
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceColloidal FAPbBr3 perovskite nanocrystals for light emission: what's going on?
Harshita
Bhatia
a,
Biplab
Ghosh
 b and
Elke
Debroye
*a
b and
Elke
Debroye
*a
aDepartment of Chemistry, KU Leuven, Celestijnenlaan 200F, B-3001 Leuven, Belgium. E-mail: elke.debroye@kuleuven.be
bcMACS, Department of Microbial and Molecular Systems, KU Leuven, Celestijnenlaan 200F, B-3001 Leuven, Belgium
First published on 9th June 2022
Abstract
Semiconducting nanomaterials have been widely explored in diverse optoelectronic applications. Colloidal lead halide perovskite nanocrystals (NCs) have recently been an excellent addition to the field of nanomaterials, promising an enticing building block in the field of light emission. In addition to the notable optoelectronic properties of perovskites, the colloidal NCs exhibit unique size-dependent optical properties due to the quantum size effect, which makes them highly attractive for light-emitting diodes (LEDs). In the past few years, perovskite-based LEDs (PeLEDs) have demonstrated a meteoritic rise in their external quantum efficiency (EQE) values, reaching over 20% so far. Among various halide perovskite compositions, FAPbBr3 and its variants remain one of the most interesting and sought-after compounds for green light emission. This review focuses on recent progress in the design and synthesis protocols of colloidal FAPbBr3 NCs and the emerging concepts in tailoring their surface chemistry. The structural and physicochemical features of lead halide perovskites along with a comprehensive discussion on their defect-tolerant properties are briefly outlined. Later, the prevalent synthesis, ligand, and compositional engineering strategies to boost the stability and photoluminescence quantum yield (PLQY) of FAPbBr3 NCs are extensively discussed. Finally, the fundamental concepts and recent progress on FAPbBr3-based LEDs, followed by a discussion of the challenges and prospects that are on the table for this enticing class of perovskites, are reviewed.
 Harshita Bhatia | Harshita Bhatia received her Master's degree in Nanotechnology from Amity University, India. She joined Karlsruhe Institute of Technology, Germany, for her Master's thesis, where she developed solid-state electrolytes for advanced fluoride-ion batteries under the supervision of Prof. Maximilian Fichtner. She is currently a PhD candidate, focusing on the synthesis, surface chemistry, and optical properties of perovskite nanocrystals for LED devices, under the guidance of Prof. Elke Debroye and Prof. Johan Hofkens. In addition, she also carried out a research placement during her PhD at BASF (Germany), in the department of Performance Materials, with Dr. Peter Erk. |
 Biplab Ghosh | Biplab Ghosh is a PDM Research Fellow at Membrane Separations, Adsorption, Catalysis, and Spectroscopy for Sustainable Solutions (cMACS) in KU Leuven, Belgium. He holds a BE from Bengal Engineering and Science University (2011), a Mtech from the Academy of Scientific and Innovative Research, Council of Scientific and Industrial Research (2013), and a PhD from Nanyang Technological University in Materials Science and Engineering (2019). From 2020 to 2021, he was a postdoctoral fellow at the Energy Research Institute @ NTU (ERIAN). His research interests include energy materials, photovoltaics, and optoelectronics devices based on halide perovskites as well as other semiconducting materials. |
 Elke Debroye | Elke Debroye obtained her PhD in 2013, in developing MRI/optical contrast agents in the Laboratory of Bioinorganic Chemistry, KU Leuven. After postdoctoral positions with Prof. Liz-Marzán in San Sebastian (Spain), Prof. Majima at Osaka University (Japan) and Prof. Bonn at MPI Mainz (Germany), she returned to the Laboratory of Photochemistry and Spectroscopy, KU Leuven. Currently, she is an assistant Professor exploring new perovskite structures with advanced structural and optical characterization techniques, consequently optimizing their performance toward their application in new-generation optoelectronics. |
1. Introduction
Perovskites originally refer to a class of oxides with the chemical formula ABO3, in which A and B are divalent and tetravalent cations, respectively. The structure was named after the Russian mineralogist Lev Perovski, and the term ‘perovskite’ was first coined by Gustav Rose, who discovered the very first perovskite compound, CaTiO3.1 Later, an extensive variety of inorganic metal oxides were discovered and actively studied due to their unique opto-physical properties such as magnetism, ferroelectricity, and electronic conductivity.Over the past few years, lead halide perovskites (LHPs) with the ABX3 structure (A = monovalent cation, B = Pb2+, X = Br, Cl or I) have gained noticeable attention in a series of optoelectronic devices, e.g., solar cells, light-emitting diodes (LEDs), and photodetectors.2–7 These materials exhibit intriguing properties such as easy solution processibility, performant charge-carrier dynamics, unprecedented defect tolerance, tunable bandgaps, excellent color purity (a narrow full-width at half-maximum (FWHM)) of emission (∼20 nm), high photoluminescence quantum yield (PLQY) values (∼100%) and a wide color gamut (∼140%).5,8 In addition, one of the leading features that makes them versatile for various applications is their flexible composition space, which enables the diverse substitution of cations or anions, thereby offering opportunities for fine-tuning of the optoelectronic properties.9,10 Consequently, a large number of breakthroughs have been witnessed in the field of optoelectronics in the past decade. Specifically, single-junction solar cells with a power conversion efficiency (PCE) of over 25% have been achieved, gaining a strong foothold in the next-generation solar industry.11 Encouraged by this unprecedentedly rapid progress, attention has also extended to light-emission applications. In 2014, Friend's group demonstrated the first CH3NH3PbX3-based LEDs at room temperature, which exhibited external quantum efficiency (EQE) values of 0.76% and 0.1% in the near-infrared (754 nm) and green (517 nm) emission ranges, respectively.12 Since then, impressive progress has been reported in perovskite-based LEDs (PeLEDs) (especially in the green, red, and near-infrared emission ranges), with EQEs of over 20% so far.13–15
The performances of PeLEDs are currently comparable to the leading organic LEDs (OLEDs) and quantum-dot LEDs (QLEDs), based mainly on CdSe, InP, etc., semiconducting materials.16–22 Furthermore, compared with QLEDs (FWHM ≈ 30 nm) and OLEDs (FWHM > 40 nm), PeLEDs exhibit superior spectral purity (FWHM ≈ 20 nm) and high brightness, which are desirable for low-cost, high-end LEDs.23–25 Importantly, the turn-on voltage, which is the drive voltage at a luminance of 1 cd m−2, can be as low as 2 V.26,27 Therefore, all these exceptional properties make perovskites a potential candidate for future LED devices. Nevertheless, despite the meteoritic rise in EQE values of PeLEDs, a few substantial challenges still exist, such as (i) the efficiency of blue and deep-blue LEDs, which reaches close to 12% and 5.5%, respectively, remains inferior compared with green or red LEDs.28–31 This is attributed to the wide optical bandgap of blue emission, which possesses high sub-bandgap trap states. Such shortcomings markedly impede the realization of electroluminescence (EL) covering the whole visible region.32,33 (ii) The short PeLED lifetime under operating conditions (ranging from T50 = 59 h (for blue)34 to 682 h (for near-infrared emissions)).35 These numbers are very far away from the current high-performing OLEDs, which have a lifetime of over 6 × 106 h.35–37
Until now, the device degradation mechanism has not been well understood; however, studies have shown that the existence of defects in the perovskite emissive layer may lead to ion migration, hysteresis, non-radiative charge recombination, etc., which are detrimental to the device performance. Typically, ion migration is inevitable in all LHPs due to the application of a high external field across the film, which consequently enhances the amount of defects that serve as degradation initiators.38–40 Thus far, considerable efforts have been devoted to addressing the stability concerns. There have been multiple studies, which demonstrate that ion migration can be impeded by applying various strategies, such as post-treatments, or the introduction of different kinds of functional molecules, Lewis acids, and bases, hydrophobic polymers, and alkylammonium halide, which tend to ameliorate the properties of the perovskite layer, thereby mitigating the ion-migration pathways.20,41–43 However, despite this considerable progress, stability remains a bottleneck for the development of EL devices.
Although most of the studies have focused on thin-film LEDs, polycrystalline films are often sensitive to the processing conditions. These can lead to the development of randomly oriented grains, the generation of secondary non-perovskite phases, and heterogeneity in the optical properties, resulting in poor device stability.44 In addition, stringent control over the perovskite compositions is required to reach a specific emission wavelength, which again complicates the fabrication process. Recently, highly luminescent colloidal nanocrystals (NCs) have gained significant attention due to their size-dependent optical properties that arise from the quantum confinement effect. In the past two decades, the quantum confinement effect has been largely explored in compound semiconductors (such as CdSe, InP, etc.) for a variety of applications.45–49 In colloidal NCs, this effect is observed when the size of the NCs is smaller or comparable to the exciton (electron–hole pair) Bohr radius of the compound, resulting in a blue shift of the emission peak and absorption edge. Such confined systems present a different angle from which the optoelectronic properties can be fine-tuned. Specifically, the energy states in these materials do not remain continuous, as in the case of the bulk, but become discrete, leading to an increase in the bandgap. Thus, the precise control of size enables fine-tuning of the emission color over a very broad spectral region within a single composition, which makes colloidal NCs very attractive for light-emission applications.50–53 As such, perovskite-based colloidal NCs also offer added benefits in luminescent properties over their polycrystalline counterparts, such as high PLQY values (close to unity) with narrow emission linewidths and high exciton binding energy (Eb) values due to the confinement effect. Furthermore, the preparation methods for colloidal NCs are comparatively facile, with the possibility of obtaining a monodisperse and phase-pure system using solution-processing routes.54,55
In general, organic compounds exhibit interesting properties such as structural diversity, high luminescence and ease of processing; by contrast, inorganic counterparts provide advantages, such as thermal stability and high mobility. The combination of the two, i.e., organic–inorganic hybrid materials, offer the opportunity to combine the best of both worlds in a single compound. Specifically, hybrid perovskites consist of inorganic anions, which form corner-sharing octahedra, alternating with organic cations, which give rise to unusual structural and optical properties.56 Motivated by these tunable optoelectronic properties, a considerable amount of attention has been devoted to LHP nanocrystals and their applications in light emission.
Over the past few years, all-inorganic CsPbX3 perovskites have stimulated enormous attention in the field of optoelectronics due to their superior thermal and moisture stability compared with their organic–inorganic lead halide perovskite counterparts. However, they do encounter the substantial challenges of facile fabrication and phase stability. On the other hand, formamidinium lead bromide (FAPbBr3) remains one of the most stable Pb-based halide perovskite materials, which offers a higher thermodynamic stability that originates from the stronger ionic interaction with the halogen anions in the perovskite crystal. Consequently, FAPbBr3 exhibits improved stability against heat, light, and moisture.57,58 Moreover, FAPbBr3-based NCs can easily maintain their PL emission of ≥520 nm with narrow FWHM and high PLQY values, providing the opportunity to realize the pure green color in the NTSC chromaticity diagram.59,60 However, as for perovskites based on methylammonium (MA) and Cs, the emission wavelength is commonly <520 nm, even for nanoparticles with a size larger than their exciton Bohr diameter (>10 nm), making them unsuitable for obtaining the ultrapure green emission. These outstanding properties make FAPbBr3 a promising candidate for LED applications.61
As the field of perovskites is advancing rapidly, it is of great importance to feature the novel developments, challenges, and progress made in the past few years. This review focuses on the development and emerging concepts in tailoring the properties of FAPbBr3 nanocrystals for light-emission applications. We begin with discussing the general structural and physicochemical features of lead halide perovskite, followed by an overview of the unique optoelectronic properties of nanocrystals arising from the synergistic effect of quantum confinement and dimensionality engineering. Hereafter, the mainstream synthesis methodologies will be discussed briefly and numerous potential approaches to improving the performance of FAPbBr3 NCs will be evaluated. Furthermore, we highlight the gradual positive impact of these strategies on the external quantum efficiency of LEDs. Finally, a brief outlook on the pending challenges and possible advancements for further research is provided.
1.1 Crystal structure of LHPs
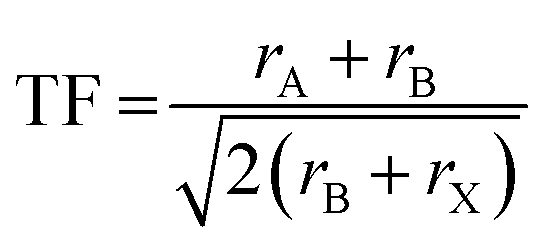 | (1) |
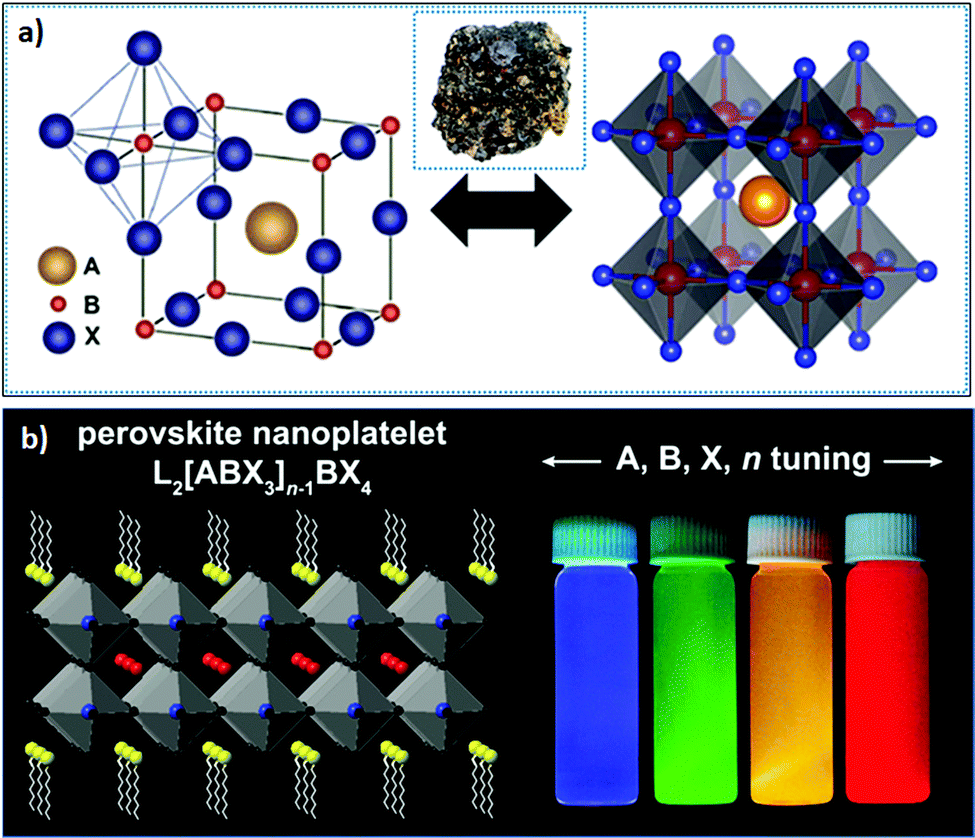 | ||
| Fig. 1 (a) Bulk structure of perovskites; the inset shows the perovskite mineral CaTiO3; and (b) layered 2D perovskite (n = 2) (left), where the emission color can be tuned upon varying X or n (right). panel (a) reprinted with permission from ref. 65. Copyright 2016 American Chemical Society. Panel (b) reprinted with permission from ref. 66 Copyright 2021 Elsevier. | ||
Notably, the aforementioned TF is computed by assuming that the ionic radii of the ions are constant. However, it is only a good zeroth order approximation to estimate the structural stability of the perovskites as its accuracy is often insufficient. Bartel et al. proposed a revised TF, τ, with a much improved accuracy:70
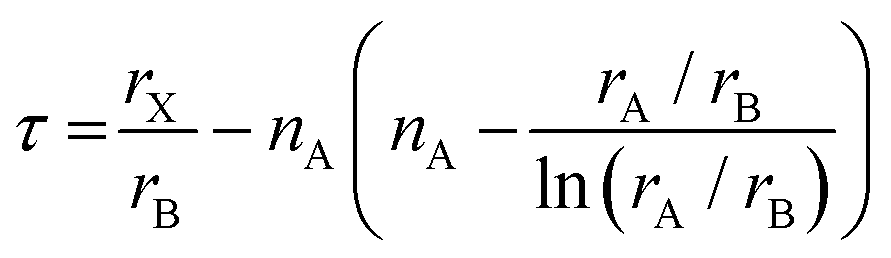 | (2) |
Although the ‘ideal’ perovskite structure is cubic, there are several distorted perovskite structures available; in fact, there are considerably more distorted structures than cubic ones. As a matter of fact, CaTiO3, the perovskite mineral itself, is structurally orthorhombic instead of cubic.72 The deviation from the ideal cubic structure could be due either to (1) cation displacements within the octahedron, (2) distortions of the octahedron, or (3) tilting of the octahedron. Among these, octahedral tilting is the most common type of distortion and can be realized by tilting the rigid BX6 octahedra along with one or more of their symmetry axes, while strictly maintaining the corner-sharing connectivity.73,74
1.2 Defects in LHPs
Compared with classical oxide-based perovskites, the larger and more polarizable ionic components in LHPs lead to weaker coulombic interactions and hence a soft crystal lattice. These ionic interactions result in fast crystallization, making them prone to structural disorders, which are associated with the formation of defects.77 Halide perovskites belong to that rare class of defect-tolerant materials where intrinsic defects are either shallow or have high formation enthalpies, thus remaining benign.78 The term ‘defect tolerance’ here indicates that the energy levels of the intrinsic defects lie close to or within the VBM or CBM, leaving the center of the bandgap defect-free and, thereby, benefitting perovskites in retaining their original properties. The superior defect tolerance in a semiconductor is assumed to stem from having the bonding orbitals at the conduction band minimum (CBM) and antibonding orbitals at the valence band maximum (VBM), which is unlikely in conventional semiconductors (bonding VBM and antibonding CBM) (Fig. 2).79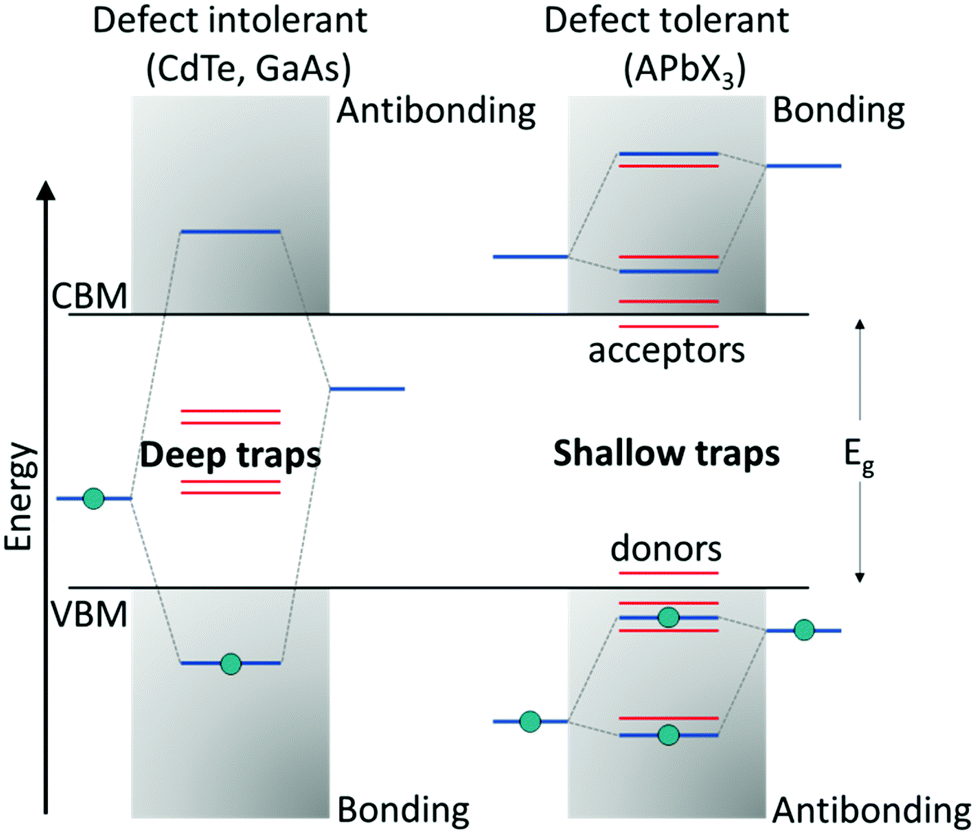 | ||
| Fig. 2 Band structure of (left) defect-intolerant conventional semiconductors (i.e., group III–V and II–VI) and (right) defect-tolerant lead halide perovskites. Figure adapted from ref. 82. Copyright 2020 Royal Society of Chemistry. | ||
The defect tolerance in LHPs could be attributed to a variety of factors. First, the VBM of APbX3 is formed via antibonding coupling between the Pb 6s and halide X np orbitals (n = 3 for Cl, n = 4 for Br, and n = 5 for I), while the CBM is composed of mostly Pb 6p atomic orbitals. The strong antibonding interaction between the Pb 6s and halide X np orbitals raises the VBM, indicating that the trap states are located within or close to the VB edge. Differently, the interaction between the Pb 6p and X np orbitals is weak, and therefore the CBM remains unaffected. Second, theoretical calculations on the unusual defect science in MHPs indicate that the dominant point defects, such as vacancies (mainly A- and X-site vacancies) that are characterized by the lowest formation energies, introduce only shallow trap states. Other remaining point defects, like antisites or interstitials, which result in deep traps, are mainly absent due to higher formation energies. Finally, on account of the large nuclear charge in MHPs, the spin–orbit coupling becomes significant, resulting in the lowering of the CB, thereby favoring the formation of shallow trap states.78,80,81 The combination of these features leads to the outstanding optoelectronic performance of perovskites. Although the intrinsic defects in the bulk are benign, surface defects still can act as non-radiative recombination centers.82,83 This is predominant in colloidal NCs, which possess an orders of magnitude higher surface-to-volume ratio than the bulk.
