
Electrochemical transformation of D,L-glutamic acid into acrylonitrile†
Justus
Kümper
 ,
Jérôme
Meyers
,
Rebecca
Sebers
,
Nils
Kurig
and
Regina
Palkovits
*
,
Jérôme
Meyers
,
Rebecca
Sebers
,
Nils
Kurig
and
Regina
Palkovits
*
Institut für Technische und Makromolekulare Chemie, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany. E-mail: palkovits@itmc.rwth-aachen.de
First published on 11th July 2023
Abstract
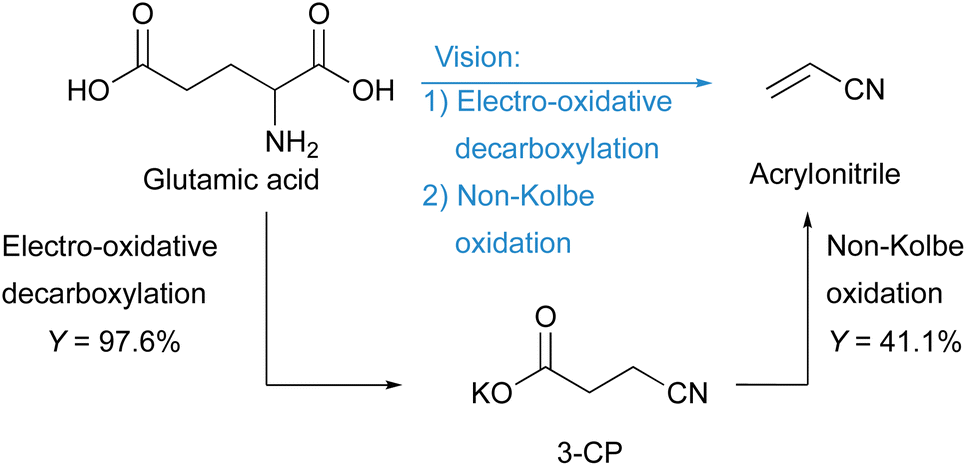
Acrylonitrile is an important industrial platform chemical mainly produced by the SOHIO process using fossil propylene, ammonia, and air as substrates. In view of climate change, ingenuity is needed to become independent of fossil resources and to achieve sustainable production of basic chemicals such as acrylonitrile. One opportunity is to use biogenic substrates that are converted by electrolysis. Using electrons as oxidants is an environmentally friendly alternative to stochiometric amounts of molecular oxidising agents that generate waste after usage. Here, we present a new route for the electrochemical synthesis of green acrylonitrile using the biogenic amino acid D,L-glutamic acid as a substrate by utilising electro-oxidative decarboxylation and non-Kolbe electrolysis. By optimising the electro-oxidative decarboxylation, the number of steps was successfully reduced, resulting in a two-step process for the formation of the monomer. Acrylonitrile was synthesised with a maximum yield of 41.1%.
Introduction
Even after more than 180 years of research, the field of electrosynthesis still holds unexplored aspects and provides novel solutions to global challenges. Versatile techniques such as (non-)Kolbe electrolysis provide novel synthesis routes to industrially relevant chemicals based on renewable feedstocks.1–3 Several industrially relevant chemicals contain nitrogen, e.g. in the form of a nitrile group.2,4,5 Since the post-functionalisation of a hydrocarbon with ammonia is very energy-intensive, the use of biomass-derived nitrogen-containing substrates such as amino acids for their production presents a sustainable alternative.6 Amino acids play an increasingly important role in industrial processes due to their availability and attractive price.7 They are widely used as additives in food and feed or as precursors in the production of flavours and taste enhancers. The amino acid with the highest worldwide annual production capacity is L-glutamic acid with 2.5 Mt a−1.8L-Glutamic acid is mainly produced by fermentation based on glucose or sucrose under the addition of ammonium sulphate. The microbial conversion of sugars into L-glutamic acid is realised by the bacterium Corynebacterium glutamicum.9 Research in this field has improved the production of L-glutamic acid from 10 g L−1 (wildtype) to titers in the range 120–150 g L−1.9–12 Furthermore, yields of at least 54% L-glutamic acid per gram of glucose are possible.13 However, alternative production routes such as chemical synthesis or enzymatic conversion are also being used for production.7Besides using L-glutamic acid as a nutritional supplement, it can also serve as a substrate to produce industrially relevant substances. For example, Lammens et al.14 published the synthesis of succinonitrile by homogeneous catalysis from L-glutamic acid. After hydrogenation of succinonitrile, 1,4-diaminobutane is formed with potential application in the production of polyamides. Alternatively, L-glutamic acid can also be converted via electrosynthesis, as Dai et al.15 have demonstrated. They synthesised adiponitrile, a precursor of hexamethylenediamine used mainly for the fabrication of polyamide 6.6. For this purpose, Kolbe electrolysis was utilised. Beyond these examples, L-glutamic acid was rarely studied as a substrate in (non-)Kolbe research.14,15
In this study, we investigated the electrochemical production of acrylonitrile from D,L-glutamic acid. Acrylonitrile had an annual production capacity of over 7 Mt in 2017.16,17 Due to their high elasticity, thermal stability and low density, homopolymers of acrylonitrile (PAN) offer ideal properties for composite structures of aircraft or for the production of sails. In addition, copolymers of acrylonitrile with 1,3-butadiene (as nitrile rubber) or butadiene/styrene (ABS resins) are widely used in the automotive, construction, or electrical industries as well as in the textile sector. Due to the broad fields of application, the demand for nitrile-containing intermediates is rising and this explains the current high production capacities. Particularly due to the interest in PAN-derived carbon fibres for weight reduction of vehicles and aircrafts, production is predicted to grow further by 11–18% annually.16,17 In addition to the use of acrylonitrile for the formation of PAN, it is also used as a substrate in the Baizer process for the production of adiponitrile via a cathodic hydrocoupling reaction.18 The Baizer process itself is a prominent example of a successfully operated electrosynthetic reaction on an industrial scale. Since the 1970s, about 0.3 Mt a−1 of adiponitrile have been produced via this route.19
