
Vat 3D printing of full-alginate hydrogels via thiol–ene reactions towards tissue engineering applications†
Michael
Zanon
ab,
Laura
Montalvillo-Jiménez
b,
Raquel
Cue-López
bd,
Enrique
Martínez-Campos
 bd,
Marco
Sangermano
c,
Annalisa
Chiappone
*e and
Paula
Bosch
b
bd,
Marco
Sangermano
c,
Annalisa
Chiappone
*e and
Paula
Bosch
b
aCenter for Sustainable Future Technologies (CSFT)@Polito, Istituto Italiano di Tecnologia, Via Livorno 60, Torino, 10144, Italy
bDepartamento de Química Macromolecular Aplicada, Instituto de Ciencia y Tecnología de Polímeros, Consejo Superior de Investigaciones Científicas (CSIC), C/Juan de la Cierva 3, Madrid, 28006, Spain
cDipartimento di Scienza Applicata e Tecnologia, Politecnico di Torino, C.so Duca degli Abruzzi 24, 10129 Turin, Italy
dGrupo de Síntesis Orgánica y Bioevaluación, Instituto Pluridisciplinar (UCM), Unidad Asociada al ICTP, IQM (CSIC), Paseo de Juan XXIII 1, Madrid, 28040, Spain
eDipartimento di Scienze Chimiche e Geologiche, Università degli studi di Cagliari, Cittadella Universitaria Blocco D, S.S. 554 bivio per Sestu 09042, Monserrato, CA, Italy. E-mail: annalisa.chiappone@unica.it
First published on 10th October 2023
Abstract
The rise of 3D printing has given an important impulse to the medical field, envisaging the possibility of creating artificially engineered tissues/organs perfectly suiting the tissue defects of patients using their own cells; this approach could in the future overcome the lack of tissue donors and decrease the possible dangerous tissue rejection. Different 3D printing technologies can be considered for the building of scaffolds; despite these promises, very few inks for light-induced 3D printing are nowadays available on the market. Herein, for the first time, the alginate backbone is completely functionalized with thiol and alkene groups (separately) to create an innovative full-alginate ink for digital light processing (DLP) printers. The alginate hydrogel is produced with the more biocompatible thiol–ene reaction instead of the most commonly used radical photopolymerization based on (meth)acrylates and without any addition of small crosslinkers to the printable formulation. Simple synthetic “two-reactions” or “one-pot” strategies are explored to functionalize alginate with thiol/alkene groups that are able to undergo click reactions. High levels of reproducibility of the modification strategy are obtained. The hydrogels are characterized by studying their formulation reactivity, mechanical properties, swelling kinetics and morphological appearance, placing the resulting hydrogel into the stiffer scaffold category. The selected hydrogel formulation, tested as the ink for DLP 3D printing, demonstrates good processability and geometry fidelity with the possibility of forming 3D suspended structures. In the end, cell attachment and proliferation are evaluated on the hydrogel, certifying the possible use of the ink for the creation of tissue/organ substitutes (e.g., intestines or tendons) in tissue engineering applications.
Introduction
Photopolymerization is a well-known crosslinking method that keeps gaining increasing interest in a large number of fields, such as paints and coatings,1,2 sealings,3,4 electronics5–7 or orthodontics.8–10 In particular, in the biomedical field, photopolymerization has emerged as one of the most efficient and versatile processes to produce chemically crosslinked polymers.11,12 Rapid reaction times upon light exposure (UV or visible), good spatiotemporal control of the forming polymer and the possibility of performing the reaction under physiological conditions (e.g., pH or temperature) make the technology highly attractive for the biomedical field.13,14Indeed, tissue engineering applications are currently trying to explore the creation of photo-curable hydrogels that are able to replace damaged or failed soft tissues/organs. 3D printing, as the natural evolution of photopolymerization processes, might assist in accomplishing this challenge by forming tailored scaffolds based exactly on the patient's needs.14–16 Starting from their computer-aided design (CAD) customized fabrication, 3D printing techniques are commonly more cost-efficient and faster than traditional manufacturing technologies.17,18 These technologies can be divided into two main categories: extrusion-based and lithography-based 3D printers.19 While extrusion-based technologies are known for their enhanced versatility (e.g., fused direct ink writing and fused deposition modeling printers), lithography-based techniques permit the creation of complex and interconnected architectures with the best resolution overall.20 In particular, digital light processing (DLP) printers are able to create every entire layer all at once, thanks to a micro-mirror system,14 reducing the production time.
Hydrogels are among the most studied materials in tissue engineering21 and their shaping by 3D printing is still an open challenge. Aiming to mimic the protein–glycosaminoglycan blend of the human extracellular matrix (ECM),22 polysaccharides are generally accepted as valid candidates.23 In detail, alginate has been extensively studied and employed for a large number of biomedical applications, due to its biocompatibility, low toxicity and relatively low cost.24 It is typically extracted from brown algae and is composed of regions of sequential (1–4)-linked β-D-mannuronic acid (M-block) monomers, regions of sequential α-L-guluronic acid (G-block) monomers, and regions of not tactically organized M and G units. The structure (i.e., composition of repetitive units), abundance and length of the different blocks are extremely important, as they will determine the physical properties of the hydrogel.21,25–27 In any case, both blocks comprise carboxylic moieties that are able to deprotonate under physiological conditions, resulting in a perfectly water-soluble natural polymer (the minimum solubility is around a pH value of 3–3.5 due to the protonation of the carboxylic groups and the onset of polar interactions).26
Aiming to combine the appealing properties of alginate with the advantages of photopolymerization and lithography-based 3D printing, photocurable reactive groups must be grafted on the polymer chain. Even if many natural polymers have been modified with acrylic/methacrylic groups to perform fast radical chain growth photopolymerization reactions,28,29 step growth propagation reactions are nowadays gaining increasing interest. In fact, the main advantages of these reactions are lower stress accumulation once the 3D hydrogel is formed, lower cytotoxicity and enhanced chemoselectivity of the reacting moieties.30–32 Under these circumstances, especially “click chemistry” reactions are employed because of their rapidity, versatility, regiospecificity, easy usability, and high yields achievable under mild conditions.33–35 Despite the fact that thiol–ene reactions are known to be biocompatible photoinduced reactions,36–38 thiomers have gained increased popularity in the last decade in the biomedical field mainly for drug delivery applications (due to their mucoadhesive properties)39–41 but not much for tissue engineering applications.42–44 Within this framework, many synthetic protocols have been reported in the last few years to functionalize alginate with thiol moieties,45–47 but not much attention has been paid to the maximization of the degree of functionalization (essential if pursuing a photoactivated crosslinking via thiol–ene reactions, also suitable for DLP printing). Strictly concerning alginate photocrosslinking, to the best of our knowledge, just a few publications focused on the creation of photocured thiol–ene/yne alginate hydrogels are available and these report the use of ene/yne-modified alginate crosslinked with the addition of synthetic dithiol molecules.31,35
Herein, aiming to enlarge the palette of 3D printable hydrogels, sodium alginate was selected considering its easy modification due to its high quantity of carboxylic moieties (compared with chitosan, hyaluronic acid, cellulose, etc.).48 Different polysaccharide batches were modified separately with alkene and thiol groups investigating different functionalization procedures and different degrees of functionalization toward the creation of hydrogels. Indeed, the intention of this work resides in the exploration of simple synthetic routes to functionalize alginate with thiol/alkene groups that are able to undergo click reactions (employing both “one-pot” and “two reaction” strategies) for the development of alginate photocurable inks presenting suitable characteristics in terms of viscosity, reactivity and mechanical properties, and the DLP printing of 3D structures. In this way, a full alginate network can be created via the thiol–ene reaction without the addition of any external crosslinking molecule. It is shown that the straightforward one-pot strategy allows the achievement of suitable degrees of functionalization on the alginate backbone both for thiol and ene functionalities: appropriate crosslinking is achieved to meet the mechanical properties required for both tissue engineering and DLP printing. Furthermore, good values of cell adhesion and proliferation ensure enhanced biocompatibility of the hydrogels, resulting in appropriate candidates for the production of 3D scaffolds in tissue engineering applications.
Experimental
Materials
Alginic acid sodium alginate from brown algae (SA, low viscosity), cysteamine hydrochloride (CSA, ≥98%), L-cysteine (CYS, 97%), sodium periodate (ACS reagent, ≥99.8%), sodium borohydride (powder, ≥98%), sodium nitrate (ACS reagent, ≥99.0%), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC, ≥98%), N-hydroxysuccinimide (NHS), lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP, ≥95%), hydrochloric acid solution (37%), hydroxylamine hydrochloride (ACS reagent, 98.0%), methyl orange (for microscopy), phosphate buffer solution tablets (PBS buffer, BioUltra, pH 7.4), 2-morpholinoethanesulfonic acid monohydrate (MES buffer, Millipore), sodium phosphate monobasic (ReagentPlus, ≥99.0%), sodium phosphate dibasic (ReagentPlus, ≥99.0%), ethylenediaminetetraacetic acid (EDTA, BioUltra, anhydrous, ≥99%), and 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB or Ellman's reagent, suitable for the determination of sulfhydryl groups, ≥98%) were all purchased from Sigma-Aldrich and used as received without further purification. Sodium hydroxide pellets were purchased from PanReac, pre-wetted dialysis membranes (MWCO 3500 Da Spectra/Por6) from Spectrum Laboratories and 5-norbornene-2-methylamine (NOR, mixture of isomers) from TCI Europe N.V.Two-reaction thiolation strategy
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10:1, and 20:1) for 3 h, following the procedure described by Huamani-Palomino et al.45 Briefly, in a round bottom flask, 0.5 g of sodium alginate was solubilized in 25 mL of distilled water (DI water) by stirring overnight at RT (2% w/v). Then, 25 mL of sodium periodate solution was added to the alginate solution while stirring at room temperature in darkness. The concentration of this solution was fixed in each case to obtain the desired molar ratio of SA and NaIO4. The reaction was quenched after 30 min with a 10% v/v solution of ethylene glycol in DI water while stirring.48 The product was isolated by dialysis against water for 6 days with a 3.5 kDa membrane and dried by rotary evaporation at 37 °C. The synthesis procedure was repeated three times and the obtained results are the average of different batches.
