
Dissipative control of the fluorescence of a 1,3-dipyrenyl calix[4]arene in the cone conformation†
Emanuele
Spatola‡
 a,
Francesco
Rispoli‡
b,
Daniele
Del Giudice‡
a,
Roberta
Cacciapaglia
a,
Alessandro
Casnati
b,
Luciano
Marchiò
b,
Laura
Baldini
*b and
Stefano
Di Stefano
*a
a,
Francesco
Rispoli‡
b,
Daniele
Del Giudice‡
a,
Roberta
Cacciapaglia
a,
Alessandro
Casnati
b,
Luciano
Marchiò
b,
Laura
Baldini
*b and
Stefano
Di Stefano
*a
aDipartimento di Chimica Università di Roma La Sapienza and ISB-CNR Sede Secondaria di Roma - Meccanismi di Reazione, P.le A. Moro 5, I-00185 Roma, Italy. E-mail: stefano.distefano@uniroma1.it
bDipartimento di Scienze Chimiche, della Vita e della Sostenibilità Ambientale, Università degli Studi di Parma, Parco Area delle Scienze 17/A, 43124 Parma, Italy. E-mail: laura.baldini@unipr.it
First published on 12th November 2021
Abstract
The temporal control (ON/OFF/ON) of the fluorescence of a dichloromethane/acetonitrile 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of calixarene 3 decorated with two pyrenyl moieties at the upper rim is attained by the addition of CCl3CO2H used as a convenient chemical fuel.
:1 solution of calixarene 3 decorated with two pyrenyl moieties at the upper rim is attained by the addition of CCl3CO2H used as a convenient chemical fuel.
Introduction
Biochemical systems often exploit the energy derived from the consumption of a chemical fuel to accomplish their function under a dissipative regime.1 For this reason, in recent years a great effort has been devoted to the design of stimuli-responsive artificial systems, which operate under dissipative conditions, achieving impressive results in the fields of self-assembly,2 synthetic DNA nanotechnology,3 and fully abiotic molecular machines.4 Lately, a number of dissipative systems have been developed, whose operation was made possible by transient variations of the solution pH. Such examples involve (i) hydrolysis of activated lactones,5 (ii) biocatalytic enzymatic reaction networks,6 (iii) the thiosulfate/sulfite redox oscillator,7 (iv) irradiation of merocyanine-based metastable photoacids,8 and (v) decarboxylation of activated carboxylic acids, such as 2-cyano-2-phenylpropanoic acid9 and its derivatives,10 trichloroacetic acid,11 and nitroacetic acid.12 For instance, in 2020 we employed 2-cyano-2-phenylpropanoic acid 2 as a chemical fuel4 to control over time the “breathing” movement13 between the two pinched-cone conformations of 1,3-diaminocalix[4]arene 1 (Fig. 1).10c In the presence of the fuel acid, monoprotonation of 1 forces the calixarene conformation in a “locked” geometry with the two aniline rings slightly convergent to share the proton (proton-bridged pinched-cone conformation). When the fuel is exhausted, the calixarene reverts to its original “unlocked” state. Here we show that trichloroacetic acid 4 (Fig. 1) can be conveniently used to control, in a dissipative manner, the geometry of 1,3-dipyrenyl-2,4-diaminocalix[4]arene 3, and, consequently, its fluorescence emission.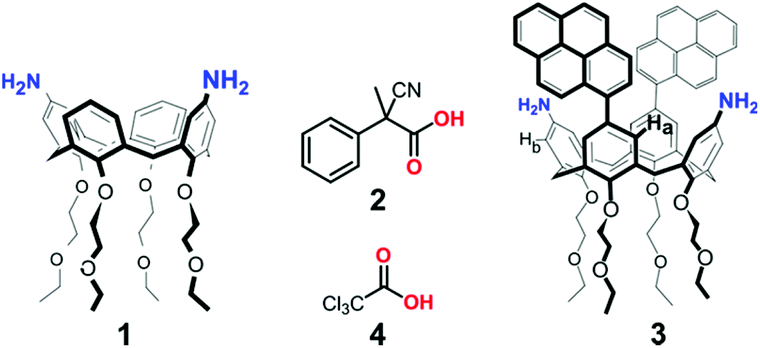 | ||
| Fig. 1 Structures of fuel acids 2 and 4 used to dissipatively drive the “breathing” movement of 1,3-diaminocalix[4]arenes 1 (see ref. 10c) and 3. | ||
The rationale behind our hypothesis is that when the calixarene conformation is “unlocked” (state A, Scheme 1), the two pyrene units are allowed to get close together and the fluorescence spectrum would be characterized by the typical excimer emission. In contrast, the addition of a fuel4 acid RCO2H would trigger a first conformational change to the “locked” pinched-cone structure (state B′). In this state, the two chromophores are forced far apart in consequence of the convergence of the aniline rings to share a proton, and should display only the monomer fluorescence (quenching of the excimer emission). Then, the just formed carboxylate anion would slowly lose CO2 to give the ion pair 3H+·R− (state B′′). A final back proton transfer from protonated calix[4]arene 3H+ to carbanion R− would restore the initial state A, leading to the “unlocked” calix[4]arene 3 and the waste product RH. Thus, the addition of the fuel with the consequent locking of the proton-bridged pinched-cone conformation would switch off the excimer fluorescence intensity of 3, due to the separation of the pyrenyl units. Only when the fuel is exhausted, the initial fluorescence intensity would be recovered.
