
Monitoring hydrogen transport through graphene by in situ surface-enhanced Raman spectroscopy†
Younghyun
Wy‡
a,
Jaesung
Park‡
b,
Sung
Huh
a,
Hyuksang
Kwon
b,
Bon Seung
Goo
a,
Jung Young
Jung
a and
Sang Woo
Han
 *a
*a
aCenter for Nanotectonics, Department of Chemistry and KI for the NanoCentury, KAIST, Daejeon 34141, Korea. E-mail: sangwoohan@kaist.ac.kr
bKorea Research Institute of Standards and Science (KRISS), Daejeon 34113, Korea
First published on 28th December 2022
Abstract
Exploring the atomic or molecular transport properties of two-dimensional materials is vital to understand their inherent functions and, thus, to expedite their use in various applications. Herein, a surface-enhanced Raman spectroscopy (SERS)-based in situ analytical tool for the sensitive and rapid monitoring of hydrogen transport through graphene is reported. In this method, a reducing agent, which can provide hydrogen species, and a Raman dye self-assembled on a SERS platform are separated by a graphene membrane, and the reduction of the Raman dye by hydrogen species transferred through graphene is monitored with SERS. For validating the efficacy of our method, the catalytic reduction of surface-bound 4-nitrothiophenol by sodium borohydride was chosen in this study. The experimental results distinctly demonstrate that the high sensitivity and rapid detection ability of SERS can allow the effective analysis of the hydrogen transport properties of graphene.
Atomic or molecular transport through two-dimensional materials has been of immense interest as it can be applied to various separation technologies.1–6 Indeed, various two-dimensional materials, such as graphene and its derivatives,3,7–10 covalent organic frameworks,11,12 zeolites,13–15 Mxenes,16 and metal dichalcogenides,17 have been investigated as promising platforms for water desalination,4,17–19 ion or molecular sieving,10,11,20,21 and gas separation.8,12,15,16 Among two-dimensional materials, graphene has been extensively studied because of its chemical stability, affordability, and light weight. Since defect-free monolayer graphene has been considered to be impermeable to most atoms and molecules under ambient conditions,1,3 perforated graphene or small-sized graphene aggregates have been used as separation membranes.10,19,20 Regarding the transport properties of graphene, it is noteworthy that the Geim group revealed that protons can pass through defect-free monolayer graphene by using an electrical method.1 This work implies that monolayer graphene can be employed as a proton membrane. Recently, the same group further demonstrated the discernible permeability of defect-free monolayer graphene toward hydrogen by showing the bulging up of a graphene membrane under exposure to hydrogen gas.2 The hydrogen permeation was attributed to the dissociative adsorption of molecular hydrogen on the graphene surface, followed by the flipping of hydrogen atoms through the graphene. These works indicate that the development of an efficient analytical tool is crucial to unveil the transport properties of graphene and, hence, to expedite its use in separation technologies.
As an ultra-sensitive real-time analytical method, surface-enhanced Raman spectroscopy (SERS) has been of enormous attention in numerous research fields.22,23 For instance, chemical events in the course of catalytic reactions have been monitored in real time by using in situ SERS techniques.24–26 This prompted us to explore the possibility of using SERS in the examination of the transport properties of graphene. Although the transfer of hot electrons at Au/graphene interfaces has been studied with an in situ SERS technique,27 analyzing the atomic or molecular transport properties of two-dimensional materials with SERS has rarely been reported.
Here, we introduce a SERS-based in situ analytical method that allows the sensitive and rapid detection of the transport of hydrogen species through defect-free monolayer graphene. The SERS-based method has been designed based on our proposition that the analysis of hydrogen species transport through graphene will be possible by separating a Raman dye and a reducing agent, which can provide hydrogen species, with a graphene membrane and then monitoring the reduction of the Raman dye induced by transferred hydrogen species with SERS. For this study, the catalytic reduction of surface-bound 4-nitrothiophenol (4-NTP) to 4-aminothiophenol (4-ATP) by sodium borohydride (NaBH4) was chosen, which has been intensively explored by in situ SERS.24,28,29 The reduction reaction of 4-NTP molecules adsorbed on a metal substrate proceeds upon the treatment of NaBH4 as BH4− ions transfer hydrogen species to the metal surface and then the surface hydrogen species provide protons and electrons to 4-NTP molecules, resulting in the reduction of 4-NTP to 4-ATP through the 6H+/6e− process. The metal substrate can indeed catalyze the reduction reaction by providing binding sites for the hydrogen species from BH4− ions and relaying electrons to 4-NTP molecules. For SERS detection, an analytical device was built by covering an Au substrate, on which 4-NTP molecules were self-assembled, with defect-free monolayer graphene. Then, the transfer of hydrogen species through graphene was tracked by SERS monitoring of the reduction reaction of 4-NTP upon the treatment of NaBH4 on the surface of graphene. The Au substrate can act as both a SERS substrate and a catalyst for the 4-NTP reduction. Thanks to the high sensitivity and rapid detection capability of SERS, the transport properties of graphene could be effectively analyzed with the present method.
