
Latest progress in asymmetrically functionalized Anderson-type polyoxometalates
Qinghe
Zhuang†
,
Zeqian
Sun†
,
Chang-Gen
Lin
 *,
Bo
Qi
* and
Yu-Fei
Song
*
*,
Bo
Qi
* and
Yu-Fei
Song
*
State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: linchg@mail.buct.edu.cn; bqi@mail.buct.edu.cn; songyf@mail.buct.edu.cn
First published on 8th February 2023
Abstract
Anderson-type polyoxometalates (POMs) are one of the most important and widely developed groups of the POM family. The covalent functionalization of Anderson POMs has attracted extensive attention and facilitated broad applications of the resultant POM hybrids in catalysis, biology, energy materials and medicine. Among the various synthetic methods for Anderson hybrids, asymmetric functionalization has been one of the hottest and unique topics in the last decade. In the structure of asymmetric Anderson hybrids, two different organic components are anchored onto each side of the Anderson cluster or only one side of the cluster is functionalized. Asymmetric functionalization provides complexity to POM assemblies and merges multiple functions into one hybrid molecule, meanwhile, bringing challenges of rational design and controllable synthetic strategies. In this review, the latest progress in the synthetic methods and applications of asymmetrically functionalized Anderson-type POMs is summarized according to the central heteroatom of the cluster, which includes Mn-, Cr-, Al- and other metal-templated Anderson POMs.
 Qinghe Zhuang | Qinghe Zhuang received his bachelor's degree from Beijing University of Chemical Technology in 2020. He is currently a master's degree candidate in Prof. Yu-Fei Song's group at the State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology. His research interests are the design and synthesis of new polyoxometalates. |
 Zeqian Sun | Zeqian Sun is currently a master's degree candidate in Prof. Yu-Fei Song's group at the State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology. His current research interest is the structural design and application of covalently modified polyoxometalates. |
 Chang-Gen Lin | Chang-Gen Lin received his BS degree from Beijing University of Chemical Technology (BUCT) in 2010, and completed his PhD under the supervision of Prof. Yu-Fei Song at the same university in 2015. After a two-year postdoc at BAIC-SM, he was appointed as a lecturer at BUCT. In 2019, he was awarded a CSC scholarship to work with Prof. Leroy Cronin at the University of Glasgow on 3D printing and automated chemical synthesis. One year later he returned to BUCT, where he is now an associate professor of chemistry. His research interests include supramolecular self-assembly, organic–inorganic polyoxometalate hybrids, and photo-/electro-responsive materials. |
 Bo Qi | Bo Qi received his bachelor's and PhD degrees in chemistry from Dalian University of Technology (supervisor: Prof. Chunying Duan). In 2015, he worked as a visiting scholar at the University of Akron (supervisor: Prof. Tianbo Liu). In 2018, he joined Prof. Yu-Fei Song's group at Beijing University of Chemical Technology. His research interests include self-assembly and heterogeneous chiral/electro/photo catalysis in polyoxometalate chemistry. |
 Yu-Fei Song | Dr Yu-Fei Song received his BS (1997) and PhD (2002) degrees from Shanxi University. After postdoc research in Leiden University, the Max-Planck Institute of Bio-inorganic Chemistry, and the University of Glasgow, he joined Beijing University of Chemical Technology (BUCT) in 2008. He is currently holding a full professor position in BUCT. His research directions mainly focus on polyoxometalate-based molecular assemblies and multifunctional materials. He has published over 280 research papers in journals such as Nat. Commun., Nat. Protoc., Angew. Chem. Int. Ed., J. Am. Chem. Soc., Energy. Environ. Sci., etc. He was awarded “National Science Foundation for Distinguished Young Scholars of China” (2016). |
1. Introduction
Polyoxometalates (POMs) are a class of anionic metal oxide clusters mainly consisting of early transition metal elements such as Mo, W, V, etc. in their highest oxidation states.1–3 POMs have shown versatile molecular structures and attractive physical–chemical properties, making them widely applied in many fields, such as energy storage,4,5 catalysis,6 molecular magnetism,7,8 biology9 and optical devices.10,11 Inorganic POMs can be covalently modified with functional organic moieties to generate POM-based organic–inorganic hybrids, which therefore enrich the diversity of POM structures and expand their application area. The POM hybrids not only introduce the advantages of organic groups (high compatibility in organic media, good processability, and diverse optical and electronic properties),12–14 but also exert unexpected synergistic effects for strengthening molecular stability15 and improving photochromic,10 electronic storage,16 and catalytic properties.17 Therefore, covalent functionalization of POMs has become one of the most important research directions in POM chemistry.Anderson-type POMs are one of the most important groups of the POM family.18–20 They play an important role in electrocatalytic oxygen evolution,21 oxidative desulfurization,22,23 dye degradation,24 antibacterial activity9 and other application areas.25 The structure of Anderson POMs was proposed by J. S. Anderson in 1937, and then confirmed by H. T. Evans using X-ray. Therefore, this kind of structure is called the “Anderson–Evans” structure, or the Anderson structure for short. The Anderson structure is composed of a central {XO6} octahedron and six surrounding {MO6} octahedra with shared edges. There are three types of oxygen atoms in the cluster, including six triple-bridged oxygens (μ3-O) coordinated to the central heteroatom, six double-bridged oxygens (μ2-O) connected to two addenda atoms, and twelve terminal oxygens (Ot) (Fig. 1). The general formula of the Anderson anion is [Hy(XO6)M6O18]n−, where y = 0–6, n = 2–8, X = central heteroatom, and M = addenda atoms (MoVI or WVI). Among the Anderson-type POMs reported so far, there are many types of elements that can serve as central heteroatoms, including the first transition system elements (Mn,26 Cr,27 V,28etc.29,30), the second transition system elements (Rh,31 Pd,32etc.), the third transition system elements (Pt,33etc.) and the main group elements (Al,34 Ga,35,36 Te,37,38 I,39etc.40).
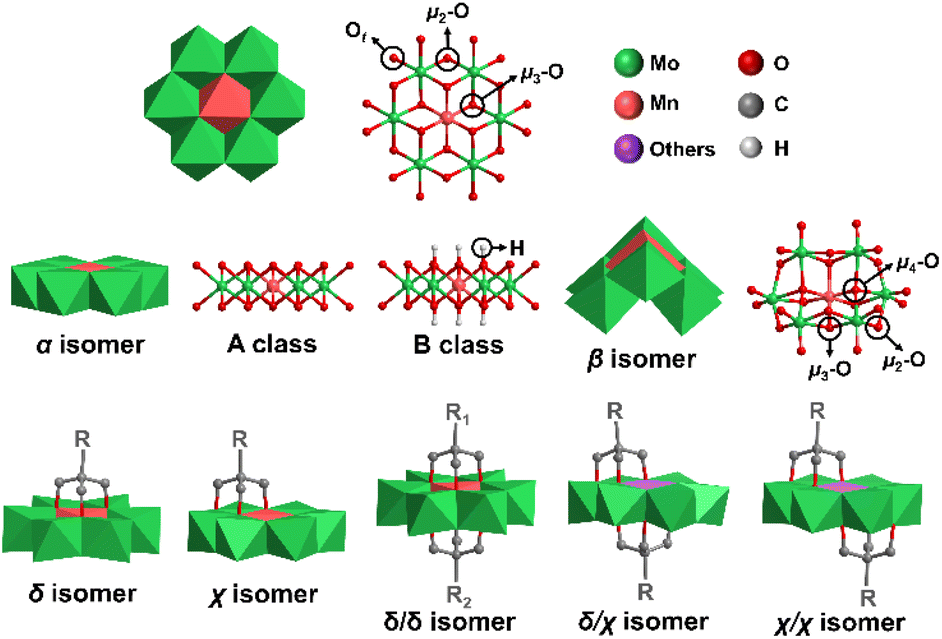 | ||
| Fig. 1 Structures and isomeric species of Anderson-type POMs, and isomeric species after modification with triol ligands. Color code: {MoO6}, green octahedron; {MnO6}, red octahedron; {XO6}, violet octahedron (X = others). Hydrogen atoms in organic ligands have been omitted for clarity. | ||
The Anderson structure has two isomers, namely, α and β isomers. The α isomer possesses an octahedral planar topology while the β isomer shows a non-planar curved structure, featuring two μ4-O atoms coordinated to three addenda atoms and the central heteroatom, two μ3-O atoms coordinated to two addenda atoms and the central heteroatom, two types of μ2-O atoms (two coordinated to one addenda atom and the central heteroatom and six coordinated to two addenda atoms) and twelve Ot. According to the protonation of μ3-O, the α isomer of the Anderson structure can be divided into A and B classes. In the A class, six μ3-O are not protonated, and the central heteroatom is in a high oxidation state. The general formula is [Xn+M6O24](12−n)− (X = TeVI, IVII, etc.). In the B class, six μ3-O are protonated and the central heteroatom is in a low oxidation state. The general formula is [Xn+(OH)6M6O18](12−n)− (X = MnIII, AlIII, etc.). The average size of the α isomer is about 8.6 × 8.6 × 2.7 Å.
The Anderson-type POMs can be functionalized by triol ligands, such as tris(hydroxymethyl)aminomethane (Triol-NH2), resulting in the formation of strong metal–oxygen–carbon bonds (M–O–C). So far, the covalent modification of Anderson-type POMs is mainly focused on symmetric systems, in which both sides of the planar Anderson-type POMs are modified with the same organic triol ligands.
Asymmetric modification represents one of the most unique research topics since it reflects the controlled assembly of metal-oxo units, which is still a long-sought task of POM chemistry. Asymmetric modification, on the one hand, can provide structural diversity and complexity. For instance, asymmetric Mn-Anderson POMs could be covalently linked to form monodisperse linear cluster oligomers by a click reaction, ranging in size from 2 to 5 Anderson units.41 On the other hand, the asymmetric modification can be precisely controlled through the rational design of anchoring ligands, making the resulting hybrids more applicable in various research fields than the symmetric ones. To name a few, the self-assembly behavior of POMs on a hydrophilic surface could be regulated by carefully controlling the non-covalent interactions between anchoring ligands42 and the covalent functionalization of the Au surface with asymmetric Anderson hybrids allowed for selective cell adhesion.43
The asymmetrically triol-functionalized Anderson-type POMs can be divided into single-sided isomers (δ isomer and χ isomer) and double-sided isomers (asymmetric δ/δ isomer, helical symmetric χ/χ isomer and δ/χ isomer at malpositions) (Fig. 1).44 For the δ isomer, three μ3-O atoms on the Anderson cluster are substituted with the triol group, while in the case of the χ isomer, two μ3-O atoms and one μ2-O atom are substituted instead. For the double-sided asymmetric isomers, the δ/δ isomers are commonly obtained with two different triol ligands grafting onto each side of the Anderson cluster. The χ/χ isomers are found in POMs with Cu, Co and Ni as the central heteroatoms,44–47 and the δ/χ isomers are found in POMs with Cu, Co and Zn as the central heteroatoms.44–46,48
In recent years, due to the rapid development of POM chemistry, remarkable reviews about covalent modification of POMs have been published.12,18,49–53 However, there are few reports on asymmetrically functionalized Anderson-type polyoxometalates. Here in this review, we concentrate on the synthetic methodologies of asymmetric Anderson POMs and the functionalities of the resulting hybrids. According to the difference of the central heteroatom, this review is divided into sections of Mn-Anderson, Cr-Anderson, Al-Anderson and others (Table 1).
