcohol content showed obvious mesopores after calcination treatment.68 Hu et al. found that the porous carbon material obtained by the calcination of MOF-5 without the addition of other carbon sources could be identified to include both microporous and small mesoporous (2–3 nm) regions, and after adding carbon tetrachloride and ethylenediamine as additional carbon sources, the obtained porous carbon material had obvious microporous characteristics.69 After the addition of phenolic resin, the pore size range from micropore to macropore was observed.
In order to achieve more diverse MOF derivative materials, heteroatoms are introduced during pyrolysis. Metal heteroatom sources such as Fe, Co, and Cu and non-metal sources such as N, P, S, and Se are introduced externally. One of its most widespread applications is the preparation of single-site and multi-site catalysts using external metal sources; at the same time, the introduction of heteroatom doping is also conducive to the regulation of the electronic structure of the material.71–74 For example, when the synthesized ZIF-8 was dispersed in n-hexane, the aqueous solution of Ni ions was introduced by the double solvent method in which the Ni precursor entered the pores of ZIF-8; when pyrolyzed at 1000 °C, Zn evaporated, leaving N-rich defects as anchor sites occupied by neighboring Ni ions, resulting in atomically dispersed single Ni sites catalyst.75
Bi-MOF and dicyandiamide (DCD) was also used to prepare highly dispersed Bi single-site catalyst.76 Compared with the catalyst without the addition of DCD, Bi clusters were formed on the support (Bi Cs/NC) at the pyrolysis temperature of 1000 °C. Further studies on the electronic structure and coordination environment of Bi single-site catalyst suggested that the local structure of the Bi atom was coordinated by four N atoms. As a nitrogen source, DCD decomposed and released ammonia during pyrolysis, which increased the doping of N species in the carbon network, and the coordination of N with Bi further promoted the atomization of Bi NPs, which played an important role in the synthesis of Bi single-site catalyst.
In order to improve the catalytic activity of single-site catalysts, it is necessary to avoid the aggregation of active sites while increasing the density of active sites. He et al. used surfactant F127 block copolymer as a capping agent to assist bimetallic MOF to prepare Co–Nx with high Co loading,77 wherein Zn2+ and Co2+ in Co–Zn-ZIF coordinated with the hydrophilic groups of the surfactant, MOF nanocrystals were wrapped by the surfactant to slow down the growth rate of the crystal for controlling the size and morphology, and the size of the particle decreased when the molecular weight of the surfactant increased. During the pyrolysis process, the surfactant was firstly carbonized and coated on the surface of the Co–Zn-ZIF polyhedron, and the Co–Zn-ZIF was carbonized with increasing temperature. The cohesive interaction between them led to a unique confinement effect, which effectively inhibited the agglomeration of Co atoms and the collapse of the microporous carbon structure in the MOF during pyrolysis; thus, the final product had a higher density of Co–N4 active sites than that prepared without the surfactant.
The core–shell structure is assembled by chemical bonds or other forces between different materials. It can often integrate various materials together and thus exhibit synergistic effects, with products formed at one catalytic site and passed to another. Sometimes the core and shell interact to produce new functions. At the same time, the shell plays a protective role to improve the stability of the catalyst and prevent the catalyst from aggregating, and the wrapping of the shell can also enrich the reactants inside.1,78 Pyrolysis after the introduction of the second component on MOF is an effective synthesis strategy for the preparation of core–shell structure catalysts; as a classic example, Liu et al. epitaxially grew ZIF-67 on ZIF-8 polyhedral nanoparticles to obtain ZIF-8@ZIF-67 composites with a core–shell structure (Fig. 3a), in which ZIF-8 acted as the crystal seeds of ZIF-67, with similar isoreticular structure and cell parameters. The composite was carbonized and acid-treated to obtain an inner shell of ZIF-8-derived N-doped microporous carbon and an outer shell of ZIF-67-derived Co-modified N-doped mesoporous graphitic carbon, which was denoted as NC@Co-NGC DSNCs.79 In the hollow structure of Co-NGC, the inner pore of the catalyst had high accessibility to the reactants, which ensured their rapid reaction and diffusion rather than aggregation, which lead to the high utilization of the active sites. Meanwhile, graphitic carbon also endowed the material with excellent conductivity.
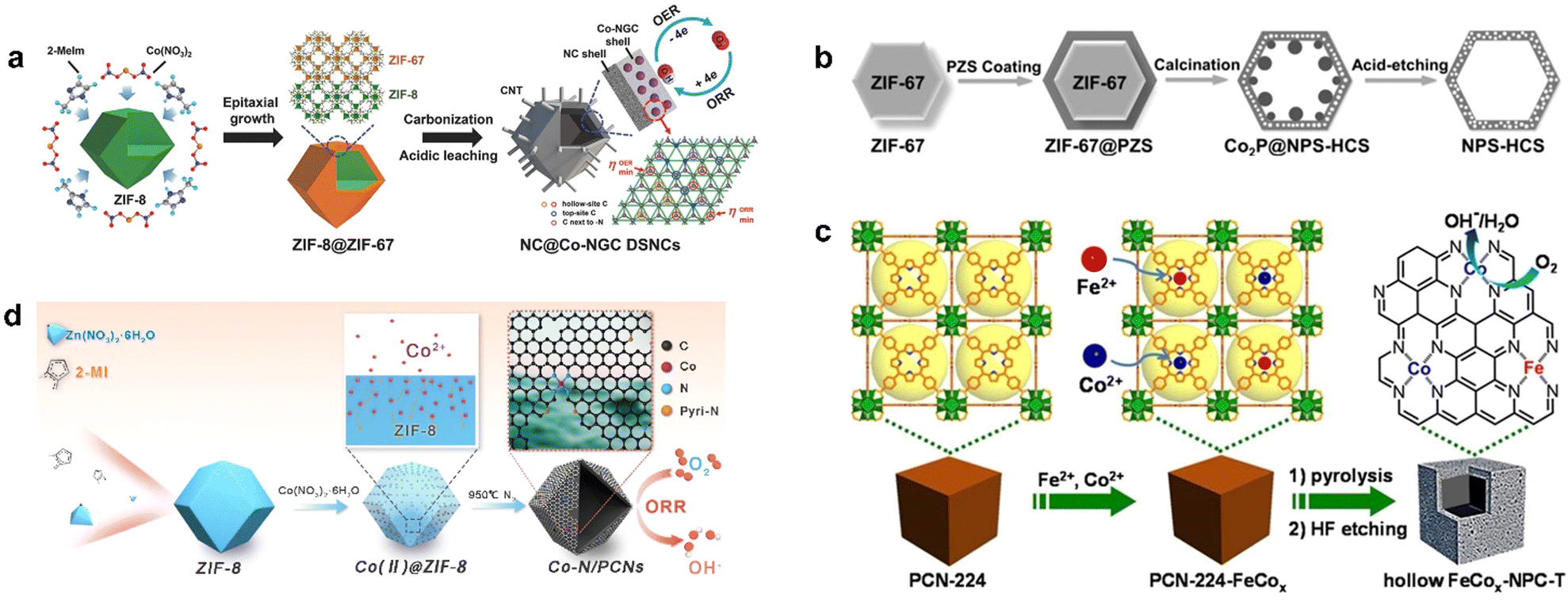 | ||
| Fig. 3 The synthetic routes of (a) NC@Co-NGC DSNCs. Reproduced from ref. 79 with permission from John Wiley and Sons, copyright 2017. (b) NPS-HCS. Reproduced from ref. 80 with permission from John Wiley and Sons, copyright 2016. (c) Hollow FeCox-NPC-T. Reproduced from ref. 81 with permission from John Wiley and Sons, copyright 2017. (d) Co–N/PCNs. Reproduced from ref. 82 with permission from Elsevier, copyright 2020. | ||
Poly(cyclotriphosphazene-Co-4,4′-sulfonyldiphenol) (PZS) was used as carbon, nitrogen, phosphorus, and sulfur source to coat the surface of ZIF-67 to form a core–shell composite ZIF-67@PZS (Fig. 3b).80 Nitrogen, phosphorus, and sulfur atoms were dispersed in the PZS at the atomic scale so that the atomic-level doping of the three heteroatoms was achieved in the carbon shell with ZIF-67 as an additional N doping source, which decomposed during pyrolysis. Finally, metal species were removed by acid etching to synthesize a nitrogen, phosphorus, and sulfur co-doped hollow carbon shell (NPS-HCS) with a high specific surface area of 1020 m2 g−1. The synthesis of this hollow structure was based on the core–shell structure of MOF-guest as a template, which provided a basis for the further preparation of more core–shell materials.
Porous PCN-224 with high surface area was used as a precursor for the synthesis of FeCo-NPC, in which Co2+ and Fe2+ were introduced by metallization to obtain cubic PCN-224-FeCo and prepared FeCo–N-doped hollow porous carbon nanocubes FeCo-NPC at high temperature (Fig. 3c). Due to the large diffusion coefficients of Co, Fe, and the pyrolysis of organic ligands, the collapse of the structure was well suppressed when the thickness of the carbon shell reached a critical thickness, thus forming a hollow structure of porous carbon.81 Zhu et al. introduced Co ions into the precursor of ZIF-8 to form highly hollow and defect-rich porous Co–N-doped C nanomaterials (Co–N/PCNs). When calcined at high temperature, small particles of Co first formed in the pores of ZIF-8 and migrated to the outside of the carbon layer (Fig. 3d).82
2.3 Post-modification of MOF-derived materials
Post-modification treatment is also an effective strategy to improve the structure of the catalysts after the pyrolysis of MOF to improve the catalytic performance. For example, final samples with various pore diameters were obtained by activation. KOH is often used to activate MOF-derived porous carbon to further regulate the channels and surface doping. ZIF-8 powder was subjected to heat treatment at 900 °C for 2 h under N2 atmosphere, followed by acid treatment to obtain the nanoporous carbon NPC; then, NPC and KOH with the mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 were added into deionized water and sonicated for 1 h.83 After heating and drying at 750 °C for a certain time, the resultant porous carbon material had a rougher surface compared with the parent NPC nanomaterial, and the roughness increased with the increase in the KOH activation time. After 1 h of KOH activation, the micropores (1.2 nm) increased and mesopores (2.3 nm) formed in the NPC-1 sample, and some micropores (<0.5 nm) appeared. When the KOH activation time increased to 2 h, the pore size range increased from 1 to 4 nm compared to the NPC-1 sample. When the KOH activation time increased to 5 h, the pore size (<1 nm) decreased. Analysis of the results confirmed that KOH activation firstly generated micropores on the surface of NPC through the etching reaction between KOH and carbon. The size of the newly generated micropores increased with the etching reaction, and then the unconnected micropores of the NPC became larger or merged into new mesopores as the activation time increased. The mechanism of KOH activation for the enlarged pore structure in the NPC material was considered as the etching reaction of carbon in the ZIF-8 derived NPC. The activation of KOH greatly increased the pore size of the material and produced abundant defects, thereby providing more exposed active sites to facilitate mass transport and enhance the catalytic activity (Fig. 4a and b). However, it is worth noting that a very long activation time could also lead to the collapse of the material structure.
:1 were added into deionized water and sonicated for 1 h.83 After heating and drying at 750 °C for a certain time, the resultant porous carbon material had a rougher surface compared with the parent NPC nanomaterial, and the roughness increased with the increase in the KOH activation time. After 1 h of KOH activation, the micropores (1.2 nm) increased and mesopores (2.3 nm) formed in the NPC-1 sample, and some micropores (<0.5 nm) appeared. When the KOH activation time increased to 2 h, the pore size range increased from 1 to 4 nm compared to the NPC-1 sample. When the KOH activation time increased to 5 h, the pore size (<1 nm) decreased. Analysis of the results confirmed that KOH activation firstly generated micropores on the surface of NPC through the etching reaction between KOH and carbon. The size of the newly generated micropores increased with the etching reaction, and then the unconnected micropores of the NPC became larger or merged into new mesopores as the activation time increased. The mechanism of KOH activation for the enlarged pore structure in the NPC material was considered as the etching reaction of carbon in the ZIF-8 derived NPC. The activation of KOH greatly increased the pore size of the material and produced abundant defects, thereby providing more exposed active sites to facilitate mass transport and enhance the catalytic activity (Fig. 4a and b). However, it is worth noting that a very long activation time could also lead to the collapse of the material structure.