:4 of the alginate units and cysteine) were solubilized in 10 mL of water and added to the OSA solution. Finally, the mixture was stirred at room temperature for 24 h in darkness under a nitrogen atmosphere.49 Then, 0.4 g of sodium borohydride NaBH4 was added to the solution and the reaction was stirred in darkness for another 15 h at room temperature under a nitrogen atmosphere. The product (TOSA, thiolated oxidized sodium alginate) was isolated by dialysis against water (6 days) in darkness at 10° C with a 3.5 kDa membrane against saline acidic DI water (pH 4 and 0.1 M NaCl). The synthesis procedure was repeated three times and the obtained results are the average of different batches.
:1, 10:1, and 20:1) for 3 h, following the procedure described by Huamani-Palomino et al.45 Briefly, in a round bottom flask, 0.5 g of sodium alginate was solubilized in 25 mL of distilled water (DI water) by stirring overnight at RT (2% w/v). Then, 25 mL of sodium periodate solution was added to the alginate solution while stirring at room temperature in darkness. The concentration of this solution was fixed in each case to obtain the desired molar ratio of SA and NaIO4. The reaction was quenched after 30 min with a 10% v/v solution of ethylene glycol in DI water while stirring.48 The product was isolated by dialysis against water for 6 days with a 3.5 kDa membrane and dried by rotary evaporation at 37 °C. The synthesis procedure was repeated three times and the obtained results are the average of different batches.
:4 of the alginate units and cysteine) were solubilized in 10 mL of water and added to the OSA solution. Finally, the mixture was stirred at room temperature for 24 h in darkness under a nitrogen atmosphere.49 Then, 0.4 g of sodium borohydride NaBH4 was added to the solution and the reaction was stirred in darkness for another 15 h at room temperature under a nitrogen atmosphere. The product (TOSA, thiolated oxidized sodium alginate) was isolated by dialysis against water (6 days) in darkness at 10° C with a 3.5 kDa membrane against saline acidic DI water (pH 4 and 0.1 M NaCl). The synthesis procedure was repeated three times and the obtained results are the average of different batches.
One-pot synthesis of sodium alginate–cysteine and sodium alginate–cysteamine conjugates (SA–CYS and SA–CSA)
0.5 g of SA was solubilized previously in 33 mL of DI water in a round bottom flask (1.5% w/v). A few drops of HCl solution were added until pH = 4. Then, a solution of EDC/NHS (1.568 g/1.163 g, 4 equivalents to SA carboxylic groups) in DI water was added dropwise. Then, the pH was adjusted to a value of 4. The solution was allowed to stir for 2 h at RT and under an N2 atmosphere to activate the carboxylic groups of alginates. The pH was then adjusted to 4 if needed. 1.223 g of CYS or 1.148 g of CSA (4 equivalents to SA carboxylic groups) were solubilized in DI water and added to the solution with a syringe. The reaction was maintained in darkness under stirring and an N2 atmosphere at RT for 24 h. The products were then dialyzed for 6 days in darkness at 10 °C with a 3.5 kDa membrane against saline acidic DI water (pH 4 and 0.1 M NaCl) to avoid thiol oxidation and disulfide bond creation.50,51 The reaction involving SA–CSA was also performed to produce 3 g of material at once. The synthesis procedure was repeated three times for each compound and the obtained results are the average of different batches.One-pot functionalization of alginate with alkene groups (SA–NOR)
First, 0.5 g of SA was solubilized in MES buffer (0.1 M) in a round bottom flask (2% w/v). The pH was then lowered to a value of 4 with the HCl solution. In a vial, EDC and NHS were solubilized together in MES buffer31 (0.940 g/0.232 g, 2.4/0.8 equivalents to SA carboxylic groups, respectively) and slowly added dropwise to alginate solution. Then, the pH was adjusted with the HCl solution until a value of 4 was achieved. The reaction was stirred for 2 h at RT to activate the carboxylic groups of alginates. The pH was then raised to 8.5 with a 0.1 M solution of NaOH. NOR was added directly to the flask under an argon atmosphere (0.311 g, 1 equivalent to SA carboxylic groups). The reaction was performed in darkness under stirring and an argon atmosphere at RT for 24 h. At the end of the reaction, the color of the solution was yellowish. The product was then dialyzed for 6 days against DI water at RT in darkness with a 3.5 kDa membrane. The reaction involving SA–NOR was also performed to produce 3 g of material at once. The synthesis procedure was repeated three times for each compound and the obtained results are the average of different batches.Preparation of the photocurable hydrogel
SA–CSA was solubilized in DI water to obtain a 10 wt% solution; separately, SA–NOR was also solubilized to obtain a second 10 wt% solution. The two solutions were then slowly mixed until a homogeneous formulation was obtained. Then, 0.99 wt% of LAP (corresponding to 1 phr i.e. per hundred resin: 0.01 g of PI per gram of alginate used) was added to the solution and stirred until complete dissolution in darkness. The formulation was then cast into molds of PDMS (≈H = 3 mm and D = 5 mm) and irradiated for 5 min at 50 mW cm−2 with a visible light lamp (Hamamatsu LC8) furnished with a cut-off filter for λ < 400 nm.Vat 3D-printing
3D printing was performed with an Asiga PICO 2 DLP-3D printer (Asiga, Australia) equipped with an LED light source emitting at 405 nm with a light intensity of 35 mW cm−2 (nominal XY pixel resolution is 39 μm and achievable Z-axis control is 1 μm). After printing, the 3D geometries were immersed in distilled water for 1 min and post-cured with a mercury lamp provided by Robotfactory (10 min, light intensity of 10 mW cm−2).Characterization
Thiol content evaluation (Ellman's reagent titration)
The content of free thiol groups was evaluated by the Ellman's reagent method.53,54 A pH 8 phosphate buffer (0.1 M) solution was prepared while EDTA (0.001 M) was added to impede metal chelation of the thiol groups. Separately, 4 mg of DTNB (i.e., Ellman's reagent) were solubilized manually in 1 mL of phosphate buffer. In the meantime, various quantities of TOSA/SA–CSA/SA–CYS (around 10 mg) were added to a 5 mL vial of buffer. Then, two UV-VIS cuvettes (blank and sample) were prepared with the indicated proportion:(1) Blank: 2.5 mL of buffer + 50 μL of Ellman's reagent solution + 250 μL of buffer
(2) Sample: 2.5 mL of buffer + 50 μL of Ellman's solution + 250 μL of TOSA/SA–CSA/SA–CYS solution
This procedure was performed in triplicate measuring the light absorption at 412 nm (see the ESI† for details). For each test, three aliquots of the same synthesis batch were withdrawn and measured, and the results were averaged. The result of the degree of functionalization is the average of three different synthesis batches.