| Asymmetric compound | Type of isomer | Single-sided (S), double-sided (D) | Synthetic method | Application | Ref. |
|---|---|---|---|---|---|
| TBA = tetrabutylammonium, TMA = tetramethylammonium, GDM = guanidinium, DMF = N,N-dimethylformamide, KA oil = mixtures of cyclohexanone and cyclohexanol. | |||||
| Mn-Anderson | |||||
| [TBA]3{MnMo6O18[(OCH2)3CNH2][(OCH2)3CNO2]} | δ/δ | –NH2, –NO2 (D) | Fractional crystallization | — | 54 |
[TBA]3{MnMo6O18[(OCH2)3CN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHC5H4N][(OCH2)3CNO2]} CHC5H4N][(OCH2)3CNO2]} |
δ/δ | –NCHC5H4N, –NO2 (D) |
Post-modification | ||
| [TBA]3{MnMo6O18[(OCH2)3CNCHC13H9][(OCH2)3CNO2]} |
δ/δ | –NCHC13H9, –NO2 (D) |
Post-modification | ||
| [TBA]3{MnMo6O18[(OCH2)3CNCHC6H9OH][(OCH2)3CNO2]} |
δ/δ | –NCHC6H9OH, –NO2 (D) |
Post-modification | ||
| [TBA]3{MnMo6O18[(OCH2)3CNCHC14H9][(OCH2)3CNO2]} |
δ/δ | –NCHC14H9, –NO2 (D) |
Post-modification | ||
| [TBA]3{MnMo6O18[(OCH2)3CNH2][(OCH2)3CNHCH2C16H9]} | δ/δ | –NH2, –NHCH2C16H9 (D) | Fractional crystallization | Cell adhesion | 43 |
| [TBA]3{MnMo6O18[(OCH2)3CC9H17][(OCH2)3CNHCH2C16H9]} | δ/δ | –C9H17, –NHCH2C16H9 (D) | Fractional crystallization | Self-assembly on hydrophilic surfaces | 42 |
| [TBA]3{MnMo6O18[(OCH2)3CNH2][(OCH2)3CNHC21H19N2O4]} | δ/δ | –NH2, –NHC21H19N2O4 (D) | Post-modification | Photochromism/electrochromism | 60 |
| [TBA]3{MnMo6O18[(OCH2)3CNH2][(OCH2)3CNHCOC14H9]} | δ/δ | –NH2, –NHCOC14H9 (D) | Fractional crystallization | — | 55 |
| [TBA]3{MnMo6O18[(OCH2)3CNHCO(CH2)2COOH][(OCH2)3CNHCOC15H31]} | δ/δ | –NHCO(CH2)2COOH, –NHCOC15H31 (D) | Fractional crystallization | ||
| [TBA]3{MnMo6O18[(OCH2)3CNH2][(OCH2)3CNHCOC15H31]} | δ/δ | –NH2, –NHCOC15H31 (D) | Fractional crystallization | ||
| [TBA]3{MnMo6O18[(OCH2)3CNH2][(OCH2)3CNHCO2C14H9]} | δ/δ | –NH2, –NHCO2C14H9 (D) | Fractional crystallization | ||
| [TBA]3{MnMo6O18[(OCH2)3CNH2][(OCH2)3CNHCOC2H5]} | δ/δ | –NH2, –NHCOC2H5 (D) | Post-modification | ||
| [TBA]3{MnMo6O18[(OCH2)3CNHCO(CH2)2OCONC2H4O2][(OCH2)3CNHCO2C14H9]} | δ/δ | –NHCO(CH2)2OCONC2H4O2, –NHCO2C14H9 (D) | Post-modification | POM integrated peptides | 56 |
| [TBA]3{MnMo6O18[(OCH2)3CNHC21H35O7N4][(OCH2)3CNHC11H21O2N2]} | δ/δ | –NHC21H35O7N4, –NHC11H21O2N2 (D) | Post-modification | ||
| [TBA]3{MnMo6O18[(OCH2)3CNHC24H21N2O2][(OCH2)3CNHC21H19N2O4]} | δ/δ | –NHC24H21N2O2, –NHC21H19N2O4 (D) | Post-modification | Photochromism/electrochromism | 62 |
| [TBA]7{MnMo6O18[(OCH2)3CNH2][(OCH2)3CNMo6O18]} | δ/δ | –NH2, –NMo6O18 (D) | Other | — | 67 |
| [TBA]3{MnMo6O18(OH)3[(OCH2)3CNH2]} | χ | –NH2 (S) | Single-side modification | — | 68 |
| [TBA]3{MnMo6O18(OH)3[(OCH2)3CNH2]} | δ | –NH2 (S) | Single-side modification | — | 82 |
| [TBA]3{MnMo6O18[(OCH2)3CNH2][(OCH2)3CC2H5]} | δ/δ | –NH2, –C2H5 (D) | Step-by-step | — | |
| [TBA]3{MnMo6O18[(OCH2)3CNH2][(OCH2)3CNHCSNHC6H5]} | δ/δ | –NH2, –NHCSNHC6H5 (D) | Post-modification | — | 20 |
| [TBA]3{MnMo6O18[(OCH2)3CNH2][(OCH2)3CNHCOC6H4N3]} | δ/δ | –NH2, –NHCOC6H4N3 (D) | Fractional crystallization | Metal oxide oligomers | 41 |
[TBA]3{MnMo6O18[(OCH2)3CNH2][(OCH2)3CNHCOC3H6C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH]} CH]} |
δ/δ | –NH2, –NHCOC3H6CCH (D) |
Post-modification | ||
| [TBA]6{(MnMo6O18)2[(OCH2)3CNH2]2[(OCH2)3CNHCOC6H4C2HN3C3H6OCNHC(CH2O)3]} | δ/δ | –NH2, –NHCOC6H4C2HN3C3H6OCNH– (D) | Post-modification | ||
| [TBA]6{(MnMo6O18)2[(OCH2)3CNHCOC3H6CCH]2[(OCH2)3CNHCOC6H4C2HN3C3H6OCNHC(CH2O)3]} |
δ/δ | –NHCOC3H6CCH, –NHCOC6H4C2HN3C3H6OCNH– (D) |
Post-modification | ||
| [TBA]12{(MnMo6O18)4[(OCH2)3CNH2]2[(OCH2)3CNHCOC6H4C2HN3C3H6OCNHC(CH2O)3]3} | δ/δ | –NH2, –NHCOC6H4C2HN3C3H6OCNH– (D) | Post-modification | ||
| [TBA]3{MnMo6O18[(OCH2)3CNHCOC2H4C6H4N3][(OCH2)3CNHC21H19N2O4]} | δ/δ | –NHCOC2H4C6H4N3, –NHC21H19N2O4 (D) | Post-modification | Photochromism | 63 |
| [TBA]3{MnMo6O18[(OCH2)3CNHCOC2H4C6H4N3C2HCH2OC6H4C17H22N2BF2][(OCH2)3CNHC21H19N2O4]} | δ/δ | –NHCOC2H4C6H4N3C2HCH2OC6H4C17H22N2BF2, –NHC21H19N2O4 (D) | Post-modification | ||
| [TBA]3{MnMo6O18[(OCH2)3CNHCOC15H31][(OCH2)3CNHC21H19N2O4]} | δ/δ | –NHCOC15H31, –NHC21H19N2O4 (D) | Post-modification | Light- and solvent-controlled self-assembly | 65 |
| [TBA]3{MnMo6O18[(OCH2)3CNHCOC3H5][(OCH2)3CNHC21H19N2O4]} | δ/δ | –NHCOC3H5, –NHC21H19N2O4 (D) | Post-modification | Photochromism | 64 |
| [TBA]3{MnMo6O18[(OCH2)3CNHC20H29S8O][(OCH2)3CNHC21H19N2O4]} | δ/δ | –NHC20H29S8O, –NHC21H19N2O4 (D) | Post-modification | Non-linear-optical properties | 66 |
| NH4{MnMo6O18[(OCH2)3CNH3]2} | β | –NH3, –NH3 (S) | Single-side modification | Cyclohexanone, cyclohexanol and KA oil oxidation | 69 |
| K3Na3{MnW6O22[(OCH2)2C(CH2OH)2]} | β | –(OCH2)2C(CH2OH)2 (S) | Single-side modification | — | 70 |
| K3.5Na1.5H{MnW6O22[(OCH2)2C(CH2OH)(NH2)]} | β | –(OCH2)2C(CH2OH)(NH2) (S) | Single-side modification | ||
| K4Na2{MnW6O22[(OCH2)2C(CH2CH3)(CH2OH)]} | β | –(OCH2)2C(CH2CH3)(CH2OH) (S) | Single-side modification | ||
| K4NaH{MnW6O22[(OCH2)2C(CH3)(NH2)]} | β | –(OCH2)2C(CH3)(NH2) (S) | Single-side modification | ||
| [TBA]3{MnMo6O18[(OCH2)3CNH2][(OCH2)3CNHCOC2H4COOH]} | δ/δ | –NH2, –NHCOC2H4COOH (D) | Post-modification | — | 59 |
| [TBA]3{MnMo6O18[(OCH2)3CNH2][(OCH2)3CNHC33H43O7N4]} | δ/δ | –NH2, –NHC33H43O7N4 (D) | Post-modification | Inhibition β-amyloid fiber aggregation | |
| [TBA]3{MnMo6O18[(OCH2)3CNHC6H12ON][(OCH2)3CNHC27H32O6N3]} | δ/δ | –NHC6H12ON, –NHC27H32O6N3 (D) | Post-modification | ||
| [TBA]3{MnMo6O18[(OCH2)3CNHC11H21O2N2][(OCH2)3CNHC22H23O5N2]} | δ/δ | –NHC11H21O2N2, –NHC22H23O5N2 (D) | Post-modification | ||
| [TBA]3{MnMo6O18[(OCH2)3CNHC20H30O3N3][(OCH2)3CNHC13H14O4N]} | δ/δ | –NHC20H30O3N3, –NHC13H14O4N (D) | Post-modification | ||
| Na3{MnMo6O18[(OCH2)3CNH2][(OCH2)3CNHC57H72O12N9]} | δ/δ | –NH2, –NHC57H72O12N9 (D) | Post-modification | Switching a β sheet to a β turn of a POM peptide | |
| Na3{MnMo6O18[(OCH2)3CNH2][(OCH2)3CNHC38H66O9N7]} | δ/δ | –NH2, –NHC38H66O9N7 (D) | Post-modification | Enhancement of binding with the DnaK protein | |
| Cr-Anderson | |||||
| [TBA]5{H7CrMo6O24[(OCH2)3CCH2OH]2} | δ′ | –CH2OH (S) | Single-side modification | — | 75 |
| [TBA]3{CrMo6O18(OH)3[(OCH2)3CCH2OH]}·CH3COOH·NH(C2H5)3Cl | δ | –CH2OH (S) | Single-side modification | ||
| [TBA]3[CrMo6O18(OH)3C{(OCH2)3CH2OH}] | δ | –CH2OH (S) | Single-side modification | Oxidative esterification of alcohols | 76 |
| [TBA]3[CrMo6O18(OH)3C(OCH2)3CH3] | δ | –CH3 (S) | Single-side modification | N-Formylation of amines | 77 |
| [TBA]3{CrMo6O18(OH)3[(OCH2)3CCH2OH]}·12H2O | δ | –CH2OH (S) | Single-side modification | — | 81 |
| [TBA]3{CrMo6O18(OH)3[(OCH2)3CCH3]}·11H2O | δ | –CH3 (S) | Single-side modification | ||
| [TBA]3{CrMo6O18[(OCH2)3CCH3][(OCH2)3CCH2OH]} | δ | –CH2OH, –CH3 (D) | Step-by-step | ||
| [TBA]3{CrMo6O18(OH)3[(OCH2)3CNH2]}·[TBA]Br·2H2O | δ | –NH2 (S) | Single-side modification | Spontaneous chiral resolution | 83 |
| [TBA]3{CrMo6O18(OH)3[(OCH2)3CCH3]}·[TBA]Br | δ | –CH3 (S) | Single-side modification | ||
| [TBA]3{CrMo6O18(OH)3[(OCH2)3CC2H5]}·[TBA]Br·NH4Br | δ | –C2H5 (S) | Single-side modification | ||
| [TBA]2H{CrMo6O18(OH)3[(OCH2)3CNH2]}·3DMF·2H2O | χ | –NH2 (S) | Single-side modification | — | 68 |
| [TBA]2H{CrMo6O18(OH)3[(OCH2)3CCH3]}·DMF·[TBA]Br·CH3CN·2EtOH | χ | –CH3 (S) | Single-side modification | ||
| [TBA]2H{CrMo6O18(OH)3[(OCH2)3CC2H5]}·5DMF·[TBA]Br | χ | –C2H5 (S) | Single-side modification | ||
| [TBA]2H{CrMo6O18(OH)3[(OCH2)3CCH2OH]}·3DMF·H2O | χ | –CH2OH (S) | Single-side modification | ||
| [TBA]3{CrMo6O18(OH)3[(OCH2)3CNH2]}·[TBA]Br·2H2O | δ | –NH2 (S) | Single-side modification | — | 82 |
| [TBA]3{CrMo6O18(OH)3[(OCH2)3CC2H5]}·[TBA]Br·NH4Br | δ | –C2H5 (S) | Single-side modification | ||
| [TBA]6{CrMo6O18[(OCH2)3CNH2][(OCH2)3CC2H5]}2·[TBA]Br | δ/δ | –NH2, –C2H5 (D) | Step-by-step | ||
| [TBA]4{CrMo6O18(OH)4[(OCH2)2(CH2OH)CNH3]}2·4[TBA]Br·2NH4Br·15H2O | ψ | –(OCH2)2(CH2OH)CNH3 (S) | Single-side modification | — | 79 |
| [TBA]4{CrMo6O18(OH)4[(OCH2)2CH3CNH3]}2·4[TBA]Br·2NH4Br·14H2O | ψ | –(OCH2)2CH3CNH3 (S) | Single-side modification | ||
| [TBA]3{CrMo6O18(OH)4[O(CH2)2CHOH]}·3H2O | ψ | –(OCH2)2CHOH (S) | Single-side modification | ||
| [TBA]3{CrMo6O18(OH)3[(OCH2)3CC5H4N]}·[TBA]Br·3H2O | δ | –(CH2O)3CC5H4N (S) | Single-side modification | High nuclear metal halide cluster | 78 |
| [TBA]3{CrMo6O18(OH)3[(OCH2)3CCH2OCH2C(CH2OH)3]}·2H2O | δ | –CH2OCH2C(CH2OH)3 (S) | Single-side modification | — | 72 |