Fig. 1a shows how a SERS device for monitoring hydrogen species transport through graphene was fabricated (see the Experimental details and Fig. S1†). The colloidal clusters of Au nanoparticles (Au NPCs), which showed stable and remarkable SERS performance in our previous works,30–33 were chosen as a SERS substrate. Au NPCs consisting of ∼30 nm Au nanoparticles were synthesized based on our reported method (see the Experimental details and Fig. S2a†).31 The prepared Au NPCs were spin-coated on a polymethyl methacrylate (PMMA)-coated Si wafer, which was pre-patterned by electron-beam lithography to form an array of 5 μm × 5 μm square-shaped holes. PMMA was then removed through a lift-off process, leaving Au NPCs patterns on the Si wafer (Fig. S2b†). The produced Au NPCs-patterned Si wafer was immersed in a 10−4 M ethanol solution of 4-NTP to form the self-assembled monolayers of 4-NTP on the surface of the Au NPCs. Defect-free monolayer graphene was prepared by a mechanical exfoliation method and transferred onto the Au NPCs pattern. Then, a metal mask, Au/Ti, with a thickness of 20 nm/2 nm and a width of 3 μm was deposited around the transferred graphene. The metal mask was deposited to prevent the scrolling of graphene and to exclude any other atomic or molecular permeation through the gap between the Si wafer and the edge of the graphene. Onto the prepared device, an aqueous NaBH4 solution was dropped to fully cover the device, and in situ SERS monitoring for the reduction reaction of 4-NTP was performed with a micro-Raman spectrometer (Fig. 1b).
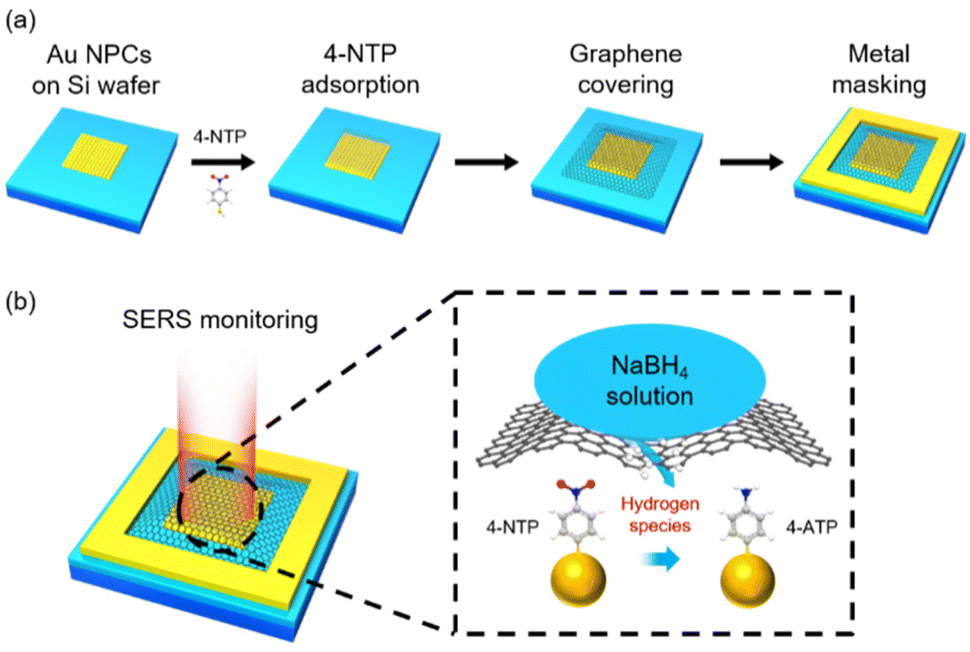 | ||
| Fig. 1 Schematic illustrations of (a) device fabrication and (b) in situ SERS monitoring of the reduction reaction of 4-NTP. The color representation of atoms: white, hydrogen; gray, carbon; blue, nitrogen; red, oxygen; yellow, sulfur. | ||
Fig. 2a shows the representative optical microscopy image of a prepared device. The presence of 4-NTP and graphene in the device was revealed by Raman spectra taken at the center of the device with the excitations of 633 and 532 nm, respectively, which distinctly exhibit the characteristic peaks of 4-NTP (Fig. 2b) and graphene (Fig. 2c). Notably, the 2D Raman band of graphene is well fitted by a single Lorentzian function with a full-width at half-maximum of 34 cm−1, indicating that the device is covered with monolayer graphene.34 Considering the monolayer coverage of 4-NTP on the Au NPCs, the observed Raman spectrum of 4-NTP is indeed a SERS spectrum. The two-dimensional Raman mapping images of the NO2 stretching peak (ν(NO2)) of 4-NTP and the 2D band of graphene measured inside the device further confirm the existence of 4-NTP molecules throughout the Au NPCs pattern and the full coverage of graphene inside the device, respectively (Fig. 2d and e). The different Raman mapping signal intensities of graphene between inside and outside the Au NPCs pattern can be attributed to the change in the relative height of the graphene layer owing to the pattern as well as to the electric field enhancement around the Au NPCs.30