Today, the most relevant route for acrylonitrile production is the SOHIO (Standard Oil of Ohio) process with about 6 Mt a–1. Fossil propylene, ammonia and air are used and an overpressure of 50–200 kPa is applied. The reaction takes place at 400–510 °C in a fluidised bed reactor and is catalysed by Bi9PMo12O52 on silica. By adding cobalt, nickel, and iron molybdates, the yield can reach about 80%. It is a single-pass process with a propene conversion of 98%. However, environmentally harmful and highly toxic hydrocyanic acid as well as acetonitrile are produced in significant amounts as by-products.5,20 In general, feedstock costs account for 67% of production costs. The current price of acrylonitrile of €2000 per t is strongly linked to the fluctuating price of propylene, which is currently around €900 per t.21
Accordingly, intensive research is devoted to alternative syntheses based on biomass in order to reduce the dependence on fossil raw materials. The highest yields of acrylonitrile (60%) were achieved with glycerol as a biomass-based feedstock. Since glycerol partly competes with the food industry, its use for industrial purposes is not unrestricted.17 Le Nôtre et al. showed that acrylonitrile can be synthesised from glutamic acid via homogeneous catalysis.22 In the first step, they synthesised 3-cyanopropanoic acid from glutamic acid with a yield of 70%. NaBr was used in catalytic amounts, while 3 eq. of NaOCl were added. In the second step, a palladium catalysed decarbonylation–elimination reaction was carried out. By microdistillation for purification, they obtained acrylonitrile with an overall yield of 17%. Although it is currently economically difficult to compete with the fossil-based SOHIO process, research on alternative reaction pathways may well become relevant for the future.22
In this work, we present an electrochemical pathway to the versatile monomer acrylonitrile starting from D,L-glutamic acid as a potentially more sustainable alternative to the SOHIO process. Our idea to produce acrylonitrile from D,L-glutamic acid is based on a synthesis reported by Dai et al.15 In 2012, they presented an environmentally benign route to adiponitrile using electrochemistry.15 Starting from L-glutamic acid 5-methyl ester, which is synthesised from L-glutamic acid with HCl and MeOH according to Baldwin et al.,23 3-cyanopropanoic acid methyl ester (CME) was generated via electro-oxidative decarboxylation with sodium bromide as the mediator (Scheme 1).15 During this electrolysis, the bromide anion was converted into the hypobromite species at the anode, which then selectively oxidised the α-amino acid moiety into the nitrile (Fig. S1†).4,15,24 Subsequently, saponification of CME to potassium 3-cyanopropanoate (3-CP) by the electrolyte KOH took place before two carbon radicals underwent a classical Kolbe dimerisation. Saponification and Kolbe coupling were carried out in a one-pot synthesis resulting in an adiponitrile yield of 78%.15
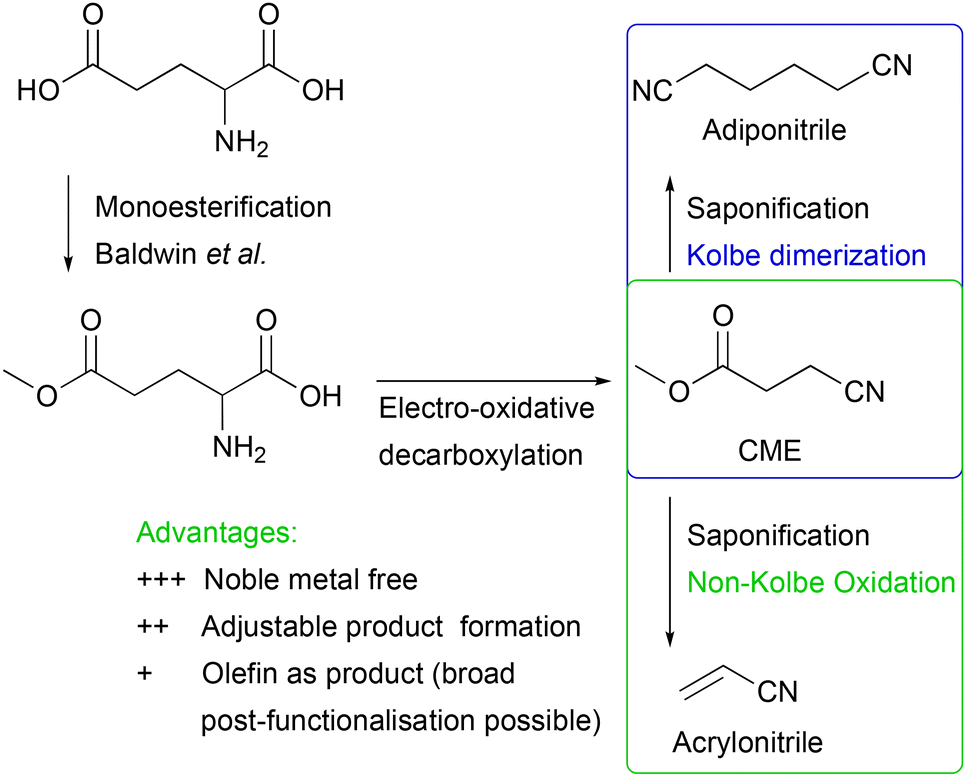 | ||
| Scheme 1 An environmentally friendly pathway to produce adiponitrile from glutamic acid via 3 steps according to Dai et al. (blue box)15 and applying non-Kolbe conditions to potassium 3-cyanopropanoate (3-CP), obtained by saponification of 3-cyanopropanoic acid methyl ester (CME) by KOH to synthesise acrylonitrile (green box). | ||
Inspired by this study, we aimed at changing the electrolysis conditions of the third step, including electrode material (platinum (Pt) to graphite (Cgr)) and current density (high to low) to oxidise the radical intermediate further to a carbocation. Subsequent deprotonation of the α-position with respect to the carbocation should then lead to the desired acrylonitrile.25,26 Along this line, we investigated nitrile formation by electro-oxidative decarboxylation followed by subsequent non-Kolbe electrolysis (Scheme 1).