| K6{[CrMo6O18(OH)3]2[(OCH2)3CCH2OCH2C(CH2O)3]}·14H2O | δ | –CH2OCH2– (S) | Single-side modification | ||
| (NH4){CrMo6O18[(OCH2)3CNH3]2} | β | –NH3 (S) | Single-side modification | — | 80 |
| [TBA]2(NH4){CrMo6O18[(OCH2)3CC2H5]2}·2H2O | β | –C2H5 (S) | Single-side modification | ||
| K3Na3{CrO3W6O18[(OCH2)3CCH2OH]} | δ | –CH2OH (S) | Single-side modification | — | 90 |
| (NH4)2{CrMo6O18(OH)3[(OCH2)3CNH3]}·5H2O | δ | –NH3 (S) | Single-side modification | — | 73 |
| Al-Anderson | |||||
| [TBA]3{AlMo6O18(OH)3[(OCH2)3CCH2OH]}·13H2O | δ | –CH2OH (S) | Single-side modification | — | 84 |
| [TBA]3{AlMo6O18(OH)3[(OCH2)3CNH2]}·7H2O | δ | –NH2 (S) | Single-side modification | ||
| [TBA]3{AlMo6O18(OH)3[(OCH2)3CCH2CH3]}·11H2O | δ | –CH2CH3 (S) | Single-side modification | ||
| [TBA]3{AlMo6O18(OH)3[(OCH2)3CNHCH2COOH]}·10H2O | δ | –NHCH2COOH (S) | Single-side modification | ||
| [TBA]6{Al2Mo12O36(OH)6[(OCH2)3CCH2OCH2C(OCH2)3]}·13H2O | δ | –CH2OCH2– (S) | Single-side modification | ||
| [TBA]3{AlMo6O18(OH)3[(OCH2)3CNHCOCH2C6H4NNC6H5]} | δ | –NHCOCH2C6H4NNC6H5 (S) | Post-modification | Chiral migration | 85 |
| [TBA]3{AlMo6O18(OH)3[(OCH2)3CC2H5]}·[TBA]Br | δ | –C2H5 (S) | Single-side modification | Spontaneous chiral resolution | 82 |
| [TBA]6{AlMo6O18[(OCH2)3CC2H5][(OCH2)3CNH2]}2·3DMF | δ/δ | –C2H5, –NH2 (D) | Step-by-step | ||
| [TBA]3{AlMo6O18(OH)3[(OCH2)3CNHC11H11S8O]} | δ | –NHC11H11S8O (S) | Single-side modification | Non-linear-optical properties | 66 |
| Na3K3{AlW6O21[(OCH2)3CCH2OH]}·16H2O | δ | –CH2OH (S) | Single-side modification | — | 90 |
| [TBA]3{AlMo6O18(OH)3[(OCH2)3CCH3]} | δ | –CH3 (S) | Single-side modification | Alcohol oxidation | 88 |
| [TBA]3{AlMo6O18(OH)3[(OCH2)3CCH3]·Cl} | δ | –CH3 (S) | Single-side modification | ||
| [TBA]3{AlMo6O18(OH)3[(OCH2)3CCH3]·Br} | δ | –CH3 (S) | Single-side modification | ||
| [TBA]3{AlMo6(OH)3[(OCH2)3CNHCOC20H19N2O3]} | δ | –NHCOC20H19N2O3 (S) | Single-side modification | Photochromism/photoluminescence | 86 |
| [TBA]3{AlMo6(OH)3[(OCH2)3CNHCOC21H19N2O]} | δ | –NHCOC21H19N2O (S) | Single-side modification | ||
| [TBA]3[AlMo6O18(OH)3(OCH2)3CNHCOC11H23]·9H2O | δ | –NHCOC11H23 (S) | Single-side modification/post-modification | Binding with human serum albumin | 87 |
| [TBA]4{AlMo6O18(OH)3[(OCH2)3CNH2]Cl} | δ | –NH2 (S) | Single-side modification | — | 89 |
| [TBA]3{AlMo6O18[(OCH2)3CCH2OH][(OCH2)3CC6H4NO2]} | δ | –CH2OH, –C6H4NO2 (D) | Step-by-step | Metal-oxo-cluster oligomers | 92 |
| [TBA]3{AlMo6O18[(OCH2)3CNH2][(OCH2)3CC6H4NO2]} | δ | –NH2, –C6H4NO2 (D) | Step-by-step | ||
| Others | |||||
| [TBA]3{GaMo6O18(OH)3[(OCH2)3CCH2OH]}·12H2O | δ | –CH2OH (S) | Single-side modification | Inversion of the protein surface charge | 35 |
| [TMA]2{GaMo6O18(OH)3[(OCH2)3CNH3]}·7H2O | δ | –NH3 (S) | Single-side modification | ||
| Na[TMA]2{FeMo6O18(OH)3[(OCH2)3CNH3]} (OH)·6H2O | δ | –NH3 (S) | Single-side modification | ||
| [TMA]3{GaMo6O18(OH)3[(OCH2)3CCH2OH]}·nH2O | δ | –CH2OH (S) | Single-side modification | ||
| [GDM]3{GaMo6O18(OH)3[(OCH2)3CCH2OH]}·nH2O | δ | –CH2OH (S) | Single-side modification | ||
| [TBA]3{HCuMo6O18[(OCH2)3CCH3]2}·[(HOCH2)3CCH3]·CH3CN | δ/χ | –CH3 (D) | Other | — | 45 |
| Na2[TMA]2[NiW6O18(OH)3(OCH2)3CCH2OH]·9H2O | δ | –CH2OH (S) | Single-side modification | Binding with human serum albumin | 91 |
| Na2[NH3C(CH2OH)3][NiMo6O18(OH)3(OCH2)3CNH3]·11.75H2O | δ | –NH3 (S) | Single-side modification | — | 93 |
| [TBA]3{CoMo6O17(OH)[(OCH2)3CCH3]2}·DMF·CH3CH2OH | δ/χ | –CH3 (D) | Other | — | 44 |
| [TBA]3{CoMo6O18(OH)3[(OCH2)3CCH3]}·10H2O | δ | –CH3 (S) | Single-side modification | ||
| [TBA]3{CoMo6O18(OH)2(CH3COO)[(OCH2)3CCH3]} | δ | –CH3 (S) | Single-side modification | ||
| [TBA]2{CoMo6O17(OCH3)[(OCH2)3CCH3]2} | δ/δ | –CH3, –OCH3 (D) | Other | ||
| [[TBA]3{FeMo6O18(OH)3[(OCH2)3CNH2]} | δ | –NH2 (S) | Single-side modification | Aerobic oxidation of aldehydes in water | 94 |
| [TBA]3{CuMo6O17(CH3O)[(OCH2)3CCH3]2}·2C3H7NO | δ/χ | –CH3 (D) | Other | — | 46 |
| Na3K3{CoW6O21[(OCH2)3CCH2OH]}·14H2O | δ | –CH2OH (S) | Single-side modification | — | 90 |
| Na3K3{CoW6O21[(OCH2)3CCH3]}·16H2O | δ | –CH3 (S) | Single-side modification | ||
| (NH4)4{ZnMo6O18(OH)3[(OCH2)3CNH2]}·4H2O | δ | –NH2 (S) | Single-side modification | CO2 cycloaddition | 48 |
| (NH4)4{CuMo6O18(OH)3[(OCH2)3CNH2]}·4H2O | δ | –NH2 (S) | Single-side modification | ||
| [TBA]3{ZnMo6O17(OH)[(OCH2)3CCH3]2}·10H2O | δ/χ | –CH3 (D) | Other | ||
| (NH4)3{CuMo6O18(OH)3[(OCH2)3CNH3]}·6H2O | δ | –NH3 (S) | Single-side modification | — | 47 |
2. Mn-Anderson
2.1 Double-sided asymmetric Mn-Anderson
As the earliest Anderson structure reported in the field of asymmetric modification, Mn-Anderson [MnMo6O24]9− (MnMo6) has always been widely employed by researchers. The first reported asymmetric Mn-Anderson structure NH2–MnMo6–NO2 was separated by Song et al. in 2008.54 In this work, two different triol ligands tris(hydroxymethyl)aminomethane (Triol-NH2) and tris(hydroxymethyl)nitromethane (Triol-NO2) were mixed and reacted with [TBA]4Mo8O26·2H2O and Mn(CH3COO)3 in anhydrous acetonitrile, as shown in Fig. 2a. Predictably, after the reaction, three products were obtained: asymmetric NH2–MnMo6–NO2 and two symmetric NH2–MnMo6–NH2 and NO2–MnMo6–NO2 clusters. Because the crystallization efficiency and polarity of the asymmetric NH2–MnMo6–NO2 were different from those of the symmetric ones, it was possible to separate it from the mixed mother liquor. To obtain the pure target asymmetric hybrid, the filtered mother liquor was left for evaporation. The obtained orange single crystals were collected every 20 min, and each batch was analyzed by electrospray ionization mass spectrometry (ESI-MS) to determine the purity. As shown in Fig. 2b, when the molecular ion peaks appeared at m/z = 1669 (asymmetric product), and the ion peaks at m/z = 1640 and 1700 (symmetric products) disappeared, the batch allocated to pure asymmetric crystals was kept. Although this fractional crystallization method required mass spectrometry to examine the purity, it pointed out a feasible way for researchers to obtain asymmetric Mn-Anderson POMs. It was worth noting that the –NH2 group of the asymmetric NH2–MnMo6–NO2 hybrid was nucleophilic, while the –NO2 group was relatively inert from a reactivity perspective. Therefore, the NH2–MnMo6–NO2 hybrid could further react with aldehydes to form a series of new POM asymmetric hybrids, which provided a good opportunity to construct a new set of asymmetric Mn-Anderson POMs. For instance, when 4-pyridylcarboxyaldehyde was selected, a new asymmetric Mn-Anderson POM, NO2–MnMo6–NCHC5H4N, was obtained and fully characterized by mass spectrometry and single-crystal X-ray crystallography (Fig. 2c).
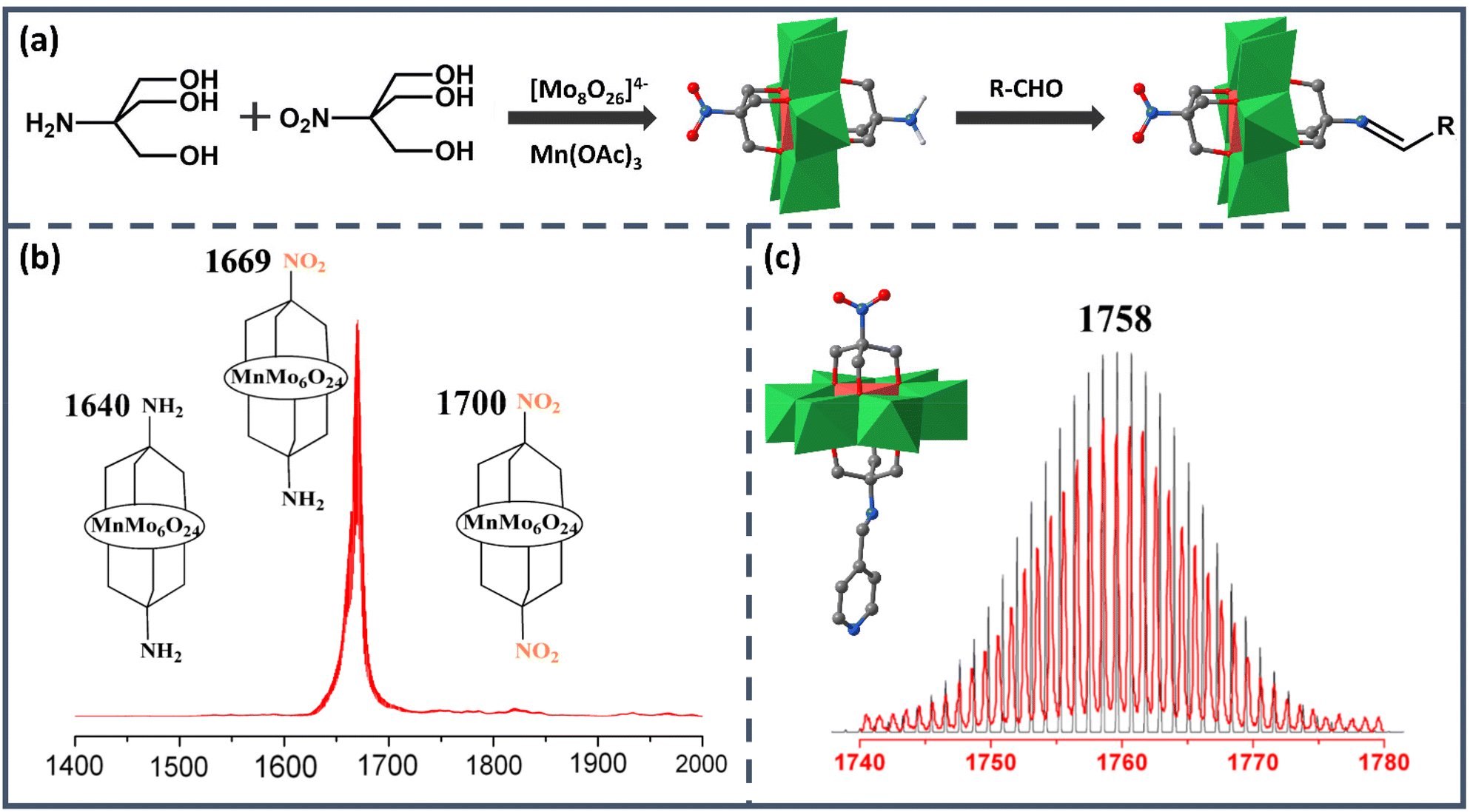 | ||
| Fig. 2 (a) Schematic route of the synthesis process of [TBA]3[NH2–MnMo6–NO2] and further reaction with aldehydes. (b) ESI-MS spectrum of the molecular ion peak of [TBA]3[NH2–MnMo6–NO2] without the observation of other symmetric products. (c) X-ray crystal structure and the ESI-MS spectrum of the molecular ion peak of [TBA]3[NO2–MnMo6–NCHC5H4N]. Color code: {MoO6}, green octahedron; {MnO6}, red octahedron; C, gray; O, deep red; N, blue; H, light grey. | ||
In 2009, Song et al. anchored the asymmetrically functionalized Anderson hybrids onto the Au surface via self-assembled monolayers (SAMs).43 The modified surface showed selective fibroblast cell adhesion properties. Interestingly, the cells could specifically adhere to the patterned areas containing aromatic pyrene-modified MnMo6 platforms, while no adhesion was observed in the patterned areas of NH2–MnMo6 or pure pyrene platforms. The different cell responsive behavior to SAM systems with different terminal groups provided the opportunity to use different functional model substrates to manipulate cell adhesion. In 2010, Cronin and co-workers synthesized a novel asymmetric Mn-Anderson hybrid [TBA]3[MnMo6O18((OCH2)3CC9H17)((OCH2)3CNHCHC16H9)], which possessed a long alkyl chain and highly conjugated pyrene units on both sides of the Anderson cluster, using the same separation method.42 This asymmetric hybrid exhibited intriguing self-assembly behaviour on a hydrophilic silicone surface, and formed a protein-like fibrous nanostructure with a high aspect ratio and anisotropy. Such behaviour was thought to be caused by the synergistic effects between the aromatic π–π interaction and the hydrophobic interaction of alkyl chains.