The surface of a semiconductor is the area where the periodicity of the crystal lattice is terminated and electronic states specific to the surface are found. These states can have a considerable effect on the electronic behavior and band structure, adversely impacting the stability and optoelectronic performance of the materials and devices.84,85 In the case of the bulk, only a small fraction of atoms are present at the surface and, therefore, the dangling bonds on the exterior contribute insignificantly to the properties of the materials. On the other hand, the role of the surface becomes dominant at the nanoscale, significantly altering the optical properties.84 Generally, when the periodicity is hindered or disrupted in LHPs, the crystal surface terminates with either organic cations or metal halide units or both, depending on the synthesis and processing conditions. As a result, under-coordinated atoms are formed, creating surface traps. The dominant surface-terminating group will have certain surface properties, which will also influence the nature of the entire material. It has been reported that defect states responsible for non-radiative recombination are mainly located toward the surface of LHPs.81 Moreover, theoretical calculations have also demonstrated that the structural defects at the surface can engender deep trap states. Uratani et al. reported a comprehensive theoretical study, investigating the chemistry of surface defects in MAPbI3 with varied stoichiometry. It was suggested that the Pb-rich condition is thermodynamically less favorable than the I-rich condition, owing to the higher formation energy of carrier trapping under the Pb-rich scenario.86 Other theoretical calculations on CsPbX3, which possesses halide vacancies on the surface, have revealed that halide vacancies are shallow in I-based perovskites, slightly deeper in Br-based perovskites, and close to a mid-gap depth in Cl-based perovskites.85,87,88 The consequence of this is also evident in the PLQY values when changing the halide from I− to Cl−.89
Nevertheless, the defects in Br-based perovskites also remain shallow, owing to the low formation energies. Overall, surface-related defects in LHP NCs are an important subject of concern and this has encouraged researchers to seek surface-defect passivation techniques.
2. Formamidinium lead halide perovskite nanocrystals: structure, synthesis, and optoelectronic properties
2.1 Polymorphism in FAPbBr3 perovskites
From a structural point of view, the rotation of corner-shared octahedra (i.e., the octahedral tilt) within perovskite structures results in different polymorphic phases (crystal structures) upon varying the temperature and pressure. The temperature-dependent crystal structures of perovskites are listed in the Bärnighausen family tree, where the space groups (having lower symmetry) derived from the aristotype (space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) exhibit different kinds of distortions. In the bulk, three polymorphs are mainly realized – cubic, tetragonal, and orthorhombic – in descending order of symmetry. These polymorphs mainly exhibit distinct degrees of tilting of the corner-sharing octahedra. At elevated temperature, all LHPs acquire a perovskite aristotype, that is, a high-symmetry cubic structure (having an X–Pb–X angle of 180°); however, as the temperature decreases, the BX6 octahedra tilt, owing to the differences in the size of the octahedral framework and A-site cation, resulting in structures with lower symmetry (hettotypes, i.e., structures that are similar to the aristotype but which possess low symmetry). Specifically, FAPbBr3 crystallizes into a stable cubic structure (Pmm space group) at room temperature (no octahedral tilting is present), transiting into a tetragonal phase (P4/mbm) between ∼275 and 250 K, followed by an orthorhombic transition (Pnma) upon decreasing the temperature to between ∼150 and 125 K.90,91 FA+ cations are quite disordered in the cubic phase and can rotate freely within the octahedral framework. As the temperature decreases, the cation in the tetragonal phase rotates between the preferred orientations, which are influenced by the octahedral tilting. However, after the material transitions to the orthorhombic phase, the rotation of FA+ cation is restricted, and a disordered state (glass-like dynamics) is exhibited. This is in contrast to the MA+ cation, which exhibits an ordered arrangement of the cation in a low-temperature orthorhombic phase.92
m) exhibit different kinds of distortions. In the bulk, three polymorphs are mainly realized – cubic, tetragonal, and orthorhombic – in descending order of symmetry. These polymorphs mainly exhibit distinct degrees of tilting of the corner-sharing octahedra. At elevated temperature, all LHPs acquire a perovskite aristotype, that is, a high-symmetry cubic structure (having an X–Pb–X angle of 180°); however, as the temperature decreases, the BX6 octahedra tilt, owing to the differences in the size of the octahedral framework and A-site cation, resulting in structures with lower symmetry (hettotypes, i.e., structures that are similar to the aristotype but which possess low symmetry). Specifically, FAPbBr3 crystallizes into a stable cubic structure (Pmm space group) at room temperature (no octahedral tilting is present), transiting into a tetragonal phase (P4/mbm) between ∼275 and 250 K, followed by an orthorhombic transition (Pnma) upon decreasing the temperature to between ∼150 and 125 K.90,91 FA+ cations are quite disordered in the cubic phase and can rotate freely within the octahedral framework. As the temperature decreases, the cation in the tetragonal phase rotates between the preferred orientations, which are influenced by the octahedral tilting. However, after the material transitions to the orthorhombic phase, the rotation of FA+ cation is restricted, and a disordered state (glass-like dynamics) is exhibited. This is in contrast to the MA+ cation, which exhibits an ordered arrangement of the cation in a low-temperature orthorhombic phase.92
Furthermore, besides the temperature-induced phase transition, the FAPbBr3 structure can also be modulated by applying pressure, which changes the atomic distance between the organic cation and inorganic framework, offering many rich phase structures. An extensive high-pressure study on FAPbBr3 crystals revealed the evolution of optical properties upon varying the pressure. Fig. 3 shows the optical absorption and PL spectra of the FAPbBr3 crystal at various levels of pressure. Notably, the FA+ cation exhibits two phase transitions at 0.53 GPa (from Pmm to Im3) and 2.2 GPa (from Im3 to Pnma) during compression, followed by amorphization above 4 GPa, and, finally, a full reversion to its original cubic phase under decompression.93 These novel structural reorganizations can unlock many unique properties that are difficult to attain in normal phase transitions, thus opening a new frontier in optoelectronic applications.
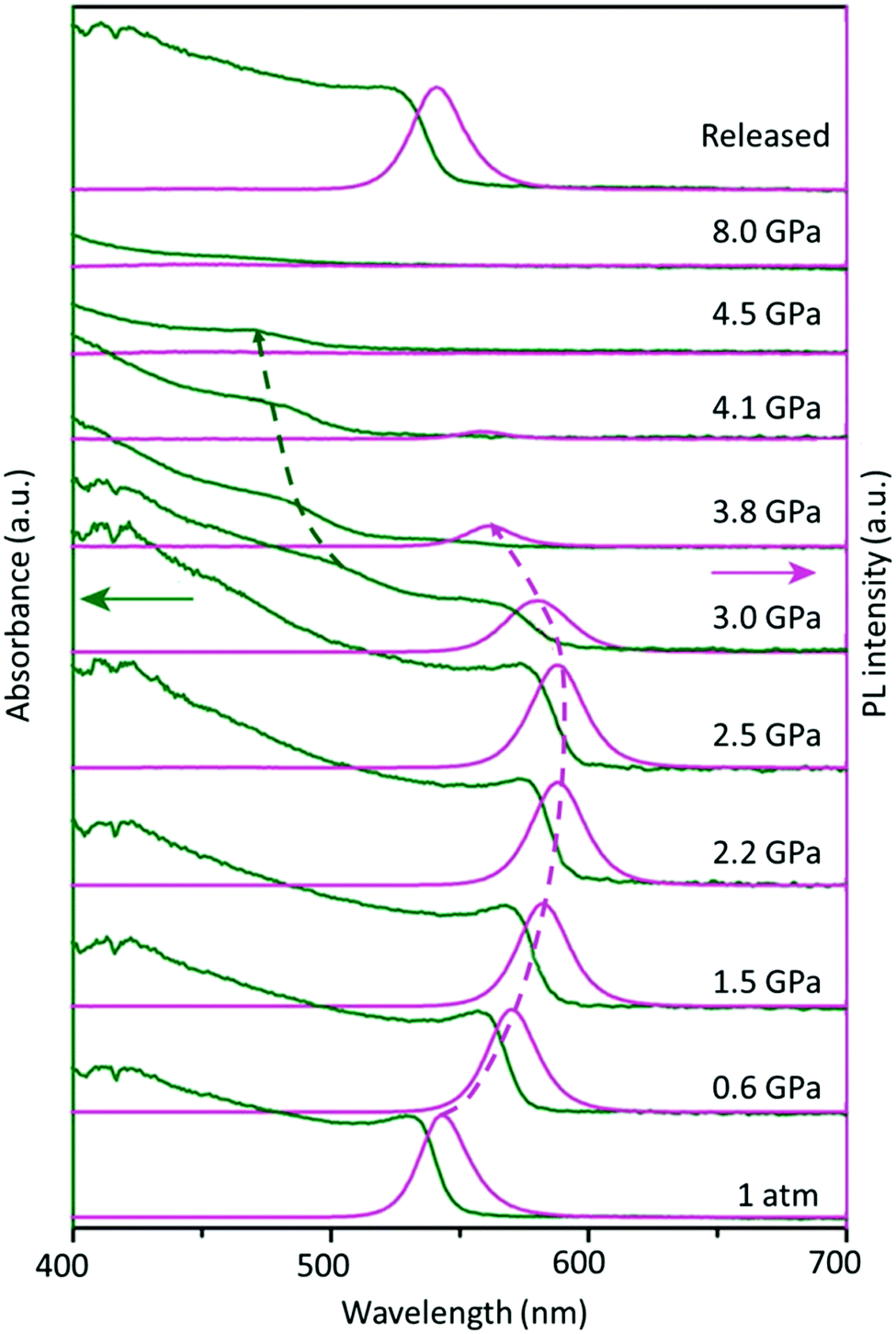 | ||
| Fig. 3 Changes in the optical absorption (green arrow) and PL spectra (pink arrow) of FAPbBr3 under pressure. Figure adapted from ref. 93. Copyright 2016 American Chemical Society. | ||
2.2 Synthesis of FAPbBr3 nanocrystals
Nanocrystals comprise hundreds to many thousands of atoms. Such particles are typically synthesized in solution and consist of surface ligands, i.e., molecules that bind to the surface, with the head group attached to the crystal surface and the hydrocarbon tail oriented in the opposite direction. The commonly used head groups are amino, carboxyl, thiol, etc.94–97 Surface ligands form a ‘capping’ layer that saturates the dangling bonds at the surface, aiding the colloidal stability and preventing the aggregation of NCs. Importantly, the synthesis of uniformly sized NCs is crucial, especially for application in optical devices, because size distribution can be directly related to the homogeneity of their chemical and physical properties. In particular, for an ensemble of NCs, a size discrepancy may lead to inhomogeneous properties, such as broadening of the optical spectra.98The synthesis of high-quality and uniformly sized NCs with excellent control over the shape, size, and composition is critical for the fabrication of high-performing LEDs as non-uniformity leads to broad emission peaks and poor color purity.98,99 Currently, the most commonly used techniques for the synthesis of FAPbBr3 NCs are the ligand-assisted reprecipitation (LARP)100,101 and hot injection (HI)59,102 methods. Schematic illustrations of these methods are shown in Fig. 4. Both of these methods are considered a bottom-up approach, which is effective for controlling the morphology of the FAPbBr3 NCs, such as nanowires,103,104 nanocubes,101,105,106 and nanosheets (Fig. 4, blue block).107–110 These techniques broadly follow the classical models of nucleation and growth mechanisms. Typically, in the nucleation step, the precursors react to form the supersaturated solution of monomers followed by a burst of nucleation for the NCs.
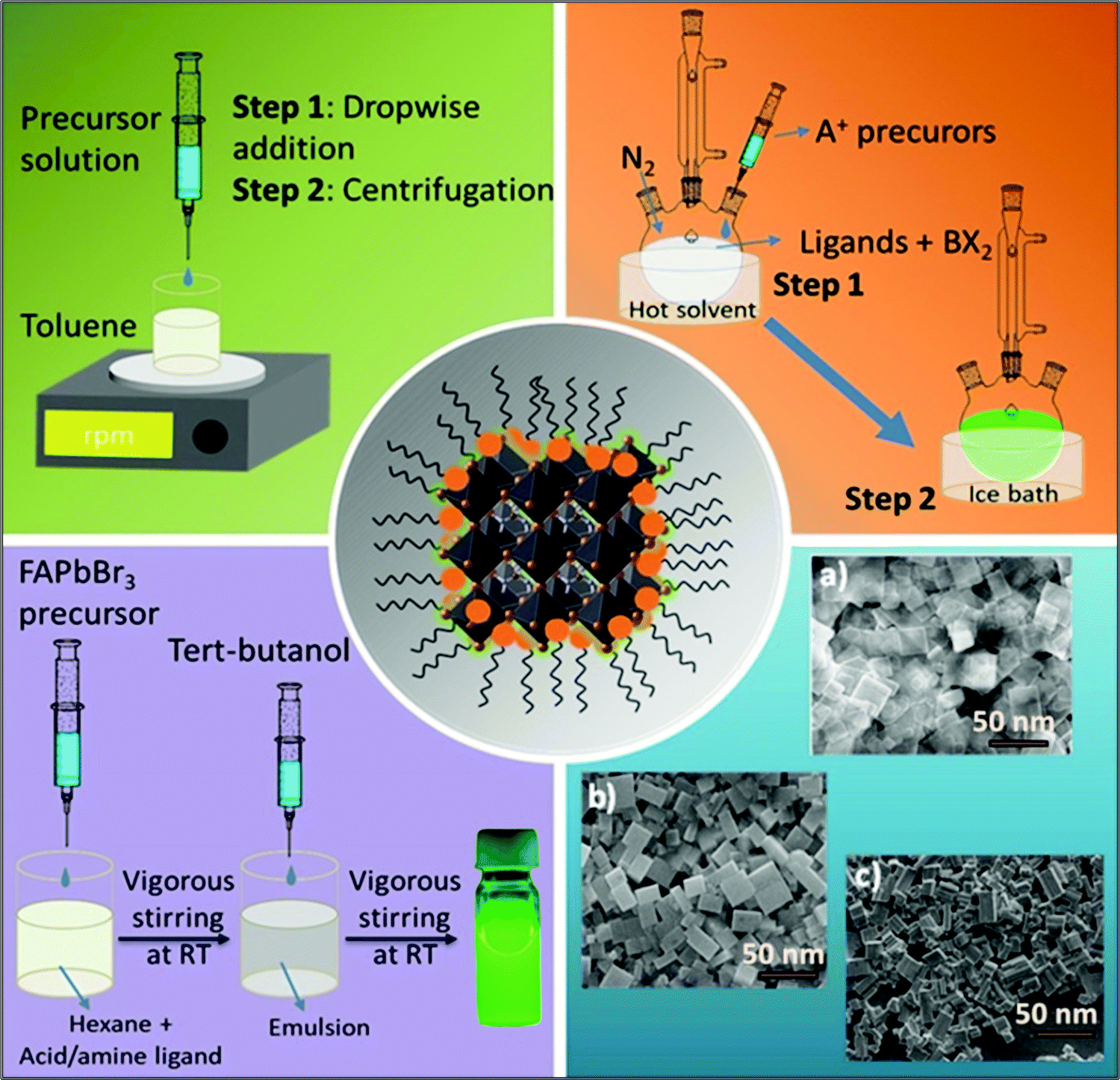 | ||
| Fig. 4 Schematic representation of the reaction system for the synthesis of colloidal perovskite NCs via ligand-assisted reprecipitation (LARP) (green), hot injection (HI) (orange), the emulsion method (violet), and SEM images of the various morphologies (blue), such as (a) nanoplatelets, (b) nanocubes and (c) nanorods, that are realizable using the different synthesis protocols. | ||
The growth of the nuclei is then followed by consuming the residual monomers present in the reaction medium.111,112 Zhang and co-workers were the first to report the LARP method for synthesizing LHP QDs (CH3NH3PbX3).101 Typically for this approach, the formation of NCs is accomplished at room temperature by first dissolving the precursors in a polar solvent (DMF, DMSO, or acetonitrile) along with organic ligands. The precursor solution is then injected into a non-polar solvent (usually toluene or hexane) to promote the nucleation and growth of the NCs (Fig. 4, green block).113 They found that long-chain ligands are necessary to achieve high-quality MAPbBr3 NCs. Following that, Perumal et al. communicated the first report on FAPbBr3 NCs synthesized via the LARP method utilizing long-chain surface ligands, octylamine (OTA), and oleic acid (OAc), thereby promoting the precipitation process. The obtained NCs had an average particle size of 10–15 nm and exhibited a green emission at 530 nm, which is blue-shifted by 10–20 nm compared with that of bulk FAPbBr3. In the solution-phase, the NCs exhibited a PLQY of 55–65% with a narrow FWHM of ∼22 nm.100
The HI technique, on the other hand, was first reported by Murray et al. in 1993 for the synthesis of cadmium chalcogenide NCs and was later adopted to synthesize a variety of NCs.116–118 This route requires the rapid injection of a precursor solution into a hot coordinating organic solvent, initiating the formation of nuclei. As soon as the temperature of the solvent drops, the formation of new nuclei is interrupted, promoting the growth process of monodisperse nuclei (Fig. 4, orange block). Notably, this approach enables the synthesis of small NCs with a narrow size distribution range. Protesescu et al. synthesized highly monodisperse cubic-shaped FAPbBr3 QDs using the HI method (at 130 °C). By controlling reaction temperature and the amount of injected oleylammonium bromide (OAmBr), they were able to tune the size of the QDs from 5 to 50 nm, which resulted in a wide range of emission wavelengths from 470 nm to 535 nm, where the smallest size corresponds to the largest bandgap.59 Currently, most of the synthesis routes for FAPbBr3 NCs are variants of HI and LARP methods. By contrast, the HI method needs a relatively high temperature (70–165 °C), an inert atmosphere, and a long preparation time, which makes it costly and difficult to control the reaction rate. On the other hand, the LARP method is a low-temperature process; however, it enables less control over the uniformity of the NCs morphology. Nevertheless, both techniques require perovskite precursor salts to be dissolved in polar solvents, such as DMF and DMSO, which leads to fast degradation of the perovskite NCs. A polar-solvent-free method would be ideal to avoid this issue. Apart from these two techniques, the non-aqueous emulsion method has recently been explored for the preparation of LHP NCs.119 Using this approach, a pair of immiscible polar and non-polar solvents, along with surface ligands, is mixed to form an emulsion. Next, a demulsifier (such as tert-butanol or acetone) is added to initiate the mixing of the precursors and the crystallization process. The resulting precipitate can be redispersed in a non-polar solvent to form a colloidal solution. Finally, by adding more demulsifier into the colloidal solution, the QDs can be precipitated and dried to a powder form (Fig. 4, violet block). Thus, control over the crystallization process and size can be achieved by varying the amount of demulsifier.119 However, they have not been many reports so far on the synthesis of FAPbBr3 NCs.