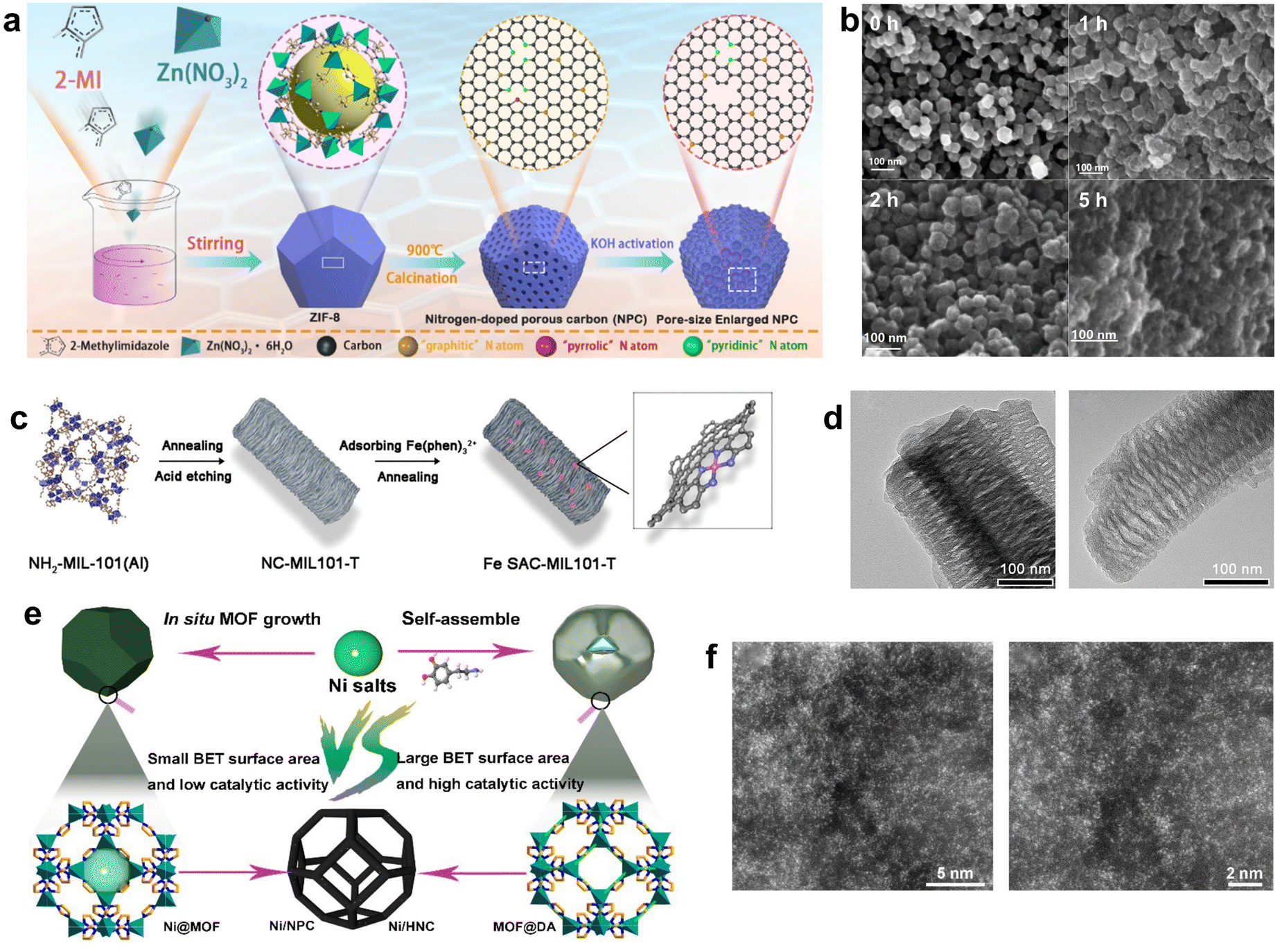 | ||
| Fig. 4 (a) Synthetic route of ZIF-8-derived NPC, (b) SEM for 0 h, 1 h, 2 h, and 5 h KOH activation, respectively. Reproduced from ref. 83 with permission from American Chemical Society, copyright 2019. (c) Schematic illustration of the preparation of Fe SAC-MIL101-T, (d) TEM images of Fe NC-MIL101-1000 (left) and SAC-MIL101-1000 (right). Reproduced from ref. 84 with permission from John Wiley and Sons, copyright 2021. (e) Schematic illustration of the synthetic strategy for Ni/NPC and Ni/HNC, (f) HAADF-STEM images of Ni/HNC. Reproduced from ref. 26 with permission from Springer Nature, copyright 2023. | ||
Besides, MOF derivatives can be used as supports for catalysts. NH2-MIL-101(Al), an MOF with a mesoporous cage structure, was used as a precursor to fabricate a series of N-doped carbon supports (NC-MIL101-T) with well-defined mesoporous structures. The NC-MIL101-T support was then impregnated with the Fe(II)–phenanthroline complex and heated again to produce the Fe SAC-MIL101-T catalyst. The catalyst was rich in accessible Fe–Nx active sites and had better mass transfer efficiency, which optimized the catalytic performance.84 Fe-SAC-MIL101-T has a similar rod shape and pore structure compared to the original MOF, and it had more Fe–Nx sites compared to Fe-MIL-101 pyrolysis alone (Fig. 4c and d). Yun et al. used Zn-MOF as a precursor to form a core–shell structure MOF@DA by adding dopamine hydrochloride and obtained hollow N-doped nanocages (HNC) by calcining at a temperature close to the boiling point of Zn in an Ar atmosphere.26 The addition of DA provided more N sites, thereby maximizing the active site density. The subsequent addition of nickel salts prevented them from aggregating during the second step of calcination. The solution containing Ni2+ was immersed in HNC and calcined again to obtain Ni-doped HNC with N sites (Ni/HNC). The hollow structure made most of the metal sites easily accessible, while the STEM images also proved that the Ni sites were highly loaded and distributed in isolation (Fig. 4e and f). Thus, it exhibited a faradaic efficiency of up to 97.2% in CO generation (−0.7 V vs. RHE), better than that of Ni/NPC.
2.4 Other synthesis strategies
Although pyrolysis is widely used in the synthesis of MOFs derivatives, it has the disadvantages of high energy consumption, difficulty in the precise control of nanostructure, and easy agglomeration of active sites. Therefore, researchers have developed some synthesis strategies for the preparation of MOF derivatives under mild conditions.85Prussian blue analogue PBA (Co3[Fe(CN)6]2) was a simple MOF material formed by the coordination of iron and cobalt ions with CN, which hydrolyzed in alkaline solution to obtain CeOx nanocubes. The reaction principle was based on the ion exchange between OH− and Fe(CN)63−. This low-temperature synthesis route could retain the morphology of the original MOF under certain conditions. OH− in alkaline solution underwent ion exchange with Fe(CN)63− firstly at the solid–liquid interface between the PBA nanocube and the solution, and a thin layer of Co(OH)2 formed on the surface of PBA. Secondly, OH− exchanged with Fe(CN)3−, resulting in the precipitation of Co(OH)2 clusters on the pre-formed Co(OH)2 layer and the formation of a cavity inside the cube. With the increasing in the thickness of Co(OH)2, the ion diffusion rate decreased and finally formed hollow CoOx nanocubes with porous shells.86 The surface of the CoOx nanocube became concave and uneven compared to PBA. The OH− concentration in the alkali solution was found to affect the CoOx nanocube morphology: high OH− concentration promoted the reaction, yet a very high concentration caused the collapse of the nanocubes. In situ TEM characterization monitored the effect of reaction time on the morphology of the derivatives. After 5 min of chemical treatment, the cubic shape of PBA remained but the surface became rough, and after 10 min, the nanocubic morphology became rough truncated quadrilateral, and some nanocubes collapsed in 30 min (Fig. 5a).
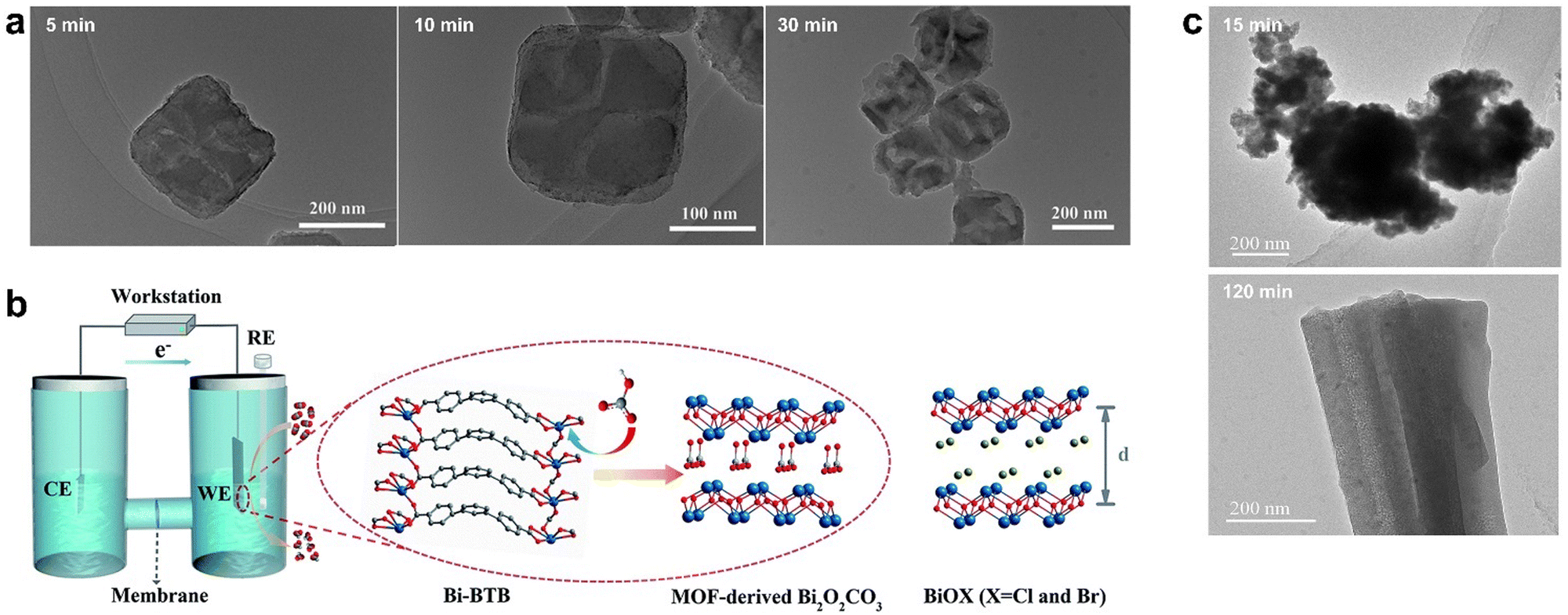 | ||
| Fig. 5 (a) TEM image of CoOx obtained by alkali etching for 5 min, 10 min, and 30 min respectively. Reproduced from ref. 86 with permission from Elsevier, copyright 2015. (b) Right: The electrochemical cell for the electrolysis experiments and proposed mechanism for the formation of MOF-derived Bi2O2CO3. RE is the reference electrode (Ag/AgCl electrode, saturated KCl), CE is the counter electrode (Pt plate, 1 × 1 cm2), and WE is the working electrode (Bi-BTB deposited on carbon paper electrode). Left: The structure of BiOX (X = Cl and Br). Red is O, grey is C, white is H, blue is N, purplish blue is Bi, and olive is halogen (d is the interlayer distance). Reproduced from ref. 87 with permission from Royal Society of Chemistry, copyright 2020. (c) TEM of HKUST-1 electrolyzed for 15 min and 120 min, respectively. Reproduced from ref. 89 with permission from John Wiley and Sons, copyright 2021. | ||
In addition to alkaline solution hydrolysis, electrochemical conversion is also used to synthesize high performance metal-based catalysts for CO2RR. Yuan et al. developed an electrochemical method that successfully converted Bi-BTB into Bi2O2CO3, which showed excellent catalytic activity in CO2RR.87 According to the HSAB theory (Hard–Soft–Acid–Base), Bi3+ ions and carboxylates belong to intermediate acids and hard bases, respectively. In view of the good stability of Bi-BTB in water and alkali, the Bi–O bond is broken by HCO3−. The derivative of Bi-TATB was the same as that of Bi-BTB, and the reaction did not occur when the electrolyte solution was changed to Na2SO4; thus, the leading role of HCO3− was further verified. The derivative Bi2O2CO3 has a layered structure similar to BiOX (X = Cl, Br) composed of [Bi2O2]2+ and CO32− but had a more stable structure than the latter, which was barely changed during electrolysis, while BiOX was transformed into Bi0 (Fig. 5b).
Structural rearrangement is often used in the synthesis of CO2RR electrocatalysts, in which the original MOF was converted into a more active catalyst. The obtained MOF derivatives usually have higher catalytic activity and stability. For example, Bi-BTB as a pre-catalyst underwent structural rearrangement at a reduction potential to form highly active and selectively catalytic Bi nanoparticles uniformly dispersed in a porous organic matrix.88 Bi-BTB decomposed at a specific potential and gradually reduced Bi3+ to metal Bi, and the prepared Bi nanoparticles overcame the inherent poor conductivity of MOF and showed better CO2RR performance.
Since the electrochemical conversion time affects the morphology and structure of MOF-derived materials, it is crucial to carefully choose the electrolysis duration to achieve enhanced electrocatalytic CO2RR properties. Han et al. prepared HKUST-1 films on porous conductive fiber carbon paper (CFP) using a simple electrodeposition method.89 When HKUST-1 was electrolyzed at a certain reduction potential, it was found that HKUST-1 reconstructed into two structures, in which 3D nanospheres composed of a large number of small fragments at 15 min of electrolysis was obtained and yielded 3D nanonetworks composed of cross-linked nanoribbons with different orientations after 120 min of electrolysis (Fig. 5c). The 3D nanosphere structure exhibited that the faradaic efficiency (FE) of C2 products (ethylene and ethanol) reached 58.6% at −0.98 V (vs. RHE) due to the more active sites, lower charge transfer resistance, and higher CuI/Cu0 ratio.
3. Application of MOF-derived materials in the electroreduction of CO2
The electrochemical reduction of CO2 in aqueous solution is essentially the reduction of CO2 coupled with the electrolysis of water. By converting CO2 into valuable chemicals and fuels, including C1 products (CO, formic acid, CH3OH and CH4), C2 products, and C2+ products, it can reduce greenhouse gas emissions, mitigate climate change, and promote circular economy. Electrolysis powered by renewable energy sources (green electricity) can provide a sustainable and carbon-neutral approach to CO2 reduction. The design of catalysts is crucial for determining the product selectivity and yield by altering the reaction kinetics/mechanism. However, several challenges and issues persist in the field of electrocatalytic CO2RR.90–92One of the primary challenges is achieving high selectivity for desired products while suppressing the generation of side products. Additionally, improving the energy efficiency of the CO2 reduction and minimizing electrode degradation are ongoing concerns. Developing stable and efficient catalysts remains a considerable challenge. Continued research and efforts are essential to overcome these obstacles and enable this technology in a sustainable and economically viable manner. In the following sections, we summarize the application of MOF-derived materials in electrocatalytic CO2RR according to the reduction products of C1, C2, and C2+, respectively.
