
Tunable functional groups on MXene regulating the catalytic property of anchored cobalt phthalocyanine for electrochemical CO2 reduction†
Zhixiang
Zhou
,
Fengshou
Yu
 ,
Yang
You
,
Jiayu
Zhan
and
Lu-Hua
Zhang
*
,
Yang
You
,
Jiayu
Zhan
and
Lu-Hua
Zhang
*
Tianjin Key Laboratory of Chemical Process Safety, School of Chemical Engineering and Technology, Hebei University of Technology, Tianjin 300130, P. R. China. E-mail: luhuazhang@hebut.edu.cn
First published on 22nd July 2023
Abstract
The regulation of the intermediate adsorption strength is the key for boosting the catalytic performance, while it is still a challenge to achieve this in a facile manner. Herein, CoPc complex was anchored on an MXene support bearing different terminal groups (–F or –OH) to systematically investigate the effect of the terminal groups of the MXene support on the performance of the electrochemical CO2 reduction reaction (ECRR). By forming Co–O axial coordination, the anchoring of CoPc was facilitated and the electronic states of Co were tuned. As a result, CoPc/MXene–OH shows an impressive faradaic efficiency for CO formation (FECO) of 92.4%, higher than that of CoPc/MXene–F (84.0%) under the same conditions. Mechanistic explorations show that the excellent performance is attributed to the electron-donating property of –OH enriching the electron density of Co, which optimizes the binding strength for intermediates and therefore boosts the ECRR performance. The simple synthesis method opens up a new avenue for regulating the electronic states of active sites and further the intermediate adsorption strength for efficient electrocatalyst design.
1. Introduction
The accelerated consumption of fossil fuel results in the increase of the accumulation of greenhouse gas, i.e. CO2, in the atmosphere, which causes some environmental problems.1–3 Powered by intermittent renewable electricity, the electrocatalytic carbon dioxide reduction reaction (ECRR) converts CO2 into fuel and chemical feedstock, enabling simultaneous conversion of CO2 and utilization of renewable energy.4,5 However, the fundamental challenges remain in the ECRR process, such as low faradaic efficiency (FE) due to the competitive hydrogen evolution reaction (HER).6,7 The key to solving the problems is the regulation of the adsorption of intermediates such as *COOH and *H at the active sites.8–10Molecular catalysts such as a metal phthalocyanine (M-Pc) complex can effectively convert CO2 into CO, and the tunable coordination environment of the metal site enables an effective modulation of electronic states and further the catalytic performance.11–13 The modification of peripheral functional groups of molecules with various substituents is a promising strategy to facilitate the electrocatalytic performance of molecular complexes, such as the decoration with methoxy and amino functional groups.14,15 The molecular catalyst is then mounted on a two-dimensional (2D) material by π–π accumulation for electrocatalytic testing.16–18 However, complex organic synthesis was usually involved for this purpose. Additionally, leaching problems and poor stability can be induced by the weak linking between M-Pc and the 2D materials.19 Therefore, the development of a more effective regulatory strategy for electronic regulation boosting the ECRR performance is significantly desired.
The central metal of M-Pc compounds has two supplemental coordination sites besides the four N coordination sites.20–22 By introducing an axial ligand, M-Pc can be immobilized on the supports in an axial coordination manner in addition to π–π stacking.23–25 For instance, Song et al. realized a twofold intrinsic activity enhancement of the Fe–N–C catalyst by anchoring FePc on Ti3C2Tx MXene as an oxygen reduction reaction (ORR) catalyst,26 since Ti3C2Tx MXene has abundant surface terminations, i.e. hydroxyl and fluorine, and the terminations can interact with the Fe(II) site and facilitate the reaction. However, the effect of different terminations on Ti3C2Tx MXene on the ECRR performance of M-Pc has not been studied.
