
Pyrene aggregation at unprecedented low concentrations in (lanthanide metal salt + urea) deep eutectic solvents†
Vaishali
Khokhar
,
Manish
Kumar
and
Siddharth
Pandey
 *
*
Department of Chemistry, Indian Institute of Technology Delhi, Hauz Khas, New Delhi 110016, India. E-mail: sipandey@chemistry.iitd.ac.in; haridasv@chemistry.iitd.ac.in
First published on 1st December 2022
Abstract
We report intermolecular excimer formation by a common and popular polycyclic aromatic hydrocarbon pyrene at very low concentrations (low μM) when dissolved in deep eutectic solvents constituted of hydrated lanthanide salts and urea. Pyrene is not known to aggregate at such low concentrations in isotropic solvents and common liquids under similar conditions.
Deep eutectic solvents (DESs) are eutectic mixtures of molecular or ionic compounds present in liquid form at room temperature.1–4 Based on the precursors used and the characteristics of the ensuing media, DESs have been categorized into five types. While types I and II DESs are prepared by mixing quaternary ammonium salts with metal chlorides and hydrated metal salts, respectively, type III constitutes the mixtures of a H-bond donor (HBD) and a quaternary ammonium salt.5,6 The type IV DESs are prepared by mixing transition metal halides or hydrated metal salts with a wide array of HBDs and type V are hydrophobic DESs composed of a quaternary ammonium salt and a hydrophobic HBD.7–11 The type IV DESs composed of lanthanide metal salt and a HBD have demonstrated novel features and can act as precursors for pure lanthanide phosphor materials which have several applications in luminescence devices, fluorescent lamps, magnets, catalysis, lasers, superconductors, data storage devices, sensor, electronics, and optoelectronics.12–15
We have found that type IV DESs composed of hydrated lanthanide salts and urea induce solute–solute interaction at very low solute concentration under ambient conditions. Specifically, we demonstrate herein that a common and popular fluorescence probe pyrene [a polycyclic aromatic hydrocarbon (PAH) with four fused benzene rings; Fig. 1] shows emission corresponding to intermolecular excimer when solubilized in eutectic mixtures of lanthanum nitrate hexahydrate and gadolinium nitrate hexahydrate, respectively, with urea at several different compositions at solute concentrations where pyrene is not known to self-aggregate in common solvents. Fluorescence emission spectra of 20 μM pyrene dissolved in DESs constituted of (lanthanum nitrate hexahydrate + urea), named (La![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :urea), and (gadolinium nitrate hexahydrate + urea), named (Gd:urea), at 1:3.5 molar ratio under ambient conditions are presented in Fig. 1A. It is surprising that, in (La:urea) and (Gd:urea) DESs, along with the usual high energy (in the vicinity of 370–420 nm) vibronically-resolved structured emission features characterizing solute pyrene, a broad structureless low-energy band between 450–550 nm (with band maxima ca. 475 ± 2 nm) appears as well. Such emission feature usually occurs at milli-molar (mM) or higher concentrations of pyrene in organic solvents due to the formation of intermolecular excimer by pyrene.16–19 This intermolecular excimer formation by some PAHs when dissolved in organic solvents at >mM concentration under ambient conditions mostly takes place in the excited-state; the excited-dimer (or the excimer) formation is usually a direct consequence of excitation of the PAH that results in favourable energetics (leading to necessary solute diffusion) for such intermolecular aggregation process. It is clear that while 20 μM pyrene does not show any excimer in other solvents, it does aggregate intermolecularly at such low concentrations in (La:urea) and (Gd:urea) DESs.