 | ||
| Scheme 1 Expected conformational cycle of 3 controlled in a dissipative manner by means of activated carboxylic acids 2 and 4. | ||
Results and discussion
Calix[4]arene 3 was synthesized in two steps from 5,17-diiodo-11,23-dinitro-25,26,27,28-tetrakis(2-ethoxyethoxy)calix[4]arene 514 (Scheme 2a). First, the pyrenyl moieties were introduced by a Suzuki coupling of 5 with pyrene-1-boronic acid pinacol ester 6, obtaining 7 in 60% yield, and then the nitro groups were reduced to amines with hydrazine and Pd/C (80% yield).
 | ||
| Scheme 2 (a) Synthesis of 1,3-dipyrenyl-2,4-diaminocalix[4]arene 3. (b) Three views of the X-ray molecular structure of 1,3-dipyrenyl-2,4-dinitrocalix[4]arene 7. | ||
The 1H NMR spectra at room temperature of 7 both in CDCl3 and in toluene-d8 are characterized by relatively sharp peaks for the calixarene protons and broad ones for the protons of the pyrenyl units, indicative of a hindered rotation of these groups around the calixarene–pyrene C–C bond. Moreover, the chemical shifts of the aromatic protons of the calixarene are diagnostic for the pinched-cone conformation of the scaffold,15 with the pyrene substituted phenol rings parallel and the nitrophenol rings diverging. Taken together, these spectral features suggest attractive π−π interactions between the pyrene groups and a molecular geometry presumably similar to the conformation adopted by 7 in the solid state (Scheme 2b, see the ESI† for a description of the X-ray structure). In the 1H NMR spectra of diamino–dipyrenyl calixarene 3 in different solvents (CDCl3, CD2Cl2, CD3OD and toluene-d8) the signals of the pyrene protons are broad as well, while the calixarene peaks remain sharp, pointing also for this compound to a π−π interaction of the pyrene units. The presence of a strong excimer-like§ band (Fig. S16†) in the fluorescence spectrum of 3 confirms that the two pyrene units are close together and that the calixarene conformation is in the unlocked state A (see Scheme 1). As in the case of 1,10c the 1H NMR titration of 2.0 mM 3 with TFA (Fig. 2a, CD2Cl2/CD3CN 1:1, 25 °C) shows that the protonation of 3 initially causes an up-field shift of the signal related to the aromatic protons ortho to the NH2 groups from 6.59 ppm to lower values, which is a characteristic feature of the “locked” pinched-cone conformation with the aniline rings slightly convergent compared to the pyrene-substituted ones, which are downfield shifted and divergent. This unusual up-field shift of the protons ortho to the NH2 upon catching the proton is due to the shielding cone that the divergent aromatic rings exert on the convergent ones. In addition, such protonation dramatically affects the aromatic signals of the pyrenyl moieties, which are shifted to lower fields and become sharper, suggesting a break-up of the π–π interaction and a higher conformational freedom. Upon further addition of TFA (more than 2 mol equiv.), the signal of the NH2-substituted aromatic rings tends to revert to the original chemical shift and becomes broader; the same, although much more mildly, holds for the pyrene-substituted aromatic rings signal. Also, the pyrene signals are again slightly shifted to higher fields and become broader. The latter pieces of evidence point to a second protonation of the calixarene structure to give 3H22+, with one proton on each of the two amino groups, whose electrostatic repulsion would cause a partial rapprochement of the pyrenyl moieties. Although less basic than 1 (two molar equiv. of TFA are needed to reach the highest concentration of mono-protonated 3H+, under the explored conditions, while mono-protonation of 1 was complete upon the addition of just one molar equivalent of TFA),10c3 behaves similar to 1. Thus, in the mono-protonated form 3H+ the proton turns out to be shared between the two amino groups, which, consequently, are tilted inwards in the “locked” conformation.