Herein, to systematically investigate the effect of various terminal groups of the MXene support on ECRR performance, we anchored the CoPc catalyst on the MXene support bearing different terminal groups (–F or –OH). The –OH terminal group is beneficial for CoPc anchoring as it forms Co–O axial coordination, through which the electronic states of the Co center are tuned, thereby realizing an efficient ECRR performance. The theoretical calculation results show that the excellent performance is attributed to the electron-donating group –OH enriching the electron density of Co, which is conducive to the formation and conversion of intermediates for the ECRR.
2. Experimental
2.1. Synthesis
2.2. Electrochemical measurements
The electrochemical performance of the ECRR was evaluated in a typical H-type cell with a classic three-electrode system on a workstation (CHI760E). The compartment of the H-type cell was separated by an ion exchange membrane, with Ag/AgCl as the reference electrode, platinum wire as the counter electrode, and 1 cm2 carbon paper with the catalyst as the working electrode. For the preparation of the working electrode, 4.0 mg of the catalyst and 50 μL of Nafion solution (117, 5 wt%) were added to 475 μL of anhydrous ethanol and 475 μL of deionized water, followed by sonication to form a homogeneous solution. The working electrode was obtained by dropping the catalyst ink on carbon paper achieving a loading of 0.2 mg cm−2.3. Results and discussion
Fig. 1a schematically shows the process of preparation of CoPc/MXene–X composites. The aluminum layer etching was carried out by adding Ti3AlC2 powder to HF solution, and the following alkalization was conducted in NaX solutions to obtain MXene–X. During the alkalization treatment, Na+ was inserted towards the bulk Ti3C2Tx MXene and the terminal groups were formed by the replacement of OH− or F− ions.27 The CoPc complex was evenly dispersed onto MXene–X through a self-assembly strategy by adding CoPc and MXene–X to dimethylformamide (DMF) solution with the assistance of sonication.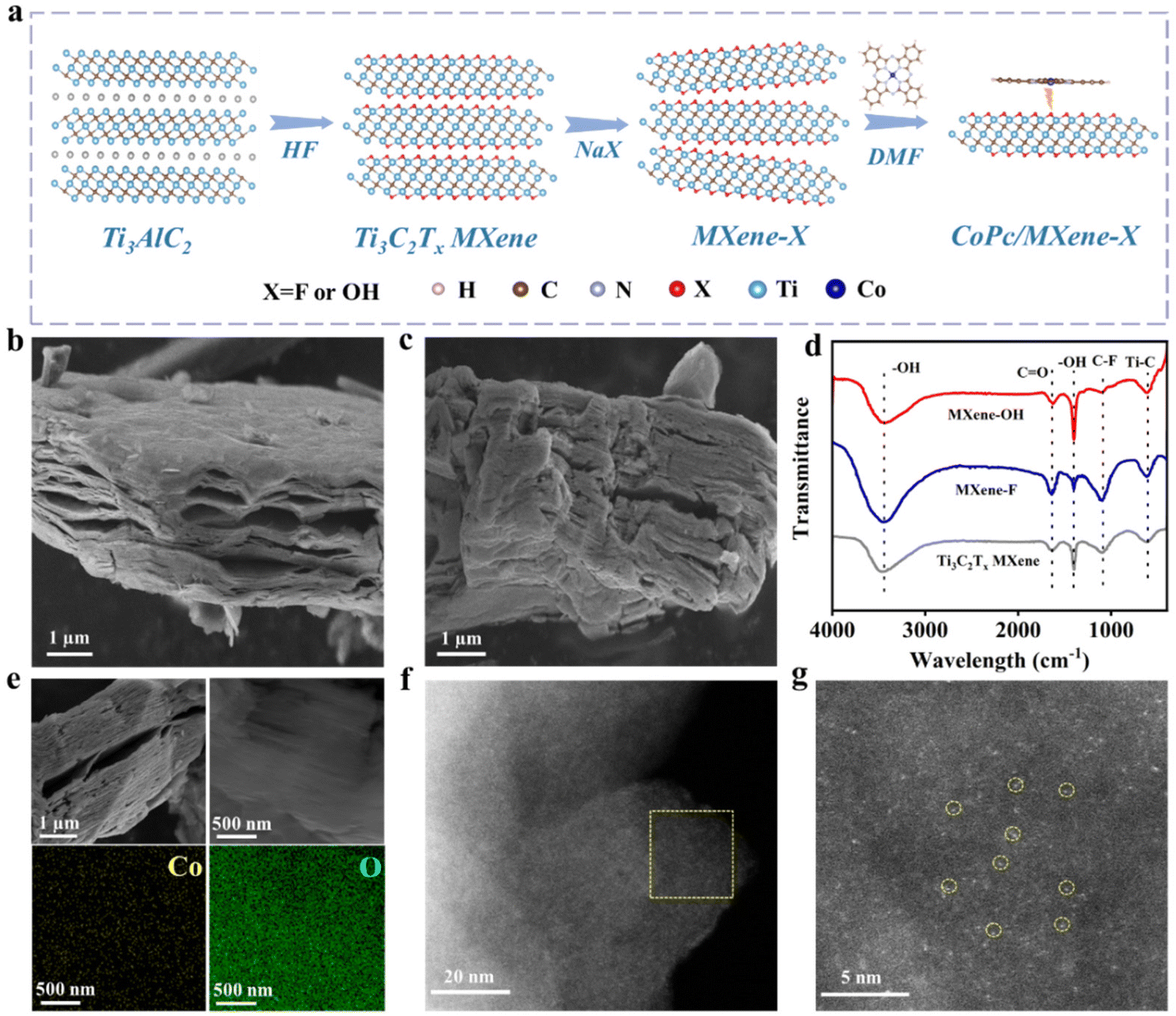 | ||
| Fig. 1 (a) Schematic illustration for CoPc/MXene–X synthesis. (b) SEM image of MXene–F. (c) SEM image of MXene–OH. (d) FTIR spectra of Ti3C2Tx MXene, MXene–F and MXene–OH. (e) SEM and the corresponding EDS mapping images of CoPc/MXene–OH. (f and g) AC-STEM images of CoPc/MXene–OH. | ||
The size and morphology of the prepared samples were characterized by SEM. A classic accordion-like structure of Ti3C2Tx MXene was obtained by etching with HF with an average size of 10 μm (Fig. S1 and S2†). After the terminal group replacement, the morphology and size of the 2D structure were maintained (Fig. 1b and c). The phase and surface functional groups of the samples were analyzed by XRD and FTIR. After etching with HF solution, the main XRD peak (103) of Ti3AlC2 disappeared, indicating that the Al layer was etched (Fig. S3†).28 The main peak (002) of MXene–OH showed the obvious shift from 9° to 7° compared with the pristine Ti3C2Tx MXene, which indicated that the –F group was replaced by the –OH group in the strong alkali solution.29 In the FTIR spectra, the pristine Ti3C2Tx MXene, MXene–F and MXene–OH exhibited the same stretching vibration at about 620, 1650 and 3420 cm−1, belonging to Ti–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –OH, respectively (Fig. 1d).30 For MXene–F, the C–F signal at 1100 cm−1 was significantly enhanced compared with the pristine Ti3C2Tx MXene, indicating the successful –F terminal group replacement. For MXene–OH, the C–F signal at 1100 cm−1 disappeared, and the stretching vibration signal of –OH at 1395 cm−1 was enhanced, which implies that the –F group was replaced by the –OH group after alkalization.31 Raman spectra of different samples showed the same characteristic Raman bands at 209, 276, 391, 579, 627 and 726 cm−1 (Fig. S4†), respectively, indicating that MXene–F and MXene–OH still maintain the original Ti3C2Tx MXene configuration.32 X-ray photoelectron spectroscopy (XPS) was performed to examine the electronic state and surface chemical composition of the samples. The presence of surface –F and –OH was verified by the presence of the C–Ti–F bond in the F 1s spectrum of MXene–F and the Ti–OH bond in the O 1s spectrum of MXene–OH (Fig. S5†). The O 1s peak strength of MXene–OH was significantly enhanced compared with Ti3C2Tx MXene (Fig. S6 and Table S1†), while the F 1s peak strength was decreased. The change of peak intensity of MXene–F was opposite.
