
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural ordering enhances highly selective production of acetic acid from CO2 at ultra-low potential†
Shreya
Sarkar
 ab,
Jithu
Raj
ab,
Debabrata
Bagchi
ab,
Arjun
Cherevotan
ab,
C. P.
Vinod
c and
Sebastian C.
Peter
*ab
ab,
Jithu
Raj
ab,
Debabrata
Bagchi
ab,
Arjun
Cherevotan
ab,
C. P.
Vinod
c and
Sebastian C.
Peter
*ab
aNew Chemistry Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bangalore, 560064, India. E-mail: sebastiancp@jncasr.ac.in
bSchool of Advanced Materials, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bangalore, 560064, India
cCatalysis and Inorganic Chemistry Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pune, 411008, India
First published on 15th December 2022
Abstract
Electrochemical reduction of CO2 to value-added chemicals and fuels using renewable energy technologies is known to facilitate the creation of an artificial carbon cycle. Although the practical use of most conventional electrocatalysts is curbed by the low efficiency and poor stability of the catalyst there is also the need of large input energy in the form of potential. In this work, a family of bismuth-based transition metal chalcogenides was designed to enable multi-electron transfer for selectively reducing CO2 to acetic acid at ultra-low potential of −0.1 V (vs. RHE). The structural design in AgBiS2, CuBiS2 and AgBiSe2 facilitated an optimized CO adsorption accounting for the production of acetic acid at low potential. The disordered arrangement of Ag and Bi in AgBiS2 also favors CO hydrogenation, which leads to the formation of a large amount of methanol in addition to acetic acid. However, an induced structural ordering of these atoms upon selected substitution enhanced the lattice strain in CuBiS2 and AgBiSe2 favoring only C–C coupling and 100% acetic acid is produced at lower potential with stability up to 100 hours. The origin of the CO2 reduced product has been validated by 13CO2 isotopic experiments and the mechanistic pathway has been proposed with the support of in situ IR experiments. Finally, a 4 times improvement in the current density of the best catalyst, AgBiSe2, was achieved in a flow cell configuration, which produced the highest ever acetic acid yield at lower potential with a faradaic efficiency of 49.81%. This work provides a novel strategy to improve electrochemical performance towards the formation of high value-added chemicals selectively at ultra-low potential.
Broader contextThis manuscript focuses on how structural ordering can facilitate the formation of acetic acid at ultra-low potential, which is a critical challenge in the electrochemical CO2RR. As per our knowledge, this is the first report of the production of acetic acid at ultra-low potential. Additionally, the manuscript provides an in-depth insight into the possible reaction mechanism using in situ IR along with deep understanding of how the crystal structures of ABiX2 electrocatalysts tune the product selectivity and activity. This is the first work reporting long-term durability to produce acetic acid via eCO2RR. The origin of the CO2 reduced product has been validated by 13CO2 isotopic experiments. Finally, the current density of the best catalyst has been improved by 4 times by performing the CO2 reduction experiments in a flow cell configuration, which produced the highest ever acetic acid yield at lower potential with a faradaic efficiency of 49.81%. This work introduces a rational design of the catalyst to facilitate the C–C coupling reaction, and is expected to motivate researchers working in the area and can be a good guideline to rationally design and develop catalysts for a desired product from CO2. |
The expedited technological advancement of modern society has led to severe environmental problems with global warming being the most pivotal amongst all others. Conversion of CO2 into value-added chemicals and fuels appears to be one of the most attractive pathways to mitigate CO2 accumulation in the atmosphere.1,2 The electrochemical CO2 reduction reaction (eCO2RR) facilitated by the usage of renewable energy resources is a sustainable alternative to large-scale dependence on fossil fuels and is advantageous since it can occur under ambient conditions and is solely dependent on potential bias.3–6 Additionally, this strategy enables the on-site production of a wide variety of value-added chemicals and fuels, which includes C1 and C2+7 hydrocarbons, mitigating their limitations in terms of distribution and storage specifically for toxic gases like CO.8 However, the wide-scale application of the eCO2RR technology requires significant improvements in energy efficiency, catalyst stability and current density. The current most pivotal constraint lies with the high thermodynamic barrier required for the conversion of CO2 to CO2˙− radicals, which is accompanied by the disruption of stable sp-hybridization to bend the linear CO2 molecule.9 Hence, CO2 activation requires large activation energy barriers, which results in large overpotential requirements to drive the forward reaction. This makes it critical to design electrocatalysts that can facilitate stabilization of the intermediate to reduce the overpotential for the overall electrochemical process in addition to inhibiting the competitive hydrogen evolution reaction (HER).10 Additionally the competition between the HER and eCO2RR stimulates low faradaic efficiencies (FEs) of certain products, which consumes most of the charge for this competitive reaction.11,12 Therefore, it is desirable to develop electrocatalysts with low overpotential, fast kinetics and high efficiency for the conversion of CO2 into multi-carbon products. Importantly, the production of C1 hydrocarbons13,14 (CH4, HCOOH and CH3OH) is easier in comparison to C2 hydrocarbons15 (C2H4, CH3COOH, C2H5OH), which have high volumetric energy densities and are the building blocks for the synthesis of long-chain hydrocarbon fuels.16,17 The C2 product involves C–C bond formation which competes with the formation of C–H and C–O bonds under similar reaction conditions, which makes its production even more strenuous.18 Hence, developing electrochemical systems that can mediate multi-proton transfer reactions at low overpotential still remains one of the most significant challenges. Amongst the C2 products, acetic acid is one of the most highly desired ones due to its immense applications in the food, pharma, chemical, textile, polymer, medicinal, and cosmetics sectors.19,20
However, as discussed earlier the eCO2RR to acetic acid involves a C–C coupling reaction along with adequate surface coverage of *CO, *CH2 and *CH3 intermediates, which requires large overpotentials with much lower FE.21Fig. 1 and Table S1 (ESI†) summarize the FE for acetic acid as a function of potential, to date. Rational selection and design of the catalysts is very crucial for efficient and selective eCO2RR. Transition metal-based catalysts are often required for the eCO2RR in order to lower the activation energy barrier and drive the process at acceptable rates and at specific potentials.22 Amongst the transition metals, Cu and its oxide derivatives have emerged as a unique class of electrocatalysts23–27 that can efficiently convert CO2 into a wide variety of hydrocarbons and their oxygenated products (CH3OH, CH4, C2 and C2+).28,29 Other transition metals like Au,30 Ag,31 and Zn32 delivered high FE towards the formation of CO.33 Additionally, P-block elements like Sn, Bi, In, Pb and their oxides have been known to catalyze the eCO2RR to produce formate or formic acid as the major product with FE as high as 90%.34–38 The presence of lone pair electrons in Bi3+ accelerates the adsorption and activation of CO2 molecules due to the Bi 6s and O 2p hybridization, which favors the formation of stereo chemically active lone pairs that promote electron donation to acidic adsorbed species such as CO2.39–41 Chalcogens have been found to accelerate water activation. The promotional effect of sulfur on the indium surface to accelerate the reduction of CO2 to formate has already been demonstrated by Ma et al.42 The presence of negative charge on the chalcogen makes it nucleophilic and hence, traps CO2.43 It is often wondered how these individual performances could be tuned selectively to a desired product upon a compound formed with a selected combination of these elements, which requires several controlled design strategies. In this work we tried to address three fundamental challenges in the eCO2RR: low CO2 solubility, large overpotential and poor selectivity. To study these, we developed a series of MBiX2 catalysts (M = Ag/Cu and X = S/Se). AgBiS2 was chosen as the pristine catalyst for the eCO2RR and Ag was replaced with Cu to develop CuBiS2 and S with Se to form AgBiSe2.