 | ||
| Fig. 2 (a) Titration of 2.0 mM 3 with TFA (1H NMR monitoring; CD2Cl2/CD3CN 1:1, 25 °C). For the assignment of the marked signals see Fig. 1, other signals belong to pyrenyl moieties. (b) Titration of 0.15 mM 3 with TFA (fluorescence monitoring, λexc = 385 nm; CH2Cl2/CH3CN 1:1, 25 °C). (c) Additional fluorescence spectra obtained after the addition of further TFA (7, 30 and 50 mol equiv.) with respect to (b). The excimer band again increases due to re-approaching of the pyrenyl groups. | ||
Fluorescence titration of 0.15 mM 3 with TFA (Fig. 2b, λexc = 385 nm, CH2Cl2/CH3CN 1:1, 25 °C) shows a decrease of the excimer emission band at 475 nm and a corresponding increase of the band at 408 nm due to the monomeric emission of the pyrenyl units, confirming the adoption of the pinched-cone conformation with the two pyrenyl units pointing far from one another in the mono-protonated form 3H+. Conversely, the addition of a larger excess of TFA (up to 50 mol equiv., see Fig. 2c), causes the excimer band at 475 nm to increase again. This is due to the protonation of both the amino- to ammonium groups, whose repulsion translates into a re-approaching of the pyrenyl groups.
Having demonstrated that the protonation is effective in producing the expected conformational variations of 3, we then started to explore the use of a chemical fuel to achieve a temporal control of the geometry of the calixarene. Initially, 2-cyano-2-phenylpropanoic acid was employed as the fuel. Unfortunately, while the 1H NMR monitoring of the reaction corroborated our hypothesis (pages S8 and S9†), we were unable to observe the expected fluorescence quenching of the excimer emission during the reaction, probably due to an unexpected interposition of the aromatic portion of the fuel 2 between the two pyrenyl moieties of 3. We then resorted to trichloroacetic acid 4, although it was found less manageable due to its deliquescence. The addition of 2.3 mol equiv. of trichloroacetic acid to 2.0 mM 3 (Fig. 3a, trace b) results in a series of shifts that closely resemble those observed in the titration with TFA, i.e. a larger separation of the aromatic signals corresponding to the calixarene scaffold (red and green stripes, Fig. 3a), and the down-field shift of the signals of the pyrenyl moieties that become more resolved, proving the adoption of the “locked” pinched-cone conformation of 3 (state B′ in Scheme 1, compare traces a and b).¶ In the following 12–13 hours, no substantial variations can be detected in the 1H NMR spectrum except for the building up of the signal at 7.49 ppm corresponding to CHCl3 produced during the decarboxylation of 4 (traces c and d, blue stripe on Fig. 3a). From this point (t > 12 h), all the signals corresponding to the calix[4]arene scaffold begin to revert to the initial chemical shift values, and at the end, after 21 h, the 1H NMR spectrum turns to be superimposable to the initial one (cfr. Fig. 3a traces f and a), demonstrating that the whole cycle A → B′ → B′′ → A has occurred. In other words, the “locked” pinched-cone structure persists as long as an excess fuel is present. Consequently, it is possible to extend the time in which the calix[4]arene scaffold remains in the “locked” state by increasing the amount of the added fuel, increasing from 13 h with 2.3 mol equiv. to 32 hours with 4.0 mol equiv. of added 4 (Fig. 3b, see pages S10 and S11†).