Results and discussion
Nitrile formation by electro-oxidative decarboxylation
Before acrylonitrile can be produced, synthesis of the intermediate 3-CP from glutamic acid is necessary. The overall goal was to produce the intermediate 3-CP in a sustainable way, minimising the number of synthesis steps as much as possible. Thus, the direct conversion of glutamic acid to 3-CP without the use of CME is highly desirable. Nevertheless, CME was initially used as a substrate, since Dai et al. had already demonstrated the general feasibility of converting CME to 3-CP via electro-oxidative decarboxylation followed by saponification, respectively.15As the first starting point, electrolysis of CME using the parameters shown in Scheme 2 was chosen.15 MeOH-d4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D2O (4:1) served as a solvent mixture to enable direct 1H-NMR measurements of the product solution for quantification. Compared to the study of Dai et al., the electrode pair was changed from Pt–Pt to Pt–Ti. However, using 5.2 Faraday equivalents (Feq), a current density of 80 mA cm−2, a reaction temperature of 0 °C and Pt–Ti as the electrode pair resulted in a CME yield of only 62% (±3%) (Fig. 1) compared with 91% in the literature.15 Stirring the solution led to a small increase in the yield (68.2% (±0.1%)), but did not increase the yield to the value reported in the literature. Moreover, an inorganic colourless solid deposition occurred during electrolysis when NaBr was applied as a mediator (Fig. S2†). Solid deposits are undesirable, since they can cause blockage of the electrode surface.
:D2O (4:1) served as a solvent mixture to enable direct 1H-NMR measurements of the product solution for quantification. Compared to the study of Dai et al., the electrode pair was changed from Pt–Pt to Pt–Ti. However, using 5.2 Faraday equivalents (Feq), a current density of 80 mA cm−2, a reaction temperature of 0 °C and Pt–Ti as the electrode pair resulted in a CME yield of only 62% (±3%) (Fig. 1) compared with 91% in the literature.15 Stirring the solution led to a small increase in the yield (68.2% (±0.1%)), but did not increase the yield to the value reported in the literature. Moreover, an inorganic colourless solid deposition occurred during electrolysis when NaBr was applied as a mediator (Fig. S2†). Solid deposits are undesirable, since they can cause blockage of the electrode surface.
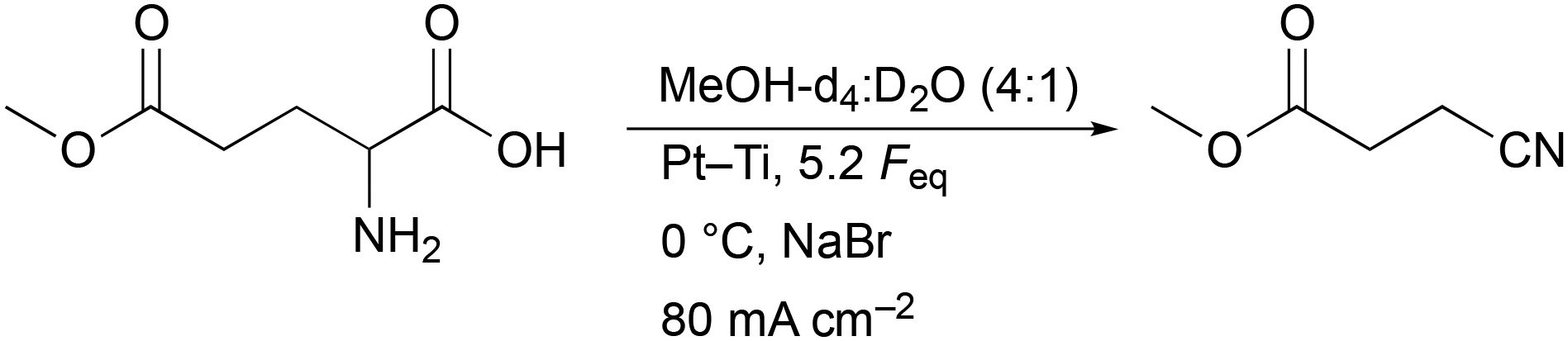 | ||
| Scheme 2 Conditions of the first experiment on the electro-oxidative decarboxylation of L-glutamic acid 5-methyl ester to CME. The concentration of L-glutamic acid 5-methyl ester was 0.2 M and the concentration of NaBr was 0.3 M. The studies of Dai et al. served as the basis for designing this experiment.15 | ||
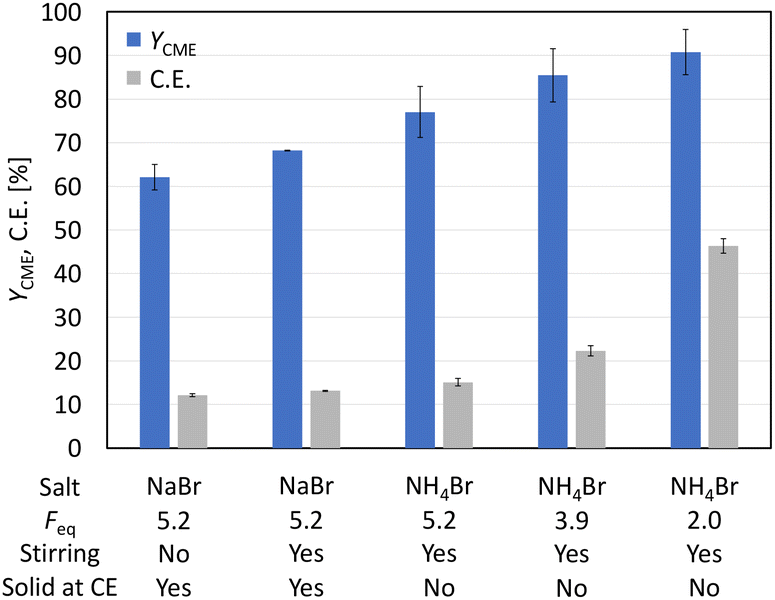 | ||
| Fig. 1 Variation of the reaction conditions and their influence on yield (YCME) and current efficiency (C.E.) for the synthesis of CME. Conditions: Pt–Ti; solvent: MeOH-d4:D2O (4:1); L-glutamic acid 5-methyl ester: 0.2 M (1 eq.); mediator salt: 0.3 M (1.5 eq.); j = 80 mA cm−2; and T = 0 °C. | ||
Matthessen et al. investigated the electro-oxidative decarboxylation of different amino acids and proposed NH4Br as an excellent mediator due to its acidity, which suppresses the oxidation of water by favouring the oxidation of the bromide species.4 The change from NaBr to NH4Br not only led to an increase in the yield from 68.2% (±0.1%) to 77% (±6%) at full conversion but also avoided solid deposits. In the next step, the amount of current that passed through the cell was reduced from 5.2 Feq to 2.0 Feq, assuming that product decomposition also contributed to the reduced yield. Reducing the Feq to 2.0 increased the yield from 77% (±6%) to 91% (±5%) at full conversion. Under the optimised conditions, a current efficiency (C.E.) of 46% (±2%) was obtained.