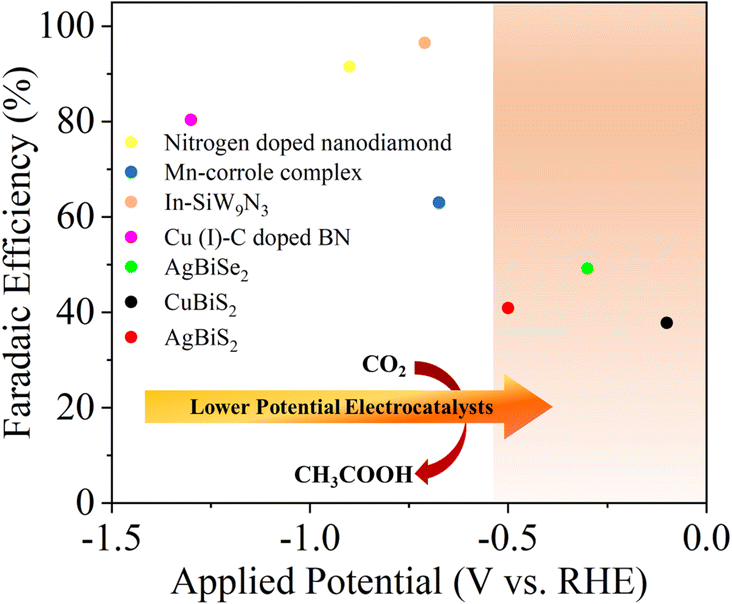 | ||
| Fig. 1 Reported faradaic efficiency for the conversion of CO2 to acetic acid as a function of applied potential on various catalysts.12,44–46 | ||
AgBiS2, CuBiS2 and AgBiSe2 were synthesized using a colloidal method with oleylamine as the solvent and reducing agent. PXRD patterns (Fig. S1, ESI†) demonstrated that AgBiS2 crystallizes in the cubic phase (space group: Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), CuBiS2 in the orthorhombic phase (space group: Pnma) and AgBiSe2 in the rhombohedral phase (Rmh). Fig. S2 (ESI†) shows the TEM images of AgBiS2, CuBiS2 and AgBiSe2. The atomic coordinates and Wyckoff sites of all the catalysts have been tabulated in Table S2 (ESI†). As seen from Fig. S2a (ESI†), AgBiS2 formed agglomerated spherical nanoparticles with an average particle size of 30–40 nm, while AgBiSe2 showed the presence of irregular shaped nanostructures with an average size of 20–25 nm (Fig. S2c, ESI†). CuBiS2 on the other hand formed a honey-comb like structure with a particle size of less than 10 nm. Selected area elemental mapping of the MBX2 catalysts depicted uniform distribution of the respective elements i.e. M = Ag/Cu, Bi and X = S/Se throughout the nanoparticles (Fig. S3–S5, ESI†). EDAX taken on an ensemble of nanoparticles is in close agreement with these measurements confirming the expected stoichiometry of 1
m), CuBiS2 in the orthorhombic phase (space group: Pnma) and AgBiSe2 in the rhombohedral phase (Rmh). Fig. S2 (ESI†) shows the TEM images of AgBiS2, CuBiS2 and AgBiSe2. The atomic coordinates and Wyckoff sites of all the catalysts have been tabulated in Table S2 (ESI†). As seen from Fig. S2a (ESI†), AgBiS2 formed agglomerated spherical nanoparticles with an average particle size of 30–40 nm, while AgBiSe2 showed the presence of irregular shaped nanostructures with an average size of 20–25 nm (Fig. S2c, ESI†). CuBiS2 on the other hand formed a honey-comb like structure with a particle size of less than 10 nm. Selected area elemental mapping of the MBX2 catalysts depicted uniform distribution of the respective elements i.e. M = Ag/Cu, Bi and X = S/Se throughout the nanoparticles (Fig. S3–S5, ESI†). EDAX taken on an ensemble of nanoparticles is in close agreement with these measurements confirming the expected stoichiometry of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 in all three compounds (Fig. S6, ESI†). The ABiX2 catalysts were further characterized by XPS and XANES to understand their electronic structure (Fig. S7–S10 and Notes S1 and S2, ESI†).
:1:2 in all three compounds (Fig. S6, ESI†). The ABiX2 catalysts were further characterized by XPS and XANES to understand their electronic structure (Fig. S7–S10 and Notes S1 and S2, ESI†).
The electrochemical CO2RR was carried out on the MBiX2 catalysts in 0.5 M KHCO3 with continuous CO2 purging at 20 sccm flow rate (Fig. S11 and S12, ESI†). Fig. S13 (ESI†) depicts the observed current density at each applied potential on different MBiX2 catalysts. At relatively lower overpotential (−0.1 V, −0.3 V vs. RHE), the current densities for all three chalcogenides are found to be similar. Chronoamperometry (CA) curves on different chalcogenide (Fig. S13b–d, ESI†) catalysts have been shown at different potentials in CO2-saturated 0.5 M KHCO3 solution, indicating good electrochemical stability of our catalyst. Fig. S14 (ESI†) shows LSV polarization curves in which the increase in current density when the atmosphere was changed from N2 to CO2 is apparent. The positive shift in onset potential upon saturating the electrolyte with CO2 indicates the dominance of the CO2 reduction process relative to the HER.
ECSA calculations demonstrate that AgBiSe2 has the highest electrochemically active surface area (Fig. S15, ESI†). Fig. 2 shows the FEs and eCO2RR product distributions as a function of applied potential from −0.1 V to −1.1 V over all three catalysts, which were calculated combining GC, HPLC and NMR analyses (Fig. S16–S20, and Notes S3, ESI†). The product distribution and their corresponding FEs are given in Table S3 (ESI†). AgBiS2 produced CH3OH at all the potentials with maximum FE (60.39%) at −0.3 V (Fig. 2a and Table S3, ESI†). On the other hand, acetic acid was formed at extremely low overpotentials (−0.1 V vs. RHE) with FE of 12.55%, which gradually increased to its maximum value of 35.97% at −0.3 V. AgBiSe2 shows a similar trend of production of CH3COOH (Fig. 2b) where the FE of CH3COOH is higher at lower overpotential (−0.1 V to −0.3 V) and decreased at higher overpotential (−0.5 V and −1.1 V). Compared to AgBiS2, AgBiSe2 does not produce any detectable amount of CH4 or CH3OH in the entire potential window. Only acetic acid was formed as the liquid product at a potential of −0.1 V and −0.3 V with 21.5% and 37.18% FE, respectively, which is higher than that observed in the case of AgBiS2. The production of CO and HCOOH was observed as similar in both catalysts. Similarly, CuBiS2 also yields CH3COOH as the only liquid product at an extremely low overpotential of −0.1 V (vs. RHE), but the maximum FE observed was 28.26%, which is less than that of Ag-based systems (Fig. 2c). To further verify that the product was derived from CO2 reduction, an isotope labelled 13CO2 study was performed on the AgBiS2 catalyst since it produced both acetic acid and methanol. 1H NMR spectra (Fig. S21, ESI†) demonstrated the H signal due to 13CH3 groups on acetic acid and methanol. For both acetic acid and methanol, the H signal splits into two peaks due to coupling with 13C atoms. The isotope labelled 13CO2 study using HPLC on AgBiS2 (Fig. S22, ESI†) further confirmed that acetic acid was produced from CO2 and not from any other chemical. Fig. S23 (ESI†) shows the formation rate of different CO2 reduced products as a function of potential, which is in accordance with the observed FE trend. From this controlled design strategy of the catalysts, it is very clear that a disordered system favored methanol formation in addition to acetic acid but the ordered nature of the atoms in CuBiS2 and AgBiSe2 facilitated C–C coupling and helped in the selective production of acetic acid at lower potential.
