
Understanding the n → π* non-covalent interaction using different experimental and theoretical approaches
Prakash
Panwaria
 and
Aloke
Das
*
and
Aloke
Das
*
Department of Chemistry, Indian Institute of Science Education and Research (IISER) Pune, Dr. Homi Bhabha Road, Pashan, Pune-411008, India. E-mail: a.das@iiserpune.ac.in
First published on 29th June 2022
Abstract
Herein, a perspective on the recent understanding of weak n → π* interaction obtained using different experimental and theoretical approaches is presented. This interaction is purely an orbital interaction that involves the delocalization of the lone pair electrons (n) on nitrogen, oxygen, and sulfur to the π* orbitals of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CN, and aromatic rings. The n → π* interaction has been found to profoundly influence the stabilization of peptides, proteins, drugs, and various small molecules. Although the functional properties of this non-covalent interaction are still quite underestimated, there are recent demonstrations of applying this interaction to the regulation of synthetic chemistry, catalysis, and molecular recognition. However, the identification and quantification of the n → π* interaction remain a demanding task as this interaction is quite weak and based on the electron delocalization between the two orbitals, while hyperconjugation interactions between neighboring atoms and the group involved in the n → π* interaction are simultaneously present. This review provides a comprehensive picture of understanding the n → π* interaction using different experimental approaches such as the X-ray diffraction technique, and electronic, NMR, microwave, and IR spectroscopy, in addition to quantum chemistry calculations. A detailed understanding of the n → π* interaction can help in modulating the strength of this interaction, which will be further helpful in designing efficient drugs, synthetic peptides, peptidomimetics, etc.
O, CN, and aromatic rings. The n → π* interaction has been found to profoundly influence the stabilization of peptides, proteins, drugs, and various small molecules. Although the functional properties of this non-covalent interaction are still quite underestimated, there are recent demonstrations of applying this interaction to the regulation of synthetic chemistry, catalysis, and molecular recognition. However, the identification and quantification of the n → π* interaction remain a demanding task as this interaction is quite weak and based on the electron delocalization between the two orbitals, while hyperconjugation interactions between neighboring atoms and the group involved in the n → π* interaction are simultaneously present. This review provides a comprehensive picture of understanding the n → π* interaction using different experimental approaches such as the X-ray diffraction technique, and electronic, NMR, microwave, and IR spectroscopy, in addition to quantum chemistry calculations. A detailed understanding of the n → π* interaction can help in modulating the strength of this interaction, which will be further helpful in designing efficient drugs, synthetic peptides, peptidomimetics, etc.
 Prakash Panwaria | Prakash Panwaria received his BSc Hons in Chemistry from Dayalbagh Educational Institute (DEI), Agra, India, in 2016. He joined the integrated PhD program in 2016 at the Indian Institute of Science Education and Research (IISER), Pune, India. He completed his MS research project with Prof. Aloke Das in 2018 and continued working with Prof. Aloke Das's group for his PhD program. His research interests focus on understanding weak n → π* non-covalent interactions in small molecules and clusters using gas-phase laser spectroscopy and quantum chemical calculation. This also involves the study of intra- and inter-molecular modulation of the n → π* interaction by varying the substituent effect on n → π* donor/acceptor sites. |
 Aloke Das | Aloke Das received his BSc (1994) and MSc (1996) degrees from Jadavpur University, Kolkata, India. He earned his PhD degree in the area of gas phase laser spectroscopy from the Indian Institute of Technology Kanpur in 2002 under the supervision of Professor Tapas Chakraborty. Subsequently, he moved to Purdue University for doing his postdoctoral research with Professor Timothy S. Zwier and worked on conformation-specific UV and IR spectroscopy of flexible aromatic hydrocarbons, which are isomeric products in fuel combustion processes (2002–2004). Later, he worked with Professor Erwin D. Poliakoff at Louisiana State University to study vibrationally resolved Vacuum-Ultraviolet (VUV) photoelectron spectroscopy of polyatomic molecules in the gas phase using synchrotron radiation at the Advanced Light Source of the Lawrence Berkeley National Laboratory (2004–2007). In 2007, he joined the Indian Institute of Science Education and Research (IISER) Pune as an Assistant Professor in the Department of Chemistry. Presently, he is a Professor at the Indian Institute of Science Education and Research (IISER) Pune. His research includes molecular level understanding of various types of non-covalent interactions through gas phase laser spectroscopy of isolated molecules and complexes relevant to biomolecules and materials using thermal heating as well as laser desorption as the vaporization source. Currently, the major focus of his research is on understanding n → π* non-covalent interaction, sulfur and selenium (Se) hydrogen-bonding, sequence dependent folding motifs of peptides, conformation-specific electronic circular dichroism spectroscopy, etc. |
Introduction
Non-covalent interactions play a crucial role in determining the specific structures and functions of biomolecules, drugs, supramolecular assemblies, materials, and molecular complexes.1–12 Various non-covalent interactions such as hydrogen bonding,1–20 halogen bonding,21–25 chalcogen bonding,25–29 π–π stacking,30–33 cation–π,34–37 and anion–π38–41 interactions have been extensively studied in the literature. Hydrogen bonding is ubiquitous and well-established among all the non-covalent interactions reported in the literature.1,2 However, another non-covalent interaction termed the n → π* interaction, which is quite analogous to the hydrogen bond in terms of electron delocalization, has been explored only recently.42–44 The n → π* interaction generally involves delocalization of the lone pair electrons (n) on nitrogen, oxygen, and sulfur to the π* orbitals of CO, CN, and aromatic rings.42–44 Despite the widespread presence of the n → π* interaction between the two neighboring CO groups in the backbone of proteins, this interaction was not appreciated earlier by the scientific community, probably due to its weak strength and counterintuitive nature. This interaction is also present in polyesters, peptides, peptoids, several drugs, and other materials.42–44
The n → π* interaction is categorized into two basic classes depending on the π* orbital of the acceptor group, i.e., n → 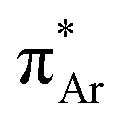 and n →
and n → 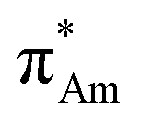 (Fig. 1). The n →
(Fig. 1). The n → 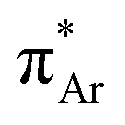 (aromatic) interaction involves the overlap of the lone-pair electrons with the π* orbital of the aromatic ring, while the n →
(aromatic) interaction involves the overlap of the lone-pair electrons with the π* orbital of the aromatic ring, while the n → 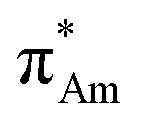 (amide) interaction involves the overlap of the lone-pair electrons with the π* orbital of the carbonyl group. A few geometrical parameters which characterize this non-covalent interaction in a molecular system or complex are the distance between the donor and acceptor atoms (d), the angle involving the donor and acceptor groups (θ and α), and the pyramidalization (Δ) of the carbon atom of the acceptor group (Fig. 1). For the n →
(amide) interaction involves the overlap of the lone-pair electrons with the π* orbital of the carbonyl group. A few geometrical parameters which characterize this non-covalent interaction in a molecular system or complex are the distance between the donor and acceptor atoms (d), the angle involving the donor and acceptor groups (θ and α), and the pyramidalization (Δ) of the carbon atom of the acceptor group (Fig. 1). For the n → 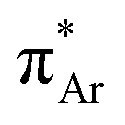 interaction, the distance (d) from a donor atom to the center of the aromatic ring should be in the range of 2.8–3.8 Å, and the angle between the plane containing the donor atom and acceptor aromatic ring should be less than 90°.42 The n →
interaction, the distance (d) from a donor atom to the center of the aromatic ring should be in the range of 2.8–3.8 Å, and the angle between the plane containing the donor atom and acceptor aromatic ring should be less than 90°.42 The n → 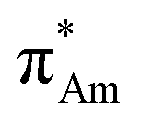 interaction follows the Bürgi–Dunitz trajectory of a nucleophilic attack to an electrophilic center. In this case, the distance (d) from the donor atom (Y = O, N, and S) to the carbon atom of the acceptor CO, i.e., Y⋯CO, should be less than 3.2 Å, and the ∠Y⋯C–O (θ) should be in the range of 109 ± 10°.42
interaction follows the Bürgi–Dunitz trajectory of a nucleophilic attack to an electrophilic center. In this case, the distance (d) from the donor atom (Y = O, N, and S) to the carbon atom of the acceptor CO, i.e., Y⋯CO, should be less than 3.2 Å, and the ∠Y⋯C–O (θ) should be in the range of 109 ± 10°.42
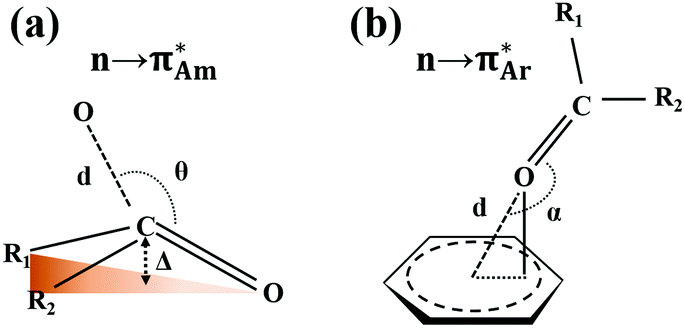 | ||
| Fig. 1 Classification of the n → π* interaction with the representation of the geometrical parameters such as the distance (d), angle (θ and α), and pyramidalization (Δ) used to characterize this interaction. | ||
This interaction was first observed in the protopine molecule by Mottus and co-workers, although they did not name it n → π* interaction.45 Around the same time, Leonard and co-workers studied this interaction in detail using infrared (IR) spectroscopy, ultraviolet-visible absorption (UV-Vis) spectroscopy, and chemical reactions and put forth the foundation of this non-covalent interaction.46,47 Later, Bürgi et al. resurfaced this interaction while studying the reaction coordinates of nucleophilic attack on the carbonyl group.48,49 Various types of n → π* interaction involving nitrogen, oxygen, sulfur, and selenium as n → π* donors and CO, CN, ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CC
CC![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) , and aromatic rings as n → π* acceptors have been explored in the literature.42,44,50 The prevalence of this interaction in proteins and small molecules has been shown by analyzing their crystal structures in the Protein Data Bank (PDB) and Cambridge Structural Database (CSD), respectively.51,52 Most of the preliminary investigations on this interaction involve the study of the stability of the collagen triple helix having three polyproline strands made of repeated –X–Y–Gly– units, where X is proline (Pro), and Y is either proline or 4(R)-hydroxyproline (Hyp).53–55 The helix is primarily stabilized due to CO⋯CO n → π* interaction without intra-strand hydrogen bonding. PDB analysis shows that ∼34% of residues of proteins involve the CO⋯CO n → π* interaction.52 Raines and co-workers contributed significantly to understanding the n → π* interaction using several model systems containing proline derivatives employing nuclear magnetic resonance (NMR) spectroscopy and quantum chemical calculations. Later, Das and co-workers presented isolated gas-phase IR spectroscopic evidence of the n → π* interaction.42,56–58
, and aromatic rings as n → π* acceptors have been explored in the literature.42,44,50 The prevalence of this interaction in proteins and small molecules has been shown by analyzing their crystal structures in the Protein Data Bank (PDB) and Cambridge Structural Database (CSD), respectively.51,52 Most of the preliminary investigations on this interaction involve the study of the stability of the collagen triple helix having three polyproline strands made of repeated –X–Y–Gly– units, where X is proline (Pro), and Y is either proline or 4(R)-hydroxyproline (Hyp).53–55 The helix is primarily stabilized due to CO⋯CO n → π* interaction without intra-strand hydrogen bonding. PDB analysis shows that ∼34% of residues of proteins involve the CO⋯CO n → π* interaction.52 Raines and co-workers contributed significantly to understanding the n → π* interaction using several model systems containing proline derivatives employing nuclear magnetic resonance (NMR) spectroscopy and quantum chemical calculations. Later, Das and co-workers presented isolated gas-phase IR spectroscopic evidence of the n → π* interaction.42,56–58
Previous reviews include a nice compendium of the literature on the n → π* interaction demonstrating its presence in biomolecules, materials, and small molecules or showing the spectroscopic evidence of this interaction.42,44,50 However, still, there is a lack of detailed understanding of this weak interaction. Herein, we have tried to understand the n → π* interaction with the help of divergent experimental techniques and theoretical analyses. We will be interpreting the n → π* interaction using various techniques such as X-ray crystallography, and electronic, IR, and NMR spectroscopy. We have also incorporated results obtained from isolated gas-phase spectroscopy, which allows studying the molecular systems free from the perturbation caused by neighboring molecules and solvents. Various quantum chemistry calculations, such as natural bond orbital (NBO), atoms in molecules (AIM), non-covalent interaction index (NCI), molecular electrostatic potential (MESP), and energy decomposition analysis (EDA), play a significant role in understanding this non-covalent interaction, which will also be discussed.
Prelude to the n → π* interaction using solution phase UV-Vis and IR spectroscopy
In 1953, Mottus and co-workers discovered the n → π* interaction in the name of the nitrogen bond in protopine (Fig. 2), which comprises two safrole rings (chromophores) separated by a ten-membered ring with a nitrogen atom and a carbonyl group.45 In the UV-Vis spectrum of protopine, observation of only one absorption band indicated that both the safrole rings were similar. It was possible only when the CO group was not in the conjugation with the safrole ring. The IR spectrum of protopine showed the absorption feature in the CO region at 1658 cm−1, red-shifted by 32 cm−1 from acetophenone (1690 cm−1).45 It was found that the IR spectrum of the perchlorate salt of the protopine was transparent in the CO stretching frequency region and showed a strong absorption band in the O–H region at 3385 cm−1, which was absent in the IR spectrum of protopine. It was inferred that there was a covalent bond formation between the nitrogen atom and the carbon atom of the carbonyl group. Thus, there was O–H bond formation during the salt formation of the protopine (Fig. 2). When the protopine molecule was treated with methyl iodide, no IR absorption band was observed in either the O–H or CO region due to the formation of the methoxy group (Fig. 2). This confirms the close contact between the N and carbonyl carbon atoms in the ten-membered ring.45
 | ||
| Fig. 2 Molecular structures of protopine and its perchlorate and iodide salt. | ||
Leonard and co-workers have studied the N⋯CO interaction (nitrogen bond) in detail using UV-Vis and IR spectroscopy as well as analyzing the chemical properties such as pKa values in different solvents of cyclic aminoacyloin.46,47 It is a class of cyclic compounds with tertiary amines and ketones with a hydroxyl group present at the α-carbon atom of the ketone group. The researchers have studied a series of 1-alkyl-1-azacyclononan-5-ol-6-ones (1 shown in Fig. 3) to study the steric effect on the N⋯CO interaction using IR spectroscopy and pKa values in different solvents.46,47 The CO stretching frequencies undergo bathochromic shift upon an increase in the bulkiness of the alkyl group attached to the nitrogen atom of the molecules, indicating the decrease in the strength of the N⋯CO interaction (Table 1). Furthermore, it was assumed that due to the N⋯CO interaction, R1R2R3N⋯CO would exist as charge-separated species like R1R2R3N+–C–O− similar to enols and acids (R1R2R3C–CO ↔ R1R2R3C+C–O−).46 Hence, these molecules should also show a decrement in the pKa values upon varying the solvent from 66% dimethylformamide (DMF) to water. A similar trend in the pKa values was observed in cyclic aminoacyloin having the N⋯CO interaction upon varying the solvent from 66% DMF to water, confirming the presence of the charge-separated species (Table 1).46
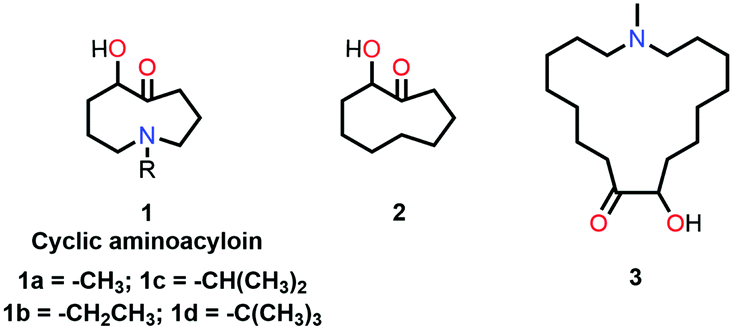 | ||
| Fig. 3 Molecular structures of 1, 2, and 3. | ||
O stretching region of the neutral form and perchlorate salts of 1-alkyl-1-azacyclononan-5-ol-6-ones along with their pKa values in different solvents for 1-alkyl-1-azacyclononan-5-ol-6-ones
| Compound | CO frequencies (cm−1) |
pKa values in solvents | |||
|---|---|---|---|---|---|
| Neutral | Perchlorate | 66% DMF | H2O | ΔpKa | |
| 1a | 1666 | — | 10.6 | 9.2 | −1.4 |
| 1b | 1671 | — | 10.2 | 9.2 | −1.0 |
| 1c | 1691 | — | 6.9 | 8.1 | +1.2 |
| 1d | 1698 | 1706 | 6.4 | 7.6 | +1.2 |
When the bulkiness of the alkyl group (Fig. 3) was increased from methyl (1a) to ethyl (1b) and iso-propyl (1c), the N⋯CO interaction decreased gradually. Interestingly, this interaction disappeared completely when the alkyl group was replaced with a tert-butyl group (1d).46 This was also inferred from the observed increment in the pKa values upon varying the solvent from 66% DMF to water.