 | ||
| Fig. 3 (a) 1H NMR monitoring of the reaction between 2.0 mM 3 (trace a) and 2.3 mol equiv. of 4. Spectra from b to f were recorded after 5 min, 7 h, 12.8 h, 18 h, and 21.2 h from the addition of 4, respectively (CD2Cl2/CD3CN 1:1, 25 °C). The blue stripe highlights the CHCl3 signal, which increases during the reaction, while red and green stripes highlight the signals related to the aromatic protons ortho to the pyrenyl units and to the NH2 groups, respectively. (b) Percentage of 3 in its locked conformation in function of time for the reactions carried out in the presence of 2.3 mol equiv. (orange trace) and 4.0 mol equiv. (azure trace) of 4 (raw data at pages S10 and S11,† as detected by 1H NMR spectroscopy). | ||
Then, we monitored the reaction using a spectrofluorometer, recording over time the emission intensity at 475 nm (λexc = 385 nm) of a 0.15 mM 3 solution to which 5.0 mol equiv. of fuel 4 were added (Fig. 4a). After the addition, an immediate decrease of the intramolecular excimer fluorescence emission takes place, witnessing the unpairing of the pyrenyl moieties (Scheme 1) and the formation of the locked conformation. The reaction proceeds and, after approximately 3 h, the emission intensity at 475 nm is recovered since the cycle A → B′ → B′′ → A is over and the pyrenyl units are again spatially paired. The observation that the reaction rate is faster at these concentrations (0.15 mM of 3) than that observed at higher concentrations (2.00 mM of 3, 1H NMR experiments), is likely due to a weaker ion pairing between 3H+ and RCO2− (see Scheme 1) at lower concentrations, which destabilizes the RCO2− anion and facilitates the decarboxylation step. As a matter of fact, decarboxylation does not proceed at any extents if dichloromethane alone is used as a solvent instead of dichloromethane:acetonitrile 1:1, both at high and low concentrations, suggesting an important role of the solvent that influences the character of ion pairs involved. Eventually, Fig. 4b shows that the system is robust and reversible. Four subsequent A → B′ → B′′ → A cycles have been achieved, with consequent loss and recovery of the fluorescence at 475 nm, by means of four successive shots of fuel 4.