2.3 Tuning the optoelectronic properties of FAPbBr3 NCs
Since the two early reports on the LARP and HI methods, tremendous efforts have been devoted to synthesizing FAPbBr3 NCs with improved optoelectronic properties for light-emission applications. Delicate chromaticity engineering using the HI and LARP methods can be achieved by optimizing multiple factors such as the precursor dose, temperature, ligand species, reaction time, etc.51,120–124 In general, bulk FAPbBr3 has a direct bandgap of 2.34 eV with a corresponding emission wavelength of 535 nm. Upon excitation of an electron, the strong coulombic interaction between the electron–hole pair results in a large average distance of 8 nm between them (the exciton Bohr radius).100 Reducing the FAPbBr3 crystallite dimensions can confine these charge carriers within a de Broglie wavelength, which is less than twice the exciton Bohr radius. Such a reduction in size leads to a strong confinement of both the electron and hole wavefunctions (as they have a similar effective mass), giving rise to discrete quantized energy states, which can be observed in optical absorption and emission spectra.126 Moreover, with the aim of light-emission applications, several parameters generally need to be optimized for obtaining high-quality NC-based thin films, such as (i) small crystallite sizes (∼1–10 nm) to strongly confine the excitons and spatially limit the exciton diffusion length, which reduces the possibility of dissociation of the exciton into free charge carriers; (ii) pinhole-free uniform film fabrication, comprising uniformly distributed grains and a smooth surface to avoid the substantial leakage current; (iii) a high Eb to strongly confine the excitons; and (iv) good emissive properties. It is critical to impede the energetic losses and curtail non-radiative recombination to further boost the efficiency of LEDs. To address these requirements, perovskite NCs can be tuned in several ways, such as by blending perovskites with a dielectric polymer, engineering the precursor stoichiometry, incorporating a bulky organic molecule with a low dielectric constant to promote the dielectric confinement inside the low-dimensional perovskites, and use of additives and surface ligands.127–131In an early report, Protesescu et al. showed that OAmBr provides a narrow size distribution compared with oleylamine (OAm), which promotes the deprotonation of FA+ in the synthesis. The bromide-containing OAmBr ligand provides an additional bromide source, which helps in providing better stability and a uniform size distribution, owing to the formation of oleylammonium oleate. Furthermore, the size of the NCs was further tuned by varying either the amount of OAmBr (a higher amount yielded smaller NCs) and the reaction temperature (a higher temperature yielded larger NCs). Consequently, the FAPbBr3 NCs exhibited a wide range of emission wavelengths, ranging from 470 to 550 nm, depending on their size.59 Using a similar approach, Paul et al. utilized N-bromosuccinimide (NBS) as a bromide source to obtain uniformly sized FAPbBr3 nanocubes with a near-unity PLQY. Specifically, the reaction of NBS with organic acids is known to generate HBr, which reacted with OAm to form OAmBr (Fig. 5a). Interestingly, the nanocubes retained nearly 85% of their initial PL after 60 days of storage in an ambient atmosphere and 80% of the initial PL after 24 h of continuous illumination. The excellent stability was attributed to the bromide-rich environment and a partial replacement of FA+ with alkylammonium cations, forming an oleylammonium lead bromide-terminated surface.114 Similarly, Mahankudo and co-workers employed three precursor HI methods to synthesize FAPbBr3, wherein 3-dibromo-5,5-dimethylhydantoin (DBDMH) was employed as an additional bromide source. The obtained NCs yielded a high PLQY with excellent stability under ambient conditions.135
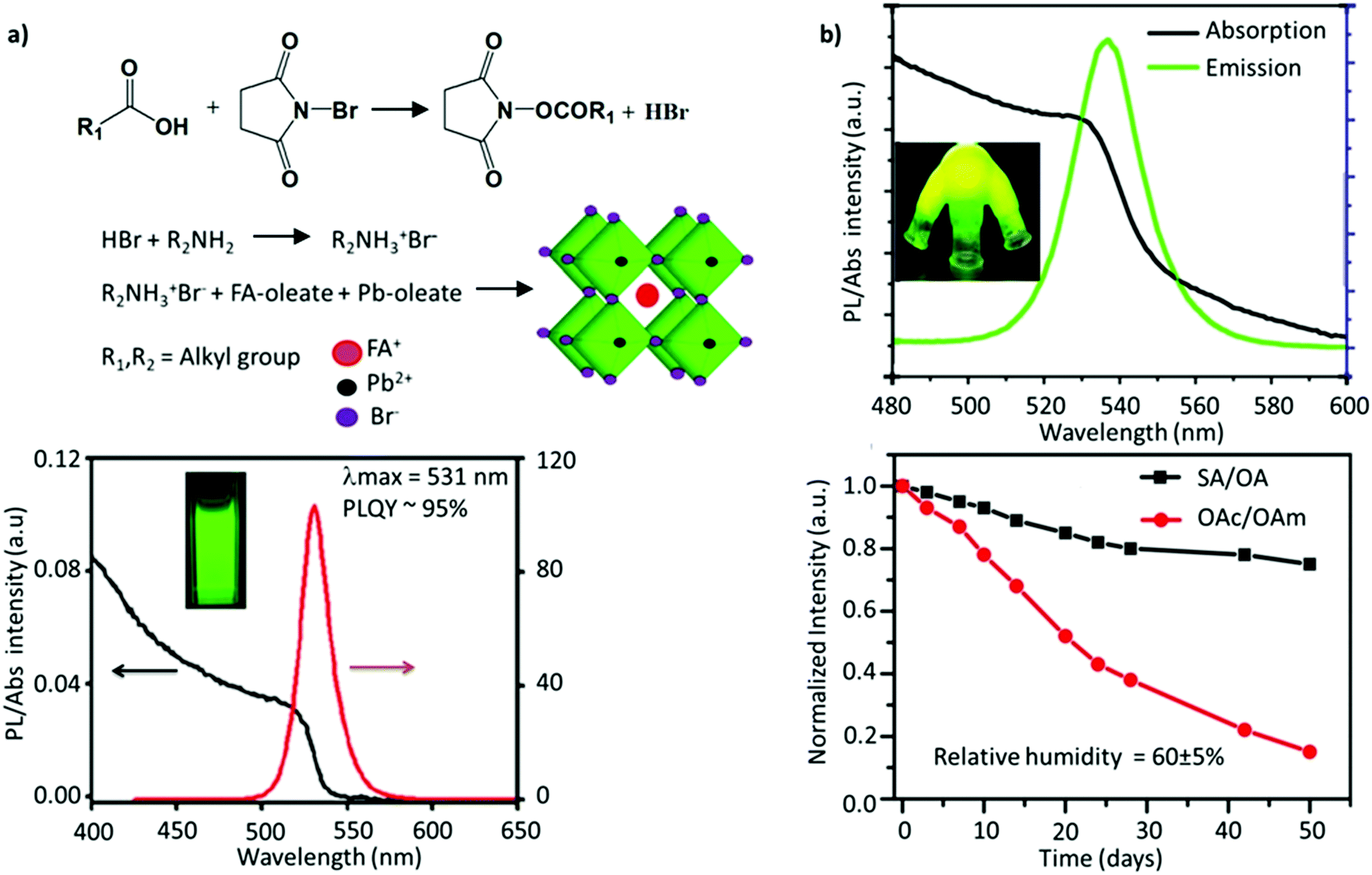 | ||
| Fig. 5 (a) Reaction scheme of NBS-mediated synthesis of FAPbBr3 NCs (top), and PL emission and absorption spectra of FAPbBr3 NCs (bottom). (b) PL and absorption spectra of FAPbBr3 quantum dots (top), where the inset shows the flask containing the solid-state ligands irradiated under UV light, and the change in PL intensity as a function of the storage time for FAPbBr3 (bottom). SA and OA denote stearic acid and octadecylamine, respectively. OAc and OAm denote oleic acid and oleylamine, respectively. Panel (a) reprinted with permission from ref. 114. Copyright 2019 American Chemical Society. Panel (b) adapted from ref. 115. Copyright 2017 American Chemical Society. | ||
Kim and co-workers investigated the effect of different lengths of the hydrocarbon chain in the amine ligand (n-butylamine, n-hexylamine, or n-octylamine) upon the formation and optoelectronic properties of NCs.134 They observed that the suppression effect of the ligands on the perovskite crystal growth was saturated when the chain length was longer than that of n-hexylamine. In fact, NCs prepared with a shorter chain ligand, n-butylamine, yielded the highest PLQY. Notably, ligand lengths shorter than n-butylamine yielded a weak luminescence and stability, while longer ones exhibited (1) steric effects, limiting the full passivation of the surface, and (2) surface effects, which intensify on decreasing the size of the NCs, leading to an increase in the defect density. The role of ligands in tuning the shape and size was further shown by Patra et al.109 They employed α-bromoketones (phenacyl bromide) as a source of both the ligand and halide, which when reacted with OAm yielded the formation of tertiary ammonium bromide ligands. In addition, a smaller amount of FA+ relative to Pb2+ was demonstrated to be crucial for obtaining a wide morphology range. Specifically, upon varying the ratio of FA+ and tertiary ammonium ions, cubic, dodecahedral, and quasi-spherical shapes of the FAPbBr3 nanocrystals were obtained with excellent monodispersity. While the emission peak remains the same, the quasi-spherical particles exhibited the highest PL lifetime among these shapes.109 The common issue with these monodentate ligands is that they are not tightly bound and usually undergo exchange between their bound and free states. This implies that the ligands are prone to undergo a complex protonation–deprotonation process that may induce detachment of the ligands during cleaning or long-time storage, engendering stability issues.136–140 To address this, adopting ligands with multiple coordinating groups is anticipated to resolve this issue. Recently, studies have demonstrated that bidentate ligands, such as dicarboxylic acid141 and phosphonic acid,142 can tightly bind to the surface, minimizing the ligand loss and resulting in high NC stability and a high PLQY. Phosphonic acid ligands are known to bind strongly to metal ions, like Pb, as has previously been shown for improving the stability of the PbSe surface against oxidation.143 Li et al. demonstrated that phosphonic acid ligands could form stable, strong hydrogen bonds with the halide ions of perovskites, P–OH⋯X.144 Recently, Ashton et al. synthesized FAPbBr3 NCs through a low-temperature, less energy-intensive, phosphine oxide route, where the traditional amine ligand is replaced by trioctylphosphine oxide (TOPO).145 This is the first study that investigated the impact of the A-site cation, FA+, on the acid–base equilibrium of the ligands, TOPO and OAc. Due to the presence of labile protons, there are two competing interactions between the FA-oleate:TOPO and OAc:TOPO complex, leading to an overall exchange of OAc:TOPO with FA-oleate:TOPO, furthering the formation of FAPbBr3 NCs. Unfortunately, the intercalation of TOPO between the layers of FAPbBr3 resulted in modest PLQY values and less uniform nanocubes.146 A similar intercalation mechanism has previously been observed for DMSO, forming FA+–DMSO–FA+ organic spacers and resulting in mixed-dimensional structures. Notably, the binding group in DMSO is similar to that of TOPO.147 Ambroz et al. excluded all the downsides of the conventional synthesis route, such as the use of polar solvents, amine ligands, or high-temperature synthesis. They synthesized FAPbBr3 NCs by replacing the traditional pair of ligands with phosphine-based alternatives, TOPO and di-iso-octyl phosphinic acid, and using octadecene (ODE) as the solvent. Although the PLQY was around 45%, the Pb–O–P bond on the surface could be easily removed after anti-solvent treatment.148 Sun and co-workers employed solid-state, high-melting-point (50–70 °C) ligands, such as stearic acid (SA) and octadecylamine (OA) to cap the FAPbBr3 QDs. This approach eliminates the absorption–desorption of ligands, as commonly observed when using conventional liquid ligands (OAc/OAm), which have a melting point of 15–25 °C. The stronger adhesion of these ligands to the perovskite surface resulted in a PL emission at 537 nm, a high PLQY of 88%, and a high color purity (FWHM ≈ 21 nm) (Fig. 5b). Moreover, the perovskites remained stable in the air for at least 50 days.115 Besides, the ligands also help in tuning the exotic optoelectronic properties. For instance, the partial replacement of oleylamine with (R)-2-octylamine during the synthesis of FAPbBr3 NCs resulted in monodisperse chiral perovskite NCs that emit circularly polarized luminescence (CPL) with a luminescence dissymmetry (g) factor of 6.8 × 10−2, which is one of the highest values among perovskite materials. Table 1 summarizes the impact of ligand engineering and the synthesis routes on the optoelectronic performance.
| Composition | Synthesis route | Ligands | PLQY (%) | Emission maximum (nm) | FWHM (nm) | Ref. |
|---|---|---|---|---|---|---|
a Values obtained for FABr![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PbBr2 = 2.2:1.
b Values obtained for rod-shaped FAPbBr3 NCs.
c Values obtained for FAPbBr3 NPLs + PMMA composite. :PbBr2 = 2.2:1.
b Values obtained for rod-shaped FAPbBr3 NCs.
c Values obtained for FAPbBr3 NPLs + PMMA composite.
|
||||||
| Ligand engineering | ||||||
| 3D FAPbBr3 QDs | HI | OAmBr/OAc | 85 | 530 | 22 | 59 |
| 3D FAPbBr3 NCs | HI | OAm/OAc | 95 | 531 | 20 | 114 |
| 3D FAPbBr3 NCs | Emulsion (RT) | Butylamine/OAc | 72 | 528 | 27 | 134 |
| Hexylamine/OAc | 69 | 522 | ||||
| Octylamine/OAm | 64 | 522 | ||||
| 3D FAPbBr3 NCs | HI | TOPO/OAc | 70 | 518 | — | 145 |
| 3D FAPbBr3 QDs | HI | SA/OA | 88 | 537 | 21 | 115 |
| 3D FAPbBr3 NCs | HI | OAm/OAc | ∼91–95% | 532 | ∼22 | 135 |
| 3D FAPbBr3 NCs | HI | OAmBr/OAc | 85.8 | 525 | 24 | 2 |
| Compositional engineering | ||||||
| 3D FAPbBr3 NCs | LARP | OTA/OAc | 74a | 532 | 22 | 125 |
| FAPbBr3 NCs | LARP | OAm/OAc | 70b | 496, 530 | — | 122 |
| FA0.8Cs0.2PbBr3 NCs | LARP | OTA/OAc | 65 | 525 | ∼20 | 153 |
| FA0.8Cs0.2Pb0.7Br3 NCs | LARP | OAm/OAc | 70–85 | 522 | ∼20 | 154 |
| FAPb0.8Sn0.2Br3 NCs | LARP | OAm/OAc | 97.5 | ∼530 | ∼23 | 155 |
| FAPb(Cl0.8Br0.2)3 NCs | Microfluidic | OAm and OAc | 56 | ∼512 | ∼27 | 136 |
| FAPbX3 NCs | One-step homogeneous (RT) | Tetraoctylammonium bromide/OAc | 76 (FAPbBr3) | 518 | 20 | 156 |
| FAPbBr3 NCs | Emulsion | OTA/OAc | 88 | 538 | 22 | 157 |
| FAPbBr3/FAxCs1−xPbBr3/CsPbBr3 | HI | OAc/OAm | 82 | 532 | 21 | 8 |
| (FAPbBr3) | 504 | 18 | ||||
| 93 (x = 1) | ||||||
| FA0.9GA0.1PbBr3 | LARP | Decylamine/OAc | 93.3 | ∼533 | — | 158 |
| Dimensional engineering | ||||||
| 2D FAPbBr3 NPLs (n = 7–10) | LARP | OTA/OAc | 92c | 530 | 22.6 | 159 |
| 2D FAPbBr3 NPLs (n = 7–10) | HI | 2-Methyl-1-pentanol | 92 | 531 | 22 | 160 |
| 2D FAPbBr3 NPLs | Ion-exchange-mediated self-assembly | OA | 85 | 525 | 25.3 | 161 |
| 2D FAPbBr3 NPLs (n = 1–4) | LARP | OTA/OAc | 91 | 532 | 21 | 60 |
| 2D FAPbBr3 NCs (n = 4) | LARP | SBE-18 | 90.6 | 534 | 20.5 | 162 |
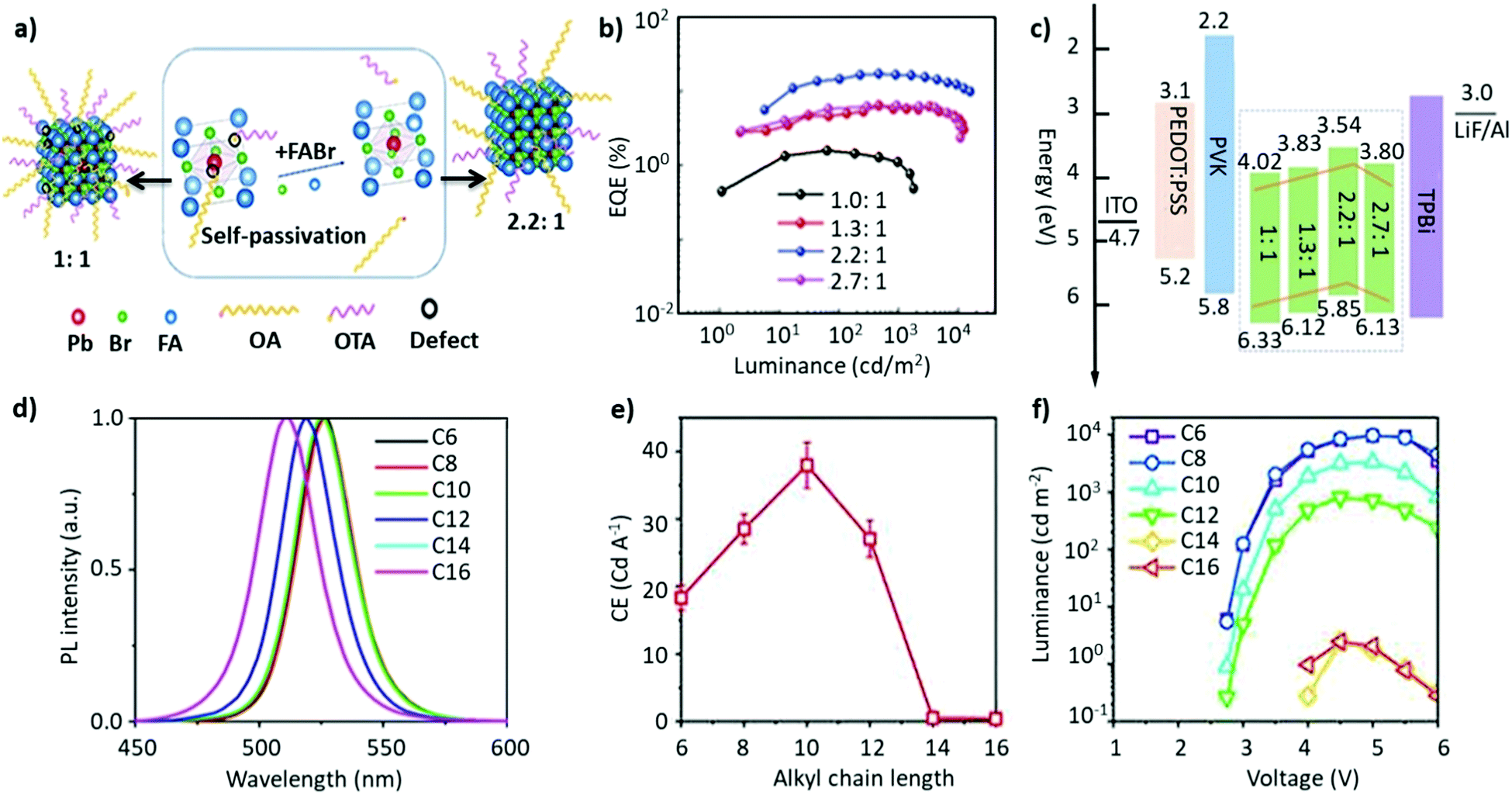
