
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoreaction of biomass burning brown carbon aerosol particles†
Carolyn
Liu-Kang
 a,
Peter J.
Gallimore
b,
Tengyu
Liu
c and
Jonathan P. D.
Abbatt
*a
a,
Peter J.
Gallimore
b,
Tengyu
Liu
c and
Jonathan P. D.
Abbatt
*a
aDepartment of Chemistry, University of Toronto, 80 St. George Street, M5S 3H6, Toronto, ON, Canada. E-mail: jonathan.abbatt@utoronto.ca
bDepartment of Earth and Environmental Sciences, University of Manchester, Manchester, M13 9PS, UK
cJoint International Research Laboratory of Atmospheric and Earth System Sciences, School of Atmospheric Sciences, Nanjing University, Nanjing, 210023, China
First published on 1st February 2022
Abstract
The light-absorbing fraction of atmospheric organic particles, known as brown carbon (BrC) aerosol, can affect climate by influencing global radiative forcing. Regional effects arising from biomass burning BrC pollution are of particular interest, as they can have very high optical depth close to their sources. Due to the numerous fuel types, combustion conditions and reaction pathways encountered during the emission and lifetime of BrC, significant uncertainty arises from the impact that atmospheric aging can have on aerosol chemical composition and optical properties. Here, we investigate short-term aging processes driven by exposure to ultraviolet light (≈360 nm) that occur with primary BrC particles generated by smoldering pine wood. Suspended particles were aged in a chamber for a duration of 30 min. The single scattering albedo (SSA) at 405 nm decreased significantly by approximately 0.02 units (for example, from 0.98 to 0.96) during that time, due to an increase in absorption relative to scattering. This change was associated with an increase in oxygenation, represented by the signal fraction at m/z = 44 (f44) from the aerosol mass spectrometer (AMS). Surprisingly, the SSA continued to decrease after light exposure stopped, pointing to the presence of long-lived reactive species, perhaps radicals, that we hypothesize to form from photosensitized reactions. While relative humidity (RH) had only a minor impact on the rate of aging during light exposure, the aging rate in the dark may have occurred faster under dry conditions than at 45% RH.
Environmental significanceBrown carbon (BrC) aerosol particles from biomass burning events such as wildfires can significantly impact Earth's energy balance due to their ability to absorb light. The extent of this contribution, however, is not well understood, in part because of the complexity of BrC's composition and the various aging mechanisms that occur in the atmosphere. This work focuses on understanding one of these processes, which is short-term (30 min) light exposure of suspended particulate BrC. Improved understanding of the multiple aging processes of brown carbon will lead to more accurate modelling predictions of climate change. |
1. Introduction
Biomass burning injects combustion compounds in enormous quantities into the atmosphere, including those that contribute to brown carbon (BrC) aerosol particles. Along with black carbon (BC), BrC is a component of carbonaceous aerosol that can significantly impact the Earth's radiative balance by its ability to absorb ultraviolet and visible light.1,2 It was estimated that the magnitude of BrC absorption is up to 25% the magnitude of BC absorption (0.1–0.25 W m−2).3 Regional, near-source absorption is also important for UV fluxes and photochemistry. Ground-based measurements have found varying contributions of BrC's absorption relative to BC depending upon proximity to the source.4 Global model predictions show large regional variations.5The effects of BrC on climate remain uncertain, due to the complexity and variability of its chemical composition, and reactivity in the atmosphere.1 Different fuels, including plant types from various locations (e.g., litter, canopy), contribute to the complexity, as do burn conditions (i.e., flaming vs. smoldering).6–10 All these factors lead to uncertainties in radiative forcing estimates. Thus, BrC aerosol optical properties and atmospheric behavior need to be better defined.11
BrC molecules can exist in the atmosphere in the particulate phase or, via cloud scavenging, in the aqueous phase.1,12–14 Particulate BrC can be subject to a variety of aging mechanisms, including light exposure, and heterogeneous oxidation by OH radicals in daylight, nighttime NO3 radicals, and ozone.12,15–19 All of these reactants can change the ability of BrC to absorb light, and consequently, its impact on climate. To constrain particulate BrC's impact and lessen uncertainties in our model predictions, it is important to study each aging process to understand its relative contribution to BrC's overall light-absorption behavior. Of importance is determining the processes that occur both close to the source, often referred to as in the near-field, and those that occur as the particles reside in the atmosphere for much longer times in the far-field.
Photochemical aging of primary brown carbon has been investigated in the laboratory in a variety of media and over different timescales. These experiments have been performed using solvent extracts of BrC filter samples, particles collected on filters, and aerosol particles suspended in air. As described below, condensed-phase photochemistry can lead to either enhanced absorption for relatively short time exposures, decreased absorption, or increased absorption followed by decreases over long experiments. This wide range of behavior is not surprising given that both direct and photosensitized reactions can lead to individual chromophore formation and loss.20 Direct light exposure frequently leads to photolysis, fragmenting chromophores into smaller, less absorbing molecules.6,19 Photosensitized reactions can also cause fragmentation, however, they also have the ability to promote oxidation, functionalization, and oligomerization by radical formation.20–22
Specifically for BrC, Fleming et al. recently described the evolution of specific chromophores during the course of ultraviolet light exposure to filter-suspended biomass burning particles. Total UV-visible absorption measurements after long irradiation times showed an overall decrease in absorption for all wavelengths on timescales of 10–41 days, although individual absorbing molecules decayed more rapidly.6 In the aqueous phase, Hems et al. observed an increase in the water-soluble fraction of the wood smoke BrC mass absorption coefficient by a factor of 2 at 400 nm, during 6 hours of light exposure. This effect was attributed to the initial formation of aromatic dimers.13 Wong et al. also conducted experiments on water-soluble BrC and observed an increase in absorption in the initial 15 hours of aging at visible wavelengths associated with high molecular weight species (≥400 Da) followed by photobleaching for the remaining 10 hours. The lower molecular weight species, however, experienced rapid photobleaching.14 Zhong & Jang looked at suspended aerosols generated from hickory wood smoke when exposed to natural sunlight. They observed an initial increase in absorption by 11–64% from secondary organic aerosol formation, followed by a decrease by 19–68% from sunlight photobleaching.23 Jones et al. observed a redshift in the absorption peak from simultaneous UV illumination and drying of a water droplet containing wood smoke BrC.24 Upon UV exposure, Saleh et al. observed enhanced absorption in the near-UV wavelengths compared to longer wavelengths. They measured the formation of SOA, however, in addition to aging by primary particles.25 Finally, Hinks et al. explored the dependence of the rates of photoreactions of 2,4-nitrophenol upon environmental parameters, such as relative humidity (RH), temperature and organic matrices, finding that these reactions occur more slowly under drier, colder, and more viscous conditions.26
Direct comparison between these different laboratory experiments is challenging, as the reaction medium, light intensity and wavelength range, BrC source, and combustion conditions all vary.6–10 Nonetheless, a consistent observation is that an increase in absorption occurs at short timescales followed by a decrease at longer timescales. This behavior is qualitatively similar to what has been observed with aging of aqueous wood smoke BrC using dissolved OH radicals,13,14 and heterogeneous OH aging of both primary and secondary BrC surrogates.12,15,17,18 This raises the question whether similar processes are involved in both photoreactions and OH oxidative aging. In particular, since the formation of OH radicals is usually initiated by photolysis reactions, OH oxidation as well as direct photochemistry are potentially both occurring in the reaction systems.