For subsequent Kolbe electrolysis, Dai et al. performed an in situ saponification reaction of CME to 3-CP (Scheme 1). The synthesis of acrylonitrile by non-Kolbe electrolysis also requires 3-CP as a substrate, but so far, the reaction pathway from glutamic acid to 3-CP is neither atom-efficient nor step-efficient due to the esterification and saponification reactions (Scheme 3). The efficiency of the reaction sequence can be increased if glutamic acid can be used directly as a substrate for the synthesis of 3-CP. Indeed, the electro-oxidative decarboxylation of glutamate, the sodium salt of glutamic acid, has already been shown by Matthessen et al.4
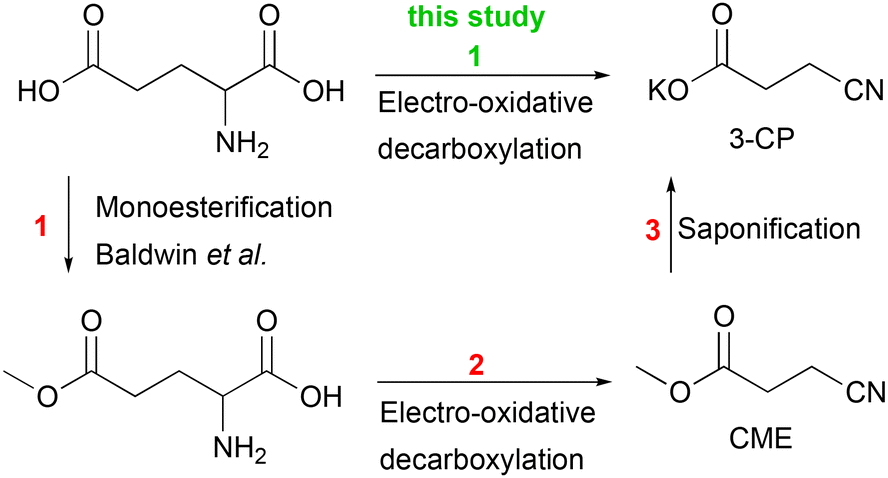 | ||
| Scheme 3 The current pathway from D,L-glutamic acid to 3-CP requires 3 steps (red) and thus is neither step-efficient nor atom-efficient.15 The synthesis of 3-CP can be more sustainable if D,L-glutamic acid is decarboxylated without pre-modification (green, this study). | ||
Applying the initial solvent mixture of MeOH-d4:D2O (4:1) to glutamic acid (0.2 M) resulted in a suspension, which was also present after electrolysis using the optimised parameters shown in Fig. 1 (Fig. S3†). The sediment was identified as non-converted substrate. The reason for the decreased solubility is the use of methanol. Various amino acids show reduced solubility in non-aqueous solvents.27 Therefore, an increase in polarity of the whole solvent mixture leads to an improved solubility of amino acids such as glutamic acid.28 Using D2O also resulted in an initial suspension, but this turned into an orange solution during electrolysis. For the follow-up step, the non-Kolbe electrolysis, the potassium species is required, and an ion exchange was performed. This was achieved by the addition of 2 eq. KOH to the product solution, which was verified by 1H-NMR measurements (Fig. S4†). The resulting 1H-NMR spectrum confirmed the successful reduction of the number of steps for the synthesis of 3-CP from glutamic acid. Since full conversion (X = 95% (±5%)) was not achieved and 2 eq. of KOH were required to obtain the characteristic signals of 3-CP in the 1H-NMR spectrum, 2 eq. of KOH were added to the substrate solution. This allowed a better solubility of glutamic acid and the initial pH changed from 3.1 to 9.3. Electrolysis was then performed, but this acidified the product solution (pH 3.9). Therefore, 2 eq. of KOH were added as before to convert 3-cyanopropanoic acid to 3-CP. This variation led to full conversion and improved the yield of 3-CP further from 67% (±7%) to 97.6% (±0.6%) (Fig. 2). With the optimised conditions, an 8% higher yield of 3-CP can be achieved than that published by Matthessen et al. for the electro-oxidative decarboxylation of glutamate to sodium 3-CP (Y = 91%).4 In addition, the C.E. reached 48.8% (±0.3%) using the optimised reaction conditions.