The IR spectra of the perchlorate salts of compounds 1a, 1b, and 1c were found to be similar to that of the perchlorate salt of protopine in terms of the absence of the CO stretching band and the presence of the strong absorption band of the O–H groups.45,46 It was concluded that a covalent bond was formed between the nitrogen and carbon atoms of the carbonyl group (R1R2R3N+–C–OH species) due to the presence of the N⋯CO interaction in 1a, 1b, and 1c.46 However, the N⋯CO interaction is absent in 1d (tert-butyl) due to an excessive increase in the sterics, and there is no covalent bond formation between the nitrogen and carbon atoms of the carbonyl group. As a result, the IR spectrum of the perchlorate salt of 1d shows the peak for the CO stretching region.46,47 The CO stretching frequencies and variations in the pKa values for different alkyl substitutions are provided in Table 1.
Leonard and co-workers further tried to find the effect of the ring size on the N⋯CO interaction in cyclic aminoacyloin compounds using similar approaches. They found that a nine-membered ring is favorable for the presence of the N⋯CO interaction as the nitrogen atom and carbonyl group are diametrically opposite and have better orbital overlap (Fig. 3).59 They used UV-Vis spectroscopy to probe the N⋯CO interaction in the aminoacyloin compounds with respect to a reference compound, 2-hydroxycyclononanone (2 shown in Fig. 3), which does not have any N⋯CO donor and shows two bands at 217 and 264 nm in the UV spectrum.47 On the other hand, a new electronic band was observed at 228 nm for compound 1a having N⋯CO interaction, while the band at 264 nm was missing.46 When the ring size was increased to make the interaction unfavorable in 1-methyl-1-azacycloheptadecan-9-ol-10-one (3 shown in Fig. 3), bands at 216 and 270 nm similar to compound 2 were observed in the UV-Vis spectrum.47 The UV absorption maxima (nm) for all bands in molecules 1a, 2, and 3 are given in Table 2.
| Compound | Band 1 | Band 2 | Band 3 | n → π* |
|---|---|---|---|---|
| 1a | 217 | 228 | — | Yes |
| 2 | 217 | — | 264 | No |
| 3 | 216 | — | 270 | No |
In a recent review, Sarma and co-workers provided a detailed discussion of the literature reports on the N⋯CO interaction in a series of aminoacyloin compounds studied through UV-Vis, IR, and NMR spectroscopy.50 The bands at 217 and 264/270 nm for compounds 2 and 3 were assigned as the π → π* and n → π* electronic transitions of the CO group, respectively. In the case of compound 1a, the n → π* transition band was shifted to 228 nm due to the N⋯CO orbital interaction, which was termed the n → π* interaction by Sarma and co-workers.50 Leonard and co-workers also studied the O⋯CO and S⋯CO types of n → π* interactions.60
Presence of the n → π* interaction in crystal structures of proteins and other small molecules
X-ray crystallography has become a crucial tool for studying the intramolecular and intermolecular non-covalent interactions in molecular systems. The n → π* interaction has also been identified by studying the crystal structures of various molecular systems in detail. Bürgi et al. observed the N⋯CO short contact in crystal structures such as cryptopine, protopine, and clivorine (Fig. 4) while finding the reaction coordinates for the nucleophilic attack to the carbonyl group.48 These short contacts follow a particular angle of ∼107° (N⋯CO) and the acceptor carbon atom deviates from the plane formed by R1, R2, and O atoms, and the degree of deviation decreases as the N⋯C distance increases.48
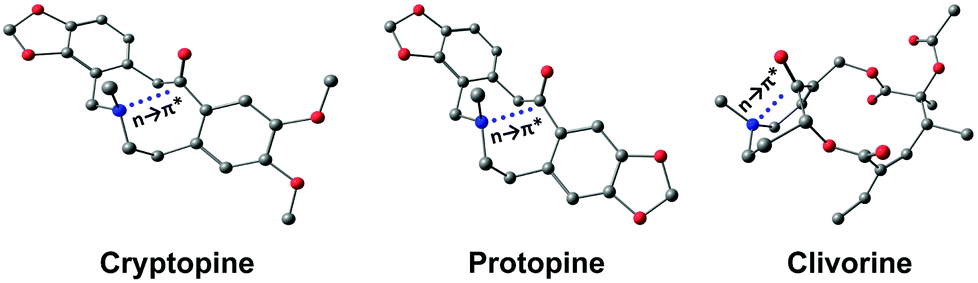 | ||
| Fig. 4 Optimized structures of cryptopine, protopine, and clivorine calculated at the B3LYP-D3/Def2-TZVP level of theory. | ||
This interaction is present in various biologically relevant molecules such as 4S-aminoproline,61 oligoproline,62 poly(lactic acid),63 homoserine lactones,64 aspirin,65,66 salicin,67 and metal complexes.42,68 Breton and co-workers observed the n → π* interaction in a series of 2-(dimethylamino)biphenyl-2′-carboxaldehydes.69 Wallis and co-workers observed N⋯CO n → π* interaction in a series of 1-dimethylaminonaphthalene-8-ketones (shown in Fig. 5(a)) and found that the N⋯C distance increases (Fig. 5(b)) as the R group increases from methyl to t-butyl.70 The alkyl group pushes the electron density towards the carbonyl carbon atom (+I effect), which reduces the electrophilicity of the carbon atom and hence reduces the strength of the n → π* interaction.70 On the other hand, when the R group is substituted with the –CF3 group, the N⋯C separation (Fig. 5(b)) decreases, which proves that the strength of the n → π* interaction increases due to the electron-withdrawing groups at the n → π* donor and vice versa.70
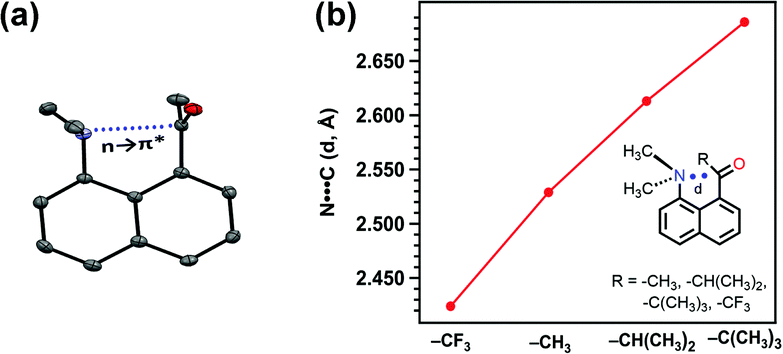 | ||
| Fig. 5 (a) Crystal structure of 1-dimethylaminonapthalene-8-ethanone and (b) plot of the distance between N and C atoms of the carbonyl group for various 1-dimethylaminonaphthalene-8-ketones in their crystal structures. Reproduced from ref. 70 with permission from [Royal Society of Chemistry], copyright [2014]. | ||
The n → π* interaction is widely present in crystal structures of proteins. Raines and co-workers performed a detailed PDB survey to find the prevalence of the n → π* interaction in proteins.43 They chose search criteria of distance ≤3.2 Å (sum of van der Waals radii of oxygen and carbon atoms) between the oxygen atom of the donor carbonyl group and the carbon atom of the (O⋯C) acceptor carbonyl group and the angle O⋯CO was chosen in the range of 99° ≤ θ ≤ 119° (Bürgi–Dunitz trajectory) for performing the CSD and PDB analyses.43 They found that the n → π* interaction is present in all 1731 protein crystal structures with 5–97% of residues.43 It has also been observed that the carbonyl groups in the backbone of proteins can act as both n → π* donors and acceptors simultaneously, termed reciprocal n → π* interactions. Sarma and co-workers carried out CSD analysis for the reciprocal n → π* interaction with the search parameters O⋯C distance ≤3.2 Å and a separation of at least three covalent bonds in the two carbonyl groups (1,5-type interaction).51 They found the reciprocal n → π* interaction in 249 crystal structures of the CSD database with the C⋯O distance ≤3.0 Å. In 117 crystal structures, a chiral carbon atom or heteroatoms were present similar to peptides and proteins. Out of the 2269 protein crystal structures in the protein database, 2184 structures showed reciprocal n → π* interaction, and most of the crystal structures were found to be in the polyproline II (PP II) or turn regions.51
Sarma and co-workers also studied the reciprocal n → π* interaction in substituted N,N′-diacylhydrazine.51 It should be mentioned that both the amide bonds in the N,N′-diacylhydrazine were found in the trans–trans configuration. It has been found that the electron-withdrawing group (EWG) at the X position enhances the electrophilicity of the 2nd CO group and increases the 1st n → π* interaction. Henceforth, it increases the electron donor capability of the 2nd CO; hence, the 2nd n → π* interaction also increases.51 Similarly, the electron-donating group (EDG) at the Y position pushes the electron density towards the 1st carbonyl group, making the 1st n → π* interaction more feasible. Fig. 6(b) and (c) show the plots for the O⋯CO distances and NBO 2nd order perturbation energies [E(2)], respectively, for the reciprocal n → π* interactions in compounds 4–11.51
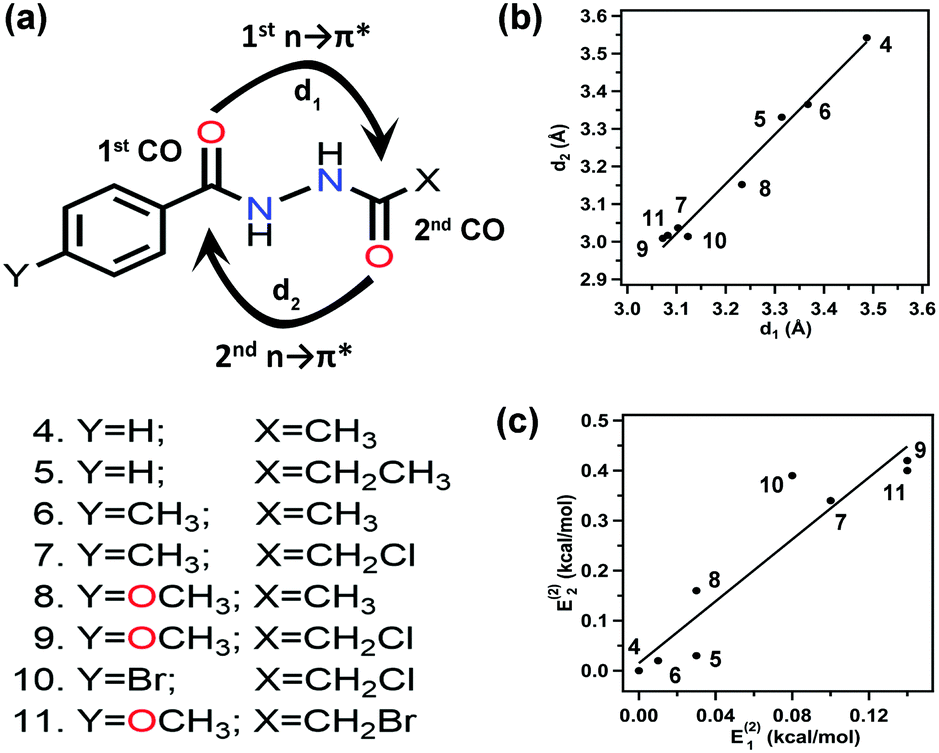 | ||
| Fig. 6 (a) Various substituted N,N′-diacylhydrazines; and plots showing the correlation between (b) O⋯C distances (d1 and d2), and (c) E(2) energies of the 2nd and 1st n → π* interactions in compounds 4–11. The E(2) energies in the crystal structures were calculated from NBO analysis at the B3LYP/6-311+G(2d,p) level of theory. Reproduced from ref. 51 with permission from [Springer Nature], Copyright [2017]. | ||
Observation of the n → π* interaction using NMR spectroscopy
Collagen protein consists of three polypeptide chains, forming a right-handed triple helix. Each helical strand consists of a sequence of (–X–Y–Gly)n, where X = Pro and Y = Hyp. The Pro residue undergoes post-translational enzymatic modification and forms a Hyp residue in the presence of the prolyl hydroxylase enzyme.71 Peptide bonds of Pro and Hyp residues are found in the trans (Z) and cis (E) configurations (Fig. 7). Interconversion between the trans and cis conformation along the C–N bond is considered the rate-limiting step. It has been proved by studying 4(R)-fluoro-L-proline (Flp) and –Pro–Hyp–Gly– that stereoelectronic inductive effects at the 4-position of proline rings significantly influence the conformation of these residues and favors the trans conformation of the prolyl peptide bond.72 NMR spectroscopy assisted in revealing the reason behind the stability of the collagen triple helix and showed that the trans conformation is favored due to the CO⋯CO n → π* interaction over the cis conformation.