:urea), and (gadolinium nitrate hexahydrate + urea), named (Gd:urea), at 1:3.5 molar ratio under ambient conditions are presented in Fig. 1A. It is surprising that, in (La:urea) and (Gd:urea) DESs, along with the usual high energy (in the vicinity of 370–420 nm) vibronically-resolved structured emission features characterizing solute pyrene, a broad structureless low-energy band between 450–550 nm (with band maxima ca. 475 ± 2 nm) appears as well. Such emission feature usually occurs at milli-molar (mM) or higher concentrations of pyrene in organic solvents due to the formation of intermolecular excimer by pyrene.16–19 This intermolecular excimer formation by some PAHs when dissolved in organic solvents at >mM concentration under ambient conditions mostly takes place in the excited-state; the excited-dimer (or the excimer) formation is usually a direct consequence of excitation of the PAH that results in favourable energetics (leading to necessary solute diffusion) for such intermolecular aggregation process. It is clear that while 20 μM pyrene does not show any excimer in other solvents, it does aggregate intermolecularly at such low concentrations in (La:urea) and (Gd:urea) DESs.
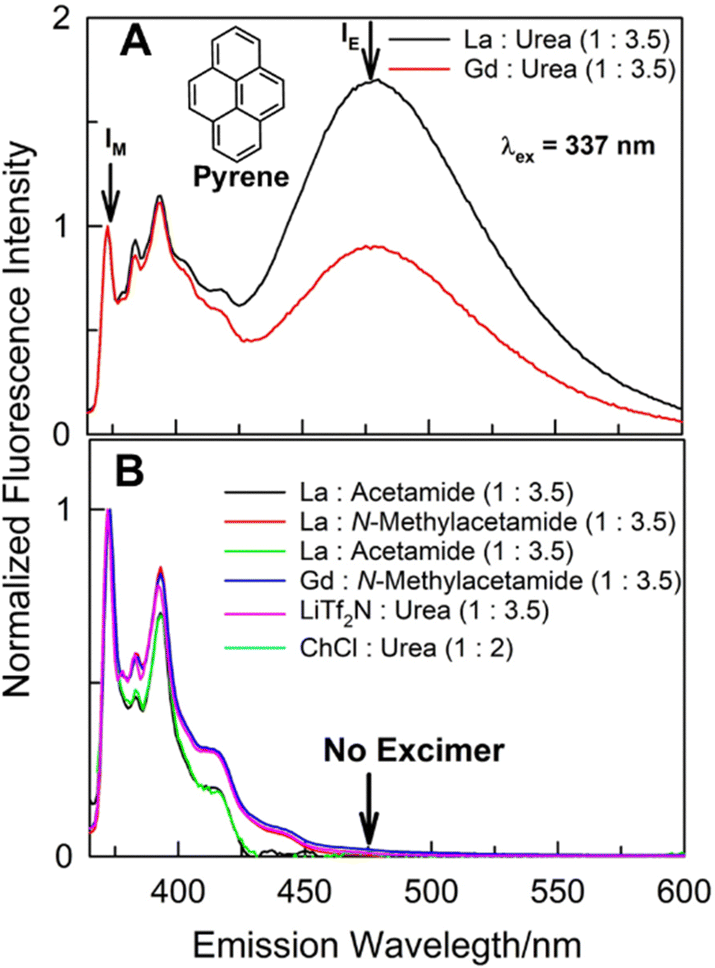 | ||
| Fig. 1 Fluorescence emission spectra of 20 μM pyrene dissolved in various DESs under ambient conditions. | ||
It appears that this intermolecular aggregation at such low concentration is specific to the solubilizing milieu afforded by (lanthanide salt + urea) DESs. In order to gain further insight to the role of DES constituents and composition in such unprecedented intermolecular pyrene aggregation process, several other DESs having one constituent (either the lanthanide salt or the urea) the same were investigated under identical conditions (Fig. 1B). Specifically, two common DESs composed of urea – (choline chloride:urea::1:2) and (LiTf2N:urea::1:3.5), respectively, do not support such pyrene–pyrene aggregation at 20 μM solute concentration; the aggregation was not observed even in (La:acetamide), (Gd:acetamide), (La:N-methylacetamide), and (Gd:N-methylacetamide) eutectic systems under identical conditions [pyrene aggregation at such low concentrations was not observed in other common solvents and ionic liquids under identical conditions as well (Fig. S1), ESI†]. It appears the presence of both urea and La(NO3)3·6H2O or Gd(NO3)3·6H2O as constituents of the DES is required. Attempt to investigate another such DES composed of Ce(NO3)3·6H2O and urea [(Ce:urea)] was unsuccessful due to the absence of any pyrene fluorescence in this DES [apparently, in comparison with La(III) and Gd(III), Ce(III) completely quenches the fluorescence from pyrene (Fig. S2, ESI†)]. At 20 μM concentration, intermolecular pyrene aggregation was found to be absent in common ‘hydrophobic’ DESs constituted of capric acid with thymol, eugenol, linalool, geraniol, and β-citronellol, respectively, and those composed of thymol and menthol at several compositions (Fig. S3, ESI†).