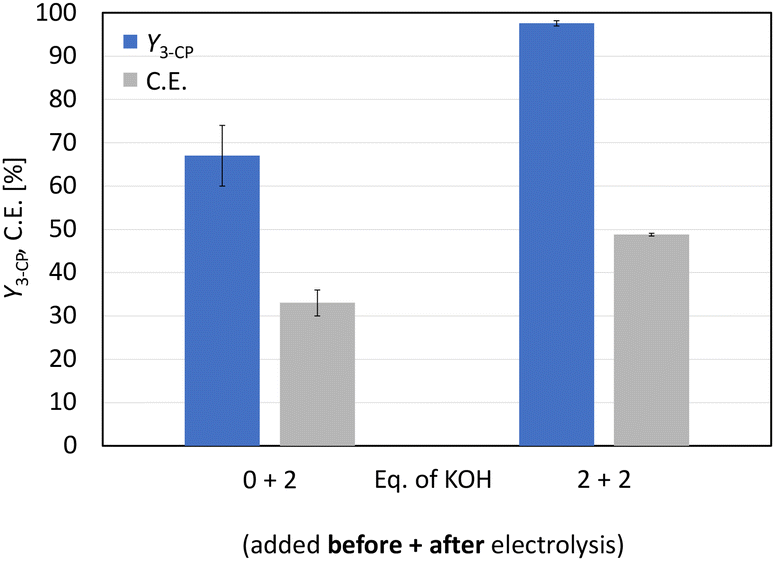 | ||
| Fig. 2 Electro-oxidative decarboxylation of D,L-glutamic acid to 3-CP. Investigating the impact of the addition of 0 eq. or 2 eq. of KOH to the substrate solution on the yield of 3-CP and the current efficiency. Conditions: Pt–Ti; solvent: D2O; D,L-glutamic acid: 0.2 M (1 eq.); NH4Br: 0.3 M (1.5 eq.); KOH (after electrolysis): 2 eq.; Feq = 2.0; j = 80 mA cm−2; and T = 0 °C. | ||
Non-Kolbe electrolysis to acrylonitrile
In contrast to Kolbe electrolysis, non-Kolbe electrolysis uses graphite anodes, low substrate concentrations, an alkaline electrolyte, and low current densities (<100 mA cm−2).25,26,29–32 Based on this, the first experiment was designed (Scheme 4), which resulted in an acrylonitrile yield of 24% (±3%) (Fig. 3). The conversion under these conditions was 61% (±8%), implying a selectivity for acrylonitrile formation of 39% (±10%). The explanation for the low selectivity of acrylonitrile formation is given by the corresponding 1H-NMR spectrum (Fig. S5†), emphasising that the Hofer–Moest side products 3-hydroxypropanenitrile (YA = 3.9% (±0.5%)) and 3-methoxypropanenitrile (YB = 8.9% (±1.5%)) were formed. In addition, the formation of oligomers of acrylonitrile was observed. Determination of the oligomer yield was not possible with the available analytical methods. A stability test of acrylonitrile in the stirred reaction solution (pH 8.6) without current supply showed no oligomer formation (Fig. S6†), so, an anionic oligomerisation mechanism cannot be the source for oligomer formation. Therefore, we suggest that the oligomers are formed by free-radical oligomerisation under electrolysis conditions (Fig. S7†). It is assumed that this radical oligomerisation is initiated by current-induced oxidation of the substrate molecule to the corresponding acyloxy radical that can react with olefins like acrylonitrile in a free radical oligomerisation reaction until termination occurs (Fig. S8†).25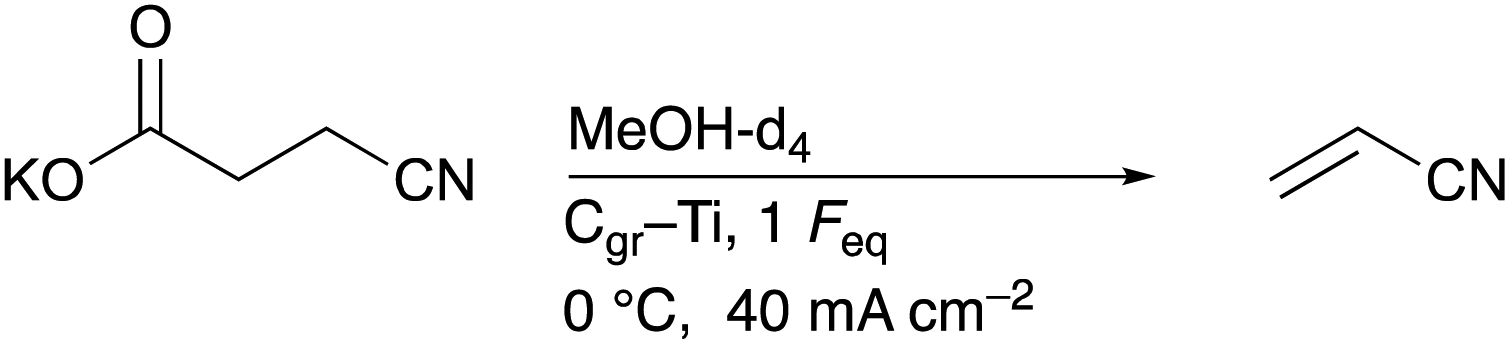 | ||
| Scheme 4 Initial conditions for the non-Kolbe electrolysis of 3-CP to acrylonitrile. The substrate concentration was 0.071 M. This experiment served as the standard experiment for the following variations. | ||
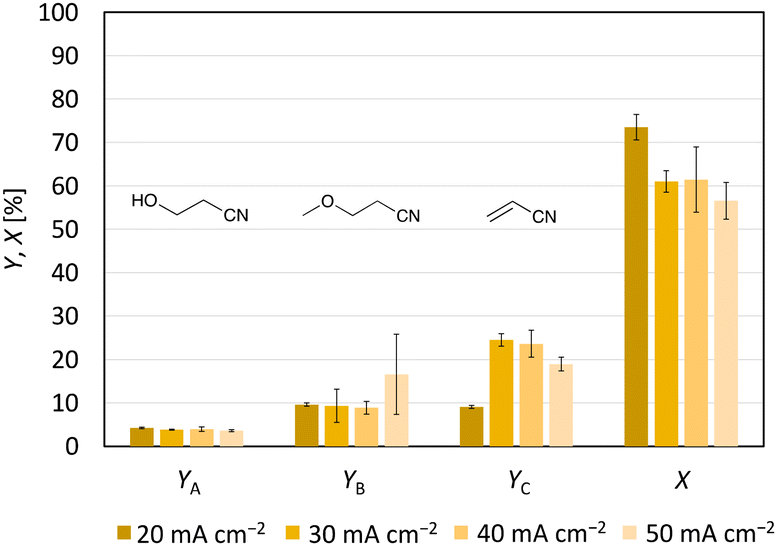 | ||
| Fig. 3 Effects of the current density on the yield of acrylonitrile (YC) and the two Hofer–Moest side products 3-hydroxypropanenitrile (YA) and 3-methoxypropanenitrile (YB). Furthermore, the conversion of the substrate (X) is visualised. Conditions: Cgr-Ti; solvent: MeOH-d4; 3-CP: 0.071 M; Feq = 1.0; stirring; and T = 0 °C. | ||
Since the yield of the first experiment (24% (±3%)) showed potential for optimisation, the impact of the current density on product formation was investigated, as this affects the number of radicals on the anode surface.25 Therefore, current densities of 20, 30, 40 and 50 mA cm−2 were used and their influence on product formation was studied (Fig. 3). Theoretically, in addition to using graphite as an anode, low current densities increase the chance of consecutive oxidation yielding the non-Kolbe product via a cationic pathway.25 With a decreasing current density, the concentration gradient at the anode is lower, so that the substrate is supplied more slowly.33 In this way, intermolecular reactions become less likely. However, at a current density of 20 mA cm−2 acrylonitrile formation decreased to 9.1% (±0.4%) although the conversion reached 74% (±3%). An increase of both Hofer–Moest products was not observed (Fig. 3). Instead, the 1H-NMR spectrum of the product solution produced at 20 mA cm−2 showed a stronger oligomer formation than at a current density of 40 mA cm−2 (Fig. S9†). The increased oligomer formation at 20 mA cm−2 could be explained by the kinetics. The current density as a measure of the (over)potential can influence the adsorption time of the organics. If uncharged acrylonitrile is not displaced quickly from the anode surface at low current densities for migration reasons, the probability to oligomerise increases. No difference in conversion and acrylonitrile formation was measured for current densities of 30 and 40 mA cm−2. Increasing the current density from 40 mA cm−2 to 50 mA cm−2 resulted in a lower conversion (from 61% (±8%) to 57% (±4%)) and acrylonitrile formation (from 24% (±3%) to 19.0% (±1.6%)). Instead, the conditions favoured the formation of 3-methoxypropanenitrile. Based on these results, the applied current density should neither be too small nor too high for the formation of acrylonitrile.