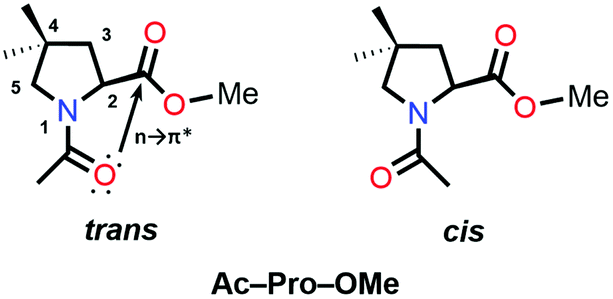 | ||
| Fig. 7 trans and cis conformations of Ac-Pro-OMe. | ||
The 1H-NMR spectrum shows two different signals corresponding to the cis and trans conformations of the prolyl peptide bond at room temperature. The upfield shift of the 1H-NMR signal corresponds to the trans conformation, whereas the downfield shift of the 1H-NMR signal corresponds to the cis conformation. The integration of both signals gives the population of both the conformations, which will provide the equilibrium constant (Ktrans/cis) for the cis–trans isomerization by taking the ratio of the populations of the trans and cis conformations.73–75 The equilibrium constant of the cis–trans isomerization determines the strength of the n → π* interaction in the trans conformers as this interaction is absent in the cis conformation. A van’t Hoff/Eyring plot can be drawn by taking these ratios at different temperature values, which can be linearly fitted in order to calculate the ΔH‡ and ΔS‡,
| ln(Ktrans/cis) = (−ΔH‡/R)(1/T) + (ΔS‡/R) |
The stereoelectronic effect at the 4-position determines the exo and endo puckering of the proline ring; the exo puckering brings both the carbonyl groups closer in favor of the CO⋯CO n → π* interaction and increases the Ktrans/cis ratio.76 Various Ac-Yaa-OMe proline substitutions (Table 3) have been studied to determine the influence of the stereoelectronic effect on Ktrans/cis of the proline peptide bond where Yaa is Flp, Hyp, Pro, 4(S)-fluoro-L-proline (flp), and 4(S)-hydroxy-L-proline (hyp). The order of the Ktrans/cis values in various Yaa proline substitutions is Flp (6.7) > Hyp (6.1) > Pro (4.6) > flp (2.5) > hyp (2.4). The melting temperature, which dictates the stability of collagen, is around 45 °C for (Pro–Flp–Gly)7 and <2 °C for (Pro–flp–Gly)7, can be correlated with the Ktrans/cis values of Ac-Flp-OMe and Ac-flp-OMe residues. It shows that the Ktrans/cis values correlate with the n → π* interaction and have an important role in collagen stability. It has been proved by the NMR studies that there is little effect of the hydrogen bond on the stability of collagen due to the presence of the Hyp residue by studying Ac-Mop-OMe [Mop = 4(R)-methoxyproline].77 The Ktrans/cis ratio of the Ac-Mop-OMe (6.7) is more than that of the Ac-Hyp-OMe (6.1), measured in 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 D2O/CD3OD solvent. It crystallizes in the trans conformation with the exo-puckering of the proline ring.77
:6 D2O/CD3OD solvent. It crystallizes in the trans conformation with the exo-puckering of the proline ring.77
| Compound | K trans/cis |
|---|---|
| Ac-Flp-OMe | 6.7 |
| Ac-Hyp-OMe | 6.1 |
| Ac-Pro-OMe | 4.6 |
| Ac-flp-OMe | 2.5 |
| Ac-hyp-OMe | 2.4 |
| Ac-Mop-OMe | 6.7 |
Raines and co-workers made a seminal contribution to elucidating the nature of n → π* interaction.78 They replaced the n → π* amide donor with thioamide for differentiating the charge–charge interaction with the n → π* interaction, as the former one will be decreased due to the less electronegative sulfur atom than the oxygen atom, and the n → π* interaction will be enhanced due to the soft base nature of the sulfur atom.78 Thus, the sulfur atom of thioamide serves as a better n → π* donor (compounds 12 and 13 shown in Fig. 8). The possibility of the dipole–dipole interaction was also ruled out by inducing a stereoelectronic effect at the 4R-position (4S) of the proline ring, which resulted in exo (endo) puckering and brought both the carbonyl groups close to (away from) each other (compounds 14–17 shown in Fig. 8).78 The Ktrans/cis ratios of various substitutions proved that the CO⋯CO interaction is due to the n → π* interaction, not the charge–charge or dipole–dipole interaction.78
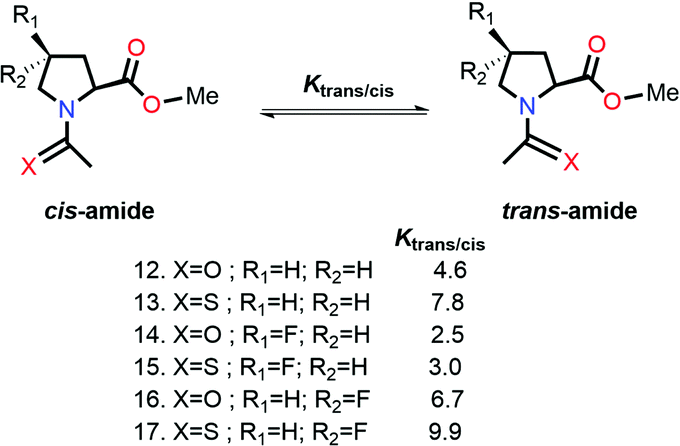 | ||
| Fig. 8 cis and trans amides of various substituted Ac-Yaa-OMe proline compounds. | ||
Raines and co-workers also studied the n → π* interaction using amides as n → π* acceptors, as amino acid residues are linked with the –C(O)–N linkage rather than the –C(O)–O– linkage.79 The electrophilicity of esters is more than that of amides, whereas the dipole moment of the esters is less than that of the amides. Greater Ktrans/cis values (Fig. 9) for the esters than the amides suggest that the n → π* interaction is not dipolar; instead, it is an orbital overlap interaction.79 We have discussed earlier that replacing the amide donor with the thioamide donor results in an increment in the Ktrans/cis ratio due to the enhanced orbital overlap. In the case of the amide as the n → π* acceptor, a similar trend was observed by replacing the amide donor with the thioamide donor. When both the donor and acceptor were replaced with thioamide, the Ktrans/cis ratio was the highest.
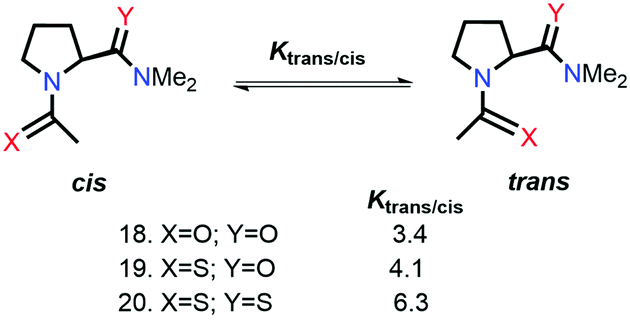 | ||
| Fig. 9 n → π* interaction with –C(O)–N as the n → π* acceptor. | ||
So far, we have discussed that the trans configuration is favorable in the exo puckering of the proline ring due to the n → π* interaction.
However, intramolecular hydrogen bond formation by the group at the 4S position with the oxygen atom of the carbonyl group of the methyl ester makes the trans configuration stable even in the endo puckering of the proline ring (Fig. 10(b)).61,80 Hydrogen-bond donating substituents at the 4S-position which induce the endo puckering of the proline ring, such as –NH3+ (5.7/3.5 in D2O), –NHAc (5.8/5.2 in CDCl3), –NHBOC (5.5/3.5 in CDCl3), and –OH (4.7/4.2 in CDCl3), have more Ktrans/cis (the values of 4S/4R substitutions are provided in parentheses) due to the hydrogen bond, whereas for the –N3 (1.9/3.9 in CDCl3) substitution which cannot form a hydrogen-bond has a lower Ktrans/cis at the 4S-position.
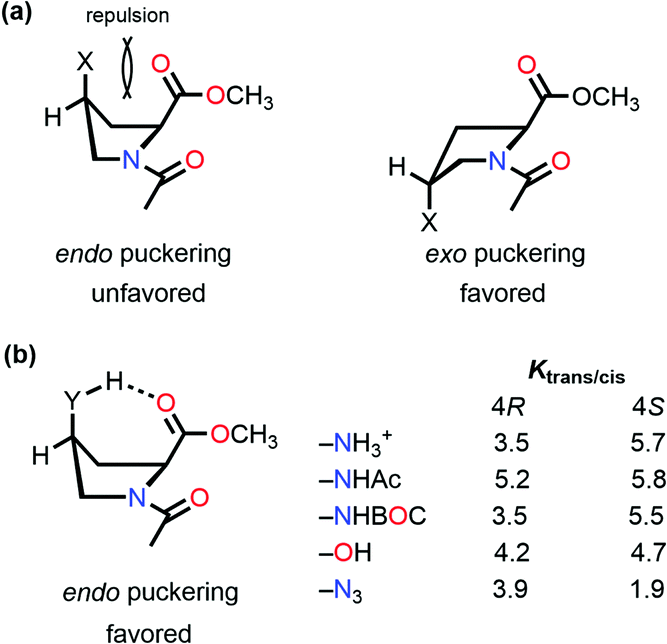 | ||
| Fig. 10 (a) endo and exo puckering in Ac-Pro-OMe; and (b) Ktrans/cis values of various 4R and 4S substitutions in Ac-Pro-OMe. | ||
It has been proved that the n → π* interaction can dictate the conformational preference of the molecular systems. Sarma and co-workers studied N,N′-diacylhydrazine, which is used as an insect growth regulator.51 This compound exists in four rotameric configurations [trans–trans (t–t), trans–cis (t–c), cis–cis (c–c), and cis–trans (c–t)] based on the orientation of the amide bond. It has been discussed in the previous section that the t–t configuration of diacylhydrazines (R3 = R4 = H) is stable among others due to the reciprocal CO⋯CO n → π* interaction (Fig. 11(a)).51
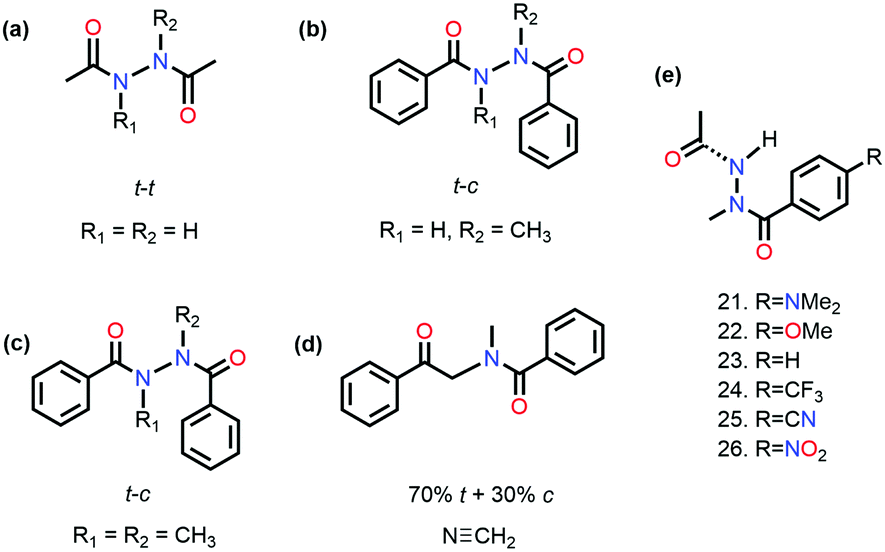 | ||
| Fig. 11 Chemical structures of various N,N′-diacylhydrazines. | ||
On the other hand, the preference of the amide bond changes drastically from t–t to t–c due to the carbon bonding (N⋯C–X) interaction when one of the N–H groups is methylated.51 A question should be raised about the fate of the amide bonds on the preference of the t–c configuration due to the n → 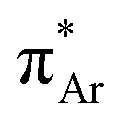 interaction if C–X is substituted with an aromatic ring. An exclusive presence of the t–c configuration was observed through 1H-NMR spectroscopy and X-ray crystallography when one of the N–H groups was methylated (Fig. 11(b)).81 The probability of stabilization of this configuration due to the N–H⋯π hydrogen bond was ruled out when the second N–H was also methylated (Fig. 11(c)), and the t–c configuration was still favored.81 However, when the nitrogen atom (n → π* donor) was replaced with the –CH2 group (Fig. 11(d)), a mixture of 70% trans and 30% cis configurations was observed. Thus, the importance of the n → π* interaction in stabilizing the t–c configuration of N,N′diacylhydrazine was established.81
interaction if C–X is substituted with an aromatic ring. An exclusive presence of the t–c configuration was observed through 1H-NMR spectroscopy and X-ray crystallography when one of the N–H groups was methylated (Fig. 11(b)).81 The probability of stabilization of this configuration due to the N–H⋯π hydrogen bond was ruled out when the second N–H was also methylated (Fig. 11(c)), and the t–c configuration was still favored.81 However, when the nitrogen atom (n → π* donor) was replaced with the –CH2 group (Fig. 11(d)), a mixture of 70% trans and 30% cis configurations was observed. Thus, the importance of the n → π* interaction in stabilizing the t–c configuration of N,N′diacylhydrazine was established.81
Sarma and co-workers also tuned the strength of the n → π* interaction by modifying various electronic substituents at the para position (21–26 in Fig. 11(e)) and observed the spectroscopic signature of this interaction.81 The nitrogen atom of the n → π* donor has also been found to be involved in the –NH–C(O)– conjugation of the amide bond along with the n → π* interaction. Hence, the increase in the strength of the n → π* interaction makes the nitrogen lone-pair less available for the –NH–C(O)– conjugation, and thus, the N–H stretching frequency gets red-shifted.81 It has been found from the NBO analysis that the E(2) energy for this interaction decreases as the distance between the nitrogen atom and carbon increases from EWGs to EDGs (Fig. 12(a)). It has been observed that the N–H stretching frequency is red-shifted for the –NO2 substituent having more E(2) values for the n → π* interaction (Fig. 12(b)). Moving from the EWGs to the EDGs, the strength of this interaction decreases, and thus, the N–H stretching frequency gets blue-shifted.81
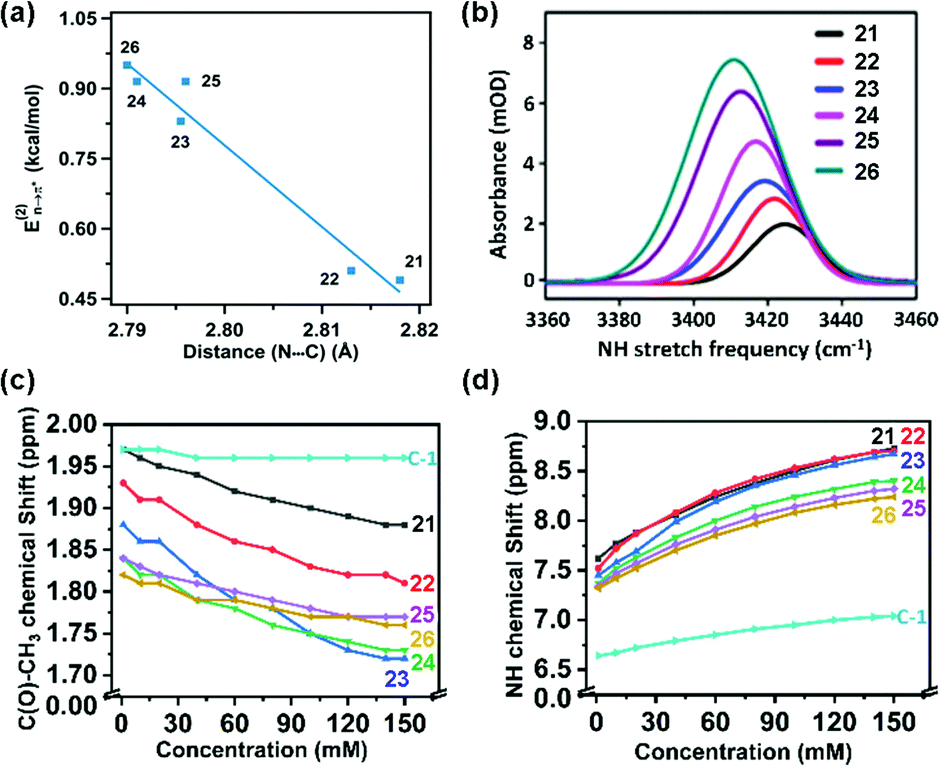 | ||
| Fig. 12 (a) Plot of E(2) energies versus N⋯C distances of 21–26; (b) N–H stretching frequencies of 21–26; and (c) and (d) plots of the chemical shifts of CH3 and NH protons versus concentrations of 21–26 and acetohydrazide. Reproduced from ref. 81 with permission from [American Chemical Society], Copyright [2021]. | ||
Based on NMR spectroscopy, the chemical shift values of the N–H and –CH3 protons of the hydrazine compounds were found to show upfield shifts.81 The conjugation in the –NH–C(O)– group, which decreases with the increment of the n → π* interaction, weakens the anisotropic effect of the –NC– double bond and leads to the upfield of the chemical shift of the N–H and –CH3 protons (Fig. 12(c) and (d)).81 The strength of the n → π* interaction was found to increase along with the upfield shift of the –CH3 protons, while the chances for the involvement of the N–H group in the intermolecular hydrogen bond formation were enhanced by increasing the concentration of the compound.81 Moreover, in the 1H-NMR spectrum of acetohydrazide (lack of n → π* interaction), the absence of the upfield shift of the –CH3 protons confirmed that the observed chemical shift of the protons in the hydrazine compounds was due to the n → π* interaction.81
Understanding of the n → π* interaction from gas-phase studies
In the previous sections, we have discussed the n → π* interactions studied in the solution phase and crystal structures of molecules and complexes where perturbations due to the solvent or crystal packing forces are present. In contrast, isolated gas-phase spectroscopy, which is free from any perturbation arising from the surrounding environment, is an ideal technique to understand the intrinsic nature and strength of this weak interaction. In the NMR spectroscopy, the n → π* interaction in the trans compared to the cis conformer of proline derivatives was elucidated based on their Ktrans/cis values.44,50 However, the NMR spectroscopy data do not directly tell the quantitative strength of the n → π* interaction. Gas-phase calculations and experiments go hand in hand to directly probe the strength and nature of this interaction. It would not be an exaggeration to say that it would be very difficult to have a comprehensive idea of various non-covalent interactions and the relative stability of different conformations of the molecules or complexes present in the gas-phase experiments without the assistance of quantum chemical calculations. Thus, it is necessary to discuss the theoretical approaches generally used to understand the n → π* interaction.(a) Quantum chemical calculations
Quantum chemical calculations such as AIM, NCI, NBO, MESP, and EDA analyses assist significantly in understanding the weak n → π* non-covalent interaction. AIM analysis helps in the identification of the specific non-covalent interaction present in a molecular system by generating the bond-critical points (BCPs) between the two atoms involved in the interaction.82 It also generates the ring/cage critical points whenever a ring/cage is formed via covalent or non-covalent bonding. At the critical point, various electronic properties such as the electron density [ρ(r), a.u.], the Laplacian of the electron density [∇2ρ(r), a.u.], and the second eigenvalue of the Hessian matrix (λ2, a.u.) signify the strength of the non-covalent interaction. ρ(r) at the BCP determines the accumulation of electron density due to the non-covalent interaction and hence determines the strength of the interaction. The sign of λ2 determines whether the interaction is attractive (negative) or repulsive (positive) in nature, and the numerical value of λ2 determines the strength of the interaction. Sometimes, AIM analysis fails to generate the BCPs for the non-covalent interaction despite its presence in the molecular systems. However, it has been proved that the absence of BCPs does not guarantee the absence of the non-covalent interaction.83NCI analysis offers qualitative ideas about the non-covalent interaction present in molecular systems.84,85 The NCI isosurface plot follows the color code from blue to red for the attractive to repulsive interaction. At the same time, the NCI plot of the reduced density gradient (RDG) of the electron density versus [sign (λ2)ρ(r)] generates spikes for each non-covalent interaction, whether it is attractive or repulsive. Spikes corresponding to the right side (≥0) of the RDG plot are for repulsive interaction, and spikes toward the left (≤0) are for attractive interaction. Spikes where both the RDG and ρ(r) are small correspond to the region of the non-covalent interaction.