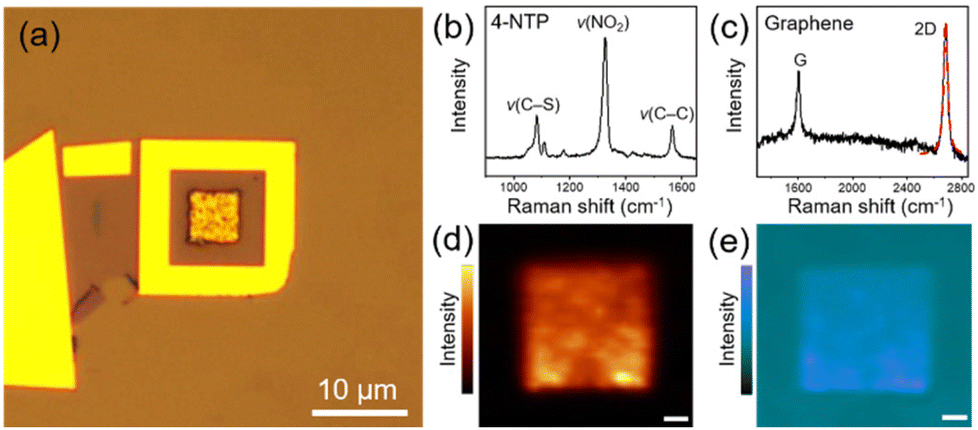 | ||
| Fig. 2 (a) Optical micrograph of a fabricated device. Raman spectra of (b) 4-NTP and (c) graphene taken at the center of the device. The red dashed curve in c shows the single Lorentzian fitting of the 2D band. Two-dimensional Raman mapping images of (d) the ν(NO2) peak of 4-NTP and (e) the 2D band of graphene measured inside the device. Scale bars indicate 1 μm. | ||
Fig. 3a and b show, respectively, the time-dependent SERS spectra of 4-NTP and the corresponding color-coded intensity map of the SERS spectra obtained after the treatment of an aqueous solution of NaBH4 (10 mM, 0.2 mL) onto the fabricated device. The amount of NaBH4 used in the experiments was optimal for the stable SERS measurements. The SERS spectra were taken every 10 s with an acquisition time of 1 s. For convenience, representative SERS spectra obtained at six different reaction times are displayed in Fig. 3a. The temperature of the device was maintained at 15 °C to avoid the thermal decomposition of BH4− ions. In fact, when a 10 mM NaBH4 aqueous solution was placed at 15 °C for 10 min, its pH was maintained at 8. Given that the pH of an aqueous NaBH4 solution increases as NaBH4 thermally decomposes,35 this result indicates that the thermal decomposition of NaBH4 was insignificant under our experimental conditions. Notably, the peak intensities at 1323 and 1566 cm−1, which are associated with the ν(NO2) and C–C stretching (ν(C–C)) modes of 4-NTP, respectively, decreased with time, while the intensity of a peak at 1587 cm−1, which can be assigned to the ν(C–C) mode of 4-ATP,36 increased. SERS measurements on the device for 10 min without the treatment of NaBH4 showed no noticeable changes in the SERS spectrum of 4-NTP (Fig. S3†). Furthermore, the formation of 4,4′-dimercaptoazobenzene, which can be generated through the photocatalysis of 4-NTP,37 was not detected. Taken together, the spectral changes of 4-NTP during the measurements can be attributed to the progress of 4-NTP reduction induced by hydrogen species, which could be formed from BH4− ions on the graphene surface and then transferred to the underlying 4-NTP-adsorbed substrate through the graphene layer. The identity of produced hydrogen species will be discussed later. The rapid progress of the 4-NTP reduction distinctly demonstrates that the monolayer graphene can indeed effectively transport hydrogen species.
