
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An electrochemical Hofmann rearrangement on acrylamide copolymers†
Muzhao
Wang
and
Paul
Wilson
 *
*
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: p.wilson.1@warwick.ac.uk
First published on 20th June 2023
Abstract
The primary amide side-chain of acrylamide copolymers has been utilised as an isocyanate surrogate. The isocyanate is formed under mild conditions via an electrochemical Hofmann rearrangement resulting in the formation of O-alkyl carbamate side-chains in alcohol solvents. This represents a new strategy for the modification of amide-functionalised (co)polymers.
Carbamides (ureas), carbamates (urethanes), carbamothioates and amides are all functional groups consisting of a carbonyl group with an α-N-substituent. They are ubiquitous in nature while synthetic analogues are widely applied in medicinal chemistry and polymer/materials science.1–5 These functional groups are typically accessed from carboxylic acids, or derivatives thereof, via isocyanate intermediates.6
In the context of polymer synthesis and functionalisation, the importance of isocyanates cannot be understated. For example, diisocyanates are required for the synthesis of polyureas and polyurethanes which have global market values in the region $1 billion and $75 billion USD respectively.7,8 Isocyanates are also attractive intermediates for polymer functionalisation because they undergo clean addition reactions with amine,9 alcohol10 and thiol-based11 nucleophiles with 100% atom economy. Despite this there are several limitations associated with the use of isocyanates. Traditional synthetic routes require multi-step reactions, employ hazardous reagents (e.g. azides, phosgene) and have poor overall atom economy.12,13 Alternative strategies for generating isocyanates at polymer chain-ends and/or side-chains have been developed. The common strategy requires the incorporation of ‘masked’ isocyanates or isocyanate surrogates.14–17 One prominent approach involves the incorporation of carbonyl-azide functional groups that can undergo a Curtius rearrangement forming isocyanates in situ.18 This has been exploited for polymer–polymer ligation,19 polymer–peptide conjugation20 and post-polymerisation modification.21 Despite the efficiency of these reactions, it is still necessary to synthesise carbonyl-azide derivatives pre- or post-polymerisation.
The Hofmann rearrangement22 provides access to isocyanates from primary amide precursors. The classical reaction employs stoichiometric amounts of halogen reagents in the presence of the NaOH resulting in highly corrosive and toxic reaction conditions. Alternative strategies employ stoichiometric amounts of iodine(III) or N-bromosuccinimide.23–25
Electrochemical intervention in (macro)molecular synthesis has received renewed interest over the last decade.26–28 Through control of the electric field, the thermodynamics and/or kinetics of electron transfer processes and therefore reaction mechanisms can be precisely interrogated, controlled and understood.29,30 Furthermore, the use of electrons as reagents, derived from sustainably generated power (solar, wind, water, etc.), represents a more environmentally-benign approach to synthesis compared to analogous chemically-driven processes.
The practical limitations associated with the Hofmann rearrangement have been overcome through the development of an electrochemical Hofmann (eHofmann) rearrangement. These methods replace stoichiometric halogens with catalytic halide salts (e.g. KBr, NaBr) and highly basic reaction media with neutral alcohol or mixed organic/alcohol solvent systems.31,32 The halogen and base required to promote the formation of the isocyanate intermediate are generated in situ by anodic oxidation (halide to halogen) and cathodic reduction (alcohol to alkoxide). The most recent strategy employs constant current electrolysis in acetonitrile/MeOH at 50 °C, using 50 mol% NaBr as mediator in the absence of any other reagents or electrolytes. Under these conditions aryl and alkyl amides are converted to O-methyl carbamates in good yields.33
Herein, we build on our recent work on the electrochemically triggered/mediated synthesis of polymers (by eATRP)34–37 by adopting the eHofmann rearrangement to achieve mild and efficient modification of acrylamide containing copolymer scaffolds for the first time. Overall we demonstrate that the primary amide side-chains can serve as ‘masked’ isocyanates which can be captured by alcohol-based nucleophiles forming O-alkyl carbamate side chains in the process.