NBO analysis calculates the E(2) values for the delocalization of the electron density from the lone-pair (n) orbital to the π* orbital involved in the n → π* interaction.86 This interaction is indeed about the overlap of the two orbitals, i.e., the n and π* orbitals. The effect of the substituents on the n → π* interaction can be understood qualitatively with the help of the NBO analysis. Moreover, energy decomposition analysis provides a qualitative idea about the nature of the non-covalent interaction present in the molecular complexes. This analysis involves the decomposition of the binding energy of the complex in various contributing factors such as electrostatic, polarization, dispersion, charge transfer, and Pauli repulsion.
Altamirano and co-workers reported from quantum chemical calculations that the n → π* interaction competes with a strong hydrogen bond and can beat the hydrogen bond in 3-nitrophthalic acid (3NFAc).87 3NFAc has several possible conformations because of the flexible dihedral angles (Fig. 13(b)). We will discuss only the two lowest energy conformers, A1 and E1. It has been found from the NBO analysis that the E(2) energy for the O13–H14⋯O8 hydrogen bond in E1 is 9.74 kcal mol−1, while the same for the n(O19) → π*(C7O8) interaction in E1 is 2.29 kcal mol−1 calculated at the MP2/cc-pVTZ level of theory. On the other hand, conformer A1 forms dual n → π* interactions, i.e., n(O19) → π*(C7O8) and n(O12) → π*(C7O8) with 2.30 and 2.47 kcal mol−1 strengths, respectively.87 Conformer E1 is expected to be the global minimum due to the strong O–H⋯O hydrogen bond. However, it is found that A1 is the most stable conformer, which is 2.27 kcal mol−1 lower in energy than E1.87
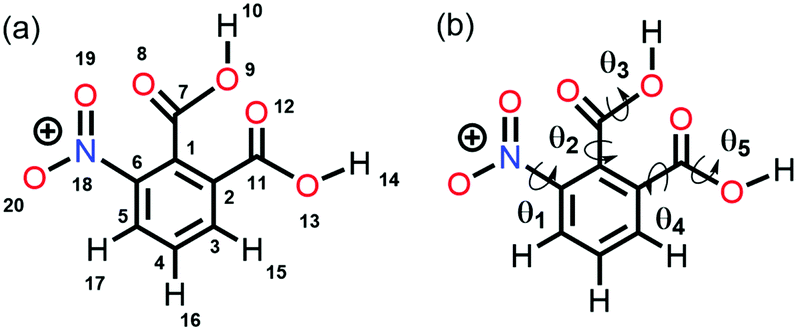 | ||
| Fig. 13 (a) Atom numbering in 3NFAc and (b) flexible dihedral angles in 3NFAc. Reproduced from ref. 87 with permission from [Springer], Copyright [2020]. | ||
The authors explored the unusual stability pattern of these two conformers. They calculated the pairwise steric contributions to the stability of these conformers.87 It was found that the E1 conformer is engaged in a steric repulsion of 15.48 kcal mol−1, which is much larger than that present in conformer A1.87 Thus, the relatively larger steric repulsion present in the E1 conformer subdues the stabilization caused by the hydrogen bond. It has been reported that the QTAIM analysis sometimes fails to generate the BCP due to the weak nature of the n → π* interaction.87 However, the presence of the BCPs in 3NFAc due to the n → π* interaction suggests that this interaction is strong enough in this compound. Fig. 14(a) and (b) show the QTAIM molecular graphs and NCI RDG plots displaying the n → π* interactions for conformers A1 and E1.87
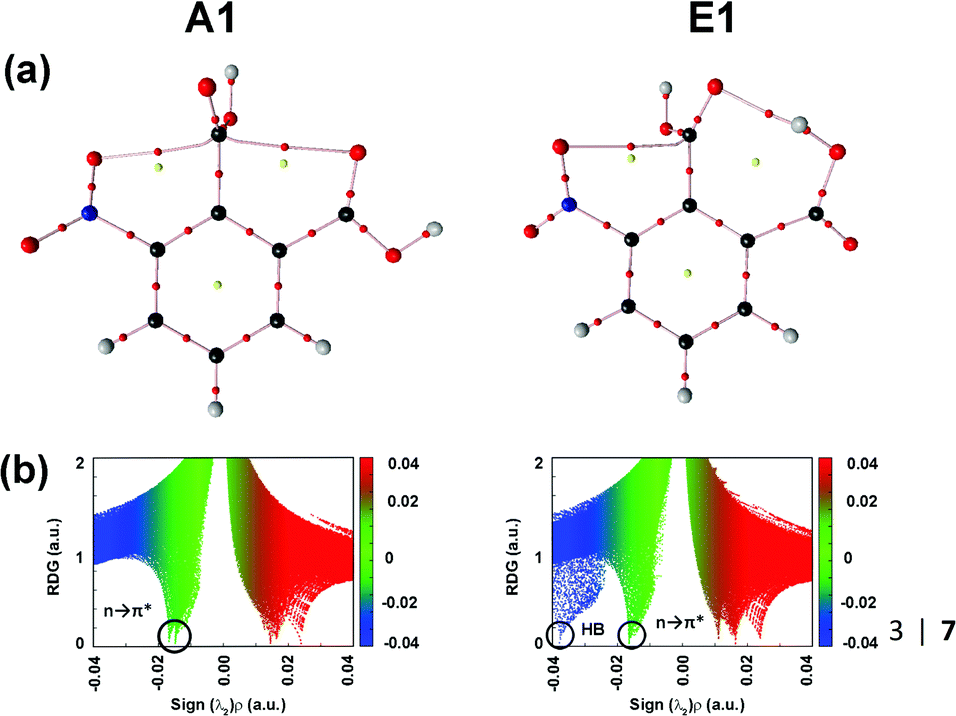 | ||
| Fig. 14 (a) QTAIM molecular graphs and (b) NCI RDG plots of conformers A1 and E1, calculated at the MP2/cc-pVTZ level of theory. Reproduced from ref. 87 with permission from [Springer], Copyright [2020]. | ||
(b) Microwave spectroscopy
Herein, the rotational spectra of the molecules or complexes having the n → π* interaction measured with the help of microwave spectroscopy are discussed. Various parameters such as rotational constants (A, B, and C) and centrifugal distortion constants (DJ, DJK, DK, d1, and d2) are extracted from this spectrum. At the same time, information obtained from the quantum chemical calculations such as rotational constants, centrifugal distortion constants, and dipole moments helps in assigning the rotational transitions observed in the microwave spectra and determining the structures of various conformers of the molecules or complexes observed in the experiment. Thus, microwave spectroscopy is an important tool for accurately determining the structures of molecules or complexes and various non-covalent interactions. Alonso and co-workers studied neurotransmitters such as β-alanine88 and γ-aminobutyric acid (GABA),89 biologically essential amino acids such as 4-hydroxyproline,90 and drugs such as aspirin66 using gas-phase using microwave spectroscopy. They found that the lowest energy conformers of all these molecules observed in the experiment are stabilized mostly by the n → π* interaction. They also observed the n → π* interaction in a Gly–Pro dipeptide studied in the gas phase.91 Interestingly, it was found that the conformer having n → π* interaction is much more abundant than the conformer having the O–H⋯O hydrogen bond.91 This finding shows the importance of the weak n → π* interaction in the conformational preference of molecular systems. The structure of the dipeptide conformer observed in the experiment was found to have a similarity with that of the Gly–Pro unit present in the crystals of collagen mimetic peptides.91Pratt and co-workers showed that the n → π* interaction can fold the amino acids by bringing the –NH2 and –COOH groups together to form the lactam (C–N bond) by demonstrating the transformation of δ-aminovaleric acid to δ-valerolactam (Fig. 15).92 So far, we have discussed the molecular systems that have intramolecular n → π* interaction, i.e., both the n → π* acceptor and donor are present in the same molecule. Pate and co-workers observed intermolecular n → π* interaction for the first time by studying β-propiolactone⋯(water)2 and β-propiolactone⋯(water)3 [BPL⋯2W and BPL⋯3W shown in Fig. 16(a)] complexes, where the oxygen atom of water molecule acts as the n → π* donor.93 It should be mentioned here that the n → π* interaction is not solely responsible for stabilizing these complexes. These complexes are stabilized primarily by the O–H⋯O hydrogen bond and have cooperativity effects with the n → π* interaction.93
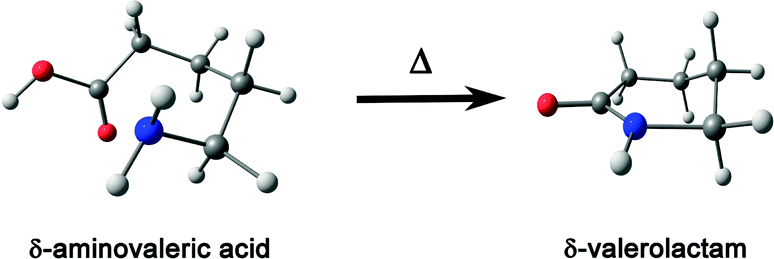 | ||
| Fig. 15 Conversion of δ-aminovaleric acid to δ-valerolactam. Reproduced from ref. 92 with permission from [Elsevier], Copyright [2012]. | ||
 | ||
| Fig. 16 Optimized geometries of (a) BPL⋯2W and BPL⋯3W complexes. Reproduced from ref. 93. (b) Py⋯HCHO and Py⋯CH3CHO complexes calculated at the B3LYP-D3/Def2-TZVP level of theory. Reproduced from ref. 94 and 95 with permission from [American Chemical Society], copyright [2018], and [Royal Society of Chemistry], copyright [2019], respectively. | ||
Lopez and co-workers studied pyridine⋯formaldehyde and pyridine⋯acetaldehyde (Py⋯HCHO and Py⋯CH3CHO; Fig. 16(b)) complexes using a combination of microwave spectroscopy and quantum chemistry calculations.94,95 The n → π* interaction was a major stabilizing force in these complexes. Here, the lone-pair orbital of the nitrogen atom of pyridine, which is in the plane of the ring, acts as the n → π* donor, while the π* orbital of the CO group of the aldehyde acts as the acceptor.94,95Fig. 17(a) shows the BCPs for the n → π* interaction and the C–H⋯O hydrogen bond in both the complexes. It can be inferred from the NCI RDG plots (Fig. 17(b)) that the n → π* interaction in Py⋯HCHO is stronger than that in the Py⋯CH3CHO complex.94,95 The n → π* interaction values calculated from the NBO analysis at the RHF/aug-cc-pVTZ level of theory for the structures optimized at the CCSD/6-311++G(2d,p) level of theory are 28.15 and 8.9 kJ mol−1 for the Py⋯HCHO and Py⋯CH3CHO complexes, respectively.94,95 The strength of the n → π* interaction in the Py⋯CH3CHO complex is weaker as the methyl group in CH3CHO pushes the electron density towards the carbonyl group and makes it less susceptible to this interaction.94,95
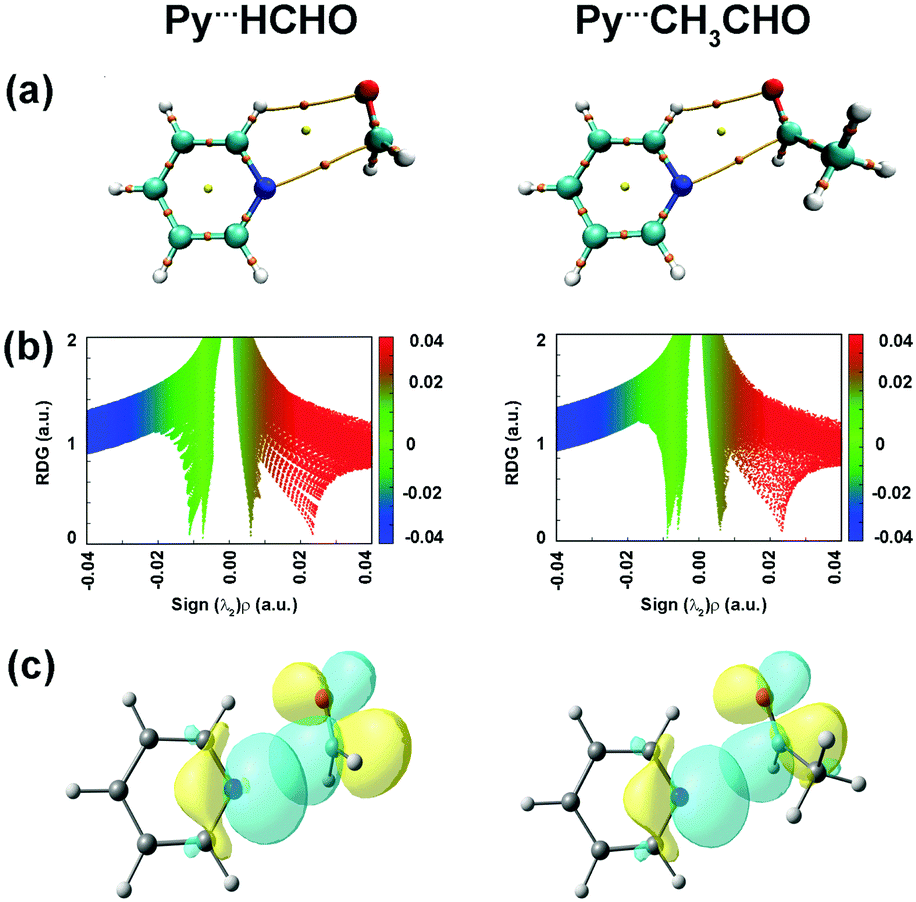 | ||
| Fig. 17 (a) BCPs; (b) NCI RDG plots; and (c) NBO orbital overlap for the n → π* interactions calculated at the RHF/aug-cc-pVTZ level for the structures optimized at the CCSD/6-311++G(2d,p) level of theory. Reproduced from ref. 94 and 95 with permission from [American Chemical Society], copyright [2018], and [Royal Society of Chemistry], copyright [2019], respectively. | ||
(c) Electronic and vibrational spectroscopy
Microwave spectroscopy provides information about the presence and strength of any non-covalent interaction from the geometry of the molecules or complexes observed in the experiment. Further, mass-selective conformation-specific IR spectroscopy in the gas phase can provide complementary information on the strength of the n → π* interaction by probing the donor or acceptor groups involved in the interaction. Generally, a red-shift in the vibrational frequency of the groups involved in the non-covalent interaction and enhancement in the intensity or broadening of the IR peaks indicate the presence of this interaction, especially the hydrogen bond. As the n → π* interaction is generally very weak, it is not always straightforward to monitor the presence of this interaction through IR spectroscopy.In fact, there are a handful of reports of IR spectroscopy studies on the n → π* interaction in the gas-phase. Das and co-workers provided the first indirect spectroscopic evidence of the weak n → π* interaction in the 7-azaindole⋯2,6-difluoropyridine (7AI⋯2,6-DFP) and 7-azaindole⋯2,3,5,6-tetrafluoropyridine (7AI⋯2,3,5,6TTFP) complexes by probing the N–H stretching frequencies in the complexes (Fig. 18).96 Here, it is called indirect IR spectroscopic proof for the presence of the n → π* interaction, as the N–H group is not directly involved in the interaction. In these complexes, the lone-pair electron density of the nitrogen atom of the 7AI molecule is delocalized into the π* orbital of the fluorosubstituted pyridines. At the same time, the N–H group of 7AI is hydrogen-bonded with the nitrogen atom of the pyridine ring.96 Das and co-workers measured mass-selected electronic spectra of these complexes using one-color resonant two-photon ionization spectroscopy (1C-R2PI).10 In this technique, the first photon of a tunable laser excites the molecules from the ground state (S0) to the excited state (S1). Subsequently, another photon ionizes the molecules to the ionization continuum (D0), and the ionized molecules are detected as a function of the wavelength of the laser.10 Conformation-specific IR spectra of the complexes were measured using resonant ion-dip infrared spectroscopy (RIDIR) by fixing the UV laser at a particular transition of the electronic spectrum and scanning the IR laser throughout the N–H stretching region.10
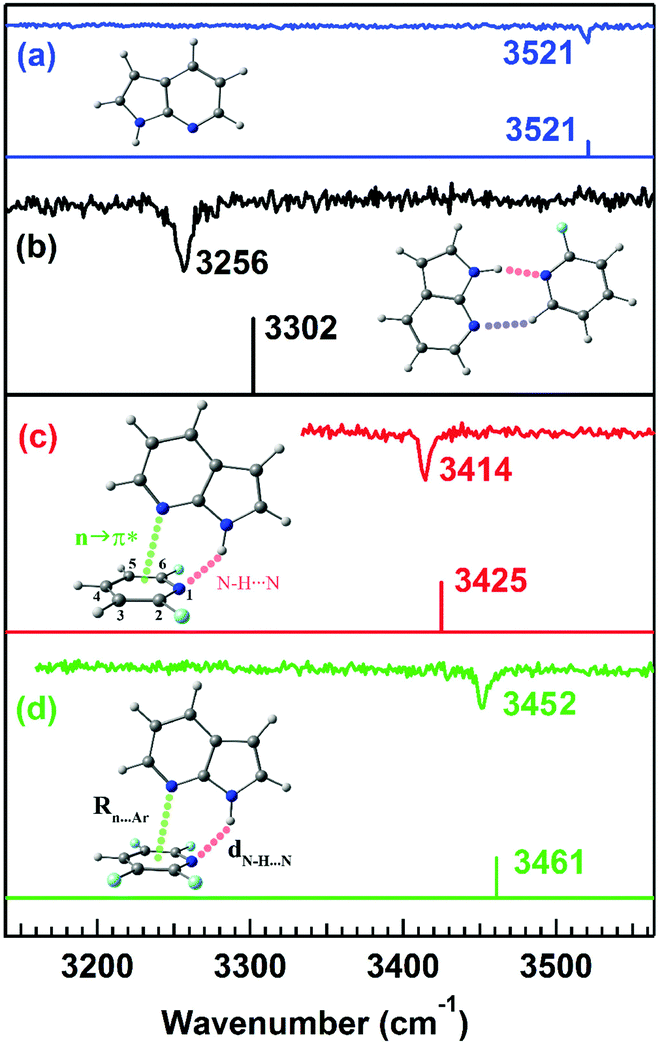 | ||
| Fig. 18 IR spectra of the (a) 7AI monomer, (b) 7AI⋯2-FP complex, (c) 7AI⋯2,6-DFP complex, and (d) 7AI⋯2,3,5,6-TTFP complex in the N–H stretching frequency region with the stick spectra calculated at the M05-2X/cc-pVTZ level of theory. Reproduced from ref. 96 with permission from [American Institute of Physics], Copyright [2016]. | ||