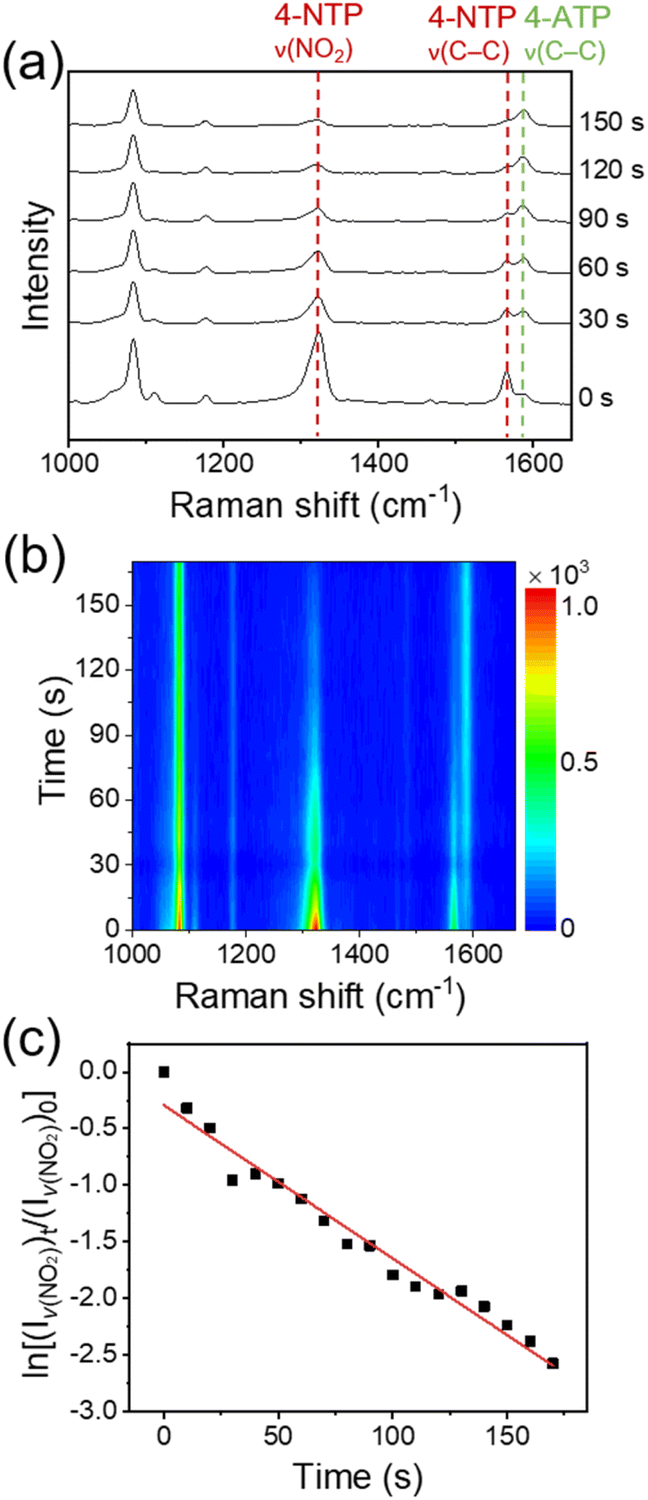 | ||
| Fig. 3 (a) Time-dependent SERS spectra of 4-NTP and (b) corresponding color-coded intensity map of the SERS spectra obtained after the treatment of an aqueous solution of NaBH4 onto the device. (c) Plot of the logarithm of the intensity of ν(NO2) peak relative to that of the initial stage versus the reaction time. | ||
Apparently, a good linear relationship was found between the logarithm of the intensity of ν(NO2) peak relative to that of the initial stage and the reaction time (Fig. 3c). Considering the largely excess amount of NaBH4 compared to the adsorbed 4-NTP molecules (∼106 times, see the Experimental details†), the linear correlation indicates that the 4-NTP reduction reaction in our system follows the pseudo-first-order kinetics. This is in line with previous reports on the metal-catalyzed reduction of aromatic nitro compounds by NaBH4.28,38 From the slopes of linear fits obtained from multiple measurements, the average rate constant of the 4-NTP reduction was estimated to be 9.47 × 10−3 s−1.