To increase the yield of acrylonitrile, the formation of the oligomers should be inhibited. However, the addition of common polymerisation inhibitors or retarders such as phenothiazine (PTZ) or monomethyl ether hydroquinone (MEHQ) did not show any increase in the yield of acrylonitrile, while maintaining high conversion rates (Fig. S12†).
Since no full conversion was obtained, the next step was to study the impact of the amount of charge (Feq) on the acrylonitrile formation. Increasing the Feq from 1.0 to 3.0 raised the conversion from 61% (±8%) to 95% (±3%) (Fig. 4). The yield of acrylonitrile increased from 24% (±3%) to 27% (±1%) when the amount of electricity applied to the reaction solution was increased from 1.0 Feq to 2.0 Feq. A further increase to 3.0 Feq resulted in a decrease in the yield to 2.8% (±0.2%). Besides, the current efficiency also decreased strongly. While it was 24% for 1.0 Feq, it only reached 13.5% for 2.0 Feq, even though a slightly higher yield of acrylonitrile was obtained with 2.0 Feq. The constantly growing gap between conversion and yield of acrylonitrile can be explained by an enhanced oligomer formation with more current passed through the cell, since the Hofer–Moest product 3-hydroxypropanenitrile showed constant values below 10%, while 3-methoxypropanenitrile was produced in yields between 8.9% (±1.5%) (1.0 Feq) and 18% (±8%) (2.5 Feq). 1H-NMR spectra of the different product solutions confirmed increasing oligomer signals, showing that with increasing Feq, not only the conversion was higher but also the formation of these oligomers (Fig. S13†). In summary, using 2.0 Feq resulted in the highest yield of acrylonitrile, while applying 1.0 Feq led to the highest C.E.
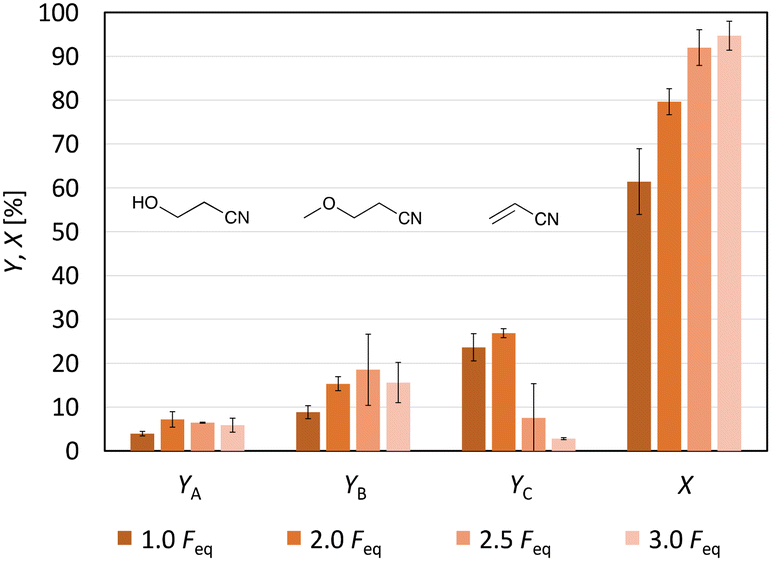 | ||
| Fig. 4 Effects of the Faraday equivalents on the yield of acrylonitrile (YC) and the two Hofer–Moest side products 3-hydroxypropanenitrile (YA) and 3-methoxypropanenitrile (YB). Furthermore, the conversion of the substrate (X) is visualised. Conditions: Cgr-Ti; solvent: MeOH-d4; 3-CP: 0.071 M; j = 40 mA cm−2; stirring; and T = 0 °C. | ||
To increase the yield of acrylonitrile by preventing the formation of the oligomers, it is necessary to stop or reduce bimolecular reactions. In (non-)Kolbe electrolysis, the addition of perchlorates reduces the probability of bimolecular reactions during electrolysis of carboxylates. The anions block parts of the anode surface, which reduces the radical concentration at the electrode and increases the yield of the non-Kolbe product.25 As the oligomer formation is also a bimolecular reaction, applying foreign anions to suppress oligomer formation was tested.
Adding tetrabutylammonium perchlorate (0.1 M) to a 0.071 M 3-CP solution (solvent: MeOH-d4) resulted in the formation of a sediment (Fig. S14†). Electrolysis of the suspension was performed, but no homogeneous solution was subsequently found. 1H-NMR analysis allowed identification of the sediment as unconverted substrate (Fig. S15†). Since the sedimentation made quantitative analysis difficult, varying the substrate concentration was identified as another way to reduce the number of radicals at the anode electrode. To study the impact of 3-CP concentration on product distribution, the initial concentration was continuously halved from 0.142 M to 0.009 M. Decreasing the concentration from 0.142 M to 0.018 M resulted in a constant increase in the yield and C.E. from 19% (±4%) to 41.1% (±1.2%) (Fig. 5). Thus, high substrate concentrations counteract the stabilisation and/or formation of acrylonitrile with respect to C.E. (1.0 Feq was used). The highest selectivity for acrylonitrile was obtained with a 0.018 M 3-CP solution (68% (±2%)). A further decrease in the substrate concentration did not improve yield or selectivity. The yield of the identified side products 3-hydroxypropanenitrile and 3-methoxypropanenitrile remained constant over the whole concentration range. Indeed, screening the influence of substrate concentration on product formation confirmed the assumption that the initial substrate concentration has an impact on the outcome of non-Kolbe electrolysis and is therefore consistent with the literature.25,34,35 If less substrate is present, the radical concentration at the anode surface is lower, leading to a higher yield of the corresponding olefins.25 Furthermore, this study has shown an alternative to the addition of foreign anions if bimolecular reactions should be circumvented.