A large red-shift of 265 cm−1 in the N–H stretching frequency due to the N–H⋯N interaction of the doubly hydrogen-bonded complex of 7-azaindole⋯2-fluoropyridine (7AI⋯2-FP) was observed with respect to that in the 7AI monomer (Fig. 18(a) and (b)).96 In contrast, a red-shift of 107 and 69 cm−1 in the N–H stretching frequency was observed in the 7AI⋯2,6-DFP and 7AI⋯2,3,5,6 TTFP complexes, respectively, which are stabilized by N–H⋯N and n → π* interactions (Fig. 18(c) and (d)).96 The 7AI⋯2-FP complex is planar due to the N–H⋯N and C–H⋯N hydrogen bonds, while the N–H⋯N hydrogen bond in the 7AI⋯2,6-DFP and 7AI⋯2,3,5,6-TTFP complexes is bent to accommodate the n → π* interaction.96 The result shows the influence of the n → π* interaction in governing the geometries of the complexes and the balance between the n → π* interaction and the N–H⋯N hydrogen bond.96 The weakening of the strength of the N–H⋯N hydrogen bond and thus the reduction in the IR red-shift in the N–H stretching frequency is affected by the presence of the n → π* interaction.96
Later, Das and co-workers provided direct spectroscopic evidence of the n → π* interaction by probing the CO stretching frequency of phenyl formate (PhOF) in the gas-phase.56 Two conformers of PhOF were observed in the experiment. The cis-PhOF conformer having n → π* interaction between the lone-pair electrons on the oxygen atom of the carbonyl group and the π* orbital of the phenyl ring is more stable by 1.32 kcal mol−1 than trans-PhOF lacking the n → π* interaction.56 The gas-phase IR spectra (Fig. 19) show that the CO stretching frequency in cis-PhOF is red-shifted by 31 cm−1 than that in trans-PhOF.56 The observed CO frequency red-shift in cis-PhOF is interpreted in terms of the presence of the n → π* interaction (Fig. 19) in the cis conformer as well as a subtle difference in various hyperconjugative interactions involving the CO group in both the conformers.56 A similar amount of red-shift in the CO stretching frequency of cis-PhOF with respect to that in trans-PhOF was also reported based on 2D IR spectroscopy experiments.97
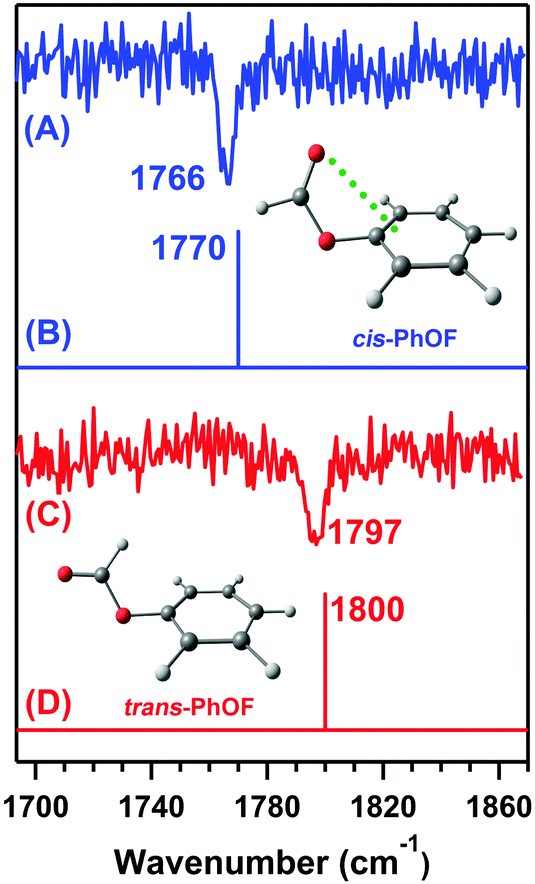 | ||
| Fig. 19 IR spectra of the (A) cis-PhOF and (C) trans-PhOF conformers. Theoretical IR stick spectra of the (B) cis-PhOF and (D) trans-PhOF calculated at the M05-2X/aug-cc-pVDZ level of theory. The calculated IR frequency has been scaled with respect to the reported experimental carbonyl stretching frequency of methyl lactate. Reproduced from ref. 56 with permission from [Wiley VCH], Copyright [2016]. | ||
Various hyperconjugative interactions present in PhOF are shown in Scheme 1 by arrow representation, and the E(2) values in both the conformers are provided in Table 4.56 The R1 interaction, which reduces the CO stretching frequency, involves the delocalization of the electron density mostly from the lone-pair orbital of the ethereal oxygen atom to the σ* and π* orbitals of the CO group.56 However, there is also a relatively small contribution to the R1 interaction by the delocalization of the electron density from the σ orbital of the C–H bond to the σ* orbitals of the CO group. The R2 interaction augments the CO stretching frequency by delocalizing the electron density from the lone-pair orbital of the carbonyl oxygen atom to the σ* orbital of the O–C and C–H bonds.56 The R3 interaction, which involves the delocalization of the lone pair electron density on the ethereal oxygen atom to the σ* and π* orbitals of the aromatic ring, opposes the R1 interaction and hence favors the increase in the CO stretching frequency.56 It can be found from Table 4 that the R1, R2, and R3 values are favorable for the reduction of CO frequency in the cis conformer compared to the trans conformer. The n → π* interaction (R4), which is present only in the cis conformer, is found to be 1.12 kcal mol−1 from the NBO calculations.56
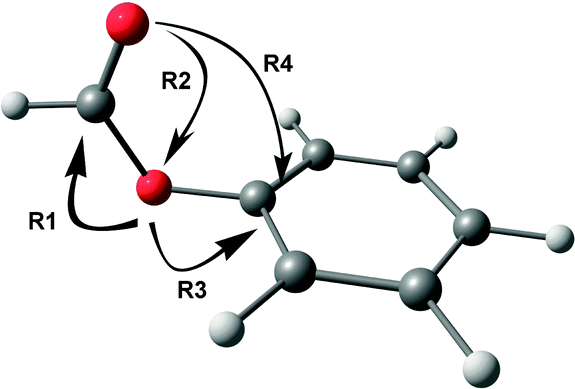 | ||
| Scheme 1 Arrow representation of various hyperconjugative interactions in one of the conformers of PhOF. | ||
| PhOF | PhOAc | |||
|---|---|---|---|---|
| cis | trans | cis | trans | |
| R1 | 66.34 | 49.30 | 64.02 | 54.92 |
| R2 | 70.86 | 72.21 | 72.64 | 71.56 |
| R3 | 30.41 | 36.43 | 29.78 | 34.59 |
| R4 (n → π*) | 1.12 | — | 1.23 | — |
| Sterics | — | 6.05 | — | 8.34 |
It should be noted here that the hyperconjugative interaction values are quite larger than the n → π* interaction values. In contrast, the hyperconjugative interactions are present in both the conformers, and the n → π* interaction is present only in the cis conformer. While it is not straightforward to normalize the NBO values for different types of hyperconjugative interactions, it can be concluded that both hyperconjugative and n → π* interactions contribute to the observed red-shift in the CO frequency of cis-PhOF compared to trans-PhOF. Further, it has been shown with the help of a detailed NBO deletion analysis that the hyperconjugative interactions, which favor the red-shift in the CO stretching frequency of cis-PhOF, are affected by the n → π* interaction.56Table 5 shows the occupancy of the p-type lone-pair on the carbonyl oxygen atom after the deletion of various interactions present in the cis conformer having the n → π* interaction. NBO deletion analysis deletes a specific interaction from a molecular system and recalculates the occupancies of the orbitals, assuming the absence of that particular interaction there. The occupancy of the donor and acceptor orbitals increases and decreases upon deletion of interaction, respectively. Herein, we will be discussing specifically the effect of deletion of individual R2 and R4 interactions as well as both of these together, as these two interactions involve the delocalization of the lone-pair electron density of the carbonyl oxygen atom. It can be noticed from Table 5 that the occupancy of the p-type lone-pair on the carbonyl oxygen atom increases when R4, i.e., the n → π* interaction, is deleted (Case II) in cis-PhOF, and these data confirm the presence of this interaction. It should be noted here that the occupancy on the carbonyl oxygen atom increases, while both the R2 and R4 interactions are deleted with respect to the deletion of only the R2 interaction. This shows that the presence of the R4 interaction reduces the availability of the lone-pair electron density for the R2 interaction, which is favorable for the red-shift in the CO stretching frequency.56
| Interactions | cis-PhOF | cis-PhOAc | |
|---|---|---|---|
| Case I | R1, R2, R3, R4 (not deleted) | 1.845 | 1.850 |
| Case II | R4 (deleted) | 1.846 | 1.851 |
| Case III | R2, R4 (deleted) | 1.988 | 1.989 |
| Case IV | R2 (deleted) | 1.987 | 1.987 |
Subsequently, in their other work, Das and co-workers showed that the steric and n → π* interactions dictate the conformational preference of phenyl acetate (PhOAc).58 They tried to tune the strength of the interaction by substituting the methyl group near the n → π* donor, i.e., the carbonyl group. It was anticipated that the methyl group would push the electron density toward the carbonyl group and make the oxygen atom of the carbonyl group better n → π* donor, and increase the n → π* interaction in the cis-PhOAc conformer.58 Herein, the authors observed only one conformer of PhOAc in the experiment, while two conformers were observed in the case of PhOF.56 The electronic spectrum was also measured in the 30%He–70%Ne gas mixture to check the possibility of conformational relaxation due to the heavy carrier gas (Ar) used in the experiment.58 No additional peak was observed in the spectrum measured in the 30%He–70%Ne gas mixture. The gas-phase IR spectra measured in the CO stretching frequency region confirmed the presence of the cis-PhOAc in the experiment (Fig. 20).58Cis-PhOAc is more stable than trans-PhOAc by 3.23 kcal mol−1 at the M05-2X/aug-cc-pVDZ level of theory. It is important to mention that cis-PhOF is more stable than its trans counterpart by only 1.32 kcal mol−1.58
 | ||
| Fig. 20 (a) RIDIR spectrum of cis-PhOAc; and (b) and (c) theoretical IR scaled spectra of the cis and trans conformers of PhOAc calculated at the M05-2X/aVDZ level of theory. The theoretical CO stretching frequencies are scaled by a factor of 0.9556 with respect to the trans-PhOF. Reproduced from ref. 58 with permission from [Wiley VCH], Copyright [2019]. | ||
It has been found that the NBO 2nd-order perturbative energy of the n → π* interaction in cis-PhOAc (1.23 kcal mol−1) is slightly higher than that in cis-PhOF (1.12 kcal mol−1).58 At the same time, there is an increase in the steric effect in trans-PhOAc by ∼2 kcal mol−1 due to the bulkiness of the methyl group as compared to the hydrogen atom present in PhOF near the n → π* donor (Table 4). Interestingly, the relative energy difference between the trans and cis conformers in PhOAc is ∼2 kcal mol−1 higher than PhOF. Hence, the larger steric effects in trans-PhOAc direct the selectivity towards the cis-PhOAc conformer having the n → π* interaction.58 The red-shift in the νCO of cis-PhOAc compared to trans-PhOAc can be understood with the help of NBO analysis, as explained earlier in the case of PhOF.58 It could be noted here that the red-shift of νCO is less in the case of PhOAc (11 cm−1) than that in PhOF (31 cm−1) despite the slight enhancement in the n → π* interaction in the former with respect to the latter one.58 The R1, R2, and R3 interactions in the cis and trans conformers of PhOAc with respect to those in PhOF favor the smaller amount of CO frequency red-shift in the former one.58 NBO deletion analysis (Table 5) confirmed the role of the n → π* interactions in the red-shift of νCO of cis-PhOAc.58
Das and co-workers also observed this interaction in a drug called salicin, which is an analog of aspirin.67 The chemical name of aspirin is 2-(hydroxymethyl)phenyl β-D-glucopyranoside, which is used as an analgesic, antipyretic, and anti-inflammatory drug.67 The authors observed three conformers (Fig. 21) of salicin in the gas-phase experiments using UV-UV hole-burning spectroscopy. Structural assignment of the experimentally observed conformers of salicin was done by recording their IR spectra in the O–H stretching frequency region and calculating their simulated Franck–Condon spectra.67 It can be noticed that all three conformers differ mostly in the orientation of the hydroxyl group of the –CH2OH of the glucopyranoside ring.