Although exfoliated graphene is known to be defect-free,7,20 we conducted control experiments to definitely exclude the possibility that BH4− ions or hydrogen species formed from them were infiltrated through defects that may exist in the graphene membrane during the reaction. Based on the fact that CN− ions can effectively etch Au,39 the device was treated with a 20 mM KCN methanol solution. Notably, after 15 min, all Au NPCs patterns that were not covered with graphene were etched severely, while the Au NPCs pattern covered with graphene was intact (Fig. 4a and b). Indeed, the graphene-covered Au NPCs pattern showed no considerable change in its SERS spectrum after the KCN treatment (Fig. S4†). Furthermore, when the device was left under the reaction conditions for 3 h, the graphene membrane swelled like a balloon, which is presumably due to hydrogen gas formed from the transported hydrogen species (Fig. 4c and d). In fact, the graphene membrane did not swell at all before the NaBH4 treatment (Fig. S5†). The results of the control experiments collectively confirm that the graphene membrane in the device is defect-free, and the 4-NTP reduction is indeed proceeded by hydrogen species transported through the graphene.
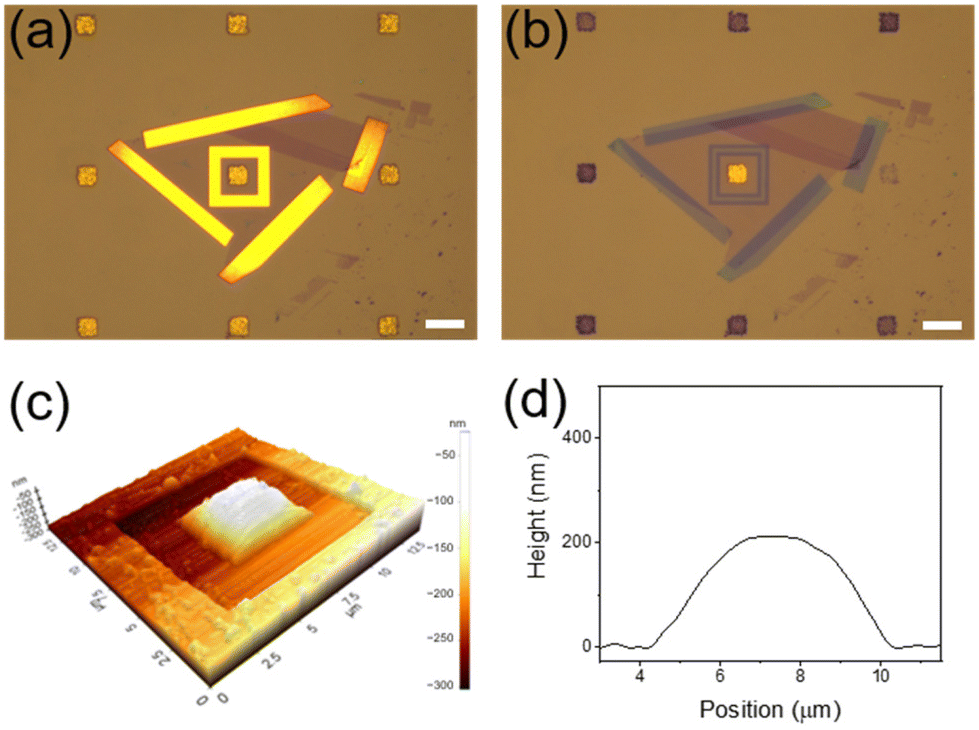 | ||
| Fig. 4 Optical micrographs of Au NPCs patterns (a) before and (b) after the KCN treatment. Scale bars indicate 10 μm. (c) Atomic force microscopy image and (d) corresponding height profile of the device left under the reaction conditions for 3 h. | ||
As abovementioned, the reaction temperature was set to 15 °C to avoid the thermal decomposition of BH4− ions. Accordingly, the hydrogen species passing through the graphene membrane could only be derived from BH4− ions adsorbed on the graphene surface. Molecular hydrogen could be formed through the hydrolysis of adsorbed BH4− ions.40 However, given that it takes several days for hydrogen gas to pass through graphene,2 molecular hydrogen cannot be the hydrogen species transported through the graphene layer in our system, where 4-NTP can be reduced in a few minutes. In this regard, we infer that atomic hydrogen or hydride ion that can be produced through the dissociative adsorption of BH4− ions on the surface of graphene might be responsible for the reduction of 4-NTP.41 To identify the hydrogen species formed from BH4− ions, we performed density functional theory (DFT) calculations for the products of the dissociative adsorption of a BH4− ion on a model graphene surface. The details of computational methods are described in the Experimental details.