Initially we adapted the reaction conditions reported to Zhang et al.33 to reproduce the eHofmann rearrangement of benzamide using our electrosynthesis apparatus (see ESI, Fig. S1†). In an undivided cell equipped with graphite working and counter electrodes quantitative conversion of benzamide into the methyl N-phenyl carbamate was achieved in the presence of NaBr (50 mol%) in MeCN/MeOH (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v) within 8 h at 60 °C under constant current electrolysis (CCE) at Iapp = 50 mA (Table S1 and Fig. S2†).
:2 v/v) within 8 h at 60 °C under constant current electrolysis (CCE) at Iapp = 50 mA (Table S1 and Fig. S2†).
With a view to applying this chemistry to acrylamide-containing copolymers, the eHofmann rearrangement of butyramide was attempted under the same conditions. Quantitative conversion to methyl N-propyl carbamate was achieved within 6 h (Table 1). The structure of the product was confirmed by 1H NMR through the appearance of the O-methyl protons at δH = 3.65 ppm and a shift in the methylene protons (Hc) from δH = 2.35 to 3.12 ppm (Fig. S3†). Considering potential solubility issues of acrylamide copolymers in MeCN, the eHofmann rearrangement of butyramide was repeated in pure MeOH. Pleasingly, quantitative conversion to methyl N-propyl carbamate was achieved within 3 h (Table 1). Reducing the reaction temperature to 50 °C had little effect on conversion, however when the reaction was run at 40 °C the rate of reaction decreased with only 68% conversion achieved within 3 h. Reactions performed on acrylamide-containing (co)polymer scaffolds were subsequently performed at 60 °C.
|
|
||||
|---|---|---|---|---|
| Time (h) | Conv. (%) MeCN/MeOH = 8:2 |
Conv. (%) MeOH 60 °C | Conv. (%) MeOH 50 °C | Conv. (%) MeOH 40 °C |
| 0 | 0 | 0 | 0 | 0 |
| 1 | 14 | 36 | 21 | 15 |
| 2 | 20 | 77 | 62 | 44 |
| 3 | 36 | >99 | 97 | 68 |
| 4 | 58 | |||
| 5 | 84 | |||
| 6 | 99 | |||
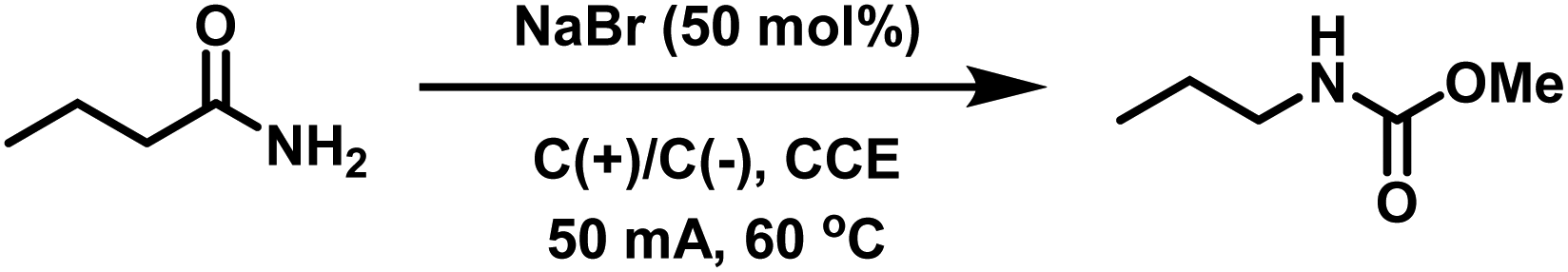
Acrylamide-containing (co)polymer scaffolds were prepared via free radical polymerization (FRP) of acrylamide (Am) and dimethylacrylamide (DMAm) in MeOH targeting PAm and PDMAm homopolymers and P(Am-co-DMAm) copolymer scaffolds with Am feed ratios between 10–50 mol% (Table S2†). The chemical composition of all the polymers was determined by 1H/13C NMR in D2O and Fourier-transform infra-red (FTIR) spectroscopy with comparison to PAm and PDMAm homopolymers. Using 1H NMR (Fig. S4†), integration of the protons corresponding to the N,N-dimethyl group of the DMAm side chain (δH = 2.78–3.15 ppm), against the entire copolymer backbone (δH = 1.19–2.77 ppm), with correction for the contribution to the backbone from DMAm, yielded Am compositional ratios of 7, 17 and 50 mol% (Table S3†). The 13C NMR (Fig. S5†) and FTIR spectra (Fig. S6†) show two distinct C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups assigned to the amide groups of Am (δC = 180 ppm) and DMAm (δC = 176 ppm) side chains respectively. Molecular weight analysis performed using aqueous size exclusion chromatography (SEC, Fig. S7†) revealed the PAm homopolymer to have an Mn,SEC = 7800 g mol−1 and Mw,SEC = 38000 g mol−1 (Table S3,† entry 5) whilst the PDMAm homopolymer had Mn,SEC = 21400 g mol−1 and Mw,SEC = 71700 g mol−1 (Table S3,† entry 1). The copolymer scaffolds gave comparable molecular weights with Mn,SEC = between 13700–20000 g mol−1 and Mw,SEC = between 71200–78800 g mol−1 (Table S3,† entries 2–4).