O and –OH, respectively (Fig. 1d).30 For MXene–F, the C–F signal at 1100 cm−1 was significantly enhanced compared with the pristine Ti3C2Tx MXene, indicating the successful –F terminal group replacement. For MXene–OH, the C–F signal at 1100 cm−1 disappeared, and the stretching vibration signal of –OH at 1395 cm−1 was enhanced, which implies that the –F group was replaced by the –OH group after alkalization.31 Raman spectra of different samples showed the same characteristic Raman bands at 209, 276, 391, 579, 627 and 726 cm−1 (Fig. S4†), respectively, indicating that MXene–F and MXene–OH still maintain the original Ti3C2Tx MXene configuration.32 X-ray photoelectron spectroscopy (XPS) was performed to examine the electronic state and surface chemical composition of the samples. The presence of surface –F and –OH was verified by the presence of the C–Ti–F bond in the F 1s spectrum of MXene–F and the Ti–OH bond in the O 1s spectrum of MXene–OH (Fig. S5†). The O 1s peak strength of MXene–OH was significantly enhanced compared with Ti3C2Tx MXene (Fig. S6 and Table S1†), while the F 1s peak strength was decreased. The change of peak intensity of MXene–F was opposite.
The CoPc/MXene–X composite catalyst was prepared through a self-assembly strategy in DMF solution under ultrasonication at room temperature. The morphology of the composite catalyst was revealed by SEM. CoPc/MXene–F and CoPc/MXene–OH showed the same accordion morphology as the pristine Ti3C2Tx MXene, indicating that the structural morphology of the substrate underwent no obvious change during the loading process (Fig. 1e and S7†). The element map showed the uniform distribution of Co and F, and Co and O elements throughout the sample. Individual scattered Co atoms in CoPc/MXene–OH can be clearly observed through AC-STEM (Fig. 1f and g, yellow circles highlighted). By means of inductively coupled plasma mass spectrometry (ICP-MS) analysis, the mass percentage of Co atoms in CoPc/MXene–OH and CoPc/MXene–F was about 3.86 wt% and 3.75 wt%, respectively.
The FTIR spectra of the composite catalysts were in good agreement with that of CoPc, indicating that CoPc was successfully loaded on MXene substrates (Fig. 2a). After loading CoPc, the signal at 533.4 eV was assigned to the Co–O bond in the O 1s spectrum of CoPc/MXene–OH, which implies the axial coordination interaction between the O atom and electron-deficient Co (Fig. 2b).33 The Co 2p3/2 and Co 2p1/2 peaks of CoPc/MXene–OH and CoPc/MXene–F moved 0.24 eV and 0.12 eV in the direction of lower binding energy, respectively (Fig. 2c), indicating an obvious electron transfer between CoPc and MXene–X. Moreover, the electronic communication between MXene–OH and CoPc was stronger, and more electrons were obtained at the metal Co site for CoPc/MXene–OH. UV-vis analysis further illustrated the interaction between CoPc and MXene vectors with different end groups (Fig. 2d). For the original CoPc molecule, an obvious Q-band was detected at 659 nm, and the Q-band of CoPc/MXene–OH showed a larger redshift than that of CoPc/MXene–F, which was caused by the increase of the electron density of Co atoms due to the coordination of axial O atoms. However, the –F terminal group with large electronegativity was not conducive to the formation of additional coordination with Co atoms.34,35
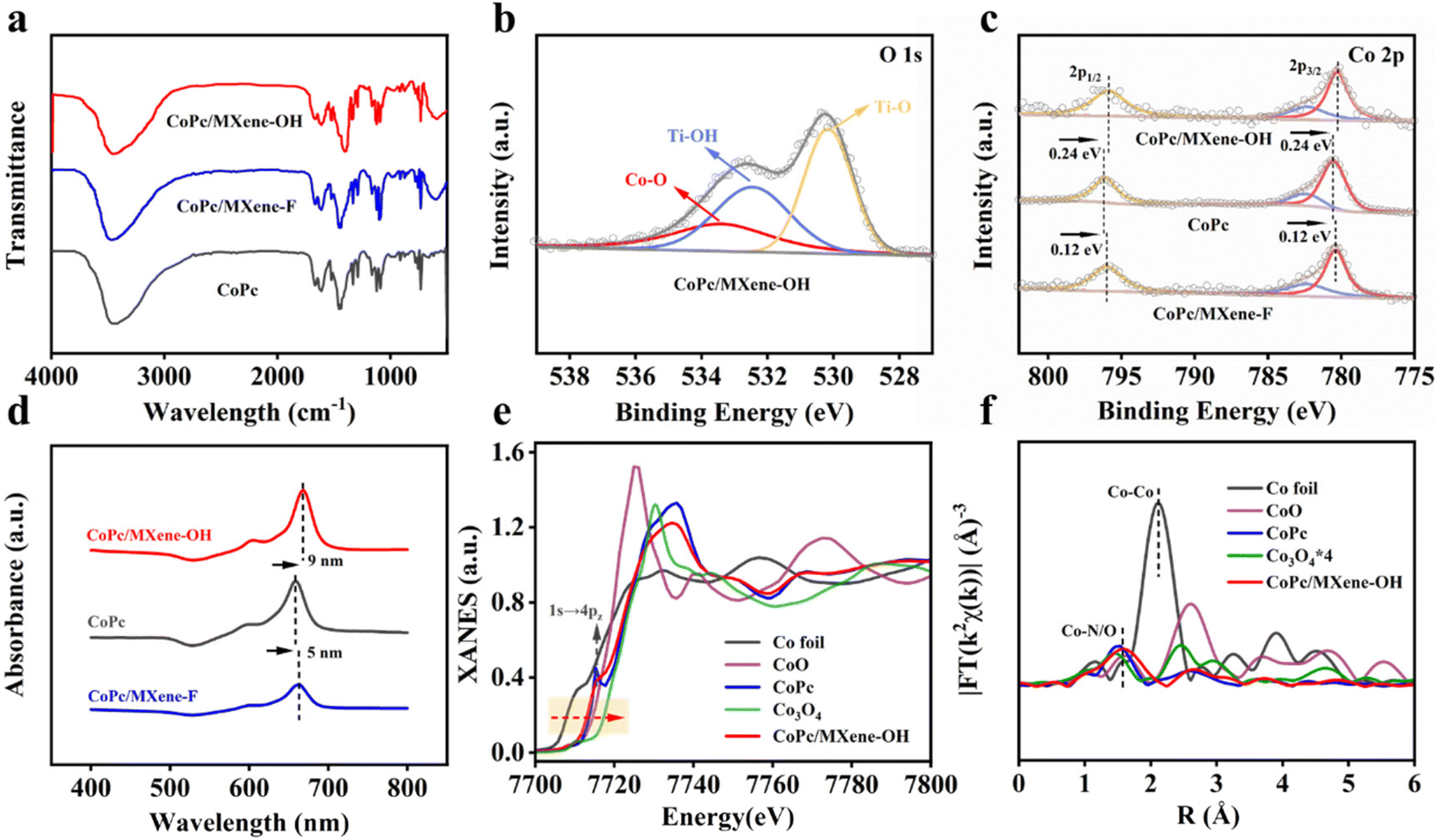 | ||
| Fig. 2 (a) FTIR patterns of CoPc, CoPc/MXene–F and CoPc/MXene–OH. (b) O 1s XPS spectrum of CoPc/MXene–OH. (c) Co 2p XPS spectra of CoPc, CoPc/MXene–F and CoPc/MXene–OH. (d) UV-vis spectra of CoPc, CoPc/MXene–F and CoPc/MXene–OH. (e) XANES spectra of CoPc/MXene–OH and comparison samples (Co foil, CoO, Co3O4 and CoPc). (f) EXAFS spectra of CoPc/MXene–OH and comparison samples. | ||