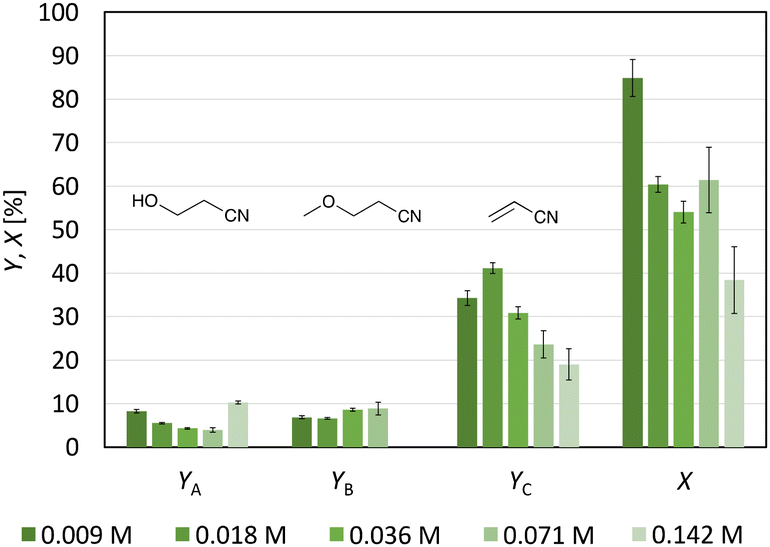 | ||
| Fig. 5 Effects of the initial 3-CP concentration on the yield of acrylonitrile (YC) and the two Hofer–Moest side products 3-hydroxypropanenitrile (YA) and 3-methoxypropanenitrile (YB). Furthermore, the conversion of the substrate (X) is visualised. Conditions: Cgr-Ti; solvent: MeOH-d4; Feq = 1.0; j = 40 mA cm−2; stirring; and T = 0 °C. | ||
The overall vision of the one-pot synthesis from D,L-glutamic acid is desirable (Scheme 5). The direct synthesis of 3-CP from D,L-glutamic acid was demonstrated in this study. Therefore, electrolysis of 3-CP in the presence of 1.5 eq. NH4Br was tested. Applying 0.93 Feq only resulted in a low conversion (2% (±4%)) and no acrylonitrile was formed. Under these conditions, a carbon balance of 98% (±4%) was measured. Increasing the Feq to 2.76 increased the conversion to 5.3% and a carbon balance of 94.7% was present, but still no product formation was observed. In conclusion, non-Kolbe electrolysis does not appear to be possible in the presence of 1.5 eq. NH4Br, which is explainable by an assumed BrO− layer at the electrochemical active surface that blocks non-Kolbe electrolysis (Fig. S16†). Consequently, it seems necessary to remove the bromide species when performing non-Kolbe electrolysis after electrochemical decarboxylation of D,L-glutamic acid.
 | ||
| Scheme 5 Vision for the synthesis of acrylonitrile using electro-oxidative decarboxylation and non-Kolbe electrolysis (light blue). So far, the one-pot synthesis has not been realised. However, it was shown that in principle acrylonitrile can be produced from glutamic acid using electrochemistry. | ||
Conclusions
We presented a new two-step concept for the synthesis of acrylonitrile starting from D,L-glutamic acid by electrosynthesis. In the first step, mediated electro-oxidative decarboxylation was utilised to synthesise the intermediate 3-cyanopropanoate (3-CP). The highest yield of 97.6% (±0.6%) was obtained after electrolysis of a reaction solution containing D,L-glutamic acid (0.2 M), NH4Br (1.5 eq.) as mediator and 2 eq. of KOH. It was shown that it is not necessary to synthesise 3-CP viaL-glutamic acid 5-methyl ester (CME), the protected version of L-glutamic acid, to achieve high yields. Furthermore, the impact of the mediator salt, applied charge, and stirring of the reaction mixture on the mediated electro-oxidative decarboxylation was investigated using CME as a model compound. Here, decreasing the Feq from 5.2 to 2.0 showed the highest impact on the yield, as it increased from 77% (±6%) to 91% (±5%).In the second part, 3-CP was converted into acrylonitrile by non-Kolbe electrolysis. Here, the effects of current density, applied charge, and substrate concentration on acrylonitrile formation were studied. Oligomer formation was identified as an undesirable side reaction that reduces the yield of acrylonitrile. Avoiding oligomer formation by using polymerisation inhibitors or retarders (PTZ or MEHQ) or low current densities (20 or 30 mA cm−2) showed no effect. In contrast, a higher yield of acrylonitrile was achieved by the reduction of the substrate concentration. By decreasing the initial 3-CP concentration to 0.018 M the yield of acrylonitrile increased to 41.1% (±1.2%) using a current density of 40 mA cm−2, a graphite anode and 1.0 Feq at 0 °C.
Data availability
The complete dataset can be found at https://zenodo.org/record/8033581. It comprises the experimental data used as the basis of the electrochemical transformation of D,L-glutamic acid into acrylonitrile: yields and faradaic efficiencies of the transformations under different reaction conditions.Author contributions
Justus Kümper: conceptualisation, formal analysis, investigation, methodology, validation, visualisation, and writing of the original draft. Jérôme Meyers: conceptualisation, data curation, validation, discussion, methodology, supervision, and writing and review of the original draft. Rebecca Sebers: conceptualisation, formal analysis, investigation, validation, visualisation, and writing and review of the original draft. Nils Kurig: conceptualisation, methodology, discussion, supervision, and writing and review of the original draft. Regina Palkovits: conceptualisation, methodology, discussion, resources, supervision, funding acquisition, and writing and review of the original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge funding by the Cluster of Excellence Fuel Science Center (EXC 2186, ID: 390919832) funded by the Excellence Initiative by the German federal and state governments to promote science and research at German universities as well as funding by German Research Foundation within PA1689/17-1. Nils Kurig acknowledges funding by Studienstiftung des deutschen Volkes e.V. We thank Ines Bachmann-Remy and Dr Meike Emondts for performing the NMR measurements.References
- M. C. Leech, A. D. Garcia, A. Petti, A. P. Dobbs and K. Lam, React. Chem. Eng., 2020, 5, 977–990 RSC.