† Through the Löwdin charge analysis, we found that a very little positive charge is allocated to hydrogen dissociated from the BH4− ion (Fig. S6†). Based on the DFT result, we can assume that the hydrogen species passing through the graphene layer is atomic hydrogen. Even if hydride ions are generated from the dissociative adsorption of BH4− ions on the surface of graphene, their charges will readily be dissipated by the conductive sp2-conjugated carbon atoms of the graphene.42 Based on the calculated number of 4-NTP molecules present in the device (see the Experimental details†) and the experimental finding that half of the 4-NTP molecules were reduced in 30 s, the lower-bound flux of hydrogen atoms transported through the graphene membrane could be estimated to be 7.2 × 106 s−1 μm−2. This value is comparable to that of protons passing through graphene, ∼107 s−1 μm−2.1
Conclusions
We devised a SERS-based analytical tool for monitoring hydrogen transport through defect-free monolayer graphene. Separating a Raman dye (4-NTP) and a reducing agent (NaBH4) with a graphene membrane and then tracking the reduction of 4-NTP by hydrogen atoms transferred through graphene with SERS could allow the rapid and in situ analysis of hydrogen transport through monolayer graphene. Since the present SERS-based method can be applied to a wide range of transported species (ions, atoms, or molecules), if a suitable Raman dye that can react with a transported species is chosen, it is expected that this method can readily be utilized to study the transport properties of various two-dimensional materials. We also foresee that the further enhancement of the precision and detection capability of this method in conjunction with the development of efficient SERS platforms will make it possible to reveal the unprecedented transport function of two-dimensional materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2015R1A3A2033469, 2018R1A5A1025208). This work was also supported by Characterization platform for advanced materials funded by Korea Research Institute of Standards and Science (KRISS–2022–GP2022-0013).References
- S. Hu, M. Lozada-Hidalgo, F. C. Wang, A. Mishchenko, F. Schedin, R. R. Nair, E. W. Hill, D. W. Boukhvalov, M. I. Katsnelson, R. A. W. Dryfe, I. V. Grigorieva, H. A. Wu and A. K. Geim, Nature, 2014, 516, 227–230 CrossRef CAS.
- P. Z. Sun, Q. Yang, W. J. Kuang, Y. V. Stebunov, W. Q. Xiong, J. Yu, R. R. Nair, M. I. Katsnelson, S. J. Yuan, I. V. Grigorieva, M. Lozada-Hidalgo, F. C. Wang and A. K. Geim, Nature, 2020, 579, 229–232 CrossRef CAS PubMed.
- L. Tsetseris and S. T. Pantelides, Carbon, 2014, 67, 58–63 CrossRef CAS.
- J. R. Werber, C. O. Osuji and M. Elimelech, Nat. Rev. Mater., 2016, 1, 16018 CrossRef.
- G. Liu, W. Jin and N. Xu, Angew. Chem., Int. Ed., 2016, 55, 13384–13397 CrossRef.
- L. Cheng, G. Liu, J. Zhao and W. Jin, Acc. Mater. Res., 2021, 2, 114–128 CrossRef.