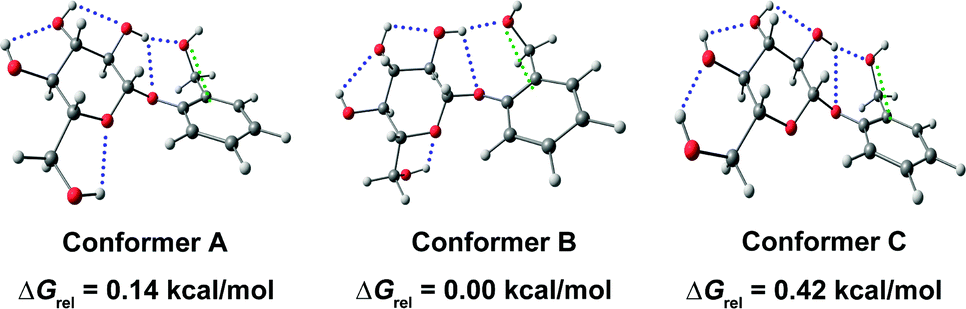 | ||
| Fig. 21 Experimentally observed conformers of salicin with their relative Gibbs free energies at the 10 K of the conformers calculated at the M06-2X/6-311++G(d,p) level of theory. Reproduced from ref. 67 with permission from [Royal Society of Chemistry], copyright [2018]. | ||
Here, the electron density of the lone-pair orbital of the benzylic oxygen atom delocalizes into the π* orbital of the aromatic ring. The strength of the n → π* interaction calculated from the NBO analysis was almost the same in all three conformers (∼1.1 kcal mol−1).67 It should be mentioned here that all of the three conformers of salicin have a subtle interplay with the hydrogen-bonding interaction of ∼9 kcal mol−1 strength.67 NBO orbital overlap pictures for both the n → π* interaction and hydrogen bonding are shown in Fig. 22(a) and (b), respectively.67
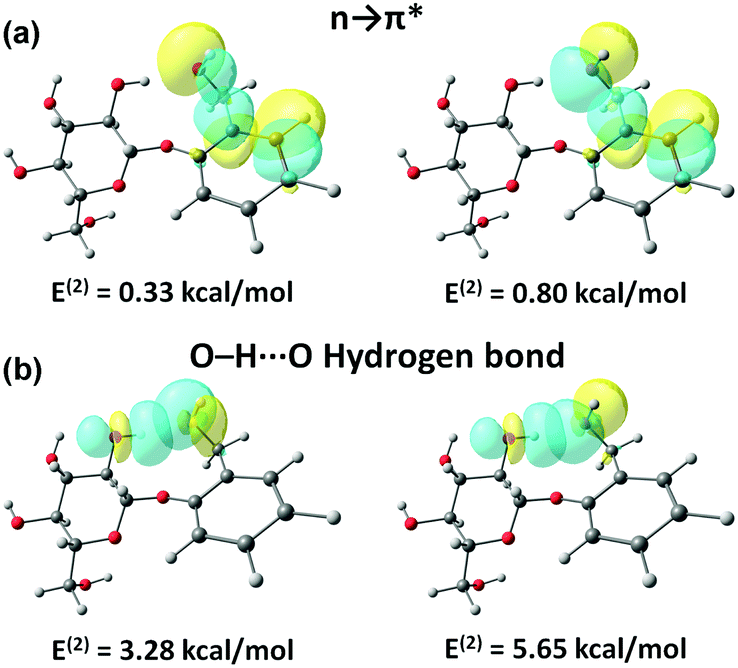 | ||
| Fig. 22 NBO orbital overlap of conformer B of the salicin for (a) n → π* interaction and (b) O–H⋯O hydrogen bonding calculated at the M06-2X/6-311++G(d,p) level of theory. Reproduced from ref. 67 with permission from [Royal Society of Chemistry], copyright [2018]. | ||
As discussed earlier, the presence of n → π* interaction in the trans conformer of proline derivatives is demonstrated by determining the equilibrium constant values for the cis–trans isomerization using NMR spectroscopy. Das and co-workers studied the CO⋯CO n → π* interaction in a capped 4R-hydroxyproline (2S,4R)-N1-(benzyloxycarbonyl)-4-hydroxyproline methyl ester (Cbz-Hyp-OMe).57Fig. 23 shows the classification of the conformers of the compound in four different groups (P, Q, R, and S) based on the orientation of the amide bond and the benzene ring. The structures of all the conformers are calculated at the M05-2X/aug-cc-pVDZ level of theory.57
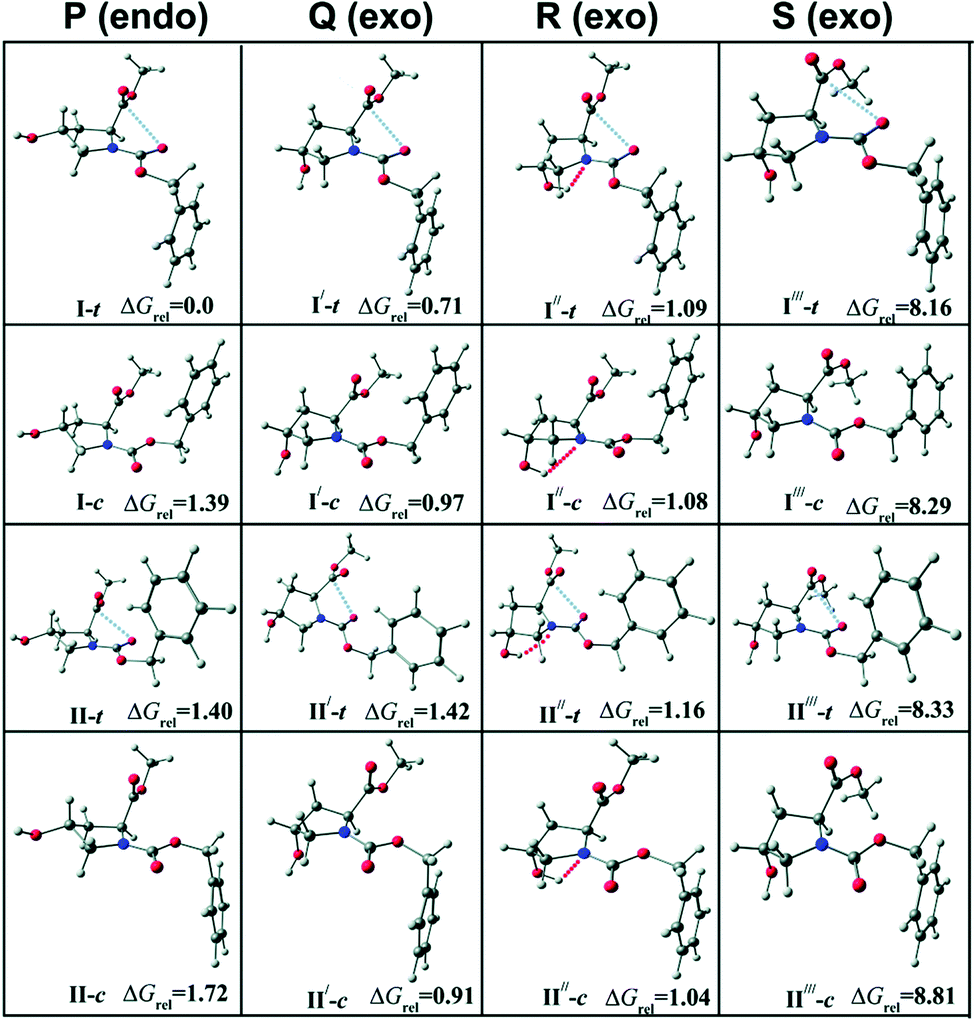 | ||
| Fig. 23 Optimized structures of low energy conformers of Cbz-Hyp-OMe calculated at the M05-2X/aug-cc-pVDZ level of theory with the relative Gibbs free energies (ΔGrel, kcal mol−1) of various conformers at a temperature of 298 K with respect to the most stable conformer (I-t). “t” stands for trans and “c” stands for cis. The change in the orientation of the amide bond generates trans and cis conformers. Reproduced from ref. 57 with permission from [Royal Society of Chemistry], copyright [2019]. | ||
They observed four conformers of the capped hydroxyproline in the experiment using UV-UV hole-burning spectroscopy.57 Two of the conformers were assigned as trans and another two as cis based on the IR stretching frequencies of the carbonyl (Fig. 24(a)) and O–H groups (Fig. 24(b)).57 The theoretical IR spectra of the conformers obtained from the calculations are provided with the experimental IR spectra in Fig. 24. It can be observed that conformers A and C have a similar stretching frequency in the CO region (amide and ester). However, conformer C shows a 35 cm−1 red-shift in the O–H frequency with respect to conformer A. The CO frequencies of conformers B and D are similar but different compared to the CO frequencies of conformers A and C. Similarly, the O–H frequency of conformer D is red-shifted by 34 cm−1 with respect to conformer B.57 Thus, the O–H groups in conformers C and D are hydrogen-bonded, while they are free in conformers A and B. Further, the amide CO (n → π* donor) frequency of conformers A and C is red-shifted by 14 cm−1 from that of conformers B and D. Similarly, the ester CO frequency (n → π* acceptor) of conformers A and C is red-shifted by 5 cm−1 from that of conformers B and D.57 It should be mentioned here that a particular structure could not be assigned to any of the experimentally observed conformers due to the similar energetics and IR stretching frequencies of a group of conformers obtained from the calculations.57 The IR spectroscopy results, along with NBO analysis, demonstrate that the observed red-shifts in the amide and ester CO frequencies in conformers A and C are due to the presence of the n → π* interaction as well as the hyperconjugative interactions involving the CO groups present there.57
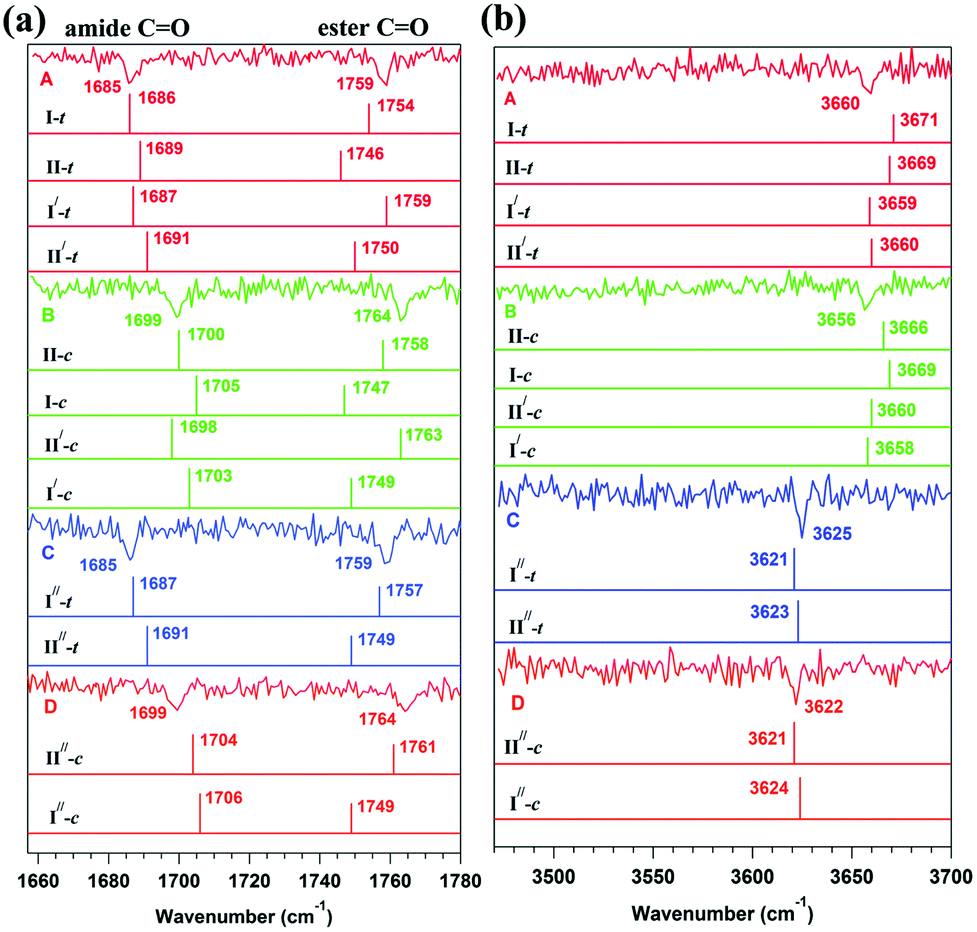 | ||
| Fig. 24 Experimental RIDIR spectra of conformers A, B, C, and D in the (a) CO and (b) O–H stretching frequency regions with the theoretical stick IR spectra of group P, Q, and R structures of Cbz-Hyp-OMe shown in Fig. 23 calculated at the M05-2X/aug-cc-pVDZ level of theory. I-t, II-c, etc., correspond to the trans and cis conformers of Cbz-Hyp-OMe. Reproduced from ref. 57 with permission from [Royal Society of Chemistry], copyright [2019]. | ||
The CO⋯CO n → π* interaction present in the trans conformers of the capped hydroxyproline is ∼2 kcal mol−1.57 It has been found that the presence of the n → π* interaction in the trans conformers reduces the lone-pair electron density of the amide carbonyl oxygen atom and thus enhances the hyperconjugation effect R1amide, which favors the red-shift in the amide CO stretching frequency.57 The R1amide interaction involves the overlap between the lone pair orbitals on the ethereal oxygen and the nitrogen atom in the proline ring with the σ* and π* orbitals of the amide CO group.57 The overlap between the oxygen lone pair orbital on the amide CO with the σ* orbitals of the neighboring C–O and C–N bonds is termed the R2amide interaction.57
The R1ester and R2ester interactions involve the overlap between the lone pair orbital of the neighboring oxygen atom with the σ* and π* orbitals of the ester CO group, while the R2ester interaction involves the overlap between the oxygen lone pair electrons on the ester CO group and the σ* orbitals of the neighboring C–O bond, respectively.57 It can be noticed from Fig. 25 that the E(2) values for R1amide are more for the trans conformers (having n → π* interaction) than that for the cis conformer (lacking n → π* interaction). It should also be noted here that the values for R2amide, R1ester, and R2ester are almost similar for the trans and cis conformers of the capped hydroxyproline.57
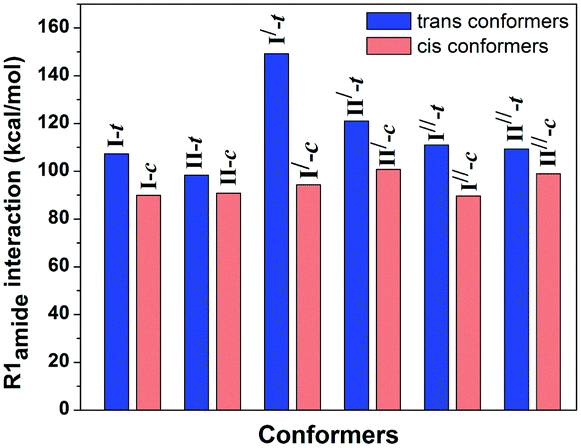 | ||
| Fig. 25 Bar diagram of E(2) values of R1 interaction for the amide CO group in the trans and cis conformers of Cbz-Hyp-OMe calculated at the M05-2X/aug-cc-pVDZ level of theory. Reproduced from ref. 57 with permission from [Royal Society of Chemistry], copyright [2019]. | ||
Applications of the n → π* interaction
Very recently, it has been demonstrated that the n → π* interaction can have potential applications in different areas. These applications are discussed briefly in this section to establish the importance of this non-covalent interaction. One of the applications of the n → π* interaction has been found in dynamic covalent chemistry, which has been utilized previously for studying the C–H⋯π and π⋯π stacking interactions.98,99 You and co-workers studied the modulation of reactivity in a series of reactions of 2-X-2′-formylbiphenyl [1(X); X = OMe, SMe, NMe2, H, F, Cl, Br, and I] with an amine (Fig. 26) to form imines [2(X)] by exploiting the n → π* interaction present there between the lone-pair electrons on the heteroatom of the X group and the CHO.100 The formation of the imine molecules is stabilized by the n → π* interaction as the electron density over imine carbon enhances and stabilizes the imine bond. Furthermore, You and co-workers studied the aldehyde exchange in the reaction of imine and aldehyde to understand the impact of the n → π* interaction (Exchange 1 shown in Fig. 27(a)).100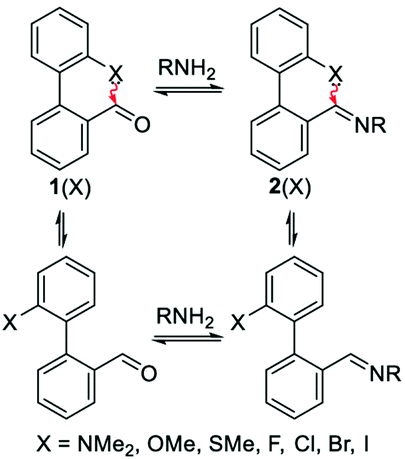 | ||
| Fig. 26 Representation of dynamic imine chemistry. Reproduced from ref. 100 with permission from [American Chemical Society]. Copyright [2019]. | ||
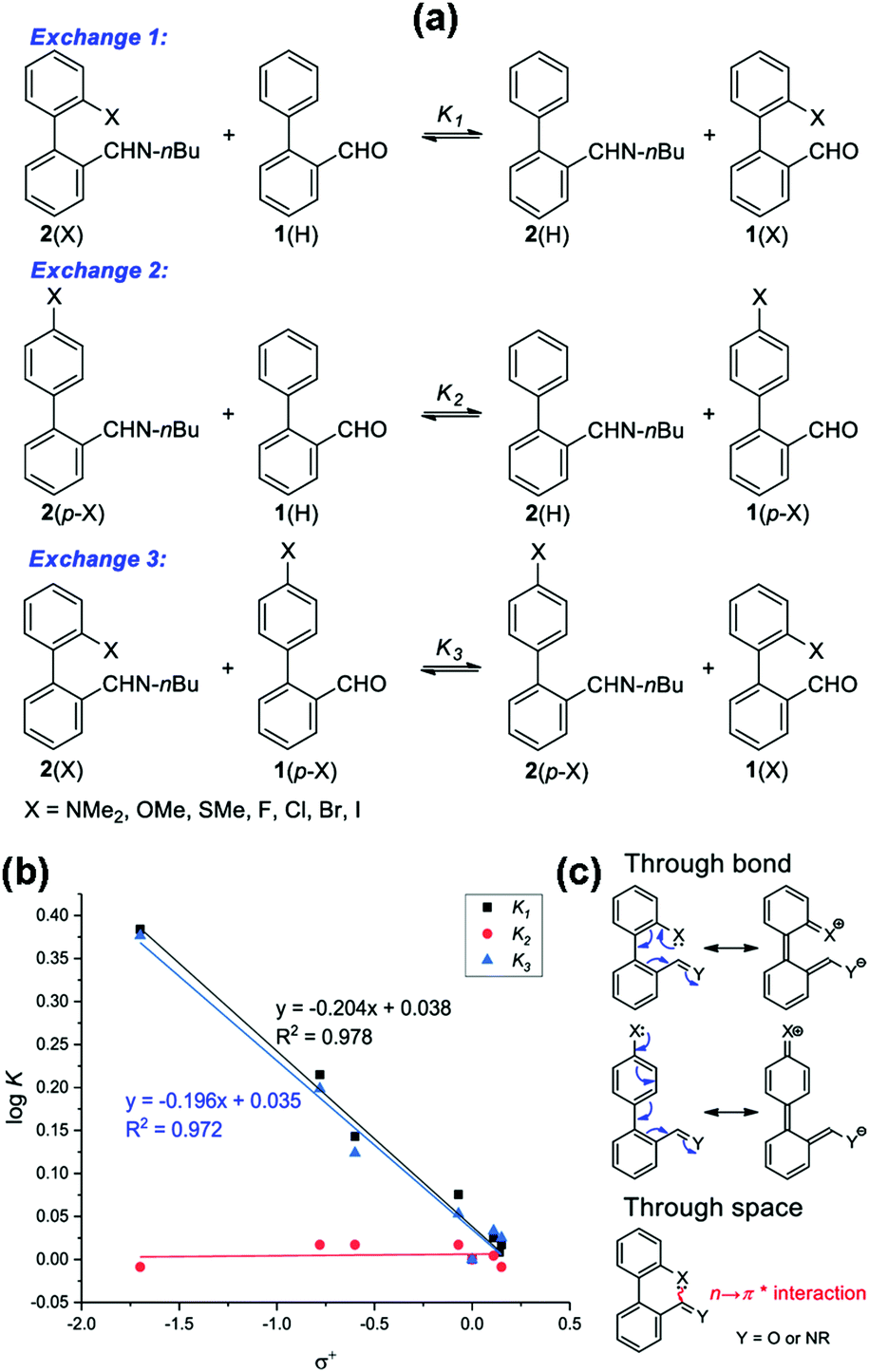 | ||
| Fig. 27 (a) Different aldehyde exchange mechanisms; (b) plot of logK versus σ+; and (c) representations of through space (n → π*) and through bond (resonance effect) interactions. Reproduced from ref. 100 with permission from [American Chemical Society], Copyright [2019]. | ||
The n → π* interaction weakens the nucleophilic attack of water on the imine [2(X)] by reducing its electrophilicity. It was found that the aldehyde exchange process in the step-wise formation of imine [2(X)] followed by the addition of aldehyde [1(H)] was prolonged.100 However, this exchange process (Exchange 1 shown in Fig. 27(a)) was extremely fast when both the aldehydes [1(X) and 1(H)] were added simultaneously.