Non-stoichiometric composition. Although an equimolar ratio of FABr and PbBr2 is desirable to form a stoichiometric FAPbBr3 composition, it was found that a non-stoichiometric composition can play an important role in the thin-film morphology and defect characteristics in NCs. For example, it has been found that the presence of metallic lead (Pb0), which negatively impacts the optoelectronic performance of perovskites by creating non-radiative decay pathways, is prevalent when an equimolar precursor ratio is used.149–152 This unintended loss of halide atoms can be easily mitigated through the use of excess halide precursors, which can prevent the formation of metallic Pb atoms in the final perovskite product. Chen and co-workers varied the ratio of FABr/PbBr2 from 1 to 2.7 during the synthesis of FAPbBr3 NCs. With increasing FABr/PbBr2 ratios from 1.0 to 2.2, the emission peak shifted from 524 to 532 nm, respectively. However, when the ratio was further increased to 2.7, the emission peak shifted back to 529 nm with the PLQY increasing from 62% to 78%. The high PLQY is attributed to the self-passivation effect of FA, while the shift in the emission peak was attributed to the different sizes of the NCs (Fig. 6a).125 Despite there being no information regarding the 2D structure, the reader can notice a small absorption band around 430 nm, which could be an indication of the 2D structure. Moreover, the addition of higher molar amounts of FABr did not cause any change in the morphology and crystal structure, as we noticed in our study.122 The observed discrepancies could stem from the different synthesis recipes. However, this strategy is advantageous as the use of a great quantity of insulating ligands could be avoided as FABr itself heals and stabilizes the structure. Similarly, we have demonstrated a novel morphology-tunable synthesis of FAPbBr3 NCs utilizing different stoichiometric ratios of the precursors.122 An excess of FABr yielded a rod-shaped morphology, which possesses a quasi-2D structure, while an equimolar ratio of the precursors yielded a typical cube-shaped morphology with a cubic crystal structure. The study revealed that in the presence of an excess of FA+, proton exchange occurs within amine-based systems via homoconjugation (FAH+⋯OAm), forming the oleylammonium cation and promoting the formation of an additional 2D structure (Fig. 6b). The presence of the quasi-2D component significantly improved the PLQY from 30% to 70%. The improvement in PLQY is attributed to the presence of an abundance of Br at the surface and also to the quasi-2D crystal structure. Moreover, for the first time, a monoclinic phase was observed for this type of FAPbBr3 nanorod.122
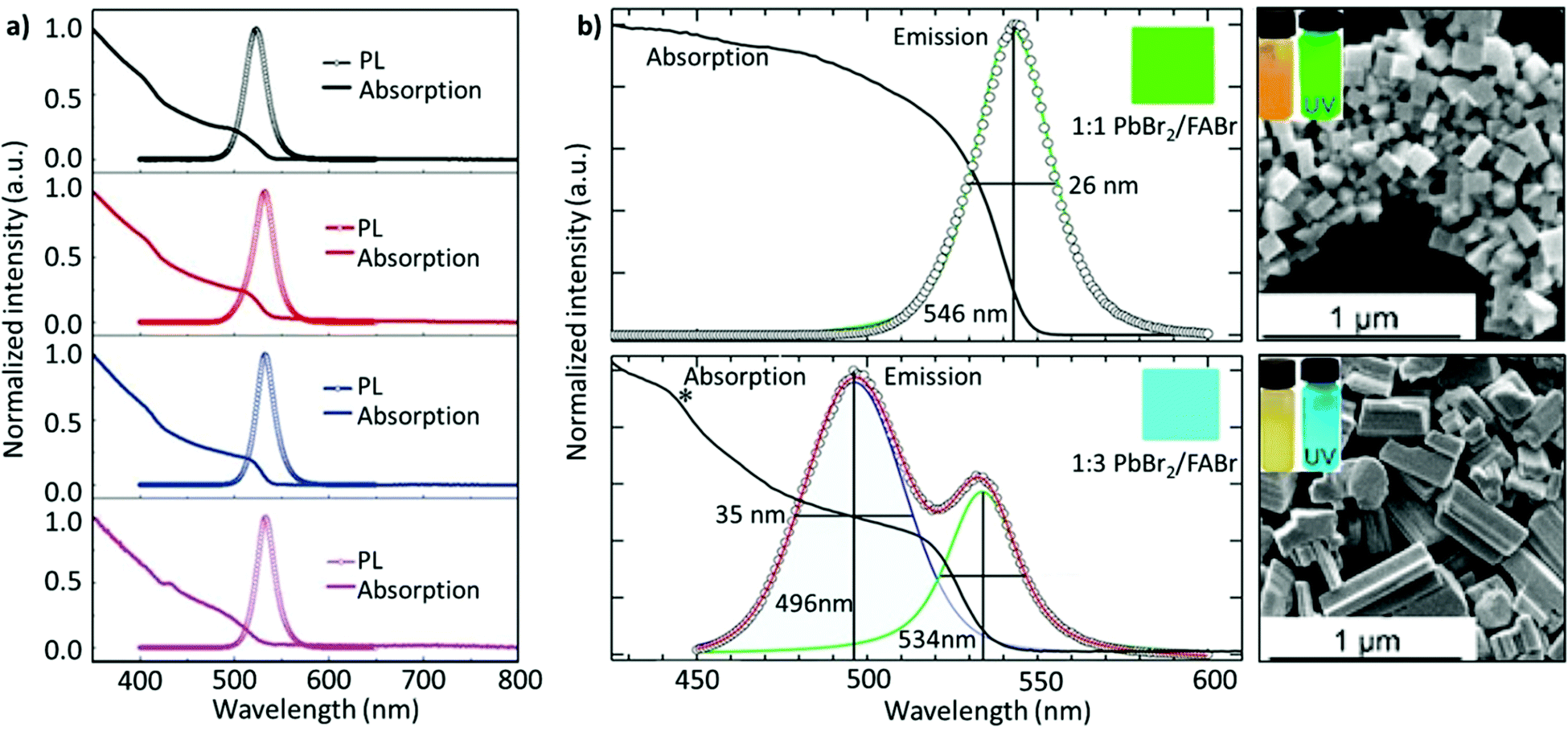 | ||
| Fig. 6 (a) PL and absorption spectra of FAPbBr3 with different FABr/PbBr2 ratios, which exhibit a red shift on increasing the FABr amount (in the spectra from top to bottom). (b) PL and absorption spectra of the corresponding nanocubes, prepared using 1:1 PbBr2/FABr (top) and of the corresponding nanorods prepared using 1:3 PbBr2/FABr (bottom). The insets in SEM images are pictures of the suspensions under visible (left) and 365 nm (right) UV illumination. Panel (a) reprinted with permission from ref. 125. Copyright 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. Panel (b) adapted from ref. 122. Copyright 2019 American Chemical Society. | ||
A-site mixing. In addition to applying FABr-rich conditions, the mixed A-site cation strategy has also helped in boosting the stability and optoelectronic performance of LHP NCs.163–169 For instance, an incremental partial replacement of FA+ cations with Cs+ yielded a blue shift in the peak position (CsxFA1−xPbBr3), from 531 to 519 nm, with a narrow FWHM of ∼19–23 nm (Fig. 7a). The highest PLQY value of 73% was obtained for x = 0.1, which gradually decreased upon further increasing the amount of Cs+ cations. Structurally, a gradual increase in Cs content brought about a slight shift in the XRD reflections towards higher 2θ values, indicating the shrinking of the lattice, owing to the smaller ionic radius of Cs. In addition, doping Cs in the FAPbBr3 crystal structure imparted a higher stability in the structure. Typically, the pristine FAPbBr3 NC film, when drop cast freshly under ambient conditions, would degrade quickly (visually turning green to yellow to orange); however, the Cs-incorporated films remained stable in air. Unfortunately, an excess amount of Cs (x > 0.2) increased the non-radiative energy transfer rate and drastically deteriorated the film morphology.153 This work was later extended, wherein, the concentration of lead was varied in the FA0.8Cs0.2PbxBr3 composition (x = 1.0, 0.8, 0.7, and 0.6). As the lead content was decreased from 1.0 to 0.6, a blue shift in the emission peak, from 526 to 522 nm, was observed (Fig. 7b). This kind of stoichiometric modification, having cation-rich and lead-deficient precursors (x = 0.6), introduced a secondary phase of FA4PbBr6/Cs4PbBr6 in the final product, which could potentially impact the radiative recombination rate. Alongside this, an interesting observation was made via an XPS study in which it was revealed that the peak intensity of metallic lead (∼137 eV) was drastically reduced for the lead-deficient samples (x = 0.8, 0.7, and 0.6). This way the non-radiative recombination caused by uncoordinated Pb atoms is inhibited.154 Recently, Kim et al. explored the substitution of FA+ in FAPbBr3 by single guanidinium (GA+) cations, where the extra amino group in GA+ interacts with lattice halides through hydrogen bonding, promoting surface stabilization. It was found that this strategy not only created an entropy-stabilized phase but also yielded smaller monodisperse NCs.158
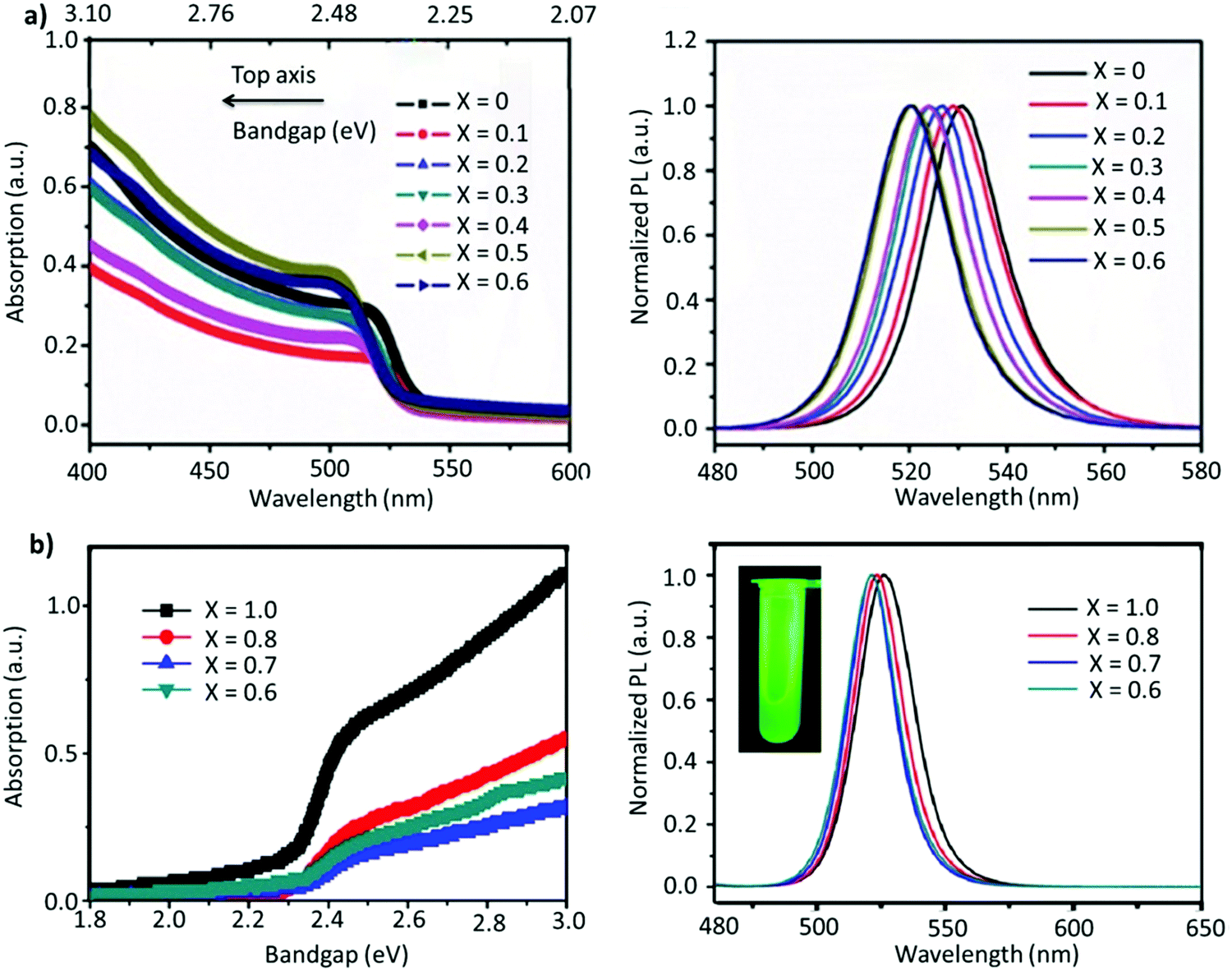 | ||
| Fig. 7 (a) Absorption (left) and PL spectra (right) of FA(1−x)CsxPbBr3 (x = 0–0.6). (b) Absorption (left) and PL spectra of perovskite (right) with variation of the proportion of Pb in FA0.8Cs0.2PbxBr3. The inset is the photo of the as-formed perovskite nanocrystals under ultraviolet excitation. Panel (a) reproduced with permission from ref. 153. Copyright 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. Panel (b) reproduced with permission from ref. 154. Copyright 2018 American Chemical Society. | ||
B-site mixing. In view of the toxicity of lead, tin (Sn) has been considered a promising substitute for lead, since both of them belong to the same group in the periodic table and possess a similar ionic radius, i.e., Pb2+ ≈ 119 pm and Sn2+ ≈ 118 pm.170 However, Sn-based perovskites are remarkably prone to oxidation, converting from Sn2+ to Sn4+ and thus deteriorating the optoelectronic properties.171 While earlier studies were focused on the Cs-based172,173 or MA-based174,175 perovskites for the Pb–Sn mixed composition, FAPb1−xSnxBr3 NCs176,177 have also gained momentum recently, along with partial Pb replacement by other metals in the perovskite structure.178–181 Cai et al. reported the facile synthesis of FAPb1−xSnxBr3 NCs at RT using a modified LARP approach.155 They found that the crystal structure remained cubic until 50% of Pb had been replaced by Sn; however, on further increasing the Sn concentration, the cubic phase partially lost its high symmetry structures. Interestingly, as the x content was increased from 0 to 0.5, the emission spectra were blue-shifted, with peak maxima at 534 nm (x = 0) and 516 nm (x = 0.5), and a high PLQY of up to 97% was yielded for the x = 0.2 composition before it plunged with a higher x content. Furthermore, the decay time also increased from 161 ns (x = 0) to 182 ns (x = 0.2) and then drastically reduced to 40 ns (x = 0.5), indicating a rise in non-radiative trap states.
X-site mixing. Another prevalent way to tune the optoelectronic properties of LHPs is to blend the halide ions. However, this approach usually exhibits unwanted phase segregation into domains with different ion contents and bandgaps, under electric field or light irradiation, which is deleterious to optoelectronic applications. Kovalenko's group studied the growth mechanism, shape evolution, and halide-ion segregation of FAPbX3 (X = Br/Cl) NCs and NPLs by employing an automated droplet-based microfluidic method.136 This route of synthesis enables in situ absorption and PL spectroscopy that assists in understanding and fine-tuning the optimal synthesis parameters. Specifically, they varied the ratio of FA and Pb, the ratio of OAm and OAc, the reaction time, and the temperature to optimize the synthesis parameters. Their study revealed that a high content of Cl atoms in the FAPbBr3 structure (above 40%) results in fast halide-ion segregation, causing instability in the optical properties.136 Next, an ion-exchange reaction was performed by blending the as-prepared FAPbBr3 NCs with PbX2 (X = Cl and I) as a post-synthetic transformation. This strategy yielded a series of NCs with tunable emission wavelengths ranging from 452 to 646 nm. The XRD patterns remained unchanged upon varying the composition. However, the mixed-halide composition exhibited a lower PLQY compared with FAPbBr3, indicating degradation and phase separation.156 Pisanu et al. utilized the mixed-cation system FA0.8Cs0.2PbBr3 to perform a halide-exchange reaction, using PbI2 and PbCl2 as a post-synthetic step at room temperature.182 They could modulate the emission wavelength simply by varying the stirring time, starting from 5 to 30 minutes, and covering a wide range of the visible spectrum from 473 nm to 605 nm. This way the high quality of the NCs was preserved, exhibiting very narrow FWHM values and, thus, serving as an alternative to complex halide-exchange reactions. The impact of mixed-cation and -anion compositions on the optoelectronic properties can be seen in Table 1.
:1 to 2.5:1, the PL emission was blue-shifted from 536 to 440 nm, which is a very large shift as compared with the previously mentioned study. Notably, in contrast to OAm,188 the thickness of the NPLs increased with an increasing amount of OTA during the synthesis. Bright-green PL emission was exhibited at 532 nm with a narrow FWHM of ∼21 nm and a high PLQY of 91% (Table 1).60
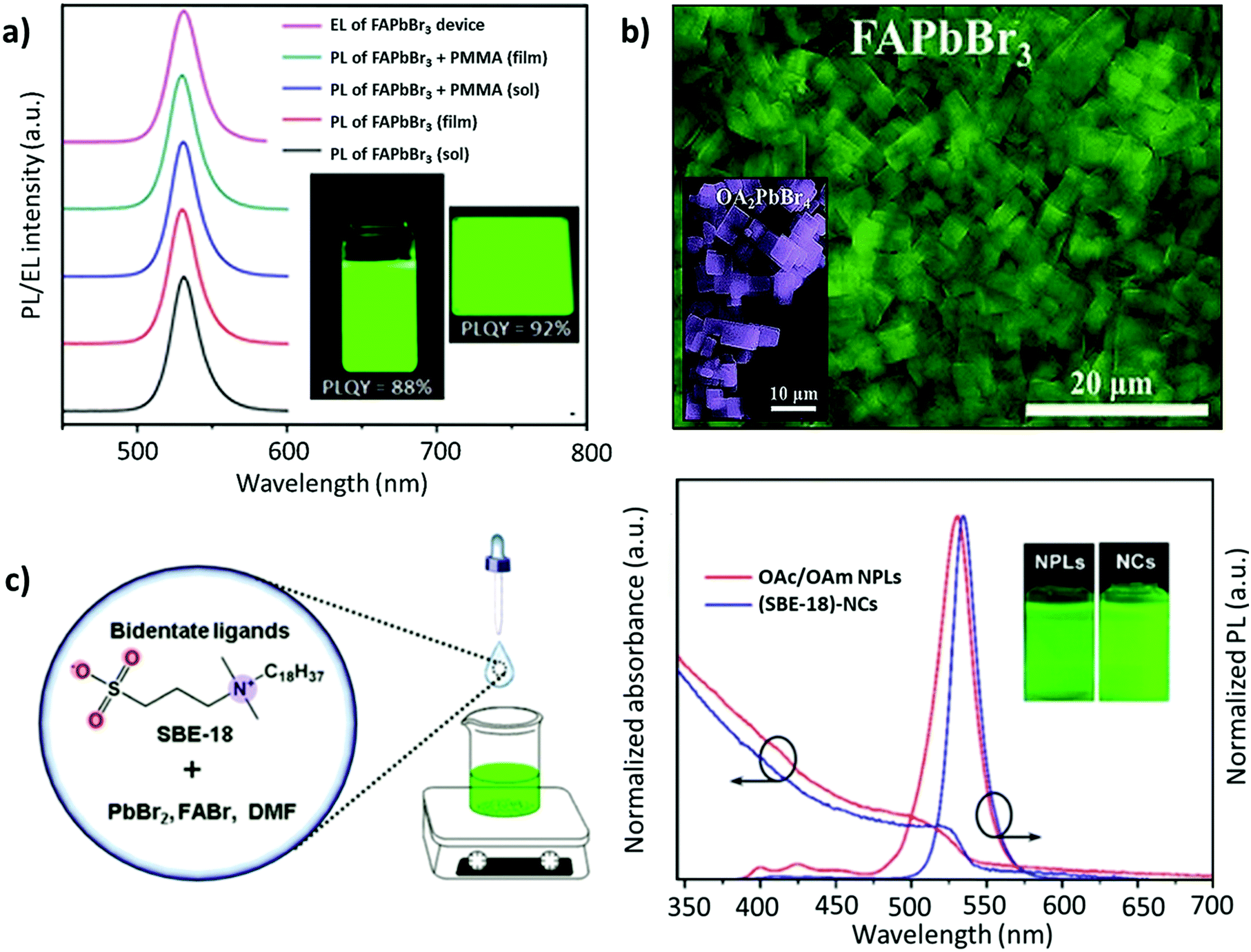 | ||
| Fig. 8 (a) PL spectra for the colloidal 2D FAPbBr3 perovskite and its composite with PMMA, where the inset depicts the 2D FAPbBr3 perovskite colloidal solution (left) and spin-coated thin film (right) under UV excitation in toluene solution and thin films. (b) SEM image of FAPbBr3 NPLs, where the inset depicts the SEM image of OA2PbBr4. (c) Schematic illustration of the synthesis process of FAPbBr3 NCs (left) and the PL absorption and emission spectra of (SBE-18)-capped FAPbBr3 NCs and OA/OAM-capped FAPbBr3 NPLs (right), with insets showing their colloidal solutions under 365 nm UV light. Panel (a) adapted from ref. 159. Copyright 2017 American Chemical Society. Panel (b) adapted from ref. 161. Copyright 2018 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. Panel (c) reproduced with permission from ref. 162. Copyright 2019 American Chemical Society. | ||
With a similar objective, Zhang and co-workers established a ligand system, containing OAc, hexanoic acid (HexAc), hexylamine (HexAM), and di-n-hexylamine (DHA) to prepare micron-sized 2D NPLs (n = 2). This study also demonstrated the impact of the ligand concentration on the morphology of NCs. Specifically, FAPbBr3 nanosheets were formed when the acid-to-amine ratio was 2:1. Notably, the nanosheets became much less stable with high amounts of OAc, HexAc, or HexAM. The high amount of DHA also accelerated the crystallization process, resulting in NCs with a heterogeneous morphology.110 Later, Zu and co-workers employed sulfobetaine-18 (SBE-18), which contains amino and sulphonate functional groups.162 This ligand was demonstrated to adhere more strongly to the surface of the NCs compared with FAPbBr3 NPLs capped with OAm/OAc, such that the PL emission did not change even after storing the NCs for 10 days. However, for the OAm/OAc-capped NPLs, the PL intensity dropped by about 48% of its initial value, while exhibiting a red shift of about 7 nm (Fig. 8c). The higher stability of the SBE-18-capped NCs is attributed to their high binding energy with NCs (40–45 kcal mol−1) and the chelating effect. Importantly, the synthesis approach yielded NCs with a high color purity (FWHM ≈ 20.5 nm) and a PLQY of up to 90.6%. Furthermore, they also explored the possibility of using the shorter carbon chain of the SBE-8 ligand; however, the passivation was not adequate and resulted in a low PLQY (55.9%).