A challenge related to performing laboratory aging of BrC samples is the ability to reproduce realistic atmospheric conditions in an indoor, controlled environment. For example, Wong et al. and Hems et al. worked with bulk aqueous media, Fleming et al. conducted experiments on a filter, and Zhong and Jang worked with suspended aerosols with secondary particle formation occurring. The goals of this project are to complement past work by focusing on the optical properties of suspended wood smoke particles when exposed to UV radiation centered at 360 nm. We purposefully scrub most semi-volatile gases that can be rapidly denuded so that we can isolate the photochemical aging behavior associated with primary, low volatility wood smoke aerosol particles, and we avoid using light sources with intensity below 300 nm. This prevents formation of secondary organic aerosol. We track both the scattering and absorption of the suspected particles, in order to measure the single scattering albedo of the particles that is the best measure of their intrinsic ability to absorb light. We use aerosol mass spectrometry to gain some indication of the nature of the chemical changes that occur, and we vary the relative humidity in the chamber to explore the impacts of this environmental parameter.
2. Methods
2.1 Generation of wood smoke
A representation of the experimental setup is shown in Fig. 1. Primary brown carbon aerosols were generated based on an experimental setup previously described.12,27 A 4 g piece of pine wood of approximately 8 × 2 × 1 cm dimensions was heated in a quartz tube (inner diameter of 2.2 cm) using a furnace at 400 °C (Thermo, Lindberg Blue M). Clean air (Linde, Grade Zero 0.1) was flowed through the tube at a rate of 2 L min−1. After the first signs of smoldering from an orange glow, but with no flame, the wood smoke was connected to a 1 m3 Teflon chamber. Before entering the chamber, the smoke passed through an impactor (457 μm diameter, 1 L min−1) and cyclone (2.5 μm cutpoint) as well as a buffer volume of approximately 0.024 m3, which removed the largest particles. Two denuders with activated charcoal (Sigma-Aldrich, 4–14 mesh) were used after the buffer volume to remove gas-phase compounds released from the wood smoke. These denuders are sufficiently effective that no secondary organic aerosol was observed in past studies, when high OH concentrations (≈107 molecules per cm3) were present in the chamber.12,15 Wood smoke was added until the optical scattering signal as well as the total mass loading (typically a few hundred μg m−3) were stable. The particles entered a chamber that had been prepared at the desired experimental condition (<15% or 45% RH) with flow from a clean air generator through an H2O bubbler.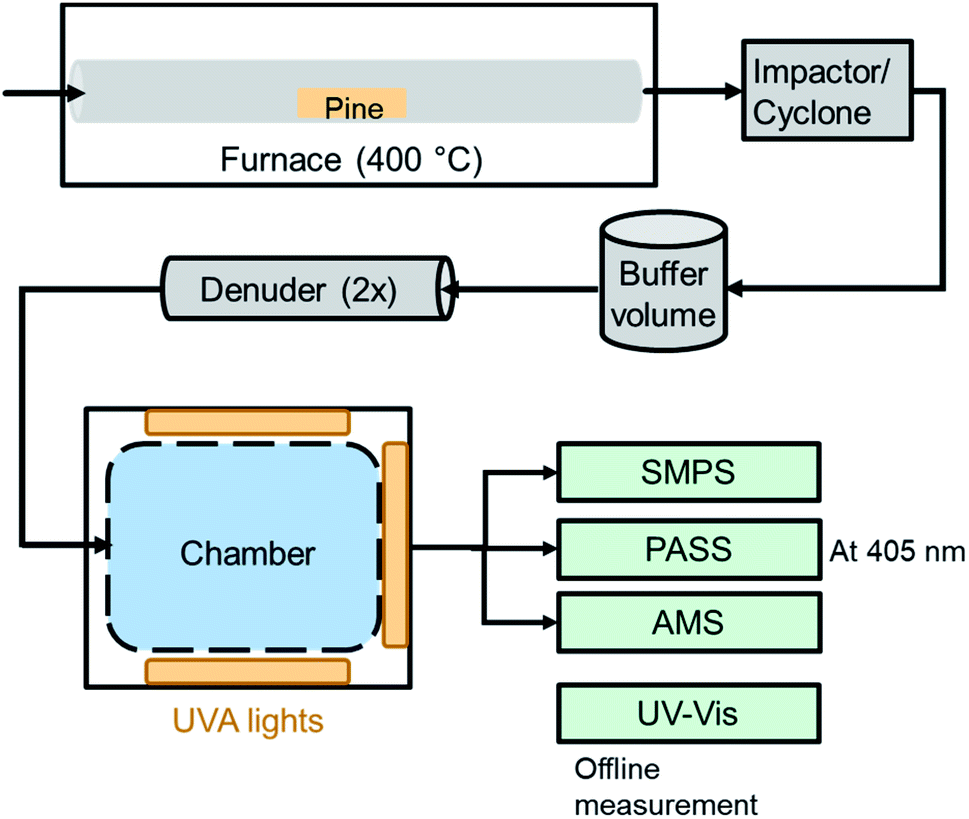 | ||
| Fig. 1 Experimental setup for the generation, aging and detection of BrC photoreaction. UV-vis was used as an offline technique and required a separate experiment to the online techniques. The AMS was not used for every experiment. The denuders were composed of activated charcoal. The UVA lights were centered at approximately 360 nm (see Fig. S1†). SMPS: scanning mobility particle sizer. PASS: photoacoustic soot spectrometer. AMS: aerosol mass spectrometer. | ||
2.2 Photoreaction aging of brown carbon
Particles were aged in the chamber during 30 min by exposure to 24 UVA lights (centered at 360 nm, Fig. S1†). Light levels measurements in the chamber (StellarNet Inc. BLACK-comet spectrometer) indicate that they are approximately the same when integrating from 300–400 nm as the actinic flux (see Fig. S1†). The experimental timescales are therefore roughly comparable to atmospheric timescales. We note, however, that there is variability in the estimations of clear-sky actinic flux depending on location and time. Additionally, light exposure varies greatly with different biomass burning plumes and within a single plume, where the darkest areas are located at the center. A dilution flow of approximately 4 L min−1 was continually flowing into the chamber. After 30 min, the lights were turned off and continuous measurements were taken for approximately 15 more minutes.Connected to the chamber was an instrument outlet for measurements by a scanning mobility particle sizer (SMPS) equipped with a differential mobility analyzer (DMA; TSI, 3080), an X-ray neutralizer (TSI, 3087), as well as a condensation particle counter (CPC; TSI, 3776). The flow rate from the CPC was set to 0.3 L min−1, and the sheath flow of the DMA was 2 L min−1. The measurements were taken during 135 s scans. A photoacoustic soot spectrometer (PASS; Droplet Measurement Technologies) was also connected to the chamber. It measured the aerosol scattering and absorption coefficients at 405 nm. The flow rate through the PASS was 1 L min−1. During specific experiments, an aerosol mass spectrometer (HR-ToF-AMS; Aerodyne; data analysis performed with SQUIRREL and PIKA softwares on Igor) was sampling as well. The AMS had a flow rate of approximately 0.07 L min−1. The reported f44 and f60 fractions were determined using high-resolution spectral peak fitting.
For offline measurements performed with UV-vis, the air from the chamber was pulled through a filter holder containing borosilicate glass filters (47 mm diameter; Pall) at a rate of approximately 15 L min−1 for 1 hour. The filter collection started as soon as the lights were turned off. For the collection of fresh wood smoke, the wood smoke was taken from the chamber without any light exposure. The mass collected on the filters was around 0.3 mg. The samples were extracted with 10 mL of dimethyl sulfoxide (DMSO; Sigma-Aldrich) and sonicated for 1 min. We have previously demonstrated that DMSO is the best solvent to remove colored wood smoke material from the filters.27 Analysis by UV-vis (Ocean Optics) was performed in a 1 cm cuvette, with an integration time of 300 ms and 3 scans per average.
3. Results
Absorption, scattering and single scattering albedo (SSA) measurements at 405 nm from a single light exposure experiment are shown in Fig. 2. The SSA is defined using the following equation: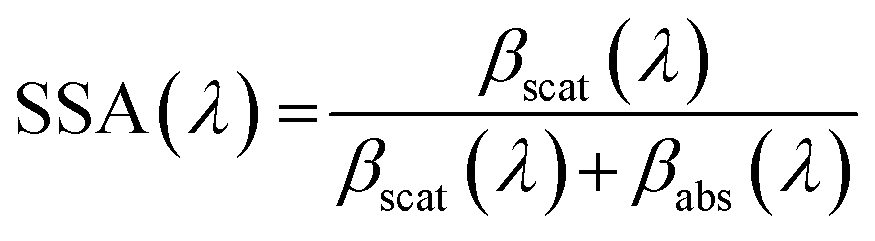 | (1) |
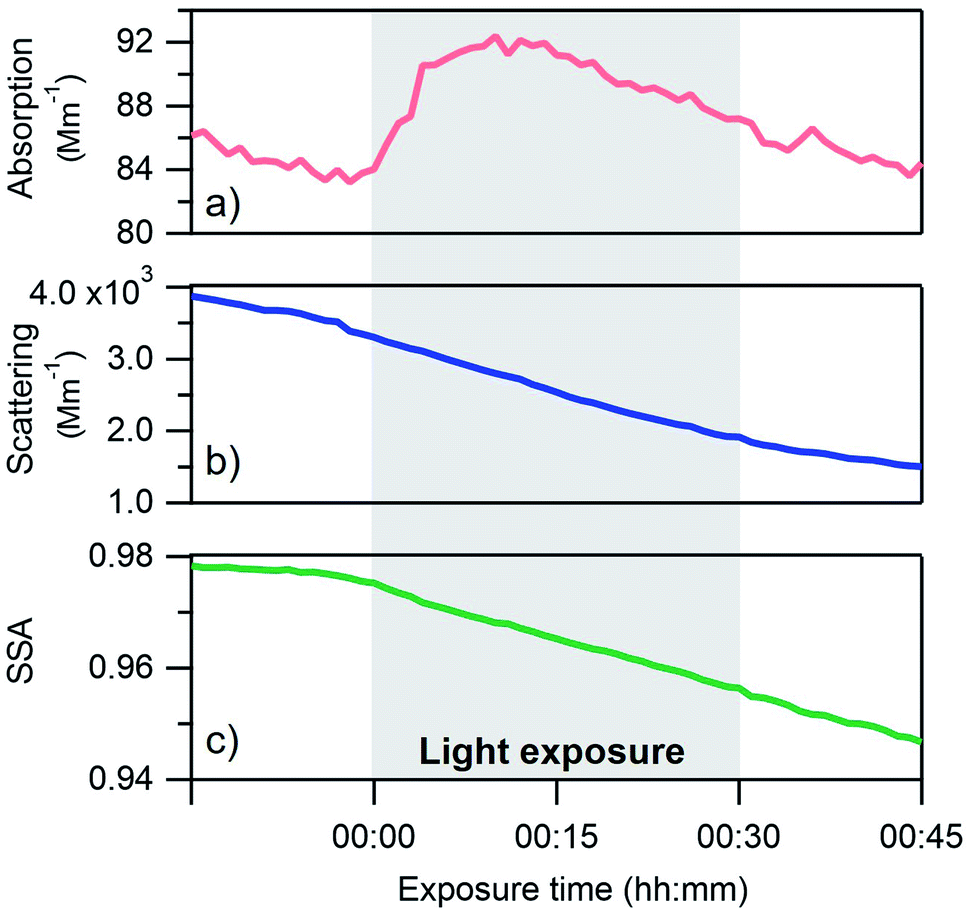 | ||
| Fig. 2 Absorption (a), scattering (b) and single scattering albedo (SSA) (c) results from a single photoreaction aging experiment of BrC. Data shown are from the PASS at 405 nm and for dry conditions (<15% RH). | ||
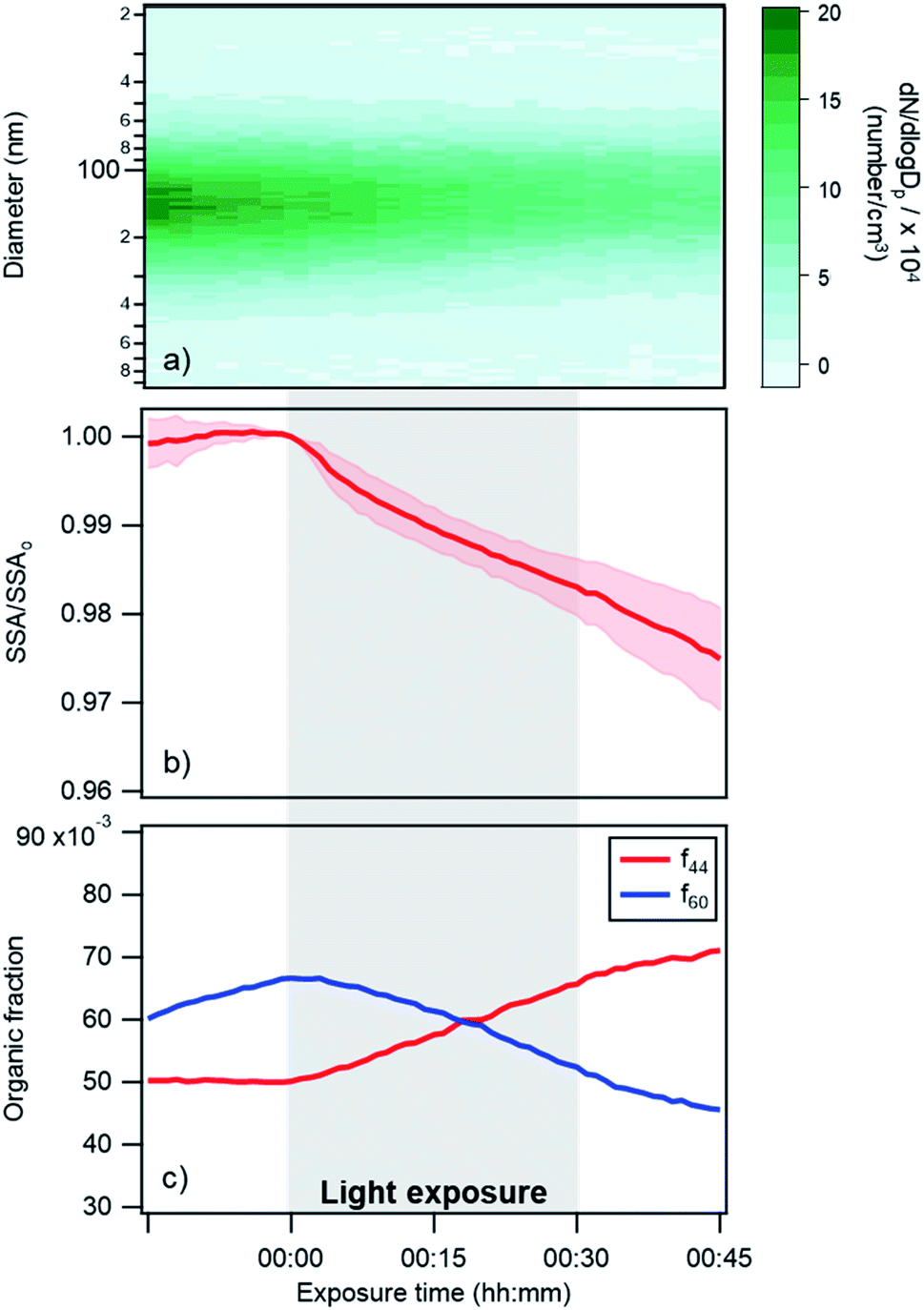 | ||
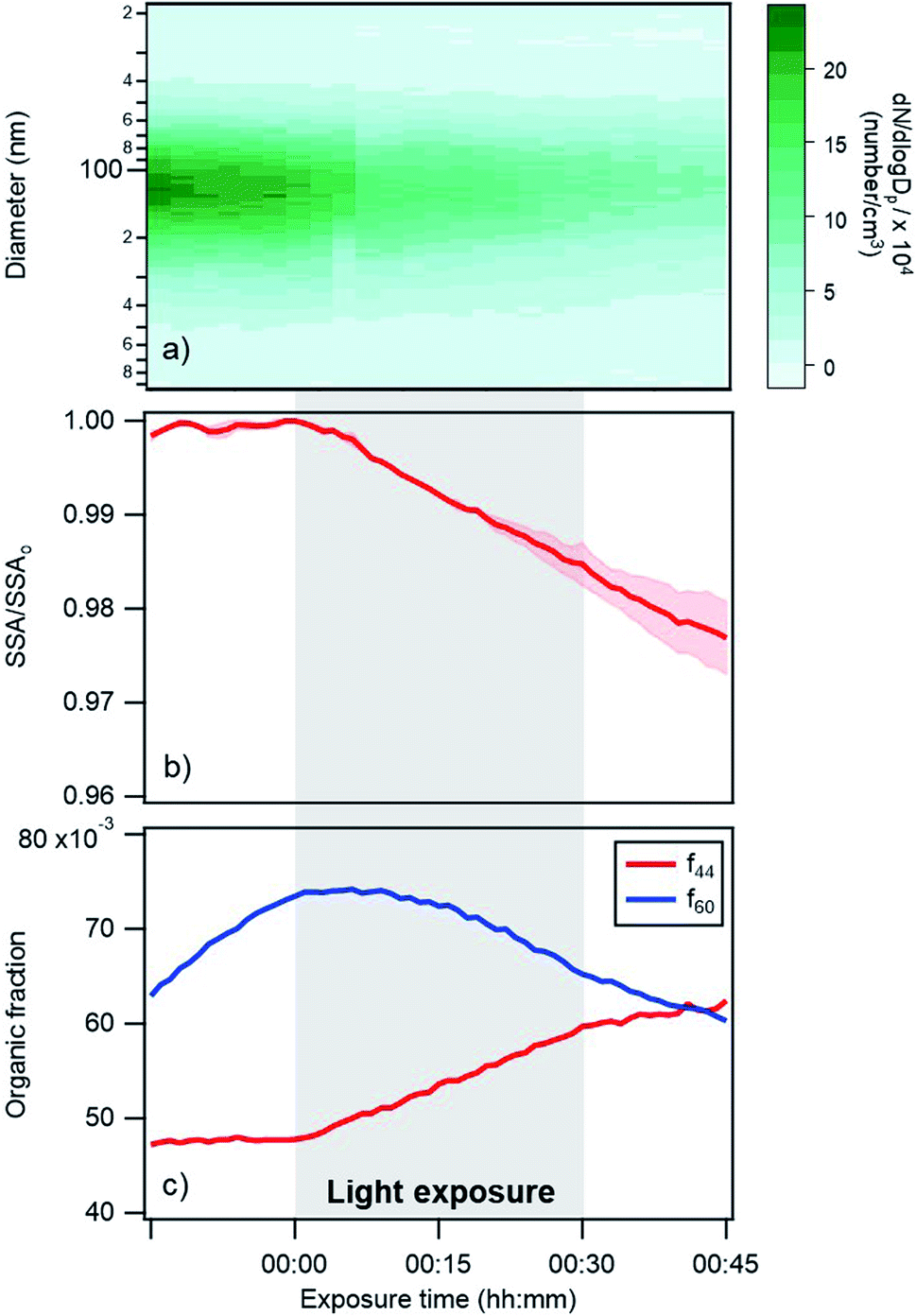
| Fig. 3 Changes in (a) particle size distribution, (b) single scattering albedo at 405 nm, and (c) AMS f44 and f60 fractions over time for a typical photoreaction experiment in dry conditions. Darker shaded regions in panel (a) represent a higher number concentration of particles. In panel (b) the SSA is represented as the ratio of experimental SSA over the SSA at exposure time = 0, in order to directly compare different experiments. The shaded pink region in (b) represent the standard deviation (for 4 different replicates). Note that the changes in f44 and f60 during a control experiment (Fig. S4†) were much smaller than the changes shown in this figure; (a) and (c) are results for one replicate. | ||
 | ||
| Fig. 4 Changes in (a) particle size distribution, (b) single scattering albedo at 405 nm, and (c) AMS f44 and f60 fractions over time for a typical photoreaction experiment at 45% RH. Darker shaded regions in panel (a) represent a higher number concentration of particles. In panel (b) the SSA is represented as the ratio of experimental SSA over the SSA at exposure time = 0 in order to compare multiple experiments. The shaded pink region represent the standard deviation (for 2 replicates). Note that the changes in f44 and f60 during a control experiment (Fig. S4†) were much smaller than the changes shown in this figure. (a) and (c) Are results for one replicate. | ||
Changes in the optical properties of primary brown carbon aerosols were investigated under 2 conditions, with average results presented for dry (Fig. 3b and S5†) and 45% RH (Fig. 4b and S5†) conditions. In these figures, in order to compare the single scattering albedo from different experiments, a normalized SSA was determined by initializing the SSA value at zero exposure time (SSAo) to have a value of 1, to account for the inherent experimental variability in starting SSA values. The average absolute starting SSAs with their standard deviations are shown in Table S1.† While initially stable, the SSA starts to decrease upon light exposure with a total decrease of approximately 0.02 during 30 min of light exposure. An additional observation is that, surprisingly, the SSA continues to decrease after the lights were turned off. This continued aging persists for at least 30 minutes (Fig. S7†). As discussed below, this suggests that light induces the formation of sufficiently long-lived reactive species that are responsible for driving chemistry in the dark.
Also, shown in the figures are aerosol particle size distributions and select quantities from the AMS measurements. In particular, the particle size distributions (Fig. 3a and 4a) as a function of time show decreasing particle number concentrations due to losses to the chamber walls but they do not exhibit any abrupt changes in the shape of the distribution upon exposure or removal of light. However, the increase in absorption at exposure time zero is associated with an increase in a measure of the particle oxidation state, represented by the fraction of the AMS organic mass spectrum (f44) (Fig. 3c and 4c). This m/z 44 mass spectral fragment arises from peroxides, esters and acids, often indicative of oxidation.7
The SSA is an important metric used to estimate aerosol absorption capability by taking into account scattering as well. To quantitatively compare the extent of SSA decrease for different experimental conditions and during the course of a photoreaction experiment, the linear regression slopes of the changing SSA values versus time are presented in Fig. 5. The more negative the slope, the faster and more significant the intrinsic absorption increase, i.e. increasing absorption relative to scattering.
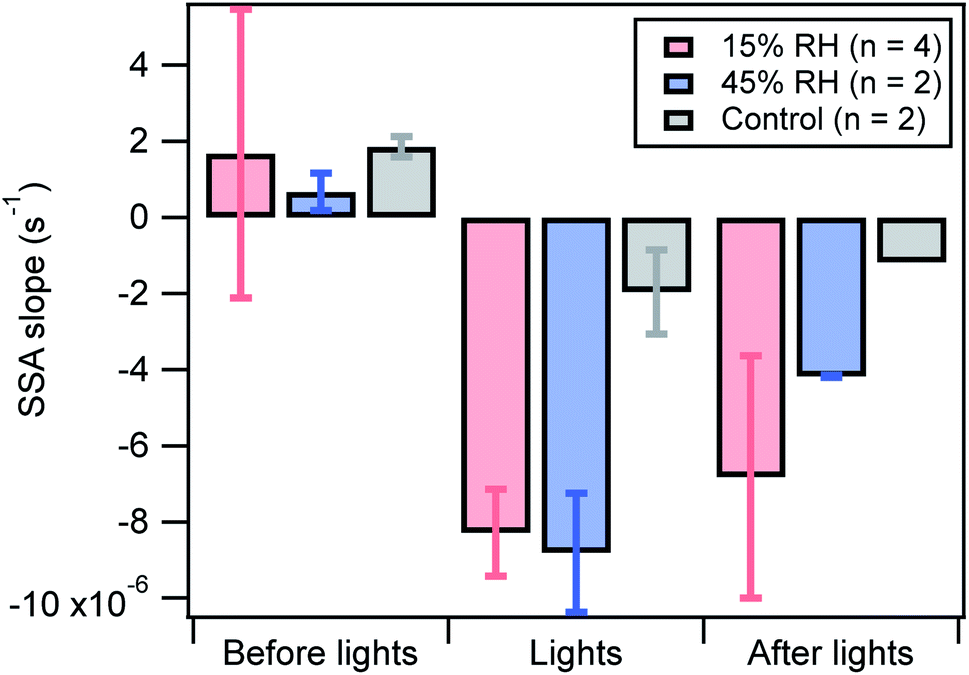 | ||
| Fig. 5 Single scattering albedo slopes before, during, and after light exposure for dry (pink) and 45% RH (blue). The slopes were recorded for 15 min both before and after the lights were turned on. The slope during light exposure was for a duration of 30 min. The error bars represent the standard deviation. The control experiments are the SSA obtained from adding BrC aerosols to the chamber without any light exposure, where the data before lights were recorded for 10 min instead of 15 min. The absence of error bars for the control conditions after lights is due to absence of data for one of the two control experiments (Fig. S6†). | ||
In general, the SSA slopes before light exposure are close to zero for all conditions, including during a control experiment during which the lights were not turned on (Fig. S6†). During light exposure, the slopes were consistently negative, signifying a decrease in SSA. Two of the dry and 45% RH experiments were performed in pairs, one immediately after the other. While the results in Fig. 5 suggest that the relative humidity doesn't significantly affect the results when the lights are on, in the paired experiments the SSA slope was consistently more negative for 45% RH. This suggests that there is potentially a small influence of RH on the optical properties.
When the lights are turned off, the photo-chemically initiated aging continues in the dark, but at a slower rate. Different environmental conditions yield different aging rates in the dark, where the higher relative humidity conditions may exhibit a slower decay than drier conditions with the caveat that the error bars are overlapping.
As described in the Methods section, filter samples were collected after light exposure for UV-vis analysis. The UV-vis absorption spectra were then measured of the DMSO extracts of the brown carbon material. Although these measurements require sample extraction, they allow us to observe shifts in the overall absorption spectrum whereas the absorption measurements described above were at a single wavelength (405 nm). In order to account for experimental variability and compare the shapes of the spectra from different experiments, the absorption was normalized to 1 at 275 nm which is approximately the maximum of the absorption spectrum. Results are shown in Fig. 6. In particular, it is shown that 30 minutes of photoreaction significantly increased the absorption at wavelengths > 350 nm.
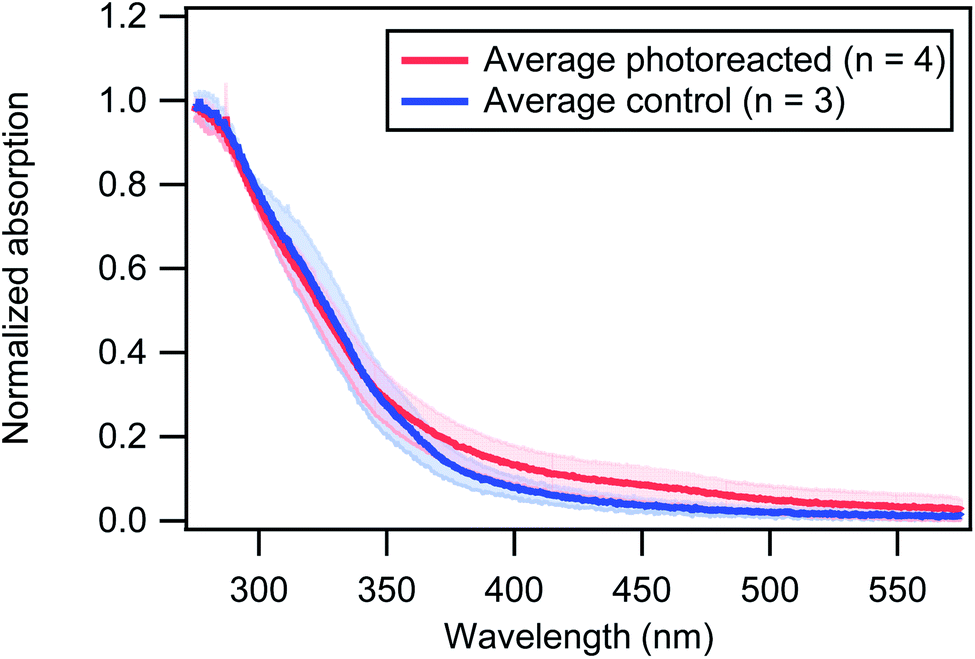 | ||
| Fig. 6 Averaged normalized absorption spectra for photoreacted wood smoke (red) and control (blue) experiments. The absorption spectra were normalized to 1 at 275 nm in order to directly compare different experiments. The shaded regions represent the standard deviation obtained from the different replicates. The samples were taken at dry conditions (<15% RH). | ||
4. Discussion
The major finding in this work is that primary wood smoke particles exhibit enhanced ultraviolet and visible absorption relative to scattering when exposed to atmospherically relevant wavelengths of ultraviolet light. This is in agreement with prior results from the literature, as discussed in the Introduction.13,14,26,28 This behavior is exhibited not only with the in situ measurements at 405 nm but also with the DMSO extracts, across the near-UV and visible parts of the spectrum. These changes occur without noticeable changes in the particles' size. As well, an intriguing observation is that the changes observed at 405 nm continue after the light exposure stops.The results indicate that oxidation is occurring in the particles during the photoreactions. In particular, when the lights are turned on, we observe a pronounced increase in the m/z 44 fraction of the organic mass spectrum, which is indicative of oxidation (see Fig. 3c and 4c). The f60 AMS fraction also shows signs of oxidative aging during the experiment. This signal is representative of sugars such as levoglucosan and is a common biomass burning tracer. While f60 is rising before lights are turned on (likely because the increasing mass loading in the chamber at this time is driving increased partitioning of semi-volatile species to the particles), the signal abruptly starts declining when the lights are turned on. This collective behavior of a decrease in m/z 60 and an increase in m/z 44 (see also Fig. S2†) is characteristic of oxidative aging of biomass burning particles.29 As well, levoglucosan decay from light exposure has been previously observed in chamber experiments and was attributed in part to OH oxidation arising from particles.23 We note that levoglucosan does not absorb 360 nm light.30
The mechanisms of formation of reactive species that lead to the f44 increase and f60 decrease are unclear. We do not believe that gas-phase OH radicals are being formed in substantial quantities, leading to heterogeneous oxidation. In particular, the flow from the wood smoke source passes through two diffusion denuders which will remove most potential gas-phase precursors, the wavelength of the UV light is high (≈360 nm), and we do not flame the wood to form NOx. Most importantly, the character of the oxidation processes does not change after the light exposure is removed, i.e. removal of light would have instantly removed gas-phase OH sources.
When the lights are on, one possibility is that OH radicals are being generated within the particles. In particular, there is a report in the literature that OH radicals are present in the aqueous phase during light exposure of wood smoke extracts using UVB wavelengths.14 More generally, for many decades, there have been indications that indirect or photosensitized condensed-phase photochemistry can drive the formation of reactive species, such as singlet molecular oxygen.20,31 However, even if such short-lived species are forming during light exposure, such generation mechanisms would stop after light exposure.
To discuss potential light-driven mechanisms in more detail, we note that there is evidence for the formation of light-induced radicals in a variety of atmospheric particle samples.20,22,32 These radicals are believed to form through excited triplet states of specific, light-absorbing BrC molecules, referred to as photosensitizers. Once excited to their triplet state, these BrC compounds can generate radicals and catalyze radical chain reactions, thus affecting chemical composition and potentially inducing a change in absorption. For example, an excited photosensitizer may react with a neighboring organic compound to form a carbon-centered organic radical, or with O2 to form reactive peroxy radicals.33,34 As well, in laboratory studies, reaction of a photosensitizer (specifically, imidazole-2-carboxaldehyde) with a fatty alcohol (1-octanol) revealed the possibility of forming a carbon centered radical, previously also observed to occur with limonene.34–36 Dimerization of organic radicals may give rise to larger, more absorbing species.21,22 If OH arises from higher levels of HOx, then it may add to aromatic rings, increasing their electron densities and ability to absorb light.15,21,37 Observations of aqueous phase reactivity of aromatic carbonyl photosensitizers with phenolic compounds have also been reported. Mechanistic insight into the reactivity of the excited triplet state revealed scavenging of the hydrogen atom from the alcohol functional group, forming an oxygen-centered radical.38 Overall, we believe that the potential formation of both C- and O-centered radicals, and of OH radicals, all contribute to increased reactivity in the BrC particles, degradation of sugars such as levoglucosan, and the formation of more absorbing molecules through dimerization or functionalization. It is possible that the tight molecular confines of an aerosol particle promote intermolecular chemistry over the rate at which it occurs in a solvent extract. However, future experiments are needed to determine the details of the dominant mechanism.
We note that, during a light exposure event, we did not observe evidence in the AMS spectrum of higher molecular weight species forming (see Fig. S3†), only oxidation from the f44 fraction (Fig. 3c and 4c). However, this does not rule out dimerization of specific precursors to form stronger chromophores, given that the AMS is not well suited to the identification of low concentrations of higher molecular weight absorbing species. However, we do observe evidence of fragmentation, which has been previously demonstrated to occur as samples age.21,22
As noted above, a novel observation in our experiments is the continued aging of the particles in the dark. While the lifetimes of excited triplet state photosensitizers are fractions of a second,39,40 the lifetime of some radicals formed in the condensed phase can be much longer. Indeed, the term “environmentally persistent free radicals” has been used to describe such species. There is considerable evidence in the literature for the presence of such radicals in biomass burning and combustion aerosol.41–44 As one example, Qin et al. recently determined a 100 day half-life of some radicals produced from light exposure of catechol, a common biomass burning aerosol component after mixing with ambient PM2.5 particle material.44 Yang et al. also observed the lifetime of radicals from PM1 to be many days, with biomass burning being one of the sources of particulate matter.43 While we cannot confidently say whether such long-lived radicals are driving continued oxidation in the particles, it is clear that some reactive intermediate with a lifetime of at least tens of minutes is present. It is interesting to note that experiments performed with the same chamber did not see evidence for continued oxidation in the dark when studying the photoreactions of water-soluble SOA generated from α-pinene ozonolysis.45 Thus, the effect appears to be specific to biomass burning aerosol, perhaps related to its reactive, aromatic composition.
Another important parameter related to the conditions in which aging occurs is the relative humidity. Within the experimental uncertainty, the results obtained in Fig. 5 show that the SSA slope is just marginally (6%) larger at higher relative humidity than in dry conditions, indicating either no impact or a slightly more rapid aging process perhaps reflecting more rapid reactive intermediate generation or reaction. Past studies of atmospheric aerosol material surrogates have shown that photoreactions proceed more rapidly at higher RH, presumably due to lower viscosity in the material and more molecular interactions.26 Other photooxidation studies have been shown to occur more rapidly at higher RH values. For example, Zhong & Jang determined that photooxidation of hickory wood smoke, when exposed to sunlight, occurred at a faster rate at higher RH.23 This was also demonstrated by Wong et al. where light exposure of SOA from α-pinene decreases the mass of total organics more rapidly at higher RH (85%) relative to mid-RH (60%) and low-RH (5%).45 As well, Schnitzler et al. observed a more rapid and higher peak absorption at 60% RH relative to 15% RH during OH oxidation of primary pine wood smoke particles.12
By contrast to these photo-oxidation studies, aging may have continued in the dark more rapidly at lower RH than at high RH after light exposure was terminated. This suggests that particle viscosity may affect the sources and sinks of reactive intermediates in the particles in a different manner. With the sources of such species turned off in the dark, it is possible that the lifetimes of the reactive intermediates are lengthened when molecular motion is suppressed at low RH. There is also potential for concentration effects, given that low RH leads to more concentrated solutes.33,46 We note that previous studies on the effects of RH on radical production from photosensitized reactions have observed complex behavior, which may be driven by a variety of factors, including concentration and viscosity.33
5. Atmospheric implications
The focus of this study was on aging by photoreaction of primary wood smoke brown carbon particles that will occur on a short timescale after injection into a sunlit atmosphere. Strong effects across the near UV and visible parts of the spectrum were observed after only 30 minutes with light levels lower than those typically present in the daytime atmosphere. While this aging effect has previously been investigated in the aqueous phase,13,14,24 the scope of this project differs from past studies by aging suspended aerosol particles using a UVA light source centered at 360 nm. Other studies have either employed stronger UV sources or performed aging measurements offline with solvent extracts or filters. Thus, we have demonstrated that darkening photoreactions occur in suspended particles that are held under steady environmental conditions.We note that this project focuses on the aging of primary particles by removing the gas-phase compounds generated from wood burning prior to aging. It is known that the formation of secondary brown carbon aerosols from gas-phase precursors generates additional absorption.23,25 By removing the effects of secondary gas-to-particle conversion, we are able to constrain the effects of light on primary particles only. We note that there is possibility of evaporation from semi-volatile compounds within the chamber, however, there was no observation of significant changes to size distribution (Fig. 3a and 4a) and no impact on the optical properties.
We demonstrate that this photo-initiated effect persists in the dark, albeit at a slower rate than when light was present. This points to an aging mechanism that involves long-lived radicals or reactive intermediates.44 This stands in contrast to OH-driven aging which only occurs in the light.1,6 The persistent photo-initiated aging that we observe in the dark is relevant for atmospheric conditions where light levels are low. For example, particles generated from a biomass burning event can rapidly shift from direct sunlight exposure to a hazy or cloudy environment.
When placing this darkening photoreaction into an atmospheric context, it is important to consider the timescales involved. The aging mechanism studied here is relevant to short-term light exposure to primary BrC particles. A longer aging timescale would likely induce significant photobleaching subsequent to the photoenhancement observed at short times.14 For example, Wong et al. saw the initial increase of absorption from high molecular weight water-soluble BrC during the first 15 hours of aging, following by bleaching for 10 hours. Fleming et al. reported the decay lifetimes of BrC chromophores from different fuels to be on the order of days to a week.6,14 Furthermore, competition between other daytime aging processes, such as OH and O3 heterogeneous oxidation, is likely to change the overall aging timescale as well.
Author contributions
CLK, PJG, JPDA conceptualized the project. CLK, PJG, TL conducted the measurements and analyzed the data. CLK, JPDA wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from NSERC and the prior work of Drs R. Hems and E. Schnitzler for developing the wood smoke source.References
- A. Laskin, J. Laskin and S. A. Nizkorodov, Chemistry of Atmospheric Brown Carbon, Chem. Rev., 2015, 115(10), 4335–4382 CrossRef CAS PubMed.
- H. Brown, X. Liu, Y. Feng, Y. Jiang, M. Wu, Z. Lu, C. Wu, S. Murphy and R. Pokhrel, Radiative effect and climate impacts of brown carbon with the Community Atmosphere Model (CAM5), Atmos. Chem. Phys., 2018, 18(24), 17745–17768 CrossRef CAS.
- Y. Feng, V. Ramanathan and V. R. Kotamarthi, Brown carbon: a significant atmospheric absorber of solar radiation?, Atmos. Chem. Phys., 2013, 13(17), 8607–8621 CrossRef.
- Y. Zhang, Y. Peng, W. Song, Y.-L. Zhang, P. Ponsawansong, T. Prapamontol and Y. Wang, Contribution of brown carbon to the light absorption and radiative effect of carbonaceous aerosols from biomass burning emissions in Chiang Mai, Thailand, Atmos. Environ., 2021, 260, 118544 CrossRef CAS.
- D. S. Jo, R. J. Park, S. Lee, S. W. Kim and X. Zhang, A global simulation of brown carbon: implications for photochemistry and direct radiative effect, Atmos. Chem. Phys., 2016, 16(5), 3413–3432 CrossRef CAS.
- L. T. Fleming, P. Lin, J. M. Roberts, V. Selimovic, R. Yokelson, J. Laskin, A. Laskin and S. A. Nizkorodov, Molecular composition and photochemical lifetimes of brown carbon chromophores in biomass burning organic aerosol, Atmos. Chem. Phys., 2020, 20(2), 1105–1129 CrossRef CAS.
- P. Lin, P. K. Aiona, Y. Li, M. Shiraiwa, J. Laskin, S. A. Nizkorodov and A. Laskin, Molecular Characterization of Brown Carbon in Biomass Burning Aerosol Particles, Environ. Sci. Technol., 2016, 50(21), 11815–11824 CrossRef CAS PubMed.
- M. Xie, X. Chen, M. D. Hays and A. L. Holder, Composition and light absorption of N-containing aromatic compounds in organic aerosols from laboratory biomass burning, Atmos. Chem. Phys., 2019, 19(5), 2899–2915 CrossRef CAS PubMed.
- J. Martinsson, A. C. Eriksson, I. E. Nielsen, V. B. Malmborg, E. Ahlberg, C. Andersen, R. Lindgren, R. Nyström, E. Z. Nordin, W. H. Brune, B. Svenningsson, E. Swietlicki, C. Boman and J. H. Pagels, Impacts of Combustion Conditions and Photochemical Processing on the Light Absorption of Biomass Combustion Aerosol, Environ. Sci. Technol., 2015, 49(24), 14663–14671 CrossRef CAS PubMed.
- C. N. Jen, L. E. Hatch, V. Selimovic, R. J. Yokelson, R. Weber, A. E. Fernandez, N. M. Kreisberg, K. C. Barsanti and A. H. Goldstein, Speciated and total emission factors of particulate organics from burning western US wildland fuels and their dependence on combustion efficiency, Atmos. Chem. Phys., 2019, 19(2), 1013–1026 CrossRef CAS.
- T. Takemura, T. Nakajima, O. Dubovik, B. N. Holben and S. Kinne, Single-scattering albedo and radiative forcing of various aerosol species with a global three-dimensional model, J. Clim., 2002, 15(4), 333–352 CrossRef.
- E. G. Schnitzler, T. Liu, R. F. Hems and J. P. D. Abbatt, Emerging investigator series: heterogeneous OH oxidation of primary brown carbon aerosol: effects of relative humidity and volatility, Environ. Sci.: Processes Impacts, 2020, 22(11), 2162–2171 CAS.
- R. F. Hems, E. G. Schnitzler, M. Bastawrous, R. Soong, A. J. Simpson and J. P. D. Abbatt, Aqueous Photoreactions of Wood Smoke Brown Carbon, ACS Earth Space Chem., 2020, 4(7), 1149–1160 CrossRef CAS.
- J. P. S. Wong, M. Tsagkaraki, I. Tsiodra, N. Mihalopoulos, K. Violaki, M. Kanakidou, J. Sciare, A. Nenes and R. J. Weber, Atmospheric evolution of molecular-weight-separated brown carbon from biomass burning, Atmos. Chem. Phys., 2019, 19(11), 7319–7334 CrossRef CAS.
- E. G. Schnitzler and J. P. D. Abbatt, Heterogeneous OH oxidation of secondary brown carbon aerosol, Atmos. Chem. Phys., 2018, 18(19), 14539–14553 CrossRef CAS.
- C. Li, Q. He, A. P. S. Hettiyadura, U. Käfer, G. Shmul, D. Meidan, R. Zimmermann, S. S. Brown, C. George, A. Laskin and Y. Rudich, Formation of Secondary Brown Carbon in Biomass Burning Aerosol Proxies through NO3 Radical Reactions, Environ. Sci. Technol., 2020, 54(3), 1395–1405 CrossRef CAS PubMed.
- E. C. Browne, X. Zhang, J. P. Franklin, K. J. Ridley, T. W. Kirchstetter, K. R. Wilson, C. D. Cappa and J. H. Kroll, Effect of heterogeneous oxidative aging on light absorption by biomass burning organic aerosol, Aerosol Sci. Technol., 2019, 53(6), 663–674 CrossRef CAS.
- B. J. Sumlin, A. Pandey, M. J. Walker, R. S. Pattison, B. J. Williams and R. K. Chakrabarty, Atmospheric Photooxidation Diminishes Light Absorption by Primary Brown Carbon Aerosol from Biomass Burning, Environ. Sci. Technol. Lett., 2017, 4(12), 540–545 CrossRef CAS.
- R. F. Hems, E. G. Schnitzler, C. Liu-Kang, C. D. Cappa and J. P. D. Abbatt, Aging of Atmospheric Brown Carbon Aerosol, ACS Earth Space Chem., 2021, 5(4), 722–748 CrossRef CAS.
- C. George, M. Ammann, B. D'Anna, D. J. Donaldson and S. A. Nizkorodov, Heterogeneous Photochemistry in the Atmosphere, Chem. Rev., 2015, 115(10), 4218–4258 CrossRef CAS PubMed.
- M. V. Misovich, A. P. S. Hettiyadura, W. Jiang, Q. Zhang and A. Laskin, Molecular-Level Study of the Photo-Oxidation of Aqueous-Phase Guaiacyl Acetone in the Presence of 3C*: Formation of Brown Carbon Products, ACS Earth Space Chem., 2021, 5(8), 1983–1996 CrossRef CAS.
- W. Jiang, M. V. Misovich, A. P. S. Hettiyadura, A. Laskin, A. S. McFall, C. Anastasio and Q. Zhang, Photosensitized Reactions of a Phenolic Carbonyl from Wood Combustion in the Aqueous Phase—Chemical Evolution and Light Absorption Properties of AqSOA, Environ. Sci. Technol., 2021, 55(8), 5199–5211 CrossRef CAS PubMed.
- M. Zhong and M. Jang, Dynamic light absorption of biomass-burning organic carbon photochemically aged under natural sunlight, Atmos. Chem. Phys., 2014, 14(3), 1517–1525 CrossRef.
- S. H. Jones, P. Friederich and D. J. Donaldson, Photochemical Aging of Levitated Aqueous Brown Carbon Droplets, ACS Earth Space Chem., 2021, 5(4), 749–754 CrossRef CAS.
- R. Saleh, C. J. Hennigan, G. R. McMeeking, W. K. Chuang, E. S. Robinson, H. Coe, N. M. Donahue and A. L. Robinson, Absorptivity of brown carbon in fresh and photo-chemically aged biomass-burning emissions, Atmos. Chem. Phys., 2013, 13(15), 7683–7693 CrossRef.
- M. L. Hinks, M. V. Brady, H. Lignell, M. Song, J. W. Grayson, A. K. Bertram, P. Lin, A. Laskin, J. Laskin and S. A. Nizkorodov, Effect of viscosity on photodegradation rates in complex secondary organic aerosol materials, Phys. Chem. Chem. Phys., 2016, 18(13), 8785–8793 RSC.
- A. Trofimova, R. F. Hems, T. Liu, J. P. D. Abbatt and E. G. Schnitzler, Contribution of Charge-Transfer Complexes to Absorptivity of Primary Brown Carbon Aerosol, ACS Earth Space Chem., 2019, 3(8), 1393–1401 CrossRef CAS.
- J. P. S. Wong, A. Nenes and R. J. Weber, Changes in Light Absorptivity of Molecular Weight Separated Brown Carbon Due to Photolytic Aging, Environ. Sci. Technol., 2017, 51(15), 8414–8421 CrossRef CAS PubMed.
- A. M. Ortega, D. A. Day, M. J. Cubison, W. H. Brune, D. Bon, J. A. de Gouw and J. L. Jimenez, Secondary organic aerosol formation and primary organic aerosol oxidation from biomass-burning smoke in a flow reactor during FLAME-3, Atmos. Chem. Phys., 2013, 13(22), 11551–11571 CrossRef.
- R. Zhao, A. K. Y. Lee, L. Huang, X. Li, F. Yang and J. P. D. Abbatt, Photochemical processing of aqueous atmospheric brown carbon, Atmos. Chem. Phys., 2015, 15(11), 6087–6100 CrossRef CAS.
- J. N. Pitts Jr, A. U. Khan, E. B. Smith and R. P. Wayne, Singlet oxygen in the environmental sciences. Singlet molecular oxygen and photochemical air pollution, Environ. Sci. Technol., 1969, 3(3), 241–247 CrossRef.
- Q. Chen, H. Sun, M. Wang, Y. Wang, L. Zhang and Y. Han, Environmentally Persistent Free Radical (EPFR) Formation by Visible-Light Illumination of the Organic Matter in Atmospheric Particles, Environ. Sci. Technol., 2019, 53(17), 10053–10061 CrossRef CAS PubMed.
- P. Corral Arroyo, T. Bartels-Rausch, P. A. Alpert, S. Dumas, S. Perrier, C. George and M. Ammann, Particle-Phase Photosensitized Radical Production and Aerosol Aging, Environ. Sci. Technol., 2018, 52(14), 7680–7688 CrossRef CAS PubMed.
- S. Rossignol, K. Z. Aregahegn, L. Tinel, L. Fine, B. Nozière and C. George, Glyoxal Induced Atmospheric Photosensitized Chemistry Leading to Organic Aerosol Growth, Environ. Sci. Technol., 2014, 48(6), 3218–3227 CrossRef CAS PubMed.
- M. Teich, D. van Pinxteren, S. Kecorius, Z. Wang and H. Herrmann, First Quantification of Imidazoles in Ambient Aerosol Particles: Potential Photosensitizers, Brown Carbon Constituents, and Hazardous Components, Environ. Sci. Technol., 2016, 50(3), 1166–1173 CrossRef CAS PubMed.
- B. You, S. Li, N. T. Tsona, J. Li, L. Xu, Z. Yang, S. Cheng, Q. Chen, C. George, M. Ge and L. Du, Environmental Processing of Short-Chain Fatty Alcohols Induced by Photosensitized Chemistry of Brown Carbons, ACS Earth Space Chem., 2020, 4(4), 631–640 CrossRef CAS.
- R. F. Hems and J. P. D. Abbatt, Aqueous Phase Photo-oxidation of Brown Carbon Nitrophenols: Reaction Kinetics, Mechanism, and Evolution of Light Absorption, ACS Earth Space Chem., 2018, 2(3), 225–234 CrossRef CAS.
- L. Yu, J. Smith, A. Laskin, C. Anastasio, J. Laskin and Q. Zhang, Chemical characterization of SOA formed from aqueous-phase reactions of phenols with the triplet excited state of carbonyl and hydroxyl radical, Atmos. Chem. Phys., 2014, 14(24), 13801–13816 CrossRef.
- E. Woods, O. T. Harris, W. E. Leiter, N. E. Burner, P. Ofosuhene, A. Krez, M. A. Hilton and K. A. Burke, Lifetime of Triplet Photosensitizers in Aerosol Using Time-Resolved Photoelectric Activity, ACS Earth Space Chem., 2020, 4(8), 1424–1434 CrossRef CAS.
- T. Felber, T. Schaefer, L. He and H. Herrmann, Aromatic Carbonyl and Nitro Compounds as Photosensitizers and Their Photophysical Properties in the Tropospheric Aqueous Phase, J. Phys. Chem. A, 2021, 125(23), 5078–5095 CrossRef CAS PubMed.
- W. A. Pyror, Biological effects of cigarette smoke, wood smoke, and the smoke from plastics: The use of electron spin resonance, Free Radic. Biol. Med., 1992, 13(6), 659–676 CrossRef.
- T. M. Lachocki, D. F. Church and W. A. Pryor, Pesistent free radicals in woodsmoke: An ESR spin trapping study, Free Radic. Biol. Med., 1989, 7(1), 17–21 CrossRef CAS PubMed.
- L. Yang, G. Liu, M. Zheng, R. Jin, Q. Zhu, Y. Zhao, X. Wu and Y. Xu, Highly Elevated Levels and Particle-Size Distributions of Environmentally Persistent Free Radicals in Haze-Associated Atmosphere, Environ. Sci. Technol., 2017, 51(14), 7936–7944 CrossRef CAS PubMed.
- L. Qin, L. Yang, J. Yang, R. Weber, K. Ranguelova, X. Liu, B. Lin, C. Li, M. Zheng and G. Liu, Photoinduced formation of persistent free radicals, hydrogen radicals, and hydroxyl radicals from catechol on atmospheric particulate matter, iScience, 2021, 24(3), 102193 CrossRef CAS PubMed.
- J. P. S. Wong, S. Zhou and J. P. D. Abbatt, Changes in Secondary Organic Aerosol Composition and Mass due to Photolysis: Relative Humidity Dependence, J. Phys. Chem. A, 2015, 119(19), 4309–4316 CrossRef CAS PubMed.
- N. Y. Kasthuriarachchi, L.-H. Rivellini, X. Chen, Y. J. Li and A. K. Y. Lee, Effect of Relative Humidity on Secondary Brown Carbon Formation in Aqueous Droplets, Environ. Sci. Technol., 2020, 54(20), 13207–13216 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ea00088h |
| This journal is © The Royal Society of Chemistry 2022 |