The coordination environment and electronic states of Co atoms in CoPc/MXene–OH samples were further studied by K-edge X-ray absorption near-edge structure (XANES) and Fourier transformed extended X-ray absorption fine structure (FT-EXAFS) analyses. The K-edge XANES spectra of Co show that the near side line of CoPc/MXene–OH is lower than that of Co3O4, which is similar to the near side line of CoPc and is located between the contrast sample CoO and Co foil, indicating that the average valence of Co in CoPc/MXene–OH is between 0 and +2 (Fig. 2e). For the FT-EXAFS analysis, CoPc shows an obvious Co–N peak at 1.48 Å, and the CoPc/MXene–OH peak has a slight right shift (Fig. 2f) implying an additional coordination. According to the fitting data, the Co–N peak and Co–O peak exist simultaneously. These observations indicate that Co atoms are directly coordinated with N and O atoms in CoPc/MXene–OH samples, which is consistent with the results of XPS analysis. According to the EXAFS fitting curve in Table S2,† the coordination number of the Co atom is around five with direct coordination of one O atom and four N atoms.
To investigate the effect of various end groups of MXene supports on the ECRR activity of CoPc, linear sweep voltammetry (LSV) was carried out in an H-cell with N2- or CO2-saturated 0.1 M KHCO3 solution. All potentials obtained in the experiment have been converted into the vs. RHE. CoPc/MXene–OH exhibits higher current density compared with CoPc/MXene–F throughout the measured potential range (Fig. 3a). On quantitative analysis by gas chromatography, the products of the ECRR are CO and H2 over the samples (Fig. 3b). The faradaic efficiency for CO formation (FECO) reaches 92.4% for CoPc/MXene–OH at −1.0 V, which is better than that of CoPc/MXene–F (84.0% at −1.0 V). Furthermore, CoPc/MXene–OH shows a FECO above 80% over a broad potential range from −0.9 to −1.2 V. Control experiments show that pure carriers with different functional groups are inactive for the ECRR, indicating that CoPc provides the active sites for the ECRR (Fig. 3c and Fig. S8†). In addition, no other gaseous or liquid products are generated (Fig. S9†). For comparison, Ti3C2Tx MXene is also used for anchoring CoPc (CoPc/Ti3C2Tx MXene). The current densities and FECO of CoPc/MXene–OH at the measured potential are significantly higher than those of CoPc/Ti3C2Tx MXene (Fig. S10†). The outstanding activity and selectivity result in excellent partial current density for CO formation (JCO) (Fig. 3d). CoPc/MXene–OH shows a JCO of −3.6 mA cm−2 at −1.0 V, which is 1.5-fold higher than that of CoPc/MXene–F (−2.4 mA cm−2). The advantage of CoPc/MXene–OH in JCO is also obviously throughout the whole potential range. In addition, JCO and –OH content show a certain linear relationship in the range of the measured potential (Fig. S11†). To evaluate the intrinsic activity, the catalytic current was normalized by the electrochemical active surface area (ECSA) (Fig. S12†). The CoPc/MXene–OH sample also shows a higher ECSA normalized activity than the control samples (Fig. S11d†), indicating that CoPc/MXene–OH shows a higher intrinsic activity for the ECRR. In addition, CoPc/MXene–OH shows excellent stability over 10 h of electrolysis at −1.0 V with a FECO more than 80% (Fig. 3e). The reaction kinetics of these as-prepared samples for the ECRR is described by the Tafel plot (Fig. S13†).36 CoPc/MXene–OH has a Tafel slope of 162 mV dec−1, lower than that of CoPc/MXene–F (182 mV dec−1), further suggesting kinetics enhancement by the MXene–OH modification.