Although the fractional crystallization method has proved its feasibility in purifying asymmetric hybrids, the tedious separation workup and poor reproducibility limit its routine use. In 2013, Cronin et al. found that when the affinities of the two triol ligands for the stationary phase were significantly different, the asymmetric Mn-Anderson compound could be separated from two corresponding symmetrical by-products by C18 RP-HPLC (reverse phase-high performance liquid chromatography).55 Using this method, they separated an asymmetric precursor: NH2–MnMo6–Fmoc (Fmoc = 9-fluorenylmethyloxycarbonyl), as shown in Fig. 3a. It was envisioned that the NH2–MnMo6–Fmoc compound could be used as a “universal” asymmetric precursor to synthesize almost any asymmetric organic–inorganic Mn-Anderson hybrids. For example, this “universal” precursor could react with propionic anhydride to give asymmetric C2H5CONH–MnMo6–Fmoc (Fig. 3b), which was able to be treated with piperidine to remove the –Fmoc group, therefore leaving the deprotected –NH2 group for further modification (Fig. 3c). Theoretically, it was possible to prepare any kind of asymmetric Mn-Anderson hybrid using this “universal” precursor. To this end, this “universal” precursor was incorporated into a solid-phase peptide synthesis approach by Cronin et al. to successfully prepare unnatural amino acids, laying the foundation for the combinatorial synthesis of inorganic amino acids and their potential application in biomedical and nanoscience research.56
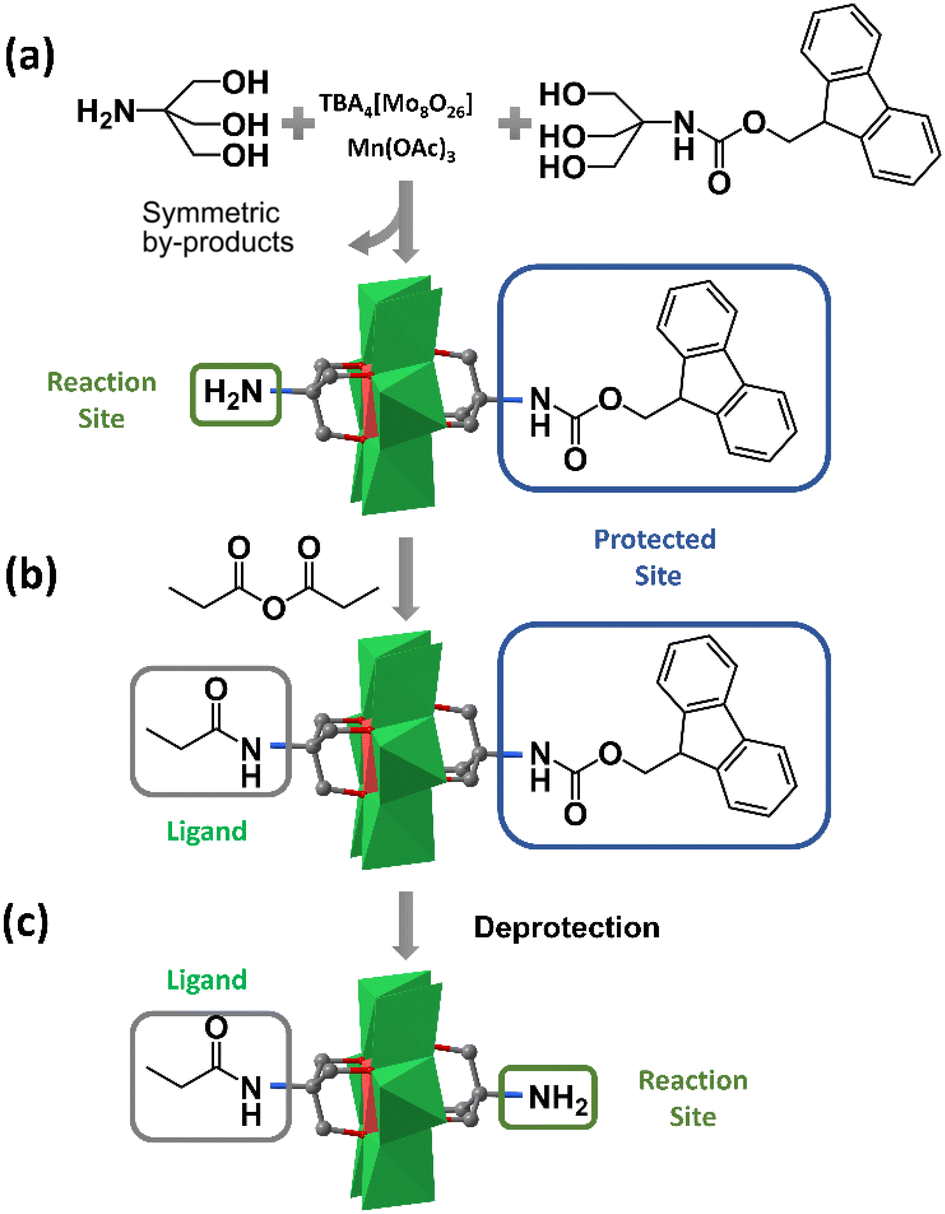 | ||
| Fig. 3 (a) Synthesis method of the “universal” asymmetric Mn-Anderson precursor: NH2–MnMo6–Fmoc. (b) Synthesis method of C2H5CONH–MnMo6–Fmoc. (c) Removal of the –Fmoc group. Color code: {MoO6}, green octahedron; {MnO6}, red octahedron; C, gray. | ||
Taking advantage of this “universal” precursor, the Cronin group subsequently synthesized a series of asymmetric Mn-Anderson hybrids bearing azide and alkyne end groups.41 These hybrids could be used as building blocks to precisely synthesize metal oxide oligomers with designed molecular structures and cluster numbers via a Cu-catalyzed alkyne–azide cycloaddition (CuAAC) reaction. Compared with the previously reported POM coupling method,57,58 this CuAAC method allowed for modular synthesis and sequential coupling of POM oligomers.
Recently, an automated inorganic amino acid synthesis system was developed by Cronin et al.59 This system permitted the automatic coupling of asymmetric Anderson NH2–MnMo6–COOH into standard amino acids with tunable peptide sequences and optimal combinations. Such POM-incorporated amino acids exhibited fascinating functions, such as significant inhibition of the aggregation of amyloid Aβ17–20, switching of the β sheet of amphiphilic KFE8 into a β turn, and enhancement of binding with the bacterial chaperone DnaK protein.
The asymmetric products could also be obtained using a post-modification method, which was performed to selectively modify one –NH2 group of the Mn-Anderson cluster NH2–MnMo6–NH2, leaving the other group intact for further functionalization. Oms and co-workers found that the asymmetrically functionalized compound could be synthesized by controlling the reaction ratio of SPCOOH (SP = spiropyran) and NH2–MnMo6–NH2.60 When the ratio was controlled to be 0.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, only one –NH2 group was modified with the SP entity, leading to the asymmetric product of NH2–MnMo6–SP. Similarly, Floquet and Cadot et al. found that when the stoichiometric ratio between the highly reactive cluster [B10H9CO]− and NH2–MnMo6–NH2 was 1:1, a powder corresponding to a mixture containing 80% of symmetric products and 20% of asymmetric products was obtained.61 Unfortunately, they were unable to separate the asymmetric structure from the mixture. Cronin et al. also examined the post-modification method and found that when the ratio of succinic anhydride and NH2–MnMo6–NH2 was fixed to 1.1:1, asymmetric products could be obtained solely.59 From the abovementioned cases, it can be concluded that the feeding ratio of the post-modification method towards asymmetric hybrids varies from one to another, and highly depends on the anchoring organic components and reaction conditions.
:1, only one –NH2 group was modified with the SP entity, leading to the asymmetric product of NH2–MnMo6–SP. Similarly, Floquet and Cadot et al. found that when the stoichiometric ratio between the highly reactive cluster [B10H9CO]− and NH2–MnMo6–NH2 was 1:1, a powder corresponding to a mixture containing 80% of symmetric products and 20% of asymmetric products was obtained.61 Unfortunately, they were unable to separate the asymmetric structure from the mixture. Cronin et al. also examined the post-modification method and found that when the ratio of succinic anhydride and NH2–MnMo6–NH2 was fixed to 1.1:1, asymmetric products could be obtained solely.59 From the abovementioned cases, it can be concluded that the feeding ratio of the post-modification method towards asymmetric hybrids varies from one to another, and highly depends on the anchoring organic components and reaction conditions.
Based on the successful preparation of NH2–MnMo6–SP, Oms and co-workers have largely extended their works on preparing novel photo- and electro-chromic asymmetric Anderson hybrids. As shown in Fig. 4, the double-sided asymmetric SN–MnMo6–SP (SN = spironaphthoxazine) hybrid was prepared by post-functionalization of NH2–MnMo6–SP with SNCOOH.62 The asymmetric SN–MnMo6–SP hybrid showed a multi-state colorization process (from deep blue to red-purple) upon UV irradiation, and a much slower decolorization process in the dark, when compared with the symmetric SN–MnMo6–SN hybrids. This was mainly due to the fact that the zwitterionic merocyanine (MC) form of the SP group anchored onto the Anderson core was more stable. A distinguished multi-state colorization process of SN–MnMo6–SP was also observed in solution under an electric field.
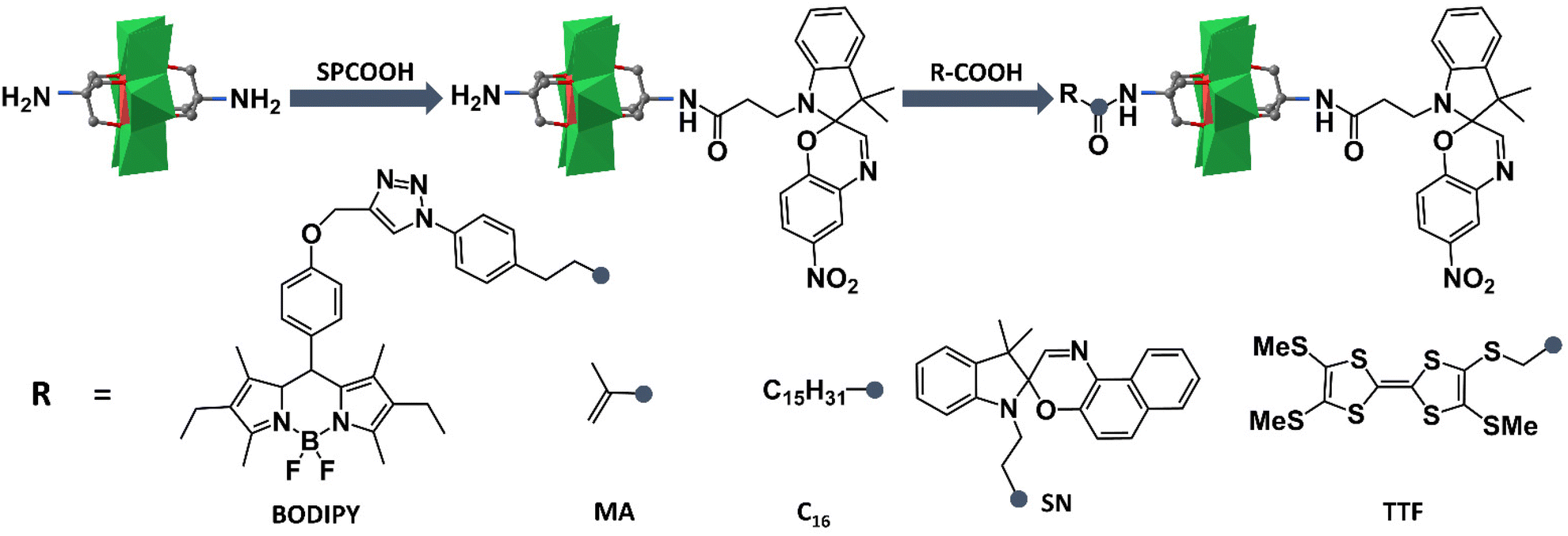 | ||
| Fig. 4 Schematic route of the synthesis process of NH2–MnMo6–SP and further reaction with acids. Color code: {MoO6}, green octahedron; {MnO6}, red octahedron; C, gray. | ||
Following a similar synthetic strategy, fluorescent BODIPY was also tethered onto the pre-synthesized NH2–MnMo6–SP.63 The resulting BODIPY–MnMo6-SP hybrid exhibited interesting photo-coupling phenomena between the two different organic components. Upon UV irradiation, the isomerization of SP to the MC form in the structure of BODIPY–MnMo6-SP could lead to a gradual decrease of the fluorescence of the BODIPY part, while the inversion of MC to the SP form could fully restore the emission intensity of the BODIPY moiety. Such a photo-coupling process may be caused by the efficient intramolecular energy transfer between the BODIPY and SP components facilitated by covalent bonding. The photochromic properties of the asymmetric NH2–MnMo6–SP hybrid could also be introduced into a polymer matrix.64 Post-functionalization of NH2–MnMo6–SP with a polymerizable MA moiety (MA = methacrylate) could lead to a novel organic–inorganic monomer, MA–MnMo6–SP. Copolymerization of MA–MnMo6–SP with methyl methacrylate (MMA) could generate ultra-sensitive polymer materials even at a very low SP dosage (1.1 wt%). Liu and Mialane et al. investigated the self-assembly behaviour of an asymmetric Anderson hybrid upon photo-irradiation.65 They designed and synthesized a new asymmetric Anderson hybrid, C16–MnMo6–SP, which bore a photochromic SP unit on one side of the Anderson cluster and a long hydrophobic alkyl chain on the other. It was observed that the asymmetric C16–MnMo6–SP hybrid self-assembled into vesicles in a polar solvent under UV irradiation, and de-assembled upon visible light irradiation. In 2018, Dolbecq and Ruhlmann et al. reported a tetrathiafulvalene (TTF) functionalized asymmetric Anderson hybrid, TTF–MnMo6–SP.66 Hyper-Rayleigh scattering measurements showed that due to the remarkable electro-attractive effects of the MnMo6 cluster, strong enhancement of the β values of the TTF moiety was observed. In addition, the oxidation of the TTF moieties by Fe3+ ions could also increase the NLO response because of the generation of TTF+˙ free radicals, which induced new absorption bands in the visible and near-infrared regions.