Nuclear magnetic resonance spectroscopy analysis (1H-NMR and solid-state 13C-NMR)
1H-NMR spectra were recorded on Bruker Avance 400 MHz and Varian Mercury 400 MHz spectrometers with samples dissolved in D2O at room temperature. The reference for the integration has always been the whole signal of the sodium alginate skeleton, considering that it corresponds to 4 protons of the pyranose ring. The solid-state NMR spectra were obtained on a Bruker Avance 400 spectrometer equipped with an 89 mm wide bore, 9.4 T super-conducting magnet (proton Larmor frequency at 400.14 MHz). The reported data were recorded at room temperature using cross-polarization (CP), magic-angle spinning (MAS), high-power 1H decoupling and a rotor spinning rate of 5 kHz. The contact time was set to 3 ms and the recycling time between subsequent acquisitions was set to 3 s. The spectral width was 35 kHz and adamantane was used as the external chemical shift reference. In both cases, the obtained spectra were evaluated using MestReNova software.Gel permeation chromatography (GPC)
The molecular weight of commercial SA and its derivatives (oxidized sodium alginate, OSA) was determined by size exclusion chromatography (GPC) using a Shimadzu modular system comprising a DGU-20A3 solvent degasser, an LC-20AD pump, a column oven, an HT-autosampler 20A HT, and an RID-10A refractive index detector. The samples were dissolved (2 mg mL−1) in the mobile phase based on Milli-Q water with NaNO3 (0.2 M).Thermogravimetric analysis (TGA)
A TGA Q500 thermal analyzer (TA Instruments) was used to perform thermogravimetric measurements under dynamic mode using a nitrogen atmosphere (flow rate of 60 mL min−1). Samples were heated from room temperature to 700 °C at a heating rate of 10 °C min−1. The initial degradation temperatures (T0) were obtained at 5% of mass loss and temperatures at the maximum degradation rate (Tmax) were obtained from the first derivative of the TGA curves (DTG).Hydrogel characterization
Real-time photorheological measurements were performed using an Anton PAAR Modular Compact Rheometer (Physica MCR 302, Graz, Austria) in parallel-plate mode (25 mm diameter) and the visible-light source was provided by positioning the light guide of the visible Hamamatsu LC8 lamp under the bottom plate, and light intensity was set at 35 mW cm−2 to mimic the printing process. During the measurements, the gap between the two glass plates was set to 0.2 mm and the sample was kept under a constant shear frequency of 1 Hz. The irradiating light was switched on after 60 s to allow the system to stabilize before the onset of polymerization. According to preliminary amplitude sweep measurements, all the tests were carried out in the linear viscoelastic region at a strain amplitude of 5%. The photorheology was studied as a function of the changes in the shear modulus (G′) and the loss modulus (G′′) of the sample versus the exposure time. Amplitude sweep tests were performed on the cured hydrogels in the range between 1 and 1000% of strain with a frequency of 1 Hz. The mechanical properties were measured by a dynamic compression test. Measurements were performed on swelled 3D printed cylindrical scaffolds (≈h = 3 mm, d = 5 mm) at 25 °C and using a universal test system, MTS QTest1/L Elite, a uniaxial testing machine equipped with a 100 N load cell in compression mode. The samples were placed between the compression plates. Each sample was subsequently deformed at 1 mm min−1. The storage modulus E′ was calculated as the slope of the stress–strain curve for the strain range below 10%. All measurements were performed in triplicate. The different photocured samples (≈h = 3 mm and d = 5 mm) were washed and dried overnight. Once dried, the samples were weighed and soaked in DI water to evaluate the swelling capability and kinetics. The samples were taken out at different time intervals and weighed once the surface droplets were wiped off with wet paper until constant weight. The swelling ratio (Sw%) was calculated as: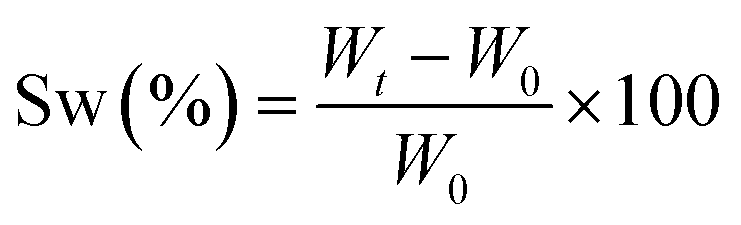 | (1) |
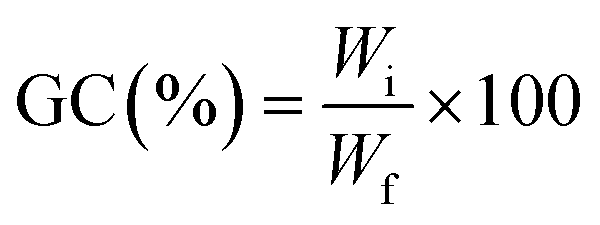 | (2) |
The morphological characterization of the samples was carried out by field emission scanning electron microscopy (FESEM, Zeiss Supra 40, Oberkochen, Germany). The hydrogel samples were first frozen, sectioned in half, and lyophilized before being coated with a 5 nm thick, thin film of Pt/Pd.
Cell viability and proliferation
Before the cell viability and proliferation assays, all the hydrogels were sterilized in a 48-well plate (Corning). The hydrogels were stored in 70% ethanol for a week and then carefully rinsed with PBS (phosphate buffer solution, Thermo Fisher) and sterilized with ultraviolet germicidal irradiation (UVGI) for 40 min. After a final rinse with PBS, the hydrogels were covered with DMEM 1× (Gibco) supplemented with 10% FBS (fetal bovine serum, Thermo Scientific) and antibiotics 100 U mL−1 penicillin and 100 μg mL streptomycin sulfate (Sigma-Aldrich). After 24 h of contact between the culture medium and the hydrogels at 37 °C, the media containing soluble extracts were collected and kept in a freezer until further use. Cell assays were performed using the C166-GFP mouse endothelial cell line (ATCC CRL-2583™, USA): 20000 cells per mL were seeded in a 12-well culture plate and allowed to adhere and grow for 24 h. Then, the media was changed to a mixture (1:1 and 1:5) of complete DMEM and the medium that had been in contact with the hydrogels. Inverted fluorescence microscopy (Olympus IX51, FITC filter λex/λem = 490/525 nm) was used daily to evaluate any changes in the cell culture morphology and proliferation that could indicate the leaching of toxins from the hydrogels. After 48 h, when the cell cultures reached confluency, the metabolic activity of the cells was measured using the Alamar Blue assay by following the instructions of the manufacturer (Biosource). This method is non-toxic and uses the natural reducing power of living cells, generating a quantitative measure of cell viability and cytotoxicity. Briefly, the Alamar Blue dye (10% of the culture volume) was added to each well containing living cells and incubated for 90 minutes. Then, the fluorescence of each well was measured using a Synergy HT plate reader (BioTek) at 535/590 nm. Finally, DNA quantitation of the cells was performed using a FluoReporter® Blue Fluorometric dsDNA Quantitation Kit. This method is based on the ability of the bisbenzimidazole derivative Hoechst 33258 to bind to the A-T-rich regions of double-stranded DNA. After binding to DNA, Hoechst 33258 exhibits an increase in fluorescence, which is measured at a 360 nm excitation wavelength and 460 nm emission using a microplate reader (BioTek, Synergy HT).
Results and discussion
Alginate functionalization reactions
Several routes of functionalization have been proposed to modify alginate (including thiolation reactions). Among them, we selected two general strategies that target different chemical species of the alginate backbone: carboxylic groups (based on carbodiimide chemistry31,39) or oxidation of the ring.45,47 So, different thiolation protocols were first explored as summarized in Fig. 1; instead, the thiol–ene chemical crosslink reaction is reported in Fig. S1.†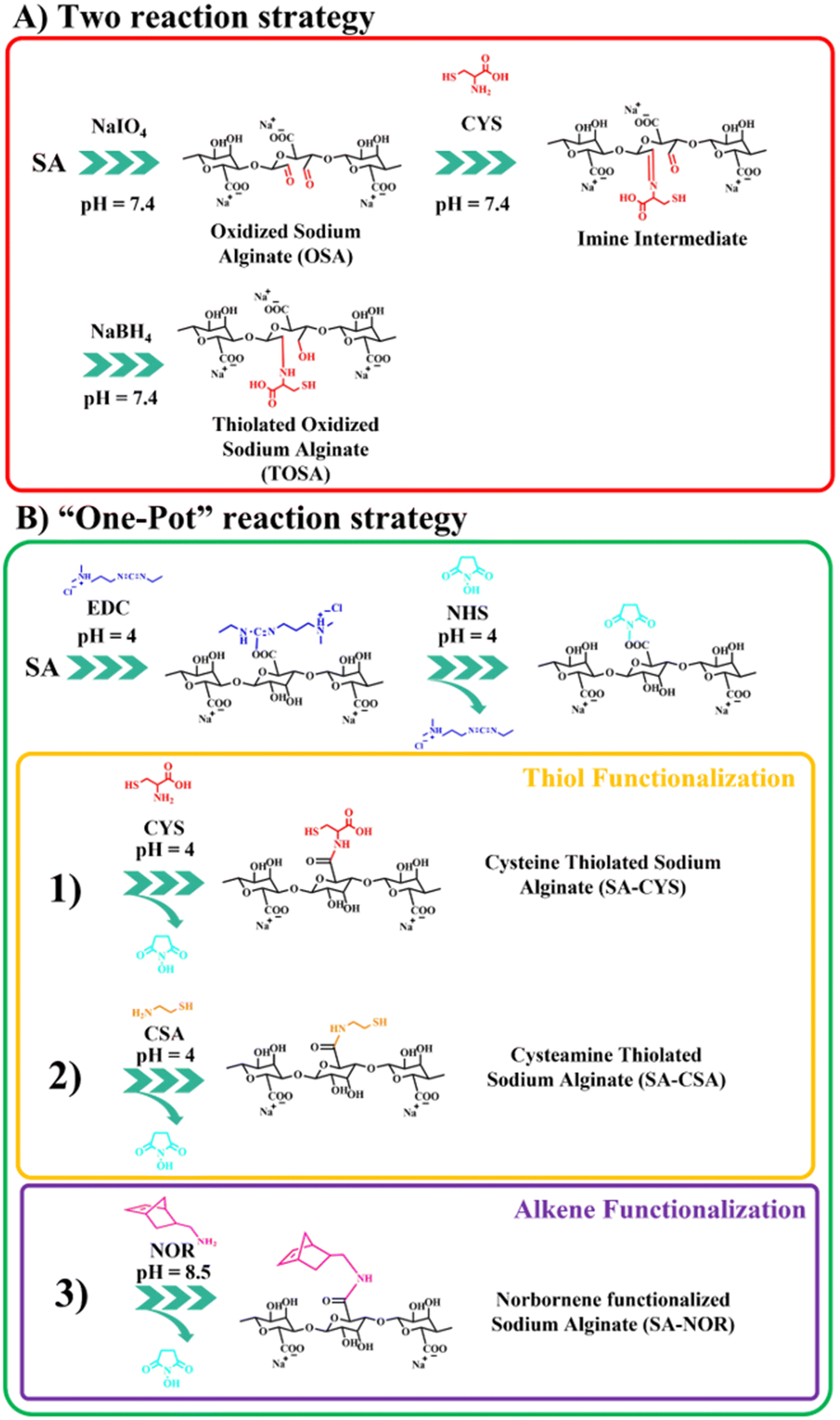 | ||
| Fig. 1 Schematic representation of the alginate functionalization reactions studied in this work. (A) Two-reaction strategy to obtain thiolated oxidized sodium alginate (TOSA). (B) Carbodiimide chemistry-based “one-pot” strategy to obtain thiolated alginate SA–CYS (1), SA–CSA (2) or alkene–alginate SA–NOR (3). | ||
The first proposed way to functionalize alginate with thiol groups involves a “two-reactions” strategy which includes an initial step of oxidation. Then, alginate is functionalized with cysteine by a spontaneous reaction between the aldehydes and the cysteine secondary amine imine, forming an imine intermediate subsequently reduced by the addition of sodium borohydride (scheme in Fig. 1A). The oxidative modification with sodium periodate of alginate is expected to take place on the adjacent C-2 and C-3 hydroxyl groups on the glucose ring, creating a couple of aldehyde moieties on the alginate backbone.46,48 For this step, three different molar ratios of the total carbohydrate rings and the oxidizer (sodium periodate) were investigated, resulting in different aldehyde content evaluated by potentiometric titration (see the ESI†). Table 1 reports the molar ratio investigated and the degree of functionalization obtained for this synthesis step. The degrees of functionalization of 33, 10 and 4% for OSA 1, OSA 2 and OSA 3 (respectively) were obtained with respect to the total alginate rings. However, considering that each cleavage on the alginate backbone forms two aldehyde moieties, the theoretical degree of functionalization for the three reaction conditions would be 40, 20 and 10% (on OSA 1, OSA 2 and OSA 3, respectively). This lower value can be explained by considering that aldehydes, once formed, are highly reactive and can react with surrounding water molecules giving the more stable hemiacetal form. In particular, this reaction can decrease the total number of the available aldehyde moieties.55 In addition, oxidation reactions could also lead to cleavages in the alginate skeleton, lowering the molecular weight of the polymer (while affecting the final hydrogel stiffness).45,56 Thus, the three oxidized alginates and the commercial sodium alginate were subjected to gel permeation chromatography to track the molecular weight after the reaction.