The slow aldehyde exchange process was attributed to the absence of water molecules in the reaction environment, and this was confirmed by performing the kinetics in CD3CN (180 days), 95:5 CD3CN/D2O (100 days), and 90:10 CD3CN/D2O (55 days).100 However, this reaction was completed within one day when both the aldehydes [1(X) and 1(H)] were reacted simultaneously.100 To concretize the role of n → π* interaction (through space interaction shown in Fig. 27(c)), they studied three sets of exchange reactions for different p-substituted imines and p-substituted aldehydes (Fig. 27(a)).100 It was found that the logK values for the exchange 2 type reactions (Fig. 27(b)) were nearly zero for all the derivatives, and hence these results show the undeniable role of the n → π* interaction in the aldehyde exchange.100 These results were further corroborated by the NBO analysis as the relative Gibbs free energy values of aldehyde exchange showed a linear correlation with the second-order perturbative energy values for the n → π* interaction.100
Raines and co-workers showed the role of the n → π* interaction in the catalysis of hydrolysis of the Michaelis complex using HIV-1 protease.101 Herein, Asp25 of one monomer abstracts the proton of the water molecule and attacks the carbonyl carbon atom of the scissile peptide bond, and the oxygen atom of the same carbonyl group abstracts a proton from another Asp25′ monomer, resulting in the regeneration of Asp25 and the formation of a gem–diol intermediate (Fig. 28).101 There is a presence of weak n → π* interaction (0.66 kcal mol−1) in the Michaelis complex, and the formation of a tetrahedral intermediate (gem-diol), upon attack of the water molecule, which is stabilized by stronger n → π* interaction (2.01 kcal mol−1).101 The n → π* interaction assists in delocalizing the negative charge developed upon the attack of the water molecule and helps in the catalysis.101
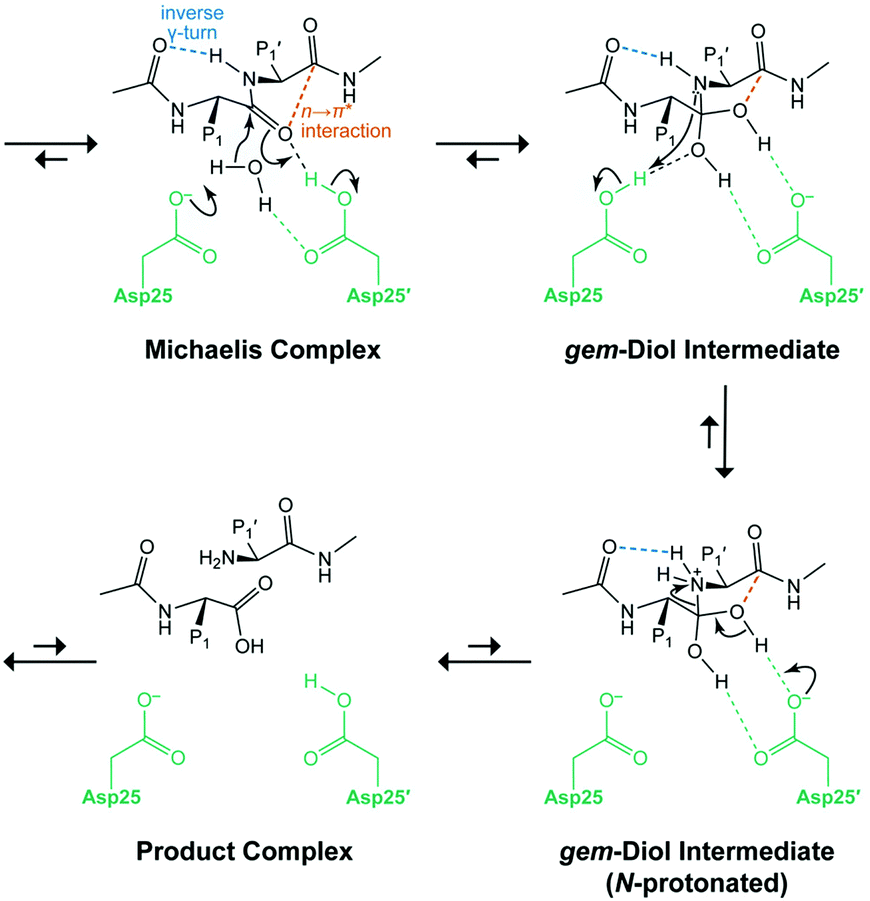 | ||
| Fig. 28 Mechanism of catalysis with HIV-1 protease. Reproduced from ref. 101 with permission from [American Chemical Society], Copyright [2019]. | ||
Shimizu and co-workers showed the utilization of the n → π* interaction in stabilizing the transition state of the rotation of the Caryl–Nimide bond in N–phenylimide molecular rotors (Fig. 29), resulting in the existence of these rotors in syn and anti conformers.102
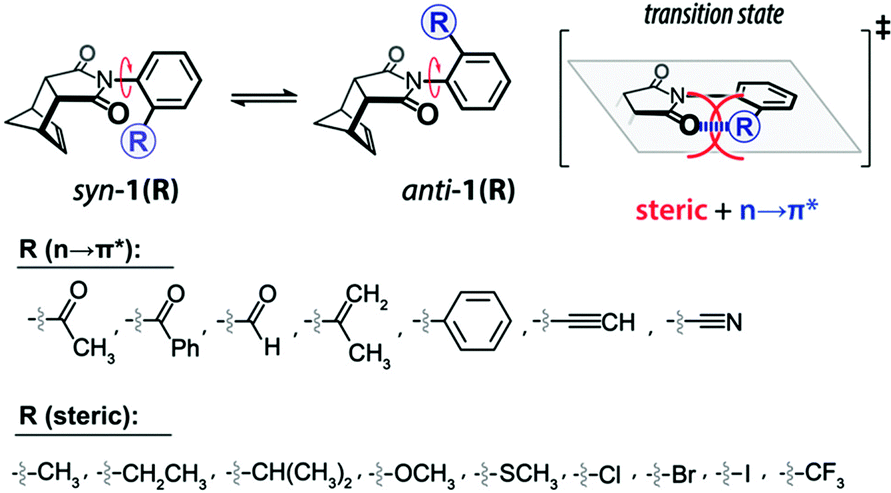 | ||
| Fig. 29 Equilibrium of syn–anti conformers of molecular rotors arising due to N-phenyl ring rotation. Reproduced from ref. 102 with permission from [American Chemical Society], Copyright [2019]. | ||
They measured the energy barrier of interconversion using 1H NMR exchange spectroscopy in the temperature range of −60 to 130 °C so that distinct peaks corresponding to the syn and anti forms can be obtained.102 The free energy for the rotational barriers was calculated with the help of Eyring plots which provided the enthalpy and entropy for the molecular rotors. There was a decrease in the energy barrier for the interconversion between the syn and anti forms when the n → π* interaction was found between the oxygen atom of the imide and the π* orbital involving the ortho group of the aromatic moiety (Fig. 30(A)).102 These energy barriers were further compared with the rotors lacking the acceptor π* orbital (Fig. 30(A)). Moreover, they took special care to maintain the bulkiness/sterics of the ortho group while comparing the different rotors with or without the acceptor π* orbital.102
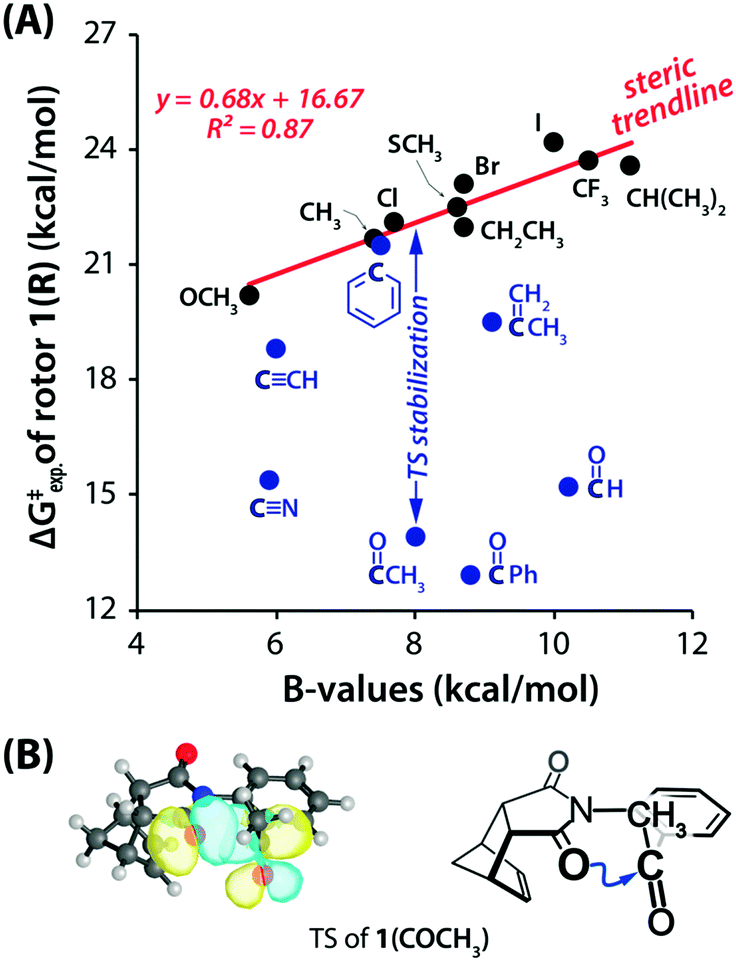 | ||
| Fig. 30 (A) Rotational barriers for various rotors versus the steric B-values of the R groups. Blue and black circles correspond to the rotors that can form n → π* interactions and cannot form n → π* interactions, respectively. (B) NBO overlap for n → π* interactions in the calculated TS (B3LYP-D3/6-311G*) of 1(COCH3) and the ChemDraw presentation of TS (blue arrows show the n → π* interaction). Reproduced from ref. 102 with permission from [American Chemical Society], Copyright [2019]. | ||
A good linear correlation of the plot of energy barrier versus steric values (B) for the R group (calculated from the Mazzanti steric B-value parameter)103,104 was observed for the rotors lacking n → π* interaction where only steric effects were present and named it as a steric trendline, shown in Fig. 30(A).102 The energy barrier is lowered from the steric trendline for the rotors having the π* orbital in the ortho group, forming the n → π* interaction, deviating from the steric trendline.102 Moreover, for the rotors having the CO group, the energy barrier is very low as the CO group is polar and acts as a better acceptor for the electron density. The NBO orbital overlap for the n → π* interaction in the transition state for one of the molecular rotors is shown in Fig. 30(B).102
Furthermore, Shimizu and co-workers performed the NBO analysis of the transition states of the molecular motors at the ωB97M-V/6-311G* level of theory.102 They found a linear correlation of the experimentally calculated rotational barriers with the second-order perturbative energy values of the n → π* interaction (Fig. 31(A)).102 The overall results suggest that the orbital–orbital interaction stabilizes the transition states. A linear correlation was also observed in the plot of the rotational barrier with the distance between the oxygen atom (n → π* donor) and the acceptor carbon atom of the π* orbital (Fig. 31(B)).102 This observation confirms that the orbital–orbital overlap stabilizes the transition states of the molecular rotors.102 The authors also observed the pyramidalization of the acceptor carbon atom, which is a signature of the presence of the n → π* interaction. Hence, these rotors were successfully employed to lower the rotational barrier by ∼9 kcal mol−1.102
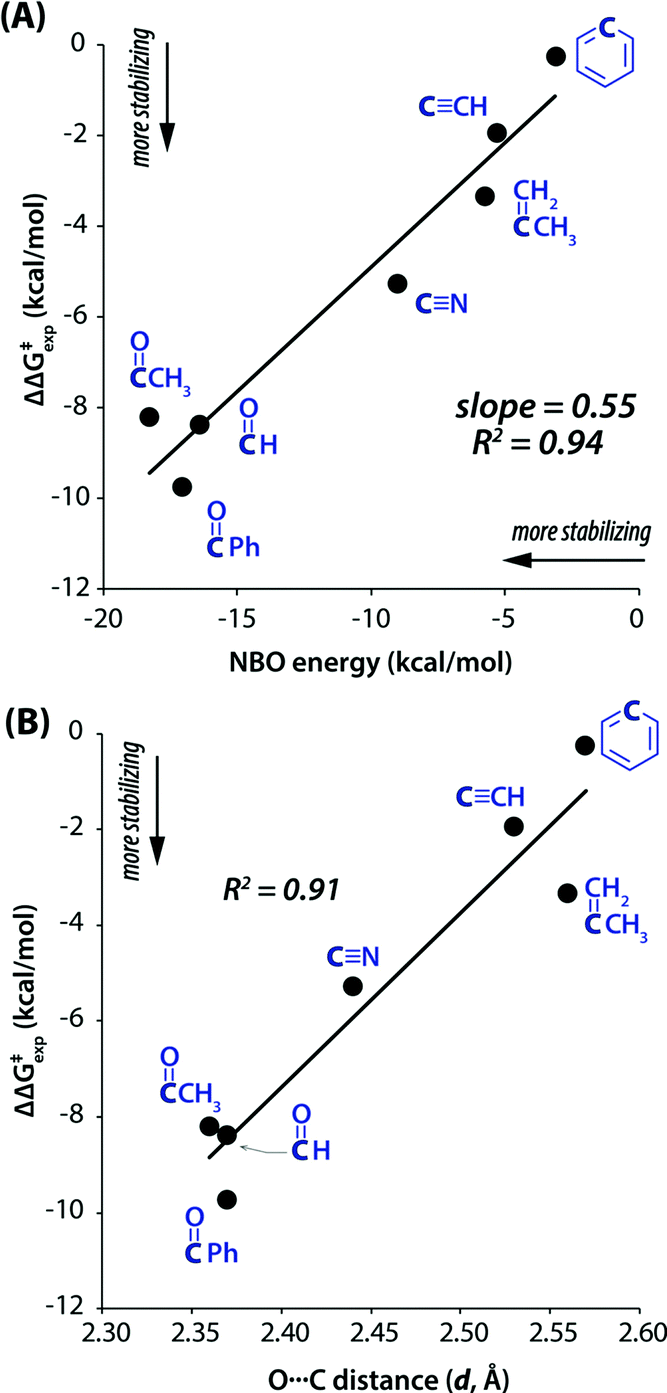 | ||
| Fig. 31 Rotational barriers for various rotors versus (A) calculated NBO energies for the TS n → π* interaction; and (B) calculated donor-to-acceptor distances (d) in the TS, calculated at the ωB97M-V/6-311G* level of theory. Reproduced from ref. 102 with permission from [American Chemical Society], Copyright [2019]. | ||
Conclusions and future outlook
The n → π* interaction, which is one of the emerging non-covalent interactions reported in the literature, is similar to hydrogen bonding in terms of the delocalization of electron density between the donor and acceptor orbitals. The n → π* interaction is prevalent between the two neighboring CO groups in the backbone of proteins. This interaction is also widely present in peptides, peptoids, several drugs, and many small molecule systems. The extensive study of the n → π* interaction is not present in the literature due to its weak and counterintuitive nature. Thus, this interaction is not well understood yet in the literature. In this review, we have tried to discuss the understanding of this non-covalent interaction using several experimental and theoretical approaches. Raines and co-workers demonstrated that the n → π* interaction is purely an orbital–orbital interaction.78 However, electrostatic and dispersion interactions can facilitate the optimum overlap between the two orbitals. Though this interaction is not fully understood yet, its importance has already been demonstrated in several fields such as catalysis, reaction chemistry, and stabilization of transition states and intermediates. This review provides an encyclopedic understanding of the n → π* interaction based on solution-phase UV-Vis, IR, and NMR spectroscopy, X-ray diffraction, and quantum chemistry calculations. Understanding this interaction using isolated gas-phase microwave, electronic, and IR spectroscopic techniques allows for studying the molecular system free from perturbation from solvent or other surrounding molecules. The strength of the n → π* interaction can be determined by measuring the red-shift in the vibrational frequency of the functional groups involved in the n → π* interaction. Modulation of the strength of the n → π* interaction by introducing substituents with the donor or acceptor groups can also be probed by monitoring the vibrational frequencies. In a nutshell, this review allows the reader to understand the n → π* interaction using solution-phase, solid-phase, and gas-phase studies.