3. Application of FAPbBr3 NCs in light-emitting diodes (LEDs)
For a typical PeLED device architecture, the emitter layer is sandwiched between electron and hole charge-transport layers, maximizing the charge confinement within the emissive layer. Under an applied voltage, electrons and holes are injected from the anode and cathode, which are transported through the charge-selective layers and then radiatively recombine inside the emitting layer to emit photons.189 The performance of LEDs depends on three major parameters: (i) the PLQY of the NC thin films, (ii) the injection efficiency of the charge-selective layers, and (iii) the interfacial recombination. Since NCs offer a combination of excellent optoelectronic and physical properties, they are considered potent candidates as emitter layers for next-generation displays and LEDs. By tuning the size and composition of the NCs, the emission wavelength can be conveniently tuned for a wide range of emission spectra. However, the realization of these properties in NC thin films is far from ideal. Due to the loss of ligands, solid-state thin films often suffer from poor surface coverage and morphology, which increases the non-radiative recombination and even promotes fast degradation of the devices. Moreover, the presence of ligands also impedes charge transport inside the NC thin films, resulting in non-radiative Auger recombination. Apart from the NC thin films, high-performing devices also require efficient charge injection from the charge-selective layers to the emitter layer. This can be achieved through proper band alignment of the charge-selective layers and utilizing additional carrier-blocking layers to effectively confine charges within the emitter layer. Recently, several outstanding advances in terms of the device architecture, EQE and stability have been realized, as summarized in Table 2.| Device architecture | EL (nm) | Voltage (V) | Luminous efficiency (cd A−1) | Maximum luminance (cd m−2) | EQE (%) | Stability | Ref. |
|---|---|---|---|---|---|---|---|
| a Device performance recorded for n-octylamine ligand. b Device performance recorded for n-butylamine ligand.TFB = poly[9,9-dioctylfluorene-co-N-[4-(3-methylpropyl)]diphenylamine], PVK = poly-N-vinylcarbazole, 3TPYMB = tris(2,4,6-trimethyl-3-(pyridin-3-yl)phenyl)borane, TAPC = 1,1-bis[(di-4-tolylamino)phenyl]cyclohexane, B3PYMPM = 4,6-bis(3,5-di(pyridin-3-yl)phenyl)-2-methylpyrimidine. | |||||||
| PEDOT:PSS/FAPbBr3/B3PYMPM/CsCO3/Al | 530.7 | 2.3 | 5.4 | 2714 | — | — | 100 |
| PEDOT:PSS/FAPbBr3/TPBi/CsCO3/Al | 533.4 | 2.8 | 6.4 | 465 | 100 | ||
| ITO/buffer hole injection layer/FAPbBr3/TPBi/LiF/Al (hole-only device) | 530 | 4.5 | 0.46a | 3.348a | 0.16a | — | 134 |
| 9.16b | 142.56b | 2.05b | |||||
| ITO/PEDOT:PSS/PVK/FAPbBr3/TPBi/LiF/Al | 536 | 3.6 | 76.8 | 466 | 17.1 | T 50 = 18 min | 125 |
| ITO/PEDOT:PSS/PVK/FAPbBr3/TPBi/LiF/Al | 545 | 3.9 | 20.3 | 33993 |
4.07 | T 50 = 6 min | 115 |
| ITO/PEDOT:PSS/TFB/FA0.8Cs0.2PbBr3/TPBi/LiF/Al | 525 | 3.5 | 10.09 | 55005 |
2.80 | T 50 = 80 s | 153 |
| ITO/PEDOT:PSS/TFB/FA0.8Cs0.2Pb0.7Br3/TPBi/LiF/Al | ∼530 | 3.3 | 28.61 | 45440 |
6.75 | T 50 = 4 min | 154 |
| ITO/PEDOT:PSS/FA0.5MA0.5PbBr3/3TPYMB/LiF/Al | 528 | 3.37 | 38.1 | 3322 | 16.2 | L 85 = 243 min | 193 |
| ITO/PEDOT:PSS/PVK/FAPb0.7Sn0.3Br3/TPBi/LiF/Al | 528 | 9.8 | 45.99 | 7492 | 11.38 | T 50 = 1.7 min | 155 |
| ITO/PEDOT:PSS/TFB/FAPbBr3 QDs/TPBi/LiF/Al | 545 | 3.3 | — | 403 | 2.8 | — | 157 |
| ITO/PEDOT:PSS/PVK/FAPbBr3 NCs/TPBi/LiF/Al | 532 | 3.8 | 85.4 | 67115 |
19.2 | T 50 ≈20 min | 199 |
| ITO/PEDOT:PSS/poly-TPD/FAPbBr3/FAxCs1−xPbBr3/CsPbBr3 NCs (x = 1)/TPBi/LiF/Al | 508 | 2.6 | 19.75 | 1758 | 8.1 | T 50 ≈48 min | 8 |
| ITO/PEDOT:PSS/PVK/FAPbBr3 NCs/TPBi/LiF/Al | 528 | 3.6 | 16.08 | 594.1 | 3.84 | — | 2 |
| ITO/PEDOT:PSS/FAPbBr3xClx/B3PYMPM/LiF/Al | 473 | 3.9 | — | 2810 | 3.1 | T 50 ≈ 74 s | 33 |
| ITO/PEDOT:PSS/PVK:TAPC/FAPbBr3 NPLs/TPBi/Ca/Ag | 532 | 3.53 | 15.31 | 3062 | 3.53 | T 50 = 32 min | 60 |
| ITO/PEDOT:PSS/2D FAPbBr3 NPLs + PMMA/3TPYMB/LiF/Al | 529 | 2.75 | 13.02 | 2939 | 3.04 | — | 159 |
| ITO/PEDOT:PSS/poly-TPD/2D FAPbBr3 NPLs/TPBi/LiF/Al | 534 | 4 | 17.32 | 7205 | 4.5 | — | 160 |
| ITO/Buf-HIL/FA0.9GA0.1PbBr3/TBTB/TPBi/LiF/Al | 531 | — | 108 | — | 23.4 | T 50 = 132 min | 158 |
3.1 Green-emitting FAPbBr3 LEDs
The utilization of FAPbBr3 NCs in LEDs was first reported by Perumal et al.100 They examined 2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole) (TPBi) and 4,6-bis(3,5-di(pyridin-3-yl)phenyl)-2-methylpyrimidine (B3PYMPM) as electron-transport layers (ETL), and poly(3,4-ethylenedioxythiophene):poly(styrene-sulfonate) (PEDOT:PSS) as the hole-transport layer (HTL) (Fig. 9a). The device with B3PYMPM exhibited bright EL of up to 2714 cd m−2, while TPBi exhibited a higher luminous efficiency of 6.4 cd A−1, and a luminous power efficiency of 5.7 lm W−1 (Fig. 9b–d).100 While the performance was modest, it is far from the standards required for commercialization or even comparable to OLEDs and QLEDs. The poor performance of the LEDs was attributed to the excess ligands and the poor NC film morphology. Although organic ligands are essential for NC synthesis, excessive ligands are detrimental to the charge-transport properties due to their insulating nature. Therefore, it is imperative to optimize the ligand density and chain length on the surface to realize efficient charge-transport and -injection properties.127–131 In addition, PeLEDs are mostly based on a mainstream device architecture that consists of the glass/ITO/PEDOT:PSS/perovskite/TPBi/LiF/Al composition, in light of the well-aligned band with the ETL and the HTL.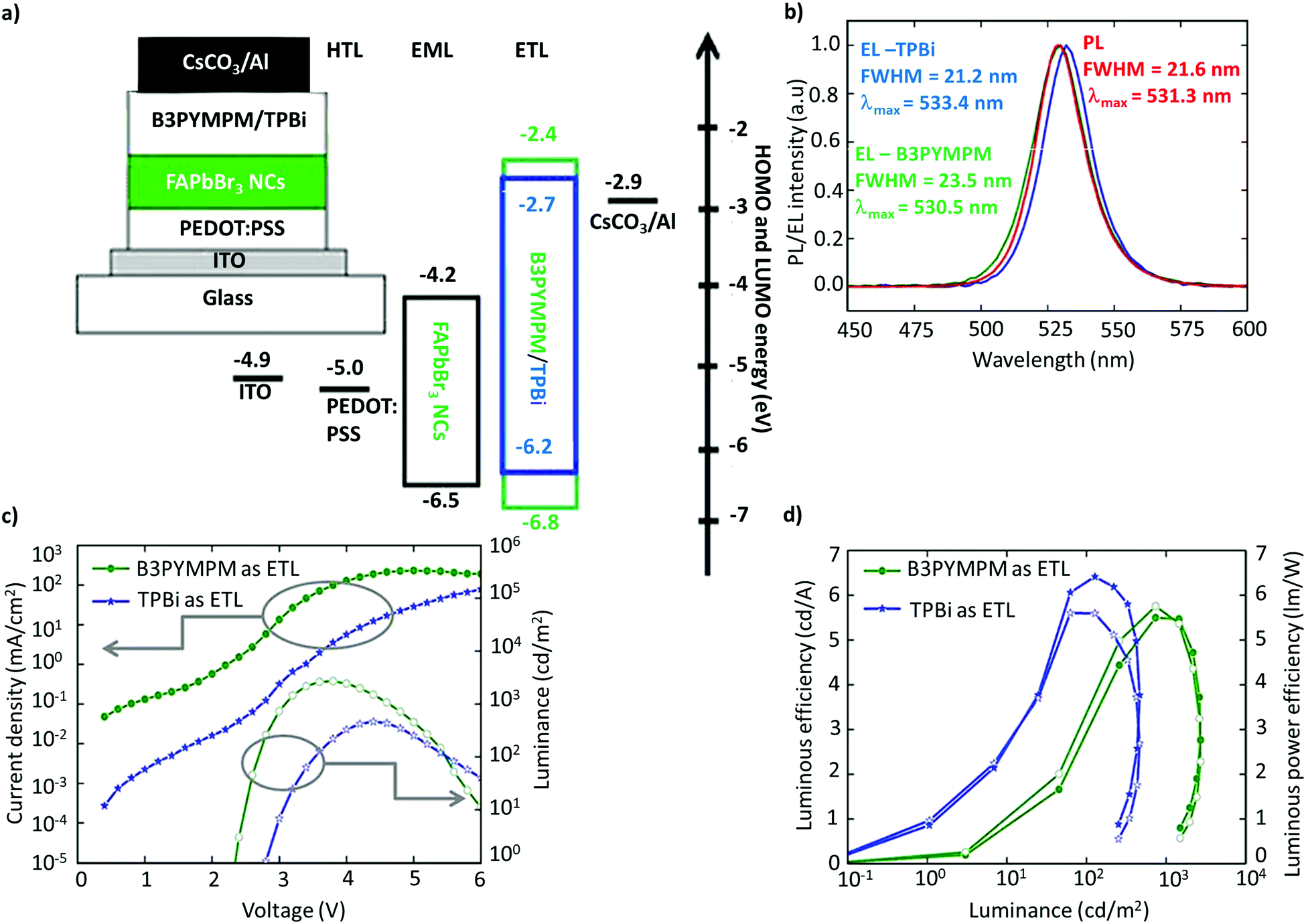 | ||
| Fig. 9 Schematic of the device architecture along with the energy levels. (b) PL spectra of FAPbBr3 NCs and the EL spectra of PeLEDs with B3PYMPM and TPBi as the ETL. (c) Current density/luminance vs. voltage characteristics of the devices with B3PYMPM and TPBi as the ETL. (d) Luminous efficiency vs. luminance and luminous power efficiency vs. luminance characteristics for PeLED devices with B3PYMPM and TPBi as the ETL. Figures from (a)–(d) are adapted from ref. 100. Copyright 2016 Springer Nature. | ||
However, the acidic nature of PEDOT:PSS and electrochemically active LiF and Al exacerbate the device instability.190,191 It has been demonstrated that the hygroscopic nature and the acidity of the PSS group cause the breakdown of the device upon exposure to oxygen and moisture.192 To this end, a tremendous amount of effort has been invested in optimizing the thin-film morphology, the ligands, the compositional engineering strategy, and the device architecture to improve the performance of PeLEDs. In an attempt to circumvent the use of excess ligands, a mixed-cation system has been considered as a potential approach for improving the device performance. Zhang et al. constructed PeLEDs based on the FA1−xCsxPbBr3 composition, where the perovskite characteristics and the device performance were strongly dependent on the Cs content. The optimized composition, with x = 2, exhibited a reasonable EQE of 2.8%, a luminance of 55005 cd m−2, and a current efficiency (CE) of 10.09 cd A−1 (Fig. 10a).153 Afterwards, Zhang et al. adopted a stoichiometric modification approach, where they utilized the aforementioned mixed A-cation composition (FA0.8Cs0.2PbxBr3), while decreasing the amount of Pb, to inhibit the non-radiative recombination caused by the presence of undercoordinated metallic lead atoms (Pb0). Interestingly, as the value of x was decreased from 1 to 0.7, the device performance and stability were greatly boosted, achieving maximum luminance, CE and EQE values of 45440 cd m−2, 28.61 cd A−1 and 6.75%, respectively (Fig. 10b). The enhanced performance is attributed to the improved hole-injection barrier at the interface between the perovskite and the HTL. Moreover, the elevated device stability is attributed to the hydrogen-bonding interaction between the organic cation and halide anions, triggered by the moderately adsorbed moisture, especially for lead-less perovskites with an organic-rich composition. Notably, a further decrease in the Pb content, from 0.7 to 0.6, deteriorated the device performance.154 Overall, the A- and B-site cations play a dominant role in affecting the device performance. For instance, a similar study based on lead-less perovskites (FAPb0.7Sn0.3Br3) exhibited a high EQE of 11.38% with a maximum luminous efficiency of 45.99 cd A−1.155 Recently, Kim et al. fabricated the most efficient green LED so far, based on FA1−xGAxPbBr3. As x was increased from 0 to 0.1, the CE increased from 61.3 to 95.7 cd A−1. With further post-treatment of the surface by introducing a bromide-vacancy-healing agent, 1,3,5-tris(bromomethyl)-2,4,6-triethylbenzene (TBTB), between the NC film and the TPBi layer, the surface defects and, hence, the non-radiative recombination were greatly suppressed, achieving a CE and an EQE of 108 cd A−1 and 23.4%, respectively. Although the mixed-cation strategy (A-site or B-site) has shown considerable improvement in device performance as well as the stability of the material towards ambient conditions, the operational stability remains far from the commercial standards needed.158 Furthermore, in addition to ligands having an insulating nature, the ligand binding to the NC surface is also highly dynamic, which can induce ligand detachment during the purification process, thus causing further instability.126 Another prevalent way to sidestep the excessive use of ligands, while maintaining the structural integrity, is by coating the structure with another material, such as an oxide, polymer, or inorganic layer, which can significantly influence the nature of the surface.194–196 However, it is important to note that such a core–shell structure is more beneficial when there is a minimum lattice mismatch between the core and the shell, reducing the interfacial energy of the hybrid structure.197,198 Zhang et al. recently synthesized stable core/alloyed-shell/shell FAPbBr3/FAxCs1−xPbBr3/CsPbBr3 NC structures. The incorporation of the alloyed layer in the core–shell structure mitigated the lattice mismatch between the core and the shell, yielding a high PLQY of 93%. The core–shell NC-based LED exhibited a higher luminance (1758 cd m−2), CE (19.75 cd A−1), and EQE (8%) compared with core-only LED devices. Moreover, the operational stability was also significantly enhanced.8 Another alternative strategy to obviate the use of excess ligands is to incorporate an excess of A-site cations, which has been demonstrated to be an effective method in promoting radiative recombination. Recently, Chen et al. synthesized FAPbBr3 NCs by varying the stoichiometry of FABr/PbBr2 in the precursor solution. As shown in Fig. 11a, excess FABr helps in self-passivating the surface defects by actively bonding with halide and FA vacancies, which would not be possible with organic ligands. While the particle size remained the same, the PLQY of the resulting thin films exhibited a considerable improvement. The LED devices showed a dramatic improvement in performance with excess FABr in the precursor solution (Fig. 11b). A molar ratio of 2.2:1 for FABr/PbBr2 yielded an EQE of 17.1%, which is a respectable value for green LEDs based on FAPbBr3 NCs (structure: ITO/PEDOT:PSS/PVK/FAPbBr3 NCs/TPBi/LiF/Al (Fig. 11c)).125 Notably, this strategy does not apply for B-site cations as an excess of Pb has been shown to deteriorate the device performance.154 Recently, mixed-cation perovskite NCs (FA0.5MA0.5PbBr3) were synthesized using a range of alkylamine ligands with various chain lengths (C6–C16).193 In this work, the authors found out that a higher degree of quantum confinement was established by increasing the hydrophobicity of the ligands (Fig. 11d), yielding smaller crystallites. Generally, it is well known that in a quantum-confined system, the electron–hole wavefunctions overlap with each other, resulting in a larger recombination rate and luminance efficiency compared with the bulk. Herein, through ligand engineering, the clear influence of the ligand length on the luminance, the turn-on voltage of the device, and the EQE were explored, and the results are depicted in Fig. 11e and f. Notably, the CE and EQE values reached a maximum of 38.1 cd A−1 and 16.2%, respectively, for C10. However, the efficiency then began to drop for ligands with a longer chain length (C12–C16), despite having a near-unity PLQY, perhaps due to their insulating nature. The exceptional device performance is attributed to a combination of the small crystallite size and dielectric confinement triggered by the ligands, resulting in short exciton lifetimes and low refractive indices in the NC thin films, enabling an improved light outcoupling efficiency.193 Similarly, Kim et al. also investigated the impact of the ligand chain length on the LED performance. Different from the abovementioned study, a gradual decrease in the chain length, from octylamine (C8) to butylamine (C4), resulted in an increase in the current density and a decrease in the turn-on voltage of FA-based LEDs, achieving a maximum EQE of 2.05%.134 The variation in the trends and performance compared with the aforementioned study could be attributed to the differences in the choice of A-site cation and the synthesis protocol. Recently, Zhao et al. replaced the conventional ligand system (OTA/OAc) with short aromatic ligands of 2-naphthalenesulfonic acid (NSA) for LED application. It was revealed that the NSA molecules not only preserved the surface properties of the NCs during the purification but also notably improved the electrical properties of the assembled emissive layers, ensuring efficient charge-injection and -transport in the devices. The champion LED exhibited EL approaching the Rec. 2020 (i.e., the ITU-R Recommendation BT.2020) green primary color, demonstrating a high brightness of 67115 cd cm−2 and an EQE of 19.2%.199 While most of the work on 3D FAPbBr3 is focused on improving the optoelectronic properties of NCs, a suitable device architecture with efficient charge injection also plays a key role in improving the performance of PeLEDs. For instance, Fan et al. discussed a strategy to effectually confine excitons in the recombination region of the perovskite emissive layer (EML) to boost the EQE of the device. Adjustment of the recombination region and film morphology was demonstrated by varying the concentration of the dispersion of FAPbBr3 NCs in toluene. They found that the parasitic emission due to the interface
gradually disappeared on increasing the concentration of NCs, indicating that exciton recombination is limited to the emissive layer. It was also noted that the concentration of the NCs also affects the surface roughness of the film, which impacts the device performance. The optimized recipe yielded a high-purity green LED, with a maximum EQE of 3.84%, Commission internationale de l’éclairage (CIE) coordinates of (0.183, 0.763), a CE of 16.08 cd A−1, and a PE of 12.95 lm W−1.2 Zhang and co-workers reported a modified non-aqueous emulsion synthesis to produce highly luminescent and color-tunable mixed-halide FAPb(BrxX1−x) QDs at room temperature, covering most of the visible region (430–690 nm). The as-prepared FAPbBr3 QDs exhibited bright-green emission at 538 nm with an FWHM of ∼22 nm and a high PLQY of 88%. However, the resulting FAPbBr3 QD-based LED could reach only a luminance of 403 cd m−2 and a maximum EQE of 2.8%. One of the possible reasons for this was the use of a polar solvent, DMF, during the synthesis, which is known to offer poor stability to the NCs.157
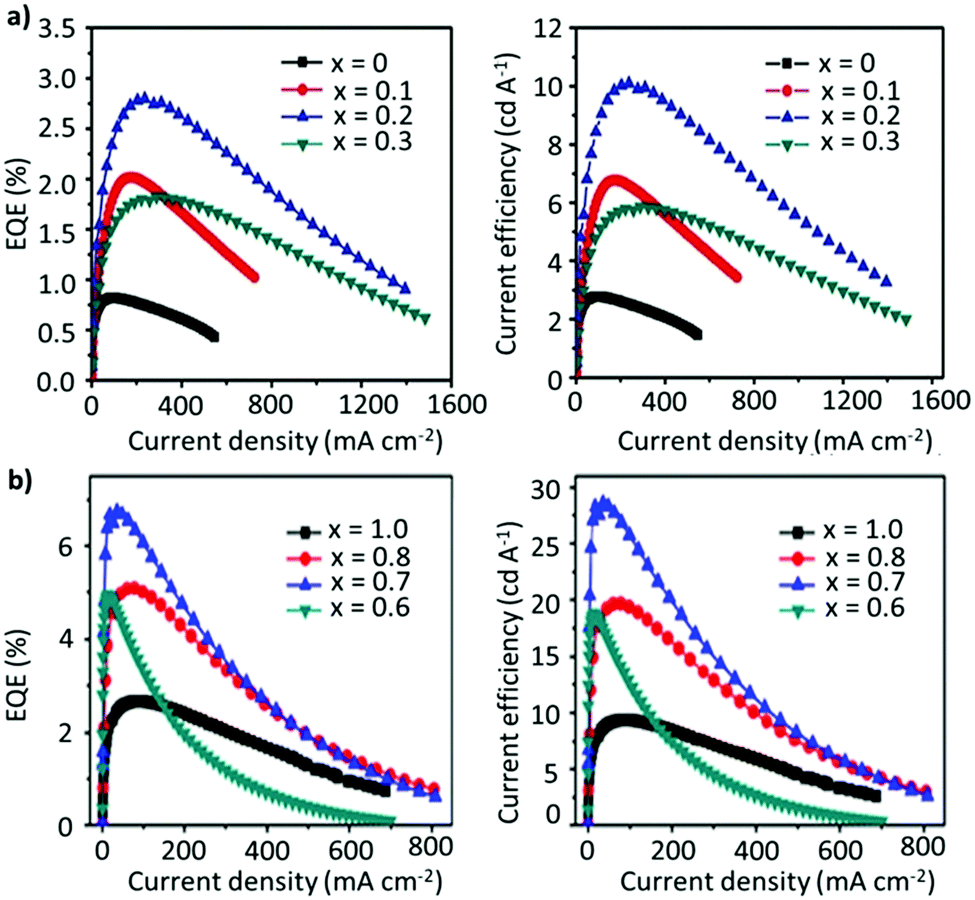 | ||
| Fig. 10 (a) EQE–current density (left), and current efficiency–current density (right) of PeLEDs based on FA1−xCsxPbBr3 (x = 0, 0.1, 0.2 and 0.3). (b) EQE–current density (left), and current efficiency-current density (right) of PeLEDs based on FA0.8Cs0.2PbxBr3 (x = 1.0, 0.8, 0.7 and 0.6). Panel (a) reproduced with permission from ref. 153. Copyright 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. Panel (b) reproduced with permission from ref. 154. Copyright 2018 American Chemical Society. | ||
 | ||
| Fig. 11 (a) Reaction illustrating the self-passivation of excess FABr. (b) EQE–luminance curves of PeLEDs based on FAPbBr3 NCs with varying molar ratios of FABr/PbBr2. (c) Energy-band diagram of FAPbBr3 NCs with varying molar ratios of FABr/PbBr2. (d) PL emission spectra of FAPbBr3 NCs synthesized using various chain lengths of alkylamine ligands, (e) current efficiency as a function of the alkyl chain length, and (f) luminance–voltage curves for PeLEDs with different chain lengths (from C6–C16). Panels (a–c) adapted from ref. 125. Copyright 2020 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. Panels (d–f) reproduced with permission from ref. 193. Copyright 2019 American Chemical Society. | ||