3.1 C1 products
Most MOF-derived materials tend to facilitate the production of C1 products in electrocatalytic CO2RR, mainly including CO, HCOOH, CH4, and CH3OH, due to the lower overpotentials and relatively simpler reaction mechanism compare to other multi-carbon products. Therefore, CO2-to-C1 process as well as the catalysts are also important to be explored and studied.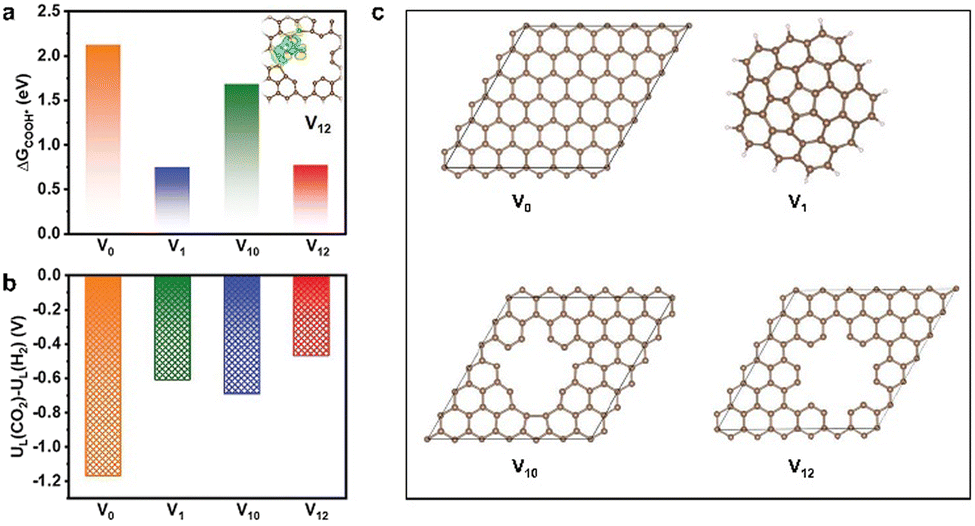 | ||
| Fig. 6 (a) Free energy diagram of K-defect-C-1100 (inset: electron density difference analysis, yellow represents charge deposition, cyan represents charge consumption), (b) The values of UL(CO2RR) − UL(HER) on V0, V1, V10, and V12 sites, (c) Schematic structural models for the catalysts involving V0, V1, V10, and V12 defect sites. Reproduced from ref. 97 with permission from John Wiley and Sons, copyright 2022. | ||
The electronegativity, charge density, and size of heteroatoms, especially non-metal atoms such as N, S, and P, usually affect the properties of host materials. Heteroatom doping can adjust the electronic structure and promote the electron transfer, thus promoting the catalytic activity. In addition, the physical and chemical properties of the material are regulated by the doping effect, which is usually considered as a favorable method to prepare the catalyst with excellent electroreduction performance of CO2.74,98,99 For example, N-doped carbon are widely used as electrocatalysts for the reduction of CO2 to CO. Wang et al. synthesized N-doped carbon materials by the direct pyrolysis of ZIF-8 at different temperatures. It was found that NC-900 prepared at 900 °C exhibited the highest FECO of 78% at −0.93 V (vs. RHE) in the electrochemical reduction of CO2, and H2 was the only by-product in the whole potential range. Meanwhile, the researchers also pyrolyzed MOF-5 under the same conditions to obtain the N-free carbon material C-900, which showed a low electroreduction activity of CO2.100
Ye et al. pyrolyzed Zn-MOF-74 and the additional nitrogen source melamine at different temperatures to obtain N-doped porous carbon NPC with a high faradaic efficiency of 98.4% toward CO at −0.55 V (vs. RHE).101 At the same time, the calcination time and temperature were controlled to adjust the content and species of N-doping in the samples, which could regulate the CO2RR performance. The highest FECO was obtained at 1000 °C, and the highest content of pyridine-N and graphite-N was 68.31% (Fig. 7a). The CO2RR performance of the samples showed that the high CO2RR activity of N-doped carbon materials were attributed to the synergistic effect of high concentration of pyridinic N, graphitic N, and porous structure. The C29H14 clusters were constructed to investigate the influence of different N species on the CO2RR; DFT theoretical studies showed that the potential limited steps of pyridine-N and graphite-N exhibited lower energy barriers to overcome, indicating that pyridine-N and graphite-N could more effectively promote CO2 electroreduction (Fig. 7b and c).
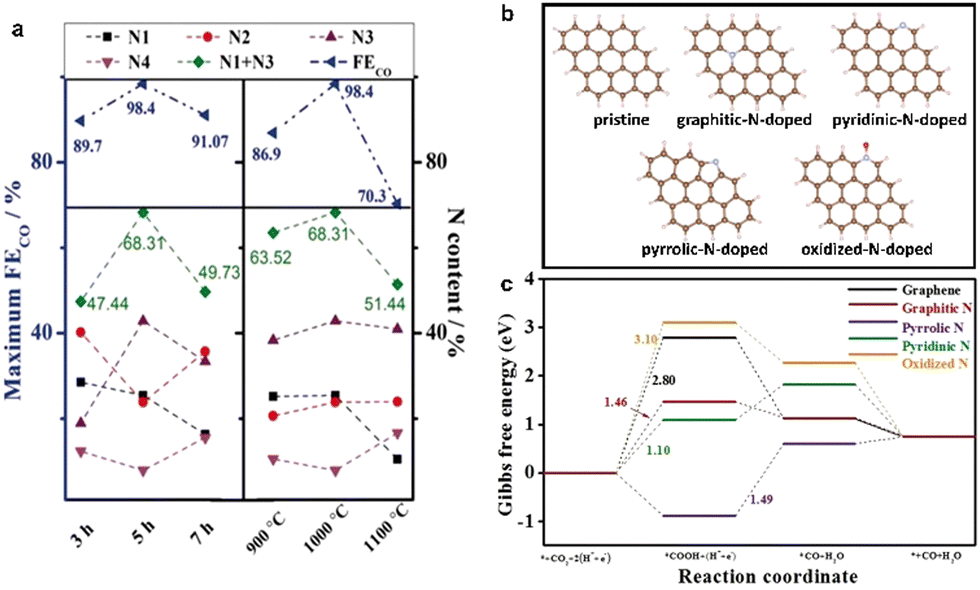 | ||
| Fig. 7 (a) Atomic contents of each N moiety, N1 + N3, and maximum FECO of NPC, (b) Optimized structures of pristine, graphitic-N-doped, pyridinic-N-doped, pyrrolic-N-doped, and oxidized-N-doped C29H14 cluster, respectively. C, H, N, and O are represented in brown, light pink, cyan, and red, respectively. (c) CO2RR Gibbs free-energy diagram for graphene, graphitic-N-doped, pyridinic-N-doped, pyrrolic-N-doped, and oxidized-N-doped C29H14 cluster. Reproduced from ref. 101 with permission from John Wiley and Sons, copyright 2020. | ||
Ag, Fe, Ni, and Co are the most commonly used metals in the electrocatalytic reduction of CO2 to CO. Taking the research in recent years as examples, Wang et al. prepared Ag-MOF-derived silver foam with 3D dendrites for CO2RR experiments.102 It was found that a high current density of 57.2 mA cm−2 was delivered at −1.15 V (vs. RHE), with a maximum FECO of 91.1% at −1.05 V (vs. RHE). Zhang et al. prepared ZIF-8-derived Ni–N-doped porous carbon and carbon nanotubes (Ni–N–C/CNTs) using the surfactant cetyltrimethylammonium bromide (CTAB) as a modifier.103 CTAB coordinated with Ni2+ generated a high content of Ni–Nx single-sites. The CO2RR performance of the catalyst was promoted together with the abundant pyrrole-N and the unique meso-microporous structure. In the H-type cell, it exhibited a high FECO of 98% at −0.7 V (vs. RHE).
Normally, the uncoordinated site is considered to be the most active site for CO2 electroreduction, and adjusting the coordination number of the metal site is an effective way to enhance the catalytic performance. The atomically dispersed Co catalyst was achieved by calcining the bimetallic CoZn-MOF. During pyrolysis, Zn was evaporated, only leaving Co anchored on the N-doped porous carbon. At that same time, the CoII-imidazolate-CoII linkages would partially decompose with increasing temperature to release CN fragments, thus leading to an increase in the uncoordinated number of Co. Co–N2, Co–N3, and Co–N4 were obtained at 800, 900, and 1000 °C, respectively.104 Performance tests showed that CO and H2 were the main products, and Co–N2 had the highest FECO of 95% at −0.68 V (vs. RHE), while Co–N3 could only obtain a maximum FECO of 63% at −0.53 V (vs. RHE). Also, the total current density was too small to accurately monitor at low overpotentials between −0.83 V and −1.03 V (vs. RHE) for Co–N4. According to the theoretical study, Co–N2 could promote the conversion of CO2RR to CO but inhibit the hydrogen evolution reaction, which provided a method to improve the efficiency of the conversion of CO2 to CO.
It is well known that N-doped carbon-supported metal materials have shown potentials to improve the performance of electrocatalysts by adjusting the electronic structures and are often widely reported as catalysts for CO2-to-CO conversion processes. In addition to the N atom, other atoms are also used to coordinate with the metal to improve the catalytic activity. For example, Zhao et al. prepared atomically dispersed Fe1O2N2/NC nanorods in which oxygen-rich Fe-doped Zn-based MOF-74 was used as a precursor and melamine as a nitrogen source.105 In the process of high-temperature pyrolysis, the ZnO5 clusters evaporated, part of the FeO5 clusters decomposed, and part of the O coordination atoms were replaced by N due to the decomposition of melamine. This catalyst showed high CO selectivity (FECO over 95% at −0.4 V to −0.8 V, 99.7% at −0.5 V vs. RHE) and long lifetime (almost no change in FECO and partial current density of CO after electrolysis for 12 h at −0.7 V). The overall performance was better than that of N-doped C and Fen/C. Similarly, O–Fe–N–C with single Fe atom distribution and O coordination was constructed using an O- and N-rich MOF.25 Through axial coordination, the binding energy of CO2RR intermediates *COOH and *CO were regulated, which was conducive to the desorption of *CO from the surface of O–Fe–N–C and inhibited the activity of HER. Compared with NC and Fe–N–C, O–Fe–N–C showed an FECO of 95% at a low applied potential of −0.5 V vs. RHE.
In addition, dual-site catalysts were used to optimize the performance of CO2RR and improve the selectivity of catalysts for specific reactions.106 Due to the interaction between adjacent active sites, the adsorption properties of intermediates could be adjusted. Jiao et al. prepared a dual-site catalyst Fe–Ni–N–C by Zn-assisted atomization method, in which Fe and Ni uniformly dispersed on the carbon support of the catalyst, and all of them were coordinated with N4 (Fig. 8a).107 There was a significant synergistic effect between Fe and Ni to achieve a high FECO of 96.2%, which was better than that of N-doped carbon modified by Fe (Fig. 8b). Although the selectivity of CO for Ni single-site N-doped carbon was similar to that of Fe–Ni, it needed a more negative potential and consumed more energy. The DFT calculation revealed that it was attributed to the interaction between Fe and Ni atoms in Fe–Ni–N–C, the free energy of the intermediate *COOH formed on Fe reduced to 0.64 eV, and it was 1.53 eV on Ni, which was lower than that on Fe–N–C (0.75 eV) and Ni–N–C (1.95 eV), thus reducing the energy barrier for the formation of the intermediate and increasing the CO2RR activity (Fig. 8c).
 | ||
| Fig. 8 (a) Synthetic route of Fe1–Ni1–N–C, (b) FECO for Fe1–Ni1–N–C, Fe1–N–C, Ni1–N–C, and N–C, respectively, (c) free energy diagrams of CO2RR for Fe1–Ni1–N–C (Fe), Fe1–Ni1–N–C (Ni), Fe1–N–C, and Ni1–N–C, respectively. Reproduced from ref. 107 with permission from American Chemical Society, copyright 2021. | ||
Previous studies have found that main group metals (In, Sn, Sb, Bi, etc.) occupy an important position as catalysts for the electroreduction of CO2 to formate.108–114 Among them, the CO2RR performance of Bi-based MOFs has also been more extensively investigated because of their simple and easy availability. Since Komatsu's electrochemical study on metallic Bi confirmed its excellent CO2RR activity, Bi-based materials are widely used as effective electrocatalysts for the electrochemical conversion of CO2 to formate.115 Li et al. prepared nanostructured Bi nanosheets (Bi NSs) by the in situ transformation of the Bi-MOF precursor, which showed remarkable electrocatalytic performance for CO2 reduction.116 The faradaic efficiency of formate was up to 98% at −0.8 V (vs. RHE), and the FEHCOOH exceeded 90% in the wide range of −0.65 to −0.96 V (vs. RHE), with no significant decrease within 40 h. This study showed that ligand-stabilized uncoordinated active sites were easily generated by the in situ transformation of metal–organic frameworks precursors and were effectively stabilized by residual ligands adsorbed on the surface, thereby achieving efficient CO2 conversion. Porous CAU-17 (Bi9(C9H3O6)9(H2O)9) sprayed on carbon paper was transformed to leaf-like nanosheets Bi NSs by in situ electroreduction. These Bi NSs were aggregated from dispersed nanoparticles of ultra-small size (<10 nm).117 The characterization results showed Bi NSs as metal/metal oxide composites with Bi/Bi–O hybrid interface. In 1 M KHCO3, the average value of FEHCOOH of Bi NSs was more than 85%, the maximum value of FEHCOOH could reach 98%, and the cathodic energetic efficiency (CEE) of Bi NSs was obviously higher than that of Bi NPs (Fig. 9a). According to the DFT calculation for CO2 reduction mechanism (Fig. 9b), the energy barrier from CO2* to *OCHO on Bi (0.46 eV) was higher than that on Bi–O (0.17 eV), indicating that the O atoms on the surface of Bi–O were conducive to reduce the energy barrier for the formation of *OCHO, which was beneficial for the electroreduction of CO2 to HCOOH. This example provided a reference for the construction of metal/metal oxide composite catalysts from MOFs.
 | ||
| Fig. 9 (a) FEs and CEEs for Bi NSs and Bi NPs. (b) Free energy diagrams of Bi NPs and Bi NSs. Reproduced from ref. 117 with permission from John Wiley and Sons, copyright 2020. (c) FEs for Bi, In and BiIn. (d) Free energy diagrams of Bi, In and BiIn. Reproduced from ref. 120 with permission from Elsevier, copyright 2022. (e) FEs for CuBi75, (f) free energy diagrams for different paths of reduction CO2 to HCOOH on BiCu75. Reproduced from ref. 121 with permission from Elsevier, copyright 2021. | ||
Similarly, the bimetallic synergy effects also promote the conversion of CO2RR to formate. The design of bimetallic sites can not only adjust the electronic structure through the cooperation of adjacent metal atoms but also provide more stable intermediates and less energy to activate CO2 molecules along the reaction path. Therefore, the construction of bimetallic sites may provide a new strategy to develop efficient catalysts for HCOOH production.118,119 For example, BiIn alloy NPs with dendritic morphology were obtained through the electroreduction of In-Bi-MOF.120 The faradaic efficiency of the electroreduction of CO2 to formate was as high as 97.2% at −0.92 V (vs. RHE) (Fig. 9c). DFT calculation results showed that the Bi–In bimetallic site promoted the formation of *OCHO by enhancing the CO2 adsorption compared with the Bi single-metal site and reduced the energy barrier from *OCHO to *HCOOH compared with the In single-metal site; thus, the cooperative effect of the Bi-In bimetallic site promoted the formation of HCOOH (Fig. 9d). Rod-like CuBiX (X is Cu content) catalysts were obtained by the calcination of bimetallic MOFs, and the molar mass ratio of Cu to Bi was determined by the amount of added Cu salt.121 CuBi75 with 75% Cu and 25% Bi had higher FEHCOOH after calcination, and the maximum FEHCOOH even reached 100% at −0.77 V (vs. RHE). At the same time, it had an ultra-wide potential window with FEHCOOH > 93.7%, from −0.57 V to −1.47 V (vs. RHE) (Fig. 9e). The characterization of CuBi showed that CuO nanoparticles were attached to the surface of the catalyst and encapsulated in the rod-like catalyst by Bi2O3 and Bi2CuO4 present at the interface. DFT calculation illustrated that the energy barrier of the HCOO* pathway was lower than that of the *COOH intermediate; thus, the HCOO* pathway was an optimal step for the formation of formic acid from CO2RR (Fig. 9f). The analysis of intrinsic activity and kinetic parameters showed that the excellent performance was attributed to the unique structure of Bi2CuO4 at the interface between Bi2O3 rod crystals and CuOx nanoparticles, which effectively strengthen the adsorption path and adsorption capacity of CO2, inhibited hydrogen evolution and C–C coupling, and thus greatly improved the selectivity of CO2RR to formate.