In order to investigate the role of the composition of the (La:urea) and (Gd:urea) DESs on the extent of intermolecular aggregation of pyrene at such low concentrations, in addition to (1:3.5), fluorescence emission spectra were acquired at (1:4), (1:5), (1:6), and (1:7) molar ratios for both the DESs under ambient conditions (DESs could not be formed beyond these molar ratio limits) (Fig. 2). The intermolecular excimer-to-monomer emission intensity ratio (IE/IM) is found to be maximum for (1:3.5) mole ratio of the DESs, and is found to decrease almost linearly with increase in the number of moles of urea per mole of the metal salt. It may imply that more the hydrated metals salt in these DESs, higher the intermolecular excimer formation efficiency although the viscosity of both the DESs becomes lower when the urea content is increased (dynamic viscosity, η, values estimated from previously reported data at 298.15 K for these DESs are also included in Fig. 2).20 The unusual outcome of decrease in IE/IM with decrease in the viscosity of the medium is discussed later.
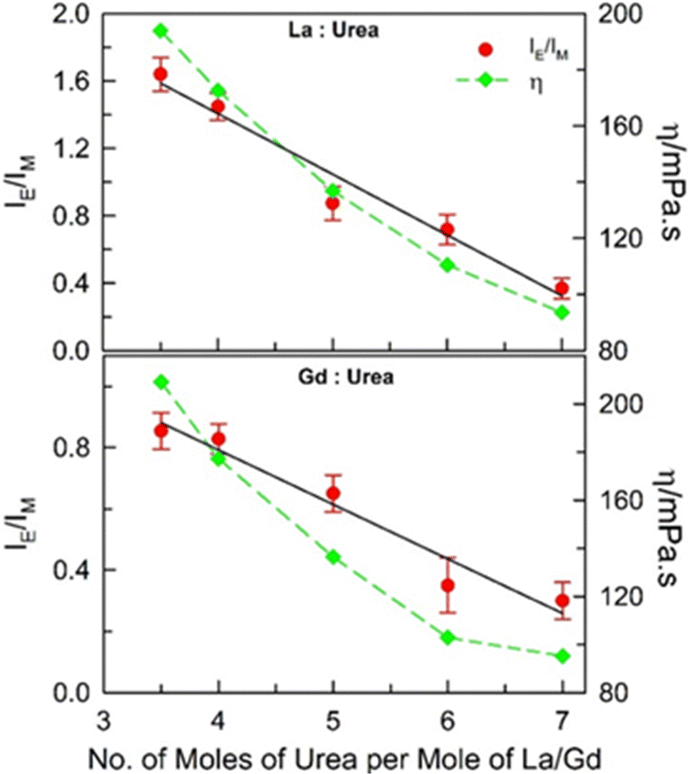 | ||
| Fig. 2
I
E/IM of 20 μM pyrene dissolved in (La:urea) and (Gd:urea) DESs of different compositions under ambient conditions. Dynamic viscosity (η) of the DESs is also plotted (error associated with η is ≤ ±2%). | ||
Steady-state fluorescence spectra (both emission and excitation) of pyrene in 1–20 μM concentration range were collected in (La:urea) DES at (1:3.5) mole ratio under ambient conditions to further decipher the aggregation mechanism (Fig. 3). As expected, fluorescence emission corresponding to both monomer and intermolecular excimer increases as pyrene concentration is increased. However, a monotonic increase in IE/IM with [pyrene] strongly hints that the process is bimolecular in nature (Fig. 3A; for a unimolecular process, e.g., intramolecular excimer formation, the IE/IM is independent of the probe concentration). For the intermolecular excimer formation by pyrene at mM or higher concentrations in organic solvents, it is inferred that two pyrene molecules are involved in such aggregation leading to excimer