O groups assigned to the amide groups of Am (δC = 180 ppm) and DMAm (δC = 176 ppm) side chains respectively. Molecular weight analysis performed using aqueous size exclusion chromatography (SEC, Fig. S7†) revealed the PAm homopolymer to have an Mn,SEC = 7800 g mol−1 and Mw,SEC = 38000 g mol−1 (Table S3,† entry 5) whilst the PDMAm homopolymer had Mn,SEC = 21400 g mol−1 and Mw,SEC = 71700 g mol−1 (Table S3,† entry 1). The copolymer scaffolds gave comparable molecular weights with Mn,SEC = between 13700–20000 g mol−1 and Mw,SEC = between 71200–78800 g mol−1 (Table S3,† entries 2–4).
Thermogravimetric analysis (TGA) of PAm revealed thermal events occurring with onset temperatures of the 278 °C and 384 °C (Fig. S8A†). Conversely, PDMAm exhibited a single thermal event with an onset temperature of 417 °C (Fig. S8B†). Both degradation profiles were consistent with literature on PAm and N-disubstituted acrylamides.38,39 The P(Am-co-DMAm) copolymers exhibited a two-step degradation profile expected for the proposed copolymer structure (Fig. S9†). Using differential scanning calorimetry (DSC) the glass transition temperatures (Tg) of PAm and PDMAm were found to be 194 °C and 125 °C respectively. For the P(Am-co-DMAm) scaffolds a single Tg was measured, supporting the targeted statistical composition of the polymers synthesised by FRP (Fig. S10†). The Tg increased with increasing mole fraction of Am. At the lowest mole fraction (7 mol%) the Tg = 140 °C which increased up to Tg = 178 °C for P(Am-co-DMAm) containing 50 mol% Am.
The P(Am-co-DMAm) scaffolds were then subjected to the conditions for eHofmann rearrangement. The scaffolds were dissolved in MeOH (1 mmol w.r.t. Am) and electrolysed via CCE at 50 mA in an undivided cell fitted with graphite working and counter electrodes with heating at 60 °C for 90 min. Structural analysis by 1H NMR revealed a significant change in the region of the polymer backbone (Fig. 1A). The broad peak at δH = 2.25 ppm of the PAm backbone was completely absent and the pattern of the peaks at δH = 1.50–1.70 ppm changed. Furthermore there was a new peak at δH = 3.62 ppm which was assigned to the O-methyl group of the new carbamate side chain. Integration of this peak against the N,N-dimethyl group of the DMAm side chain (δH = 2.78–3.15 ppm) reveals quantitative conversion of the amide side chains to carbamate side chains. These structural changes are supported by 13C NMR which shows a shift in the CO carbon of the Am side from δC = 180 ppm in P(Am-co-DMAm) to δC = 158 ppm in the rearrangement product as well as appearance of the O-methyl carbon at δC = 53 ppm. The CO signal from DMAm side chains is retained at δC = 176 ppm (Fig. 1B). Furthermore, FTIR of the scaffolds after rearrangement indicated retention of the DMAm derived CO at 1614 cm−1 whilst the CO derived from the Am side chain (1660 cm−1) shifted completely to 1700 cm−1, indicative of carbamate formation (Fig. 1C).