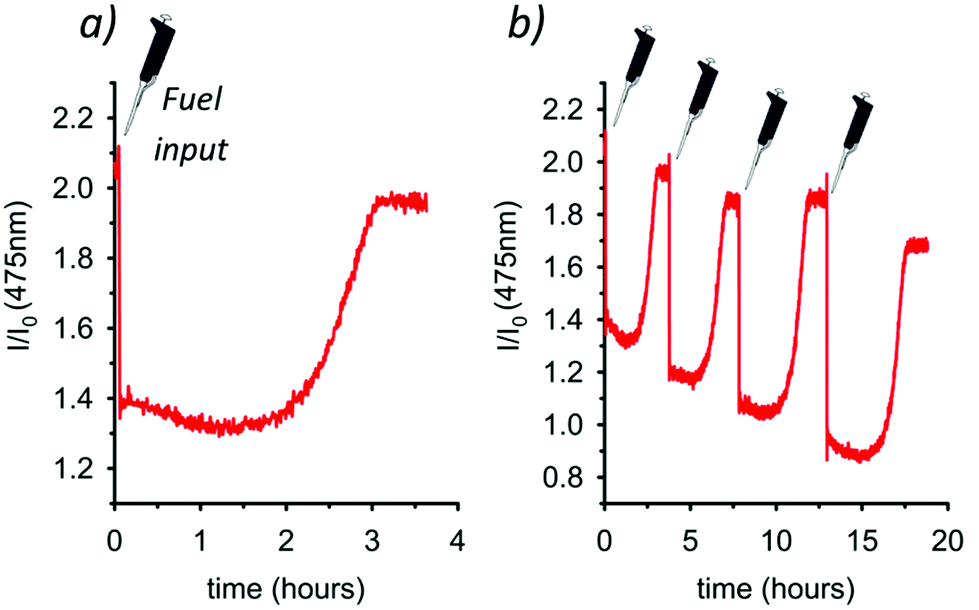 | ||
| Fig. 4 (a) Time course monitoring of the fluorescence emission at 475 nm (λexc = 385 nm) before and after the addition of 5.0 mol equiv. of 4 (CH2Cl2/CH3CN 1:1 at 25 °C). (b) Up to four dissipative conformational cycles were obtained by the subsequent additions of 5.0 mol equiv. of 4 (CH2Cl2/CH3CN 1:1 at 25 °C). | ||
Experimental section
Instrument and methods
Kinetic experiments. 1H NMR spectra were recorded on a 300 MHz spectrometer. The spectra were internally referenced to the residual proton signal of the solvent at 5.32 ppm (CD2Cl2) and 1.94 ppm (CD3CN). All the experiments were carried out in thermostated NMR tubes. Emission measurements were performed as described before, with both excitation and emission slit sets at 1 nm.
Synthesis
:5) yielded the pure product as a light-yellow solid (0.161 g, 60%). Mp 247.7–248.2 °C. 1H NMR (400 MHz, CDCl3) δ 8.21 (4H, bs, ArH), 7.68 (4H, br s, ArH(Py)), 7.50 (4H, br s, ArH(Py)), 7.25 (4H, br s, ArH(Py)), 6.94 (2H, br s, ArH(Py)), 6.84 (4H, br s, ArH), 6.56 (2H, br s, ArH(Py)), 5.98 (2H, br s, ArH(Py)), 4.87 (4H, d, ArCHHeqAr, J = 13.3 Hz) 4.75 (4H, br s, OCH2CH2O), 4.15 (4H, br s, OCH2CH2O), 4.08 (4H, br s, OCH2CH2O), 3.91 (4H, br s, OCH2CH2O), 3.68 (4H, q, OCH2CH3J = 7.0 Hz), 3.59 (4H, q, OCH2CH3, J = 7.0 Hz), 3.53 (4H, d, ArCHHaxAr, J = 13.3 Hz), 1.36 (6H, t, OCH2CH3, J = 6.5 Hz) 1.25 (6H, t, OCH2CH3, J = 7.0 Hz). 13C NMR (100 MHz, CDCl3) δ 163.57, 154.04, 142.73, 138.17, 135.76, 135.08, 132.15, 131.23, 130.57, 129.60, 129.07, 126.62, 126.03, 125.52, 124.86, 124.21, 124.02, 123.07, 122.80, 77.24, 74.79, 73.81, 70.15, 69.65, 66.64, 66.31, 31.22, 15.37. MS (ESI+-MS, m/z): 1225.64 ([M − Na]+, 100%), 1241.47 (40, [M − K]+), 1203.47 (10, M − H+).