Kim et al. chose Cu-MOF-74 as a precursor to prepare highly isolated and ordered Cu NP clusters.127 The faradaic efficiency for methane reached ca. 50% at −1.3 V (vs. RHE) in CO2RR and inhibited the synthesis of C2, while Cu-MOF-74 showed similar selectivity for CH4 (28.7%) and C2H4 (29.5%). The partial current density of CH4 was 2.3 times higher than that of Cu-MOF-74, which is probably due to the difference in the distance between the two nanoparticle clusters. Guan et al. prepared Cu, N co-doped carbon nanosheets by the high temperature calcination of the mixture of Cu-MOF and dicyandiamide. Cu–Nx is uniformly distributed on the calcined MOF-derived framework, and the loading amount of Cu is also affected by the calcination temperature.128 The Cu loading concentration was 4.9% mol at 800 °C. The high loading density of Cu enabled the adjacent Cu atoms to be close enough to each other, and the synergistic effect between adjacent Cu atoms induced C–C coupling, facilitating the dimerization of *CO intermediates to form ethylene with FEC2H4 at −1.4 V (vs. RHE) of 24.8% accompanied by CH4 (13.9%). When the temperature increased, part of the Cu atoms coordinated with N gradually changed from Cu–Nx coordination to metallic copper. At 1000 °C, the concentration of Cu decreased to 2.4%, and the Cu atoms were separated from each other, resulting in the increase in the FE of CH4 (38.6%) and the decrease in the FE of ethylene; the main product of CO2RR was CH4. Cao et al. obtained Cu2O@CuHHTP composites by in situ electrochemical treatment on conductive CuHHTP.129 The obtained Cu2O QDs were mainly Cu2O(111) plane and uniformly dispersed on CuHHTP. A large number of hydroxyl groups were exposed at the surface of Cu2O QDs, which formed hydrogen bonds with intermediates (such as *COOH, *CO, and *CHO) to reduce the energy barrier for CH4 formation and significantly improved the selectivity of CH4, with FECH4 as high as 73% and the partial current density reaching 10.8 mA cm−2 at −1.4 V (vs. RHE).
3.2 C2 products
Copper is considered to be the most effective catalyst for the formation of C2 and C2+ products. Since copper has a moderate adsorption capacity for both *CO and *H, C1 products (such as CO, formic acid) or multi-carbon compounds can be generated by regulating the adsorption of key intermediates, and the latter are more desirable products due to their higher value and higher energy density.142–144 However, the high overpotential and low selectivity for multi-carbon products of Cu catalysts hinder their practical application; thus, it is necessary to develop higher performance electrocatalysts, which require improving both the selectivity of the target product and maintaining the catalytic activity.Table 1 summarizes recent studies on the electrocatalytic conversion of CO2 to C2 and C2+ products catalyzed by MOF-derived materials. Here, we firstly discuss the catalysts for the formation of C2 product. It was reported that HKUST-1 with the partial decomposition of organic ligands electrochemically reduced CO2 to C2H4 with considerable faradaic efficiency. After controllable pyrolysis, the secondary structural unit of Cu dimer in HKUST-1 was distorted, resulting in a higher number of unpaired pairs of Cu. CO2RR experiments showed that the increase in the uncoordinated sites in Cu clusters promoted the formation of C2H4 while inhibiting the production of H2, CO, and CH4 (Fig. 10a).130 Therefore, it is a reasonable way to adjust the distribution of C2 products by regulating the coordination number of metal sites in MOFs.
| Pristine MOF | Electrocatalysts | Main product | Maximum FE | Partial current density (mA cm−2) | Stability | Ref. |
|---|---|---|---|---|---|---|
| HKUST-1 | Cu–Cu2O | C2 products | 58.6% at −0.98 V (vs. RHE) | 19.2 | 2 h | 89 |
| (C2H4 and C2H5OH) | ||||||
| HKUST-1 | HKUST-1 derived | C2H4 | 45% at −1.07 V (vs. RHE) | 262 | ∼7.7 h (H-cell) | 130 |
| Cu clusters | ∼2.3 h (flow cell) | |||||
| Cu–ASP | Porous CuO nanowires | C2 products | H-cell: 70% (C2), 43% (C2H4) | ∼141 (flow cell) | 12 h (H-cell) | 131 |
| (C2H4 and C2H5OH) | Flow cell: ∼37% (C2H4) at −1.3 V (vs. RHE) | |||||
| BEN–Cu–BTC | Cu@N doped C | C2 products | 11.2% (C2H4) | NA | 8 h | 132 |
| (C2H4 and C2H5OH) | 18.4% (C2H5OH) at −1.01 V (vs. RHE) | |||||
| Cu3(HITP)2@KB | Cu@KB | C2H4 | 70% at −1.37 V (vs. RHE) | ∼30 | 10 h | 133 |
| Cu–TCNQ | Cu@NxC | C2 products | ∼80% (C2) at −1.1 V (vs. RHE) | ∼17 | ∼2.7 h | 134 |
| (C2H4 and C2H5OH) | ||||||
| Cu–BTC | CuOx@C | C2 products | 82% (C2), 46% (C2H5OH) at | ∼315 (C2) | 50 h | 135 |
| (C2H5OH) | ∼−1.0 V (vs. RHE) | ∼166 (C2H5OH) | ||||
| CuCo-MOF-74 | M–CuCo/C | C2 products | 79.2% (C2), 29.7% (C2H4), 49.5 (C2H5OH) at −0.75 V (vs. RHE) | 10.1 | NA | 136 |
| (C2H4 and C2H5OH) | ||||||
| Co-doped Cu2(BDC)2 | CoCu single-atom alloy | C2H4 | 15.6% at −1.07 V (vs. RHE) | 282 | ∼3 h | 137 |
| HKUST-1 | Cu/C | Isopropanol | 22.5% at −2.4 V (vs. RHE) | ∼30 | NA | 138 |
| Cu-doped ZIF-8 | Cu–SA/NPC | Acetone | 36.7% at −0.36 V (vs. RHE) | NA | 5 times | 139 |
| HKUST-1 | Cu/Ag | C2+ products | ∼21% at −80 and −100 (mA cm−2) | NA | NA | 140 |
| (C2H4, C2H6, C2H5OH, C3H7OH) | ||||||
| HKUST-1 | PTFE/CuxOyCz | C2+ products | 54% at −80 | NA | NA | 141 |
| (C2H4, C2H6, C2H5OH, C3H7OH) | (mAcm−2) |
 | ||
| Fig. 10 FEs for (a) HKUST-1 (250 °C, 3 h calcined)-derived Cu compared with CuAc (200 °C, 1 h calcined)-derived Cu. Reproduced from ref. 130 with permission from American Chemical Society, copyright 2018. (b) Cu–ASP. Reproduced from ref. 131 with permission from Royal Society of Chemistry, copyright 2020. (c) Ben–Cu–BTB-derived Cu–N-doped carbon composite materials. Reproduced from ref. 132 with permission from Royal Society of Chemistry, copyright 2019. Cu3(HITP)2 (d) with and (e) without KB conductive supports. Reproduced from ref. 133 with permission from Royal Society of Chemistry, copyright 2021. (f) Cu–TCNQ-derived Cu@NC. Reproduced from ref. 134 with permission from Nature, copyright 2021. (g) CuCo-MOF-74-derived Co3O4–CuOx/C. (h)–(j) Catalytic mechanism of Co3O4–CuOx/C. Reproduced from ref. 136 with permission from Elsevier, copyright 2022. | ||
The construction of grain boundaries and defects also could enhance the selectivity of multi-carbon products in CO2RR. Yang et al. prepared porous copper oxide nanowires with abundant crystalline surface/interface by the controlled annealing of Cu–aspartic acid (Cu–ASP). Catalytic experiments of CO2RR indicated that FEC2 and FEC2H4 reached ∼70% and ∼37%, respectively, at −1.3 V (vs. RHE) in a flow cell (Fig. 10b).131Ex situ/in situ characterizations suggested that the oxide-derived metastable Cu sites at grain boundaries and defect regions could stabilize the *CO and *COH intermediates, thus inhibiting the formation of C1 products and promoting the formation of C2 products.
In addition, N-doping also promoted the CO2RR performance in metal/carbon catalysts. Cheng et al. constructed a Cu–N-doped carbon composite by N-containing benzimidazole-modified Cu-BTC.132 It was found that the high content of pyrrole-N and Cu–N doping provided abundant CO2 adsorption sites, promoted C–C coupling on the Cu surface, and improved the selectivity of C2 products (Fig. 10c). However, the high content of graphitic nitrogen and nitric oxide led to a strong adsorption of H*, which enhanced the competitive reaction HER. Therefore, the optimization of N-doping types was also of great significance for the improvement of selectivity.
Sun et al. synthesized multicrystalline Cu nano-crystallites derived from semi-conductive MOF (Cu3(HITP)2) modified with conductive support and investigated its CO2RR performance composite.133 The conductive support KB provided higher current density transport for uniform charge distribution; thus, Cu+ on MOF was rapidly reduced to Cu crystallites, while Cu species on MOF without KB tended to aggregate due to uneven surface polarization and inadequate nuclei seeding. Compared with the KB@Cu NPs prepared by mixing commercial Cu nanoparticles and KB with the same loading, the Cu crystallites obtained by electronic conversion have higher total current density in CO2RR, the FE of C2H4 reached 65% at −1.56 V (vs. RHE) (Fig. 10d and e), and the formation of H2 by-product was suppressed due to abundant grain boundaries and defects. It is indicated that the conductive carrier regulated the charge distribution, which affected the MOF reconstruction, thus regulating the product distribution in CO2RR.
Li et al. found that the surrounding environment of the active center also had an influence on the performance of catalyst. By investigating the core–shell structure of Cu@NC derived from the calcination of Cu–TCNQ, they found that the NC shell and the Cu core showed a synergistic effect on the selective reduction of CO2 to C2 products.134 For Cu@NC, the main products of CO2RR were C2H5OH and C2H4, and the total FEC2 was up to 80% at −1.1 V (vs. RHE), which was much higher than the highest value of 39% of FEC2 on Cu NP. It was found that CO2 was adsorbed and enriched at the Cu/NC interface layer and activated by the NC layer, resulting in a high coverage of the CO* intermediate, thus promoting CO–CO coupling. Meanwhile, the side reaction HER was effectively inhibited due to the hydrophobicity of the NC layer; the faradaic efficiency of C2 reached 80% and HER was suppressed as low as 8% at −1.1 V (vs. RHE) (Fig. 10f).
It has been reported that regulating the Cu oxidation state could change the product distribution, thereby increasing the content of C2 and C2+ products.145,146 For example, carbon-encapsulated CuO (CuOx@C) was obtained by the simple low temperature pyrolysis of Cu-BTC in air.135 Compared with CuOx obtained by pyrolysis at 800 °C, it was found that carbon species could stabilize Cu+, improve the electronic conductivity of the catalyst, and promote the timely conversion of reactant molecules, thus inhibiting the self-reduction of CuOx. CuOx was mainly transferred into metallic Cu after CO2RR, while CuOx@C was converted to Cu2O phase. The inhibition effect of the carbon layer on the reduction of CuOx was demonstrated, which stabilized the Cu+ species in CuOx@C under the wide current density window, and the catalyst showed only slight degradation in the 50 h stability test. In addition, the higher Cu+ ratio promoted the adsorption of *CO, thus facilitating the formation of C2+ products. It had a faradaic efficiency of 82% for the C2 product and 46% for ethanol (−1.0 V vs. RHE), while the partial current density of ethanol was as high as 166 mA cm−2. The limited yield and selectivity of the electrocatalytic reduction of CO2 to C2 products is mainly due to the difficulty of C–C coupling. Utilizing two complementary catalysts to construct a tandem catalyst is an effective way to generate C2 products in CO2RR. Tandem catalysis is divided into two steps for the formation of C2 products, namely, CO2-to-CO and CO-to-C2, which are realized on two different catalysts (the catalyst used in the CO-to-C2 step is basically the copper-based material). The main advantage is to introduce an in situ CO source in the tandem catalyst, increase the local *CO coverage on the Cu surface, and enable the rapid coupling of CO on the Cu-based catalyst as a key intermediate.147–150 The key point of tandem catalysts is that the active sites need to have independent but adjacent distribution to reduce the transfer barrier and avoid the loss of activity of intermediate products. He et al. reported a Co3O4–CuOx/C catalyst derived from a bimetallic CuCo-MOF-74 framework,136 which consisted of Co3O4 and CuOx sites encapsulated in a carbon skeleton; the faradaic efficiency of C2 reached 79.2% at −0.75 V (vs. RHE) with a generation rate of 275.6 μmol g−1 h−1 (Fig. 10g). In situ characterization and DFT showed that CO was released on oxygen-vacancy rich Co3O4, which enhanced the local CO concentration on M–CuCo/C, and then C–C coupling occurred on CuOx sites to generate C2 products (Fig. 10h–j).