 | ||
| Fig. 1 Representative 1H NMR (A), 13C NMR (B) in D2O and (C) FTIR showing the carbonyl region confirms quantitative conversion of the acrylamide side-chain to the O-methyl carbamate side chain using P(Am-co-DMAm) scaffold containing 50 mol% Am. | ||
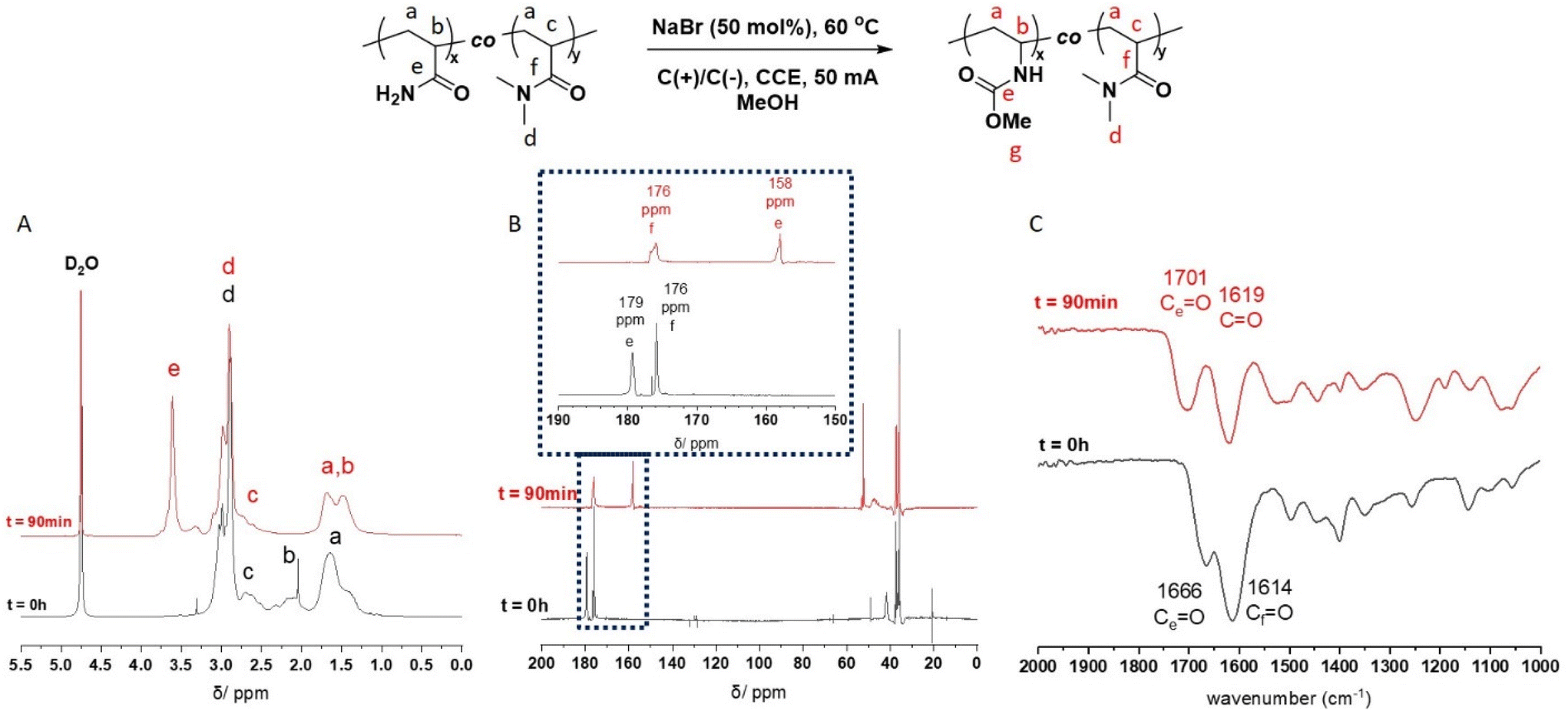
Molecular weight analysis by aqueous SEC indicated that the eHofmann rearrangement proceeded with a small decrease in molecular weight (Table S4 and Fig. S11†). Molecular weight analysis of the P(Am-co-DMAm) scaffolds and rearrangement productions was repeated using DMF GPC. The P(Am-co-DMAm) scaffold containing 50 mol% Am was not soluble in DMF. However the scaffolds contain 7 and 17 mol% Am gave Mn,SEC values of 39200 g mol−1 (Đm = 3.5) and 38500 g mol−1 (Đm = 3.3) respectively. The formation of the O-methyl carbamate rearrangement products resulted in an increase in molecular weight and little change in the dispersity (Table 2 and Fig. 2). Furthermore, whilst the P(Am-co-DMAm) scaffold containing 50 mol% Am was not soluble in DMF, the O-methyl carbamate rearrangement product derived from this scaffold was soluble with Mn,SEC = 36700 g mol−1 and Đm = 3.3.