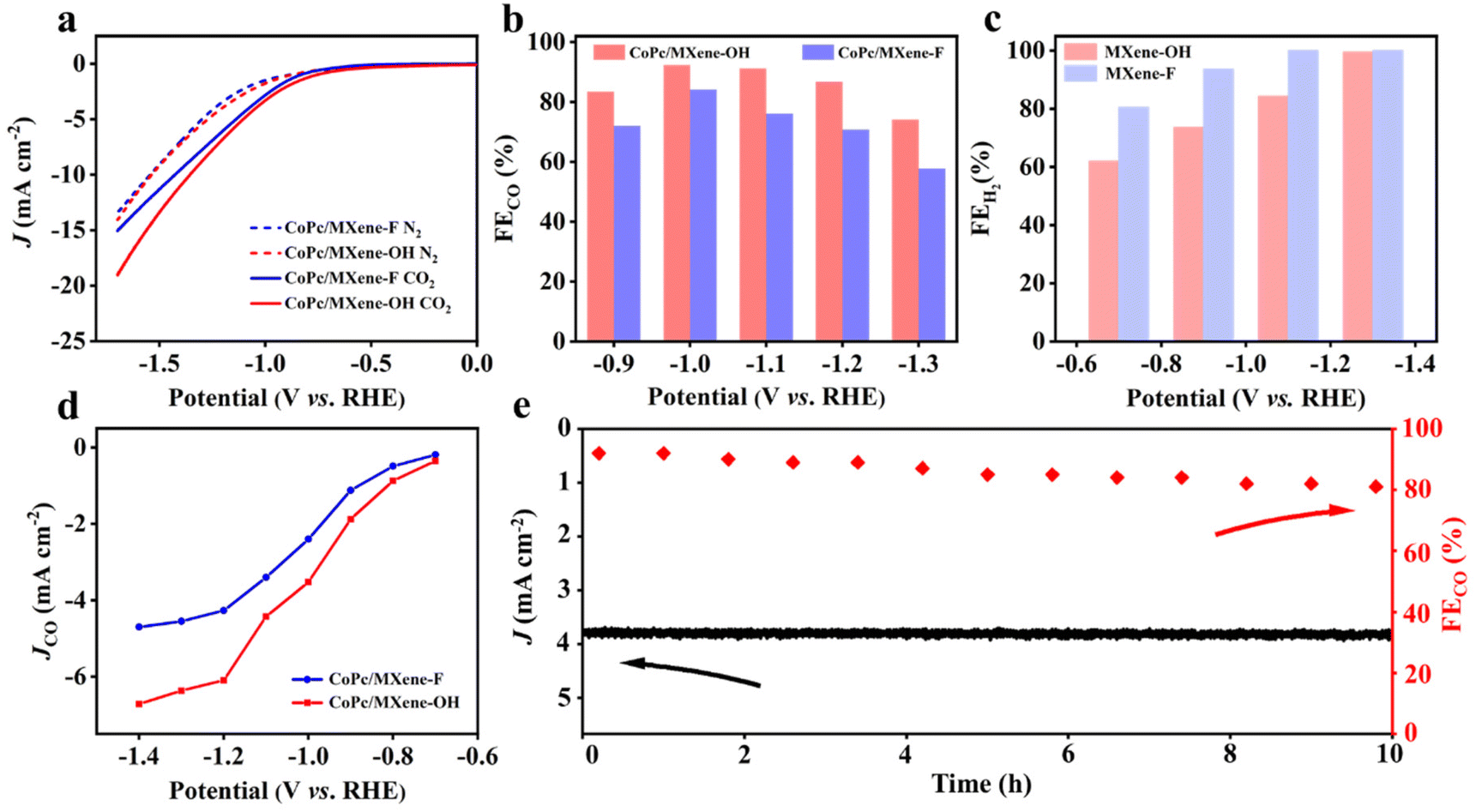 | ||
| Fig. 3 (a) LSV curves of CoPc/MXene–F and CoPc/MXene–OH in CO2- and N2-saturated 0.1 M KHCO3 solution. (b) FE of CO at various potentials of CoPc/MXene–F and CoPc/MXene–OH. (c) FE of H2 at various potentials of MXene–F and MXene–OH supports. (d) JCO at various potentials of CoPc/MXene–F and CoPc/MXene–OH. (e) Long-term stability at −1.0 V. | ||
To uncover the high performances and selectivity of the CoPc/MXene–OH catalyst for CO2-to-CO conversion, density functional theory (DFT) calculations were performed. The ECRR pathway was investigated via the computational hydrogen electrode model (Fig. 4a and Fig. S14†). The first step for *CO2 formation is exothermic on the surfaces of CoPc/MXene–F and CoPc/MXene–OH (−0.29 and −0.13 eV), with CoPc/MXene–OH exhibiting relatively weak adsorption, which is favourable for the next conversions (Fig. 4b). The steps for *COOH and *CO formation, together with CO desorption, were analysed. The *COOH formation step shows the largest energy barrier, and therefore it was assigned as the rate determining step (RDS) for the overall ECRR. CoPc/MXene–OH (0.71 eV) shows a relatively small barrier in the RDS than CoPc/MXene–F (0.85 eV), consistent with the higher electrochemical ECRR performance. As the main side-reaction, the HER has a suppressive influence on the ECRR performance.37,38 The adsorption strength for *H was weakened on CoPc/MXene–OH showing a large *H endothermic energy of the HER, the main competitive reaction of the ECRR, while the adsorption of *H by CoPc/MXene–F is stronger (Fig. 4c). Moreover, CoPc/MXene–OH shows a lower UD (CO2) − UD (H2) value (−0.44 V) than CoPc/MXene–F (−0.78 V) (Fig. 4d), which is consistent with the above analysis.39,40 To gain a deeper understanding of ECRR performance, we further investigated the partial state density (PDOS) as well as differential charge density (CCD) of the catalysts. PDOS analysis shows that the d-band center of the Co active site in CoPc/MXene–OH (εd = −2.002 eV) is more negative than in CoPc/MXene–F (εd = −1.203 eV) (Fig. S15†). The downward shift of the d-band center after axial coordination indicates a decrease in the antibonding energy state, which is beneficial for the next conversions and inhibits the undesired HER.41,42 According to the Bader charge analysis (Fig. S16†), MXene–OH (0.289 e−) provides more additional electrons to CoPc compared with MXene–F (0.237 e−). The stronger electron interaction between MXene–OH and CoPc is beneficial for electron transfer to the intermediates, promoting the ECRR performance.