formation. Evidence on whether the pyrene–pyrene aggregation takes place exclusively in the excited-state or in the ground-state as well is offered by variable-wavelength excitation scans along with the excited-state emission intensity decay [I(t)] collected at emission characterized by the monomer and the excimer, respectively. The excitation spectra collected with λem = 373 nm (monomer emission) and 473 nm (excimer emission), respectively, of 20 μM pyrene dissolved in (La:urea::1:3.5) do not overlap perfectly (Fig. 3B and Fig. S4, ESI†) suggesting the presence of ground-state heterogeneity. More importantly, the excitation scan at λem = 473 nm does show a low-energy spectral feature hinting at pyrene–pyrene aggregation in the ground-state (Fig. 3B and Fig. S4, ESI†). Further evidence for the intermolecular ground-state aggregation by pyrene at 20 μM (and lower concentrations) is obtained from the [I(t)] data (Fig. 3C and Table S1, ESI†). Specifically, change in emission intensity with time acquired at 473 nm [IE(t), corresponding to excimer emission] does not exhibit initial growth immediately after excitation which indicates presence of ground-state pyrene aggregates and that the IE(t) is due to the excitation of these aggregates. To corroborate this, the fit of the IE(t), while not acceptable to a single-exponential decay, is better to a double-exponential decay function with no appearance of a negative pre-exponential factor (pre-exponential factor is negative for excimer forming in the excited-state). The two recovered decay times, one in the vicinity of ∼1 ns while the other longer, decrease with increasing temperature thus belonging to the emissive molecular states with distinct spectral features. The presence of ground-state pyrene aggregates is further substantiated nonetheless.
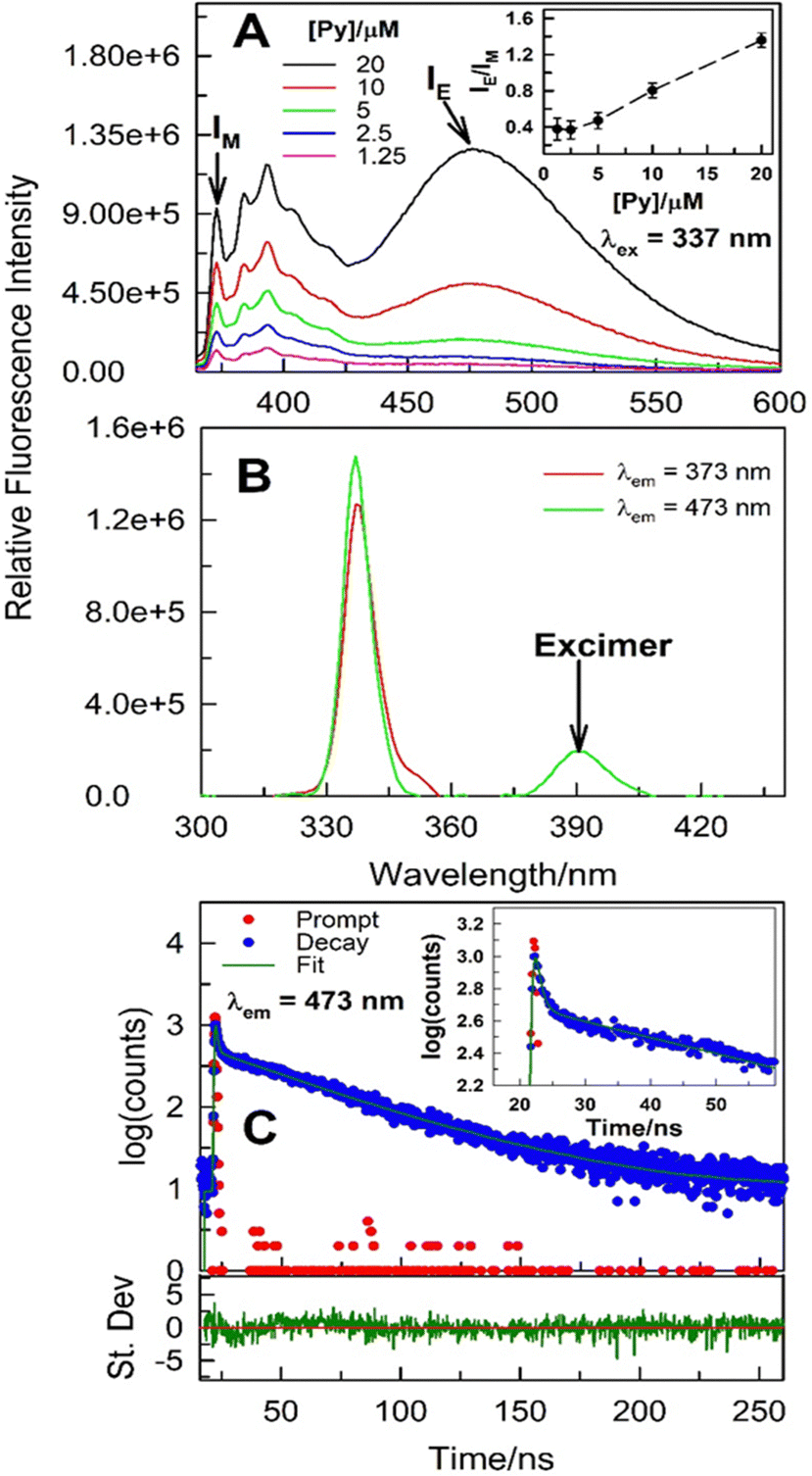 | ||
| Fig. 3 Fluorescence emission (panel A) and excitation spectra (panel B) of 20 μM pyrene dissolved in La:urea (1:3.5) DES under ambient conditions (inset in panel A shows IE/IM dependence on pyrene concentration). Excited-state intensity decay and it's fit to a second exponential decay equation for pyrene (20 μM; excitation with 340 nm NanoLED) dissolved in La:urea (1:3.5) DES. Residuals are provided below the panel C (inset shows absence of growth in the intensity immediately after excitation hinting at ground-state aggregation). | ||