Kinetic and titration experiments
Conclusions
In conclusion, we have demonstrated that trichloroacetic acid 4 can be used for driving the temporal control of the A → B′ → B′′ → A conformational cycle of calix[4]arene 3 in a dissipative fashion. Given the presence of the two pyrenyl units in the opposite positions of the upper rim of calix[4]arene 3 such control translates into the dissipative time-modulation of the solution fluorescence.16Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
FR, AC, LM and LB acknowledge the Centro Interdipartimentale Misure “G. Casnati” of Parma University, the COMP-HUB Initiative ‘Departments of Excellence’ (MIUR, 2018–2022), BacHounds project (MIUR-2017E44A9P) and Chiesi Farmaceutici SpA for the support of the D8 Venture X-ray equipment. SDS thanks the University of Rome La Sapienza (Grandi Progetti di Ricerca, Ateneo 2018).Notes and references
- (a) L. S. Kariyawasam, M. M. Hossain and C. S. Hartley, Angew. Chem., Int. Ed., 2021, 60, 12648 CrossRef CAS; (b) K. Das, L. Gabrielli and L. J. Prins, Angew. Chem., Int. Ed., 2021, 60, 20120 CrossRef CAS PubMed.
- Some selected examples are: (a) S. A. P. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519 RSC; (b) F. Della Sala, S. Neri, S. Maiti, J. L.-Y. Chen and L. J. Prins, Curr. Opin. Biotechnol., 2017, 46, 27 CrossRef CAS; (c) C. Pezzato, C. Cheng, J. F. Stoddart and R. D. Astumian, Chem. Soc. Rev., 2017, 46, 5491 RSC; (d) A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476 RSC; (e) R. Merindol and A. Walther, Chem. Soc. Rev., 2017, 46, 5588 RSC; (f) G. Ragazzon and L. J. Prins, Nat. Nanotechnol., 2018, 13, 882 CrossRef CAS; (g) R. D. Astumian, C. Pezzato, Y. Feng, Y. Qiu, P. R. McGonigal, C. Cheng and J. F. Stoddart, Mater. Chem. Front., 2020, 4, 1304 RSC; (h) B. Rieß, R. K. Grötsch and J. Boekhoven, Chem, 2020, 6, 552 CrossRef; (i) M. Weißenfels, J. Gemen and R. Klajn, Chem, 2021, 7, 23–37 CrossRef.
- (a) B. Yurke, A. J. Turberfield, A. P. Mills Jr., F. C. Simmel and J. L. Neumann, Nature, 2000, 406, 605 CrossRef CAS PubMed; (b) T. Omabegho, R. Sha and N. C. Seeman, Science, 2009, 324, 67 CrossRef CAS PubMed; (c) J. Lloyd, C. H. Tran, K. Wadhwani, C. C. Samaniego, H. K. K. Subramanian and E. Franco, ACS Synth. Biol., 2018, 7, 30 CrossRef CAS; (d) E. Del Grosso, A. Amodio, G. Ragazzon, L. J. Prins and F. Ricci, Angew. Chem., Int. Ed., 2018, 57, 10489 CrossRef CAS; (e) E. Del Grosso, G. Ragazzon, L. J. Prins and F. Ricci, Angew. Chem., Int. Ed., 2019, 17, 5582 CrossRef; (f) E. Del Grosso, I. Ponzo, G. Ragazzon, L. J. Prins and F. Ricci, Angew. Chem., Int. Ed., 2020, 59, 21058 CrossRef CAS; (g) J. Deng, D. Bezold, H. J. Jessen and A. Walther, Angew. Chem., Int. Ed., 2020, 29, 12084 CrossRef; (h) E. Del Grosso, L. J. Prins and F. Ricci, Angew. Chem., Int. Ed., 2020, 59, 13238 CrossRef CAS; (i) J. Deng and A. Walther, J. Am. Chem. Soc., 2020, 142, 685 CrossRef; (j) Z. Zhou, Y. Ouyang, J. Wang and I. Willner, J. Am. Chem. Soc., 2021, 143, 5071 CrossRef CAS.