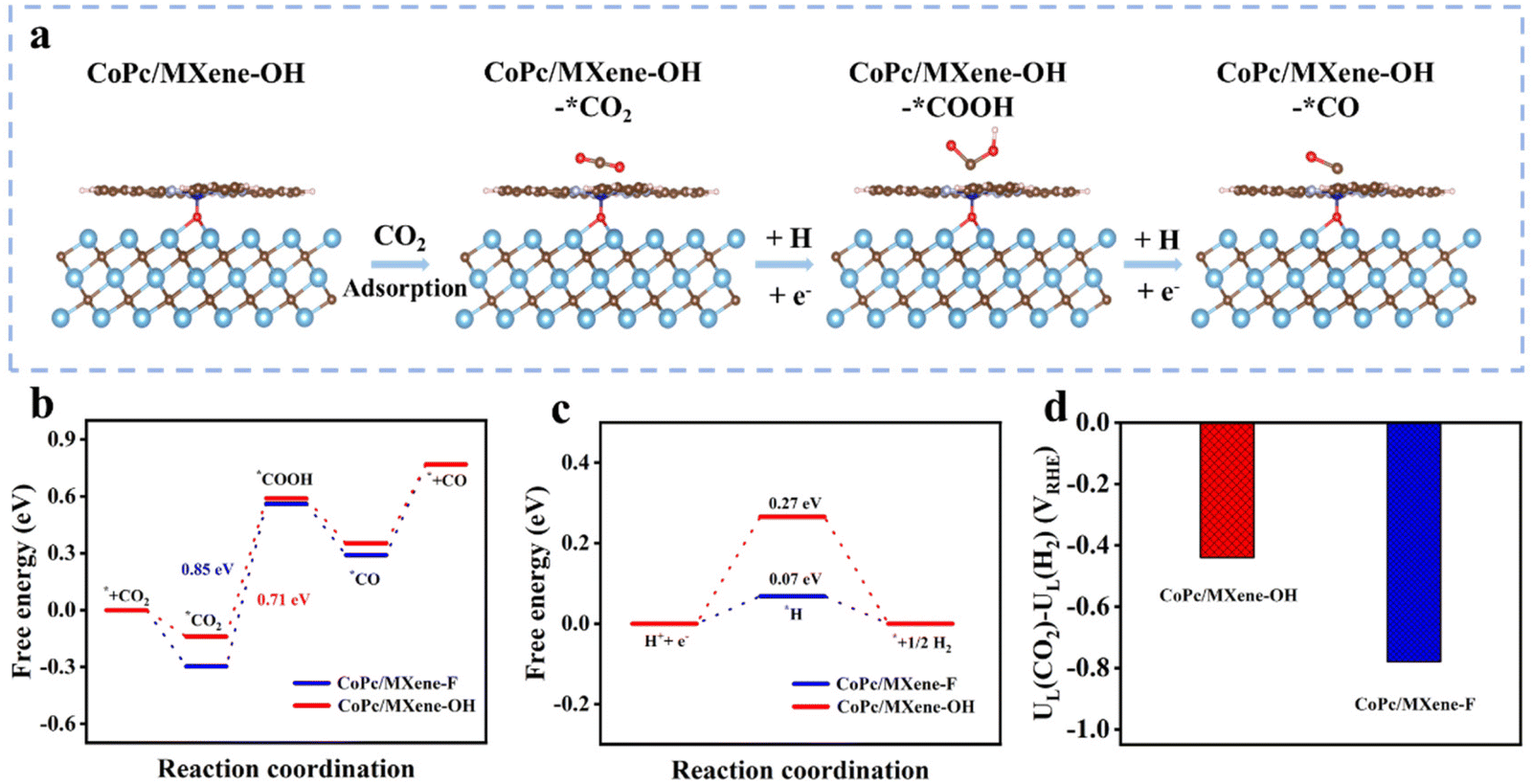 | ||
| Fig. 4 (a) The optimized configurations of the adsorption of intermediates on CoPc/MXene–OH. (b) Calculated free energy diagram of CoPc/MXene–F and CoPc/MXene–OH for the ECRR. (c) Calculated free energy diagram of CoPc/MXene–F and CoPc/MXene–OH for the HER. (d) Limiting potential differences between CO2 reduction and H2 evolution on different catalyst models at U = 0 V. | ||
4. Conclusions
In summary, we have demonstrated that the terminal groups on the MXene support play an important role in electronic state regulation of the molecular catalyst by anchoring CoPc on the MXene–X (X = –F or –OH) support. Benefitting from the Co–O axial coordination, CoPc/MXene–OH shows an impressive FECO of 92.4%, higher than that of CoPc/MXene–F (84.0%) under the same conditions. The theoretical calculation results show that the excellent performance is attributed to the electron-donating property of –OH enriching the electron density of Co, which optimizes the binding strength for intermediates and therefore boosts the ECRR performance. The simple synthesis method and excellent performance make CoPc/MXene–OH a promising electrocatalyst and open up a new avenue for regulating the electronic states of active sites and further the intermediate adsorption strength for efficient electrocatalyst design.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22278108 and 22008048), the Hundred Talents Project of Hebei Province (No. E2019050015), the Natural Science Foundation of Tianjin (22JCYBJC00250), the Natural Science Foundation for Outstanding Youth Scholars of Hebei Province (No. B2021202061), the Natural Science Foundation of Hebei Province (No. B2021202010), and the State Key Laboratory of Fine Chemicals, Dalian University of Technology (KF 2108).References
- L. Ye, X. Chen, Y. Gao, X. Ding, J. Hou and S. Cao, Ultrathin Two-dimensional Metal–organic Framework Nanosheets for Efficient Electrochemical CO2 reduction, J. Energy Chem., 2021, 57, 627–631 CrossRef CAS.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Advances and Challenges in Understanding the Electrocatalytic Conversion of Carbon Dioxide to Fuels, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang and Z. Jin, Progress and Perspective of Electrocatalytic CO2 Reduction for Renewable Carbonaceous Fuels and Chemicals, Adv. Sci., 2018, 5, 1700275 CrossRef PubMed.
- J. Guo, W. Zhang, L. H. Zhang, D. Chen, J. Zhan, X. Wang, N. R. Shiju and F. Yu, Control over Electrochemical CO2 Reduction Selectivity by Coordination Engineering of Tin Single-Atom Catalysts, Adv. Sci., 2021, 8, 2102884 CrossRef CAS PubMed.
- Y. Zhang, J. Lan, F. Xie, M. Peng, J. Liu, T.-S. Chan and Y. Tan, Aligned InS Nanorods for Efficient Electrocatalytic Carbon Dioxide Reduction, ACS Appl. Mater. Interfaces, 2022, 14, 25257–25266 CrossRef CAS PubMed.