Different from the fractional crystallization or post-modification method, asymmetric Anderson hybrids could sometimes be obtained under non-conventional conditions such as microwave irradiation. Ritchie et al. reported their discovery of microwave-assisted synthesis of an asymmetric Lindqvist–Anderson hybrid dimer, which was composed of a NH2–MnMo6–NH2 cluster connected to a Mo6 Lindqvist anion through the MoO bond.67 The synthetic parameters were almost the same as the preparation of NH2–MnMo6–NH2 except the use of microwave irradiation instead of refluxing.
2.2 Single-sided asymmetric Mn-Anderson
Single-sided asymmetric assembly means that only one side of the planar Anderson cluster was functionalized with triol ligands, whereas the other side remained unaffected. For the case of Mn-Anderson, a small number of single-sided compounds were reported. In 2015, Wei et al. synthesized single-sided δ type MnMo6–NH2 by refluxing the mixture of Triol-NH2 and the pre-synthesized [Mn(OH)6Mo6O18]3− in aqueous solution.68 A χ isomer of single-sided MnMo6–NH2 was also reported by selectively activating μ2-O through protonation.In 2018, Wei and Zhang et al. reported a very interesting butterfly-shaped β isomer of the Mn-Anderson compound (NH4){MnMo6O18[(CH2O)3CNH3]2} by a reaction of [Mn(OH)6Mo6O18] and triol-NH2 ligands in hot DMF under a N2 atmosphere.69 Different from most of the reported single-sided compounds with a planar α-structure, the two Triol-NH2 groups were grafted onto the same side of the β isomer (Fig. 5a). Due to its non-planar configuration, the active Mn3+ central heteroatom was more “uncovered” than the planar topology of the α isomer, which led to an excellent catalytic performance in the selective oxidation of a mixture of cyclohexanol and cyclohexanone to adipic acid. Besides the bi-functionalized β isomer, a series of mono-derivatized β isomers were also prepared by Wei et al. using [MnW6O24]8− (MnW6) as the starting material (Fig. 5b).70 Thanks to their butterfly-shaped structure, these types of clusters showed unprecedented affinity for coordination with metal ions and would have potential in the synthesis of more complicated transition metal frameworks.
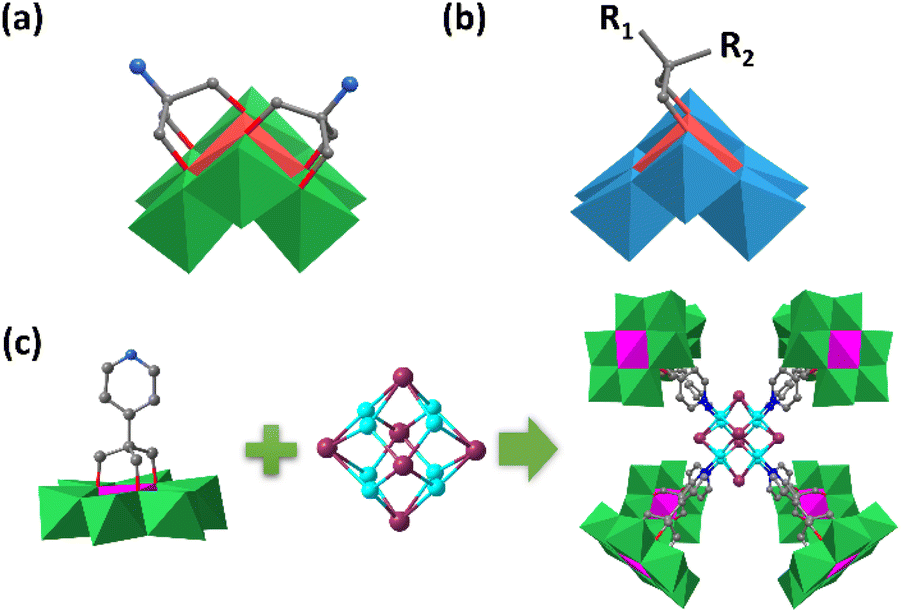 | ||
| Fig. 5 (a) Schematic diagram of the {[H3NC(CH2O)3]2MnMo6O18}− structure. (b) Schematic diagram of the [{R1R2C(CH2O)2}MnIVW6O22]6− structure. (c) Synthesis diagram of [TBA]14[Cu8I6][HCr(OH)3Mo6O18L3]8. Color code: {MoO6}, green octahedron; {MnO6}, red octahedron; {WO6}, blue octahedron; {CrO6}, pink octahedron; C, gray; N, light blue; Cu, turquoise; I, purple. | ||
3. Cr-Anderson
In 1970, Perloff first synthesized the Cr-Anderson compound Na3[Cr(OH)6Mo6O18]·8H2O (CrMo6) by refluxing the mixture of Na2MoO4 and Cr(NO3)3 in aqueous solution.71 Wei, Cronin, and Song et al. explored the asymmetric organic modification methods of Cr-Anderson. Different from Mn-Anderson which could be modified in double sides with organic ligands, Cr-Anderson could be functionalized only in single side with high yield and high selectivity, even when the ratio of triol ligands to the parent Cr-Anderson was two or much higher than two.72,73 The other side of Triol-Cr-Anderson could be further modified in a stepwise manner with other different triol ligands (e.g. Triol-CH2OH, Triol-CH3). Therefore, the double-sided asymmetric Cr-Anderson compounds were controllably synthesized with high yields without the symmetric by-products.3.1 Single-sided asymmetric Cr-Anderson
In the classical functionalization methods of asymmetric Mn-Anderson hybrids, the final products were obtained by refluxing the mixture of Mo8O26 salts, Mn(CH3COO)3, and organic triol ligands in an organic solvent.74 For the preparation of organic modified Cr-Anderson, the pre-synthesized Na3[Cr(OH)6Mo6O18] was reacted with pentaerythritol (Triol-CH2OH), resulting in the formation of single-side functionalized CrMo6–CH2OH in high yield and selectivity in aqueous solution.75 The authors indicated that the high selectivity of the single-sided product might be related to the aqueous environment. In crystals, two CrMo6–CH2OH molecules formed a very stable dimeric structure in which the unmodified POM sides were linked together by hydrogen bonds. In acetonitrile solution, the dimer structure could be split into monomers by adding triethylamine and FeCl3. It was shown that all the three μ3-O atoms on the unmodified side were protonated, inferring that these μ3-O atoms were activated for further modification.The single-side functionalized CrMo6–CH2OH had an advantage that the heteroatom Cr(III) could be exposed as a catalytically active site. CrMo6–CH2OH was suggested to be a cheap, easily prepared, and recoverable green catalyst for oxidative transformation from alcohols to esters.76 In the presence of H2O2, the center Cr(III) could be converted into a Cr(V) intermediate which served as an oxidization site for alcohol oxidation and an acid site for an addition reaction of an aldehyde and alcohol. Wei et al. used the single-sided CrMo6–CH3 as a catalyst for the formylation of amines with formic acid, which had shown excellent activity, chemoselectivity and a broad substrate scope.77 Compared with the inorganic simple Anderson POMs, the organic modified POMs exhibited more structural stability and relevant structural modification for specific catalytic reactions.
The organic ligands in functionalized CrMo6 could further coordinate with other transition metal ions forming a more complex structure. Zheng and Yang et al. grafted [2-(hydroxymethyl)-2-(pyridin-4-yl)-1,3-propanediol] (Triol-pyridine) on the single-side of CrMo6 under hydrothermal conditions (Fig. 5c).78 The assembly of the resultant CrMo6-pyridin precursor with CuI gave rise to an unprecedented composite hybrid building up from one high nuclear cationic metal halide cluster [Cu8I6]2+ core and eight anionic CrMo6-pyridine ligands. Unlike the single-sided CrMo6-pyridin, the double-side modified pyridin–MnMo6–pyridin preferred to form 2D or 3D extended frameworks with the linkage of binuclear {Cu2I2} and tetranuclear {Cu4I4} cores.
In most cases, the triol ligands were grafted onto the three μ3-O atoms around the Cr atom. The modification of unreactive μ2-O atoms became a great challenge. Wei et al. reported that the μ2-O atoms can be regioselectively activated to become μ2-OH reactive sites through proton introduction and further be controllably modified with single-sided triol ligands forming the χ isomers, in which two μ3-OH and one μ2-OH were substituted.68 After realizing the importance of additional protons, they extended the strategy to the synthesis of diol functionalized single-sided Cr-Anderson by adding excess hydrochloric acid. The diol ligands were substituted with two activated μ3-OH on one side of CrMo6. The desired diol functionalized compounds were denoted as ψ isomers and their structures were more accidental than can be theoretically foreseen.79 In 2017, Wei et al. discovered the first triol-functionalized butterfly-shaped β isomers of Cr-Anderson POMs.80 Different from the flat α isomers, the butterfly-shaped β isomers possessed two μ4-O atoms that were hidden in the concave side of the butterfly-shaped structure. The two organic ligands were modified on the same side of the “wings of butterfly”. These single-side functionalized molecules enriched the POM family and provided opportunities for the exploration of more applications based on the distorted Cr sites.
3.2 Double-sided asymmetric Cr-Anderson
Taking advantage of the high yield single-sided Cr-Anderson, the asymmetric double-sided Cr-Anderson molecules could be more controllably obtained and the tedious isolation process was avoided. Song et al. presented a stepwise method that had been adopted during the preparation of the asymmetric compound CH3–CrMo6–CH2OH, in which organic Triol-CH2OH and Triol-CH3 were separately modified on the two sides of CrMo6 (Fig. 6).81 Firstly, the single-sided compounds were prepared in the presence of Triol-CH2OH and an equivalent amount of pre-synthesized CrMo6 under hydrothermal conditions. Secondly, the pure crystals of CrMo6–CH2OH were mixed with another ligand Triol-CH3 in a molar ratio of 1:1 to obtain the asymmetric double-sided compound CH3–CrMo6–CH2OH. Under the hydrothermal conditions, the symmetric double-side modified CH3–CrMo6–CH3 and HOCH2–CrMo6–CH2OH were obtained with the molar ratio of CrMo6 to Triol ligands being 1:3 in aqueous solution.
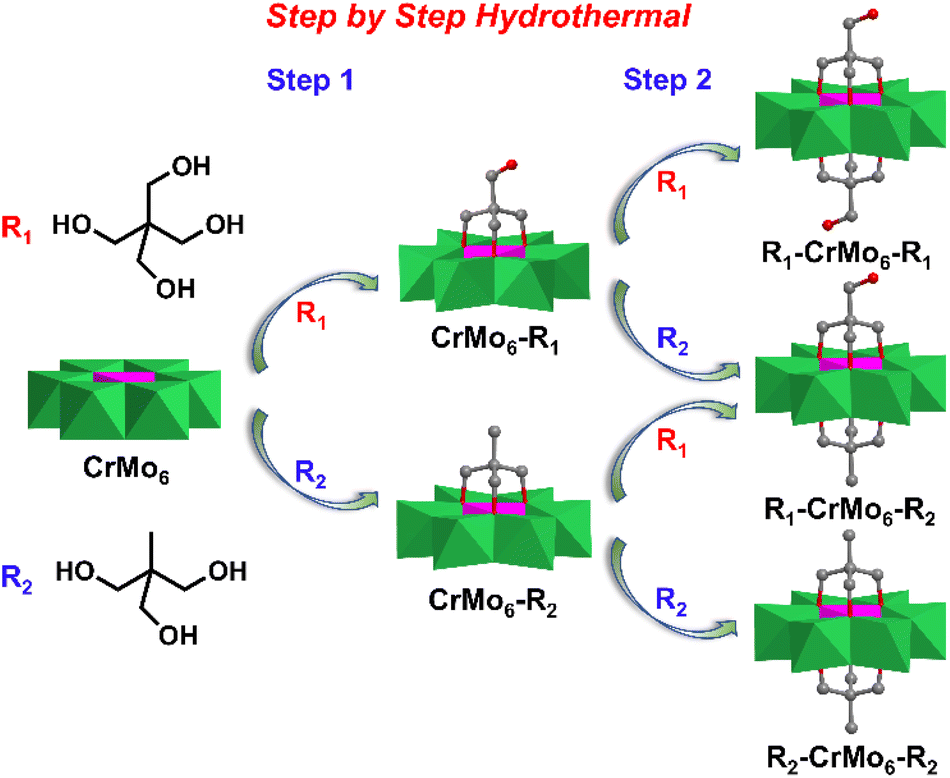 | ||
| Fig. 6 Synthesis routes of a series of tripodal alcohol substituted Anderson-type POMs under hydrothermal conditions via a pre-designed step-by-step strategy. Color code: {MoO6}, green octahedron; {CrO6}, pink octahedron; C, gray; O, deep red. | ||
Interestingly, some of these asymmetric compounds, including single-side and double-side functionalized compounds, were found to crystallize in the chiral space group although all of the precursors were achiral.82,83 Wei et al. synthesized the asymmetric compound NH2–XMo6–CH2CH3 (X = Cr, Mn, Al) via a two-step modification strategy (Fig. 7).82 They found that all these compounds crystallized in the orthorhombic chiral space group P212121 and their spontaneous chiral resolution can be achieved by tuning a 65:35 DMF/MeCN mixed solvent during the crystallization process. The circular dichroism (CD) spectra suggested that the chiroptical activity of these asymmetric hybrids was stable in the solid state while racemization was observed in the solution state. They claimed that the origin of their chirality was due to the symmetry reduction of the central Cr–O6 coordination structure. The Cr–O6 structure has the centre and mirror D3d symmetry in parent Anderson while it reduced to the centre and mirror breaking C1 symmetry in double-sided asymmetric triol functionalized Anderson clusters.