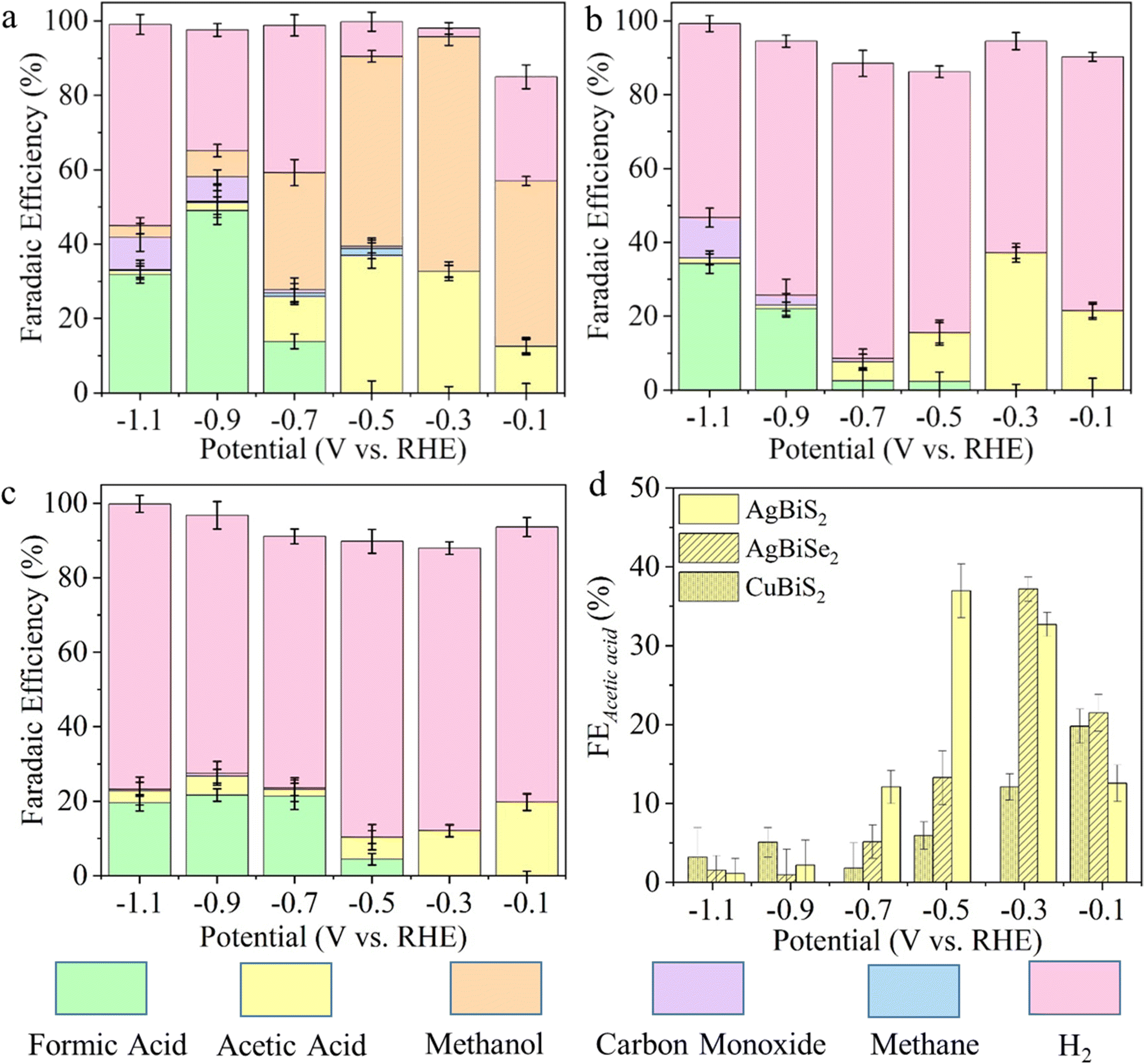 | ||
| Fig. 2 Faradaic efficiency for each CO2 reduced gaseous product (carbon monoxide, methane, hydrogen) and major liquid product (methanol, formic acid and acetic acid) as a function of potential after 1 hour of CA during the eCO2RR on (a) AgBiS2, (b) AgBiSe2 and (c) CuBiS2, and (d) comparison of FE for acetic acid at different potentials for all the catalysts. | ||
Fig. 2d depicts the trend of FE towards CH3COOH as a function of potential on each catalyst. AgBiS2 and AgBiSe2 have exhibited a volcano kind of trend where CH3COOH is formed at lower overpotential (−0.1 to −0.3 V) with large FE. On the other hand, the CH3COOH FE in the case of CuBiS2 linearly increased from higher potential to the maximum at lower potential (−0.1 V). To scrutinize the durability of the ABiX2 catalysts we performed an endurance electrolysis experiment at −0.3 V vs. RHE and monitored the liquid products generated using HPLC and NMR. Prolonged electrolysis of six hours showed negligible degradation in current density for AgBiS2, AgBiSe2 and CuBiS2 electrocatalysts indicating their high stability (Fig. S24, ESI†). Acetic acid remained as the major C2 product in the AgBiSe2, CuBiS2 and AgBiS2 electrocatalysts upon prolonged electrolysis of six hours along with formic acid while methanol formation was still observed for AgBiS2. Additionally, long-term electrolysis led to the formation of other C2 products: ethanol and diethylene glycol for all three electrocatalysts (Fig. S25, ESI†).
Additionally, we also exploited long-term electrolysis up to 100 hours for our best active catalyst AgBiSe2. AgBiSe2 demonstrated enhanced activity and performance under eCO2RR conditions. The catalyst was found to be durable up to 100 hours of electrolysis with negligible degradation in electrocatalytic activity. In addition to being durable up to 100 hours it was observed that the faradaic efficiency (FE) for acetic acid was consistent during this prolonged period with FE being 37.16% and 40.63% at the end of 1 hour and 100 hours, respectively (Fig. S26, ESI†). Post-electrochemical XRD at the end of 100 hours revealed no notable structural changes during prolonged electrolysis except that negligible Ag2O was formed at this potential of −0.3 V vs. RHE (Fig. S27, ESI†). The formation of ethanol and diethylene glycol was also observed upon long term electrolysis up to 100 hours (Fig. S28, ESI†). The FE of the obtained liquid products after 100 hours of electrolysis on the AgBiSe2 catalyst is shown in Fig. S29 (ESI†).
The reaction pathways and intermediates involved in the eCO2RR over the three chalcogenides were probed by in situ attenuated total reflection (ATR-IR) spectroscopy (Fig. 3 and Table S4, ESI†).47 Over AgBiSe2, at −0.1 V vs. RHE, a strong band appears at 1411 cm−1 (Fig. 3a) indicating the symmetric stretch mode of the carbon bound *COO− intermediate.48 (All the surface bound species will be prefixed * from hereafter.) The intensity of the COO− peak increases with time coinciding with the high FE of acetic acid. This is in line with previous reports in which *COO− serves as the intermediate for acetic acid by further proton coupled electron transfer processes and C–C coupling.49 The slight hump at 1566 cm−1 is assigned to the asymmetric stretch of COO−. The weak peak appearing at 1289 cm−1 belongs to the O–H deformation of the surface bound COOH intermediate. The intensity of this peak also rises with time indicating that it is also involved in acetic acid formation. Water consumption associated with the HER is indicated by the H–O–H bend at 1621 cm−1.50 This peak may be overlapping with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch of the *COOH intermediate as seen from the slight hump at 1660 cm−1.48 A weak band appearing at 2055 cm−1 can be ascribed to linearly adsorbed CO.47 The intensity of the peak is quite low as free CO does not form at this potential. The presence of *CO at low applied potential only on AgBiSe2 is in agreement with its high FE for acetic acid at this potential. The formed *CO may immediately turn to *HCO, another important intermediate in the CO2 reduction pathway.
O stretch of the *COOH intermediate as seen from the slight hump at 1660 cm−1.48 A weak band appearing at 2055 cm−1 can be ascribed to linearly adsorbed CO.47 The intensity of the peak is quite low as free CO does not form at this potential. The presence of *CO at low applied potential only on AgBiSe2 is in agreement with its high FE for acetic acid at this potential. The formed *CO may immediately turn to *HCO, another important intermediate in the CO2 reduction pathway.
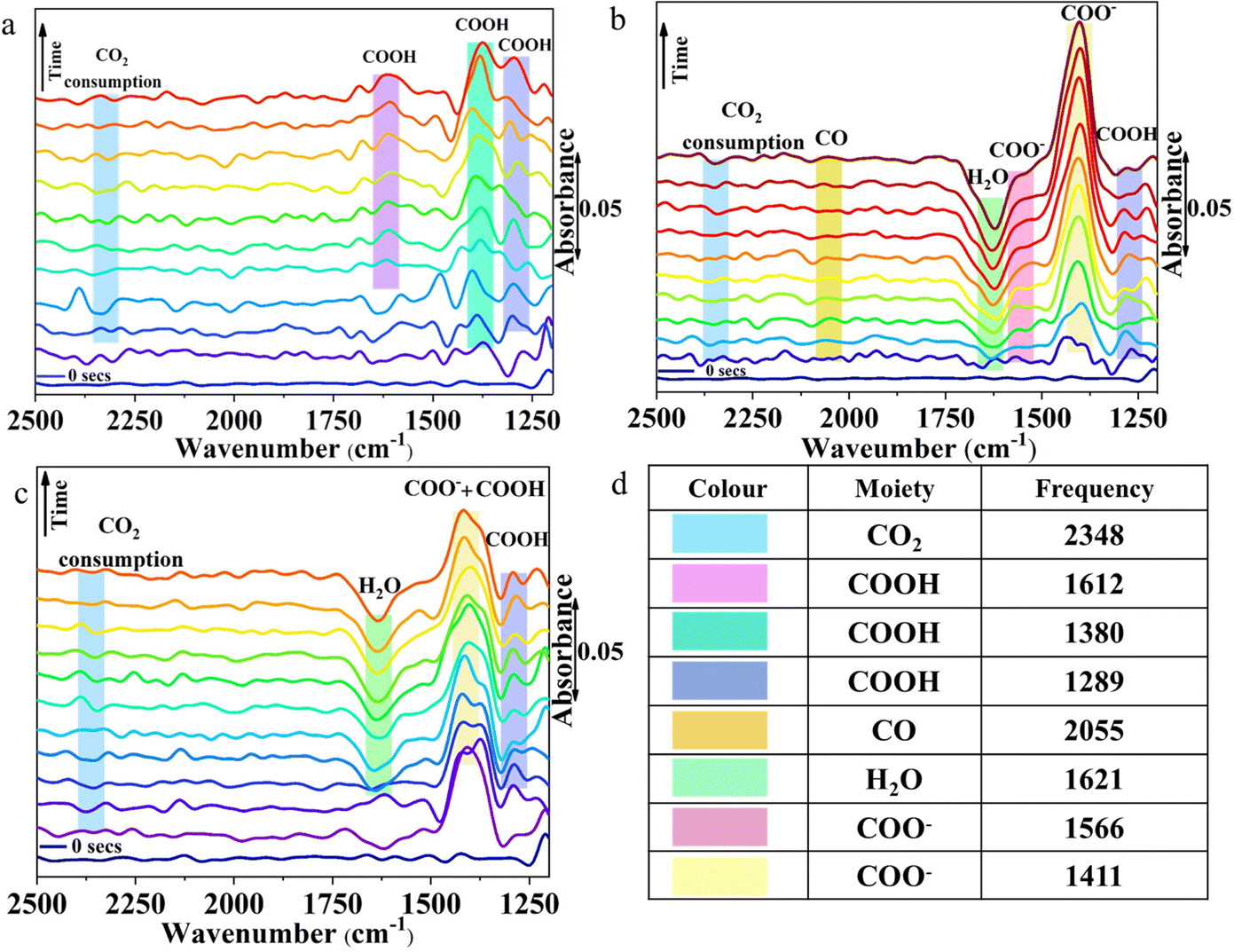 | ||
| Fig. 3 (a–c) ATR-IR spectra during electrochemical CO2 reduction on AgBiSe2, AgBiS2, and CuBiS2 electrodes, respectively, at an applied potential of −0.1 V vs. RHE and (d) adsorption frequencies of different intermediates adsorbed on the catalyst surface during CO2 reduction. | ||
The minor dip at 2340 cm−1 indicates the consumption of CO2 as evident from previous reports.51 In AgBiS2, the three vibrational modes of *COOH are clearly seen: O–H deformation, C–O stretch and CO stretch at 1289, 1380 and 1612 cm−1, respectively. The absence of the negative H–O–H bend is associated with the HER, which is in line with the high overall FE of the AgBiS2 (Fig. 3b) at low applied potentials.52 In CuBiS2, peaks associated with *COO− and *COOH groups are overlapping at around 1400 cm−1 (Fig. 3c). The relatively low intensity of the COO− peaks as compared to AgBiSe2 and the absence of a hump near the H–O–H bending region for the *COOH intermediate as in AgBiS2 is commensurate with the low overall FE of CuBiS2. The IR spectra of CuBiS2 in the range of −0.1 to −0.5 V vs. RHE (Fig. S30–S32, ESI†) also show that the intensity of the COO− and COOH peaks is quite low as compared to AgBiS2 and AgBiSe2. The declining peak intensity of COO− with applied potential (Fig. S33, ESI†) depicts that the production of formic acid at high potentials is via a separate mechanism and not through the carbon bound intermediates. To confirm that IR bands observed during in situ ATR arise due to CO2 reduction and not from bicarbonate species, we have performed the ATR studies using 0.1 M KCl (Fig. S34, ESI†). Since, the IR bands appear almost at the same stretching frequencies as those in 0.5 M KHCO3 we can confirm that CO2 reduction results in the formation of the observed *COO− and *COOH intermediates. Based on the results of the in situ ATR studies and with the aid of the previous reports, the mechanism for the conversion of CO2 to acetic acid and methanol formation over the chalcogenide catalysts may be proposed (Fig. 4). The first step involved in CO2 reduction is the activation of the CO2 molecule. Here, CO2 is adsorbed into the chalcogenide surface by accepting an electron as a carbon bound CO2˙− radical ion.53 The LUMO of the activated CO2 molecule is localized at C while the HOMO is localized at O due to the lower electronegativity of C in comparison to that of O. Hence, CO2 is liable to undergo both nucleophilic and electrophilic reactions, respectively.43 The adsorbed CO2 molecule then undergoes hydrogenation to form a *COOH moiety followed by proton coupled electron transfer processes that result in *CHO, thereby converting to acetic acid and methanol in the ensuing C–C coupling process at lower potential.49
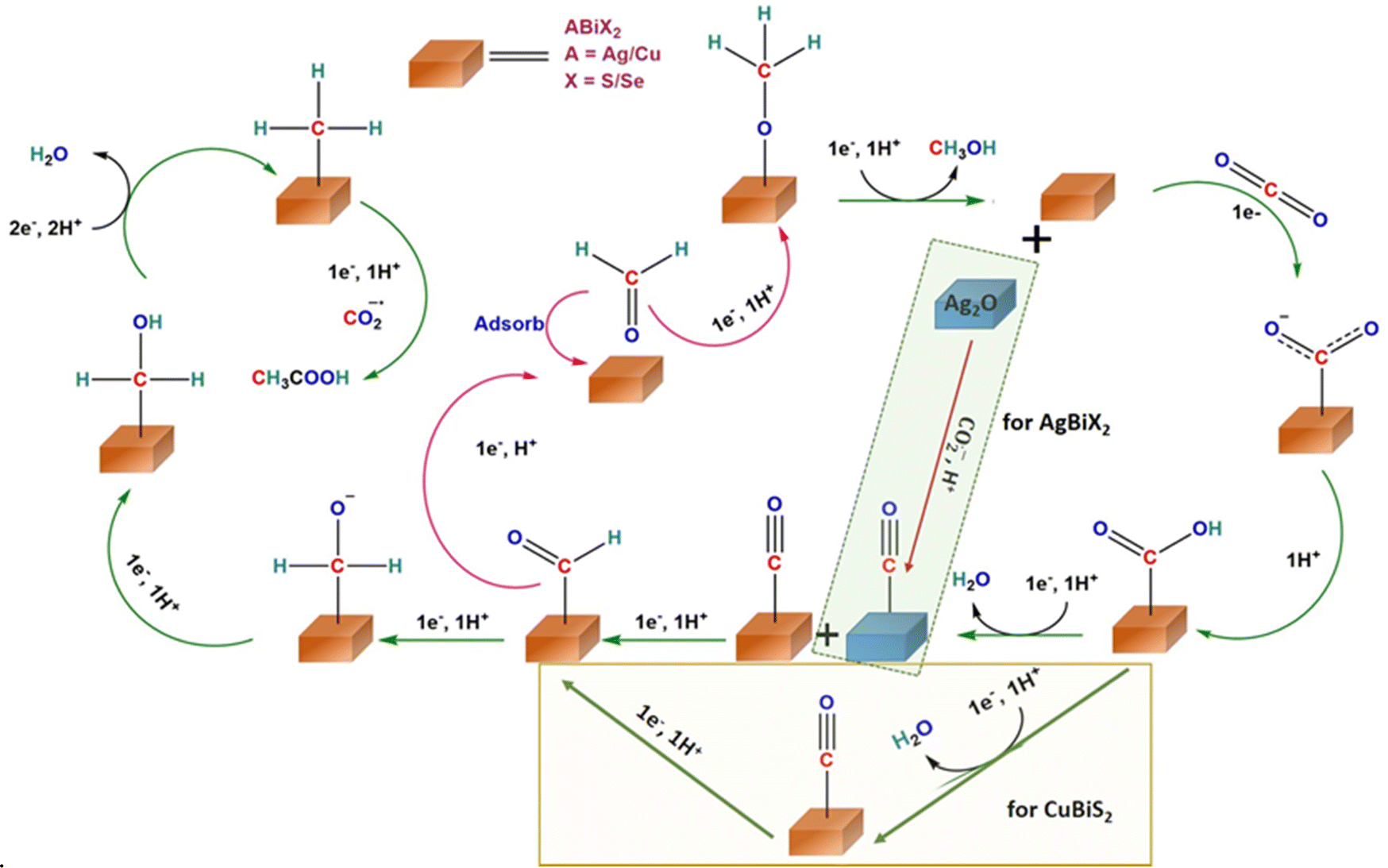 | ||
| Fig. 4 Proposed reaction mechanism of the eCO2RR for the production of CH3COOH and HCOOH on CuBiS2, AgBiSe2, and AgBiS2 predicted from in situ IR experiments. | ||
Based on earlier reports of chalcogenides with p-block elements, it can be predicted that the formation of HCOO* on the S–Bi surface is expected to be exergonic while the formation of H* from H2O is endergonic, thereby facilitating the formation of formic acid at higher potential.24 To validate the mechanism further, we explored the crystal structure of all three catalysts. As seen from Fig. 5, AgBiSe2 and CuBiS2 have ordered structures with shortest X–Bi (X = S/Se) bond distances of 3.04 Å and 3.15 Å, respectively. The shorter bond distance aids in the close proximity of adsorbed C atoms on AgBiSe2 and CuBiS2 surfaces, thereby favoring only C–C coupling at lower potential and accounting for its high selectivity towards acetic acid as compared to AgBiS2. Also, the adsorption energy of reaction intermediates plays a pivotal role in product selectivity. A lower d-band center indicates stronger adsorption of the reaction intermediate and weak desorption ability.22 Since C–C coupling is a necessity for the formation of C2 products, stronger CO adsorption will facilitate the coupling. This makes it important to analyze the local charge distribution in the lattice. To have a qualitative understanding of the variation in localized charge distribution of the three different lattice systems, XPS and XANES analyses were performed (Fig. S7–S10 and Notes S1 and S2, ESI†). Fig. S35 (ESI†) shows the valence band spectra of AgBiS2, AgBiSe2 and CuBiS2 derived from XPS, which shows that AgBiSe2 has the lowest d-band center followed by CuBiS2 and it is high in the case of AgBiS2. CO adsorption is expected to be the strongest in AgBiSe2 with weak desorption. Hence, C–C coupling in AgBiSe2 is more favored accounting for its high selectivity followed by CuBiS2. However, since AgBiS2 has the highest bond distance, CO hydrogenation will be kinetically more favored as compared to C–C coupling. CO hydrogenation results in the formation of *CHO or *COH intermediates resulting in the formation of methanol and methane. This rationale validates our experimental findings of AgBiS2 favoring the formation of methanol and methane in addition to acetic acid. XANES and XPS analyses further shed light on the activity difference between the ordered structures AgBiSe2 and CuBiS2. Fig. 5d and e shows a downshift in both binding energy and absorption edge of Bi in AgBiSe2 relative to that of CuBiS2. This indicates that Bi in AgBiSe2 has an oxidation state of 3δ− instead of 3+ which makes Se more electronegative. On the contrary Cu in CuBiS2 is expected to have an oxidation state in between 0 and +1 due to the presence of Bi in the 3δ+ state.
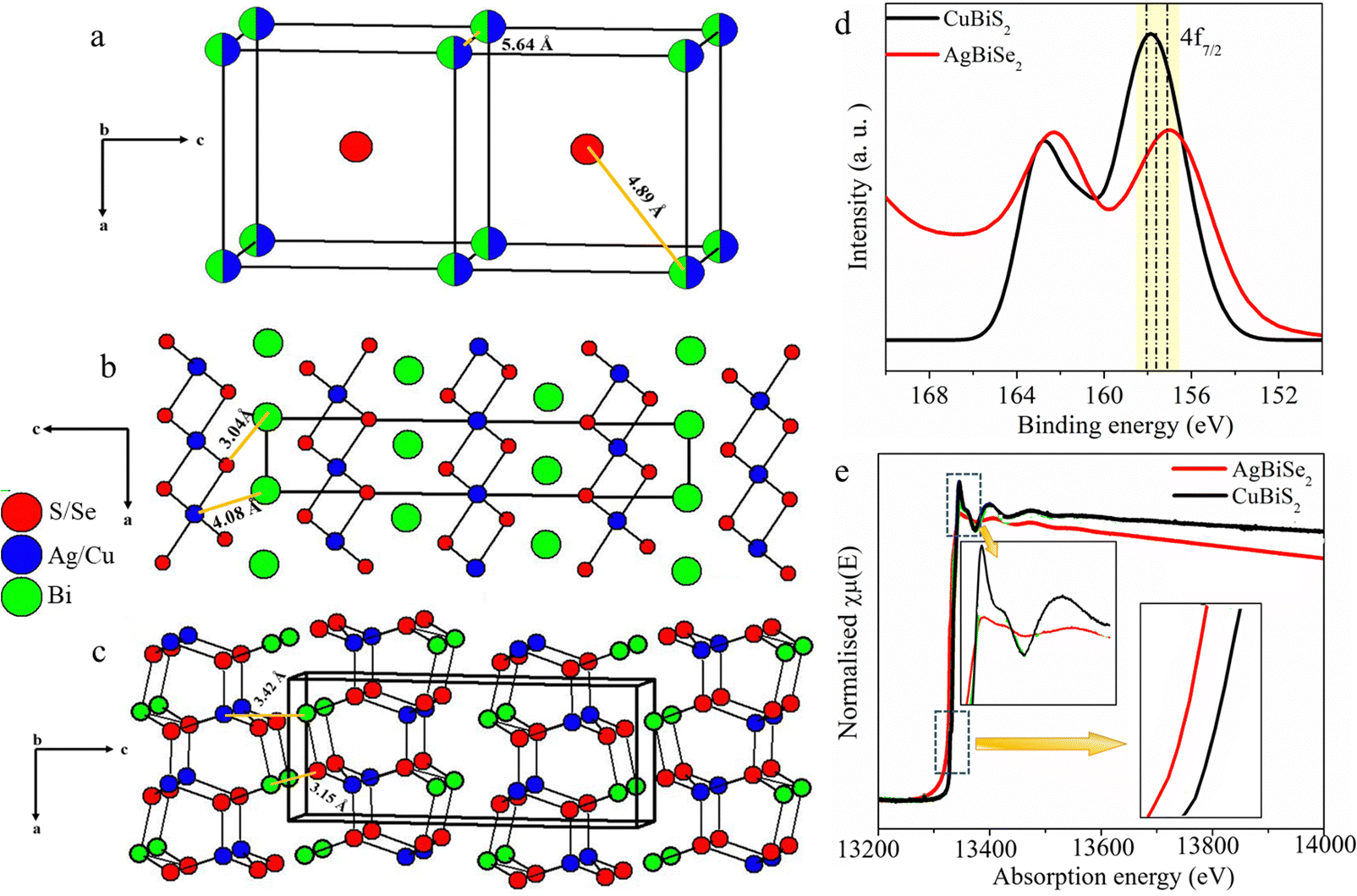 | ||
| Fig. 5 Crystal structures of (a) AgBiS2, (b) AgBiSe2 and (c) CuBiS2. Comparison of XPS spectra of Bi 4f (d) and XANES spectra of the Bi–L III edge (e) for AgBiSe2 and CuBiS2 electrocatalysts. | ||
A careful investigation of the crystal structure (Fig. 5b) demonstrates that no bond exists between Ag and Bi in AgBiSe2 while a Cu–Bi bond with bond distance of 3.4 Å exists in CuBiS2. Hence, correlating the electronic and crystal structure of both the ordered compounds, i.e. AgBiSe2 and CuBiS2, we anticipate that facile charge transfer between Bi and Se in AgBiSe2 expedites easy adsorption of the C-bound *COOH intermediate, which rationalizes the higher FE for acetic acid in AgBiSe2 relative to that of CuBiS2.54,55
Since structural changes can happen at the reducing voltage in the CO2 reduction reaction, powder XRD patterns of the working electrodes were recorded immediately after the electrolysis at each potential to understand the active crystallographic phase during that potential. During the electrochemical measurements on AgBiX2 compounds, the formation of Ag2O was observed (Fig. S36a and b, ESI†) whereas CuBiS2 did not undergo any structural change (Fig. S36c, ESI†). AgBiSe2 has not changed its structure at any potential, but AgBiS2 became structurally ordered upon increasing the applied potential.
This strongly confirms the earlier explanation that AgBiS2 favors the production of both acetic acid and methanol at lower potential because of the presence of both ordered and disordered phases. Additionally, in both cases the evolution of a small amount of Ag2O facilitates CO2 to CO conversion, which is an intermediate step in acetic acid formation. Because of this additional reaction, we have higher FE towards acetic acid in the case of Ag compounds compared to Cu compounds. Post electrolysis XPS analysis was done to understand the changes in electronic structure under CO2RR electrolysis conditions (Fig. S37 and S38, ESI†). As evident from Fig. S39 (ESI†), Ag surfaces in ABiX2 catalysts undergo a shift to lower binding energies upon application of negative potential, which is a good indication of significant reduction of metallic surfaces during the eCO2RR.56 Besides selectivity and activity, stability is another crucial descriptor while evaluating the performance of electrocatalysts for the eCO2RR.
To minimize CO2 mass transport issues and achieve high current density we explored the CO2RR activity of the MBiX2 catalysts in a flow-cell configuration (Fig. S40a–c, ESI†) as compared to the H-cell configuration. Flow cells with a gas diffusion electrode (GDE) can enable efficient transport of CO2 to the gas–electrolyte–electrode interface and minimize the diffusion layer thickness to the nanometer scale.57,58 A flow-cell configuration helped in achieving current densities which are almost 4-times as compared to that in the H-cell59,60 (Fig. S41 and Table S5, ESI†). The flow-cell led to the formation of acetic acid and formic acid as the major liquid products as was observed from the H-cell. However, interestingly, product analysis showed a considerable reduction in H2 FE while the FE of acetic acid was improved significantly (Fig. 6). Our best catalyst, AgBiSe2 demonstrated acetic acid as the CO2 reduced product with FE as high as 49.81% at an ultra-low potential of −0.3 V (Fig. S6b, ESI†). The achieved FE for acetic acid from AgBiSe2 in the flow-cell was almost 4 times that achieved from the H-cell at a current density of 15.8 mA cm−2. The increased selectivity to acetic acid and decreased selectivity to hydrogen is likely due to the higher local pH in the flow cell microenvironment.61 This accelerates the OH-mediated nucleophilic attack of the ethenone intermediate leading to acetic acid.62 The maximum achieved FE for acetic acid at a current density of 15.8 mA cm−2 was found to be stable up to 40 hours in the flow-cell configuration (Fig. S42, ESI†).
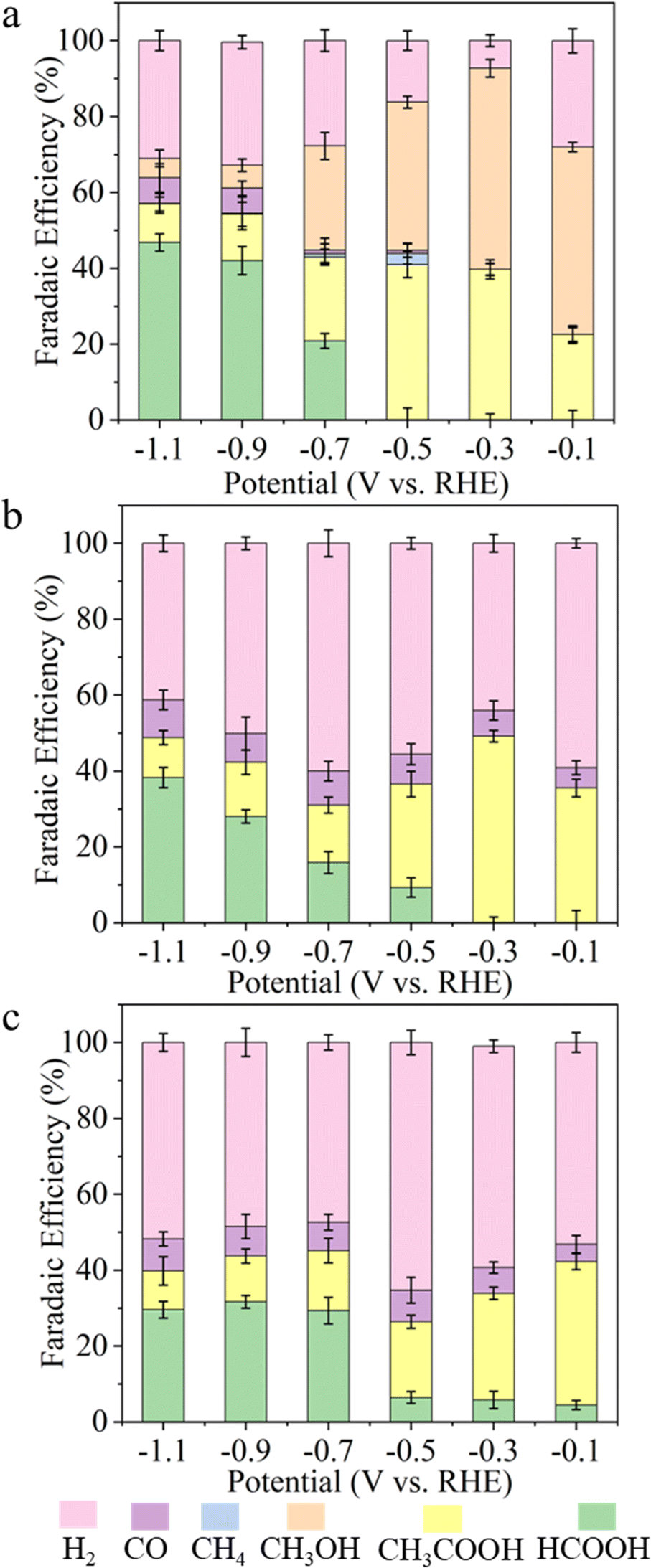 | ||
| Fig. 6 Faradaic efficiency for all the liquid and gaseous products found during CO2 reduction as a function of potential on (a) AgBiS2 and (b) AgBiSe2 and (c) CuBiS2 catalysts in the flow cell configuration in 0.5 M KHCO3. | ||
In conclusion, we have rationally designed a set of catalysts to tune the selective production of acetic acid from CO2 at lower potential. Our controlled studies clearly manifest that atomic ordering and optimized chemical bonding are very crucial parameters in controlling the reaction pathway to a desired product. We have chosen a couple of the most studied transition metals for CO2 reduction (Cu and Ag) and alloyed them with Bi and chalcogens, which helped in tuning the global and local structure. The atomically ordered AgBiS2 and AgBiSe2 compounds favored the selective production of acetic acid with significant FE at low overpotential. In situ IR studies mapped the reaction pathways with each step and important intermediates for the formation of products at various potentials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Department of Science and Technology (DST) (DST/TM/EWO/MI/CCUS/13(G) and DST/TMDEWO/CCUS/CoE/2020/JNCASR(c)), Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR) is gratefully acknowledged. SCP thanks DST for the Swarna Jayanti Fellowship (Grant Number: DST/SJF/CSA-02/2017-18). SS thanks JNCASR for her research fellowship. DB thanks UGC for his research fellowship. JR thanks IC3 for financial support.References
- S. C. Peter, ACS Energy Lett., 2018, 3, 1557–1561 CrossRef CAS.
- S. Roy, A. Cherevotan and S. C. Peter, ACS Energy Lett., 2018, 3, 1938–1966 CrossRef CAS.
- C. Xia, P. Zhu, Q. Jiang, Y. Pan, W. Liang, E. Stavitski, H. N. Alshareef and H. Wang, Nat. Energy, 2019, 4, 776–785 CrossRef CAS.
- S. Overa, B. H. Ko, Y. Zhao and F. Jiao, Acc. Chem. Res., 2022, 55, 638–648 CrossRef CAS.
- C. Chen, X. Yan, R. Wu, Y. Wu, Q. Zhu, M. Hou, Z. Zhang, H. Fan, J. Ma, Y. Huang, J. Ma, X. Sun, L. Lin, S. Liu and B. Han, Chem. Sci., 2021, 12, 11914–11920 RSC.
- D. Bagchi, S. Sarkar, A. K. Singh, C. P. Vinod and S. C. Peter, ACS Nano, 2022, 16, 6185–6196 CrossRef CAS.
- J. Han, C. Long, J. Zhang, K. Hou, Y. Yuan, D. Wang, X. Zhang, X. Qiu, Y. Zhu, Y. Zhang, Z. Yang, S. Yan and Z. Tang, Chem. Sci., 2020, 11, 10698–10704 RSC.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- Q. Zhu, X. Sun, D. Yang, J. Ma, X. Kang, L. Zheng, J. Zhang, Z. Wu and B. Han, Nat. Commun., 2019, 10, 3851 CrossRef.
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS.
- Y. Liu, S. Chen, X. Quan and H. Yu, J. Am. Chem. Soc., 2015, 137, 11631–11636 CrossRef CAS PubMed.
- A. Cherevotan, J. Raj, L. Dheer, S. Roy, S. Sarkar, R. Das, C. P. Vinod, S. Xu, P. Wells, U. V. Waghmare and S. C. Peter, ACS Energy Lett., 2021, 6, 509–516 CrossRef CAS.
- D. Bagchi, J. Raj, A. K. Singh, A. Cherevotan, S. Roy, K. S. Manoj, C. P. Vinod and S. C. Peter, Adv. Mater., 2022, 34, 2109426 CrossRef CAS.
- F. Yang, A. Chen, P. L. Deng, Y. Zhou, Z. Shahid, H. Liu and B. Y. Xia, Chem. Sci., 2019, 10, 7975–7981 RSC.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- K. D. Yang, C. W. Lee, K. Jin, S. W. Im and K. T. Nam, J. Phys. Chem. Lett., 2017, 8, 538–545 CrossRef CAS PubMed.
- S. Ajmal, Y. Yang, M. A. Tahir, K. Li, A.-U.-R. Bacha, I. Nabi, Y. Liu, T. Wang and L. Zhang, Catal. Sci. Technol., 2020, 10, 4562–4570 RSC.
- J. D. Medrano-García, R. Ruiz-Femenia and J. A. Caballero, Comput. – Aided Chem. Eng., 2019, 46, 145–150 Search PubMed.
- J. Nayak and P. Pal, A Green Process for Acetic Acid Production, 7th International Conference on Chemical, Ecology and Environmental Sciences (ICCEES’2015), Pattaya, Thailand, 2015pp. 47–51 DOI:10.13140/RG.2.1.4879.0883.
- A. D. Handoko, K. W. Chen and B. S. Yeo, ACS Energy Lett., 2017, 2, 2103–2109 CrossRef CAS.
- F. Franco, C. Rettenmaier, H. S. Jeon and B. Roldan Cuenya, Chem. Soc. Rev., 2020, 49, 6884–6946 RSC.
- Y. Hori, C. Vayenas, R. White and M. Gamboa-Aldeco, Electrochemical CO2 Reduction on Metal Electrodes, Springer, New York, 2008, pp. 89–189 Search PubMed.
- Z. Sun, Y. Hu, D. Zhou, M. Sun, S. Wang and W. Chen, ACS Energy Lett., 2021, 6, 3992–4022 CrossRef CAS.
- D. Karapinar, C. E. Creissen, J. G. Rivera de la Cruz, M. W. Schreiber and M. Fontecave, ACS Energy Lett., 2021, 6, 694–706 CrossRef CAS.
- J.-J. Lv, M. Jouny, W. Luc, W. Zhu, J.-J. Zhu and F. Jiao, Adv. Mater., 2018, 30, 1803111 CrossRef.
- Y. Xu, F. Li, A. Xu, J. P. Edwards, S.-F. Hung, C. M. Gabardo, C. P. O’Brien, S. Liu, X. Wang, Y. Li, J. Wicks, R. K. Miao, Y. Liu, J. Li, J. E. Huang, J. Abed, Y. Wang, E. H. Sargent and D. Sinton, Nat. Commun., 2021, 12, 2932 CrossRef CAS.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Phys. Chem. B, 2002, 106, 15–17 CrossRef CAS.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS.
- E. R. Cave, J. H. Montoya, K. P. Kuhl, D. N. Abram, T. Hatsukade, C. Shi, C. Hahn, J. K. Nørskov and T. F. Jaramillo, Phys. Chem. Chem. Phys., 2017, 19, 15856–15863 RSC.
- X. Zhu, J. Huang and M. Eikerling, ACS Catal., 2021, 11, 14521–14532 CrossRef CAS.
- W. Luo, J. Zhang, M. Li and A. Züttel, ACS Catal., 2019, 9, 3783–3791 CrossRef CAS.
- Y. Yang, M. Z. Ertem and L. Duan, Chem. Sci., 2021, 12, 4779–4788 RSC.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- B. Kumar, V. Atla, J. P. Brian, S. Kumari, T. Q. Nguyen, M. Sunkara and J. M. Spurgeon, Angew. Chem., Int. Ed., 2017, 56, 3645–3649 CrossRef CAS PubMed.
- A. Del Castillo, M. Alvarez-Guerra, J. Solla-Gullón, A. Sáez, V. Montiel and A. Irabien, J. CO2 Utili., 2017, 18, 222–228 CrossRef CAS.
- M.-Y. Lee, S. Ringe, H. Kim, S. Kang and Y. Kwon, ACS Energy Lett., 2020, 5, 2987–2994 CrossRef CAS.
- I. Grigioni, L. K. Sagar, Y. C. Li, G. Lee, Y. Yan, K. Bertens, R. K. Miao, X. Wang, J. Abed, D. H. Won, F. P. García de Arquer, A. H. Ip, D. Sinton and E. H. Sargent, ACS Energy Lett., 2021, 6, 79–84 CrossRef CAS.
- M. Nolan, ACS Omega, 2018, 3, 13117–13128 CrossRef CAS.
- A. Walsh, D. J. Payne, R. G. Egdell and G. W. Watson, Chem. Soc. Rev., 2011, 40, 4455–4463 RSC.
- R. J. Walker, A. Pougin, F. E. Oropeza, I. J. Villar-Garcia, M. P. Ryan, J. Strunk and D. J. Payne, Chem. Mater., 2016, 28, 90–96 CrossRef CAS.
- W. Ma, S. Xie, X.-G. Zhang, F. Sun, J. Kang, Z. Jiang, Q. Zhang, D.-Y. Wu and Y. Wang, Nat. Commun., 2019, 10, 892 CrossRef PubMed.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS.
- R. De, S. Gonglach, S. Paul, M. Haas, S. S. Sreejith, P. Gerschel, U.-P. Apfel, T. H. Vuong, J. Rabeah, S. Roy and W. Schöfberger, Angew. Chem., Int. Ed., 2020, 59, 10527–10534 CrossRef CAS PubMed.
- B. Zha, C. Li and J. Li, J. Catal., 2020, 382, 69–76 CrossRef CAS.
- X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, J. Ma, Z. Zhang, G. Yang and B. Han, Green Chem., 2017, 19, 2086–2091 RSC.
- S. Zhu, T. Li, W.-B. Cai and M. Shao, ACS Energy Lett., 2019, 4, 682–689 CrossRef CAS.
- N. J. Firet and W. A. Smith, ACS Catal., 2017, 7, 606–612 CrossRef CAS.
- C. Genovese, C. Ampelli, S. Perathoner and G. J. G. C. Centi, Green Chem., 2017, 19, 2406–2415 RSC.
- K. Kunimatsu, T. Senzaki, G. Samjeské, M. Tsushima and M. Osawa, Electrochim. Acta, 2007, 52, 5715–5724 CrossRef CAS.
- S. Zhu, B. Jiang, W.-B. Cai and M. Shao, J. Am. Chem. Soc., 2017, 139, 15664–15667 CrossRef CAS PubMed.
- M. C. Figueiredo, I. Ledezma-Yanez and M. T. M. Koper, ACS Catal., 2016, 6, 2382–2392 CrossRef CAS.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato, R. Haasch, P. Zapol, B. Kumar, R. F. Klie, J. Abiade, L. A. Curtiss and A. Salehi-Khojin, Science, 2016, 353, 467 CrossRef CAS.
- K. Chan, C. Tsai, H. A. Hansen and J. K. Nørskov, ChemCatChem, 2014, 6, 1899–1905 CrossRef CAS.
- N. Martić, C. Reller, C. Macauley, M. Löffler, A. M. Reichert, T. Reichbauer, K.-M. Vetter, B. Schmid, D. McLaughlin, P. Leidinger, D. Reinisch, C. Vogl, K. J. J. Mayrhofer, I. Katsounaros and G. Schmid, Energy Environ. Sci., 2020, 13, 2993–3006 RSC.
- Y. Luo, K. Zhang, Y. Li and Y. Wang, InfoMat, 2021, 3, 1313–1332 CrossRef CAS.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12, 136 CrossRef CAS PubMed.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- Z. Zhang, L. Melo, R. P. Jansonius, F. Habibzadeh, E. R. Grant and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 3101–3107 CrossRef CAS.
- W. Luc, X. Fu, J. Shi, J.-J. Lv, M. Jouny, B. H. Ko, Y. Xu, Q. Tu, X. Hu, J. Wu, Q. Yue, Y. Liu, F. Jiao and Y. Kang, Nat. Catal., 2019, 2, 423–430 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional characterization, CO2RR polarization curves, NMR spectra, IR spectra and table. See DOI: https://doi.org/10.1039/d2ey00081d |
| This journal is © The Royal Society of Chemistry 2023 |