3.3 C2+ products
CO2RR into multi-carbon products involves the formation and transformation of multiple intermediates, such as the formation steps of C–C bond and C–H bond. It is a complex multi-electron coupling multi-proton process, which requires not only the consumption of a large number of electrons but also the efficient transfer and storage of electrons to ensure the reaction. In addition, there are great challenges in clarifying the reaction pathways and revealing the reaction mechanisms. Finally, there are many side reactions and competing reactions in CO2RR. In this case, the electrocatalytic synthesis of C2+ products by MOF-derived materials shows great promise.146,151 By pyrolyzing HKUST-1 and PCN-6 at different temperatures, respectively, Rayer et al. obtained the Cu/C composite that was used as an ink to coat metallic nickel or copper supports to prepare MOF derivative electrodes for CO2RR.138 They found that Cu-HKUST-1 pyrolyzed at 600 °C produced isopropanol between −2.0 V and −2.5 V and had the highest faradaic efficiency of 22.5%, which was rarely obtained in the electrocatalytic reduction of CO2. Although Cu mesh and Cu foil had larger FE of isopropanol, MOF-derived catalysts contained only a small amount of uniformly distributed copper instead of bulk metals, and the utilization efficiency of active sites was higher (Fig. 11a). The formation of isopropanol mainly came from the high density of CO* on the surface of the catalyst, which enabled the C2H3O* generated by the dimerization of CO* couple with CO* to again form a new C–C bond, and finally isopropanol was obtained through the transfer of protons and electrons. However, CO2 generates C1 and C2 byproducts through other reaction pathways at high current density (Fig. 11b).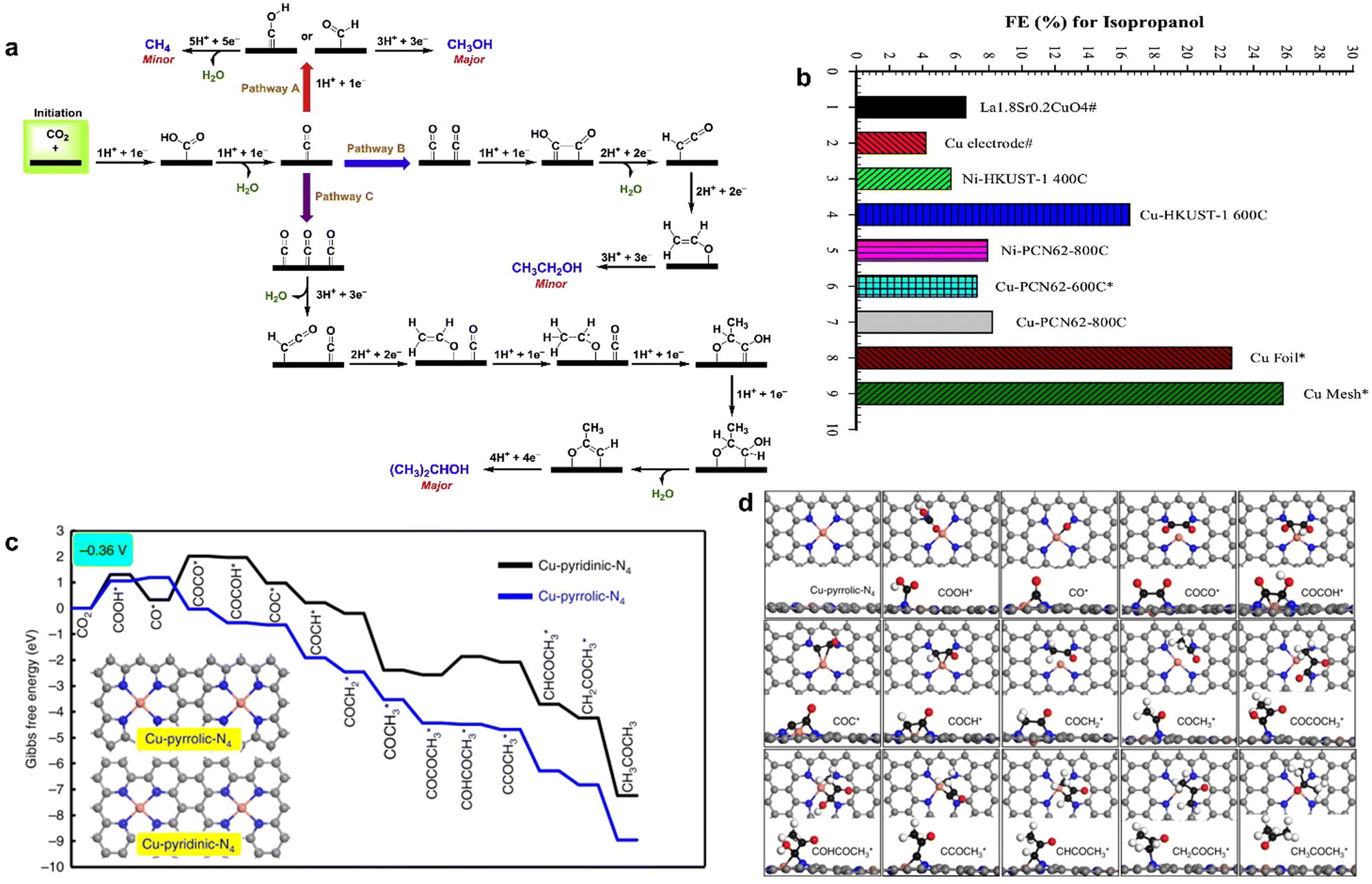 | ||
| Fig. 11 (a) FE for isopropanol. (b) Proposed reaction pathways for CO2RR. Reproduced from ref. 138 with permission from Elsevier, copyright 2020. (c) Free energy diagrams calculated at a potential of −0.36V for CO2 reduction to CH3COCH3 on Cu-pyridinic-N4 and Cu-pyrrolic-N4 sites of Cu-SA/NPC (the computational models were included in the figure). (d) Optimized structures of all reaction intermediates involved in the pathways of CO2 reduction on the Cu-pyrrolic-N4 site (gray: C of catalyst; black: C of adsorbate; red: O; orange: Cu; blue: N; white: H). Reproduced from ref. 139 with permission from Nature, copyright 2020. | ||
The species of N coordinated to single Cu atom also regulated the product distribution. Zhao et al. synthesized N-doped porous carbon-supported single copper atom material (Cu–SA/NPC) derived from Cu-doped ZIF-8.139 Cu–SA/NPC was tested in the range fro, −0.16 to −0.96 V (vs. RHE) for CO2 electroreduction; five liquid products including formic acid, acetic acid, methanol, ethanol, and acetone were detected at low overpotential, and the faradaic efficiency of acetone was 15.6–36.7%. With the negative shift of applied potential, the faradaic efficiency of acetone increased and reached a maximum of 36.7% at −0.36 V (vs. RHE). Acetone was the main reduction product at low potential. Based on DFT calculations, it was confirmed that Cu-pyrrole-N had higher acetone selectivity than Cu-pyridine-N (Fig. 11c and d); COOH* intermediate on Cu-pyrrole-N was stabler than that on Cu-pyridine-N, and the energy barrier of CO* dimerization to generate COCO* species on Cu-pyridine-N was −1.23 eV, while that of Cu-pyridine-N was 1.67 eV. Thus, C–C coupling on Cu-pyrrole-N was more likely to occur, resulting in a high selectivity for acetone formation.
Since Ag has considerable CO activity in CO2RR; therefore, it could provide enough CO to form a tandem catalyst with Cu-based catalyst to synthesize multi-carbon products. For example, an Ag–Cu bimetallic tandem catalyst prepared by the redox method successfully converted CO2 into C2+ products through the synergistic effect of bimetallic Cu–Ag.140 The catalyst with Cu-coated thin carbon layer was firstly generated by the pyrolysis of HKUST-1, and then Cu was partially replaced by atomic Ag. When the Ag loading was about 5.7% to 10.6%, the C2+ yield reached an optimal value. A very high Ag loading affected the C–C coupling step between the adjacent Cu atom. The mixed CuI/Cu0 valence state served as an active site for C2+ formation, and the CO spillover effect of the Ag site increased the local CO concentration, resulting in a higher *CO coverage at the CuI/Cu0 center. Thus, the selectivity of C2+ products was improved through the tandem pathway.
4. Conclusions
MOF-derive materials inherit the unique properties of the original MOFs, including high surface area, porosity, and uniform distribution of catalytic active sites, while providing enhanced conductivity and structural stability. These characteristics make MOF-derive materials highly promising in the field of CO2 reduction.Compared to conventional electrocatalysts, MOF-derived materials offer increased exposure of more active sites, thereby improving the efficiency of the catalytic process. Carbonized MOF-derived materials, in particular, exhibit superior electron transfer kinetics and long-term stability. This durability is crucial for practical applications as it ensures that the materials can withstand the harsh conditions and rigorous operation required for CO2 electrocatalysis. Furthermore, MOF-derived materials can be engineered to achieve new physical and chemical properties and enhanced performance through the manipulation of electron and chemical structures. This opens up possibilities for optimizing CO2 reduction electrocatalysts and providing a pathway toward optimizing CO2 reduction electrocatalysts for industrial-scale production.152–154 In this review, the synthesis strategies of main MOF derivatives including pyrolysis, precursor regulation, and post-modification as well as other synthetic strategies are summarized, and some classic examples are listed, which is helpful to understand the synthesis methods of different types of MOF-derived materials and to select suitable synthesis routes in future research. Then, we discuss the application of MOF-derived materials in the electroreduction of CO2 according to the classification of reduction products, in which most of the reduction products of MOF-derived electrocatalysts are CO and/or HCOOH with high faradaic efficiency. The selectivity of other C1 products, such as CH4 and CH3OH, is relatively low due to their complex proton/electron transfer steps. An in-depth understanding of the CH4 and CH3OH formation mechanism is needed to tune the catalyst structure and surrounding environment of the active site, ultimately improving the selectivity. In the discussion of MOF-derived electrocatalysts for C2 and C2+ products, we found that the reports of these electrocatalysts basically focused on Cu-based MOF derivatives and composites, which produce a large number of C1 by-products, and how to improve the selectivity of C2 and C2+ products is also an issue that needs to be addressed. Meanwhile, Cu-based materials are widely used as electrocatalysts for the reduction of CO2 to produce C2 and C2+ due to their promotion of C–C coupling. Nevertheless, non-copper catalysts, such as N-doped nanodiamond/Si arrays (NDD/Si RA), were also used to produce C2 and C2+ products, which converted CO2 to acetate.155 Benefiting from the high hydrogen evolution overpotential and N-doping of NDD/Si RA, the faradaic efficiency of acetate reached 77.3–77.6% under a reduction potential range of −0.8 V to −1.0 V (vs. RHE), which opened up a new way for carbon-based electrocatalytic materials to convert CO2 into multi-carbon products. The manganese molecular polymer formed by modifying Mn3+ with polyethylene glycol also had high electrocatalytic reduction selectivity for acetic acid.156 This also inspired us to use non-copper-based MOF-derived materials to produce C2 and C2+.
The solution of the above problems requires a more thorough and rigorous study on the catalytic mechanism. So far, most of the mechanisms for electrocatalytic CO2 reduction with MOF-derived materials are obtained through theoretical calculations, and it is also necessary to combine advanced in situ/operation characterization technology to clarify the actual chemical state and structure of active sites under electrochemical CO2RR conditions as well as track and grasp the specific reaction mechanism so as to better control the synthesis method and achieve the ultimate purpose of optimizing the electrocatalytic performance of the CO2RR.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by NSFC (22121005) and the Fundamental Research Funds for the Central Universities (63223012).Notes and references
- S. Das, J. Perez-Ramirez, J. Gong, N. Dewangan, K. Hidajat, B. C. Gates and S. Kawi, Core-shell structured catalysts for thermocatalytic, photocatalytic, and electrocatalytic conversion of CO2, Chem. Soc. Rev., 2020, 49, 2937–3004 RSC.
- A. Velty and A. Corma, Advanced zeolite and ordered mesoporous silica-based catalysts for the conversion of CO2 to chemicals and fuels, Chem. Soc. Rev., 2023, 52, 1773–1946 RSC.
- T. Zheng, K. Jiang and H. Wang, Recent advances in electrochemical CO2-to-CO conversion on heterogeneous catalysts, Adv. Mater., 2018, 30, e1802066 CrossRef PubMed.
- D. Xu, K. Li, B. Jia, W. Sun, W. Zhang, X. Liu and T. Ma, Electrocatalytic CO2 reduction towards industrial applications, Carbon Energy, 2022, 5, e230 CrossRef.
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Electrocatalytic reduction of CO2 and CO to multi-carbon compounds over Cu-based catalysts, Chem. Soc. Rev., 2021, 50, 12897–12914 RSC.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, What would it take for renewably powered electrosynthesis to displace petrochemical processes?, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- J. Choi, J. Kim, P. Wagner, S. Gambhir, R. Jalili, S. Byun, S. Sayyar, Y. M. Lee, D. R. MacFarlane, G. G. Wallace and D. L. Officer, Energy efficient electrochemical reduction of CO2 to CO using a three-dimensional porphyrin/graphene hydrogel, Energy Environ. Sci., 2019, 12, 747–755 RSC.
- J. Choi, J. Kim, P. Wagner, J. Na, G. G. Wallace, D. L. Officer and Y. Yamauchi, Highly ordered mesoporous carbon/iron porphyrin nanoreactor for the electrochemical reduction of CO2, J. Mater. Chem. A, 2020, 8, 14966–14974 RSC.
- J. Choi, P. Wagner, R. Jalili, J. Kim, D. R. MacFarlane, G. G. Wallace and D. L. Officer, A porphyrin/graphene framework: a highly efficient and robust electrocatalyst for carbon dioxide reduction, Adv. Energy Mater., 2018, 8, 1801280 CrossRef.
- J. B. Lin, T. T. T. Nguyen, R. Vaidhyanathan, J. Burner, J. M. Taylor, H. Durekova, F. Akhtar, R. K. Mah, O. Ghaffari-Nik, S. Marx, N. Fylstra, S. S. Iremonger, K. W. Dawson, P. Sarkar, P. Hovington, A. Rajendran, T. K. Woo and G. K. H. Shimizu, A scalable metal-organic framework as a durable physisorbent for carbon dioxide capture, Science, 2021, 374, 1464–1469 CrossRef CAS PubMed.
- Y. Cui, B. Li, H. He, W. Zhou, B. Chen and G. Qian, Metal-organic frameworks as platforms for functional materials, Acc. Chem. Res., 2016, 49, 483–493 CrossRef CAS PubMed.
- F. Saraci, V. Quezada-Novoa, P. R. Donnarumma and A. J. Howarth, Rare-earth metal-organic frameworks: from structure to applications, Chem. Soc. Rev., 2020, 49, 7949–7977 RSC.