 | ||
| Fig. 2 Molecular weight distributions from DMF SEC of the O-methyl carbamate functionalised polymer products formed via eHofmann rearrangement (Table 2). | ||
| Am (mol%) | P(Am-co-DMAm) | eHofmann product | ||
|---|---|---|---|---|
| M n,SEC (g mol−1) | Đ m | M n,SEC (g mol−1) | Đ m | |
| 7 | 39200 |
3.5 | 45200 |
3.2 |
| 17 | 38500 |
3.3 | 45300 |
3.3 |
| 50 | — | — | 36700 |
3.3 |
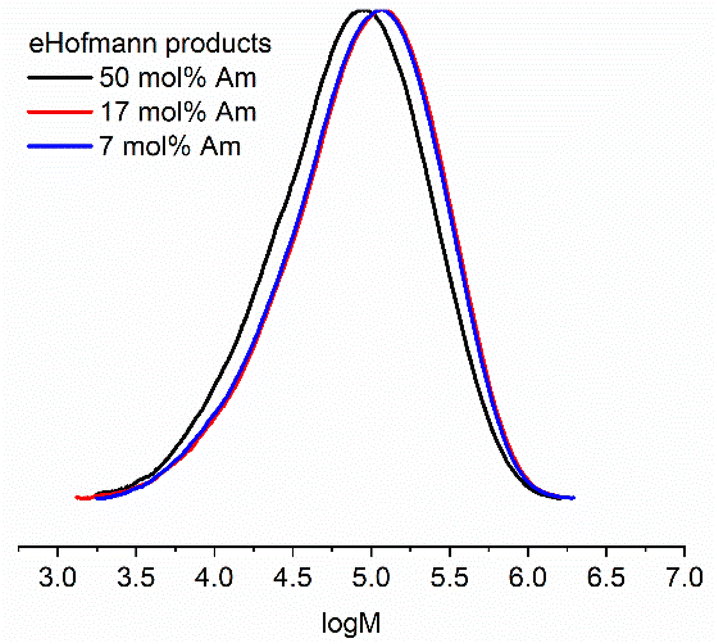
The effect of converting the pendant amide groups into carbamates on the thermal properties of the copolymer scaffolds was explored by TGA and DSC. Methyl N-alkyl/aryl carbamates are known to undergo thermal decomposition via competing pathways, one of which involves elimination of MeOH and formation of alkyl/aryl isocyanates, the proposed intermediate of the forward eHofmann reaction.40–44 The methyl carbamate-DMAm copolymers were shown to have different degradation profiles to the parent P(Am-co-DMAm) scaffolds by TGA (Fig. S9†), exhibited three meaningful thermal events (Fig. 3A). The two thermal events that occur at lower temperatures (Td,onset = 205–250 °C) can be attributed to decomposition of the O-methyl carbamate side chain. The higher temperature event (T > 330 °C) relates to degradation of the remaining polymer. Likewise, DSC revealed that the Tg of the O-methyl carbamate copolymers was lower than the parent P(Am-co-DMAm) scaffolds which is attributed to the relative hydrogen bonding ability of the primary amide and carbamate side chains respectively (Table S5,†Fig. 3B and S12–14†).
 | ||
| Fig. 3 For the O-methyl carbamate functionalised polymer product derived from P(Am-co-DMAm) containing 50 mol% Am; (A) TGA analysis showing two thermal events occurring between T = 200–350 °C and a third event at T > 350 °C; (B) DSC analysis showing that Tg = 152 °C, decreasing from 178 °C following eHofmann rearrangement (Fig. S14†). | ||
From a mechanistic point of view, cyclic voltammetry (CV) was performed to confirm that the P(Am-co-DMAm) scaffolds in MeOH were not redox active (Fig. S15†). Upon addition of NaBr (50 mol%) a significant anodic current was observed which is consistent with the reactions performed on small molecule substrates and can be attributed to the oxidation of bromide. Thus, with reference to the mechanism proposed by Zhang et al.33 we hypothesis that bromine formed via oxidation of bromide is captured by the Am side chains forming N-bromoamide intermediates. At the cathode, reduction of MeOH forming MeO− and H2 generates the base required to trigger rearrangement of the N-bromoamide to an isocyanate which subsequently reacts with MeO− to yield the O-methyl carbamate product (Fig. 4).