- Z. Wang, J. Zhao and Q. Cai, CO2 Electroreduction Performance of a Single Transition Metal Atom Supported on Porphyrin-like Graphene: A Computational Study, Phys. Chem. Chem. Phys., 2017, 19, 23113–23121 RSC.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, Monodisperse Au Nanoparticles for Selective Electrocatalytic Reduction of CO2 to CO, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- H. Li, Y. Pan, Z. Wang, Y. Yu, J. Xiong, H. Du, J. Lai, L. Wang and S. Feng, Coordination Engineering of Cobalt Phthalocyanine by Functionalized Carbon Nanotube for Efficient and Highly Stable Carbon Dioxide Reduction at High Current Density, Nano Res., 2022, 15, 3056–3064 CrossRef CAS.
- T. Zhang, X. Han, H. Liu, M. Biset-Peiró, X. Zhang, P. Tan, P. Tang, B. Yang, L. Zheng, J. R. Morante and J. Arbiol, Quasi-double-star Nickel and Iron Active Sites for High-efficiency Carbon Dioxide Electroreduction, Energy Environ. Sci., 2021, 14, 4847–4857 RSC.
- A. Álvarez, M. Borges, J. J. Corral-Pérez, J. G. Olcina, L. Hu, D. Cornu, R. Huang, D. Stoian and A. Urakawa, CO2 Activation over Catalytic Surfaces, ChemPhysChem, 2017, 18, 3135–3141 CrossRef PubMed.
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang, J. Lu and J. Wang, A Graphene-Supported Single-Atom FeN5 Catalytic Site for Efficient Electrochemical CO2 Reduction, Angew. Chem., Int. Ed., 2019, 58, 14871–14876 CrossRef CAS PubMed.
- M. Cong, X. Chen, K. Xia, X. Ding, L. Zhang, Y. Jin, Y. Gao and L. Zhang, Selective Nitrogen Reduction to Ammonia on Iron Porphyrin-based Single-site Metal–organic Frameworks, J. Mater. Chem. A, 2021, 9, 4673–4678 RSC.
- B. Mei, C. Liu, J. Li, S. Gu, X. Du, S. Lu, F. Song, W. Xu and Z. Jiang, Operando HERFD-XANES and Surface Sensitive Δμ Analyses Identify the Structural Evolution of Copper(II) Phthalocyanine for Electroreduction of CO2, J. Energy Chem., 2022, 64, 1–7 CrossRef CAS.
- A. Maurin and M. Robert, Noncovalent Immobilization of a Molecular Iron-Based Electrocatalyst on Carbon Electrodes for Selective, Efficient CO2-to-CO Conversion in Water, J. Am. Chem. Soc., 2016, 138, 2492–2495 CrossRef CAS PubMed.
- J. Choi, P. Wagner, S. Gambhir, R. Jalili, D. R. MacFarlane, G. G. Wallace and D. L. Officer, Steric Modification of a Cobalt Phthalocyanine/Graphene Catalyst To Give Enhanced and Stable Electrochemical CO2 Reduction to CO, ACS Energy Lett., 2019, 4, 666–672 CrossRef CAS.
- J. Su, J. J. Zhang, J. Chen, Y. Song, L. Huang, M. Zhu, B. I. Yakobson, B. Z. Tang and R. Ye, Building a Stable Cationic Molecule/electrode Interface for Highly Efficient and Durable CO2 Reduction at an Industrially Relevant Current, Energy Environ. Sci., 2021, 14, 483–492 RSC.
- X. M. Hu, M. H. Rønne, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Enhanced Catalytic Activity of Cobalt Porphyrin in CO2 Electroreduction upon Immobilization on Carbon Materials, Angew. Chem., Int. Ed., 2017, 56, 6468–6472 CrossRef CAS PubMed.
- F. Xu, L. Zhang, X. Ding, M. Cong, Y. Jin, L. Chen and Y. Gao, Selective Electroreduction of Dinitrogen to Ammonia on a Molecular Iron Phthalocyanine/O-MWCNT Catalyst Under Ambient Conditions, ChemComm, 2019, 55, 14111–14114 RSC.
- D. S. Jeong, H. S. Shin and J. Yang, Influence of the Molecular Structure of Metal-phthalocyanine on Electrocatalytic Reactions, Sci. China Mater., 2022, 65, 3324–3333 CrossRef CAS.
- Y. Xia, S. Kashtanov, P. Yu, L.-Y. Chang, K. Feng, J. Zhong, J. Guo and X. Sun, Identification of Dual-active Sites in Cobalt Phthalocyanine for Electrochemical Carbon Dioxide Reduction, Nano Energy, 2020, 67, 104163 CrossRef CAS.
- X. Wu, J. W. Sun, P. F. Liu, J. Y. Zhao, Y. Liu, L. Guo, S. Dai, H. G. Yang and H. Zhao, Molecularly Dispersed Cobalt Phthalocyanine Mediates Selective and Durable CO2 Reduction in a Membrane Flow Cell, Adv. Funct. Mater., 2022, 32, 2107301 CrossRef CAS.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang, Y. Liang and H. Wang, Highly Selective and Active CO2 Reduction Electrocatalysts Based on Cobalt Phthalocyanine/carbon Nanotube Hybrid Structures, Nat. Commun., 2017, 8, 14675 CrossRef PubMed.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, Design of Single-Atom Co–N5 Catalytic Site: A Robust Electrocatalyst for CO2 Reduction with Nearly 100% CO Selectivity and Remarkable Stability, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- X. Wang, Y. Wang, X. Sang, W. Zheng, S. Zhang, L. Shuai, B. Yang, Z. Li, J. Chen, L. Lei, N. M. Adli, M. K. H. Leung, M. Qiu, G. Wu and Y. Hou, Dynamic Activation of Adsorbed Intermediates via Axial Traction for the Promoted Electrochemical CO2 Reduction, Angew. Chem., Int. Ed., 2021, 60, 4192–4198 CrossRef CAS PubMed.
- M. Zhu, J. Chen, R. Guo, J. Xu, X. Fang and Y.-F. Han, Cobalt Phthalocyanine Coordinated to Pyridine-functionalized Carbon Nanotubes with Enhanced CO2 Electroreduction, Appl. Catal., B, 2019, 251, 112–118 CrossRef CAS.