From the device application point of view, besides the PLQY, the Eb is another significant parameter for the PeLED performance. Theoretically, a large Eb promotes the recombination of injected electrons and holes, which is highly desirable for LED applications. Two of the most essential attributes of low-dimensional perovskites are their large Eb values (as a result of reduced dielectric screening) and the quantum confinement effect, which promotes the formation of excitons rather than free carriers. In addition, they also take advantage of efficient energy funneling from small n to large n domains, promoting radiative recombination.200 Kumar et al. reported ultrapure green LEDs based 2D FAPbBr3 NPLs (n ≈ 7–10 and Eb ≈ 162 meV), which exhibited a maximum EQE and luminance of 3.04% and 2939 cd m−2, respectively. Notably, the efficiency of the device based on the 2D FAPbBr3–PMMA complex exhibited 3-fold better performance compared with the neat FAPbBr3, which can be attributed to the dielectric confinement effect induced by the polymer. Although the recorded device performance is fair to middling, this study demonstrated the ‘greenest’ LED ever reported, corresponding to the CIE 1931 color coordinates of (0.168, 0.773).159 Similarly, another study on FAPbBr3 NPLs (n = 1–4) demonstrated a large Eb of 287.5 meV. The PeLEDs based on these FAPbBr3 NPLs displayed a maximum CE and EQE of 15.31 cd A−1 and 3.53%, respectively, with decent air stability.60 Li et al. explored the impact of solvent polarity on the device performance. In particular, they synthesized FAPbBr3 NPLs using a low-polar solvent, which greatly improved the stability of the perovskites – such that the NPLs could be washed 3 times to remove the excess insulating ligands without losing the structural integrity. Consequently, the corresponding LED device exhibited an improved performance, having a CE and EQE of 17.32 cd A−1 and 4.5%, respectively. The high performance is attributed to the high Eb (168 meV) and the impressive PL properties of the FAPbBr3 NPLs. In addition, the adoption of the low-polar solvent also assisted in improving the stability, which certainly also contributed to the high device performance.160
3.2 Blue-emitting FAPbBr3 LEDs
The development of blue PeLEDs has not gained a similar level of success compared with either the green or red counterparts. Blue-emitting perovskites possess a wide bandgap, which significantly increases the likelihood of sub-bandgap trap states. Moreover, the larger bandgap requires a higher turn-on voltage, which again adversely impacts the stability of the material.Generally, the blue luminescent perovskites can be obtained in either of the two ways: (i) doping Cl− in Br-based perovskites, as the bandgap of pure chloride lies in the UV region;201,202 or (ii) through utilizing the quantum confinement effect in the pure Br-based perovskite NCs (QDs or NPLs).30,203 However, the mixed-halide perovskites have two major limitations: (i) the Cl−-based perovskites usually show low PLQY values, which ultimately affect the efficiency of the resulting blue emission; and (ii) they tend to exhibit phase segregation, resulting in either multiple emission peaks or a shift in the emission towards the green region.204 To mitigate inhomogeneous distribution and ion migration during operation, Zhang et al. employed the Cl-terminated amine ligand 2-(2-naphthyl)ethylamine hydrochloride to synthesize FAPbBr3−xClx NCs. Instead of using a typical NC synthesis, they formed NCs directly on the conducting substrate via an antisolvent treatment. The device architecture is shown in Fig. 12a. The resulting device showed a significantly improved performance with the EL emission centered at 474nm, a maximum luminance of 2810cd m−2, and an EQE of 3.1% along with enhanced spectral stability (Fig. 12).33 Compared with the Br-ligands, the Cl-terminated ligands demonstrated a much-improved device efficiency and stability with the smallest peak shifts with an increase in the applied voltage (Fig. 12e and f). By contrast, single halide NPLs, possess a large Eb and a large dielectric barrier, which is expected to impede ionic migration. Recently, Peng et al. synthesized deep-blue-emitting colloidal FAPbBr3 NPLs (n = 2) using the one-step TOPO-mediated reprecipitation method. Using this approach, they synthesized FAPbBr3 NPLs using both conventional pairs of ligands, OAc/OAm and OAc/OAmBr. The latter combination of ligands yielded stable blue-emission at 440 nm and a high QY of up to 55%. Due to the low solubility of PbBr2 in a polar solvent, TOPO was employed as a coordinating solvent in the reaction. Phosphines are known to possess a strong coordination ability as the functional group P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O acts as an electron-donating ligand and binds to the Pb atom, forming a PbBr2–TOPO adduct. Furthermore, the concentration of organic ligands, mainly oleic acid, at the surface could be tailored by varying the amount of TOPO, owing to the acid–base equilibrium between protic acid and TOPO. By effectively optimizing the concentration of ligands, the final product FAPbBr3 NPLs showed high chemical and structural stability in ambient air for about a week, owing to reduced trap states. Importantly, TOPO is considered here to work as an intermediate ligand, which could be removed at a later stage, thus benefitting the charge-carrier transport properties. The deep-blue emitting perovskite was employed as an emissive layer in the LED device structure, yielding an EQE of 0.14% at a low operating voltage.32
O acts as an electron-donating ligand and binds to the Pb atom, forming a PbBr2–TOPO adduct. Furthermore, the concentration of organic ligands, mainly oleic acid, at the surface could be tailored by varying the amount of TOPO, owing to the acid–base equilibrium between protic acid and TOPO. By effectively optimizing the concentration of ligands, the final product FAPbBr3 NPLs showed high chemical and structural stability in ambient air for about a week, owing to reduced trap states. Importantly, TOPO is considered here to work as an intermediate ligand, which could be removed at a later stage, thus benefitting the charge-carrier transport properties. The deep-blue emitting perovskite was employed as an emissive layer in the LED device structure, yielding an EQE of 0.14% at a low operating voltage.32
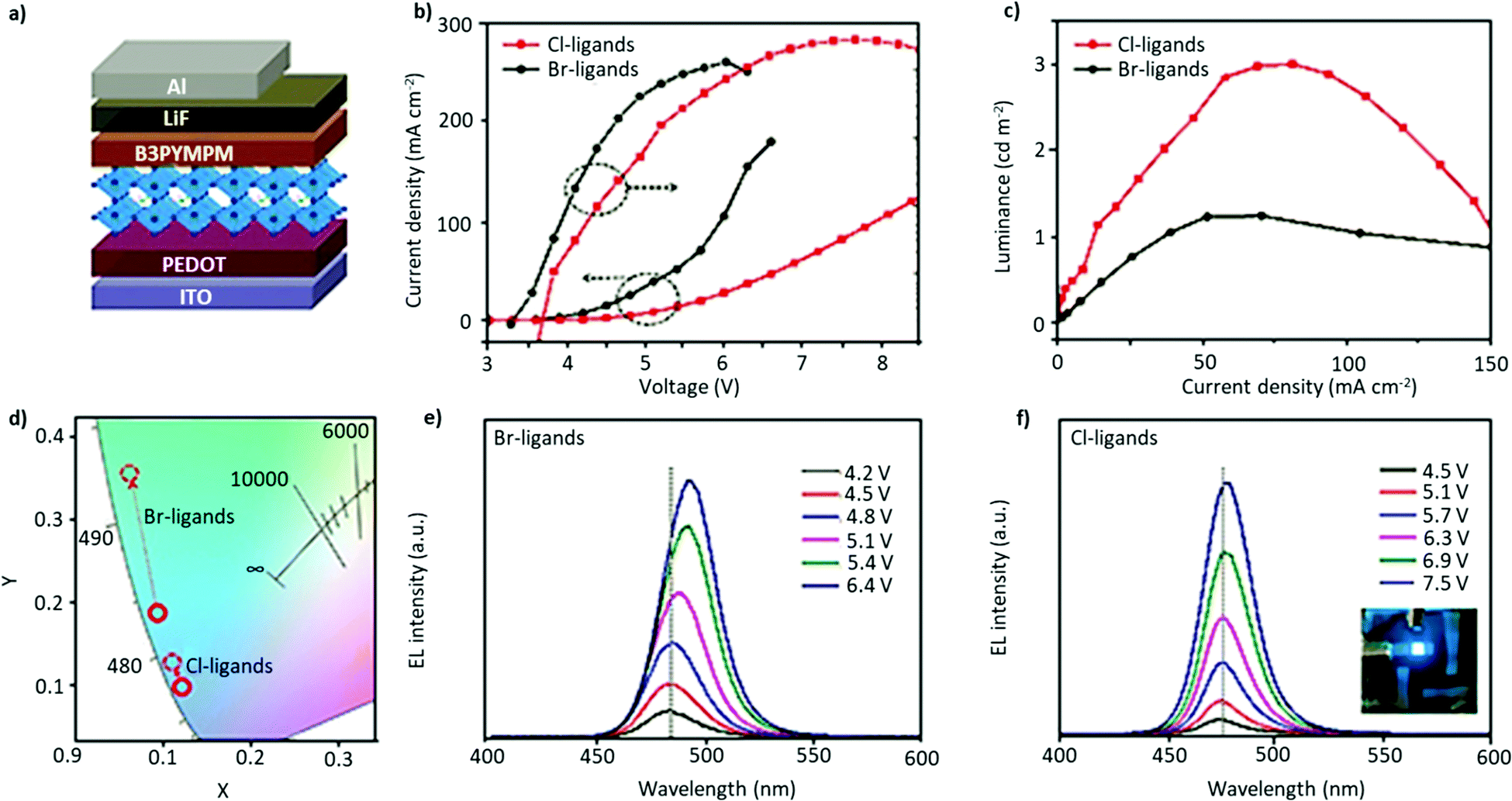 | ||
| Fig. 12 (a) Device structure of FAPbBr3−xClx thin films. (b and c) Current density–voltage curves and luminance–current density curves of the resulting PeLEDs fabricated using Br-ligands and Cl-ligands. (d) The corresponding CIE color coordinates of the EL devices based on Br-ligands and Cl-ligands. (e) EL spectra based on Br-ligand-derived devices under different bias values. (f) EL spectra of Cl-ligands derived devices under different bias values. The inset shows the optical picture of an operating device. Panels (a)–(f) adapted with permission from ref. 33. Copyright 2020 American Chemical Society. | ||
4. Looking forward
Lead halide perovskites have the potential to outperform the efficiency of traditional LED devices at a fraction of the cost. The basis of this prognosis lies in their impressive optoelectronic properties such as a high defect tolerance compared with conventionally applied semiconductor materials (e.g., CdSe, GaAs, CdTe, etc.) and facile tunability of their structural and optical properties using cost-efficient solution-processing routes. FAPbBr3, with an ideal perovskite crystal structure, remains one of the most important representative candidates of the halide perovskites. The application of FAPbBr3 NCs in light emission has been demonstrated to cover a wide range of wavelengths. In this review, we have summarized the recent developments in various approaches to engineering and fine-tuning the optoelectronic properties of FAPbBr3 NCs and their variants. The eventual impact of the composition, the surface, and the judicious defect engineering on the LED device performance has also been reviewed. Within the past decade, various strategies have been developed to improve the performance, stability, and LED characteristics of NCs. However, despite the great success achieved so far, several bottlenecks remain to be addressed before commercialization. We envisage that with a common goal of sustainability and environmental friendliness, significant challenges are waiting to be tackled for halide perovskites in general, and for FAPbBr3 NCs, specifically.The first and the most important issue of halide perovskites is their instability towards moisture, light, and oxidative environments, which is a tough nut to crack. Their lack of stability, consequently, promotes ion migration, which reduces the device lifetime, thus hindering its commercial value. Besides, it is challenging to retain the full potential of their exceptional properties when deposited in the solid state (as thin films) under operational conditions (that is, under biasing or heating) due to the presence of poorly conductive surface ligands, which are commonly employed for NCs.
With a focus on achieving optimal LED devices, the key concern is to achieve a fine balance between the stabilizing – though insulating – hydrophobic ligands and charge-transport properties. In particular, organic ligands are essential for improving the morphology and air-stability of perovskite NCs. Moreover, the incorporation of ligands also offers the potential for dimensional-dependent photophysical properties. It has been illustrated that the length of the alkyl chain greatly influences the optoelectronic properties, where short-chain ligands yield weak luminescence, and longer ones exhibit steric effects, limiting full passivation of the surface. These ligands are also labile and desorb easily upon washing, causing stability issues. To this end, multi-dentate ligands have been explored as an alternative, which have enabled better stability in perovskites. Nevertheless, stability is still far below the expectation bar. Post-treatment with inorganic passivating agents could be employed as an additional synthesis step to heal the defects formed upon washing. In addition, environmental stability could be established by coating NC surfaces with a metal oxide or hydrophobic polymer, resulting in a core–shell structure.
The size of the A-cation not only determines the crystal structure but also impacts the stability. Therefore, by carefully designing the composition of the A-site cation, strong electrostatic interactions, such as hydrogen bonding or halogen bonding, can be induced. For instance, partial substitution of the A-site cation with an inorganic substitute, such as NH4Br, could be an interesting alternative to boost stability. This substitution offers the following: (1) a stronger ionic interaction of NH4+ with the halide ion compared with FA+, owing to the differences in their ionic radii, resulting in enhanced stability; and (2) the Br-rich conditions provided by NH4Br have been demonstrated to passivate the undercoordinated Pb sites, promoting stability. More such alternatives of substituting FA+ with inorganic cations could be an interesting approach for boosting the stability of FAPbBr3 NCs. In addition, mixed A-site cations with lead-deficient compositions have also given a boost to the device performance as well as the stability. Therefore, a fusion of both may be beneficial for stability enhancement. Moreover, besides A-site mixing, a partial B-site cation-exchange reaction between Pb2+ and Mn2+ has been widely studied, especially for inorganic perovskites, and has been proved to add stability to the structure. Moreover, it also reduces the toxicity of Pb-based perovskites. However, to the best of our knowledge, no study on Mn2+-doped FAPbBr3 NCs has yet been reported. These ideas represent an important area for further investigation and may pave the way to ameliorate the balance between stability and efficiency.
Low-dimensional perovskites exhibit a quantum confinement effect, which results in a larger exciton binding energy. In addition to this, these structures provide moisture stability, which altogether makes this class a promising one for LED applications. Although many studies have been performed on (quasi-)2D FAPbBr3-based thin films for LED applications, rarely studies have been reported on the synthesis of 2D-FAPbBr3 NCs and their implementation in device applications. Together with the aid of compositional and phase engineering, it is believed that stability-related concerns can be alleviated further.
As much as ligands or the partial substitution of cations and anions is necessary for enhancing the performance of perovskite NCs, the synthesis methodologies can also have a substantial impact. Thus far, the ligand-assisted reprecipitation (LARP) and hot injection (HI) approaches have been adopted chiefly for the preparation of FAPbBr3 NCs; however, the emulsion-based method (a room-temperature re-precipitation approach) has rarely been explored for this composition. Moreover, other synthesis approaches, such as mechano-chemical methods, can limit the use of polar solvents and high temperatures, which could favor the stability concerns and processing costs. In addition, this technique is advantageous for mass production, which can be promising for large-scale commercial applications, and, thus, it is worthwhile to pursue it further.
The PL peak position is tunable by altering the composition of the perovskites as well as by tuning the size and shape of the NCs. Although size and compositional tuning have been widely explored, studies on the shape-tunable synthesis of NCs, especially for FA-based perovskites, are still lacking and require more attention. Such morphology-tunable synthesis would be intriguing to understand the effect of the shape of the NCs on the device performance. For instance, the oriented deposition of rod- or wire-like NC structures could induce directional charge mobility, thereby promoting the overall charge-transfer properties.
Overall, impressive progress has been made in the device engineering of PeLEDs, which already foreshadows the prospect of its commercialization in display-technology applications. Specifically, FAPbBr3 holds potential for the unprecedented generation of cost-effective, wide-color-gamut and flexible displays. More attractively, FAPbBr3 NCs have a PL spectrum at ≥520 nm with narrow FWHM values, providing the opportunity to achieve the newly defined critical window of ‘Rec. 2020’. However, there are some remaining challenges to address before this exciting field can be advanced further:
(1) The main constituents of LED devices are the emissive layer, charge-transport layers, and electrodes. Therefore, the operational stability not only stems from the perovskite NCs but is also significantly affected by the constituent layers. Amongst others, PEDOT:PSS is one of the most widely used hole-transport layers (HTLs) in the fabrication of FAPbBr3 NC-based LED devices, and it is well known that PEDOT:PSS tends to corrode the ITO, deteriorating the device performance. Therefore, the acidic PEDOT:PSS needs to be neutralized or undergo post-treatment. Alternatively, it may be substituted by inorganic metal oxides that have higher carrier mobilities and better thermal stability compared with organic materials.
(2) The development of blue PeLEDs, possessing high efficiency and operating lifetime, remains the “Holy Grail” for solid-state light emission and display applications (white-light generation). Therefore, further optimization via introducing additives, compositional engineering, and designing core–shell structures or post-treatment strategies is highly desirable for better stabilization. In addition, the traditional charge-transport layer (especially the HTL) also hampers efficient hole injection in the devices, owing to unsatisfactory energy-level alignment with the blue-emitting NCs (with deep valence-band energy levels).
(3) Although spin coating is a simple method for the deposition of uniform thin films, it is not suitable for large-area deposition due to its limited scalability. In an attempt to exclude this approach, several scalable techniques, such as spray deposition, inject printing, slot-die coating, and doctor blading, have recently gained momentum for the fabrication of perovskite-based solar cells. Such methods can also be adapted for the fabrication of NC-based PeLEDs, which could pave a way for future commercialization.
Author contributions
H. B. and E. D. have drafted the paper with the contribution of B. G., who provided input in restructuring the manuscript and providing constructive feedback. The work was supervised and reviewed by E. D. All authors have given their approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Research Foundation – Flanders (FWO Grant Numbers S002019N, G.0B39.15, G.0B49.15, G098319N, and ZW15_09-GOH6316), the KU Leuven Research Fund (C19/19/079, iBOF-21-085 PERSIST, STG/21/010, and PDM scholarship to B. G.), and the ERC through the Marie Curie ITN iSwitch PhD fellowship to H. B. (Grant Number 642196).References
- R. H. Buttner and E. N. Maslen, Acta Crystallogr., Sect. B: Struct. Sci., 1992, 48, 644–649 CrossRef
.
- L. Fan, K. Ding, H. Chen, S. Xiang, R. Zhang, R. Guo, Z. Liu and L. Wang, Org. Electron., 2018, 60, 64–70 CrossRef CAS
- X. Yang, X. Zhang, J. Deng, Z. Chu, Q. Jiang, J. Meng, P. Wang, L. Zhang, Z. Yin and J. You, Nat. Commun., 2018, 9, 570 CrossRef PubMed
- J. Zhang, G. Hodes, Z. Jin and S. (Frank) Liu, Angew. Chem., Int. Ed., 2019, 58, 15596–15618 CrossRef CAS PubMed
- M. Zhang, F. Zhang, Y. Wang, L. Zhu, Y. Hu, Z. Lou, Y. Hou and F. Teng, Sci. Rep., 2018, 8, 11157 CrossRef PubMed
- X. Li, J. M. Hoffman and M. G. Kanatzidis, Chem. Rev., 2021, 121, 2230–2291 CrossRef CAS PubMed
- L. Clinckemalie, D. Valli, M. B.-J. Roeffaers, J. Hofkens, B. Pradhan and E. Debroye, ACS Energy Lett., 2021, 6, 1290–1314 CrossRef CAS
- C. Zhang, S. Wang, X. Li, M. Yuan, L. Turyanska and X. Yang, Adv. Funct. Mater., 2020, 30, 1910582 CrossRef CAS
- J. Albero and H. García, J. Mater. Chem. C, 2017, 5, 4098–4110 RSC
- M. I. Saidaminov, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2017, 2, 889–896 CrossRef CAS
- Best Research-Cell Efficiency Chart, https://www.nrel.gov/pv/cell-efficiency.html, (accessed May 17, 2021).