- H. Zeng, M. Xie, T. Wang, R. J. Wei, X. J. Xie, Y. Zhao, W. Lu and D. Li, Orthogonal-array dynamic molecular sieving of propylene/propane mixtures, Nature, 2021, 595, 542–548 CrossRef CAS PubMed.
- Y. Zhao, H. Zeng, X. W. Zhu, W. Lu and D. Li, Metal-organic frameworks as photoluminescent biosensing platforms: mechanisms and applications, Chem. Soc. Rev., 2021, 50, 4484–4513 RSC.
- S. Dutta and I. S. Lee, Metal-organic framework based catalytic nanoreactors: synthetic challenges and applications, Mater. Chem. Front., 2021, 5, 3986–4021 RSC.
- J. Du, F. Li and L. Sun, Metal-organic frameworks and their derivatives as electrocatalysts for the oxygen evolution reaction, Chem. Soc. Rev., 2021, 50, 2663–2695 RSC.
- T. Qiu, Z. Liang, W. Guo, H. Tabassum, S. Gao and R. Zou, Metal–organic framework-based materials for energy conversion and storage, ACS Energy Lett., 2020, 5, 520–532 CrossRef CAS.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Metal-organic frameworks in heterogeneous catalysis: recent progress, new trends, and future perspectives, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed.
- R. Hinogami, S. Yotsuhashi, M. Deguchi, Y. Zenitani, H. Hashiba and Y. Yamada, Electrochemical reduction of carbon dioxide using a copper rubeanate metal organic framework, ECS Electrochem. Lett., 2012, 1, H17–H19 CrossRef CAS.
- B. Liu, H. Shioyama, T. Akita and Q. Xu, Metal-organic framework as a template for porous carbon synthesis, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS PubMed.
- Y. Z. Chen, R. Zhang, L. Jiao and H. L. Jiang, Metal-organic framework-derived porous materials for catalysis, Coord. Chem. Rev., 2018, 362, 1–23 CrossRef CAS.
- J. L. Liu, D. D. Zhu, C. X. Guo, A. Vasileff and S. Z. Qiao, Design strategies toward advanced MOF-derived electrocatalysts for energy-conversion reactions, Adv. Energy Mater., 2017, 7, 1700518 CrossRef.
- H. B. Aiyappa, J. Masa, C. Andronescu, M. Muhler, R. A. Fischer and W. Schuhmann, MOFs for electrocatalysis: from serendipity to design strategies, Small Methods, 2019, 3, 1800415 CrossRef.
- F. Ismail, A. Abdellah, H.-J. Lee, V. Sudheeshkumar, W. Alnoush and D. C. Higgins, Impact of nickel content on the structure and electrochemical CO2 reduction performance of nickel-nitrogen-carbon catalysts derived from zeolitic imidazolate frameworks, ACS Appl. Energy Mater., 2022, 5, 430–439 CrossRef CAS.
- T. Zhang, X. Han, H. Liu, M. Biset-Peiró, J. Li, X. Zhang, P. Tang, B. Yang, L. Zheng, J. R. Morante and J. Arbiol, Site-specific axial oxygen coordinated FeN4 active sites for highly selective electroreduction of carbon dioxide, Adv. Funct. Mater., 2022, 32, 2111446 CrossRef CAS.
- R. Yun, R. Xu, C. Shi, B. Zhang, T. Li, L. He, T. Sheng and Z. Chen, Post-modification of MOF to fabricate single-atom dispersed hollow nanocages catalysts for enhancing CO2 conversion, Nano Res., 2023, 16, 8970–8976 CrossRef CAS.
- W. Xiao, M. Cheng, Y. Liu, J. Wang, G. Zhang, Z. Wei, L. Li, L. Du, G. Wang and H. Liu, Functional metal/carbon composites derived from metal–organic frameworks: insight into structures, properties, performances, and mechanisms, ACS Catal., 2023, 13, 1759–1790 CrossRef CAS.
- T. Xie, S. Li, L. Ma, J. Li, S. Pang, C. Hu, R. Zhao and S. Tang, Bimetallic MOF-derived composites with broad electromagnetic wave absorption and strong corrosion resistance, Carbon, 2023, 208, 33–42 CrossRef CAS.
- S. Ma, W. Han, W. Han, F. Dong and Z. Tang, Recent advances and future perspectives in MOF-derived single-atom catalysts and their application: a review, J. Mater. Chem. A, 2023, 11, 3315–3363 RSC.
- F. Marpaung, M. Kim, J. H. Khan, K. Konstantinov, Y. Yamauchi, M. S. A. Hossain, J. Na and J. Kim, Metal-organic framework (MOF)-derived nanoporous carbon materials, Chem. – Asian J., 2019, 14, 1331–1343 CrossRef CAS PubMed.
- B. N. Bhadra, L. K. Shrestha and K. Ariga, Porous carbon nanoarchitectonics for the environment: detection and adsorption, CrystEngComm, 2022, 24, 6804–6824 RSC.
- S. Xu, A. Dong, Y. Hu, Z. Yang, S. Huang and J. Qian, Multidimensional MOF-derived carbon nanomaterials for multifunctional applications, J. Mater. Chem. A, 2023, 11, 9721–9747 RSC.
- H. L. Jiang, B. Liu, Y. Q. Lan, K. Kuratani, T. Akita, H. Shioyama, F. Zong and Q. Xu, From metal-organic framework to nanoporous carbon: toward a very high surface area and hydrogen uptake, J. Am. Chem. Soc., 2011, 133, 11854–11857 CrossRef CAS PubMed.
- Q. Wang, W. Xia, W. Guo, L. An, D. Xia and R. Zou, Functional zeolitic-imidazolate-framework-templated porous carbon materials for CO2 capture and enhanced capacitors, Chem. – Asian J., 2013, 8, 1879–1885 CrossRef CAS PubMed.
- S. J. Yang, T. Kim, J. H. Im, Y. S. Kim, K. Lee, H. Jung and C. R. Park, MOF-derived hierarchically porous carbon with exceptional porosity and hydrogen storage capacity, Chem. Mater., 2012, 24, 464–470 CrossRef CAS.
- M. Hu, J. Reboul, S. Furukawa, N. L. Torad, Q. Ji, P. Srinivasu, K. Ariga, S. Kitagawa and Y. Yamauchi, Direct carbonization of Al-based porous coordination polymer for synthesis of nanoporous carbon, J. Am. Chem. Soc., 2012, 134, 2864–2867 CrossRef CAS PubMed.
- F. H. Bai, Y. D. Xia, B. L. Chen, H. Q. Su and Y. Q. Zhu, Preparation and carbon dioxide uptake capacity of N-doped porous carbon materials derived from direct carbonization of zeolitic imidazolate framework, Carbon, 2014, 79, 213–226 CrossRef CAS.
- W. Zhou, Y. Tang, X. Zhang, S. Zhang, H. Xue and H. Pang, MOF derived metal oxide composites and their applications in energy storage, Coord. Chem. Rev., 2023, 477, 214949 CrossRef CAS.
- S. Guo, Y. Zhao, H. Yuan, C. Wang, H. Jiang and G. J. Cheng, Ultrafast laser manufacture of stable, efficient ultrafine noble metal catalysts mediated with MOF derived high density defective metal oxides, Small, 2020, 16, e2000749 CrossRef PubMed.
- R. R. Salunkhe, J. Tang, Y. Kamachi, T. Nakato, J. H. Kim and Y. Yamauchi, Asymmetric supercapacitors using 3D nanoporous carbon and cobalt oxide electrodes synthesized from a single metal-organic framework, ACS Nano, 2015, 9, 6288–6296 CrossRef CAS PubMed.
- B. Liu, X. B. Zhang, H. Shioyama, T. Mukai, T. Sakai and Q. Xu, Converting cobalt oxide subunits in cobalt metal-organic framework into agglomerated Co3O4 nanoparticles as an electrode material for lithium ion battery, J. Power Sources, 2010, 195, 857–861 CrossRef CAS.
- C. Li, T. Q. Chen, W. J. Xu, X. B. Lou, L. K. Pan, Q. Chen and B. W. Hu, Mesoporous nanostructured Co3O4 derived from MOF template: a high-performance anode material for lithium-ion batteries, J. Mater. Chem. A, 2015, 3, 5585–5591 RSC.
- L. Hu, N. Yan, Q. Chen, P. Zhang, H. Zhong, X. Zheng, Y. Li and X. Hu, Fabrication based on the Kirkendall effect of Co3O4 porous nanocages with extraordinarily high capacity for lithium storage, Chem. – Eur. J., 2012, 18, 8971–8977 CrossRef CAS PubMed.
- L. Wang, J. Wan, Y. Zhao, N. Yang and D. Wang, Hollow multi-shelled structures of Co3O4 dodecahedron with unique crystal orientation for enhanced photocatalytic CO2 reduction, J. Am. Chem. Soc., 2019, 141, 2238–2241 CrossRef CAS PubMed.
- T. De Villenoisy, X. Zheng, V. Wong, S. S. Mofarah, H. Arandiyan, Y. Yamauchi, P. Koshy and C. C. Sorrell, Principles of design and synthesis of metal derivatives from MOFs, Adv. Mater., 2023, 35, e2210166 CrossRef PubMed.
- Z. Deng, S. Hu, J. Ji, S. Wu, H. Xie, M. Xing and J. Zhang, Deep insight of the influence of Cu valence states in co-catalyst on CO2 photoreduction, Appl. Catal., B, 2022, 316, 121621 CrossRef CAS.
- L. Xu, J. Feng, L. Wu, X. Song, X. Tan, L. Zhang, X. Ma, S. Jia, J. Du, A. Chen, X. Sun and B. Han, Identifying the optimal oxidation state of Cu for electrocatalytic reduction of CO2 to C2+ products, Green Chem., 2023, 25, 1326–1331 RSC.
- Z.-Z. Wu, F.-Y. Gao and M.-R. Gao, Regulating the oxidation state of nanomaterials for electrocatalytic CO2 reduction, Energy Environ. Sci., 2021, 14, 1121–1139 RSC.
- X. Xu, R. Cao, S. Jeong and J. Cho, Spindle-like mesoporous α-Fe2O3 anode material prepared from MOF template for high-rate lithium batteries, Nano Lett., 2012, 12, 4988–4991 CrossRef CAS PubMed.
- W. Cho, S. Park and M. Oh, Coordination polymer nanorods of Fe-MIL-88B and their utilization for selective preparation of hematite and magnetite nanorods, Chem. Commun., 2011, 47, 4138–4140 RSC.
- L. Hou, X. Jiang, Y. Jiang, T. Jiao, R. Cui, S. Deng, J. Gao, Y. Guo and F. Gao, Facile preparation of porous rod-like CuxCo3−xO4/C composites via bimetal-organic framework derivation as superior anodes for lithium-ion batteries, ACS Omega, 2019, 4, 7565–7573 CrossRef CAS PubMed.
- X. Wang, J. Ying, Y. Mai, J. Zhang, J. Chen, M. Wen and L. Yu, MOF-derived metal oxide composite Mn2Co1Ox/CN for efficient formaldehyde oxidation at low temperature, Catal. Sci. Technol., 2019, 9, 5845–5854 RSC.
- C. Yu, Y. Wang, J. Cui, D. Yu, X. Zhang, X. Shu, J. Zhang, Y. Zhang, R. Vajtai, P. M. Ajayan and Y. Wu, MOF-74 derived porous hybrid metal oxide hollow nanowires for high-performance electrochemical energy storage, J. Mater. Chem. A, 2018, 6, 8396–8404 RSC.
- R. Wu, X. Qian, K. Zhou, J. Wei, J. Lou and P. M. Ajayan, Porous spinel ZnxCo3−xO4 hollow polyhedra templated for high-rate lithium-ion batteries, ACS Nano, 2014, 8, 6297–6303 CrossRef CAS PubMed.
- J. J. Ma, H. J. Wang, X. Yang, Y. Q. Chai and R. Yuan, Porous carbon-coated CuCo2O4 concave polyhedrons derived from metal-organic frameworks as anodes for lithium-ion batteries, J. Mater. Chem. A, 2015, 3, 12038–12043 RSC.
- Z. Jia, Y. Ma, L. Yang, C. Guo, N. Zhou, M. Wang, L. He and Z. Zhang, NiCo2O4 spinel embedded with carbon nanotubes derived from bimetallic NiCo metal-organic framework for the ultrasensitive detection of human immune deficiency virus-1 gene, Biosens. Bioelectron., 2019, 133, 55–63 CrossRef CAS PubMed.
- G. Xu, Q. Liu and H. Yan, Recent advances of single-atom catalysts for electro-catalysis, Chem. Res. Chin. Univ., 2022, 38, 1146–1150 CrossRef CAS.
- M. Ahmad, J. Chen, J. Liu, Y. Zhang, Z. Song, S. Afzal, W. Raza, L. Zeb, A. Mehmood, A. Hussain, J. Zhang, X. Z. Fu and J. L. Luo, Metal-organic framework-based single-atom electro-/photocatalysts: synthesis, energy applications, and opportunities, Carbon Energy, 2023, e382 CrossRef.
- M. Humayun, M. Israr, A. Khan and M. Bououdina, State-of-the-art single-atom catalysts in electrocatalysis: from fundamentals to applications, Nano Energy, 2023, 113, 108570 CrossRef CAS.
- Q. Yang, Y. Jiang, H. Zhuo, E. M. Mitchell and Q. Yu, Recent progress of metal single-atom catalysts for energy applications, Nano Energy, 2023, 111, 108404 CrossRef CAS.
- S. Ma, G. A. Goenaga, A. V. Call and D. J. Liu, Cobalt imidazolate framework as precursor for oxygen reduction reaction electrocatalysts, Chem. – Eur. J., 2011, 17, 2063–2067 CrossRef CAS PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Single cobalt atoms with precise N-coordination as superior oxygen reduction reaction catalysts, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- Q. L. Zhu and Q. Xu, Metal-organic framework composites, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC.
- S. Li and F. Huo, Metal-organic framework composites: from fundamentals to applications, Nanoscale, 2015, 7, 7482–7501 RSC.
- Y. Li, M. Karimi, Y.-N. Gong, N. Dai, V. Safarifard and H.-L. Jiang, Integration of metal-organic frameworks and covalent organic frameworks: design, synthesis, and applications, Matter, 2021, 4, 2230–2265 CrossRef CAS.