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC.
- G. Creusen, F. J. Holzhäuser, J. Artz, S. Palkovits and R. Palkovits, ACS Sustainable Chem. Eng., 2018, 6, 17108–17113 CrossRef CAS.
- R. Matthessen, L. Claes, J. Fransaer, K. Binnemans and D. E. D. Vos, Eur. J. Org. Chem., 2014, 6649–6652 CrossRef CAS.
- J. F. Brazdil, in Ullmann's Encyclopedia of Industrial Chemistry, 2012 Search PubMed.
- E. Scott, F. Peter and J. Sanders, Appl. Microbiol. Biotechnol., 2007, 75, 751–762 CrossRef CAS.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- V. F. Wendisch, Metab. Eng., 2020, 58, 17–34 CrossRef CAS PubMed.
- K. Drauz, I. Grayson, A. Kleemann, H.-P. Krimmer, W. Leuchtenberger and C. Weckbecker, in Ullmann's Encyclopedia of Industrial Chemistry, ed. W.-V. V. G. C. KGaA, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007 Search PubMed.
- V. Gopinath and K. M. Nampoothiri, in Encyclopedia of Food Microbiology (Second Edition), ed. C. A. Batt and M. L. Tortorello, Academic Press, Oxford, 2014, pp. 504–517 Search PubMed.
- D. Zhang, D. Guan, J. Liang, C. Guo, X. Xie, C. Zhang, Q. Xu and N. Chen, Braz. J. Microbiol., 2014, 45, 1477–1483 CrossRef CAS PubMed.
- S. Sanchez, R. Rodríguez-Sanoja, A. Ramos and A. L. Demain, J. Antibiot., 2018, 71, 26–36 CrossRef CAS.
- R. Aoki, M. Wada, N. Takesue, K. Tanaka and A. Yokota, Biosci., Biotechnol., Biochem., 2005, 69, 1466–1472 CrossRef CAS.
- T. M. Lammens, J. Le Nôtre, M. C. R. Franssen, E. L. Scott and J. P. M. Sanders, ChemSusChem, 2011, 4, 785–791 CrossRef CAS PubMed.
- J.-J. Dai, Y.-B. Huang, C. Fang, Q.-X. Guo and Y. Fu, ChemSusChem, 2012, 5, 617–620 CrossRef CAS PubMed.
- E. M. Karp, T. R. Eaton, V. Sànchez i Nogué, V. Vorotnikov, M. J. Biddy, E. C. D. Tan, D. G. Brandner, R. M. Cywar, R. Liu, L. P. Manker, W. E. Michener, M. Gilhespy, Z. Skoufa, M. J. Watson, O. S. Fruchey, D. R. Vardon, R. T. Gill, A. D. Bratis and G. T. Beckham, Science, 2017, 358, 1307–1310 CrossRef CAS PubMed.
- R. K. Grasselli and F. Trifirò, Top. Catal., 2016, 59, 1651–1658 CrossRef CAS.
- T. Fuchigami, M. Atobe and S. Inagi, Fundamentals and Applications of Organic Electrochemistry, John Wiley & Sons, Ltd, 1 edn., 2014 Search PubMed.
- J. Seidler, J. Strugatchi, T. Gärtner and S. R. Waldvogel, MRS Energy Sustain., 2020, 7(1), e42 CrossRef.
- E. Gail, S. Gos, R. Kulzer, J. Lorösch, A. Rubo and M. Sauer, in Ullmann's Encyclopedia of Industrial Chemistry, 2004 Search PubMed.
- S. J. Asadauskas, M. Benyagoub, Y. Boumghar, F. Cavani, L. Charbonneau, A. Chieregato, R. Ciriminna and J.-L. Dubois, in Industrial Green Chemistry, ed. M. J. D. Mahboub, S. García, M. Garcia-Perez, A. Grigucevičienė, S. Kaliaguine, F. Lali, R. Luther, R. Mazzoni, M. Pagliaro, G. S. Patience, S. Poulston, P. Stavárek, T. Tabanelli, E. Terrell and H. V. Thang, De Gruyter, Berlin, Boston, 2020 Search PubMed.
- J. L. Nôtre, E. L. Scott, M. C. R. Franssen and J. P. M. Sanders, Green Chem., 2011, 13, 807–809 RSC.
- J. E. Baldwin, M. North, A. Flinn and M. G. Moloney, Tetrahedron, 1989, 45, 1453–1464 CrossRef CAS.
- A. H. Friedman and S. Morgulis, J. Am. Chem. Soc., 1936, 58, 909–913 CrossRef CAS.
- H.-J. Schäfer, Electrochemistry IV, 1990 Search PubMed.
- F. J. Holzhäuser, J. B. Mensah and R. Palkovits, Green Chem., 2020, 22, 286–301 RSC.
- T. E. Needham, A. N. Paruta and R. J. Gerraughty, J. Pharm. Sci., 1971, 60, 565–567 CrossRef CAS.
- N. A. Bowden, J. P. M. Sanders and M. E. Bruins, J. Chem. Eng. Data, 2018, 63, 488–497 CrossRef CAS PubMed.
- H. J. Schäfer, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, pp. 633–658 Search PubMed.
- J. Meyers, N. Kurig, C. Gohlke, M. Valeske, S. Panitz, F. J. Holzhäuser and R. Palkovits, ChemElectroChem, 2020, 7, 4873–4878 CrossRef CAS.
- N. Kurig, J. Meyers, F. J. Holzhäuser, S. Palkovits and R. Palkovits, ACS Sustainable Chem. Eng., 2021, 9, 1229–1234 CrossRef CAS.
- E. Klocke, A. Matzeit, M. Gockeln and H. J. Schäfer, Chem. Ber., 1993, 126, 1623–1630 CrossRef CAS.
- D. Pletcher, A First Course in Electrode Processes, The Royal Society of Chemistry, 2nd edn, 2009 Search PubMed.
- J. H. P. Utley and G. B. Yates, J. Chem. Soc., Perkin Trans. 2, 1978, 395–400 RSC.
- L. Brakha and J. Y. Becker, Electrochim. Acta, 2012, 77, 143–149 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01045g |
| This journal is © The Royal Society of Chemistry 2023 |