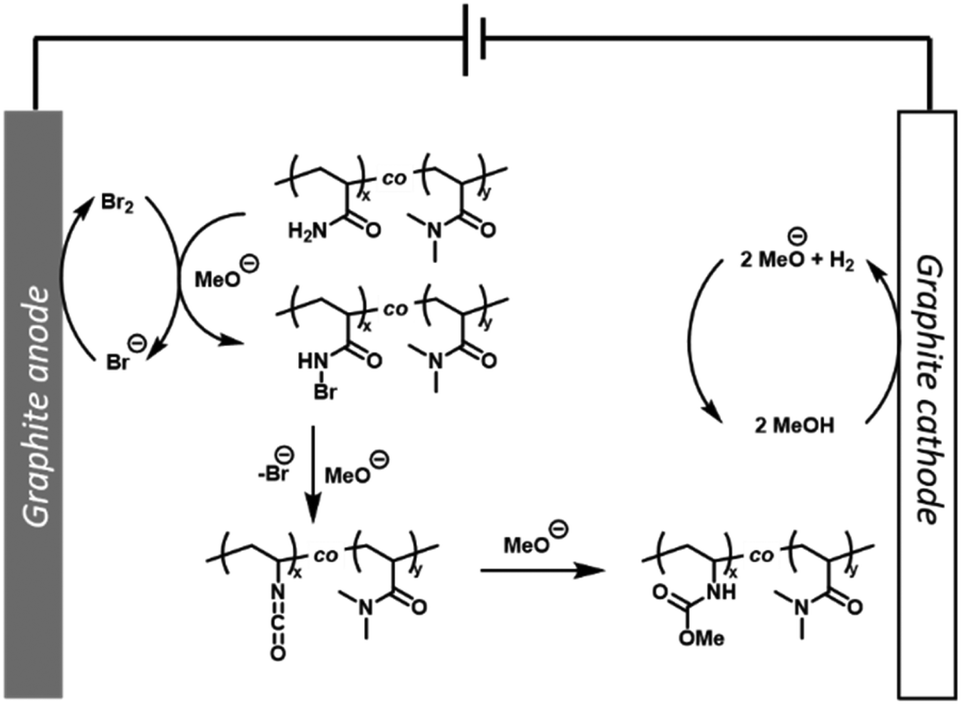 | ||
| Fig. 4 Proposed mechanism for the eHofmann rearrangement on P(Am-co-DMAm) scaffolds. | ||
Finally, the P(Am-co-DMAm) scaffold containing 50 mol% Am was subjected to the conditions for eHofmann rearrangement using diethylene glycol monomethyl ether (DEGME) as alternative alcohol solvent. The reaction was performed with the same electrochemical cell configuration. The loading of NaBr was increase (400 mol%) and the reaction temperature was increased to 90 °C to ensure full dissolution of the P(Am-co-DMAm) scaffolds and sufficient charging of the system. Pleasingly, the reaction reached near quantitative conversion in 4 h (Fig. S16†). Formation of the O-DEGME carbamate side chains was confirmed by 1H/13C and FTIR spectroscopy (Fig. S16–S18†). Likewise, the thermal properties of the O-DEGME carbamate-DMAm copolymers confirmed changes to the copolymer structure. TGA revealed a 2-step degradation profile with an onset temperature of Td = 225 °C (Fig. S19A†) while DSC revealed an expected decrease in Tg (87 °C, Fig. S19B†) relative to the parent P(Am-co-DMAm) scaffold (Fig. S10†).