- Y. Lee, J. H. Ahn, H. Jang, J. Lee, S. Yoon, D.-G. Lee, M. G. Kim, J. H. Lee and H. K. Song, Very Strong Interaction between FeN4 and Titanium Carbide for Durable 4-electron Oxygen Reduction Reaction Suppressing Catalyst Deactivation by Peroxide, J. Mater. Chem. A, 2022, 10, 24041–24050 RSC.
- M. Alhabeb, K. Maleski, B. Anasori, P. Lelyukh, L. Clark, S. Sin and Y. Gogotsi, Guidelines for Synthesis and Processing of Two-Dimensional Titanium Carbide (Ti3C2Tx MXene), Chem. Mater., 2017, 29, 7633–7644 CrossRef CAS.
- Y. Li, Y. Liu, D. Xing, J. Wang, L. Zheng, Z. Wang, P. Wang, Z. Zheng, H. Cheng, Y. Dai and B. Huang, 2D/2D Heterostructure of Ultrathin BiVO4/Ti3C2 Nanosheets for Photocatalytic Overall Water Splitting, Appl. Catal., B, 2021, 285, 119855 CrossRef CAS.
- Z. Jin, C. Liu, Z. Liu, J. Han, Y. Fang, Y. Han, Y. Niu, Y. Wu, C. Sun and Y. Xu, Rational Design of Hydroxyl-Rich Ti3C2Tx MXene Quantum Dots for High-Performance Electrochemical N2 Reduction, Adv. Energy Mater., 2020, 10, 2000797 CrossRef CAS.
- Z. Jin, Y. Fang, X. Wang, G. Xu, M. Liu, S. Wei, C. Zhou, Y. Zhang and Y. Xu, Ultra-efficient Electromagnetic Wave Absorption with Ethanol-thermally Treated Two-dimensional Nb2CTx Nanosheets, J. Colloid Interface Sci., 2019, 537, 306–315 CrossRef CAS PubMed.
- G. Liu, C. Sun, H. G. Yang, S. C. Smith, L. Wang, G. Q. Lu and H. M. Cheng, Nanosized Anatase TiO2 Single Crystals for Enhanced Photocatalytic Activity, ChemComm, 2010, 46, 755–757 RSC.
- X. Chen, X. Sun, W. Xu, G. Pan, D. Zhou, J. Zhu, H. Wang, X. Bai, B. Dong and H. Song, Ratiometric Photoluminescence Sensing Based on Ti3C2 MXene Quantum Dots as an Intracellular pH Sensor, Nanoscale, 2018, 10, 1111–1118 RSC.
- Y.-T. Liu, X. Chen, J. Yu and B. Ding, Carbon-Nanoplated CoS@TiO2 Nanofibrous Membrane: An Interface-Engineered Heterojunction for High-Efficiency Electrocatalytic Nitrogen Reduction, Angew. Chem., Int. Ed., 2019, 58, 18903–18907 CrossRef CAS PubMed.
- G. Gao, A. P. O'Mullane and A. Du, 2D MXenes: A New Family of Promising Catalysts for the Hydrogen Evolution Reaction, ACS Catal., 2017, 7, 494–500 CrossRef CAS.
- H. Bao, Y. Qiu, X. Peng, J. A. Wang, Y. Mi, S. Zhao, X. Liu, Y. Liu, R. Cao, L. Zhuo, J. Ren, J. Sun, J. Luo and X. Sun, Isolated Copper Single Sites for High-performance Electroreduction of Carbon Monoxide to Multicarbon Products, Nat. Commun., 2021, 12, 238 CrossRef CAS PubMed.
- D. Chen, L. H. Zhang, J. Du, H. Wang, J. Guo, J. Zhan, F. Li and F. Yu, A Tandem Strategy for Enhancing Electrochemical CO2 Reduction Activity of Single-Atom Cu-S1N3 Catalysts via Integration with Cu Nanoclusters, Angew. Chem., Int. Ed., 2021, 60, 24022–24027 CrossRef CAS PubMed.
- H. Wang, C. Tsai, D. Kong, K. Chan, F. Abild-Pedersen, J. K. Nørskov and Y. Cui, Transition-metal Doped Edge Sites in Vertically Aligned MoS2 Catalysts for Enhanced Hydrogen Evolution, Nano Res., 2015, 8, 566–575 CrossRef CAS.
- C. Guo, T. Zhang, X. Liang, X. Deng, W. Guo, Z. Wang, X. Lu and C. M. L. Wu, Single Transition Metal Atoms on Nitrogen-doped Carbon for CO2 Electrocatalytic Reduction: CO production or further CO reduction?, Appl. Surf. Sci., 2020, 533, 147466 CrossRef CAS.
- W. Bi, X. Li, R. You, M. Chen, R. Yuan, W. Huang, X. Wu, W. Chu, C. Wu and Y. Xie, Surface Immobilization of Transition Metal Ions on Nitrogen-Doped Graphene Realizing High-Efficient and Selective CO2 Reduction, Adv. Mater., 2018, 30, 1706617 CrossRef PubMed.
- X. Hou, J. Ding, W. Liu, S. Zhang, J. Luo and X. Liu, Asymmetric Coordination Environment Engineering of Atomic Catalysts for CO2 Reduction Nanomaterials, Nanomaterials, 2023, 13, 309 CrossRef CAS PubMed.
- Z. Chen, Y. Song, J. Cai, X. Zheng, D. Han, Y. Wu, Y. Zang, S. Niu, Y. Liu, J. Zhu, X. Liu and G. Wang, Tailoring the d-Band Centers Enables Co4N Nanosheets To Be Highly Active for Hydrogen Evolution Catalysis, Angew. Chem., Int. Ed., 2018, 57, 5076–5080 CrossRef CAS PubMed.
- H. L. Zhu, L. Zhang, M. Shui, Z.-Y. Li, J. J. Ma and Y. Q. Zheng, A Novel Manner of Anchoring Cobalt Phthalocyanine on Edge-Defected Carbon for Highly Electrocatalytic CO2 Reduction, J. Phys. Chem. Lett., 2023, 14, 3844–3852 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qi01070h |
| This journal is © the Partner Organisations 2023 |