- J. S. Bunch, S. S. Verbridge, J. S. Alden, A. M. van der Zande, J. M. Parpia, H. G. Craighead and P. L. McEuen, Nano Lett., 2008, 8, 2458–2462 CrossRef PubMed.
- H. W. Kim, H. W. Yoon, S.-M. Yoon, B. M. Yoo, B. K. Ahn, Y. H. Cho, H. J. Shin, H. Yang, U. Paik, S. Kwon, J.-Y. Choi and H. B. Park, Science, 2013, 342, 91–95 CrossRef PubMed.
- M. Lozada-Hidalgo, S. Hu, O. Marshall, A. Mishchenko, A. N. Grigorenko, R. A. W. Dryfe, B. Radha, I. V. Grigorieva and A. K. Geim, Science, 2016, 351, 68–70 CrossRef PubMed.
- L. Chen, G. Shi, J. Shen, B. Peng, B. Zhang, Y. Wang, F. Bian, J. Wang, D. Li, Z. Qian, G. Xu, G. Liu, J. Zeng, L. Zhang, Y. Yang, G. Zhou, M. Wu, W. Jin, J. Li and H. Fang, Nature, 2017, 550, 380–383 CrossRef CAS PubMed.
- Y. Li, Q. Wu, X. Guo, M. Zhang, B. Chen, G. Wei, X. Li, X. Li, S. Li and L. Ma, Nat. Commun., 2020, 11, 599 CrossRef CAS PubMed.
- Y. Ying, M. Tong, S. Ning, S. K. Ravi, S. B. Peh, S. C. Tan, S. J. Pennycook and D. Zhao, J. Am. Chem. Soc., 2020, 142, 4472–4480 CrossRef CAS.
- K. Varoon, X. Zhang, B. Elyassi, D. D. Brewer, M. Gettel, S. Kumar, J. A. Lee, S. Maheshwari, A. Mittal, C.-Y. Sung, M. Cococcioni, L. F. Francis, A. V. McCormick, K. A. Mkhoyan and M. Tsapatsis, Science, 2011, 334, 72–75 CrossRef CAS.
- M. Y. Jeon, D. Kim, P. Kumar, P. S. Lee, N. Rangnekar, P. Bai, M. Shete, B. Elyassi, H. S. Lee, K. Narasimharao, S. N. Basahel, S. Al-Thabaiti, W. Xu, H. J. Cho, E. O. Fetisov, R. Thyagarajan, R. F. DeJaco, W. Fan, K. A. Mkhoyan, J. I. Siepmann and M. Tsapatsis, Nature, 2017, 543, 690–694 CrossRef CAS PubMed.
- M. Dakhchoune, L. F. Villalobos, R. Semino, L. Liu, M. Rezaei, P. Schouwink, C. E. Avalos, P. Baade, V. Wood, Y. Han, M. Ceriotti and K. V. Agrawal, Nat. Mater., 2021, 20, 362–369 CrossRef CAS PubMed.
- L. Ding, Y. Wei, L. Li, T. Zhang, H. Wang, J. Xue, L.-X. Ding, S. Wang, J. Caro and Y. Gogotsi, Nat. Commun., 2018, 9, 155 CrossRef PubMed.
- H. Li, T.-J. Ko, M. Lee, H.-S. Chung, S. S. Han, K. H. Oh, A. Sadmani, H. Kang and Y. Jung, Nano Lett., 2019, 19, 5194–5204 CrossRef CAS PubMed.
- D. Cohen-Tanugi and J. C. Grossman, Nano Lett., 2012, 12, 3602–3608 CrossRef CAS PubMed.
- S. P. Surwade, S. N. Smirnov, I. V. Vlassiouk, R. R. Unocic, G. M. Veith, S. Dai and S. M. Mahurin, Nat. Nanotechnol., 2015, 10, 459–464 CrossRef CAS PubMed.
- S. P. Koenig, L. Wang, J. Pellegrino and J. S. Bunch, Nat. Nanotechnol., 2012, 7, 728–732 CrossRef CAS PubMed.
- Y. Peng, Y. Li, Y. Ban, H. Jin, W. Jiao, X. Liu and W. Yang, Science, 2014, 346, 1356–1359 CrossRef CAS.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392–395 CrossRef CAS.