In conclusion, an efficient electrochemical method for the rearrangement of the primary amide side-chain, present in Am-containing polymers, to an O-methyl carbamate side-chain in the presence of MeOH has been developed. A simple, undivided electrochemical cell configuration containing non-noble metal electrodes (graphite) and NaBr (50 mol%) under CCE at 50 mA was sufficient to drive an eHofmann rearrangement on P(Am-co-DMAm) scaffolds containing 7–50 mol% Am side chains. The success of the reactions was determined by quantitative conversion of the amide side chains to carbamates according to 1H/13C and FTIR spectroscopy. Furthermore, the rearrangement from amide to carbamate side chains was confirmed by changes in the thermal properties of the resulting carbamate-functionalised copolymers. The reaction is compatible with other alcohol solvents, exemplified by formation of O-DEGME carbamate containing copolymers when reactions were performed in DEGME. This represents the first report of an eHofmann reaction for the functionalisation of Am-containing polymers. Crucially, the amide plays the role of a ‘masked’ isocyanate which through electrochemical intervention can be accessed in the absence of the hazardous reagents such as phosgene. At this stage the scope and limitation of the eHofmann rearrangement in polymer synthesis and functionalisation is unknown and under investigation in our team. These preliminary results allude to the potential of this chemistry as a platform to synthesise and characterise alkyl/aryl-N-vinyl carbamate functionalised (co)polymers which are synthetically challenging by other routes and as a result have received limited attention in the field up to now.45,46 Alternative nucleophilic reacting partners including amines and thiols are also being explored as potential routes to urea and carbamothioate functionalised (co)polymers respectively. Furthermore, by exploiting the control enabled by reversible deactivation radical polymerization (RDRP), the potential of incorporating these functional groups into more complex (co)polymer compositions and architectures is also being investigated.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Polymer Characterization Research Technology Platform for maintenance, access and use of facilities. P. W. thanks the Royal Society and Tata companies for the award of a University Research Fellowship (URF\R1\180274).References
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- J. Kozakiewicz, G. Rokicki, J. Przybylski, K. Sylwestrzak, P. G. Parzuchowski and K. M. Tomczyk, Polym. Degrad. Stab., 2010, 95, 2413–2420 CrossRef CAS.
- A. M. C. Maan, A. H. Hofman, W. M. de Vos and M. Kamperman, Adv. Funct. Mater., 2020, 30, 2000936 CrossRef CAS.
- F. Xie, T. Zhang, P. Bryant, V. Kurusingal, J. M. Colwell and B. Laycock, Prog. Polym. Sci., 2019, 90, 211–268 CrossRef CAS.
- A. K. Ghosh and M. Brindisi, J. Med. Chem., 2015, 58, 2895–2940 CrossRef CAS PubMed.
- R. Ahmed, R. Gupta, Z. Akhter, M. Kumar and P. P. Singh, Org. Biomol. Chem., 2022, 20, 4942–4948 RSC.
- https://www.grandviewresearch.com/industry-analysis/polyurethane-pu-market (accessed 25/5/2023).
- https://www.grandviewresearch.com/industry-analysis/polyurea-market , (accessed 25/5/2023).
- J. Yoon and P. A. Lovell, Macromol. Chem. Phys., 2008, 209, 279–289 CrossRef CAS.
- J. D. Flores, N. J. Treat, A. W. York and C. L. McCormick, Polym. Chem., 2011, 2, 1976–1985 RSC.
- R. M. Hensarling, S. B. Rahane, A. P. LeBlanc, B. J. Sparks, E. M. White, J. Locklin and D. L. Patton, Polym. Chem., 2011, 2, 88–90 RSC.
- H. Lebel and O. Leogane, Org. Lett., 2005, 7, 4107–4110 CrossRef CAS PubMed.
- H. Babad and A. G. Zeiler, Chem. Rev., 1973, 73, 75–91 CrossRef CAS.
- M. S. Rolph, M. Inam and R. K. O'Reilly, Polym. Chem., 2017, 8, 7229–7239 RSC.
- M. S. Rolph, A. L. J. Markowska, C. N. Warriner and R. K. O'Reilly, Polym. Chem., 2016, 7, 7351–7364 RSC.
- W. Liu, S. Yang, L. Huang, J. Xu and N. Zhao, Chem. Commun., 2022, 58, 12399–12417 RSC.
- A. Petti, C. Fagnan, C. G. W. van Melis, N. Tanbouza, A. D. Garcia, A. Mastrodonato, M. C. Leech, I. C. A. Goodall, A. P. Dobbs, T. Ollevier and K. Lam, Org. Process Res. Dev., 2021, 25, 2614–2621 CrossRef CAS.