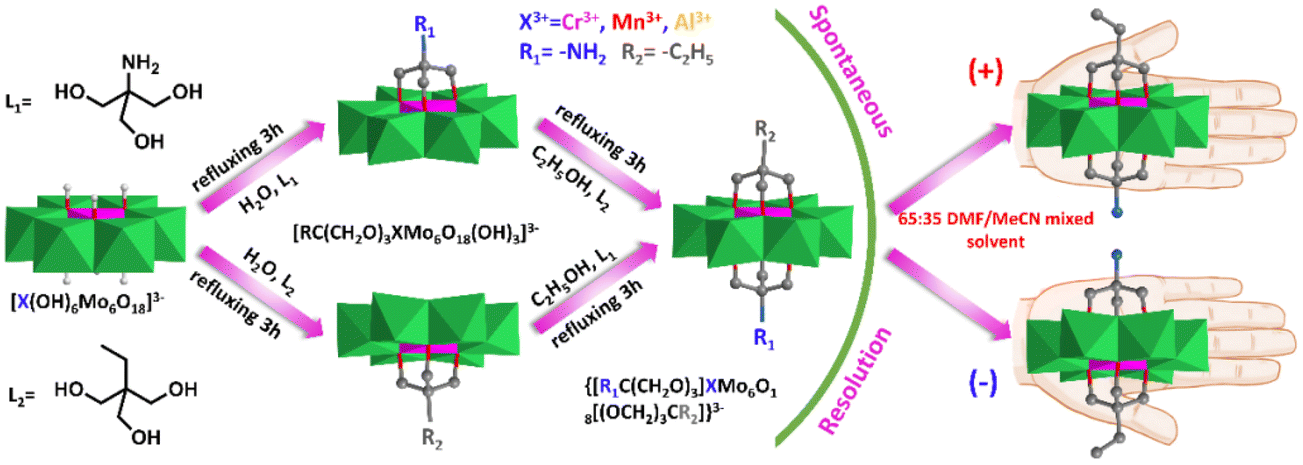 | ||
| Fig. 7 The asymmetric compound NH2–XMo6–CH2CH3 (X = Cr, Mn, Al) was synthesized by a two-step modification strategy. Its spontaneous chiral resolution could be achieved by adjusting the 65:35 DMF/MeCN mixed solvent in the crystallization process. Color code: {MoO6}, green octahedron; {CrO6}, pink octahedron; C, gray; N, light blue; H, light grey. | ||
4. Al-Anderson
Al-Anderson POMs were more inclined to form single-sided asymmetric structures than the symmetric ones in aqueous solution. This, according to Wu and Li et al., was because of the improved stability of Al-Anderson after single-side modification and the inertness of the remaining μ3-O atoms on the other side of the Anderson cluster.84 The double-side asymmetric modification of Al-Anderson POMs could be achieved by adopting a stepwise modification method.824.1 Single-sided asymmetric Al-Anderson
In 2014, Wu and Li et al. reported a series of single-sided Al-Anderson hybrids, [TBA]3[AlMo6O24{(OCH2)3CR}] (R = CH2OH, NH2, CH2CH3, NHCH2COOH, CH2OCH2C(CH2OH)3).84 These compounds were synthesized adopting a similar method as the preparation of single-sided Cr-Anderson.75 It should be noted that the asymmetric AlMo6–CH2OH hybrid could also be directly prepared by refluxing the mixture of AlCl3, Na2MoO4, and Triol-CH2OH in acidified aqueous solution despite a relatively low yield (ca. 15% based on Mo). The asymmetric AlMo6–NH2 hybrid offered a platform for further modification towards multi-functional applications. Wu et al. constructed an azobenzene (Azo) functionalized single-sided AlMo6–Azo hybrid through the amidation reaction between Azo–COOH and AlMo6–NH2.85 This AlMo6–Azo hybrid displayed interesting chirality migration properties when combined with α-cyclodextrin (α-CD) and methylene blue (MB) cations due to both host–guest and electrostatic interactions. The chirality of α-CD could be transferred and amplified into the MB dye under the bridging effect of the AlMo6–Azo hybrid.Using a similar post-modification method, Oms and co-workers synthesized two novel single-sided asymmetric Al-Anderson hybrids AlMo6–SN and AlMo6–SP.86 Both the hybrids exhibited strong solid-state photochromism under UV irradiation at room temperature. In particular, AlMo6–SN had a high light-driven “recording-erasing” potentiality and AlMo6–SP exhibited intense red emission under UV irradiation when compared with less luminescent NH2–MnMo6–SP. This could be explained by the fact that the AlMo6–NH2 unit had an absorption threshold at 350 nm, while the NH2–MnMo6–NH2 unit had an absorption band between 300 and 450 nm that partially overlapped with the absorption band of the SP group. Therefore, the Al-Anderson core would compete less with the SP unit in the AlMo6–SP hybrid when excited at 365 nm to activate the ring-opening process in SP.
In 2020, Rompel et al. reported a single-sided AlMo6–LA (LA = lauric acid) hybrid that possessed a long alkane chain and interacted with a protein.87 The AlMo6–LA hybrid could be prepared by either pre- or post-modification methods (Fig. 8). For pre-modification, the long alkyl chain was linked with triol-NH2 first, and then anchored onto the AlMo6 core. Regarding post-modification, single-sided AlMo6–NH2 was first prepared and then reacted with lauroyl chloride to form the AlMo6–LA hybrid. The interaction of AlMo6–LA with human serum albumin (HSA) was investigated by fluorescence and circular dichroism spectroscopy. Compared to the unmodified Al-Anderson hybrid, AlMo6–LA showed an increased affinity towards HSA and caused the static fluorescence quenching.
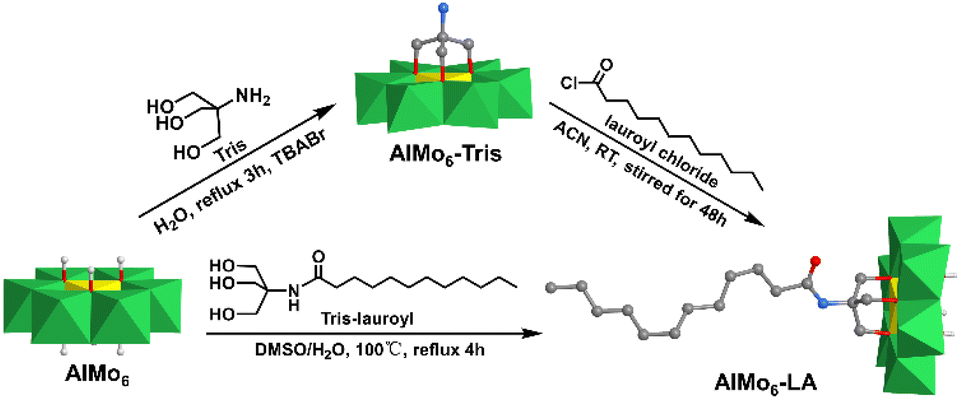 | ||
| Fig. 8 Synthesis of AlMo6–LA by pre- or post-modification methods. Color code: {MoO6}, green octahedron; {AlO6}, yellow octahedron; C, gray; O, deep red; N, light blue; H, light grey. | ||
Supramolecular binding of single-sided Al-Anderson hybrids to halide ions88,89 was performed to modulate the catalytic activities of metal oxide clusters. In 2019, Yin and Wei et al. combined Cl− or Br− halide ions with AlMo6–CH3 and investigated the catalytic activity of the resulting stable complexes.88 The halide ions tended to form hydrogen–halide bonds with the protonated μ3-O atoms. In the oxidation reaction of benzyl alcohol to benzaldehyde using AlMo6–CH3 as the catalyst, introducing halide ions could block the Al3+ catalytic site and weaken the oxidation reaction. It was also found that the catalytic activity could be restored by the addition of water.
Among various Anderson-type XM6 (X = central heteroatom, M = WVI) structures, the asymmetric modification of the XW6 clusters was rarely explored. The reason can be explained as follows: (1) the slow reaction rate of XW6 Anderson compared with its XMo6 analogue, (2) the easy precipitation of the central heteroatom X with tungstate or polyoxotungstates, and (3) the quick transformation of Anderson POMs into more stable Keggin polyanions.90,91 To overcome these obstacles, Wei et al. developed a kinetically favoured synthetic approach using triol ligands as weak complexing reagents.90 It was envisioned that the triol ligands were able to keep the heteroatom in an octahedral coordination mode with the assistance of water molecules to give a kinetically stabilized complex, X(H2O)3[(OCH2)3CR], which impeded the formation of Keggin polyanions and easily reacted with tungstates to form single-sided Anderson-type hybrids. The resulting asymmetric hybrids shared a general formula of [XW6O21{(OCH2)3}CR], where X represented heteroatoms such as Al.
4.2 Double-sided asymmetric Al-Anderson
Recently, Song et al. reported that the type of triol ligand had a profound influence on the stepwise asymmetric modification of the Al-Anderson cluster.92 It was found that commercially available triol ligands such as pentaerythritol and triol-NH2 inevitably generated symmetric by-products upon asymmetric modification, while amide-functionalized triol-derivatives could selectively form asymmetric products. The authors claimed that such phenomena were related to the stability of single-sided Anderson hybrids in ethanol upon refluxing, and the stability may be closely related to the acid–base properties of the triol ligands. To isolate the target asymmetric hybrids from symmetric by-products, the authors systematically investigated the solubility of the modified hybrids, and optimized the purification process by carefully selecting the anchoring triol ligands. As such two novel asymmetrically modified Al-Anderson hybrids, [TBA]3{AlMo6O18[(OCH2)3CCH2OH][(OCH2)3CC6H4NO2]} and [TBA]3{AlMo6O18[(OCH2)3CNH2][(OCH2)3CC6H4NO2]}, were obtained. Post-functionalization of the obtained asymmetric hybrids led to a series of versatile building blocks that could be coupled to form homo- and hetero-cluster oligomers. This work provided a promising approach for the development of functionalized asymmetric hybrids and the controlled synthesis of metal-oxo-cluster oligomers with a precise cluster number and chain length.5. Others
Besides the most frequently studied Mn-, Cr-, and Al-Anderson hybrids, the asymmetric modification of Anderson clusters with other transition metal heteroatoms (Cu, Co, Zn, etc.93,94) has also attracted attention from POM chemists. These asymmetric Anderson hybrids usually refer to the δ/χ isomer featuring the heteroatoms of Cu45,46 and Co.44 The preparation of the δ/χ isomer can be simply achieved by refluxing the mixture of heteroatom salts, triol ligands, and the primary Anderson cluster or [TBA]4Mo8O26. It should be noted that the solvent system can sometimes affect the modification product. For example, when methanol was adopted in the preparation of Co-templated Anderson hybrids, the decoration of the methyl group on μ-O was always observed. The presence of acetic acid in the solvent could cause the transformation between the asymmetric δ/χ isomer and the symmetric χ/χ isomer.446. Conclusions
In this review, we have briefly discussed the covalent modification methods of asymmetric Anderson-type POMs based on the central heteroatoms of MnIII, CrIII, AlIII, and others. The Mn-templated Anderson cluster is the most largely developed one towards asymmetric modification. The corresponding methods include fractional crystallization, RP-HPLC, and post-modification. One shared advantage of these methods lies in their simple synthetic processes that are similar to the preparations of symmetric hybrids. However, to successfully purify the asymmetric products, several factors must be taken into account. For fractional crystallization, the polarities of the triol ligands need to be considered to guarantee the different crystallization timescales between asymmetric molecules and symmetric by-products. The RP-HPLC method requires the attachment of the aromatic component onto the Tris–MnMo6–Tris cluster for sensitive UV detection, while the post-modification method needs careful control of the feed ratio. Compared with the aforementioned methods for asymmetric Mn-Anderson hybrids, the recently developed methods (i.e., the single-side and the step-by-step method) for asymmetric Cr- and Al-Anderson hybrids seem to be promising. These methods are more straightforward in obtaining asymmetric hybrids and usually do not need extra purification. However, the newly reported research by Song et al.92 reveals that the type of triol ligand, especially the synthetic ones, plays an essential role in step-by-step modification, and in this case a purification process is necessary.Although great progress has been achieved in the development of asymmetric POMs, the rational design and function-directed application of asymmetric Anderson hybrids are still highly challenging. It is envisioned that the fine structural control of asymmetric hybrids can lead to more complex self-assembly of metal-oxo clusters, and thereby facilitate the diversity of cluster functions and applications. A promising area is asymmetric photo- and electro-chromic hybrids that allow for efficient charge transfer between inorganic POM skeletons and sensitive organic components, which results in multi-state colour changes as a reflection of anti-fatigue sensing materials. Another potential application of the asymmetric clusters is that these asymmetric structures can be used as versatile building blocks to construct secondary structures and hierarchical assemblies. Besides, the asymmetric hybrids can more easily serve as giant metal-oxo ligands to perform the surface modification of graphene, metal–organic frameworks, and even proteins. It is believed that with the gradual maturity of the synthetic methods of asymmetric POM hybrids, these novel molecular metal-oxo platforms will lead to brand new research areas and a foreseeable broad future of POM chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (22178019, 21901016, 22101017, and 22288102) and the Fundamental Research Funds for the Central Universities (XK1802-6, XK1803-05, XK1902).References
- Y.-F. Song and R. Tsunashima, Recent advances on polyoxometalate-based molecular and composite materials, Chem. Soc. Rev., 2012, 41, 7384–7402 RSC
.