- Z.-K. Tan, R. S. Moghaddam, M. L. Lai, P. Docampo, R. Higler, F. Deschler, M. Price, A. Sadhanala, L. M. Pazos, D. Credgington, F. Hanusch, T. Bein, H. J. Snaith and R. H. Friend, Nat. Nanotechnol., 2014, 9, 687–692 CrossRef CAS PubMed
- K. Lin, J. Xing, L. N. Quan, F. P.-G. de Arquer, X. Gong, J. Lu, L. Xie, W. Zhao, D. Zhang, C. Yan, W. Li, X. Liu, Y. Lu, J. Kirman, E. H. Sargent, Q. Xiong and Z. Wei, Nature, 2018, 562, 245–248 CrossRef CAS PubMed
- T. Chiba, Y. Hayashi, H. Ebe, K. Hoshi, J. Sato, S. Sato, Y.-J. Pu, S. Ohisa and J. Kido, Nat. Photonics, 2018, 12, 681–687 CrossRef CAS
- X. Zhao and Z.-K. Tan, Nat. Photonics, 2020, 14, 215–218 CrossRef CAS
- W. Xu, Q. Hu, S. Bai, C. Bao, Y. Miao, Z. Yuan, T. Borzda, A. J. Barker, E. Tyukalova, Z. Hu, M. Kawecki, H. Wang, Z. Yan, X. Liu, X. Shi, K. Uvdal, M. Fahlman, W. Zhang, M. Duchamp, J.-M. Liu, A. Petrozza, J. Wang, L.-M. Liu, W. Huang and F. Gao, Nat. Photonics, 2019, 13, 418–424 CrossRef CAS
- Y. Cao, N. Wang, H. Tian, J. Guo, Y. Wei, H. Chen, Y. Miao, W. Zou, K. Pan, Y. He, H. Cao, Y. Ke, M. Xu, Y. Wang, M. Yang, K. Du, Z. Fu, D. Kong, D. Dai, Y. Jin, G. Li, H. Li, Q. Peng, J. Wang and W. Huang, Nature, 2018, 562, 249–253 CrossRef CAS PubMed
- Y. Zou, T. Wu, F. Fu, S. Bai, L. Cai, Z. Yuan, Y. Li, R. Li, W. Xu, T. Song, Y. Yang, X. Gao, F. Gao and B. Sun, J. Mater. Chem. C, 2020, 8, 15079–15085 RSC
- C. Zhang, D.-B. Kuang and W.-Q. Wu, Small Methods, 2020, 4, 1900662 CrossRef CAS
- Q. Wan, W. Zheng, C. Zoub, F. Carulli, C. Zhang, H. Song, M. Liu, Q. Zhang, L. Y. Lin, L. Kong, L. Li and S. Brovelli, 2021, arXiv:2106.01924.
- P. Tang, L. Xie, X. Xiong, C. Wei, W. Zhao, M. Chen, J. Zhuang, W. Su and Z. Cui, ACS Appl. Mater. Interfaces, 2020, 12, 13087–13095 CrossRef CAS PubMed
- W. Cao, C. Xiang, Y. Yang, Q. Chen, L. Chen, X. Yan and L. Qian, Nat. Commun., 2018, 9, 2608 CrossRef PubMed
- Z. Wei and J. Xing, J. Phys. Chem. Lett., 2019, 10, 3035–3042 CrossRef CAS PubMed
- Y.-H. Kim, H. Cho and T.-W. Lee, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11694–11702 CrossRef CAS PubMed
- A. Ren, H. Wang, W. Zhang, J. Wu, Z. Wang, R. V. Penty and I. H. White, Nat. Electron., 2021, 4, 559–572 CrossRef
- Q. Shan, J. Li, J. Song, Y. Zou, L. Xu, J. Xue, Y. Dong, C. Huo, J. Chen, B. Han and H. Zeng, J. Mater. Chem. C, 2017, 5, 4565–4570 RSC
- P. Xiao, Y. Yu, J. Cheng, Y. Chen, S. Yuan, J. Chen, J. Yuan and B. Liu, Nanomaterials, 2021, 11, 103 CrossRef CAS PubMed
- J. Yao, L. Wang, K. Wang, Y. Yin, J. Yang, Q. Zhang and H. Yao, Sci. Bull., 2020, 65, 1150–1153 CrossRef CAS
- Y. Shen, K.-C. Shen, Y.-Q. Li, M. Guo, J. Wang, Y. Ye, F.-M. Xie, H. Ren, X. Gao, F. Song and J.-X. Tang, Adv. Funct. Mater., 2021, 31, 2006736 CrossRef CAS
- Y. Liu, J. Cui, K. Du, H. Tian, Z. He, Q. Zhou, Z. Yang, Y. Deng, D. Chen, X. Zuo, Y. Ren, L. Wang, H. Zhu, B. Zhao, D. Di, J. Wang, R. H. Friend and Y. Jin, Nat. Photonics, 2019, 13, 760–764 CrossRef CAS
- Z. Chu, Y. Zhao, F. Ma, C.-X. Zhang, H. Deng, F. Gao, Q. Ye, J. Meng, Z. Yin, X. Zhang and J. You, Nat. Commun., 2020, 11, 4165 CrossRef CAS PubMed
- S. Peng, Z. Wen, T. Ye, X. Xiao, K. Wang, J. Xia, J. Sun, T. Zhang, G. Mei, H. Liu, B. Xu, X. Li, R. Chen, G. Xing, K. Wang and Z. Tang, ACS Appl. Mater. Interfaces, 2020, 12, 31863–31874 CrossRef CAS PubMed
- F. Zhang, X. Zhang, C. Wang, M. Sun, X. Luo, Y. Yang, S. Chang, D. Zhang and L. Duan, Nano Energy, 2021, 79, 105486 CrossRef CAS
- Z. Yao, C. Bi, A. Liu, M. Zhang and J. Tian, Nano Energy, 2022, 95, 106974 CrossRef CAS
- H. Li, H. Lin, D. Ouyang, C. Yao, C. Li, J. Sun, Y. Song, Y. Wang, Y. Yan, Y. Wang, Q. Dong and W. C.-H. Choy, Adv. Mater., 2021, 33, 2008820 CrossRef CAS PubMed
- Y. Zou, Z. Yuan, S. Bai, F. Gao and B. Sun, Mater. Today Nano, 2019, 5, 100028 CrossRef
- M. Xie and J. Tian, J. Phys. Chem. Lett., 2022, 13, 1962–1971 CrossRef CAS PubMed
- H. Yuan, E. Debroye, K. Janssen, H. Naiki, C. Steuwe, G. Lu, M. Moris, E. Orgiu, H. Uji-i, F. De Schryver, P. Samorì, J. Hofkens and M. Roeffaers, J. Phys. Chem. Lett., 2016, 7, 561–566 CrossRef CAS PubMed
- Y. Ma, Y. Cheng, X. Xu, M. Li, C. Zhang, S. H. Cheung, Z. Zeng, D. Shen, Y.-M. Xie, K. L. Chiu, F. Lin, S. K. So, C.-S. Lee and S.-W. Tsang, Adv. Funct. Mater., 2021, 31, 2006802 CrossRef CAS
- N. Li, Y. Jia, Y. Guo and N. Zhao, Adv. Mater., 2022, 34, 2108102 CrossRef CAS PubMed
- J. Pan, L. N. Quan, Y. Zhao, W. Peng, B. Murali, S. P. Sarmah, M. Yuan, L. Sinatra, N. M. Alyami, J. Liu, E. Yassitepe, Z. Yang, O. Voznyy, R. Comin, M. N. Hedhili, O. F. Mohammed, Z. H. Lu, D. H. Kim, E. H. Sargent and O. M. Bakr, Adv. Mater., 2016, 28, 8718–8725 CrossRef CAS PubMed
- R. K. Behera, S. Das Adhikari, S. K. Dutta, A. Dutta and N. Pradhan, J. Phys. Chem. Lett., 2018, 9, 6884–6891 CrossRef CAS PubMed
- Y. Wu, L. Qiu, J. Liu, M. Guan, Z. Dai and G. Li, Adv. Opt. Mater., 2022, 10, 2102661 CrossRef CAS
- S.-J. Woo, J. S. Kim and T.-W. Lee, Nat. Photonics, 2021, 15, 630–634 CrossRef CAS
- A. J. Nozik, M. C. Beard, J. M. Luther, M. Law, R. J. Ellingson and J. C. Johnson, Chem. Rev., 2010, 110, 6873–6890 CrossRef CAS PubMed
- Ç. Allahverdi and M. H. Yukselici, Phys. Scr., 2008, 78, 015702 CrossRef
- M. H. Yükselici and Ç. Allahverdi, Phys. Status Solidi B, 2003, 236, 694–701 CrossRef
- B. G. Potter, J. H. Simmons, P. Kumar and C. J. Stanton, J. Appl. Phys., 1994, 75, 8039–8045 CrossRef CAS
- J. M. Costa-Fernández, R. Pereiro and A. Sanz-Medel, TrAC, Trends Anal. Chem., 2006, 25, 207–218 CrossRef
- L. Polavarapu, B. Nickel, J. Feldmann and A. S. Urban, Adv. Energy Mater., 2017, 7, 1700267 CrossRef
- J. A. Sichert, Y. Tong, N. Mutz, M. Vollmer, S. Fischer, K. Z. Milowska, R. García Cortadella, B. Nickel, C. Cardenas-Daw, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2015, 15, 6521–6527 CrossRef CAS PubMed
- Y. Tong, E. Bladt, M. F. Aygüler, A. Manzi, K. Z. Milowska, V. A. Hintermayr, P. Docampo, S. Bals, A. S. Urban, L. Polavarapu and J. Feldmann, Angew. Chem., Int. Ed., 2016, 55, 13887–13892 CrossRef CAS PubMed
- B. J. Bohn, Y. Tong, M. Gramlich, M. L. Lai, M. Döblinger, K. Wang, R. L.-Z. Hoye, P. Müller-Buschbaum, S. D. Stranks, A. S. Urban, L. Polavarapu and J. Feldmann, Nano Lett., 2018, 18, 5231–5238 CrossRef CAS PubMed
- M. De Franco, M. Cirignano, T. Cavattoni, H. Bahmani Jalali, M. Prato and F. Di Stasio, Opt. Mater.: X, 2022, 13, 100124 CAS
- J. Song, T. Fang, J. Li, L. Xu, F. Zhang, B. Han, Q. Shan and H. Zeng, Adv. Mater., 2018, 30, 1805409 CrossRef PubMed
- D. B. Mitzi, J. Chem. Soc., Dalton Trans., 2001, 1–12 RSC
- F. C. Hanusch, E. Wiesenmayer, E. Mankel, A. Binek, P. Angloher, C. Fraunhofer, N. Giesbrecht, J. M. Feckl, W. Jaegermann, D. Johrendt, T. Bein and P. Docampo, J. Phys. Chem. Lett., 2014, 5, 2791–2795 CrossRef CAS PubMed
- A. A. Zhumekenov, M. I. Saidaminov, M. A. Haque, E. Alarousu, S. P. Sarmah, B. Murali, I. Dursun, X.-H. Miao, A. L. Abdelhady, T. Wu, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2016, 1, 32–37 CrossRef CAS
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Bertolotti, N. Masciocchi, A. Guagliardi and M. V. Kovalenko, J. Am. Chem. Soc., 2016, 138, 14202–14205 CrossRef CAS PubMed
- H. Fang, W. Deng, X. Zhang, X. Xu, M. Zhang, J. Jie and X. Zhang, Nano Res., 2019, 12, 171–176 CrossRef CAS
- R. Qu, M. Gao, H. Li, X. Qi, C. Liu, W. Zhao, J. Yan and Z. Zhang, Opt. Mater., 2021, 113, 110776 CrossRef CAS
- A. Krishna, S. Gottis, M. K. Nazeeruddin and F. Sauvage, Adv. Funct. Mater., 2019, 29, 1806482 CrossRef
- Y. Fan, H. Meng, L. Wang and S. Pang, Solar RRL, 2019, 3, 1900215 CrossRef
-
R. J.-D. Tilley, Perovskites: Structure-Property Relationships, John Wiley & Sons, 2016 Search PubMed
- P. Goel, S. Sundriyal, V. Shrivastav, S. Mishra, D. P. Dubal, K.-H. Kim and A. Deep, Nano Energy, 2021, 80, 105552 CrossRef CAS
- M. C. Weidman, M. Seitz, S. D. Stranks and W. A. Tisdale, ACS Nano, 2016, 10, 7830–7839 CrossRef CAS PubMed
- C.-H. Chen, Z. Li, Q. Xue, Y.-A. Su, C.-C. Lee, H.-L. Yip, W.-C. Chen and C.-C. Chueh, Org. Electron., 2019, 75, 105400 CrossRef CAS
- W. Travis, E. N.-K. Glover, H. Bronstein, D. O. Scanlon and R. G. Palgrave, Chem. Sci., 2016, 7, 4548–4556 RSC
- F. A. Roghabadi, M. Alidaei, S. Maryam Mousavi, T. Ashjari, A. Shokrolahzadeh Tehrani, V. Ahmadi and S. Mojtaba Sadrameli, J. Mater. Chem. A, 2019, 7, 5898–5933 RSC
- C. J. Bartel, C. Sutton, B. R. Goldsmith, R. Ouyang, C. B. Musgrave, L. M. Ghiringhelli and M. Scheffler, Sci. Adv., 2019, 5, eaav0693 CrossRef CAS PubMed
- L. Dou, J. Mater. Chem. C, 2017, 5, 11165–11173 RSC
- P. M. Woodward, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 32–43 CrossRef
- M. W. Lufaso and P. M. Woodward, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 10–20 CrossRef PubMed
- J. L. Knutson, J. D. Martin and D. B. Mitzi, Inorg. Chem., 2005, 44, 4699–4705 CrossRef CAS PubMed
- S. A. Kulkarni, G. Han, K. S. Tan, S. G. Mhaisalkar and N. Mathews, Curr. Opin. Electrochem., 2018, 11, 91–97 CrossRef CAS
- Y. Zhang, M. Keshavarz, E. Debroye, E. Fron, M. C.-R. González, D. Naumenko, H. Amenitsch, J. V. de Vondel, S. D. Feyter, P. Heremans, M. B.-J. Roeffaers, W. Qiu, B. Pradhan and J. Hofkens, Nano Photonics, 2020, 10, 2145–2156 Search PubMed
- S. Lee, D. B. Kim, J. C. Yu, C. H. Jang, J. H. Park, B. R. Lee and M. H. Song, Adv. Mater., 2019, 31, e1805244 CrossRef PubMed
- G.-W. Kim and A. Petrozza, Adv. Energy Mater., 2020, 10, 2001959 CrossRef CAS
- H. Huang, M. I. Bodnarchuk, S. V. Kershaw, M. V. Kovalenko and A. L. Rogach, ACS Energy Lett., 2017, 2, 2071–2083 CrossRef CAS PubMed
- L. D. Whalley, J. M. Frost, Y.-K. Jung and A. Walsh, J. Chem. Phys., 2017, 146, 220901 CrossRef PubMed
- J. Ye, M. M. Byranvand, C. O. Martínez, R. L.-Z. Hoye, M. Saliba and L. Polavarapu, Angew. Chem., Int. Ed., 2021, 133, 21804–21828 CrossRef
- H. Jin, E. Debroye, M. Keshavarz, I. G. Scheblykin, M. B.-J. Roeffaers, J. Hofkens and J. A. Steele, Mater. Horiz., 2020, 7, 397–410 RSC
- A. Merdasa, Y. Tian, R. Camacho, A. Dobrovolsky, E. Debroye, E. L. Unger, J. Hofkens, V. Sundström and I. G. Scheblykin, ACS Nano, 2017, 11, 5391–5404 CrossRef CAS PubMed
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 141–153 CrossRef CAS PubMed
- Y. Yang, M. Yang, D. T. Moore, Y. Yan, E. M. Miller, K. Zhu and M. C. Beard, Nat. Energy, 2017, 2, 1–7 Search PubMed
- H. Uratani and K. Yamashita, J. Phys. Chem. Lett., 2017, 8, 742–746 CrossRef CAS PubMed
- D. P. Nenon, K. Pressler, J. Kang, B. A. Koscher, J. H. Olshansky, W. T. Osowiecki, M. A. Koc, L.-W. Wang and A. P. Alivisatos, J. Am. Chem. Soc., 2018, 140, 17760–17772 CrossRef CAS PubMed
- X. Zheng, Y. Hou, H.-T. Sun, O. F. Mohammed, E. H. Sargent and O. M. Bakr, J. Phys. Chem. Lett., 2019, 10, 2629–2640 CrossRef CAS PubMed
- H. Jin, J. A. Steele, R. Cheng, N. Parveen, M. B.-J. Roeffaers, J. Hofkens and E. Debroye, Adv. Opt. Mater., 2021, 9, 2002240 CrossRef CAS
- E. C. Schueller, G. Laurita, D. H. Fabini, C. C. Stoumpos, M. G. Kanatzidis and R. Seshadri, Inorg. Chem., 2018, 57, 695–701 CrossRef CAS PubMed
- M. Keshavarz, M. Ottesen, S. Wiedmann, M. Wharmby, R. Küchler, H. Yuan, E. Debroye, J. A. Steele, J. Martens, N. E. Hussey, M. Bremholm, M. B.-J. Roeffaers and J. Hofkens, Adv. Mater., 2019, 31, 1900521 CrossRef PubMed
- A. Alaei, A. Circelli, Y. Yuan, Y. Yang and S. S. Lee, Mater. Adv., 2021, 2, 47–63 RSC
- L. Wang, K. Wang and B. Zou, J. Phys. Chem. Lett., 2016, 7, 2556–2562 CrossRef CAS PubMed
- J. Khan, X. Zhang, J. Yuan, Y. Wang, G. Shi, R. Patterson, J. Shi, X. Ling, L. Hu, T. Wu, S. Dai and W. Ma, ACS Energy Lett., 2020, 5, 3322–3329 CrossRef CAS
- Y. Deng, X. Li and R. Wang, Sol. Energy Mater. Sol. Cells, 2021, 230, 111242 CrossRef CAS
- L. Zhang, K. Cao, J. Qian, Y. Huang, X. Wang, M. Ge, W. Shen, F. Huang, M. Wang, W. Zhang, S. Chen and T. Qin, J. Mater. Chem. C, 2020, 8, 17482–17490 RSC
- D. Jia, J. Chen, M. Yu, J. Liu, E. M.-J. Johansson, A. Hagfeldt and X. Zhang, Small, 2020, 16, 2001772 CrossRef CAS PubMed
- M. L. Steigerwald and L. E. Brus, Annu. Rev. Mater. Sci., 1989, 19, 471–495 CrossRef CAS
- A. Dey, J. Ye, A. De, E. Debroye, S. K. Ha, E. Bladt, A. S. Kshirsagar, Z. Wang, J. Yin, Y. Wang, L. N. Quan, F. Yan, M. Gao, X. Li, J. Shamsi, T. Debnath, M. Cao, M. A. Scheel, S. Kumar, J. A. Steele, M. Gerhard, L. Chouhan, K. Xu, X. Wu, Y. Li, Y. Zhang, A. Dutta, C. Han, I. Vincon, A. L. Rogach, A. Nag, A. Samanta, B. A. Korgel, C.-J. Shih, D. R. Gamelin, D. H. Son, H. Zeng, H. Zhong, H. Sun, H. V. Demir, I. G. Scheblykin, I. Mora-Seró, J. K. Stolarczyk, J. Z. Zhang, J. Feldmann, J. Hofkens, J. M. Luther, J. Pérez-Prieto, L. Li, L. Manna, M. I. Bodnarchuk, M. V. Kovalenko, M. B.-J. Roeffaers, N. Pradhan, O. F. Mohammed, O. M. Bakr, P. Yang, P. Müller-Buschbaum, P. V. Kamat, Q. Bao, Q. Zhang, R. Krahne, R. E. Galian, S. D. Stranks, S. Bals, V. Biju, W. A. Tisdale, Y. Yan, R. L.-Z. Hoye and L. Polavarapu, ACS Nano, 2021, 15, 10775–10981 CrossRef CAS PubMed
- A. Perumal, S. Shendre, M. Li, Y. K.-E. Tay, V. K. Sharma, S. Chen, Z. Wei, Q. Liu, Y. Gao, P. J.-S. Buenconsejo, S. T. Tan, C. L. Gan, Q. Xiong, T. C. Sum and H. V. Demir, Sci. Rep., 2016, 6, 36733 CrossRef CAS PubMed
- F. Zhang, H. Zhong, C. Chen, X. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed
- M. Imran, F. Di Stasio, Z. Dang, C. Canale, A. H. Khan, J. Shamsi, R. Brescia, M. Prato and L. Manna, Chem. Mater., 2016, 28, 6450–6454 CrossRef CAS PubMed
- D. Zhang, Y. Yang, Y. Bekenstein, Y. Yu, N. A. Gibson, A. B. Wong, S. W. Eaton, N. Kornienko, Q. Kong, M. Lai, A. P. Alivisatos, S. R. Leone and P. Yang, J. Am. Chem. Soc., 2016, 138, 7236–7239 CrossRef CAS PubMed
- C. Otero-Martínez, D. García-Lojo, I. Pastoriza-Santos, J. Pérez-Juste and L. Polavarapu, Angew. Chem., 2021, 133, 26881–26888 CrossRef
- I. Levchuk, A. Osvet, X. Tang, M. Brandl, J. D. Perea, F. Hoegl, G. J. Matt, R. Hock, M. Batentschuk and C.