| Sample | Molar ratio (SA:NaIO4) |
Degree of functionalization (DF%) | M n | M w/Mn |
|---|---|---|---|---|
| OSA 1 | 5:1 |
33 ± 1 | 16228 |
2.23 |
| OSA 2 | 10:1 |
10 ± 1 | 38686 |
2.52 |
| OSA 3 | 20:1 |
4 ± 1 | 46131 |
2.83 |
| SA | — | — | 58537 |
3.33 |
Table 1 reports the number average molecular weight (Mn) and polydispersity index (Mw/Mn) of SA and the obtained oxidized product. As expected, lower molecular weights were found once a higher quantity of the oxidizer was used, confirming the reactions of chain breaking. Even though Mw decreases, OSA 1 was chosen for the further step of functionalization due to the acceptable final molecular weight, lower polydispersity and higher level of modification, needed for the next step of thiolation and in line with previously reported works.45,56
The presence of aldehyde functional groups on OSA 1 was further confirmed by ATR FT-IR, 1H-NMR and solid-state 13C-NMR spectra (Fig. 2). The FT-IR spectra showed the appearance of a new peak around 1720 cm−1, related to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbonyl stretch of saturated aliphatic aldehydes (Fig. 2A),48 while 1H-NMR (Fig. 4B) and solid-state 13C-NMR (Fig. 4C) spectra confirmed the characteristic aldehyde bands at 5.31–5.61 ppm and 92 ppm, respectively.45,57
O carbonyl stretch of saturated aliphatic aldehydes (Fig. 2A),48 while 1H-NMR (Fig. 4B) and solid-state 13C-NMR (Fig. 4C) spectra confirmed the characteristic aldehyde bands at 5.31–5.61 ppm and 92 ppm, respectively.45,57
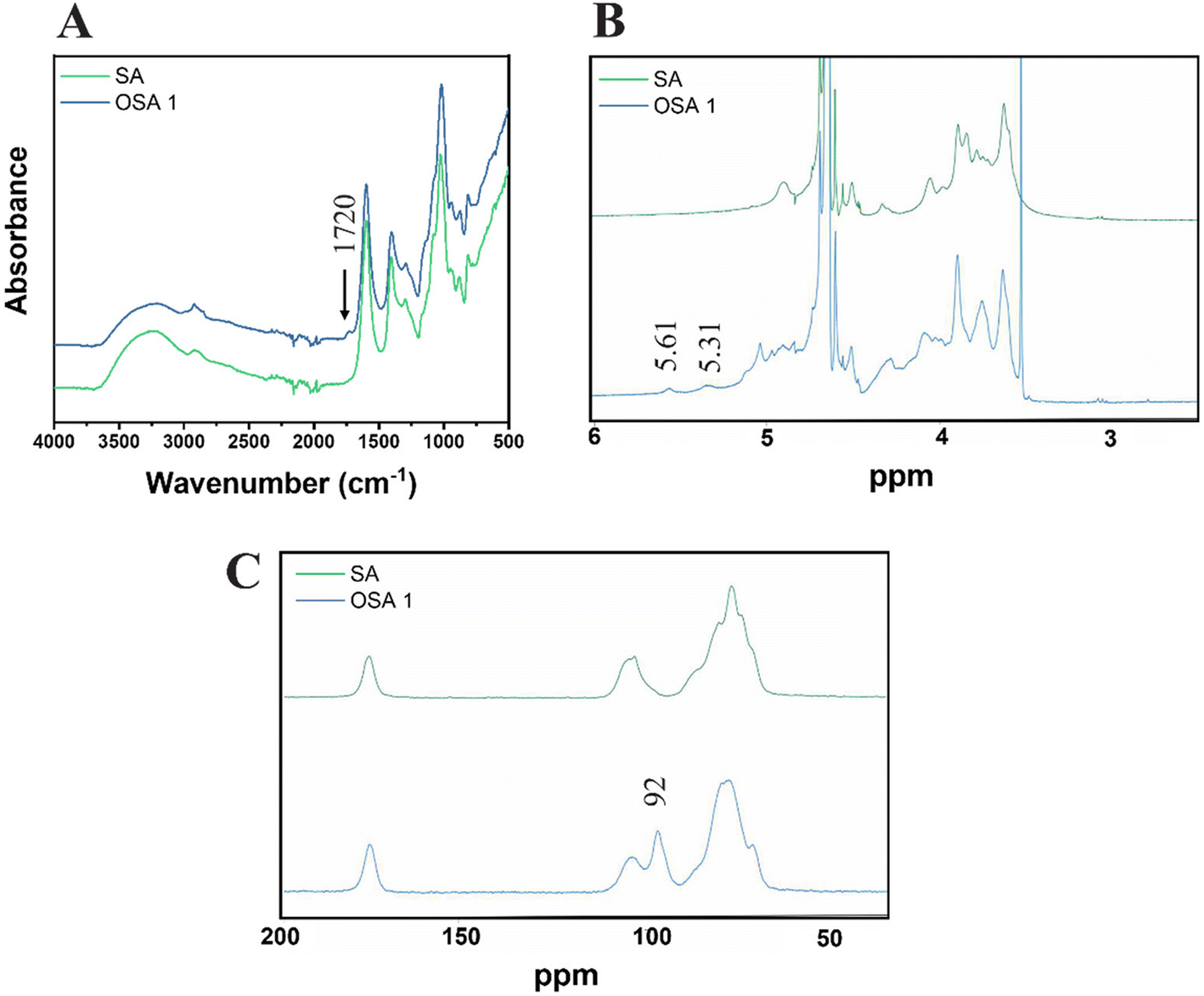 | ||
| Fig. 2 ATR FT-IR (A), 1H-NMR (B) and solid-state 13C-NMR (C) of OSA 1. | ||
In the second step, the thiolation reaction was performed using cysteine (CYS) as the reactant45,47 with a molar ratio of 1:4 of the alginate rings and cysteine. The synthesis conditions are also reported in Table S1 (see the ESI†). Briefly, the Schiff base reaction leads to a nucleophilic addition of the cysteine primary amines on the OSA aldehydes.58 Then, the carbinolamine intermediate evolves by dehydration to an imine functionality.46,59 The pH was maintained at 7.4 to ensure an effective Schiff base reaction.60 The reduction of the imine groups by sodium borohydride at a molar 1:1 ratio (compared with the carbonyl groups of OSA) produces thiolated oxidized sodium alginate (TOSA). The degree of substitution was evaluated using the Ellman's reagent protocol (see the ESI†). Unfortunately, the degree of functionalization measured by absorption on the UV-VIS spectra resulted in 0.5 ± 0.1%, which is too low for any further reaction of cross-linking. The lower degree of functionalization of TOSA may be related to an ineffective reduction amination reaction and subsequent hydrolysis of the imine group during dialysis.45,61
The second synthetic route to functionalize sodium alginate with thiol groups involves the activation of the carboxylic moieties via carbodiimide chemistry39,62,63 in a “one-pot” strategy (just by adding subsequently the reactants into the same container). The two-step amination reaction employs EDC and NHS as activating agents. In order to compare this reaction with the previously reported one, cysteine (CYS) was selected as the functional agent. In addition, to eventually boost further the degree of functionalization, cysteamine (CSA) was also used to modify alginate with thiol moieties. To compare the efficiency of the reactions, the previously used molar ratio of reactants was selected (1:4 = SA:CYS or SA:CSA). The reaction schemes are reported in Fig. 1B (1) and (2). Avoiding the oxidation step, the only requirement relates to the control of the reaction pH during different steps. In the first step (activation of carboxylic groups), an acidic pH is needed (generally around 4) in order to protonate the carboxylic moieties and to make the first conjugation with EDC (unstable intermediate) possible, followed by the reaction of NHS to form a stable ester.64,65 During thiolation, the maintenance of an N2 controlled atmosphere, darkness and acidic pH (i.e., 4) is crucial to prevent the easy oxidation of thiols in the presence of oxygen, especially in water environments. Moreover, in water solution, oxygen can also catalyze the formation of disulfide bonds, decreasing the total number of thiol groups.39,49,50 The degree of functionalization was measured by the Ellman protocol using the previously reported ε (see the ESI†); the used synthesis conditions are summarized in Table S2.† SA–CYS showed a degree of functionalization of 4.5 ± 0.3%, considerably higher than the previous reaction (0.5% = TOSA). Considering the higher degrees of functionalization and the reduction of one purification step, the carbodiimide reaction was selected to functionalize alginate. When cysteamine (SA–CSA) was used, the degree of functionalization obtained was even higher (14 ± 2%). In this case, we attribute the increase in modification to the lower steric hindrance of the CSA molecule with respect to CYS, because of the absence of the second carboxylic group.45 Considering the results, SA–CSA was chosen as the most promising candidate for the creation of hydrogels. For the sake of completeness, the functionalized alginate was also analyzed by 1H-NMR and ATR-FTIR, which indicated the presence of the grafted molecules. Furthermore, different degradation paths were observed by thermogravimetric analysis after the functionalization reaction. ATR, NMR and TGA analyses are reported and commented in the ESI (Fig. S4–S8†).