- S. Omwoma, C. T. Gore, Y. Ji, C. Hu and Y.-F. Song, Environmentally benign polyoxometalate materials, Coord. Chem. Rev., 2015, 286, 17–29 CrossRef CAS
- H. N. Miras, J. Yan, D.-L. Long and L. Cronin, Engineering polyoxometalates with emergent properties, Chem. Soc. Rev., 2012, 41, 7403–7430 RSC
- Y. Liu, G.-H. Wen, J. Liang, S.-S. Bao, J. Wei, H. Wang, P. Zhang, M. Zhu, Q. Jia, J. Ma, L.-M. Zheng and Z. Jin, Aqueous colloid flow batteries based on redox-reversible polyoxometalate clusters and size-exclusive membranes, ACS Energy Lett., 2023, 8, 387–397 CrossRef CAS
- L. Fan, M. Wang, X. Dong, G.-G. Gao, J. Yu, H. Liu and X. Liu, V-substitution function on polyoxometalate catalyst for rapid conversion of polyselenides in Li-Se batteries, Chem. Eng. J., 2022, 449, 137819 CrossRef CAS
- Y. Zhu, Y. Huang, Q. Li, D. Zang, J. Gu, Y. Tang and Y. Wei, Polyoxometalate-based photoactive hybrid: uncover the first crystal structure of covalently linked hexavanadate-porphyrin molecule, Inorg. Chem., 2020, 59, 2575–2583 CrossRef CAS PubMed
- C.-G. Lin, M. Hutin, C. Busche, N. L. Bell, D.-L. Long and L. Cronin, Elucidating the paramagnetic interactions of an inorganic-organic hybrid radical-functionalized Mn-Anderson cluster, Dalton Trans., 2021, 50, 2350–2353 RSC
- R. Pütt, P. Kozłowski, I. Werner, J. Griebel, S. Schmitz, J. Warneke and K. Y. Monakhov, {P2V3W15}-Polyoxometalates functionalized with phthalocyaninato Y and Yb moieties, Inorg. Chem., 2021, 60, 80–86 CrossRef PubMed
- H. Soria-Carrera, I. Franco-Castillo, P. Romero, S. Martín, J. M. de la Fuente, S. G. Mitchell and R. Martín-Rapún, On-POM ring-opening polymerisation of N-carboxyanhydrides, Angew. Chem., Int. Ed., 2021, 60, 3449–3453 CrossRef CAS PubMed
- A. Parrot, A. Bernard, A. Jacquart, S. A. Serapian, C. Bo, E. Derat, O. Oms, A. Dolbecq, A. Proust, R. Métivier, P. Mialane and G. Izzet, Photochromism and dual-color fluorescence in a polyoxometalate-benzospiropyran molecular switch, Angew. Chem., Int. Ed., 2017, 56, 4872–4876 CrossRef CAS PubMed
- A. Boulmier, M. Haouas, S. Tomane, L. Michely, A. Dolbecq, A. Vallée, V. Brezová, D.-L. Versace, P. Mialane and O. Oms, Photoactive polyoxometalate/DASA covalent hybrids for photopolymerization in the visible range, Chem. – Eur. J., 2019, 25, 14349–14357 CrossRef CAS PubMed
- J. Zhang, Y. Huang, G. Li and Y. Wei, Recent advances in alkoxylation chemistry of polyoxometalates: from synthetic strategies, structural overviews to functional applications, Coord. Chem. Rev., 2019, 378, 395–414 CrossRef CAS
- A. Proust, R. Thouvenot and P. Gouzerh, Functionalization of polyoxometalates: towards advanced applications in catalysis and materials science, Chem. Commun., 2008, 1837–1852 RSC
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Hybrid organic-inorganic polyoxometalate compounds: from structural diversity to applications, Chem. Rev., 2010, 110, 6009–6048 CrossRef CAS PubMed
- Z.-Q. Lu, L.-L. Zhang, Y. Yan and W. Wang, Polyelectrolytes of inorganic polyoxometalates: acids, salts, and complexes, Macromolecules, 2021, 54, 6891–6900 CrossRef CAS
- W. Yu, B. Li, Y. Zhang, Q. Yan and J. Yan, Discovery of a fullerene-polyoxometalate hybrid exhibiting enhanced photocurrent response, Inorg. Chem., 2020, 59, 5266–5270 CrossRef CAS PubMed
- Y. Liu, P. Zuo, R. Wang, Y. Liu and W. Jiao, Covalent immobilization of Dawson polyoxometalates on hairy particles and its catalytic properties for the oxidation desulfurization of tetrahydrothiophene, J. Cleaner Prod., 2020, 274, 122774 CrossRef CAS
- P. Wu, Y. Wang, B. Huang and Z. Xiao, Anderson-type polyoxometalates: from structures to functions, Nanoscale, 2021, 13, 7119–7133 RSC
- A. Blazevic and A. Rompel, The Anderson–Evans polyoxometalate: from inorganic building blocks via hybrid organic–inorganic structures to tomorrows “Bio-POM”, Coord. Chem. Rev., 2016, 307, 42–64 CrossRef CAS
- X. Zeng, C. Gong, H. Guo, H. Xu, J. Zhang and J. Xie, A new phenylthiourea grafted Mn-Anderson polyoxometalate cluster: synthesis, crystal structure and characterization, Polyhedron, 2018, 151, 37–42 CrossRef CAS
- A. Joshi, P. Sood, A. Gaur, D. Rani, V. Madaan and M. Singh, Improved OER performance of an Anderson-supported cobalt coordination polymer by assembling with acetylene black, J. Mater. Chem. A, 2022, 10, 12805–12810 RSC
- M. Chi, T. Su, L. Sun, Z. Zhu, W. Liao, W. Ren, Y. Zhao and H. Lü, Biomimetic oxygen activation and electron transfer mechanism for oxidative desulfurization, Appl. Catal., B, 2020, 275, 119134 CrossRef CAS
- Y. Zhang, X. Wang, Y. Wang, N. Xu and X.-L. Wang, Anderson-type polyoxometalate-based sandwich complexes bearing a new “V”-like bis-imidazole-bis-amide ligand as electrochemical sensors and catalysts for sulfide oxidation, Polyoxometalates, 2022, 1, 9140004 CrossRef
- R. Ma, N. Liu, T.-T. Lin, T. Zhao, S.-L. Huang and G.-Y. Yang, Anderson polyoxometalate built-in covalent organic frameworks for enhancing catalytic performances, J. Mater. Chem. A, 2020, 8, 8548–8553 RSC
- Q. Liu, Z. Chen, H. Shabbir, J. Duan, W. Bi, Z. Lu, N. Schweitzer, S. Alayoglu, S. Goswami, K. W. Chapman, R. B. Getman, Q. Wang, J. M. Notestein and J. T. Hupp, Presentation of gas-phase-reactant-accessible single-rhodium-atom catalysts for CO oxidation, via MOF confinement of an Anderson polyoxometalate, J. Mater. Chem. A, 2022, 10, 18226–18234 RSC
- K. Nomiya, T. Takahashi, T. Shirai and M. Miwa, Anderson-type heteropolyanions of molybdenum(VI) and tungsten(VI), Polyhedron, 1987, 6, 213–218 CrossRef CAS
- A. Perloff, Crystal structure of sodium hexamolybdochromate(III) octahydrate, Na3(CrMo6O24H6)·8H2O, Inorg. Chem., 1970, 9, 2228–2239 CrossRef CAS
- M. Cheng, Z. Xiao, L. Yu, X. Lin, Y. Wang and P. Wu, Direct syntheses of nanocages and frameworks based on Anderson-type polyoxometalates via one-pot reactions, Inorg. Chem., 2019, 58, 11988–11992 CrossRef CAS PubMed
- U. Lee, H.-C. Joo and J.-S. Kwon, Tetraammonium hexahydrogen hexamolybdonickelate(II) tetrahydrate, (NH4)4[H6NiMo6O24]·4H2O, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, i6–i8 CrossRef CAS
- F. Ito, T. Ozeki, H. Ichida, H. Miyamae and Y. Sasaki, Structure of tetraammonium hexahydrogenhexamolybdocuprate(II) tetrahydrate, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1989, 45, 946–947 CrossRef
- Y. Ozawa, Y. Hayashi and K. Isobe, Structure of triammonium hexahydrogenhexamolybdorhodate(III) hexahydrate, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 637–638 CrossRef
- S. Angus-Dunne, R. C. Burns, D. C. Craig and G. A. Lawrance, Synthesis and crystal structure of the palladium(IV) polyoxomolybdate, K0.75Na3.75[PdMo6O24H3.5]·17H2O, Z. Anorg. Allg. Chem., 2010, 636, 727–734 CrossRef CAS
- U. Lee and Y. Sasaki, Isomerism of the hexamolybdo-platinate(IV) polyanion. Crystal structures of K3.5[α,-H4.5PtMo6O24]·3H2O and (NH4)4[β-H4PtMo6O24]·1.5H2O, Chem. Lett., 1984, 13, 1297–1300 CrossRef
- V. Shivaiah and S. K. Das, Supramolecular assembly based on a heteropolyanion: synthesis and crystal structure of Na3(H2O)6[Al(OH)6Mo6O18]·2H2O, J. Chem. Sci., 2005, 117, 227–233 CrossRef CAS
- A. Blazevic, E. Al-Sayed, A. Roller, G. Giester and A. Rompel, Tris-functionalized hybrid Anderson polyoxometalates: synthesis, characterization, hydrolytic stability and inversion of protein surface charge, Chem. – Eur. J., 2015, 21, 4762–4771 CrossRef CAS PubMed
- S. Himeno, S. Murata and K. Eda, A route to a Keggin-type α-[(XIIIO4)Mo12O35(OH)]4− anion through an Anderson-type [XIII(OH)6Mo6O18]3− anion: X = Ga, Dalton Trans., 2009, 6114–6119 RSC
- H. T. Evans, The molecular structure of the hexamolybdotellurate ion in the crystal complex with telluric acid (NH4)6[TeMo6O24]·Te(OH)6·7H2O, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 2095–2100 CrossRef
- D. Dutta, A. D. Jana, M. Debnath, A. Bhaumik, J. Marek and M. Ali, Robust 1D open rack-like architecture in coordination polymers of Anderson POMs [Na4(H2O)14Cu(gly)2][TeMo6O24] and [Cu(en2)3TeW6O24]: synthesis, characterization and heterogeneous catalytic epoxidation of olefines, Dalton Trans., 2010, 39, 11551–11559 RSC
- M. Filowitz, R. K. C. Ho, W. G. Klemperer and W. Shum, Oxygen-17 nuclear magnetic resonance spectroscopy of polyoxometalates. 1. Sensitivity and resolution, Inorg. Chem., 1979, 18, 93–103 CrossRef CAS
- A. Ogawa, H. Yamato, U. Lee, H. Ichida, A. Kobayashi and Y. Sasaki, Structure of pentapotassium dihydrogenhexamolybdoantimonate heptahydrate, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 1879–1881 CrossRef
- A. Macdonell, N. A. B. Johnson, A. J. Surman and L. Cronin, Configurable nanosized metal oxide oligomers via precise “click” coupling control of hybrid polyoxometalates, J. Am. Chem. Soc., 2015, 137, 5662–5665 CrossRef CAS PubMed
- M. H. Rosnes, C. Musumeci, C. P. Pradeep, J. S. Mathieson, D.-L. Long, Y.-F. Song, B. Pignataro, R. Cogdell and L. Cronin, Assembly of modular asymmetric organic-inorganic polyoxometalate hybrids into anisotropic nanostructures, J. Am. Chem. Soc., 2010, 132, 15490–15492 CrossRef CAS PubMed
- Y.-F. Song, N. McMillan, D.-L. Long, S. Kane, J. Malm, M. O. Riehle, C. P. Pradeep, N. Gadegaard and L. Cronin, Micropatterned surfaces with covalently grafted unsymmetrical polyoxometalate-hybrid clusters lead to selective cell adhesion, J. Am. Chem. Soc., 2009, 131, 1340–1341 CrossRef CAS PubMed
- Y. Wang, X. Liu, W. Xu, Y. Yue, B. Li and L. Wu, Triol-ligand modification and structural transformation of Anderson-Evans oxomolybdates via modulating oxidation state of Co-heteroatom, Inorg. Chem., 2017, 56, 7019–7028 CrossRef CAS PubMed
- Y. Wang, B. Li, H. Qian and L. Wu, Controlled triol-derivative bonding and decoration transformation on Cu-centered Anderson-Evans polyoxometalates, Inorg. Chem., 2016, 55, 4271–4277 CrossRef CAS PubMed
- Y. Wang, X. Kong, W. Xu, F. Jiang, B. Li and L. Wu, Ratio-controlled precursors of Anderson-Evans polyoxometalates: synthesis, structural transformation, and magnetic and catalytic properties of a series of triol ligand-decorated
{M2Mo6} clusters (M = Cu2+, Co2+, Ni2+, Zn2+), Inorg. Chem., 2018, 57, 3731–3741 CrossRef CAS PubMed
- Y. Wang, F. Duan, X. Liu and B. Li, Cations modulated assembly of triol-ligand modified Cu-centered Anderson-Evans polyanions, Molecules, 2022, 27, 2933 CrossRef CAS PubMed
- W.-D. Yu, Y. Zhang, Y.-Y. Han, B. Li, S. Shao, L.-P. Zhang, H.-K. Xie and J. Yan, Microwave-assisted synthesis of tris-Anderson polyoxometalates for facile CO2 cycloaddition, Inorg. Chem., 2021, 60, 3980–3987 CrossRef CAS PubMed