J. Brabec, Nano Lett., 2017, 17, 2765–2770 CrossRef CAS PubMed
- J. Song, L. Xu, J. Li, J. Xue, Y. Dong, X. Li and H. Zeng, Adv. Mater., 2016, 28, 4861–4869 CrossRef CAS PubMed
- J. Shamsi, Z. Dang, P. Bianchini, C. Canale, F. Di Stasio, R. Brescia, M. Prato and L. Manna, J. Am. Chem. Soc., 2016, 138, 7240–7243 CrossRef CAS PubMed
- A. Patra, S. Bera, D. Nasipuri, S. K. Dutta and N. Pradhan, ACS Energy Lett., 2021, 6, 2682–2689 CrossRef CAS
- Y. Zhang, C. Wang and Z. Deng, Chem. Commun., 2018, 54, 4021–4024 RSC
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef CAS PubMed
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS
- E. Debroye, H. Yuan, E. Bladt, W. Baekelant, M. Van der Auweraer, J. Hofkens, S. Bals and M. B.-J. Roeffaers, ChemNanoMat, 2017, 3, 223–227 CrossRef CAS PubMed
- S. Paul and A. Samanta, ACS Energy Lett., 2020, 5, 64–69 CrossRef CAS
- Q. Li, H. Li, H. Shen, F. Wang, F. Zhao, F. Li, X. Zhang, D. Li, X. Jin and W. Sun, ACS Photonics, 2017, 4, 2504–2512 CrossRef CAS
- C. B. Murray, D. J. Norris and M. G. Bawendi, Synthesis and characterization of nearly monodisperse CdE (E = sulfur, selenium, tellurium) semiconductor nanocrystallites, https://pubs.acs.org/doi/pdf/10.1021/ja00072a025, (accessed February 13, 2021).
- Q. Guo, G. M. Ford, H. W. Hillhouse and R. Agrawal, Nano Lett., 2009, 9, 3060–3065 CrossRef CAS PubMed
- J. Tang, S. Hinds, S. O. Kelley and E. H. Sargent, Chem. Mater., 2008, 20, 6906–6910 CrossRef CAS
- H. Huang, F. Zhao, L. Liu, F. Zhang, X. Wu, L. Shi, B. Zou, Q. Pei and H. Zhong, ACS Appl. Mater. Interfaces, 2015, 7, 28128–28133 CrossRef CAS PubMed
- A. Pan, B. He, X. Fan, Z. Liu, J. J. Urban, A. P. Alivisatos, L. He and Y. Liu, ACS Nano, 2016, 10, 7943–7954 CrossRef CAS PubMed
- L. C. Schmidt, A. Pertegás, S. González-Carrero, O. Malinkiewicz, S. Agouram, G. Mínguez Espallargas, H. J. Bolink, R. E. Galian and J. Pérez-Prieto, J. Am. Chem. Soc., 2014, 136, 850–853 CrossRef CAS PubMed
- H. Bhatia, J. A. Steele, C. Martin, M. Keshavarz, G. Solis-Fernandez, H. Yuan, G. Fleury, H. Huang, I. Dovgaliuk, D. Chernyshov, J. Hendrix, M. B.-J. Roeffaers, J. Hofkens and E. Debroye, Chem. Mater., 2019, 31, 6824–6832 CrossRef CAS
- Z. Song, S. C. Watthage, A. B. Phillips, B. L. Tompkins, R. J. Ellingson and M. J. Heben, Chem. Mater., 2015, 27, 4612–4619 CrossRef CAS
- X. Ren, Z. Yang, D. Yang, X. Zhang, D. Cui, Y. Liu, Q. Wei, H. Fan and S. Liu, Nanoscale, 2016, 8, 3816–3822 RSC
- H. Chen, L. Fan, R. Zhang, C. Bao, H. Zhao, W. Xiang, W. Liu, G. Niu, R. Guo, L. Zhang and L. Wang, Adv. Opt. Mater., 2020, 8, 1901390 CrossRef CAS
-
F. Divsar, Quantum Dots: Fundamental and Applications, BoD – Books on Demand, 2020 Search PubMed
-
A. E.-T. Bonatto, G. I. Selli, P. T. Martin and F. T. Bonatto, in Environmental Applications of Nanomaterials, ed. A. Kopp Alves, Springer International Publishing, Cham, 2022, pp. 255–267 Search PubMed
- S. Ahn, M.-H. Park, S.-H. Jeong, Y.-H. Kim, J. Park, S. Kim, H. Kim, H. Cho, C. Wolf, M. Pei, H. Yang and T.-W. Lee, Adv. Funct. Mater., 2019, 29, 1807535 CrossRef
- H. Cho, J. S. Kim, C. Wolf, Y.-H. Kim, H. J. Yun, S.-H. Jeong, A. Sadhanala, V. Venugopalan, J. W. Choi, C.-L. Lee, R. H. Friend and T.-W. Lee, ACS Nano, 2018, 12, 2883–2892 CrossRef CAS PubMed
- X. Yang, Z. Chu, J. Meng, Z. Yin, X. Zhang, J. Deng and J. You, J. Phys. Chem. Lett., 2019, 10, 2892–2897 CrossRef CAS PubMed
- A. Tiwari, N. S. Satpute, C. M. Mehare and S. J. Dhoble, J. Alloys Compd., 2021, 850, 156827 CrossRef CAS
- J. Z. Zhang, J. Phys. Chem. Lett., 2019, 10, 5055–5063 CrossRef CAS PubMed
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed
- Y.-H. Kim, G.-H. Lee, Y.-T. Kim, C. Wolf, H. J. Yun, W. Kwon, C. G. Park and T.-W. Lee, Nano Energy, 2017, 38, 51–58 CrossRef CAS
- S. K. Mahankudo, A. Das, K. Mishra, M. Barai, P. Mal and S. Ghosh, ACS Appl. Nano Mater., 2022, 5, 4360–4366 CrossRef CAS
- I. Lignos, L. Protesescu, D. B. Emiroglu, R. Maceiczyk, S. Schneider, M. V. Kovalenko and A. J. deMello, Nano Lett., 2018, 18, 1246–1252 CrossRef CAS PubMed
- D. N. Minh, J. Kim, J. Hyon, J. H. Sim, H. H. Sowlih, C. Seo, J. Nam, S. Eom, S. Suk, S. Lee, E. Kim and Y. Kang, Chem. Mater., 2017, 29, 5713–5719 CrossRef CAS
- Z. Liu, Z. Hu, Z. Zhang, J. Du, J. Yang, X. Tang, W. Liu and Y. Leng, ACS Photonics, 2019, 6, 3150–3158 CrossRef CAS
- Q. Zhong, M. Cao, Y. Xu, P. Li, Y. Zhang, H. Hu, D. Yang, Y. Xu, L. Wang, Y. Li, X. Zhang and Q. Zhang, Nano Lett., 2019, 19, 4151–4157 CrossRef CAS PubMed
- Y. Tan, Y. Zou, L. Wu, Q. Huang, D. Yang, M. Chen, M. Ban, C. Wu, T. Wu, S. Bai, T. Song, Q. Zhang and B. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 3784–3792 CrossRef CAS PubMed
- H. Wang, Z. Zhang, J. V. Milić, L. Tan, Z. Wang, R. Chen, X. Jing, C. Yi, Y. Ding, Y. Li, Y. Zhao, X. Zhang, A. Hagfeldt, M. Grätzel and J. Luo, Adv. Energy Mater., 2021, 11, 2101082 CrossRef CAS
- S. R. Smock, T. J. Williams and R. L. Brutchey, Angew. Chem., Int. Ed., 2018, 57, 11711–11715 CrossRef CAS PubMed
- J. Y. Woo, S. Lee, S. Lee, W. D. Kim, K. Lee, K. Kim, H. J. An, D. C. Lee and S. Jeong, J. Am. Chem. Soc., 2016, 138, 876–883 CrossRef CAS PubMed
- X. Li, M. Ibrahim Dar, C. Yi, J. Luo, M. Tschumi, S. M. Zakeeruddin, M. K. Nazeeruddin, H. Han and M. Grätzel, Nat. Chem., 2015, 7, 703–711 CrossRef CAS PubMed
- O. J. Ashton, A. R. Marshall, J. H. Warby, B. Wenger and H. J. Snaith, Chem. Mater., 2020, 32, 7172–7180 CrossRef CAS
- G. Almeida, O. J. Ashton, L. Goldoni, D. Maggioni, U. Petralanda, N. Mishra, Q. A. Akkerman, I. Infante, H. J. Snaith and L. Manna, J. Am. Chem. Soc., 2018, 140, 14878–14886 CrossRef PubMed
- M. Shin, J. Kim, Y.-K. Jung, T. Ruoko, A. Priimagi, A. Walsh and B. Shin, J. Mater. Chem. C, 2019, 7, 3945–3951 RSC
- F. Ambroz, W. Xu, S. Gadipelli, D. J.-L. Brett, C.-T. Lin, C. Contini, M. A. McLachlan, J. R. Durrant, I. P. Parkin and T. J. Macdonald, Part. Part. Syst. Charact., 2020, 37, 1900391 CrossRef CAS
- Y. Zhang, H. Lv, C. Cui, L. Xu, P. Wang, H. Wang, X. Yu, J. Xie, J. Huang, Z. Tang and D. Yang, Nanotechnology, 2017, 28, 205401 CrossRef PubMed
- H. Yu, H. Lu, F. Xie, S. Zhou and N. Zhao, Adv. Funct. Mater., 2016, 26, 1411–1419 CrossRef CAS
- H. Cho, S.-H. Jeong, M.-H. Park, Y.-H. Kim, C. Wolf, C.-L. Lee, J. H. Heo, A. Sadhanala, N. Myoung, S. Yoo, S. H. Im, R. H. Friend and T.-W. Lee, Science, 2015, 350, 1222–1225 CrossRef CAS PubMed
- R. Lindblad, N. K. Jena, B. Philippe, J. Oscarsson, D. Bi, A. Lindblad, S. Mandal, B. Pal, D. D. Sarma, O. Karis, H. Siegbahn, E. M.-J. Johansson, M. Odelius and H. Rensmo, J. Phys. Chem. C, 2015, 119, 1818–1825 CrossRef CAS
- X. Zhang, H. Liu, W. Wang, J. Zhang, B. Xu, K. L. Karen, Y. Zheng, S. Liu, S. Chen, K. Wang and X. W. Sun, Adv. Mater., 2017, 29, 1606405 CrossRef PubMed
- X. Zhang, W. Wang, B. Xu, H. Liu, H. Shi, H. Dai, X. Zhang, S. Chen, K. Wang and X. W. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 24242–24248 CrossRef CAS PubMed
- R. Cai, X. Qu, H. Liu, H. Yang, K. Wang and X. W. Sun, IEEE Trans. Nanotechnol., 2019, 18, 1050–1056 CAS
- Y.-L. Tong, Y.-W. Zhang, K. Ma, R. Cheng, F. Wang and S. Chen, ACS Appl. Mater. Interfaces, 2018, 7 Search PubMed
- T. Zhang, H. Li, P. Yang, J. Wei, F. Wang, H. Shen, D. Li and F. Li, Org. Electron., 2019, 68, 76–84 CrossRef CAS
- Y.-H. Kim, S. Kim, A. Kakekhani, J. Park, J. Park, Y.-H. Lee, H. Xu, S. Nagane, R. B. Wexler, D.-H. Kim, S. H. Jo, L. Martínez-Sarti, P. Tan, A. Sadhanala, G.-S. Park, Y.-W. Kim, B. Hu, H. J. Bolink, S. Yoo, R. H. Friend, A. M. Rappe and T.-W. Lee, Nat. Photonics, 2021, 15, 148–155 CrossRef CAS
- S. Kumar, J. Jagielski, N. Kallikounis, Y.-H. Kim, C. Wolf, F. Jenny, T. Tian, C. J. Hofer, Y.-C. Chiu, W. J. Stark, T.-W. Lee and C.-J. Shih, Nano Lett., 2017, 17, 5277–5284 CrossRef CAS PubMed
- F. Li, L. Yang, Z. Cai, K. Wei, F. Lin, J. You, T. Jiang, Y. Wang and X. Chen, Nanoscale, 2018, 10, 20611–20617 RSC
- D. Yu, F. Cao, Y. Gao, Y. Xiong and H. Zeng, Adv. Funct. Mater., 2018, 28, 1800248 CrossRef
- Y. Zu, J. Xi, L. Li, J. Dai, S. Wang, F. Yun, B. Jiao, H. Dong, X. Hou and Z. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 2835–2841 CrossRef CAS PubMed
- L. Protesescu, S. Yakunin, S. Kumar, J. Bär, F. Bertolotti, N. Masciocchi, A. Guagliardi, M. Grotevent, I. Shorubalko, M. I. Bodnarchuk, C.-J. Shih and M. V. Kovalenko, ACS Nano, 2017, 11, 3119–3134 CrossRef CAS PubMed
- H. Si, Z. Zhang, Q. Liao, G. Zhang, Y. Ou, S. Zhang, H. Wu, J. Wu, Z. Kang and Y. Zhang, Adv. Mater., 2020, 32, 1904702 CrossRef CAS PubMed
- C. M. Sutter-Fella, Q. P. Ngo, N. Cefarin, K. L. Gardner, N. Tamura, C. V. Stan, W. S. Drisdell, A. Javey, F. M. Toma and I. D. Sharp, Nano Lett., 2018, 18, 3473–3480 CrossRef CAS PubMed
- H.-C. Hsu, S.-H. Wu, Y.-L. Tung and C.-F. Shih, Org. Electron., 2022, 101, 106400 CrossRef CAS
- A. J. Knight, J. Borchert, R. D.-J. Oliver, J. B. Patel, P. G. Radaelli, H. J. Snaith, M. B. Johnston and L. M. Herz, ACS Energy Lett., 2021, 6, 799–808 CrossRef CAS PubMed
- B. Charles, J. Dillon, O. J. Weber, M. Saiful Islam and M. T. Weller, J. Mater. Chem. A, 2017, 5, 22495–22499 RSC
- S. Cai, J. Dai, Z. Shao, M. U. Rothmann, Y. Jia, C. Gao, M. Hao, S. Pang, P. Wang, S. P. Lau, K. Zhu, J. J. Berry, L. M. Herz, X. C. Zeng and Y. Zhou, J. Am. Chem. Soc., 2022, 144, 1910–1920 CrossRef CAS PubMed
- R. D. Shannon, Acta Crystallogr., Sect. A: Found. Crystallogr., 1976, 32, 751–767 CrossRef
- W. Ke, C. C. Stoumpos and M. G. Kanatzidis, Adv. Mater., 2019, 31, 1803230 CrossRef CAS PubMed
- H.-C. Wang, W. Wang, A.-C. Tang, H.-Y. Tsai, Z. Bao, T. Ihara, N. Yarita, H. Tahara, Y. Kanemitsu, S. Chen and R.-S. Liu, Angew. Chem., 2017, 129, 13838–13842 CrossRef
- F. Liu, J. Jiang, T. Toyoda, M. A. Kamarudin, S. Hayase, R. Wang, S. Tao and Q. Shen, ACS Appl. Nano Mater., 2021, 4, 3958–3968 CrossRef CAS
- Y. Wang, D. Yang, D. Ma, D. H. Kim, T. Ahamad, S. M. Alshehri and A. Vadim, Sci. China: Mater., 2019, 62, 790–796 CrossRef CAS
- M. Coduri, T. A. Strobel, M. Szafrański, A. Katrusiak, A. Mahata, F. Cova, S. Bonomi, E. Mosconi, F. De Angelis and L. Malavasi, J. Phys. Chem. Lett., 2019, 10, 7398–7405 CrossRef CAS PubMed
- G. E. Eperon and D. S. Ginger, ACS Energy Lett., 2017, 2, 1190–1196 CrossRef CAS
- L. Dai, Z. Deng, F. Auras, H. Goodwin, Z. Zhang, J. C. Walmsley, P. D. Bristowe, F. Deschler and N. C. Greenham, Nat. Photonics, 2021, 15, 696–702 CrossRef CAS
- X. Miao, T. Qiu, S. Zhang, H. Ma, Y. Hu, F. Bai and Z. Wu, J. Mater. Chem. C, 2017, 5, 4931–4939 RSC
- D. Straus and R. Cava, ChemRxiv, 2022 Search PubMed
- W. Tang, J. Zhang, S. Ratnasingham, F. Liscio, K. Chen, T. Liu, K. Wan, E. Suena Galindez, E. Bilotti, M. Reece, M. Baxendale, S. Milita, M. A. McLachlan, L. Su and O. Fenwick, J. Mater. Chem. A, 2020, 8, 13594–13599 RSC
- Y. Gao, X. Su, J. Zhang, H. Tan, J. Sun, J. Ouyang and N. Na, Small, 2021, 17, 2103773 CrossRef CAS PubMed
- A. Pisanu, P. Quadrelli and L. Malavasi, RSC Adv., 2019, 9, 13263–13268 RSC
- D. B. Mitzi, C. A. Feild, W. T.-A. Harrison and A. M. Guloy, Nature, 1994, 369, 467–469 CrossRef CAS
- Q. Wang, X.-D. Liu, Y.-H. Qiu, K. Chen, L. Zhou and Q.-Q. Wang, AIP Adv., 2018, 8, 025108 CrossRef
- G. Xing, B. Wu, X. Wu, M. Li, B. Du, Q. Wei, J. Guo, E. K.-L. Yeow, T. C. Sum and W. Huang, Nat. Commun., 2017, 8, 14558 CrossRef CAS PubMed
- X. Hong, T. Ishihara and A. V. Nurmikko, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 6961–6964 CrossRef CAS PubMed
- M. S. de Holanda, R. F. Moral, P. E. Marchezi, F. C. Marques and A. F. Nogueira, EcoMat, 2021, 3, e12124 CrossRef CAS
- Q. A. Akkerman, S. G. Motti, A. R. Srimath Kandada, E. Mosconi, V. D’Innocenzo, G. Bertoni, S. Marras, B. A. Kamino, L. Miranda, F. De Angelis, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2016, 138, 1010–1016 CrossRef CAS PubMed
- Y. E. Panfil, M. Oded and U. Banin, Angew. Chem., Int. Ed., 2018, 57, 4274–4295 CrossRef CAS PubMed
- L. N. Quan, F. P. García de Arquer, R. P. Sabatini and E. H. Sargent, Adv. Mater., 2018, 30, 1801996 CrossRef PubMed
- J. Cameron and P. J. Skabara, Mater. Horiz., 2020, 7, 1759–1772 RSC
- K. Norrman, M. V. Madsen, S. A. Gevorgyan and F. C. Krebs, J. Am. Chem. Soc., 2010, 132, 16883–16892 CrossRef CAS PubMed
- S. Kumar, J. Jagielski, T. Marcato, S. F. Solari and C.-J. Shih, J. Phys. Chem. Lett., 2019, 10, 7560–7567 CrossRef CAS PubMed
- M. Meyns, M. Perálvarez, A. Heuer-Jungemann, W. Hertog, M. Ibáñez, R. Nafria, A. Genç, J. Arbiol, M. V. Kovalenko, J. Carreras, A. Cabot and A. G. Kanaras, ACS Appl. Mater. Interfaces, 2016, 8, 19579–19586 CrossRef CAS PubMed
- G. Li, Z.-K. Tan, D. Di, M. L. Lai, L. Jiang, J. H.-W. Lim, R. H. Friend and N. C. Greenham, Nano Lett., 2015, 15, 2640–2644 CrossRef CAS PubMed
- Q. Zhong, M. Cao, H. Hu, D. Yang, M. Chen, P. Li, L. Wu and Q. Zhang, ACS Nano, 2018, 12, 8579–8587 CrossRef CAS PubMed
- C. Tan, J. Chen, X.-J. Wu and H. Zhang, Nat. Rev. Mater., 2018, 3, 1–13 CrossRef
- Q. Zhong, M. Cao and Q. Zhang, Nanoscale, 2021, 13, 19341–19351 RSC
- H. Zhao, H. Chen, S. Bai, C. Kuang, X. Luo, P. Teng, C. Yin, P. Zeng, L. Hou, Y. Yang, L. Duan, F. Gao and M. Liu, ACS Energy Lett., 2021, 6, 2395–2403 CrossRef CAS
- M. U. Ali, W. Cai, J. Cai, J. Miao, S. Zhang, J. Chen, L. Xiao, H. Meng, C. Yan and G. Wei, Adv. Opt. Mater., 2022, 10, 2100671 CrossRef CAS
- A. Sadhanala, S. Ahmad, B. Zhao, N. Giesbrecht, P. M. Pearce, F. Deschler, R. L.-Z. Hoye, K. C. Gödel, T. Bein, P. Docampo, S. E. Dutton, M. F.-L. De Volder and R. H. Friend, Nano Lett., 2015, 15, 6095–6101 CrossRef CAS PubMed
- S. Wang, Y. Wang, Y. Zhang, X. Zhang, X. Shen, X. Zhuang, P. Lu, W. W. Yu, S. V. Kershaw and A. L. Rogach, J. Phys. Chem. Lett., 2019, 10, 90–96 CrossRef CAS PubMed
- J. Leng, T. Wang, X. Zhao, E. W.-Y. Ong, B. Zhu, J. D.-A. Ng, Y.-C. Wong, K. H. Khoo, K. Tamada and Z.-K. Tan, J. Phys. Chem. Lett., 2020, 11, 2036–2043 CrossRef CAS PubMed
- X. Zhu, L. Ge, Y. Wang, M. Li, R. Zhang, M. Xu, Z. Zhao, W. Lv and R. Chen, Adv. Opt. Mater., 2021, 9, 2100058 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2022 |