- L. Jiao, J. Wang and H.-L. Jiang, Microenvironment modulation in metal–Organic framework-based catalysis, Acc. Mater. Res., 2021, 2, 327–339 CrossRef CAS.
- S. S. A. Shah, T. Najam, M. Wen, S.-Q. Zang, A. Waseem and H.-L. Jiang, Metal–organic framework-based electrocatalysts for CO2 reduction, Small Struct., 2021, 3, 2100090 CrossRef.
- L. Radhakrishnan, J. Reboul, S. Furukawa, P. Srinivasu, S. Kitagawa and Y. Yamauchi, Preparation of microporous carbon fibers through carbonization of Al-based porous coordination polymer (Al-PCP) with furfuryl alcohol, Chem. Mater., 2011, 23, 1225–1231 CrossRef CAS.
- J. A. Hu, H. L. Wang, Q. M. Gao and H. L. Guo, Porous carbons prepared by using metal-organic framework as the precursor for supercapacitors, Carbon, 2010, 48, 3599–3606 CrossRef CAS.
- X. Sun, T. Wu, Z. Yan, W.-J. Chen, X.-B. Lian, Q. Xia, S. Chen and Q.-H. Wu, Novel MOF-5 derived porous carbons as excellent adsorption materials for n-hexane, J. Solid State Chem., 2019, 271, 354–360 CrossRef CAS.
- Z. Zhang, H. Wang, Y. Li, M. Xie, C. Li, H. Lu, Y. Peng and Z. Shi, Confined pyrolysis synthesis of well-dispersed cobalt copper bimetallic three-dimensional N-doped carbon framework as efficient water splitting electrocatalyst, Chem. Res. Chin. Univ., 2022, 38, 750–757 CrossRef CAS.
- J. Wang, G. Wang, J. Zhang, Y. Wang, H. Wu, X. Zheng, J. Ding, X. Han, Y. Deng and W. Hu, Inversely tuning the CO2 electroreduction and hydrogen evolution activity on metal oxide via heteroatom doping, Angew. Chem., Int. Ed., 2021, 60, 7602–7606 CrossRef CAS PubMed.
- F. Yang, H. Yu, X. Mao, Q. Meng, S. Chen, Q. Deng, Z. Zeng, J. Wang and S. Deng, Boosting electrochemical CO2 reduction on ternary heteroatoms-doped porous carbon, Chem. Eng. J., 2021, 425, 131661 CrossRef CAS.
- T. F. Liu, S. Ali, Z. Lian, B. Li and D. S. Su, CO2 electoreduction reaction on heteroatom-doped carbon cathode materials, J. Mater. Chem. A, 2017, 5, 21596–21603 RSC.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, Ionic exchange of metal-organic frameworks to access single nickel sites for efficient electroreduction of CO2, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- E. Zhang, T. Wang, K. Yu, J. Liu, W. Chen, A. Li, H. Rong, R. Lin, S. Ji, X. Zheng, Y. Wang, L. Zheng, C. Chen, D. Wang, J. Zhang and Y. Li, Bismuth single atoms resulting from transformation of metal-organic frameworks and their use as electrocatalysts for CO2 reduction, J. Am. Chem. Soc., 2019, 141, 16569–16573 CrossRef CAS PubMed.
- Y. H. He, S. Hwang, D. A. Cullen, M. A. Uddin, L. Langhorst, B. Y. Li, S. Karakalos, A. J. Kropf, E. C. Wegener, J. Sokolowski, M. J. Chen, D. Myers, D. Su, K. L. More, G. F. Wang, S. Litster and G. Wu, Highly active atomically dispersed CoN4 fuel cell cathode catalysts derived from surfactant-assisted MOFs: carbon-shell confinement strategy, Energy Environ. Sci., 2019, 12, 250–260 RSC.
- Z. M. Zhao, J. W. Ding, R. M. Zhu and H. Pang, The synthesis and electrochemical applications of core-shell MOFs and their derivatives, J. Mater. Chem. A, 2019, 7, 15519–15540 RSC.
- S. Liu, Z. Wang, S. Zhou, F. Yu, M. Yu, C. Y. Chiang, W. Zhou, J. Zhao and J. Qiu, Metal-organic-framework-derived hybrid carbon nanocages as a bifunctional electrocatalyst for oxygen reduction and evolution, Adv. Mater., 2017, 29, 1700874 CrossRef PubMed.
- S. Yang, L. Peng, P. Huang, X. Wang, Y. Sun, C. Cao and W. Song, Nitrogen, phosphorus, and sulfur Co-doped hollow carbon shell as superior metal-free catalyst for selective oxidation of aromatic alkanes, Angew. Chem., Int. Ed., 2016, 55, 4016–4020 CrossRef CAS PubMed.
- X. Fang, L. Jiao, S. H. Yu and H. L. Jiang, Metal-organic framework-derived FeCo-N-doped hollow porous carbon nanocubes for electrocatalysis in acidic and alkaline media, ChemSusChem, 2017, 10, 3019–3024 CrossRef CAS PubMed.
- Y. Zhu, Z. Y. Zhang, Z. Lei, Y. Y. Tan, W. Wu, S. C. Mu and N. C. Cheng, Defect-enriched hollow porous Co-N-doped carbon for oxygen reduction reaction and Zn-Air batteries, Carbon, 2020, 167, 188–195 CrossRef CAS.
- Y. Zhu, Z. Y. Zhang, W. Q. Li, Z. Lei, N. C. Cheng, Y. Y. Tan, S. C. Mu and X. L. Sun, Highly exposed active sites of defect-enriched derived MOFs for enhanced oxygen reduction reaction, ACS Sustainable Chem. Eng., 2019, 7, 17855–17862 CrossRef CAS.
- X. Xie, L. Peng, H. Yang, G. I. N. Waterhouse, L. Shang and T. Zhang, MIL-101-derived mesoporous carbon supporting highly exposed Fe single-atom sites as efficient oxygen reduction reaction catalysts, Adv. Mater., 2021, 33, e2101038 CrossRef PubMed.
- S. Mukherjee, S. J. Hou, S. A. Watzele, B. Garlyyev, W. J. Li, A. S. Bandarenka and R. A. Fischer, Avoiding pyrolysis and calcination: advances in the benign routes leading to MOF-derived electrocatalysts, ChemElectroChem, 2022, 9, e202101476 CrossRef CAS.
- Y. H. Song, J. He, H. L. Wu, X. Li, J. Yu, Y. Y. Zhang and L. Wang, Preparation of porous hollow CoOx nanocubes via chemical etching prussian blue analogue for glucose sensing, Electrochim. Acta, 2015, 182, 165–172 CrossRef CAS.
- W. W. Yuan, J. X. Wu, X. D. Zhang, S. Z. Hou, M. Xu and Z. Y. Gu, In situ transformation of bismuth metal-organic frameworks for efficient selective electroreduction of CO2 to formate, J. Mater. Chem. A, 2020, 8, 24486–24492 RSC.
- P. Lamagni, M. Miola, J. Catalano, M. S. Hvid, M. A. H. Mamakhel, M. Christensen, M. R. Madsen, H. S. Jeppesen, X. M. Hu, K. Daasbjerg, T. Skrydstrup and N. Lock, Restructuring metal–organic frameworks to nanoscale bismuth electrocatalysts for highly active and selective CO2 reduction to formate, Adv. Funct. Mater., 2020, 30, 1910408 CrossRef CAS.
- Y. Han, S. Zhu, S. Xu, X. Niu, Z. Xu, R. Zhao and Q. Wang, Understanding structure-activity relationship on metal-organic-framework-derived catalyst for CO2 electroreduction to C2 products, ChemElectroChem, 2021, 8, 3174–3180 CrossRef CAS.
- J. J. Lv, R. Yin, L. Zhou, J. Li, R. Kikas, T. Xu, Z. J. Wang, H. Jin, X. Wang and S. Wang, Microenvironment engineering for the electrocatalytic CO2 reduction reaction, Angew. Chem., Int. Ed., 2022, 61, e202207252 CrossRef CAS PubMed.
- X. She, Y. Wang, H. Xu, S. Chi Edman, S. Tsang and S. Ping Lau, Challenges and opportunities in electrocatalytic CO2 reduction to chemicals and fuels, Angew. Chem., Int. Ed., 2022, 61, e202211396 CrossRef CAS PubMed.
- X. Li, S. Wang, L. Li, Y. Sun and Y. Xie, Progress and perspective for in situ studies of CO2 reduction, J. Am. Chem. Soc., 2020, 142, 9567–9581 CAS.
- K. Fernández-Caso, G. Díaz-Sainz, M. Alvarez-Guerra and A. Irabien, Electroreduction of CO2: advances in the continuous production of formic acid and formate, ACS Energy Lett., 2023, 8, 1992–2024 CrossRef.
- B. Jia, Z. Chen, C. Li, Z. Li, X. Zhou, T. Wang, W. Yang, L. Sun and B. Zhang, Indium cyanamide for industrial-grade CO2 electroreduction to formic acid, J. Am. Chem. Soc., 2023, 145, 14101–14111 CrossRef CAS PubMed.
- L. P. Chi, Z. Z. Niu, X. L. Zhang, P. P. Yang, J. Liao, F. Y. Gao, Z. Z. Wu, K. B. Tang and M. R. Gao, Stabilizing indium sulfide for CO2 electroreduction to formate at high rate by zinc incorporation, Nat. Commun., 2021, 12, 5835 CrossRef CAS PubMed.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Advances and challenges for the electrochemical reduction of CO2 to CO: from fundamentals to industrialization, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS PubMed.
- L. L. Ling, L. Jiao, X. Liu, Y. Dong, W. Yang, H. Zhang, B. Ye, J. Chen and H. L. Jiang, Potassium-assisted fabrication of intrinsic defects in porous carbons for electrocatalytic CO2 reduction, Adv. Mater., 2022, 34, e2205933 CrossRef PubMed.
- M. Fan, J. Cui, J. Wu, R. Vajtai, D. Sun and P. M. Ajayan, Improving the catalytic activity of carbon-supported single atom catalysts by polynary metal or heteroatom doping, Small, 2020, 16, e1906782 CrossRef PubMed.
- H. Wang, Y. Shao, S. Mei, Y. Lu, M. Zhang, J. K. Sun, K. Matyjaszewski, M. Antonietti and J. Yuan, Polymer-derived heteroatom-doped porous carbon materials, Chem. Rev., 2020, 120, 9363–9419 CrossRef CAS PubMed.
- R. Wang, X. Sun, S. Ould-Chikh, D. Osadchii, F. Bai, F. Kapteijn and J. Gascon, Metal-organic-framework-mediated nitrogen-doped carbon for CO2 electrochemical reduction, ACS Appl. Mater. Interfaces, 2018, 10, 14751–14758 CrossRef CAS PubMed.
- L. Ye, Y. Ying, D. Sun, Z. Zhang, L. Fei, Z. Wen, J. Qiao and H. Huang, Highly efficient porous carbon electrocatalyst with controllable N-species content for selective CO2 reduction, Angew. Chem., Int. Ed., 2020, 59, 3244–3251 CrossRef CAS PubMed.
- D. Wang, Y. Zhu, W. T. Yu, Z. Q. He, F. L. Dong, Y. Shen, T. Zeng, X. H. Lu, J. Ma, L. Z. Wang and S. Song, Ag-MOF-derived 3D Ag dendrites used for the efficient electrocatalytic reduction of CO2 to CO, Electrochim. Acta, 2022, 403, 139652 CrossRef CAS.
- Y. Zhang, T. Sun, P. Zhang, K. Liu, F. Li and L. Xu, Synthesizing MOF-derived Ni-N-C catalyst via surfactant modified strategy for efficient electrocatalytic CO2 to CO, J. Colloid Interface Sci., 2023, 631, 96–101 CrossRef CAS PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Regulation of coordination number over single Co sites: triggering the efficient electroreduction of CO2, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed.
- D. Zhao, K. Yu, P. Song, W. Feng, B. Hu, W.-C. Cheong, Z. Zhuang, S. Liu, K. Sun, J. Zhang and C. Chen, Atomic-level engineering Fe1N2O2 interfacial structure derived from oxygen-abundant metal–organic frameworks to promote electrochemical CO2 reduction, Energy Environ. Sci., 2022, 15, 3795–3804 RSC.
- J. Fu, K. Liu, H. Li, J. Hu and M. Liu, Bimetallic atomic site catalysts for CO2 reduction reactions: a review, Environ. Chem. Lett., 2021, 20, 243–262 CrossRef.
- L. Jiao, J. Zhu, Y. Zhang, W. Yang, S. Zhou, A. Li, C. Xie, X. Zheng, W. Zhou, S. H. Yu and H. L. Jiang, Non-bonding interaction of neighboring Fe and Ni single-atom pairs on MOF-derived N-doped carbon for enhanced CO2 electroreduction, J. Am. Chem. Soc., 2021, 143, 19417–19424 CrossRef CAS PubMed.
- Y. Yang, J.-j Fu, T. Tang, S. Niu, L.-B. Zhang, J.-n Zhang and J.-S. Hu, Regulating surface In–O in In@InO core-shell nanoparticles for boosting electrocatalytic CO2 reduction to formate, Chin. J. Catal., 2022, 43, 1674–1679 CrossRef CAS.
- Y. Guan, Y. Liu, J. Yi and J. Zhang, Zeolitic imidazolate framework-derived composites with SnO2 and ZnO phase components for electrocatalytic carbon dioxide reduction, Dalton Trans., 2022, 51, 7274–7283 RSC.
- Q. Wang, X. Yang, H. Zang, F. Chen, C. Wang, N. Yu and B. Geng, Metal-organic framework-derived BiIn bimetallic oxide nanoparticles embedded in carbon networks for efficient electrochemical reduction of CO2 to Formate, Inorg. Chem., 2022, 61, 12003–12011 CrossRef CAS PubMed.
- S. Liu, Y. Fan, Y. Wang, S. Jin, M. Hou, W. Zeng, K. Li, T. Jiang, L. Qin, Z. Yan, Z. Tao, X. Zheng, C. Shen, Z. Liu, T. Ahmad, K. Zhang and W. Chen, Surface-oxygen-rich Bi@C nanoparticles for high-efficiency electroreduction of CO2 to formate, Nano Lett., 2022, 22, 9107–9114 CrossRef CAS PubMed.