- A. K. Ghosh, A. Sarkar and M. Brindisi, Org. Biomol. Chem., 2018, 16, 2006–2027 RSC.
- J. C. Brendel, G. Gody and S. Perrier, Polym. Chem., 2016, 7, 5536–5543 RSC.
- G. Gody, D. A. Roberts, T. Maschmeyer and S. Perrier, J. Am. Chem. Soc., 2016, 138, 4061–4068 CrossRef CAS PubMed.
- G. Gody, C. Rossner, J. Moraes, P. Vana, T. Maschmeyer and S. Perrier, J. Am. Chem. Soc., 2012, 134, 12596–12603 CrossRef CAS PubMed.
- H. E. Baumgarten and A. Staklis, J. Am. Chem. Soc., 1965, 87, 1141–1142 CrossRef CAS.
- N. Satoh, T. Akiba, S. Yokoshima and T. Fukuyama, Tetrahedron, 2009, 65, 3239–3245 CrossRef CAS.
- M. Iinuma, K. Moriyama and H. Togo, Tetrahedron, 2013, 69, 2961–2970 CrossRef CAS.
- X. Huang, M. Seid and J. W. Keillor, J. Org. Chem., 1997, 62, 7495–7496 CrossRef CAS PubMed.
- S. D. Minteer and P. Baran, Acc. Chem. Res., 2020, 53, 545–546 CrossRef CAS PubMed.
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358–3375 RSC.
- B. Zhao and P. Wilson, Polym. Chem., 2023, 14, 2000–2021 RSC.
- K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed.
- C. Sandford, M. A. Edwards, K. J. Klunder, D. P. Hickey, M. Li, K. Barman, M. S. Sigman, H. S. White and S. D. Minteer, Chem. Sci., 2019, 10, 6404–6422 RSC.
- S. Tatsuya, M. Yoshihiro, Y. Shin-ichiro and K. Shigenori, Chem. Lett., 1982, 11, 565–568 CrossRef.
- Y. Matsumura, T. Maki and Y. Satoh, Tetrahedron Lett., 1997, 38, 8879–8882 CrossRef CAS.
- L. Li, M. Xue, X. Yan, W. Liu, K. Xu and S. Zhang, Org. Biomol. Chem., 2018, 16, 4615–4618 RSC.
- B. Zhao, J. Cheng, J. Gao, D. M. Haddleton and P. Wilson, Macromol. Chem. Phys., 2023, 2300039 CrossRef.
- M. Mohammed, B. A. Jones and P. Wilson, Polym. Chem., 2022, 13, 3460–3470 RSC.
- B. Zhao, F. Pashley-Johnson, B. A. Jones and P. Wilson, Chem. Sci., 2022, 13, 5741–5749 RSC.
- B. Zhao, M. Mohammed, B. A. Jones and P. Wilson, Chem. Commun., 2021, 57, 3897–3900 RSC.
- M. E. S. R. e. Silva, E. R. Dutra, V. Mano and J. C. Machado, Polym. Degrad. Stab., 2000, 67, 491–495 CrossRef.
- M. J. Caulfield, G. G. Qiao and D. H. Solomon, Chem. Rev., 2002, 102, 3067–3084 CrossRef CAS PubMed.
- N. J. Daly and F. Ziolkowski, Int. J. Chem. Kinet., 1980, 12, 241–252 CrossRef CAS.
- N. J. Daly, G. Heweston and F. Ziolkowski, Aust. J. Chem., 1973, 26, 1259–1262 CrossRef CAS.
- N. Daly and F. Ziolkowski, Aust. J. Chem., 1972, 25, 1453–1458 CrossRef CAS.
- N. Daly and F. Ziolkowski, Aust. J. Chem., 1971, 24, 2541–2546 CrossRef CAS.
- C. E. Schweitzer, US2409712A, 1944.
- L. J. Guilbault and H. J. Harwood, J. Polym. Sci., Polym. Chem. Ed., 1974, 12, 1461–1467 CrossRef CAS.
- G. O. Schulz and H. J. Harwood, J. Polym. Sci., Polym. Chem. Ed., 1974, 12, 1451–1460 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00594a |
| This journal is © The Royal Society of Chemistry 2023 |