Given these findings, carbodiimide chemistry was also selected as the strategy to introduce a double bond in the alginate skeleton and norbornene methylamine (NOR) was selected as the reactant. The reaction scheme is reported in Fig. 1B (3). In this case, after a similar activation step using EDC and NHS at pH 4 (as described before), the pH was increased to 8.5 to increase the amine nucleophilicity of norbornene methylamine while making possible its reaction with the intermediate NHS-ester;61,66,67 the reaction was maintained under an argon atmosphere to prevent the self-reaction of the NOR molecules. The synthesis conditions are reported in Table S3 (see the ESI†). Once successfully dialyzed, the product (SA–NOR) was analyzed by 1H-NMR. The successful functionalization was evidenced by the characteristic peaks at 6.02 and 6.27 ppm in the 1H-NMR spectrum (Fig. 3A and Fig. S9†), corresponding to the two methylenes of the norbornene double bond.31 A degree of functionalization of 15% was calculated by integration, a value similar to its correlative SA–CSA counterpart. Lastly, the reproducibility of the reactions was evaluated by repeating the procedure in triplicate, experiencing similar values of modification for all the products obtained with the “one-pot” strategy (SA–CYS, SA–CSA and SA–NOR) and independently of the functionalization reaction. The degrees of functionalization of all the modified alginates are summarized in Fig. 3B.
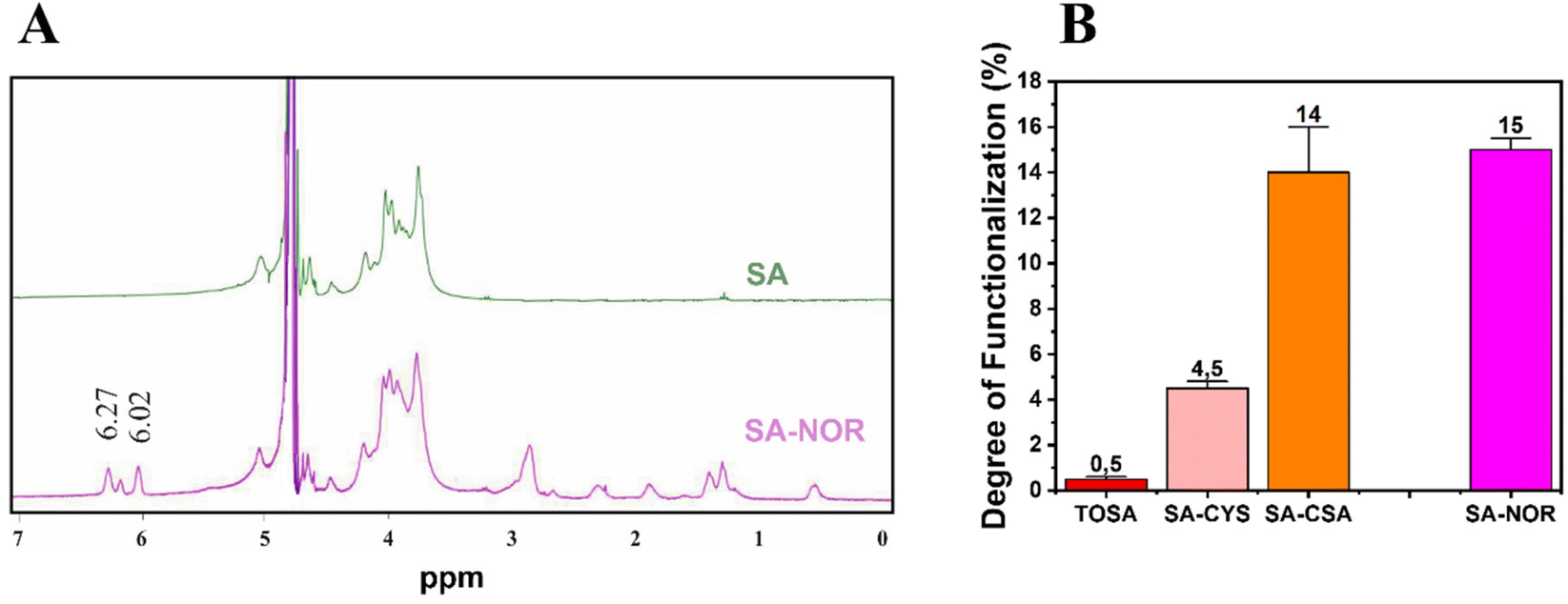 | ||
| Fig. 3 1H-NMR spectra of SA–NOR and sodium alginate SA (A). Calculated degree of functionalization, obtained by the Ellman's reagent method for the thiolated monomers and by integration of the NMR peaks for SA–NOR (B). | ||
Hydrogel production and characterization
Thiol–ene reactions are known to be highly regiospecific, reactive, and insensitive to oxygen and aqueous environments.34 Thus, the two selected products (SA–CSA and SA–NOR) showing a similar degree of substitution (∼15%) were solubilized separately in DI water to obtain two solutions with a concentration of 10 wt% each. The two solutions resulting in a similar concentration of thiol and alkene groups (0.67 and 0.68 mmol for each gram of materials, respectively), were then mixed expecting a 1:1 regioselective conversion between the two moieties. This relative ratio was chosen to try to avoid the presence of unreacted groups in the final material. Instead, the total concentration of 10 wt% was chosen according to a preliminary investigation of the viscosity of solutions prepared at different concentrations (Fig. S11 in the ESI†). Envisaging DLP printing 10 wt% resulted in a good compromise between viscosity and final mechanical properties47 in order to create stiff and self-standing hydrogels. With a homogeneous solution, the LAP photoinitiator was added to the solution and solubilized. The photoinitiator choice is mainly based on its low cytotoxic effects on living cells and visible light absorption.68
The formation of the network, the system reactivity and the irradiation time were investigated by photorheology (Fig. 4A). As can be seen, the variation of the storage modulus G′ and the loss modulus G′′ measured during the photo-crosslinking reaction indicated a high reactive system: the absence of reaction delay after light exposure and high slope of storage modulus G′. Also, thiol–ene hydrogel (TE Hydrogel) doesn't show any clear upper plateau even though the obtained G′ values (≈3 × 103 Pa) are comparable with other reported hydrogels used in tissue engineering.69–71 This behavior still suggests a slow ongoing progression of the reaction even after 500 s of irradiation. The mechanical properties of the hydrogel were tested by both amplitude sweep measurements and compression tests. The hydrogel stability over an incremental strain was measured by amplitude sweep (Fig. 4B). Herein, an important parameter used to understand the hydrogel properties is the yield point (or the maximum strain point applicable before the hydrogel collapse). This system possesses a yield point at 138% of strain, which is in line or higher with respect to other polysaccharide-based crosslinked hydrogels.31,35,69,71,72 Furthermore, the compressive elastic modulus of 44 ± 3 kPa places the resulting hydrogel among the stiff soft tissue scaffolds, with properties comparable with the human intestine or tendons (Fig. 4C).73 Moreover, the low elongation at the rupture (27%) and high ultimate strength (≈19 kPa) support the brittleness hydrogel observations during amplitude sweep and the theorized high crosslinking density.
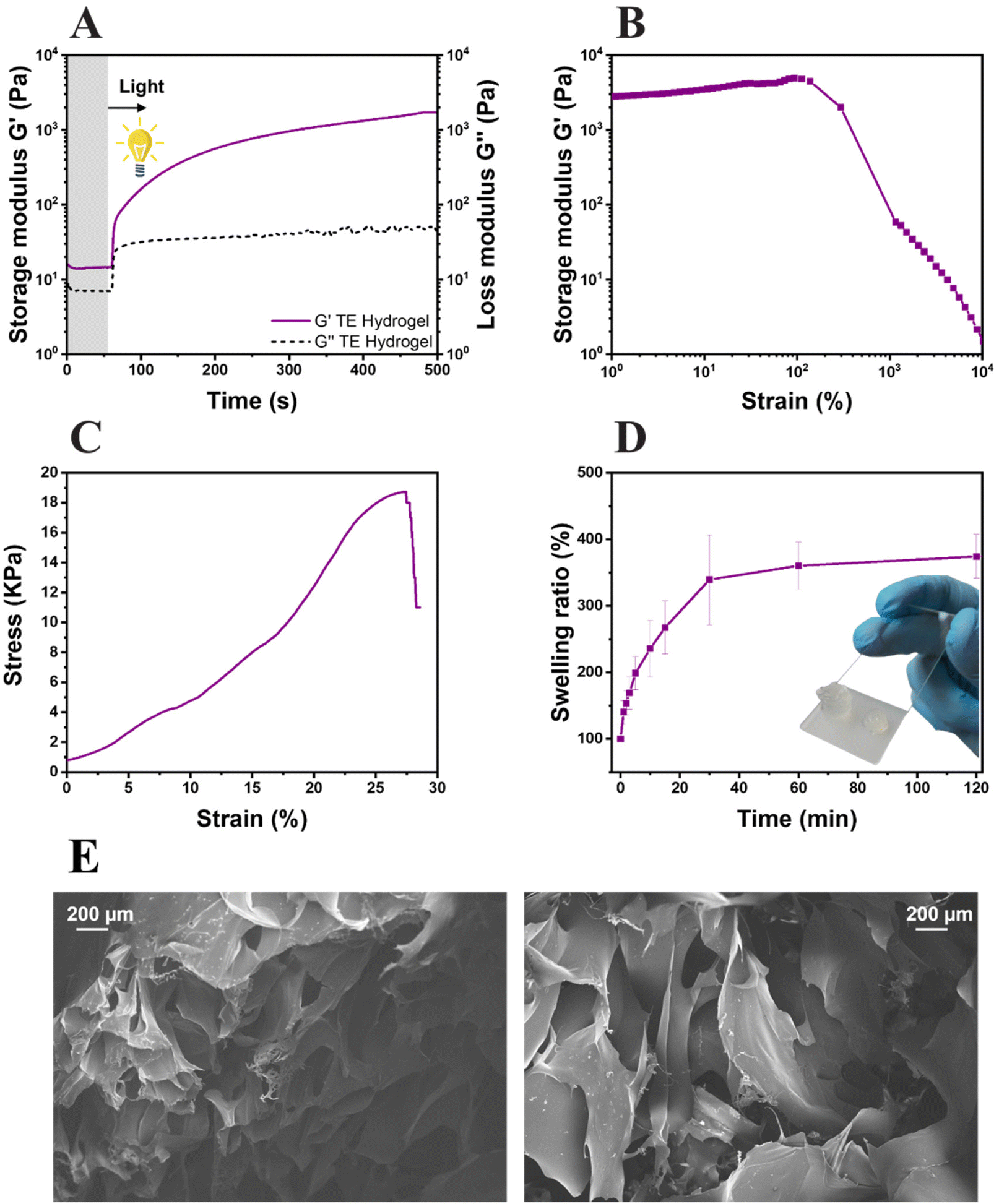 | ||
| Fig. 4 Photorheology (A), amplitude sweep (B), compression test (C) and swelling kinetics (D) of the thiol–ene hydrogel (TE hydrogel). Field Emission Scanning Electron Microscopy (FESEM) of the TE hydrogels; scale bar 200 μm (E). | ||
The swelling ability of the hydrogel was also evaluated (Fig. 4D); a swelling equilibrium of 376 ± 21% was reached after 30 min of immersion in DI water. The results are comparable with the data reported in similar studies, both in terms of values and trends of swelling kinetics.31 The gel content of the hydrogels, assessed in triplicate, showed a value of 78 ± 3%. Both results again suggest the high crosslinking density of the gels. However, as it is known that the scaffolds need to possess a certain degree of porosity to allow cells to attach and migrate inside the hydrogel, the morphology of the lyophilized hydrogels was investigated by FESEM. As visible in Fig. 4E, the hydrogels possess highly porous structures with diameters in the range of 200 μm, totally compatible with cell dimensions.74,75 With the confirmation of the high reactivity of the formulation, the hydrogel high yield point and storage modulus at compression, optimal swelling degree and acceptable porosity, the hydrogel was further studied for 3D printing and biological characterization.
Vat 3D-printing
Considering its high reactivity, the full-alginate formulation was tested for 3D printing with DLP technology. Besides reactivity, the mechanical properties of the cured material are also important during the printing process; indeed, since in DLP printers the final object is built upside-down (scheme in Fig. 5), the hydrogel requires sufficient properties of mechanical resistance under low solicitations or strains. The previously measured properties were totally comparable with values reported in the literature.69 The printing parameters were empirically optimized and are reported in Table S4 (see the ESI†). Two different 3D CAD files were prepared in order to test the printing of structures with increasing complexity. As a first attempt, a honeycomb-like structure was printed (Fig. 5). This geometry was tested to create shaped bulk structures with thin self-standing walls. As visible, small architectures (around 9.5 mm) were printed with defined angles, forming clear hexagonal cavities (with dimensions of around 3 mm). The ability to create defined structures, both in terms of fidelity and resolution, was then explored to form suspended architectures. In fact, a stable cubic geometry (named “Hollow cube”) was printed with an internal cavity in the range of 6 mm. Considering that, trying to avoid the use of different molecules, no dyes or radical scavengers were added to the formulation to limit the light scattering and increase the resolution, the obtained structures can be considered with good shape fidelity to the original file.69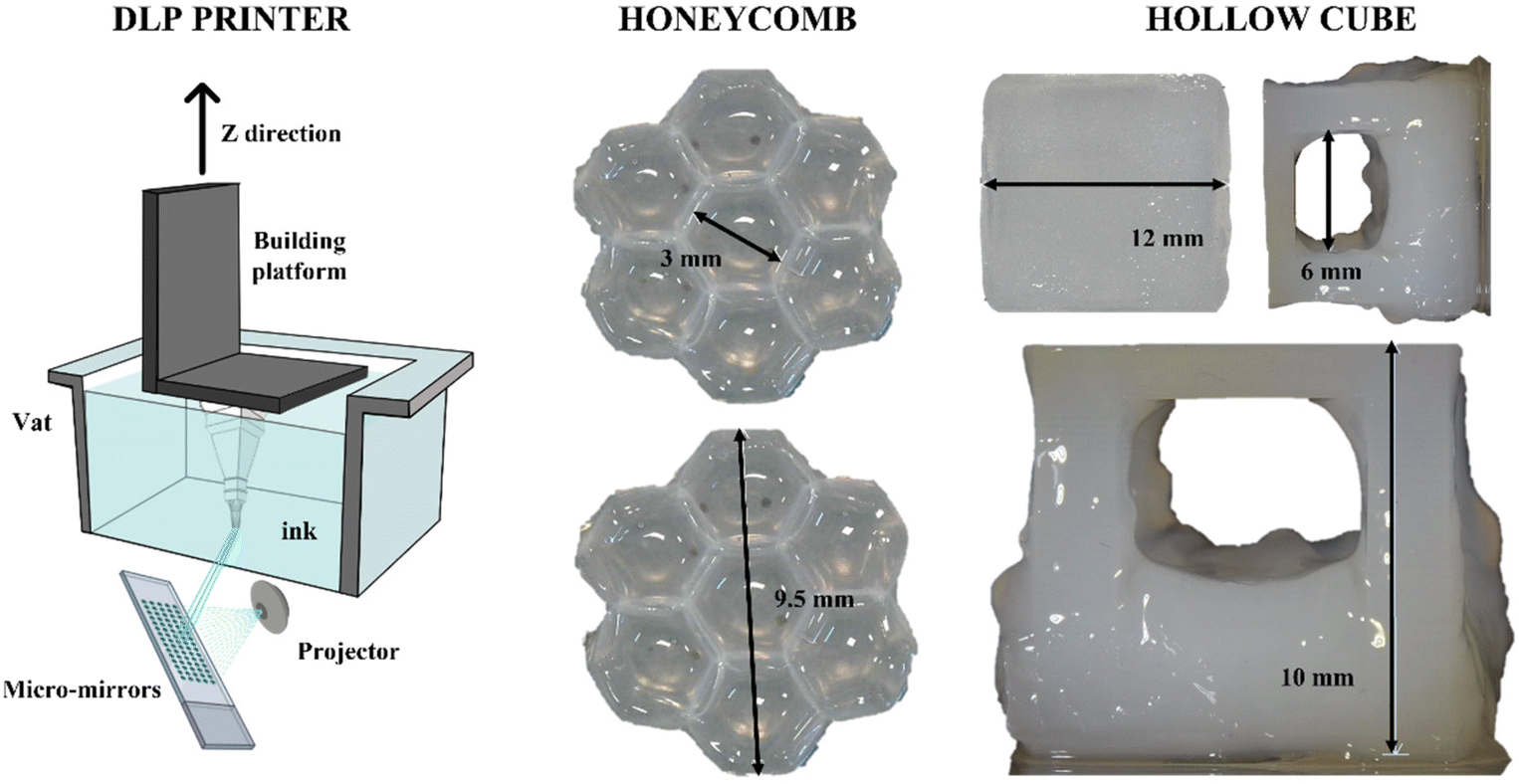 | ||
| Fig. 5 Digital light projection (DLP) printer scheme and the TE hydrogel processed geometries. | ||
Cell viability and proliferation
As described in the characterization method paragraph, a triplicate of the hydrogel was sterilized and immersed in a biological medium at 37 °C for 24 h, prior to the biological evaluation. At the same time, autofluorescent C166-GFP endothelial cells were seeded on a 12-well plate and adhered for 24 h. The biological medium extracted from the TE hydrogels was brought into contact (1:1 and 1:5 dilutions) with the endothelial cells in the 12-well plate to ensure no toxic substances are released from the hydrogels (according to ISO 10993-5 recommendations). Firstly, cell proliferation was certified over 48 h by inverted field microscopy (Fig. 6A), and then metabolic activity and DNA quantification were assessed to ensure the hydrogel cytocompatibility (Fig. 6B and C, respectively). No evidence of cell detachment, necrotic or apoptotic cell bodies was reported after the culture media replacement but rather the cultures presented healthy and confluent cell monolayers. Compared with the control without extracts, proper levels of mitochondrial metabolic activity (Alamar blue assay, Fig. 6B) and the optimal levels of DNA content were evidenced, verifying that no cytotoxic substances that could disrupt cell viability were released from the hydrogels (Fig. 6C). In summary, the fully modified alginate hydrogel obtained by the thiol–ene reaction did not present any signs of indirect in vitro cytotoxicity and may be used as stiff scaffolds for tissue engineering applications.