- H. Zhang, W.-L. Zhao, H. Li, Q. Zhuang, Z. Sun, D. Cui, X. Chen, A. Guo, X. Ji, S. An, W. Chen and Y.-F. Song, Latest progress in covalently modified polyoxometalates-based molecular assemblies and advanced materials, Polyoxometalates, 2022, 1, 9140011 CrossRef
- J. M. Cameron, G. Guillemot, T. Galambos, S. S. Amin, E. Hampson, K. M. Haidaraly, G. N. Newton and G. Izzet, Supramolecular assemblies of organo-functionalised hybrid polyoxometalates: from functional building blocks to hierarchical nanomaterials, Chem. Soc. Rev., 2022, 51, 293–328 RSC
- Z. Wei, J. Wang, H. Yu, S. Han and Y. Wei, Recent advances of Anderson-type polyoxometalates as catalysts largely for oxidative transformations of organic molecules, Molecules, 2022, 27, 5212 CrossRef CAS PubMed
- M. Aureliano, N. I. Gumerova, G. Sciortino, E. Garribba, C. C. McLauchlan, A. Rompel and D. C. Crans, Polyoxidovanadates’ interactions with proteins: an overview, Coord. Chem. Rev., 2022, 454, 214344 CrossRef CAS
- Y. Zhang, Y. Liu, D. Wang, J. Liu, J. Zhao and L. Chen, State-of-the-art advances in the syntheses, structures, and applications of polyoxometalate-based metal–organic frameworks, Polyoxometalates, 2023, 2, 9140017 CrossRef
- Y.-F. Song, D.-L. Long, S. E. Kelly and L. Cronin, Sorting the assemblies of unsymmetrically covalently functionalized Mn-Anderson polyoxometalate clusters with mass spectrometry, Inorg. Chem., 2008, 47, 9137–9139 CrossRef CAS PubMed
- C. Yvon, A. Macdonell, S. Buchwald, A. J. Surman, N. Follet, J. Alex, D.-L. Long and L. Cronin, A collection of robust methodologies for the preparation of asymmetric hybrid Mn–Anderson polyoxometalates for multifunctional materials, Chem. Sci., 2013, 4, 3810–3817 RSC
- C. Yvon, A. J. Surman, M. Hutin, J. Alex, B. O. Smith, D.-L. Long and L. Cronin, Polyoxometalate clusters integrated into peptide chains and as inorganic amino acids: solution- and solid-phase approaches, Angew. Chem., Int. Ed., 2014, 53, 3336–3341 CrossRef CAS PubMed
- J. Zhang, J. Hao, Y. Wei, F. Xiao, P. Yin and L. Wang, Nanoscale chiral rod-like molecular triads assembled from achiral polyoxometalates, J. Am. Chem. Soc., 2010, 132, 14–15 CrossRef CAS PubMed
- C. P. Pradeep, M. F. Misdrahi, F.-Y. Li, J. Zhang, L. Xu, D.-L. Long, T. Liu and L. Cronin, Synthesis of modular “inorganic-organic-inorganic” polyoxometalates and their assembly into vesicles, Angew. Chem., Int. Ed., 2009, 48, 8309–8313 CrossRef CAS PubMed
- S. She, N. L. Bell, D. Zheng, J. S. Mathieson, M. D. Castro, D.-L. Long, J. Koehnke and L. Cronin, Robotic synthesis of peptides containing metal-oxide-based amino acids, Chem, 2022, 8, 2734–2748 CAS
- O. Oms, K. Hakouk, R. Dessapt, P. Deniard, S. Jobic, A. Dolbecq, T. Palacin, L. Nadjo, B. Keita, J. Marrot and P. Mialane, Photo- and electrochromic properties of covalently connected symmetrical and unsymmetrical spiropyran-polyoxometalate dyads, Chem. Commun., 2012, 48, 12103–12105 RSC
- M. Diab, A. Mateo, J. A. Cheikh, M. Haouas, A. Ranjbari, F. Bourdreux, D. Naoufal, E. Cadot, C. Bo and S. Floquet, Unprecedented coupling reaction between two anionic species of a closo-decahydrodecaborate cluster and an Anderson-type polyoxometalate, Dalton Trans., 2020, 49, 4685–4689 RSC
- A. Saad, O. Oms, J. Marrot, A. Dolbecq, K. Hakouk, H. E. Bekkachi, S. Jobic, P. Deniard, R. Dessapt, D. Garrot, K. Boukheddaden, R. Liu, G. Zhang, B. Keita and P. Mialane, Design and optical investigations of a spironaphthoxazine/polyoxometalate/spiropyran triad, J. Mater. Chem. C, 2014, 2, 4748–4758 RSC
- A. Saad, O. Oms, A. Dolbecq, C. Menet, R. Dessapt, H. Serier-Brault, E. Allard, K. Baczko and P. Mialane, A high fatigue resistant, photoswitchable fluorescent spiropyran-polyoxometalate-BODIPY single-molecule, Chem. Commun., 2015, 51, 16088–16091 RSC
- I. Bazzan, P. Bolle, O. Oms, H. Salmi, N. Aubry-Barroca, A. Dolbecq, H. Serier-Brault, R. Dessapt, P. Roger and P. Mialane, The design of new photochromic polymers incorporating covalently or ionically linked spiropyran/polyoxometalate hybrids, J. Mater. Chem. C, 2017, 5, 6343–6351 RSC
- Y. Chu, A. Saad, P. Yin, J. Wu, O. Oms, A. Dolbecq, P. Mialane and T. Liu, Light- and solvent-controlled self-assembly behavior of spiropyran-polyoxometalate-alkyl hybrid molecules, Chem. – Eur. J., 2016, 22, 11756–11762 CrossRef CAS PubMed
- A. Boulmier, A. Vacher, D. Zang, S. Yang, A. Saad, J. Marrot, O. Oms, P. Mialane, I. Ledoux, L. Ruhlmann, D. Lorcy and A. Dolbecq, Anderson-type polyoxometalates functionalized by tetrathiafulvalene groups: synthesis, electrochemical studies, and NLO properties, Inorg. Chem., 2018, 57, 3742–3752 CrossRef CAS PubMed
- C. Ritchie and G. Bryant, Microwave assisted synthesis of a mono organoimido functionalized Anderson polyoxometalate, Dalton Trans., 2015, 44, 20826–20829 RSC
- J. Zhang, Z. Liu, Y. Huang, J. zhang, J. Hao and Y. Wei, Unprecedented χ isomers of single-side triol-functionalized Anderson polyoxometalates and their proton-controlled isomer transformation, Chem. Commun., 2015, 51, 9097–9100 RSC
- J. Luo, Y. Huang, B. Ding, P. Wang, X. Geng, J. Zhang and Y. Wei, Single-atom Mn active site in a triol-stabilized β-Anderson manganohexamolybdate for enhanced catalytic activity towards adipic acid production, Catalysts, 2018, 8, 121 CrossRef
- Q. Li and Y. Wei, Unprecedented monofunctionalized β-Anderson clusters: [R1R2C(CH2O)2MnIVW6O22]6−, a class of potential candidates for new inorganic linkers, Chem. Commun., 2021, 57, 3865–3868 RSC
- A. Perloff, Crystal structure of sodium hexamolybdochromate(III) octahydrate, Na3(CrMo6O24H6)·8H2O, Inorg. Chem., 1970, 9, 2228–2239 CrossRef CAS
- Q. Xu, S. Yuan, L. Zhu, J. Hao and Y. Wei, Synthesis of novel bis(triol)-functionalized Anderson clusters serving as potential synthons for forming organic-inorganic hybrid chains, Chem. Commun., 2017, 53, 5283–5286 RSC
- Y. Zhang, H. Jia, Q. Li, Y. Huang and Y. Wei, Synthesis and characterization of an unprecedented water-soluble tris-functionalized Anderson-type polyoxometalate, J. Mol. Struct., 2020, 1219, 128555 CrossRef CAS
- P. R. Marcoux, B. Hasenknopf, J. Vaissermann and P. Gouzerh, Developing remote metal binding sites in heteropolymolybdates, Eur. J. Inorg. Chem., 2003, 2003, 2406–2412 CrossRef
- P. Wu, P. Yin, J. Zhang, J. Hao, Z. Xiao and Y. Wei, Single-side organically functionalized Anderson-type polyoxometalates, Chem. – Eur. J., 2011, 17, 12002–12005 CrossRef CAS PubMed
- J. Wang, F. Jiang, C. Tao, H. Yu, L. Ruhlmann and Y. Wei, Oxidative esterification of alcohols by a single-side organically decorated Anderson-type chrome-based catalyst, Green Chem., 2021, 23, 2652–2657 RSC
- D. Dan, F. Chen, W. Zhao, H. Yu, S. Han and Y. Wei, Chromium-catalysed efficient N-formylation of amines with a recyclable polyoxometalate-supported green catalyst, Dalton Trans., 2021, 50, 90–94 RSC
- X.-X. Li, X. Ma, W.-X. Zheng, Y.-J. Qi, S.-T. Zheng and G.-Y. Yang, Composite hybrid cluster built from the integration of polyoxometalate and a metal halide cluster: synthetic strategy, structure, and properties, Inorg. Chem., 2016, 55, 8257–8259 CrossRef CAS PubMed
- J. Zhang, Q. Li, M. Zeng, Y. Huang, J. Zhang, J. Hao and Y. Wei, The proton-controlled synthesis of unprecedented diol functionalized Anderson-type POMs, Chem. Commun., 2016, 52, 2378–2381 RSC
- J. Zhang, Y. Huang, J. Hao and Y. Wei,
β-{Cr[RC(CH2O)3]2Mo6O18}3−: the first organically-functionalized β isomer of Anderson-type polyoxometalates, Inorg. Chem. Front., 2017, 4, 1215–1218 RSC
- C.-G. Lin, W. Chen, D.-L. Long, L. Cronin and Y.-F. Song, Step-by-step covalent modification of Cr-templated Anderson-type polyoxometalates, Dalton Trans., 2014, 43, 8587–8590 RSC
- J. Zhang, J. Luo, P. Wang, B. Ding, Y. Huang, Z. Zhao, J. Zhang and Y. Wei, Step-by-step strategy from achiral precursors to polyoxometalates-based chiral organic-inorganic hybrids, Inorg. Chem., 2015, 54, 2551–2559 CrossRef CAS PubMed
- J. Zhang, Z. Zhao, J. Zhang, S. She, Y. Huang and Y. Wei, Spontaneous resolution of polyoxometalate-based inorganic-organic hybrids driven by solvent and common ion, Dalton Trans., 2014, 43, 17296–17302 RSC
- H. Ai, Y. Wang, B. Li and L. Wu, Synthesis and characterization of single–side organically grafted Anderson–type polyoxometalates, Eur. J. Inorg. Chem., 2014, 2014, 2766–2772 CrossRef CAS
- B. Zhang, L. Yue, Y. Wang, Y. Yang and L. Wu, A novel single-side azobenzene-grafted Anderson-type polyoxometalate for recognition-induced chiral migration, Chem. Commun., 2014, 50, 10823–10826 RSC
- H. Dridi, A. Boulmier, P. Bolle, A. Dolbecq, J.-N. Rebilly, F. Banse, L. Ruhlmann, H. Serier-Brault, R. Dessapt, P. Mialane and O. Oms, Directing the solid-state photochromic and luminescent behaviors of spiromolecules with Dawson and Anderson polyoxometalate units, J. Mater. Chem. C, 2020, 8, 637–649 RSC
- A. Bijelic, A. Dobrov, A. Roller and A. Rompel, Binding of a fatty acid-functionalized Anderson-type polyoxometalate to human serum albumin, Inorg. Chem., 2020, 59, 5243–5246 CrossRef CAS PubMed
- S. She, M. Li, Q. Li, Z. Huang, Y. Wei and P. Yin, Unprecedented halide-ion binding and catalytic activity of nanoscale anionic metal oxide clusters, ChemPlusChem, 2019, 84, 1668–1672 CrossRef CAS PubMed
- R.-Y. Zhen, X.-Z. Ge, L. Zhu and J. Hao, Novel morphologies including cowry-like crystal of polyoxometalates derivatives via coupled twinning between enantiomers, J. Solid State Chem., 2020, 288, 121416 CrossRef CAS
- Q. Li and Y. Wei, A series of unprecedented triol-stabilized [H3MW6O24]n−: the missing piece between A- and B-type Anderson-Evans polyoxometalates, Chem. Commun., 2018, 54, 1375–1378 RSC
- N. I. Gumerova, A. Roller and A. Rompel, [Ni(OH)3W6O18(OCH2)3CCH2OH]4−: the first tris-functionalized Anderson-type heteropolytungstate, Chem. Commun., 2016, 52, 9263–9266 RSC
- M.-M. Zhang, Y.-A. Yin, W.-J. Chen, C.-G. Lin, Y. Wei and Y.-F. Song, Asymmetric modification of Anderson-type polyoxometalates towards organic-inorganic homo-and hetero-cluster oligomers, Inorg. Chem. Front., 2023 10.1039/d2qi02233h
- N. I. Gumerova, A. Roller and A. Rompel, Synthesis and characterization of the first nickel(II)–centered single–side tris–functionalized Anderson–type polyoxomolybdate, Eur. J. Inorg. Chem., 2016, 2016, 5507–5511 CrossRef CAS
- H. Yu, S. Ru, G. Dai, Y. Zhai, H. Lin, S. Han and Y. Wei, An efficient iron(III)-catalyzed aerobic oxidation of aldehydes in water for the green preparation of carboxylic acids, Angew. Chem., Int. Ed., 2017, 56, 3867–3871 CrossRef CAS PubMed
Footnote |
| † These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2023 |