- Y. Guan, X. Zhang, Y. Zhang, T. N. V. Karsili, M. Fan, Y. Liu, B. Marchetti and X. D. Zhou, Achieving high selectivity towards electro-conversion of CO2 using In-doped Bi derived from metal-organic frameworks, J. Colloid Interface Sci., 2022, 612, 235–245 CrossRef CAS PubMed.
- J. Zhai, Q. Kang, Q. Liu, D. Lai, Q. Lu and F. Gao, In-situ generation of In2O3 nanoparticles inside In[Co(CN)6] quasi-metal-organic-framework nanocubes for efficient electroreduction of CO2 to formate, J. Colloid Interface Sci., 2022, 608, 1942–1950 CrossRef CAS PubMed.
- C. Qiu, K. Qian, J. Yu, M. Sun, S. Cao, J. Gao, R. Yu, L. Fang, Y. Yao, X. Lu, T. Li, B. Huang and S. Yang, MOF-transformed In2O3−x@C nanocorn electrocatalyst for efficient CO2 reduction to HCOOH, Nano-Micro Lett., 2022, 14, 167 CrossRef CAS PubMed.
- S. Komatsu, T. Yanagihara, Y. Hiraga, M. Tanaka and A. Kunugi, Electrochemical reduction of CO2 at Sb and Bi electrodes in KHCO3 solution, Denki Kagaku oyobi Kogyo Butsuri Kagaku, 1995, 63, 217–224 CrossRef CAS.
- N. H. Li, P. Yan, Y. H. Tang, J. H. Wang, X. Y. Yu and H. B. Wu, In-situ formation of ligand-stabilized bismuth nanosheets for efficient CO2 conversion, Appl. Catal., B, 2021, 297, 120481 CrossRef CAS.
- J. Yang, X. L. Wang, Y. T. Qu, X. Wang, H. Huo, Q. K. Fan, J. Wang, L. M. Yang and Y. E. Wu, Bi-based metal-organic framework derived leafy bismuth nanosheets for carbon dioxide electroreduction, Adv. Energy Mater., 2020, 10, 2001709 CrossRef CAS.
- Q. Wang, X. Yang, H. Zang, C. Liu, J. Wang, N. Yu, L. Kuai, Q. Qin and B. Geng, InBi bimetallic sites for efficient electrochemical reduction of CO2 to HCOOH, Small, 2023, e2303172 CrossRef PubMed.
- X. Guo, S.-M. Xu, H. Zhou, Y. Ren, R. Ge, M. Xu, L. Zheng, X. Kong, M. Shao, Z. Li and H. Duan, Engineering hydrogen generation sites to promote electrocatalytic CO2 reduction to formate, ACS Catal., 2022, 12, 10551–10559 CrossRef CAS.
- K. L. Yao, H. B. Wang, X. T. Yang, Y. Huang, C. D. Kou, T. Jing, S. H. Chen, Z. Y. Wang, Y. C. Liu and H. Y. Liang, Metal-organic framework derived dual-metal sites for electroreduction of carbon dioxide to HCOOH, Appl. Catal., B, 2022, 311, 121377 CrossRef CAS.
- Z. X. Yang, H. Z. Wang, X. Fei, W. H. Wang, Y. Z. Zhao, X. S. Wang, X. J. Tan, Q. S. Zhao, H. P. Wang, J. X. Zhu, L. Zhou, H. Ning and M. B. Wu, MOF derived bimetallic CuBi catalysts with ultra-wide potential window for high-efficient electrochemical reduction of CO2 to formate, Appl. Catal., B, 2021, 298, 120571 CrossRef CAS.
- J. Cai, Q. Zhao, W. Y. Hsu, C. Choi, Y. Liu, J. M. P. Martirez, C. Chen, J. Huang, E. A. Carter and Y. Huang, Highly selective electrochemical reduction of CO2 into methane on nanotwinned Cu, J. Am. Chem. Soc., 2023, 145, 9136–9143 CrossRef CAS PubMed.
- C. Hu, Z. Jiang, Q. Wu, S. Cao, Q. Li, C. Chen, L. Yuan, Y. Wang, W. Yang, J. Yang, J. Peng, W. Shi, M. Zhai, M. Mostafavi and J. Ma, Selective CO2 reduction to CH3OH over atomic dual-metal sites embedded in a metal-organic framework with high-energy radiation, Nat. Commun., 2023, 14, 4767 CrossRef CAS PubMed.
- X. Wang, P. Ou, J. Wicks, Y. Xie, Y. Wang, J. Li, J. Tam, D. Ren, J. Y. Howe, Z. Wang, A. Ozden, Y. Z. Finfrock, Y. Xu, Y. Li, A. S. Rasouli, K. Bertens, A. H. Ip, M. Graetzel, D. Sinton and E. H. Sargent, Gold-in-copper at low *CO coverage enables efficient electromethanation of CO2, Nat. Commun., 2021, 12, 3387 CrossRef CAS PubMed.
- H. Bai, T. Cheng, S. Li, Z. Zhou, H. Yang, J. Li, M. Xie, J. Ye, Y. Ji, Y. Li, Z. Zhou, S. Sun, B. Zhang and H. Peng, Controllable CO adsorption determines ethylene and methane productions from CO2 electroreduction, Sci. Bull., 2021, 66, 62–68 CrossRef CAS PubMed.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, Scalable production of efficient single-atom copper decorated carbon membranes for CO2 electroreduction to methanol, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed.
- M. K. Kim, H. J. Kim, H. Lim, Y. Kwon and H. M. Jeong, Metal-organic framework-mediated strategy for enhanced methane production on copper nanoparticles in electrochemical CO2 reduction, Electrochim. Acta, 2019, 306, 28–34 CrossRef CAS.
- A. X. Guan, Z. Chen, Y. L. Quan, C. Peng, Z. Q. Wang, S. K. Sham, C. Yang, Y. L. Ji, L. P. Qian, X. Xu and G. F. Zheng, Boosting CO2 electroreduction to CH4 via tuning neighboring single-copper sites, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS.
- J. D. Yi, R. Xie, Z. L. Xie, G. L. Chai, T. F. Liu, R. P. Chen, Y. B. Huang and R. Cao, Highly selective CO2 electroreduction to CH4 by in situ generated Cu2O single-type sites on a conductive MOF: stabilizing key intermediates with hydrogen bonding, Angew. Chem., Int. Ed., 2020, 59, 23641–23648 CrossRef CAS PubMed.
- D. H. Nam, O. S. Bushuyev, J. Li, P. De Luna, A. Seifitokaldani, C. T. Dinh, F. P. Garcia de Arquer, Y. Wang, Z. Liang, A. H. Proppe, C. S. Tan, P. Todorovic, O. Shekhah, C. M. Gabardo, J. W. Jo, J. Choi, M. J. Choi, S. W. Baek, J. Kim, D. Sinton, S. O. Kelley, M. Eddaoudi and E. H. Sargent, Metal-organic frameworks mediate Cu coordination for selective CO2 electroreduction, J. Am. Chem. Soc., 2018, 140, 11378–11386 CrossRef CAS PubMed.
- F. Yang, P. L. Deng, Q. Y. Wang, J. X. Zhu, Y. Yan, L. Zhou, K. Qi, H. F. Liu, H. S. Park and B. Y. Xia, Metal-organic framework-derived cupric oxide polycrystalline nanowires for selective carbon dioxide electroreduction to C2 valuables, J. Mater. Chem. A, 2020, 8, 12418–12423 RSC.
- Y. S. Cheng, X. P. Chu, M. Ling, N. Li, K. L. Wu, F. H. Wu, H. Li, G. Z. Yuan and X. W. Wei, An MOF-derived copper@nitrogen-doped carbon composite: the synergistic effects of N-types and copper on selective CO2 electroreduction, Catal. Sci. Technol., 2019, 9, 5668–5675 RSC.
- H. Sun, L. Chen, L. Xiong, K. Feng, Y. Chen, X. Zhang, X. Yuan, B. Yang, Z. Deng, Y. Liu, M. H. Rummeli, J. Zhong, Y. Jiao and Y. Peng, Promoting ethylene production over a wide potential window on Cu crystallites induced and stabilized via current shock and charge delocalization, Nat. Commun., 2021, 12, 6823 CrossRef CAS PubMed.
- Z. Li, Y. Yang, Z. Yin, X. Wei, H. Peng, K. Lyu, F. Wei, L. Xiao, G. Wang, H. D. Abruña, J. Lu and L. Zhuang, Interface-enhanced catalytic selectivity on the C2 products of CO2 electroreduction, ACS Catal., 2021, 11, 2473–2482 CrossRef CAS.
- Y. Zang, T. Liu, P. Wei, H. Li, Q. Wang, G. Wang and X. Bao, Selective CO2 electroreduction to ethanol over a carbon-coated CuOx catalyst, Angew. Chem., Int. Ed., 2022, 61, e202209629 CrossRef CAS PubMed.
- A. B. He, Y. Yang, Q. Zhang, M. Yang, Q. Zou, J. Du, C. Y. Tao and Z. H. Liu, The enhanced local CO concentration for efficient CO2 electrolysis towards C2 products on tandem active sites, Chem. Eng. J., 2022, 450, 138009 CrossRef CAS.
- B. Kim, Y. C. Tan, Y. Ryu, K. Jang, H. G. Abbas, T. Kang, H. Choi, K.-S. Lee, S. Park, W. Kim, P.-P. Choi, S. Ringe and J. Oh, Trace-level cobalt dopants enhance CO2 electroreduction and ethylene formation on copper, ACS Energy Lett., 2023, 8, 3356–3364 CrossRef CAS.
- A. V. Rayer, E. Reid, A. Kataria, I. Luz, S. J. Thompson, M. Lail, J. Zhou and M. Soukri, Electrochemical carbon dioxide reduction to isopropanol using novel carbonized copper metal organic framework derived electrodes, J. CO2 Util., 2020, 39, 101159 CrossRef CAS.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Selective electroreduction of CO2 to acetone by single copper atoms anchored on N-doped porous carbon, Nat. Commun., 2020, 11, 2455 CrossRef CAS PubMed.
- N. Sikdar, J. R. C. Junqueira, D. Ohl, S. Dieckhofer, T. Quast, M. Braun, H. B. Aiyappa, S. Seisel, C. Andronescu and W. Schuhmann, Redox replacement of silver on MOF-derived Cu/C nanoparticles on gas diffusion electrodes for electrocatalytic CO2 reduction, Chem. – Eur. J., 2022, 28, e202104249 CrossRef CAS PubMed.
- N. Sikdar, J. R. C. Junqueira, S. Dieckhofer, T. Quast, M. Braun, Y. Song, H. B. Aiyappa, S. Seisel, J. Weidner, D. Ohl, C. Andronescu and W. Schuhmann, A metal-organic framework derived CuxOyCz catalyst for electrochemical CO2 reduction and impact of local pH change, Angew. Chem., Int. Ed., 2021, 60, 23427–23434 CrossRef CAS PubMed.
- Y. Hori, K. Kikuchi and S. Suzuki, Production of CO and CH4 in electrochemical reduction of CO2 at metal electrodes in aqueous hydrogencarbonate solution, Chem. Lett., 1985, 1695–1698 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- Y. Hori, A. Murata and R. Takahashi, Formation of hydrocarbons in the electrochemical reduction of carbon dioxide at a copper electrode in aqueous solution, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- F. Chang, M. Xiao, R. Miao, Y. Liu, M. Ren, Z. Jia, D. Han, Y. Yuan, Z. Bai and L. Yang, Copper-based catalysts for electrochemical carbon dioxide reduction to multicarbon products, Electrochem. Energy Rev., 2022, 5, 4 CrossRef CAS.
- A. R. Woldu, Z. Huang, P. Zhao, L. Hu and D. Astruc, Electrochemical CO2 reduction (CO2RR) to multi-carbon products over copper-based catalysts, Coord. Chem. Rev., 2022, 454, 214340 CrossRef CAS.
- T. Akter, H. Pan and C. J. Barile, Tandem electrocatalytic CO2 reduction inside a membrane with enhanced selectivity for ethylene, J. Phys. Chem. C, 2022, 126, 10045–10052 CrossRef CAS.
- Y. Zhu, X. Cui, H. Liu, Z. Guo, Y. Dang, Z. Fan, Z. Zhang and W. Hu, Tandem catalysis in electrochemical CO2 reduction reaction, Nano Res., 2021, 14, 4471–4486 CrossRef CAS.
- T. Zhang, Z. Li, A. K. Ummireddi and J. Wu, Navigating CO utilization in tandem electrocatalysis of CO2, Trends Chem., 2023, 5, 252–266 CrossRef CAS.
- Q. Zhu, Y. Hu, H. Chen, C. Meng, Y. Shang, C. Hao, S. Wei, Z. Wang, X. Lu and S. Liu, Graphdiyne supported Ag-Cu tandem catalytic scheme for electrocatalytic reduction of CO2 to C2+ products, Nanoscale, 2023, 15, 2106–2113 RSC.
- B. Zhang, B. Zhang, Y. Jiang, T. Ma, H. Pan and W. Sun, Single-atom electrocatalysts for multi-electron reduction of CO2, Small, 2021, 17, e2101443 CrossRef PubMed.
- L. Xiao, Z. Wang and J. Guan, 2D MOFs and their derivatives for electrocatalytic applications: recent advances and new challenges, Coord. Chem. Rev., 2022, 472, 214777 CrossRef CAS.
- X. Li and Q.-L. Zhu, MOF-based materials for photo- and electrocatalytic CO2 reduction, EnergyChem, 2020, 2, 100033 CrossRef.
- C. Li, Y. Ji, Y. Wang, C. Liu, Z. Chen, J. Tang, Y. Hong, X. Li, T. Zheng, Q. Jiang and C. Xia, Applications of metal-organic frameworks and their derivatives in electrochemical CO2 reduction, Nano-Micro Lett., 2023, 15, 113 CrossRef CAS PubMed.
- Y. Liu, S. Chen, X. Quan and H. Yu, Efficient electrochemical reduction of carbon dioxide to acetate on nitrogen-doped nanodiamond, J. Am. Chem. Soc., 2015, 137, 11631–11636 CrossRef CAS PubMed.
- R. De, S. Gonglach, S. Paul, M. Haas, S. S. Sreejith, P. Gerschel, U. P. Apfel, T. H. Vuong, J. Rabeah, S. Roy and W. Schofberger, Electrocatalytic reduction of CO2 to acetic acid by a molecular manganese corrole complex, Angew. Chem., Int. Ed., 2020, 59, 10527–10534 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2023 |