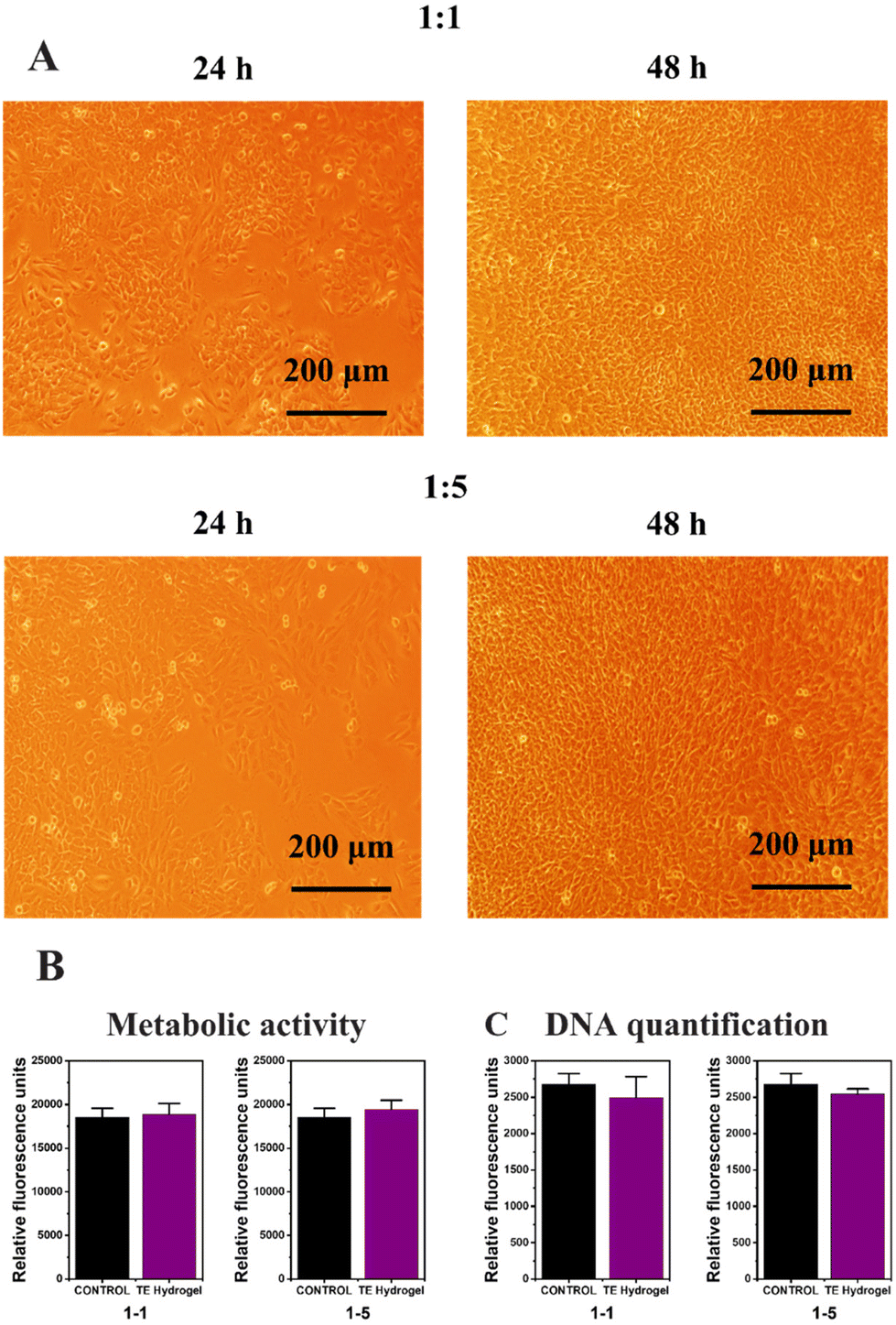 | ||
| Fig. 6 Bright field microscopy (A), metabolic activity (B) and DNA quantification (C) of the TE Hydrogel. | ||
Conclusions
Herein, a simple synthetic route was described to functionalize alginate with both thiol and alkene groups, with the aim to perform thiol–ene click reactions without the addition of any external crosslinker molecule. Concerning the thiol functionalization, a “two-reaction strategy” including the oxidation of the alginate chain and a “one-pot strategy” based on carbodiimide chemistry were evaluated. The best degree of functionalization was achieved using the “one-pot” strategy in the presence of cysteamine as the functionalizing agent (∼14%). Then, the same chemical route was selected to incorporate norbornene methylamine, obtaining products with functionalization ranges of the same degree (∼15%). More importantly, high levels of reaction reproducibility were evidenced, independently of the employed functional molecule. The formulation reactivity was studied by photorheology, which confirmed the adequacy of the ink properties for DLP 3D printers. The resulting 3D printed structures presented defined and self-standing hydrogel architectures, with well-shaped angles/surfaces and the possibility to create suspended geometries. The mechanical, swelling and morphological properties of the hydrogels were also evaluated, placing the material among the stiff soft tissue scaffolds, with properties comparable with the human intestine or tendons. Furthermore, the modified alginate hydrogel obtained by the thiol–ene reaction did not present any signs of indirect in vitro cytotoxicity, suggesting its feasible employment as stiff scaffolds for tissue engineering applications.Conflicts of interest
There are no conflicts to declare.References
- K. D. Weiss, Prog. Polym. Sci., 1997, 22, 203–245 CrossRef.
- F. Peltier and D. Thierry, Coatings, 2022, 12, 518–545 CrossRef CAS.
- M. S. Malik, S. Schlögl, M. Wolfahrt and M. Sangermano, Polymers, 2020, 12, 2146–2180 CrossRef CAS PubMed.
- B. A. Suslick, J. Hemmer, B. R. Groce, K. J. Stawiasz, P. H. Geubelle, G. Malucelli, A. Mariani, J. S. Moore, J. A. Pojman and N. R. Sottos, Chem. Rev., 2023, 123, 3237–3298 CrossRef CAS PubMed.
- U. Zschieschang, H. Klauk and J. W. Borchert, Adv. Mater. Technol., 2023, 8, 2201888–2201893 CrossRef CAS.
- A. G. MacDiarmid, Angew. Chem., Int. Ed., 2001, 40, 2581–2590 CrossRef CAS PubMed.
- Y. K. Yang, Microelectron. Int., 2006, 23, 26–32 CrossRef CAS.
- I. Kunio and E. Takeshi, Dent. Mater. J., 2010, 29, 481–501 CrossRef PubMed.
- F. Petko, A. Świeży and J. Ortyl, Polym. Chem., 2021, 12, 4593–4612 RSC.
- A. Santini, I. T. Gallegos and C. M. Felix, Prim. Dent. J., 2013, 2, 30–33 CrossRef PubMed.
- I. Chiulan, E. B. Heggset, Ş. I. Voicu and G. Chinga-Carrasco, Biomacromolecules, 2021, 22, 1795–1814 CrossRef CAS PubMed.
- C. Felipe-Mendes, L. Ruiz-Rubio and J. L. Vilas-Vilela, Emergent Mater., 2020, 3, 453–468 CrossRef CAS.
- M. Zanon, D. Baruffaldi, M. Sangermano, C. F. Pirri, F. Frascella and A. Chiappone, Eur. Polym. J., 2021, 160, 110813–110826 CrossRef CAS.
- V. S. D. Voet, J. Guit and K. Loos, Macromol. Rapid Commun., 2021, 42, 2000475 CrossRef CAS PubMed.
- M. Pagac, J. Hajnys, Q.-P. Ma, L. Jancar, J. Jansa, P. Stefek and J. Mesicek, Polymers, 2021, 13, 598 CrossRef CAS PubMed.
- D. Lei, Y. Yang, Z. Liu, B. Yang, W. Gong, S. Chen, S. Wang, L. Sun, B. Song, H. Xuan, X. Mo, B. Sun, S. Li, Q. Yang, S. Huang, S. Chen, Y. Ma, W. Liu, C. He, B. Zhu, E. M. Jeffries, F.-L. Qing, X. Ye, Q. Zhao and Z. You, Mater. Horiz., 2019, 6, 1197–1206 RSC.
- A. Salas, M. Zanatta, V. Sans and I. Roppolo, ChemTexts, 2023, 9, 1–16 CrossRef.
- B. Tosetto, M. Gastaldi, G. Renno, C. F. Pirri, C. Barolo, A. Fin and I. Roppolo, Polym. Chem., 2023, 14, 1213–1223 RSC.