- J. Langer, D. Jimenez de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguié, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. J. García de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Käll, J. Kneipp, N. A. Kotov, H. Kuang, E. C. Le Ru, H. K. Lee, J.-F. Li, X. Y. Ling, S. A. Maier, T. Mayerhöfer, M. Moskovits, K. Murakoshi, J.-M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlücker, L.-L. Tay, K. G. Thomas, Z.-Q. Tian, R. P. Van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzán, ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed.
- H. Zhang, X.-G. Zhang, J. Wei, C. Wang, S. Chen, H.-L. Sun, Y.-H. Wang, B.-H. Chen, Z.-L. Yang, D.-Y. Wu, J.-F. Li and Z.-Q. Tian, J. Am. Chem. Soc., 2017, 139, 10339–10346 CrossRef CAS PubMed.
- D. Bohra, I. Ledezma-Yanez, G. Li, W. de Jong, E. A. Pidko and W. A. Smith, Angew. Chem., Int. Ed., 2019, 58, 1345–1349 CrossRef CAS.
- H. Ze, X. Chen, X.-T. Wang, Y.-H. Wang, Q.-Q. Chen, J.-S. Lin, Y.-J. Zhang, X.-G. Zhang, Z.-Q. Tian and J.-F. Li, J. Am. Chem. Soc., 2021, 143, 1318–1322 CrossRef.
- J.-L. Yang, H.-J. Wang, Z. Zhu, M.-F. Yue, W.-M. Yang, X.-G. Zhang, X. Ruan, Z. Guan, Z.-L. Yang, W. Cai, Y.-F. Wu, F.-R. Fan, J.-C. Dong, H. Zhang, H. Xu, Z.-Q. Tian and J.-F. Li, Angew. Chem., Int. Ed., 2022, 61, e202112749 Search PubMed.
- V. Joseph, C. Engelbrekt, J. Zhang, U. Gernert, J. Ulstrup and J. Kneipp, Angew. Chem., Int. Ed., 2012, 51, 7592–7596 CrossRef.
- W. Xie, R. Grzeschik and S. Schlücker, Angew. Chem., Int. Ed., 2016, 55, 13729–13733 CrossRef PubMed.
- S. Lee, J. W. Hong, S.-U. Lee, Y. W. Lee and S. W. Han, Chem. Commun., 2015, 51, 8793–8796 RSC.
- S. Lee, S. Kang, J. Kim, Y. Wy and S. W. Han, ChemNanoMat, 2017, 3, 772–778 CrossRef.
- S. Lee, H. Hwang, W. Lee, D. Schebarchov, Y. Wy, J. Grand, B. Auguié, D. H. Wi, E. Cortés and S. W. Han, ACS Energy Lett., 2020, 5, 3881–3890 CrossRef.
- S. Lee, Y. Wy, Y. W. Lee, K. Ham and S. W. Han, Small, 2017, 13, 1701633 CrossRef.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef.
- H. I. Schlesinger, H. C. Brown, A. E. Finholt, J. R. Gilbreath, H. R. Hoekstra and E. K. Hyde, J. Am. Chem. Soc., 1953, 75, 215–219 CrossRef.
- M. Futamata, J. Phys. Chem., 1995, 99, 11901–11908 CrossRef.
- Y.-F. Huang, H.-P. Zhu, G.-K. Liu, D.-Y. Wu, B. Ren and Z.-Q. Tian, J. Am. Chem. Soc., 2010, 132, 9244–9246 CrossRef.
- K. Zhang, J. Zhao, J. Ji, Y. Li and B. Liu, Anal. Chem., 2015, 87, 8702–8708 CrossRef PubMed.
- T. A. Green, Gold Bull., 2014, 47, 205–216 CrossRef.
- U. B. Demirci and P. Miele, C. R. Chim., 2014, 17, 707–716 CrossRef.
- G. Guella, C. Zanchetta, B. Patton and A. Miotello, J. Phys. Chem. B, 2006, 110, 17024–17033 CrossRef PubMed.
- J. Song, S. W. Kang, Y. W. Lee, Y. Park, J.-H. Kim and S. W. Han, ACS Appl. Mater. Interfaces, 2017, 9, 1692–1701 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and additional data (Fig. S1–S6). See DOI: https://doi.org/10.1039/d2nr06010h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |