
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZSM-5-confined Cr1–O4 active sites boost methane direct oxidation to C1 oxygenates under mild conditions†
Mengyu
Zeng‡
ab,
Lu
Cheng‡
cd,
Qingqing
Gu‡
e,
Bing
Yang
 e,
Baiyang
Yu
ab,
Jing
Xu
f,
Ying
Zhang
ab,
Chengsi
Pan
ab,
Xiao-Ming
Cao
*cd,
Yang
Lou
*ab and
Yongfa
Zhu
g
e,
Baiyang
Yu
ab,
Jing
Xu
f,
Ying
Zhang
ab,
Chengsi
Pan
ab,
Xiao-Ming
Cao
*cd,
Yang
Lou
*ab and
Yongfa
Zhu
g
aKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi, Jiangsu 214122, China. E-mail: yang.lou@jiangnan.edu.cn
bInternational Joint Research Center for Photoresponsive Molecules and Materials, Jiangnan University, Wuxi, Jiangsu 214122, China
cKey Laboratory for Advanced Materials and Feringa Nobel Prize Scientist Joint Research Center, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai, 200237, China. E-mail: xmcao@ecust.edu.cn
dCentre for Computational Chemistry and Research Institute of Industrial Catalysis, East China University of Science and Technology, Shanghai, 200237, China
eDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, 457 Zhongshan Road, Dalian, 116023, China
fSchool of Food Science and Technology, Jiangnan University, Wuxi, Jiangsu 214122, China
gDepartment of Chemistry, Tsinghua University, Beijing 100084, China
First published on 28th November 2022
Abstract
Direct oxidation of methane (DOM) to value-added chemicals is of great significance but is still challenging under mild conditions. Herein, we report that Cr1–O4 as an active site in a ZSM-5 anchored Cr single atom catalyst (Cr1/ZSM-5 SAC) can efficiently catalyse the DOM to form value-added C1 oxygenated products with a productivity of 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 μmol gcat−1 h−1 and a selectivity of 99.8% at 50 °C within 30 min, which outperforms most reported state-of-the-art catalysts. Density functional theory (DFT) calculations and experimental results show that Cr1 atoms anchored on the wall of ZSM-5 micropores form the Cr1–O4 active sites, which boosts the formation of the reactive oxygen species (adsorbed OH species) and the activation of a C–H bond in CH4. We believe that our atomic-level design strategy on a non-noble metal offers an approach to rationally design efficient catalysts for methane conversion.
100 μmol gcat−1 h−1 and a selectivity of 99.8% at 50 °C within 30 min, which outperforms most reported state-of-the-art catalysts. Density functional theory (DFT) calculations and experimental results show that Cr1 atoms anchored on the wall of ZSM-5 micropores form the Cr1–O4 active sites, which boosts the formation of the reactive oxygen species (adsorbed OH species) and the activation of a C–H bond in CH4. We believe that our atomic-level design strategy on a non-noble metal offers an approach to rationally design efficient catalysts for methane conversion.
Broader contextTransforming methane to high-value oxygenated products (like CH3OH, HCCOH etc.) is highly attractive. Traditionally, methane conversion can be achieved through an indirect process that couples methane reforming and a Fischer–Tropsch synthesis but such an indirect route normally requires high pressures (up to 30 bar) and temperatures (1100–1300 K) with large energy inputs. Alternatively, the direct oxidation of methane (DOM) to form value-added oxygenates is considered as a potential way to efficiently and economically convert methane into commercial chemicals and fuels but remains a major challenge to simultaneously obtain high activity and selectivity. In this present work, we report a ZSM-5 supported non-noble Cr single atom catalyst as a proof-of-concept catalyst (Cr1/ZSM-5 SAC) with a unique Cr1–O4 entity as the active site to selectively and efficiently convert methane to HCOOH and other value-added C1 oxygenates under mild conditions. |
Introduction
Transforming methane (the main composition of natural gas) into high-value oxygenated products (formic acid, methanol, etc.) is highly attractive.1–3 Traditionally, methane conversion can be achieved through an indirect process that couples methane reforming and a Fischer–Tropsch synthesis4 but such an indirect route normally requires high pressures (up to 30 bar) and temperatures (1100–1300 K) with large energy inputs.5 Alternatively, the direct oxidation of methane (DOM) to value-added oxygenated products by selective oxidation of CH4 is a more economical and environmentally friendly route than that of the indirect route.6 However, the DOM remains highly challenging since it is very difficult to balance the activation of a C–H bond in CH4 with the oxidation capability of the active oxygen species. As a result, the practical application of the DOM is inhibited by the low productivity and selectivity. Hence, designing novel catalysts with high productivity and selectivity for the DOM is highly important.Single-atom catalysts (SACs) that possess fully exposed active atoms and high homogeneity of the active site enable a significant boost in the selective conversion of various chemical transformations compared with nanocluster and nanoparticle catalysts.7,8 Especially, the M1–Ox entity stabilized by the oxygen moieties of appropriate supports as active sites possesses a unique catalytic capability for methane selective conversion.9–11 For example, the Pd1–O4 entity anchored on the internal surface of microporous ZSM-5 can convert CH4 to methanol or methyl peroxide.10 The Rh1–O5 entity decorated in ZSM-5 frameworks can efficiently catalyse CH4 to CH3COOH and CH3OH by introducing CO and O2 at 150 °C, which is around 1000 times higher than that of unsupported Rh cations.11 However, non-noble metal based catalysts for the DOM with excellent catalytic performance under mild reaction are still rarely reported.12,13
Herein, we report that atomically-dispersed chromium sites anchored on ZSM-5 can efficiently catalyse the DOM and achieve a yield of 21100 μmol gcat.−1 h−1 with a selectivity of 99.8% for value-added C1 oxygenated products (HCOOH, CH3OH, CH3OOH and CH2(OH)2) with H2O2 as the oxidant at 50 °C, which outperforms most reported state-of-the-art catalysts under similar reaction conditions (Table S1, ESI†). DFT and experimental results indicate that Cr1–O4 as the active site of the Cr1/ZSM-5 SACs promotes the formation of the reactive oxygen species (adsorbed OH group) and the activation of a C–H bond in CH4, thereby enhancing the yield of the DOM.
Results and discussion
The Cr1/ZSM-5 SAC was synthesized via a modified adsorption method by finely tuning the adsorption parameters of the metal precursors in the ethanol solution (details shown in the ESI†).14 The XRD patterns of the Cr1/ZSM-5 SAC suggest that the original crystal structure of H-ZSM-5 is maintained, indicating that the metal species do not change the lattice structure of H-ZSM-5 (Fig. S1, ESI†). In addition, no characteristic peaks of Cr particles are observed from XRD, which reveals that the chromium species are highly dispersed in H-ZSM-5. To probe the morphology and structure information of the Cr1/ZSM-5 SAC at the atomic level, aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was used. The low-dose HAADF-STEM images indicate that isolated Cr atoms are uniformly dispersed on ZSM-5 (Fig. 1a). No Cr particles or clusters can be observed in the fresh Cr1/ZSM-5 SAC as shown in the low magnification HAADF-STEM images (Fig. S2, ESI†). The energy dispersive spectroscopy (EDS) elemental maps further confirm that the Cr species are atomically dispersed in selected regions of the Cr1/ZSM-5 SAC (Fig. 1c and d). By examining numerous low/high magnification HAADF-STEM images and back-scattered electron images obtained from different areas of the Cr1/ZSM-5 SAC, we conclude that the as-synthesized Cr1/ZSM-5 SAC contains only isolated Cr atoms that are anchored onto ZSM-5. For the used Cr1/ZSM-5 SAC (50 °C for 0.5 h), the atomically dispersed Cr species are still retained and no Cr clusters and nanoparticles are observed (Fig. 1b and Fig. S3, S4, ESI†), which reveals that the Cr1 atoms are stably anchored on ZSM-5 and also supports the idea that the measured catalytic activity is originally from the isolated Cr atoms rather than the Cr clusters or particles. | ||
| Fig. 1 Catalytic structural characterizations of the Cr1/ZSM-5 SAC in methane oxidation under mild conditions. HAADF-STEM images of the synthesized Cr1/ZSM-5 SAC at different magnifications (a–c) and once it had been used (b). The blue circles show single Cr atoms in the matrix of ZSM-5. Energy dispersive spectroscopy (EDS) mapping images of the Cr1/ZSM-5 SAC in figure (d). | ||
For comparison studies, a Cr/ZSM-5-NP sample composed of Cr particles dispersed on ZSM-5 was prepared (with a loading of 1.33 wt%). The average size of the Cr particles was 3.0 ± 0.6 nm, and the spatial distributions of the Cr particles in the Cr/ZSM-5-NP are shown in Fig. S5 (ESI†).
To further explore the electronic state of Cr species, UV-Vis diffuse reflectance spectroscopy (DRS) was conducted as shown in Fig. S6 (ESI†). For the Cr/ZSM-5 NP, the broad absorption bands at ∼458 nm and ∼600 nm are associated with the A2g–T1g and A2g–T2g transitions of Cr(III) in octahedral symmetry respectively, which is the typical characteristic absorption band of the octahedral Cr3+ species in CrOx clusters or in Cr2O3.15–17 The absence of such an absorption band in the Cr1/ZSM-5 SAC indicates that the Cr species are atomically dispersed on ZSM-5 (Fig. S2, ESI†), which corroborates the HAADF-STEM and elemental mapping results.
To investigate the chemical structure and electronic states of the Cr species in the Cr1/ZSM-5 SAC, Fourier transform extended X-ray absorption fine structure (FT-EXAFS) in R space was performed (Fig. 2a). To accurately reveal the local coordination environment of the Cr species in the Cr1/ZSM-5 SAC, the standard crystal model for the DFT simulations was used to fit the EXAFS data. The fitted data fits the original EXAFS data of Cr species well, which secures that the fitted results are reliable and enable us to accurately demonstrate the local structure of the Cr species in the Cr1/ZSM-5 SAC. There is only a Cr–O peak located at 1.50 Å for the Cr1/ZSM-5 SAC, without any Cr–Cr or Cr–O–Cr peaks (from 2.00 Å to 3.00 Å), which suggests that the Cr1/ZSM-5 SAC mainly contains atomically dispersed Cr atoms instead of nanoparticles or multi-core clusters, and further verifies the results of the HAADF-STEM, elemental mapping and UV-vis DRS. In addition, the detailed fitting data reveals that each Cr1 atom is coordinated to four oxygen atoms (coordination number: 3.9 ± 0.1) as shown in Fig. 2b. Therefore, the Cr1–O4 configuration describes the nearest-neighbor environment of the Cr1 atoms in the Cr1/ZSM-5 SAC. The fitting details are displayed in Table S2 (ESI†).
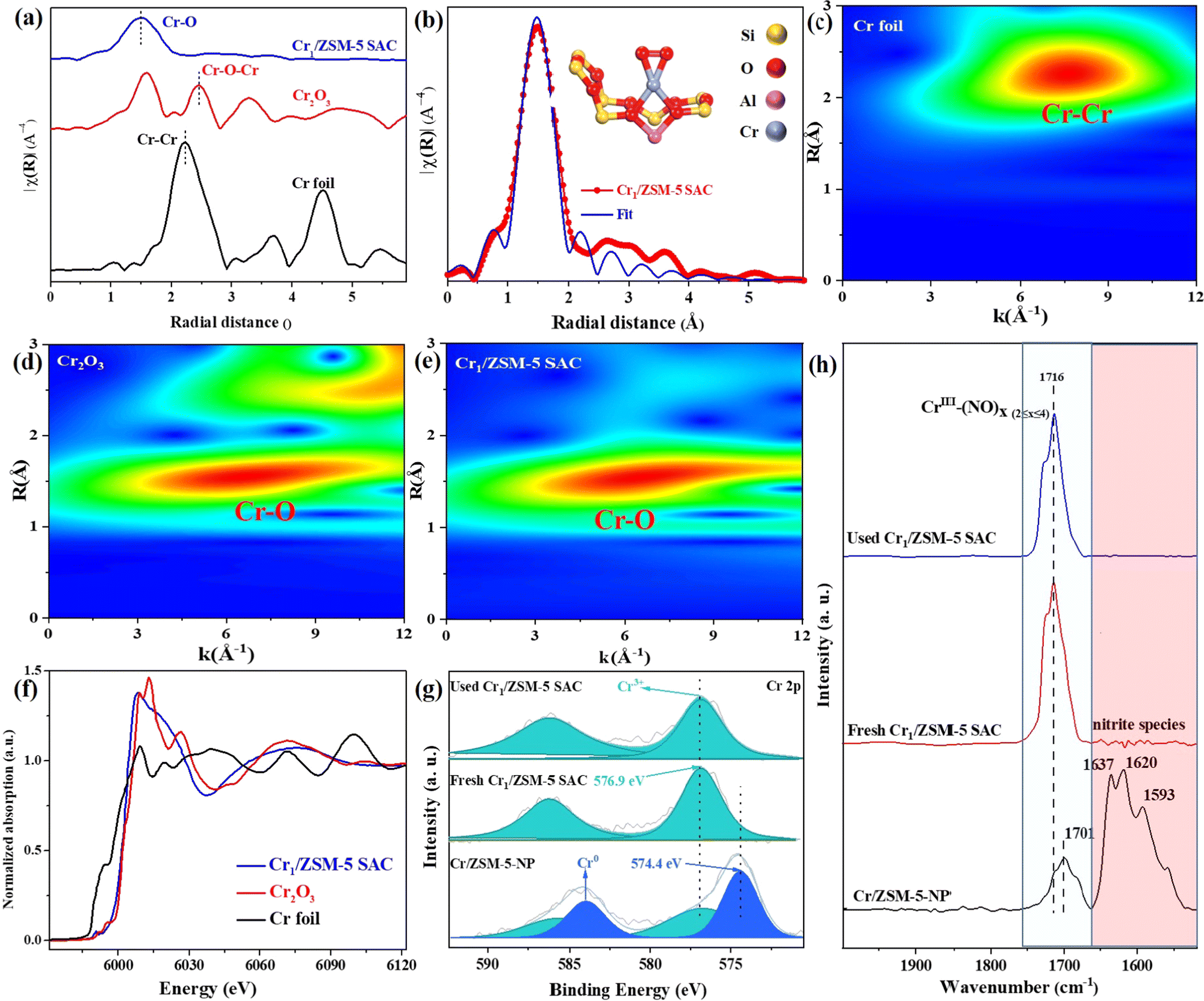 | ||
| Fig. 2 Coordination structure and chemical state of the Cr/ZSM-5 catalysts. (a) The k3-weighted Cr K-edge Fourier transform EXAFS spectra of the Cr1/ZSM-5 SAC as well as Cr foil and Cr2O3 references. (b) Experimental and fitting Cr K-edge EXAFS curves of the Cr1/ZSM-5 SAC. (c) In situ NO-DRIFTS spectra adsorbed on Cr/ZSM-5-NP, the fresh Cr1/ZSM-5 SAC and the used Cr1/ZSM-5 SAC at 30 °C. (d) Cr K-edge X-ray absorption near-edge structure (XANES) spectra. (c) Cr 2p XPS peak of the fresh Cr1/ZSM-5 SAC, the used Cr1/ZSM-5 SAC after reaction and Cr/ZSM-5 NP. The WT-EXAFS plot of Cr foil (f), Cr2O3 (g), and the Cr1/ZSM-5 SAC (h). | ||
To reveal the atomic dispersion of the Cr species in the Cr1/ZSM-5 SAC more clearly, a wavelet transform (WT) of the Cr K-edge EXAFS oscillations was analyzed. For the Cr1/ZSM-5 SAC, the strong WT maximum focuses at approximately 6.0 Å−1 (Fig. 2c–e), which clearly distinguishes it from WT contour plots of Cr foil (Cr–Cr, k = 8.0 Å−1) and Cr2O3 (Cr–O, k = 6.3 Å−1). These results further confirm that the Cr species are atomically dispersed on the Cr1/ZSM-5 SAC.
To probe the electronic states of the isolated Cr species in the Cr1/ZSM-5 SAC, the X-ray absorption near edge structure (XANES) is investigated. The white-line intensity of the Cr species in the Cr1/ZSM-5 SAC is slightly higher than that of Cr foil (Fig. 2f), which reveals that the Cr1 atoms are in an oxidative state.18 Moreover, the absorption edge of the Cr1/ZSM-5 SAC is higher than that of Cr foil and close to that of Cr2O3, which further indicates that the Cr species are in a high oxidation state (close to +3). X-ray photoelectron spectroscopy (XPS) was further used to probe the oxidation state of isolated Cr species (Fig. 2e). The detailed analysis of the XPS data indicates that the oxidation state of the Cr1 atoms in the Cr1/ZSM-5 SAC is +3 (peak centered at 576.9 eV in the spectra of Cr 2p3/2), which is in line with the XANES data. The Cr species in the Cr/ZSM-5 NP sample possess two oxidation states:18 a peak centered at 576.9 eV, assignable to Cr3+, and a peak centered at 574.4 eV, assignable to Cr0. The XPS data clearly confirms that the electronic state of the isolated Cr species in the Cr1/ZSM-5 SAC is obviously different from that of the Cr particles/clusters in Cr/ZSM-5-NP. Furthermore, the binding energy of the Cr species in the used Cr1/ZSM-5 SAC is similar to that of Cr species in the fresh Cr1/ZSM-5 SAC, which further verifies that the Cr1 atoms are stably anchored on ZSM-5.
In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) of NO adsorption was used to further identify the microscopic properties of the active sites in the Cr/ZSM-5 samples. For NO adsorption on the Cr/ZSM-5-NP sample (Fig. 2f), the band centered at 1701 cm−1 is associated with the stretching mode of NO on the isolated Cr sites (Cr1–(NO)x≥2 nitrosyl species)19–21 and the peaks centered at 1637 cm−1 (bridge nitrate), 1620 cm−1 (monodentate nitrates) and 1593 cm−1 (bidentate nitrate) are associated with the bridged adsorption mode of NO (bridging nitrate) on the multiple Cr sites.22–25 These results corroborate the TEM imaging results that the Cr species exist mainly in the form of nanometer-sized Cr particles (3.0 ± 0.6 nm) dispersed on ZSM-5 (Fig. S6, ESI†). For NO adsorption on the fresh Cr1/ZSM-5 SAC sample, the peak at 1716 cm−1 corresponds to the NO adsorption on isolated Cr sites (forming Cr1–(NO)x≥2 nitrosyl species)19–21 and the bridged adsorption mode of NO is absent, which indicates that the Cr species are mainly in the form of single atoms in the Cr1/ZSM-5 SAC. Compared with the Cr/ZSM-5-NP sample, the obvious blueshift of Cr1–(NO)x≥2 nitrosyl species by 15 cm−1 on the Cr1/ZSM–5 SAC reveals that the Cr1 atoms possess a much higher oxidation state, which is in line with the XPS and XANES data. For NO adsorption over the used Cr1/ZSM-5 SAC, there is only one peak at 1716 cm−1 associated with the NO adsorption on isolated Cr sites and the bridging nitrate associated with the NO molecules adsorbed on multiple Cr sites is not observed (Fig. 2c), which further confirms that the Cr1 atoms are stably anchored on ZSM-5.
To investigate the catalytic performance of the Cr1/ZSM-5 SAC, the DOM is conducted in a batch reactor with H2O2 as the oxidant. By optimizing the DOM reaction conditions including Cr loading, temperature, methane pressure and amounts of H2O2, the highest catalytic performance was achieved for the Cr1/ZSM-5 SAC at 3 MPa and 50 °C under the reaction conditions of 0.5 M H2O2 (Fig. S7, ESI†). For the DOM over the Cr1/ZSM-5 SAC, the productivity of the C1 oxygenates that are mainly composed of HCOOH, CH3OH, CH3OOH and CH2(OH)2 is 21, 100 μmol gcat.−1 h−1 with a total selectivity of 99.8% in all the products at 50 °C within 30 min (Fig. 3a), which surpasses most state-of-the-art catalysts reported in the literature (Table S1, ESI†). The compositions of gas-phase and liquid-phase products were analyzed and quantified by gas chromatography and nuclear magnetic resonance (Fig. S8 and S9, ESI†). The productivity of the C1 oxygenates over the Cr1/ZSM-5 SAC is around 1.5 times higher than that of the highest catalyst (Cu–Fe/ZSM-5) under similar reaction conditions (50 °C) in the literature.26
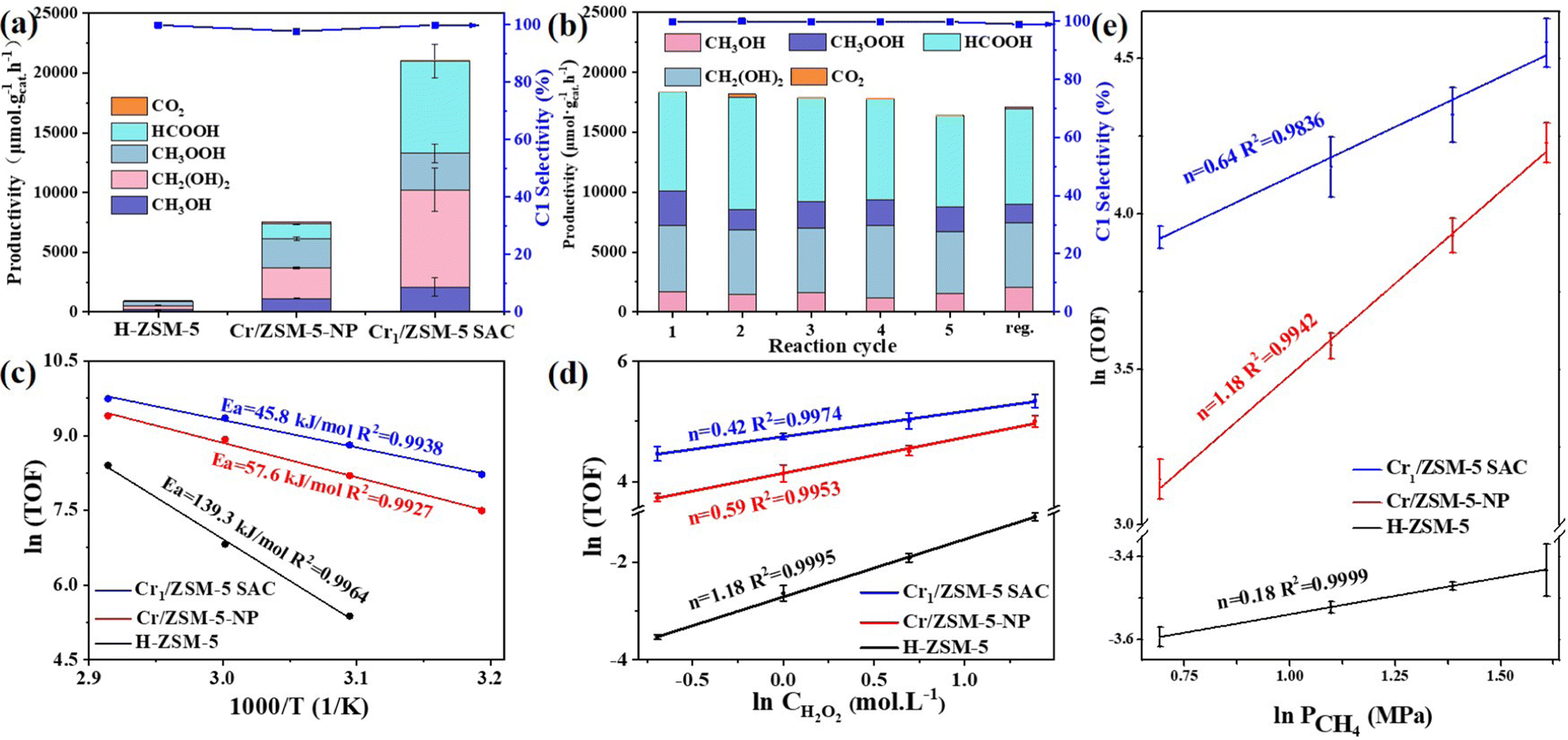 | ||
| Fig. 3 Catalytic performances and kinetic parameters of the Cr/ZSM-5 samples for the DOM. The product distribution and selectivity of the DOM on pure H-ZSM-5, Cr/ZSM-5-NP and the Cr1/ZSM-5 SAC (a); recycle performance and regeneration of the Cr1/ZSM-5 SAC (b). (c) Arrhenius plots (apparent activation energy, Ea) of the Cr1/ZSM-5 SAC (blue line), Cr/ZSM-5-NP (red line) and pure H-ZSM-5 (black line) in the temperature (T) range of 40 to 70 °C with 10 °C increments. (d) Reaction order (n) for the H2O2 on the Cr1/ZSM-5 SAC (blue line), Cr/ZSM-5-NP (red line) and pure H-ZSM-5 (black line) in a H2O2 initial concentration range of 0.5 to 4 M. (e) Reaction order (n) for the CH4 on the Cr1/ZSM-5 SAC (blue line), Cr/ZSM-5-NP (red line) and pure H-ZSM-5 (black line) in a CH4 pressure range of 20 to 50 bars with 10 bars increments. (Reaction condition: 10 mg catalysts; 40–70 °C; P(CH4) of 20–50 bars; 0.5–4 M H2O2; 0.5 h; 10 mL; 1,500 rpm.) The error bars are defined as standard deviations of three experiments. | ||
For the DOM reaction over H-ZSM-5 at 50 °C (Fig. 3a), the productivity of the C1 oxygenates is only 432 μmol gcat.−1 h−1 within 30 min and the major product is methyl hydroperoxide, around 48.9 times lower than that of the Cr1/ZSM-5 SAC. To verify the existing form of the active Cr species, the DOM was conducted on commercial Cr2O3. The productivity of commercial Cr2O3 for C1 oxygenates that are mainly composed of methyl hydroperoxide is approximately 420 μmol gcat−1 h−1 at 50 °C within 30 min (Fig. S9 and Table S1, ESI†), about 50 times lower than that of the Cr1/ZSM-5 SAC, which indicates that the Cr1 species are the main active species. For the DOM over Cr/ZSM-5-NP, the productivity of the C1 oxygenates that are mainly composed of CH3OOH and CH2(OH)2 is 7249 μmol gcat.−1 h−1 with a total selectivity of 96.8% in all of the products at 50 °C within 30 min, about 3 times lower than that of the Cr1/ZSM-5 SAC. Moreover, the turnover frequencies (TOFs) defined as the moles of the C1 oxygenates generated on one mole of Cr per hour are used to further characterize the intrinsic activity of the Cr species in the Cr/ZSM-5 samples.27 The corresponding TOFs of the Cr species in Cr/ZSM-5-NP are 7.0 times lower than that of the Cr1/ZSM-5 SAC (14.2 h−1vs. 99.8 h−1) at 50 °C within 30 min (Fig. S10, ESI†), which further indicates that the Cr1 species possess a much higher intrinsic activity for the DOM compared with nanometer-sized Cr species.
To demonstrate the efficiency of H2O2 utilization in the reaction, a gain factor defined as the molar ratio of formed C1 oxygenate products and consumed hydrogen peroxide28 was measured as the standard (Fig. S11, ESI†). The gain factor of the Cr1/ZSM-5 SAC (0.16) is 3.2-fold higher than that of Cr/ZSM-5-NP (0.05) at 50 °C. These results indicate that the Cr1–O4 as the active site in the Cr1/ZSM-5 SAC significantly enhances the activation process of the oxygen species. In addition, no significant decrease in the selectivity and yield of the C1 oxygenates on the Cr1/ZSM-5 SAC can be observed after five consecutive DOM cycles and the performance can be easily recovered after a simple calcination treatment (Fig. 3b), which indicates that the Cr1–O4 as the active site in the Cr1/ZSM-5 SAC is highly stable for the DOM (Fig. S12, ESI†).
To better understand the kinetic behavior of the Cr1/ZSM-5 SAC, the apparent activation energy (Ea) has been investigated. Under the conditions of kinetic control, the Ea of the Cr1/ZSM-5 SAC (45.8 kJ mol−1) is much lower than that of Cr/ZSM-5-NP (57.6 kJ mol−1) and pure H-ZSM-5 (139.3 kJ mol−1) as shown in Fig. 3c, which indicates that the catalytic process of the DOM is intrinsically boosted on the Cr1/ZSM-5 SAC compared with that of Cr/ZSM-5-NP and H-ZSM-5. The reaction orders of CH4 and H2O2 over the Cr1/ZSM-5 SAC (0.64 for CH4 and 0.42 for H2O2) are significantly different from that over Cr/ZSM-5-NP (1.18 for CH4 and 0.59 for H2O2) and pure H-ZSM-5 (0.18 for CH4 and 1.18 for H2O2) as shown in Fig. 3d, e and Table S3 (ESI†), which further reveals that the presence of the Cr1 atoms significantly modulates the catalytic process of the DOM compared with that of Cr/ZSM-5-NP and pure H-ZSM-5.
We further computationally investigated the reaction mechanism of the DOM over the Cr1/ZSM-5 SAC. After optimizing the structure of ZSM-5 (Fig. S13, ESI†), a series of potential mononuclear active sites coordinated with two framework oxygen anions (Ofm, Fig. S14, ESI†) were studied, including Z[CrO2]+, Z[Cr]+, Z[CrO]+, Z[Cr(OH)2]+, Z[CrOH]+, and Z[Cr]2+. The sites of Z[Cr]+, Z[Cr]2+, Z[CrO]+, and Z[CrOH]+, which are not compatible with either the 4-fold coordinated Cr as confirmed by the EXAFS data or the trivalence of Cr as confirmed by the XPS results, were firstly excluded from the list of active site. The Z[Cr(OH)2]+ was also excluded because of its low reactivity. The charge density analysis further revealed that both hydroxyls at the Cr sites of Z[Cr(OH)2]+ are OH− due to the strong electron-donating ability of the Cr species (Fig. S15 and Table S5, ESI†). The corresponding free energy barrier for the first C–H bond activation of methane is as high as 1.78 eV, which is formidable to activate a C–H bond of methane at the reaction temperature of 323 K. Moreover, even if the dissociative chemisorption of H2O2 could occur at the Z[Cr(OH)2]+ site under the reaction conditions to generate Z[Cr(OH)4]+, all of the hydroxyls at the Cr atom would be OH− (Table S5, ESI†). The corresponding free energy barrier of the first C–H bond activation of methane is also as high as 1.44 eV, which is still insurmountable to activate methane. Hence, the sites of Z[Cr(OH)2]+ and Z[Cr(OH)4]+ were excluded because of their low reactivity.
Alternatively, the Z[CrO2]+ site corresponds to an oxygen molecule chemisorbed at its Cr center via a π configuration, enabling the Cr1 atom to be coordinated with two oxygen adatoms and two Ofm to form the Cr1–O4 coordination structure as presented in the inset picture of Fig. 2b. The O–O bond length of the chemisorbed O2 is 1.424 Å, indicating that the O2 presents as peroxide O22− on Z[CrO2]+.29–32 The spin charge density analysis further confirms that the peroxide O22− and Cr3+ would exist at Z[CrO2]+ (Fig. S15b, ESI†) since [CrO2] needs to compensate a charge to the skeleton of the zeolite. It is in line with the XPS, XANES and NO-DRIFTS data. Based on the above experimental and DFT simulation results, the Z[CrO2]+ site represents the local environment of the Cr1 active site in the Cr1/ZSM-5 SAC well and a mechanistic study was performed at the Z[CrO2]+ site.
The DFT theoretical calculations prove that the reaction mechanism of the DOM by the Cr1/ZSM-5 SAC can be divided into three stages (Fig. S16, ESI†): (i) pre-activation of the reaction site, (ii) oxidation of methane, and (iii) regeneration of the active sites. The first key step of the DOM using H2O2 as oxidant at Z[CrO2]+ is the pre-activation of the reaction sites from Z[CrO2]+ to Z[Cr(O)2(OH)2]+ with H2O2 as shown in Fig. 4a. H2O2 is likely to be intrinsically activated by the Z[CrO2]+ site. It can chemisorb at the Z[CrO2]+ site with a free adsorption energy of −0.52 eV followed by its O–O bond dissociation with a free energy barrier of 0.77 eV. Meanwhile, the Z[CrO2]+ (IM1) turns into Z[CrO2(OH)2]+ (IM3). Interestingly, the presence of water molecules could significantly accelerate the H2O2 activation. As shown in Fig. 4a, in the presence of H2O, the Z[CrO2]+ site preferentially chemisorbs the H2O with a stronger free adsorption energy of −0.87 eV. Then, the adsorbed H2O can rapidly assist the O–O bond activation of H2O2 through H migration. It only needs to overcome a slight free energy barrier of 0.16 eV to generate the Z[CrO2(OH)2]+ (IM3) sites. Due to the strong electron-donating ability of Cr, the 4-fold Cr center is oxidized to the +5 valence state in the Z[CrO2(OH)2]+ configuration, where two adsorbed hydroxyl anions (OH−*) and two oxygen adatoms (O22−*) are coordinated with the isolated Cr center. After forming the Z[CrO2(OH)2]+ configuration, the O–O bond of O22−* at IM3 can be further broken by overcoming a free energy barrier of 1.07 eV (TS2). The addition of H2O2 lowers the free energy barrier by 0.14 eV (1.21 eV at IM1), indicating that the addition of H2O2 boosts the O22−* dissociation. After the O22−* dissociation, the formation of two O2− anions results in the further oxidation of the Z[CrO2(OH)2]+ configuration (IM3) to Cr6+, which forms the Z[Cr(O)2(OH)2]+ configuration (IM4). The valence states of the chromium cations in these key intermediates are further demonstrated by Bader charge analysis as listed in Table S6 (ESI†). The Raman results fully support the dynamic transformation of Cr3+ to Cr6+ during the catalytic process of the DOM. The Cr6+ species (Raman characteristic peak at 875 cm−1) would appear once the H2O2 interacts with the fresh and used Cr1/ZSM-5 SACs (Fig. S17, ESI†), which indicates that the presence of H2O2 enables the efficient oxidization of the Cr3+ species to the active Cr6+ species. The Bader charge density analysis (Fig. S15c and Table S5, ESI†) demonstrates that the formation of two O2− anions renders one OH−* at Z[CrO2(OH)2]+ (IM3) to be the radical state of OH˙* at Z[Cr(O)2(OH)2]+ (IM4), which could be the reactive oxygen species for the DOM. It is very clear that the presence of H2O2 plays a crucial role in the O–O bond dissociation of O22−* and the formation of the reactive oxygen species.
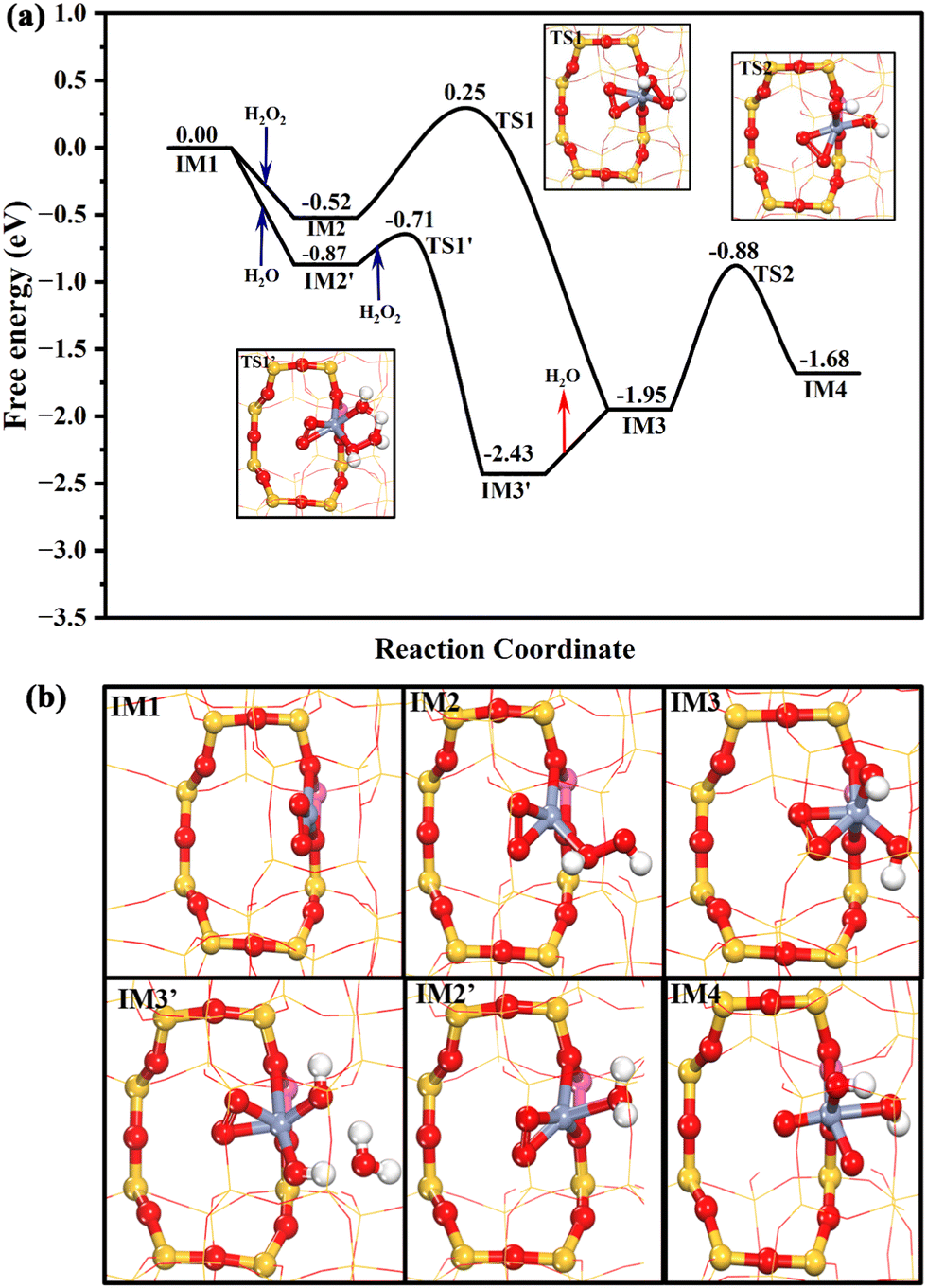 | ||
| Fig. 4 The Gibbs free energy profile for the pre-activation of the reaction site over the Cr1/ZSM-5 SAC with the geometry structures of key transition states (a); the geometry structures of all the intermediates in the whole catalytic cycle (b). The Gibbs energy profile was calculated under the reaction conditions of 30 bar CH4, 0.5 M H2O2 at 323 K. The H, O, Si, Al, and Cr atoms of the reaction sites are displayed in white, red, yellow, pink, and blue in a ball and stick style, respectively. The display settings were kept the same throughout the whole paper. | ||
Due to the formation of Cr6+, the reactive OH˙* is generated. As shown in Fig. 5, the OH˙* could significantly promote the CH4 activation. The first C–H bond of CH4 is activated by the OH˙* of IM4 through a radical-like mechanism,33,34 which only needs to climb over a free energy barrier of 0.68 eV (TS3). It is in good agreement with the previous reports that hexavalent Cr could be highly active.35,36 After methane activation, the generated free methyl radical could be readily captured by the O2−* at Z[Cr(O)2(OH)]+ (IM6) to form Z[Cr(O)(CH3O)(OH)]+ (IM7), which is significantly exothermic by 1.88 eV. Then, the OH* at IM7 could abstract one H atom of CH3O* to form H2O* and HCHO* at Z[Cr(O)(CH2O)(H2O)]+ (IM8) by overcoming a free energy barrier of 1.02 eV. Subsequently, the HCHO and H2O could be desorbed from the Cr center in sequence, which are respectively endothermic by 0.58 and 0.50 eV. Meanwhile, the Z[CrO]+ (IM10) is formed and the valence state of the Cr species returns to trivalent, which is consistent with the phenomenon detected by XPS (Fig. 2e). Hence, it is clear that the conversion of methane to formaldehyde catalyzed by the Cr1/ZSM-5 SAC depends on the formation of hexavalent Cr ions and the surface reactive oxygen species.
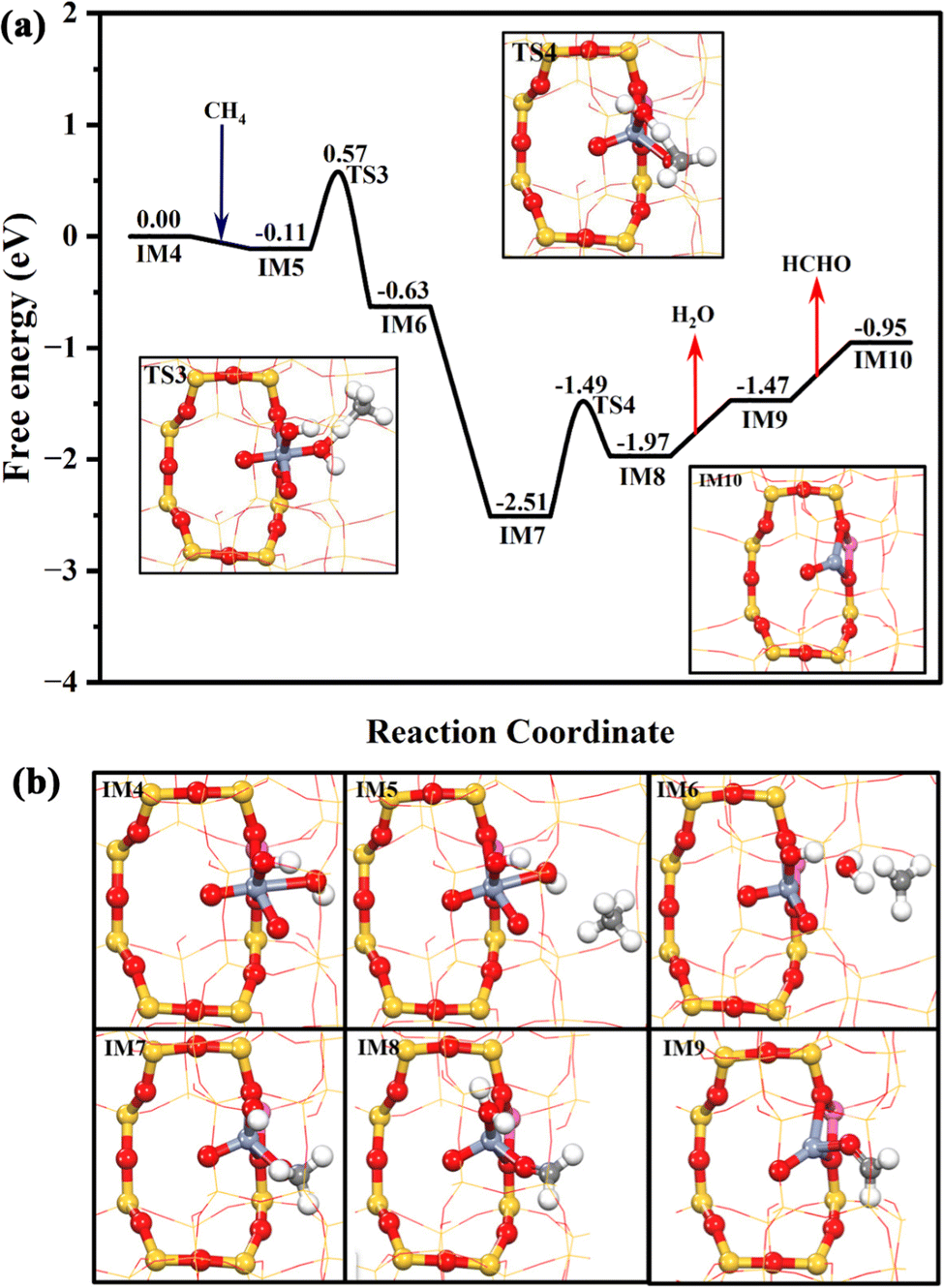 | ||
| Fig. 5 The Gibbs free energy profile of the HCHO production branch over the Cr1/ZSM-5 SAC with the geometry structures of key transition states (a); the geometry structures of all the intermediates in the whole catalytic cycle (b). The Gibbs energy profile was calculated under the reaction conditions of 30 bar CH4, 0.5 M H2O2 at 323 K. | ||
To prove the above conclusion from DFT calculations, the adsorption behavior of OH* on the surface of the Cr1/ZSM-5 SAC was explored by in situ H2O2-DRIFTS at 30 °C (Fig. S18). For the adsorption of H2O2 on the Cr1/ZSM-5 SAC sample, the band at 3751 cm−1 associated with the surface-adsorbed OH* is clearly observed as long as the H2O2 is introduced into the in situ chamber.37 Alternatively, no significant OH* adsorption was observed on Cr/ZSM-5-NP and H-ZSM-5, which further reveals that the formation of surface-adsorbed OH* was promoted on the Cr1 atoms. These results corroborate the conclusions of the DFT simulation results.
As shown in Fig. 6, Z[Cr(O)(CH2O)]+ (IM9) is generated after methane and methoxy dehydrogenation. H2O2 can be easily chemisorbed at Z[Cr(O)(CH2O)]+ (IM9) with a free adsorption energy of −0.88 eV. Interestingly, one OH of H2O2* could be readily abstracted by the neighboring HCHO* to form Z[Cr(O)(OH)(H2COOH)]+ (IM12), assisting the cleavage of the O–O bond of H2O2. This process only needs to climb over a subtle free energy barrier of 0.18 eV (TS5). Then, the HCOOH* (Z[Cr(O)(H2O)(HCOOH)]+, IM13) is formed through the oxidative dehydrogenation of H2COOH* by OH* with a free energy barrier of 1.21 eV. Then, the HCOOH is finally produced after overcoming a trivial desorption energy of 0.14 eV. The Z[CrO]+ (IM10) could also be formed in the pathway for HCOOH production after the desorption of H2O (0.65 eV) from Z[Cr(O)(H2O)]+ (IM14), which is the same as the reaction pathway of HCHO production.
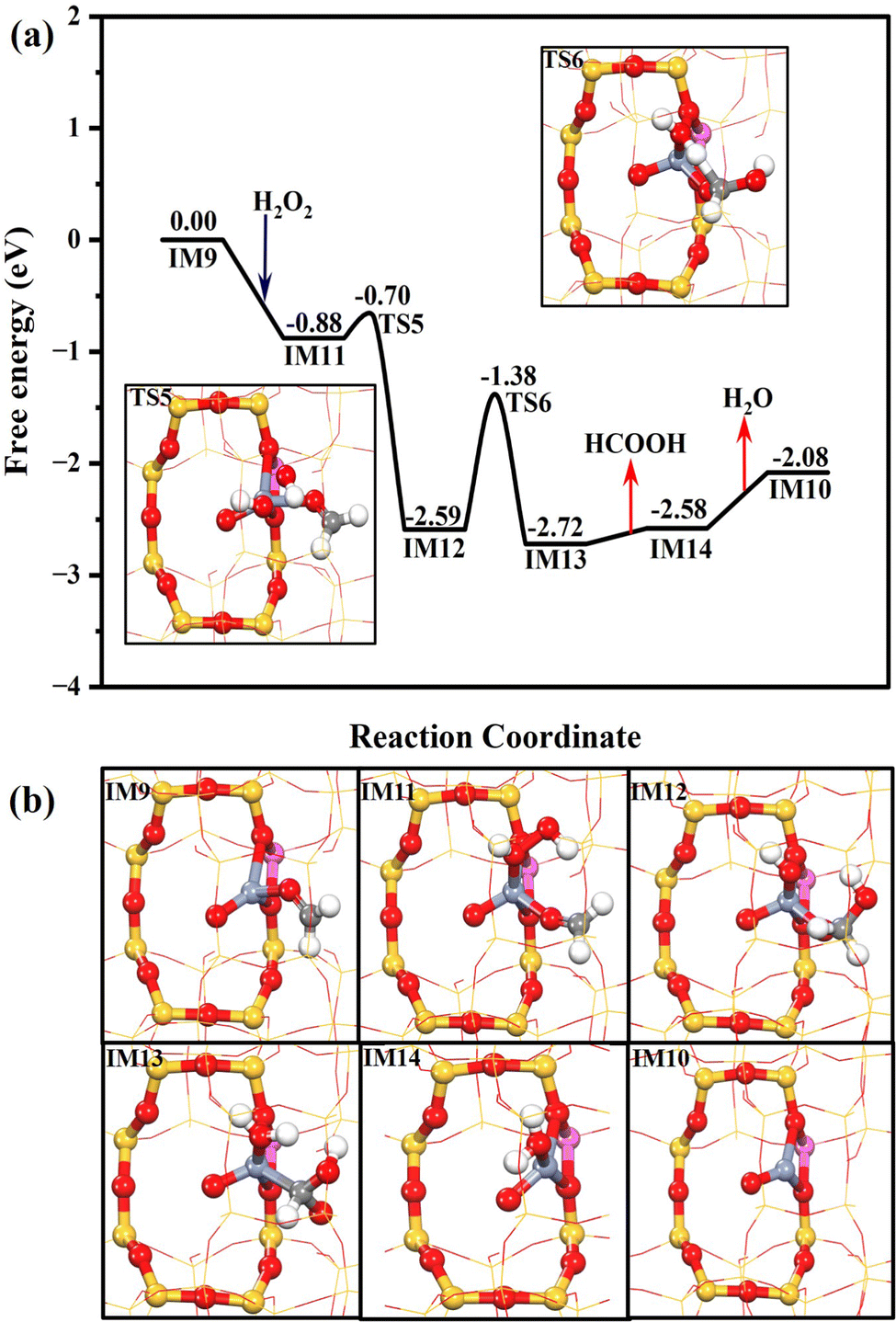 | ||
| Fig. 6 The Gibbs free energy profile of the HCOOH production branch over the Cr1/ZSM-5 SAC with the geometry structures of key transition states (a); the geometry structures of all the intermediates in the whole catalytic cycle (b). The Gibbs energy profile was calculated under the reaction conditions of 30 bar CH4, 0.5 M H2O2 at 323 K. | ||
As confirmed by the DFT simulation and experimental results, the active Cr species is hexavalent (Cr6+) in the presence of H2O2, and resulting reactive oxygen species trigger the DOM. Meanwhile, the Cr3+ species can be regenerated from the Cr6+ species after the desorption of the products, regardless of the HCHO or HCOOH production pathway. Since our Raman spectra prove that the hexavalent Cr could appear again when the used Cr1/ZSM-5 SAC is treated with H2O2 (Fig. S17, ESI†), we further computationally investigate the pathway for the regeneration of the hexavalent Cr site (Fig. 7). Similar to the results above, the electron-donating trivalent Cr ion at Z[CrO]+ could also break the O–O bond of H2O2 and form two OH−* readily (Z[Cr(O)(OH)2]+, IM15) after overcoming a subtle free energy barrier of 0.12 eV. Then, the H migration between two OH* at IM15 could produce Z[Cr(O)2(H2O)]+ (IM16) with a free energy barrier of 0.83 eV, followed by the water desorption (−0.36 eV). The formed Z[Cr(O)2]+ (IM17) could also chemisorb the H2O2 with the free adsorption energy of −0.34 eV. After its O–O bond has been broken (TS9, 1.03 eV), the hexavalent active site of Z[Cr(O)2(OH)2]+ (IM4) is regenerated. Therefore, the Cr1 center would exhibit the trivalent status in the absence of H2O2, while it is transformed to the active hexavalent Cr center in the presence of H2O2, enabling the DOM to form the C1 oxygenates.
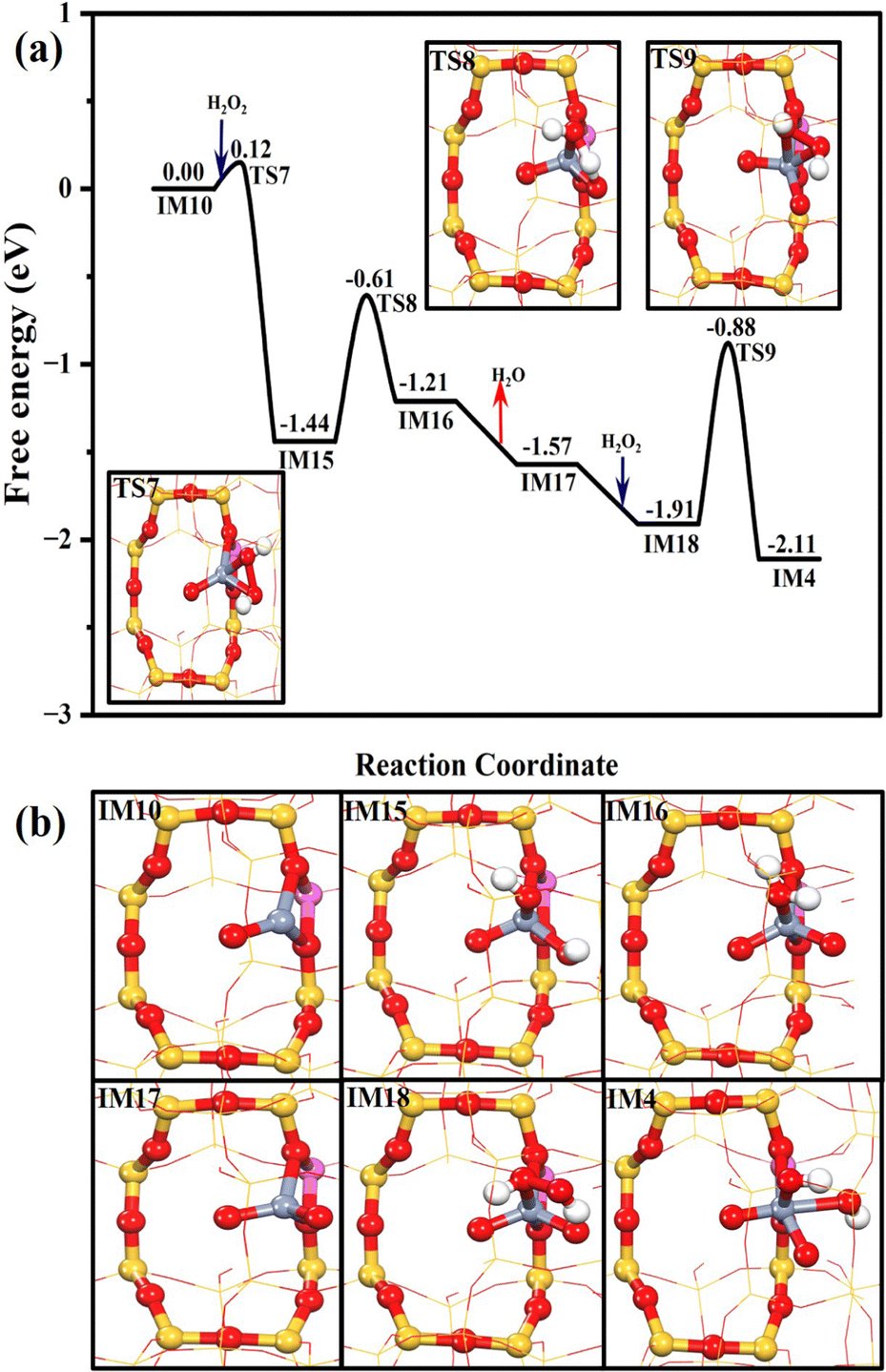 | ||
| Fig. 7 The Gibbs free energy profile of the regeneration of the active sites with the geometry structures of key transition states (a); the geometry structures of all the intermediates in the whole catalytic cycle (b). | ||
Conclusion
In summary, we demonstrate that the atomically-dispersed chromium sites anchored on ZSM-5 possess the excellent catalytic performance for the direct oxidation of methane to C1 oxygenated products with H2O2 as the oxidant at 50 °C. Compared with the low yield of C1 oxygenates from Cr/ZSM-5-NP, the yield of C1 oxygenates and selectivity are 21100 μmol gcat−1 h−1 and 99.8% on the Cr1/ZSM-5 SAC at 50 °C, which, to our knowledge, outperforms most reported state-of-the-art catalysts. The in situ characterization and experimental results confirm that the transformation of the Cr3+ to Cr6+ species is the key to the high activity of the Cr1/ZSM-5 SAC for the DOM. DFT theoretical calculations prove that the reaction mechanism of methane oxidation by the Cr1/ZSM-5 SAC can be divided into three steps: (i) pre-activation of the reaction site; (ii) oxidation of methane; (iii) regeneration of the active sites. We believe that this study not only provides a new prospect for the efficient DOM under mild conditions but also promotes the design of non-noble metal SACs in heterogeneous catalysis.
Author contributions
Mengyu Zeng: data curation, formal analysis, writing-original draft. Lu Cheng: DFT data curation, investigation, formal analysis, writing-original draft. Qingqing Gu: STEM data curation, formal analysis. Bing Yang: STEM data curation, formal analysis. Baiyang Yu: investigation. Jing Xu: methodology, formal analysis. Ying Zhang: methodology, investigation. Chengsi Pan: methodology, formal analysis. Yang Lou: conceptualization, methodology, data curation, funding acquisition, writing-original draft, review & editing. Xiaoming Cao: DFT data curation, funding acquisition, supervision, writing – review & editing. Yongfa Zhu: funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported financially by the National Key R&D Program of China (2021YFB3501900), the National Natural Science Foundation of China (21908079, U21A20326, 21872145, 21902009, 22172065, 22022302, 92045303 and 91845111), the Jiangsu Specially-Appointed Professor (1046010241211400), the Natural Science Foundation of Jiangsu Province (BK20211239, BK20201345, BK20221541), the State Key Laboratory of Fine Chemicals, the Dalian University of Technology (KF2005). We also thank the Central Laboratory, School of Chemical and Material Engineering, Jiangnan University.Notes and references
- P. Tang, Q. Zhu, Z. Wu and D. Ma, Energy Environ. Sci., 2014, 7, 2580–2591 RSC.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- W. Taifan and J. Baltrusaitis, Appl. Catal., B, 2016, 198, 525–547 CrossRef CAS.
- B. Wang, S. Albarracín-Suazo, Y. Pagán-Torres and E. Nikolla, Catal. Today, 2017, 285, 147–158 CrossRef CAS.
- B. L. Conley, W. J. Tenn, K. J. H. Young, S. K. Ganesh, S. K. Meier, V. R. Ziatdinov, O. Mironov, J. Oxgaard, J. Gonzales, W. A. Goddard and R. A. Periana, J. Mol. Catal. A: Chem., 2006, 251, 8–23 CrossRef CAS.
- K. Kwapien, J. Paier, J. Sauer, M. Geske, U. Zavyalova, R. Horn, P. Schwach, A. Trunschke and R. Schlogl, Angew. Chem., Int. Ed., 2014, 53, 8774–8778 CrossRef CAS PubMed.
- S. Ji, Y. Chen, X. Wang, Z. Zhang, D. Wang and Y. Li, Chem. Rev., 2020, 120, 11900–11955 CrossRef CAS PubMed.
- Y. Shi, Y. Zhou, Y. Lou, Z. Chen, H. Xiong and Y. Zhu, Adv. Sci., 2022, 9, 2201520 CrossRef CAS PubMed.
- K. Zhu, S. Liang, X. Cui, R. Huang, N. Wan, L. Hua, H. Li, H. Chen, Z. Zhao, G. Hou, M. Li, Q. Jiang, L. Yu and D. Deng, Nano Energy, 2021, 82, 105718 CrossRef CAS.
- W. Huang, S. Zhang, Y. Tang, Y. Li, L. Nguyen, Y. Li, J. Shan, D. Xiao, R. Gagne, A. I. Frenkel and F. F. Tao, Angew. Chem., Int. Ed., 2016, 55, 13441–13445 CrossRef CAS PubMed.
- Y. Tang, Y. Li, V. Fung, D. E. Jiang, W. Huang, S. Zhang, Y. Iwasawa, T. Sakata, L. Nguyen, X. Zhang, A. I. Frenkel and F. F. Tao, Nat. Commun., 2018, 9, 1231 CrossRef PubMed.
- Q. Shen, C. Cao, R. Huang, L. Zhu, X. Zhou, Q. Zhang, L. Gu and W. Song, Angew. Chem., Int. Ed., 2020, 59, 1216–1219 CrossRef CAS PubMed.
- P. Xie, J. Ding, Z. Yao, T. Pu, P. Zhang, Z. Huang, C. Wang, J. Zhang, N. Zecher-Freeman, H. Zong, D. Yuan, S. Deng, R. Shahbazian-Yassar and C. Wang, Nat. Commun., 2022, 13, 1375 CrossRef CAS PubMed.
- X. Tang, L. Wang, B. Yang, C. Fei, T. Yao, W. Liu, Y. Lou, Q. Dai, Y. Cai, X.-M. Cao, W. Zhan, Y. Guo, X. Q. Gong and Y. Guo, Appl. Catal., B, 2021, 285, 119827 CrossRef CAS.
- Z. Zhu, Z. Chang and L. Kevan, J. Phys. Chem. B, 1999, 103, 2680–2688 CrossRef CAS.
- B. M. Weckhuysen, I. E. Wachs and R. A. Schoonheydt, Chem. Rev., 1996, 96, 3327–3350 CrossRef CAS PubMed.
- P. J. McCarthy, J. C. Lauffenburger, P. M. Skonezny and D. C. Rohrer, Inorg. Chem., 1981, 20, 1566–1570 CrossRef CAS.
- Q. Luo, Y. Li, X. Huo, L. Li, Y. Song, S. Chen, H. Lin and N. Wang, Adv. Sci., 2022, 9, 2105346 CrossRef CAS PubMed.
- A. Penkova and K. Hadjiivanov, Catal. Commun., 2003, 4, 485–491 CrossRef CAS.
- M. F. Zhou and L. Andrews, J. Phys. Chem. A, 1998, 102, 7452–7461 CrossRef CAS.
- N. W. Felvey, M. J. Meloni, C. X. Kronawitter and R. C. Runnebaum, Catal. Sci. Technol., 2020, 10, 5069–5081 RSC.
- M. Mihaylov, A. Penkova, K. Hadjiivanov and M. Daturi, J. Mol. Catal. A: Chem., 2006, 249, 40–46 CrossRef CAS.
- D. Wang, L. Zhang, K. Kamasamudram and W. S. Epling, ACS Catal., 2013, 3, 871–881 CrossRef CAS.
- K. I. Hadjiivanov, Catal. Rev., 2000, 42, 71–144 CrossRef CAS.
- E. Gao, H. Pan, W. Zhang, Y. Li, G. Cao, M. T. Bernards, Y. He and Y. Shi, Chem. Eng. J., 2020, 386, 123956 CrossRef CAS.
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS PubMed.
- N. Yang, Y. Zhao, P. Wu, G. Liu, F. Sun, J. Ma, Z. Jiang, Y. Sun and G. Zeng, Appl. Catal., B, 2021, 299, 120682 CrossRef CAS.
- N. Agarwal, S. J. Freakley, R. U. McVicker, S. M. Althahban, N. Dimitratos, Q. He, D. J. Morgan, R. L. Jenkins, D. J. Willock, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Science, 2017, 358, 223–227 CrossRef CAS PubMed.
- X. M. Cao, H. Zhou, L. Zhao, X. Chen and P. Hu, Chin. Chem. Lett., 2021, 32, 1972–1976 CrossRef CAS.
- M. S. Palmer, M. Neurock and M. M. Olken, J. Am. Chem. Soc., 2002, 124, 8452–8461 CrossRef CAS PubMed.
- Q. Meng, W. Wang, X. Weng, Y. Liu, H. Wang and Z. Wu, J. Phys. Chem. C, 2016, 120, 3259–3266 CrossRef CAS.
- J. Wang and G. C. Wang, J. Phys. Chem. C, 2018, 122, 17338–17346 CrossRef CAS.
- J. Xu, X. M. Cao and P. Hu, J. Phys. Chem. C, 2019, 123, 28802–28810 CrossRef.
- A. A. Latimer, H. Aljama, A. Kakekhani, J. S. Yoo, A. Kulkarni, C. Tsai, M. Garcia-Melchor, F. Abild-Pedersen and J. K. Nørskov, Phys. Chem. Chem. Phys., 2017, 19, 3575–3581 RSC.
- J. Gao, Y. Zheng, Y. Tang, J. M. Jehng, R. Grybos, J. Handzlik, I. E. Wachs and S. G. Podkolzin, ACS Catal., 2015, 5, 3078–3092 CrossRef CAS.
- N. Mimura, M. Okamoto, H. Yamashita, S. T. Oyama and K. Murata, J. Phys. Chem. B, 2006, 110, 21764–21770 CrossRef CAS PubMed.
- X. Xu, L. Liu, Y. Tong, X. Fang, J. Xu, D. E. Jiang and X. Wang, ACS Catal., 2021, 11, 5762–5775 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, XRD, Raman, DRIFTs, 1H NMR, GC, HAADF-STEM, TEM, XPS, XAFS and theoretical calculations are elucidated. See DOI: https://doi.org/10.1039/d2ey00080f |
| ‡ These authors contribute equally to the article. |
| This journal is © The Royal Society of Chemistry 2023 |