Intermolecular aggregation of pyrene at such low concentrations in (hydrated lanthanide salt + urea) DESs is unprecedented. Earlier, Schlautman and Carraway have presented evidence of interaction between pyrene at sub-μM concentration in aqueous solution with Ag+ at very high concentrations (0.15 M).21 Based on small shifts in absorbance bands with support from steady-state and time-resolved fluorescence spectroscopies, presence of ground-state cation–aromatic π electron interactions were suggested. Presence of any such interaction between La3+/Gd3+ and pyrene resulting in appearance of a prominent broad and structureless bathochromically-shifted (with respect to monomer emission) emission band may be easily ruled out as this band does not appear in (La:acetamide::1:3.5) and (La:N-methylacetamide::1:3.5) eutectic mixtures. Interestingly, formation of intermolecular pyrene excimer at extremely low concentrations (50 μM) in supercritical (sc) fluids, sc CO2 and sc C2H4, were reported by Brennecke et al.22 While the intermolecular excimer formation by pyrene in sc fluids could not be entirely justified based on viscosity, it was suggested that solute–solute interactions were pronounced in asymmetric sc fluid solutions in comparison to normal liquids. Based on the overall data, it was warned that solute–solute interactions in sc fluid phase equilibria at low solute concentration should not be neglected and the concept of infinite dilution in sc fluids should be re-evaluated. Later on, Tato group presented unusual pyrene intermolecular excimer formation at 1.24 μM concentration during sodium deoxycholate gelation using steady-state and time-resolved fluorescence.23 Their results suggested that the formation of pyrene excimer was a result of the aggregation process where two clusters carrying probes formed a larger one; pyrene molecules could interact in both the ground and excited states.