- C. Biagini and S. Di Stefano, Angew. Chem., Int. Ed., 2020, 59, 8344 CrossRef CAS PubMed.
- (a) T. Heuser, A. Steppert, C. Molano Lopez, B. Zhu and A. Walther, Nano Lett., 2015, 15, 2213 CrossRef CAS; (b) L. Heinen and A. Walther, Chem. Sci., 2017, 8, 4100 RSC; (c) A. Del Giudice, C. Dicko, L. Galantini and N. V. Pavel, J. Phys. Chem. B, 2017, 121, 4388 CrossRef CAS PubMed.
- (a) X. Fan and A. Walther, Angew. Chem., Int. Ed., 2021, 60, 11398 CrossRef CAS; (b) X. Fan and A. Walther, Angew. Chem., Int. Ed., 2020, 60, 3619 CrossRef PubMed; (c) I. Maity, C. Sharma, F. Lossada and A. Walther, Angew. Chem., Int. Ed., 2021, 60, 22537–22546 CrossRef CAS.
- T. Liedl and F. C. Simmel, Nano Lett., 2005, 5, 1894 CrossRef CAS PubMed.
- (a) S. Silvi, A. Arduini, A. Pochini, A. Secchi, M. Tomasulo, F. M. Raymo, M. Baroncini and A. Credi, J. Am. Chem. Soc., 2007, 129, 13378 CrossRef CAS PubMed; (b) Y. Liao, Acc. Chem. Res., 2017, 50, 1956 CrossRef CAS PubMed; (c) C. Berton, D. M. Busiello, S. Zamuner, E. Solari, R. Scopelliti, F. Fadaei-Tirani, K. Severin and C. Pezzato, Chem. Sci., 2020, 11, 8457 RSC; (d) D. Go, D. Rommel, Y. Liao, T. Haraszti, J. Sprakel and A. J. C. Kuehne, Soft Matter, 2018, 14, 910 RSC.
- (a) J. A. Berrocal, C. Biagini, L. Mandolini and S. Di Stefano, Angew. Chem., Int. Ed., 2016, 55, 6997 CrossRef CAS PubMed; (b) A. Ghosh, I. Paul, M. Adlung, C. Wickleder and M. Schmittel, Org. Lett., 2018, 20, 1046 CrossRef CAS PubMed; (c) C. Biagini, F. Di Pietri, L. Mandolini, O. Lanzalunga and S. Di Stefano, Chem. – Eur. J., 2018, 24, 10122 CrossRef CAS; (d) C. Biagini, G. Capocasa, V. Cataldi, D. Del Giudice, L. Mandolini and S. Di Stefano, Chem. – Eur. J., 2019, 25, 15205 CrossRef CAS PubMed; (e) C. Biagini, G. Capocasa, D. Del Giudice, V. Cataldi, L. Mandolini and S. Di Stefano, Org. Biomol. Chem., 2020, 18, 3867–3873 RSC; (f) A. Ghosh, I. Paul and M. Schmittel, J. Am. Chem. Soc., 2021, 143, 5319–5323 CrossRef CAS; (g) A. Goswami, S. Saha, E. Elramadi, A. Ghosh and M. Schmittel, J. Am. Chem. Soc., 2021, 143, 14926–14935 CrossRef CAS.
- (a) C. Biagini, S. Albano, R. Caruso, L. Mandolini, J. A. Berrocal and S. Di Stefano, Chem. Sci., 2018, 9, 181 RSC; (b) P. Franchi, C. Poderi, E. Mezzina, C. Biagini, S. Di Stefano and M. Lucarini, J. Org. Chem., 2019, 84, 9364 CrossRef CAS PubMed; (c) D. Del Giudice, E. Spatola, R. Cacciapaglia, A. Casnati, L. Baldini, G. Ercolani and S. Di Stefano, Chem. – Eur. J., 2020, 26, 14954 CrossRef CAS PubMed.