- E. M. Maines, M. K. Porwal, C. J. Ellison and T. M. Reineke, Green Chem., 2021, 23, 6863–6897 RSC.
- M. Shahbazi and H. Jäger, ACS Appl. Bio Mater., 2021, 4, 325–369 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1879 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487–492 CrossRef CAS PubMed.
- W. R. Gombotz and S. F. Wee, Adv. Drug Delivery Rev., 1998, 31, 267–285 CrossRef CAS PubMed.
- J. A. Rowley, G. Madlambayan and D. J. Mooney, Biomaterials, 1999, 20, 45–53 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 CrossRef CAS PubMed.
- M. E. Furth, A. Atala and M. E. Van Dyke, Biomaterials, 2007, 28, 5068–5073 CrossRef CAS PubMed.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- X. Hu, Z. Zhang, H. Wu, S. Yang, W. Zhao, L. Che, Y. Wang, J. Cao, K. Li and Z. Qian, Biomater. Adv., 2023, 152, 213501–213521 CrossRef CAS PubMed.
- S. Vanslambrouck, R. Riva, B. Ucakar, V. Préat, M. Gagliardi, D. G. M. Molin, P. Lecomte and C. Jérôme, Molecules, 2021, 26, 1750–1774 CrossRef CAS PubMed.
- H. W. Ooi, C. Mota, A. T. ten Cate, A. Calore, L. Moroni and M. B. Baker, Biomacromolecules, 2018, 19, 3390–3400 CrossRef CAS PubMed.
- R. M. Desai, S. T. Koshy, S. A. Hilderbrand, D. J. Mooney and N. S. Joshi, Biomaterials, 2015, 50, 30–37 CrossRef CAS PubMed.
- J. C. Kloxin, F. T. Scott and N. C. Bowman, Macromolecules, 2009, 42, 2551–2556 CrossRef PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- M. Zanon, L. Montalvillo-Jiménez, P. Bosch, R. Cue-López, E. Martínez-Campos, M. Sangermano and A. Chiappone, Polymers, 2022, 14, 4709–4721 CrossRef CAS PubMed.
- H. Choi, M. Kim, J. Jang and S. Hong, Angew. Chem., Int. Ed., 2020, 59, 22514–22522 CrossRef CAS PubMed.
- Y. Liu, W. Hou, H. Sun, C. Cui, L. Zhang, Y. Jiang, Y. Wu, Y. Wang, J. Li, B. S. Sumerlin, Q. Liu and W. Tan, Chem. Sci., 2017, 8, 6182–6187 RSC.
- A. Oesterreicher, M. Roth, D. Hennen, F. H. Mostegel, M. Edler, S. Kappaun and T. Griesser, Eur. Polym. J., 2017, 88, 393–402 CrossRef CAS.
- A. Bernkop-Schnürch, Adv. Drug Delivery Rev., 2005, 57, 1569–1582 CrossRef PubMed.
- J. Iqbal, G. Shahnaz, S. Dünnhaupt, C. Müller, F. Hintzen and A. Bernkop-Schnürch, Biomaterials, 2012, 33, 1528–1535 CrossRef CAS PubMed.
- Y. Zhang, S. Zhou, F. Deng, X. Chen, X. Wang, Y. Wang, H. Zhang, W. Dai, B. He, Q. Zhang and X. Wang, Eur. J. Pharm. Biopharm., 2018, 133, 188–199 CrossRef CAS PubMed.
- P. M. Kharkar, M. S. Rehmann, K. M. Skeens, E. Maverakis and A. M. Kloxin, ACS Biomater. Sci. Eng., 2016, 2, 165–179 CrossRef CAS PubMed.
- C.-C. Lin, A. Raza and H. Shih, Biomaterials, 2011, 32, 9685–9695 CrossRef CAS PubMed.
- R. Holmes, X.-B. Yang, A. Dunne, L. Florea, D. Wood and G. Tronci, Polymers, 2017, 9, 226–251 CrossRef PubMed.
- R. G. Huamani-Palomino, B. M. Córdova, E. R. Pichilingue, L. T. Venâncio and A. C. Valderrama, Polymers, 2021, 13, 255–271 CrossRef CAS PubMed.
- M. Ø. Dalheim, A. S. T. Ulset, I. B. Jenssen and B. E. Christensen, Carbohydr. Polym., 2017, 157, 1844–1852 CrossRef CAS PubMed.
- S. Hauptstein, S. Dezorzi, F. Prüfert, B. Matuszczak and A. Bernkop-Schnürch, Carbohydr. Polym., 2015, 124, 1–7 CrossRef CAS PubMed.
- Z. Emami, M. Ehsani, M. Zandi and R. Foudazi, Carbohydr. Polym., 2018, 198, 509–517 CrossRef CAS PubMed.
- C. M. Q. Le, F. Morlet-Savary and A. Chemtob, Polym. Chem., 2021, 12, 6594–6605 RSC.
- D. Schilter, Nat. Rev. Chem., 2017, 1, 0013 CrossRef.
- S. W. Griffiths, J. King and C. L. Cooney, J. Biol. Chem., 2002, 277, 25486–25492 CrossRef CAS PubMed.
- J. S. Fritz, S. S. Yamamura and E. C. Bradford, Anal. Chem., 1959, 31, 260–263 CrossRef CAS.
- H. Peng, W. Chen, Y. Cheng, L. Hakuna, R. Strongin and B. Wang, Sensors, 2012, 12, 15907–15946 CrossRef CAS PubMed.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS PubMed.
- A. Jejurikar, X. T. Seow, G. Lawrie, D. Martin, A. Jayakrishnan and L. Grøndahl, J. Mater. Chem., 2012, 22, 9751–9758 RSC.
- K. H. Bouhadir, K. Y. Lee, E. Alsberg, K. L. Damm, K. W. Anderson and D. J. Mooney, Biotechnol. Prog., 2001, 17, 945–950 CrossRef CAS PubMed.
- C. G. Gomez, M. Rinaudo and M. A. Villar, Carbohydr. Polym., 2007, 67, 296–304 CrossRef CAS.
- C. Le-Tien, M. Millette, M. Lacroix and M. A. Mateescu, Biotechnol. Appl. Biochem., 2004, 39, 189–198 CrossRef CAS PubMed.
- E. W. Baxter and A. B. Reitz, Org. React., 2004, 1–714, DOI:10.1002/0471264180.or059.01.
- P. Di Bernardo, P. L. Zanonato, S. Tamburini, P. Tomasin and P. A. Vigato, Dalton Trans., 2006, 4711–4721, 10.1039/B604211B.
- J. Battersby, R. Clark, W. Hancock, E. Puchulu-Campanella, N. Haggarty, D. Poll and D. Harding, J. Controlled Release, 1996, 42, 143–156 CrossRef CAS.
- S. Dünnhaupt, J. Barthelmes, C. C. Thurner, C. Waldner, D. Sakloetsakun and A. Bernkop-Schnürch, Carbohydr. Polym., 2012, 90, 765–772 CrossRef PubMed.
- T. F. Palmberger, K. Albrecht, B. Loretz and A. Bernkop-Schnürch, Eur. J. Pharm. Biopharm., 2007, 66, 405–412 CrossRef CAS PubMed.
- E. Guzmán, R. G. Rubio and F. Ortega, Adv. Colloid Interface Sci., 2020, 282, 102197 CrossRef PubMed.
- A. Hansson, N. Hashom, F. Falson, P. Rousselle, O. Jordan and G. Borchard, Carbohydr. Polym., 2012, 90, 1494–1500 CrossRef CAS PubMed.
- S. Bian, M. He, J. Sui, H. Cai, Y. Sun, J. Liang, Y. Fan and X. Zhang, Colloids Surf., B, 2016, 140, 392–402 CrossRef CAS PubMed.
- J. C. Breger, B. Fisher, R. Samy, S. Pollack, N. S. Wang and I. Isayeva, J. Biomed. Mater. Res., Part B, 2015, 103, 1120–1132 CrossRef CAS PubMed.
- B. Huber, K. Borchers, G. E. Tovar and P. J. Kluger, J. Biomater. Appl., 2016, 30, 699–710 CrossRef CAS PubMed.
- M. Zanon, R. Cue-López, E. Martínez-Campos, P. Bosch, D.-L. Versace, H. Hayek, N. Garino, C. F. Pirri, M. Sangermano and A. Chiappone, Addit. Manuf., 2023, 69, 103553 CAS.
- Z. Feng, M. Hakkarainen, H. Grützmacher, A. Chiappone and M. Sangermano, Macromol. Chem. Phys., 2019, 220, 1900174 CrossRef.
- C. Noè, C. Tonda-Turo, A. Chiappone, M. Sangermano and M. Hakkarainen, Polymers, 2020, 12, 1359 CrossRef PubMed.
- S. Kim and S. Jung, Carbohydr. Polym., 2020, 250, 116934 CrossRef CAS PubMed.
- A. M. Handorf, Y. Zhou, M. A. Halanski and W. J. Li, Organogenesis, 2015, 11, 1–15 CrossRef PubMed.
- A. D. Doyle, R. J. Petrie, M. L. Kutys and K. M. Yamada, Curr. Opin. Cell Biol., 2013, 25, 642–649 CrossRef CAS PubMed.
- P. Friedl, E. Sahai, S. Weiss and K. M. Yamada, Nat. Rev. Mol. Cell Biol., 2012, 13, 743–747 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00902e |
| This journal is © The Royal Society of Chemistry 2023 |