In a recent investigation, based on neutron and X-ray diffraction data and fits for (hydrated lanthanide nitrate + urea) DESs, Edler group has suggested the presence of many oligomeric [–lanthanide–NO3−] species, such as, [Ce3(NO3)7]2+, among others.9 According to the authors, the ionic structuring of such lanthanide DESs is more similar to molten salts and halometallate ionic liquids and is less similar to most organic solvents, ionic liquids, and other type III DESs. The authors have proposed the presence of two separate but interacting nanostructures in the liquid bulk of these lanthanide-urea DESs – a charge-dense pseudophase containing lanthanide centres among others, and a mostly uncharged pseudophase comprising of H-bonded network of urea and water with networked molecular solvent-like domain associated to the lanthanide. We believe preferential interaction of the π-cloud of the pyrene with the polycationic species involving lanthanide results in bringing of pyrene molecules together in the vicinity of the urea–water H-bonded nano-domains leading to pyrene–pyrene aggregation at unprecedented low concentrations.
Support to this hypothesis is offered, in part, by the temperature-dependent fluorescence emission data. Fluorescence emission spectra of 20 μM pyrene in (La:urea) and (Gd:urea) DESs, respectively, at (1:3.5) mole ratios in the temperature range 293.15–323.15 K (Fig. 4) reveal decrease in both monomer and excimer fluorescence with increase in temperature. While decrease in IM with increasing temperature is easy to comprehend based on enhanced rates of non-radiative decay pathways (i.e., thermal quenching), the decrease in IE is seldom obvious as it is an interplay of the two factors – thermal quenching that results in decrease in fluorescence intensity with increasing temperature versus faster diffusion of the probe molecules leading to more aggregate formation with increase in temperature that brings in decrease in the viscosity. The report of increase in IE/IM by pyrene and other PAHs for both inter- and intra-molecular excimer formation on increasing temperature leading to the decrease in viscosity of the medium is abundant in literature.24–26 However, interestingly, concerning intermolecular pyrene–pyrene aggregation in (La:urea) and (Gd:urea) DESs, the IE/IM decreases with increasing temperature (Fig. 4; the decrease in dynamic viscosity, η, of the two DESs with increasing temperature is also presented). As postulated above, the interaction of the probe pyrene with the polycationic species of the DESs that results in close proximity of the pyrene molecules around the urea–water H-bonded nano-domains leading to intermolecular aggregate formation at anomalously low pyrene concentrations is disrupted by the increase in temperature that brings in the decrease in viscosity – decreased viscosity and/or increased temperature results in pyrene moieties in closer proximity to diffuse away from each other. The decrease in viscosity with increasing urea content in (lanthanide salt:urea) DESs leading to decrease in IE/IM could also be attributed to this (vide supra). Further investigation to establish the cause of such low-concentration solute aggregation within (hydrated lanthanide nitrate + urea) DESs is currently underway in our laboratory.