- (a) S. Erbas-Cakmak, S. D. P. Fielden, U. Karaca, D. A. Leigh, C. T. McTernan, D. J. Tetlow and M. R. Wilson, Science, 2017, 358, 340 CrossRef CAS PubMed; (b) C. Biagini, S. D. P. Fielden, D. A. Leigh, F. Schaufelberger, S. Di Stefano and D. Thomas, Angew. Chem., Int. Ed., 2019, 58, 9876 CrossRef CAS; (c) E. Olivieri, G. Quintard, J.-V. Naubron and A. Quintard, J. Am. Chem. Soc., 2021, 143, 12650 CrossRef CAS PubMed.
- (a) D. Del Giudice, E. Spatola, M. Valentini, C. Bombelli, G. Ercolani and S. Di Stefano, Chem. Sci., 2021, 12, 7460 RSC; (b) D. Mariottini, D. Del Giudice, G. Ercolani, S. Di Stefano and F. Ricci, Chem. Sci., 2021, 12, 11735 RSC.
- For the importance of the “breathing” motions on the reactivity of the calixarene scaffold, see for example: (a) M. Galli, J. A. Berrocal, S. Di Stefano, R. Cacciapaglia, L. Mandolini, L. Baldini, A. Casnati and F. Ugozzoli, Org. Biomol. Chem., 2012, 10, 5109 RSC; (b) M. Ciaccia, I. Tosi, R. Cacciapaglia, A. Casnati, L. Baldini and S. Di Stefano, Org. Biomol. Chem., 2013, 11, 3642 RSC; (c) J. A. Berrocal, M. B. Baker, L. Baldini, A. Casnati and S. Di Stefano, Org. Biomol. Chem., 2018, 16, 7255 RSC.
- P. Timmerman, W. Verboom, D. N. Reinhoudt, A. Arduini, S. Grandi, A. R. Sicuri, A. Pochini and R. Ungaro, Synthesis, 1994, 185 CrossRef CAS.
- I. Tosi, M. S. Centellas, E. Campioli, A. Iagatti, A. Lapini, C. Sissa, L. Baldini, C. Cappelli, M. Di Donato, F. Sansone, F. Santoro and F. Terenziani, ChemPhysChem, 2016, 17, 1686 CrossRef CAS.
- For other systems for which the fluorescence due the intramolecular excimer between two pyrenyl units is switched ON/OFF or vice versa, see: (a) Z. Jarolímová, M. Vishe, J. Lacour and E. Bakker, Chem. Sci., 2016, 7, 525 RSC; (b) Y. Deng, S. Kin-Man Lai, L. Kong and H. Y. Au-Yeung, Chem. Commun., 2021, 57, 2931 RSC . None of them operates under dissipative conditions.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data of 7, fluorescence and UV-Vis spectra, and NMR data. CCDC 2108932. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ob02096j |
| ‡ These authors contributed equally. |
| § Given the evidence of an attractive interaction between the pyrenes in their ground-states, this band can be considered as originating from a “static excimer”, see the ESI, page S17† for a detailed explanation. |
| ¶ Fig. S15 in the ESI† shows the comparison of the 1H NMR spectra of the solutions obtained (i) after the addition of 2 mol equiv. of TFA to 2.0 mM 3 and (ii) immediately after the addition of 2.3 mol equiv. of 4 to 2.0 mM 3 (CD2Cl2/CD3CN 1:1). The spectra, which correspond to the third trace from the bottom in Fig. 2a and trace b in Fig. 3a, respectively, are very similar indicating very similar conformation and protonation states of calixarene 3 in the two cases. |
| This journal is © The Royal Society of Chemistry 2022 |