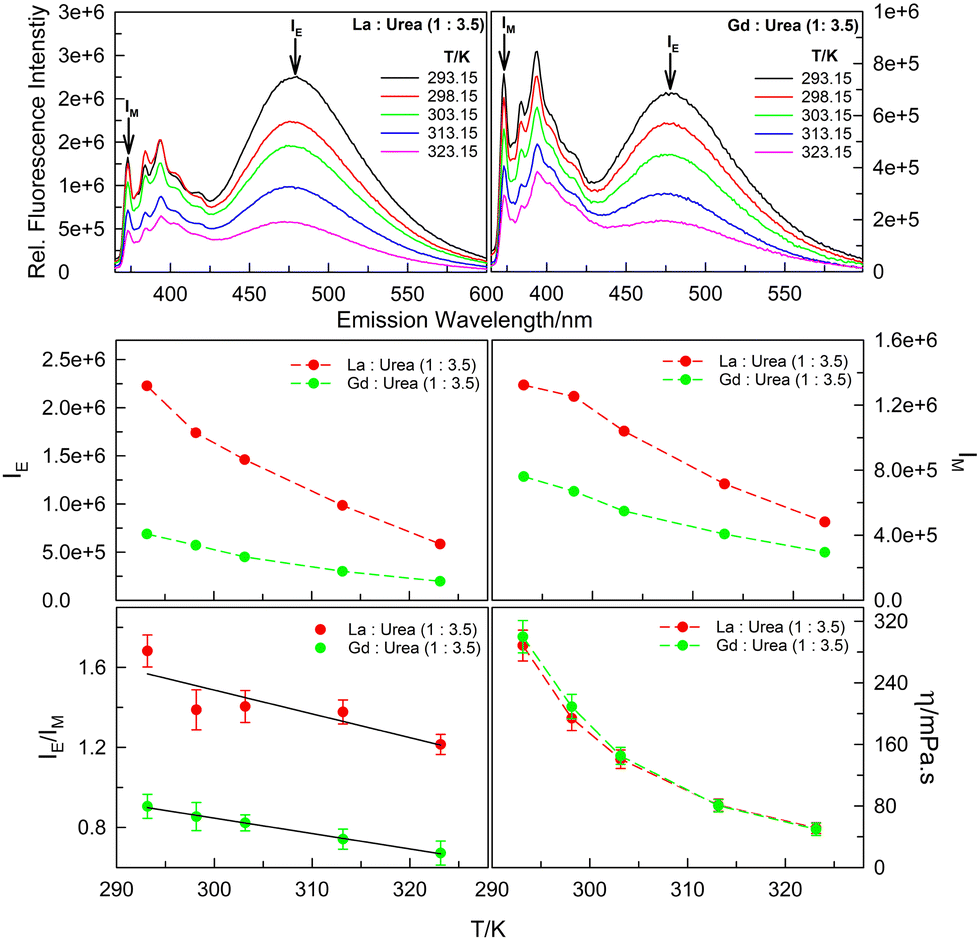 | ||
| Fig. 4 Fluorescence emission spectra (λex = 337 nm), IE, IM, IE/IM and η values of 20 μM pyrene dissolved in La:urea (1:3.5) and Gd:urea (1:3.5) DESs in (293.15–323.15) K temperature range. | ||
In conclusion, we have demonstrated intermolecular aggregation of solute pyrene within type IV DESs composed of hydrated lanthanide nitrate and urea under ambient conditions at such low solute concentration where aggregation is not known to take place in organic solvents, ionic liquids, and DESs of other types. While further investigation to pin-point the reason for this unusual observation is currently underway in our laboratory, we believe the outcomes reported here will open new avenues for application of such DESs especially in the area of material sciences, optics, and biotechnology.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is generously supported by the Council of Scientific & Industrial Research, EMR-II (CSIR-EMR-II), Government of India through a grant to Siddharth Pandey [grant number 01(3043)/21/EMR-II]. V. K. would like to thank University Grants Commission (UGC), Government of India for her fellowship. M. K. would like to thank Council of Scientific and Industrial Research (CSIR), Government of India for his fellowship.Notes and references
- T. el Achkar, H. Greige-Gerges and S. Fourmentin, Environ. Chem. Lett., 2021, 19, 3397–3408 CrossRef CAS.
- M. A. R. Martins, S. P. Pinho and J. A. P. Coutinho, J. Solution Chem., 2019, 48, 962–982 CrossRef.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2021, 121, 1232–1285 CrossRef CAS PubMed.
- A. P. Abbott, R. C. Harris, K. S. Ryder, C. D’Agostino, L. F. Gladden and M. D. Mantle, Green Chem., 2011, 13, 82–90 RSC.
- A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS PubMed.
- K. Shahbaz, I. M. Alnashef, R. J. T. Lin, M. A. Hashim, F. S. Mjalli and M. M. Farid, Sol. Energy Mater. Sol. Cells, 2016, 155, 147–154 CrossRef CAS.
- Y. Marcus, ACS Sustainable Chem. Eng., 2017, 5, 11780–11787 CrossRef CAS.
- O. S. Hammond, D. T. Bowron and K. J. Edler, ACS Sustainable Chem. Eng., 2019, 7, 4932–4940 CrossRef CAS.
- D. J. G. P. van Osch, L. F. Zubeir, A. van den Bruinhorst, M. A. A. Rocha and M. C. Kroon, Green Chem., 2015, 17, 4518–4521 RSC.
- D. J. G. P. van Osch, C. H. J. T. Dietz, J. van Spronsen, M. C. Kroon, F. Gallucci, M. van Sint Annaland and R. Tuinier, ACS Sustainable Chem. Eng., 2019, 7, 2933–2942 CrossRef CAS.
- J. Wu, K. Fujii, M. Yashima, A. Staykov, T. Akbay, T. Ishihara and J. A. Kilner, J. Mater. Chem. A, 2018, 6, 11819–11829 RSC.
- R. K. Tamrakar, D. P. Bisen and N. Brahme, Res. Chem. Intermed., 2014, 40, 1771–1779 CrossRef CAS.
- H. Guo, X. Yang, T. Xiao, W. Zhang, L. Lou and J. Mugnier, Appl. Surf. Sci., 2004, 230, 215–221 CrossRef CAS.
- E. Li, N. Wang and S. Wan, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 484, 012031 CAS.
- J. B. Birks and L. G. Christophorou, Nature, 1962, 194, 442–444 CrossRef CAS.
- T. Förster and K. Kasper, Z. Elektrochem., Ber. Bunsengesellschaft phys. Chem., 1955, 59, 976–980 Search PubMed.
- J. B. Birks, D. J. Dyson and I. H. Munro, Proc. R. Soc. Lond., 1963, 275, 575–588 CAS.
- J. B. Birks and L. G. Christophorou, Spectrochim. Acta, 1963, 19, 401–410 CrossRef.
- V. Khokhar, D. Dhingra and S. Pandey, J. Mol. Liq., 2022, 360, 119396 CrossRef CAS.
- J. H. Lee, M. A. Schlautman, E. R. Carraway, S. Yim and B. E. Herbert, J. Photochem. Photobiol., A, 2004, 163, 165–170 CrossRef CAS.
- J. F. Brennecke, D. L. Tomasko and C. A. Eckert, J. Phys. Chem., 1990, 94, 7692–7700 CrossRef CAS.
- A. Jover, F. Meijide, E. Rodríguez Núñez, J. Vázquez Tato, M. Mosquera and F. Rodríguez Prieto, Langmuir, 1996, 12, 1789–1793 CrossRef CAS.
- R. Horiguchi, N. Iwasaki and Y. Maruyama, J. Phys. Chem., 1987, 91, 5135–5139 CrossRef CAS.
- J. B. Birks, M. D. Lumb and I. H. Munro, Proc. R. Soc. Lond., 1964, 280, 289–297 CAS.
- M. J. Snare, P. J. Thistlethwaite and K. P. Ghiggino, J. Am. Chem. Soc., 1983, 105, 3328–3332 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp05155a |
| This journal is © the Owner Societies 2023 |