The study of the n → π* interaction using gas-phase IR spectroscopy is still quite limited in the literature. The n → π* interaction generally occurs through the CO groups present in proteins, peptides, amides, or esters. The IR spectroscopy experiment is done by probing the frequency of the CO groups, which are generally in conjugation with neighboring nitrogen or oxygen atoms. Thus, several types of hyperconjugative interactions, which are in play simultaneously, also affect the CO stretching frequency perturbed by the n → π* interaction. The hyperconjugative interactions are present in the conformers with and without the n → π* interaction, albeit with slightly different quantities. A detailed NBO analysis of the hyperconjugative interactions, n → π* interaction, and NBO deletion calculation can provide some qualitative idea about these interactions. However, it is not straightforward to find the exact contribution of the n → π* interaction to the red-shift in the CO frequency. The contribution of the purely n → π* interaction to the red-shift of the CO frequency in a molecular system in the absence of the hyperconjugation interaction is not explored. Hence, aldehydes and ketones, free from hyperconjugative interactions involving neighboring heteroatoms, can serve as better systems for understanding the n → π* interaction in terms of the red-shift in the IR frequency. It is also required to explore intermolecular complexes which are preferentially bound by the n → π* interaction using NMR and IR spectroscopy as well as quantum chemistry calculations.
The n → π* interaction has found its own place in terms of contributing to the stability of the structures of proteins, which is one of the most important classes of biomolecules. However, very recent applications of the n → π* interaction in catalysis and dynamic chemistry have demonstrated its importance as a non-covalent interaction. Earlier, it has been reported that the n → π* interaction plays an essential role in the structure and function of drugs such as aspirin. It is anticipated that the application of the n → π* interaction will be found in many more diverse areas of chemistry and biology in the future. Future studies should be focused more on modulating the strength of this interaction to have its further importance in synthesizing more efficient drugs and various supramolecular assemblies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Indian Institute of Science Education and Research (IISER) Pune for providing financial support to perform this research. Financial support from the Science and Engineering Research Board (SERB), India (Grant No. CRG/2020/002696), is also acknowledged. The computational support and resources provided by the “PARAM Brahma Facility” under the National Supercomputing Mission, Government of India, IISER, Pune, are also acknowledged. P. P. acknowledges IISER Pune for his research fellowship and Trimurti research grant for funding.Notes and references
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, New York, 1999 Search PubMed.
- G. A. Jeffrey and W. Saenger, Hydrogen bonding in biological structures, Springer, Berlin, 1991 Search PubMed.
- W. Saenger, Annu. Rev. Biophys. Biophys. Chem., 1987, 16, 93–114 CrossRef CAS PubMed.
- R. Wolfenden, L. Andersson, P. M. Cullis and C. C. B. Southgate, Biochemistry, 1981, 20, 849–855 CrossRef CAS PubMed.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- J. W. Steed and J. L. Atwood, Supramolecular chemistry, Wiley, New York, 2000 Search PubMed.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS PubMed.
- S. Kumar, P. Biswas, I. Kaul and A. Das, J. Phys. Chem. A, 2011, 115, 7461–7472 CrossRef CAS PubMed.
- P. Panwaria and A. Das, Exploring Non-covalent Interactions by Jet-Cooled, Electronic and Vibrational Spectroscopy in Modern Techniques of Spectroscopy: Basics, Instrumentation, and Applications, ed. D. K. Singh, M. Pradhan and A. Materny, Springer Nature, 2021, pp. 57–86 Search PubMed.
- A. Karshikoff, Non-covalent Interactions in Proteins, Imperial College Press, London, 2006 Search PubMed.
- W. Saenger, Principles of Nucleic Acid Structure, Springer-Verlag, New York, 1984 Search PubMed.
- M. Ghosh, P. Panwaria, S. Tothadi, A. Das and S. Khan, Inorg. Chem., 2020, 59, 17811–17821 CrossRef CAS PubMed.
- G. A. Jeffrey, An introduction to hydrogen bonding, Oxford University Press, New York, 1997 Search PubMed.
- S. Scheiner, Hydrogen Bonding: A Theoretical Perspective, Oxford University Press, New York, 1997 Search PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg and P. Hobza, Pure Appl. Chem., 2011, 83, 1637–1641 CrossRef CAS.
- T. Ebata, A. Fujii and N. Mikami, Int. Rev. Phys. Chem., 1998, 17, 331–361 Search PubMed.
- H. S. Biswal, S. Bhattacharyya, A. Bhattacherjee and S. Wategaonkar, Int. Rev. Phys. Chem., 2015, 34, 99–160 Search PubMed.
- A. S. Hansen, E. Vogt and H. G. Kjaergaard, Int. Rev. Phys. Chem., 2019, 38, 115–148 Search PubMed.
- C. A. Hunter, Chem. Soc. Rev., 1994, 23, 101–109 RSC.
- P. Metrangolo and G. Resnati, Halogen bonding: fundamentals and applications, Springer-Verlag, Berlin Heidelberg, 2008 Search PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- L. P. Wolters, P. Schyman, M. J. Pavan, W. L. Jorgensen, F. M. Bickelhaupt and S. Kozuch, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 523–540 CAS.
- S. Benz, A. I. Poblador-Bahamonde, N. Low-Ders and S. Matile, Angew. Chem., Int. Ed., 2018, 57, 5408–5412 CrossRef CAS PubMed.
- P. Sanz, M. Yáñez and O. Mó, Chem. – Eur. J., 2003, 9, 4548–4555 CrossRef CAS PubMed.
- M. Iwaoka, H. Komatsu, T. Katsuda and S. Tomoda, J. Am. Chem. Soc., 2002, 124, 1902–1909 CrossRef CAS PubMed.
- R. Gleiter, G. Haberhauer, D. B. Werz, F. Rominger and C. Bleiholder, Chem. Rev., 2018, 118, 2010–2041 CrossRef CAS PubMed.
- C. B. Aakeroy, D. L. Bryce, G. Desiraju, A. Frontera, A. C. Legon, F. Nicotra, K. Rissanen, S. Scheiner, G. Terraneo, P. Metrangolo and G. Resnati, Pure Appl. Chem., 2019, 91, 1889–1892 CrossRef CAS.
- C. A. Hunter and J. K. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- S. Burley and G. A. Petsko, Science, 1985, 229, 23–28 CrossRef CAS PubMed.
- C. J. Pace and J. Gao, Acc. Chem. Res., 2013, 46, 907–915 CrossRef CAS PubMed.
- J. H. Deng, J. Luo, Y. L. Mao, S. Lai, Y. N. Gong, D. C. Zhong and T. B. Lu, Sci. Adv., 2020, 6, eaax9976 CrossRef CAS PubMed.
- D. A. Dougherty, Science, 1996, 271, 163–168 CrossRef CAS PubMed.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS PubMed.
- D. A. Dougherty, Acc. Chem. Res., 2013, 46, 885–893 CrossRef CAS PubMed.
- M. R. Davis and D. A. Dougherty, Phys. Chem. Chem. Phys., 2015, 17, 29262–29270 RSC.
- D. Quiñonero, C. Garau, C. Rotger, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Angew. Chem., Int. Ed., 2002, 114, 3539–3542 CrossRef.
- P. Ballester, Acc. Chem. Res., 2013, 46, 874–884 CrossRef CAS PubMed.
- H. T. Chifotides and K. R. Dunbar, Acc. Chem. Res., 2013, 46, 894–906 CrossRef CAS PubMed.
- I. Caracelli, I. Haiduc, J. Zukerman-Schpector and E. R. Tiekink, Coord. Chem. Rev., 2013, 257, 2863–2879 CrossRef CAS.
- S. K. Singh and A. Das, Phys. Chem. Chem. Phys., 2015, 17, 9596–9612 RSC.
- G. J. Bartlett, A. Choudhary, R. T. Raines and D. N. Woolfson, Nat. Chem. Biol., 2010, 6, 615–620 CrossRef CAS PubMed.
- R. W. Newberry and R. T. Raines, Acc. Chem. Res., 2017, 50, 1838–1846 CrossRef CAS PubMed.
- E. H. Mottus, H. Schwarz and L. Marion, Can. J. Chem., 1953, 31, 1144–1151 CrossRef CAS.
- N. J. Leonard and M. Ōki, J. Am. Chem. Soc., 1954, 76, 3463–3465 CrossRef CAS.
- N. J. Leonard and M. Ōki, J. Am. Chem. Soc., 1955, 77, 6239–6240 CrossRef CAS.
- H. Bürgi, J. Dunitz and E. Shefter, J. Am. Chem. Soc., 1973, 95, 5065–5067 CrossRef.
- H. Bürgi, J. Dunitz and E. Shefter, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 1517–1527 CrossRef.
- B. Sahariah and B. K. Sarma, Phys. Chem. Chem. Phys., 2020, 22, 26669–26681 RSC.
- A. Rahim, P. Saha, K. K. Jha, N. Sukumar and B. K. Sarma, Nat. Commun., 2017, 8, 1–13 CrossRef CAS PubMed.
- R. W. Newberry and R. T. Raines, ACS Chem. Biol., 2019, 14, 1677–1686 CrossRef CAS PubMed.
- S. K. Holmgren, K. M. Taylor, L. E. Bretscher and R. T. Raines, Nature, 1998, 392, 666–667 CrossRef CAS PubMed.
- M. L. DeRider, S. J. Wilkens, M. J. Waddell, L. E. Bretscher, F. Weinhold, R. T. Raines and J. L. Markley, J. Am. Chem. Soc., 2002, 124, 2497–2505 CrossRef CAS PubMed.
- J. C. Horng and R. T. Raines, Prot. Sci., 2006, 15, 74–83 CrossRef CAS PubMed.
- S. K. Singh, K. K. Mishra, N. Sharma and A. Das, Angew. Chem., Int. Ed., 2016, 55, 7801–7805 CrossRef CAS PubMed.
- S. K. Singh, S. More, S. Kumar, K. K. Mishra, K. N. Ganesh and A. Das, Phys. Chem. Chem. Phys., 2019, 21, 4755–4762 RSC.
- S. K. Singh, P. Panwaria, K. K. Mishra and A. Das, Chem. – Asian J., 2019, 14, 4705–4711 CrossRef CAS PubMed.
- N. J. Leonard, R. C. Fox and M. Oki, J. Am. Chem. Soc., 1954, 76, 5708–5714 CrossRef CAS.
- N. J. Leonard, D. F. Morrow and M. T. Rogers, J. Am. Chem. Soc., 1957, 79, 5476–5479 CrossRef CAS.
- C. Siebler, N. Trapp and H. Wennemers, J. Pept. Sci., 2015, 21, 208–211 CrossRef CAS PubMed.
- P. Wilhelm, B. Lewandowski, N. Trapp and H. Wennemers, J. Am. Chem. Soc., 2014, 136, 15829–15832 CrossRef CAS PubMed.
- R. W. Newberry and R. T. Raines, Chem. Commun., 2013, 49, 7699–7701 RSC.
- R. W. Newberry and R. T. Raines, ACS Chem. Biol., 2014, 9, 880–883 CrossRef CAS PubMed.
- A. Choudhary, K. J. Kamer and R. T. Raines, J. Org. Chem., 2011, 76, 7933–7937 CrossRef CAS PubMed.
- C. Cabezas, J. L. Alonso, J. C. Lopez and S. Mata, Angew. Chem., Int. Ed., 2012, 51, 1375–1378 CrossRef CAS PubMed.
- S. K. Singh, P. R. Joshi, R. A. Shaw, J. G. Hill and A. Das, Phys. Chem. Chem. Phys., 2018, 20, 18361–18373 RSC.
- J. Echeverría, Chem. Commun., 2018, 54, 3061–3064 RSC.
- G. W. Breton, L. O. Davis, K. L. Martin and T. A. Chambers, Cryst. Growth Des., 2019, 19, 3895–3904 CrossRef CAS.
- N. Mercadal, S. P. Day, A. Jarmyn, M. B. Pitak, S. J. Coles, C. Wilson, G. J. Rees, J. V. Hanna and J. D. Wallis, CrystEngComm, 2014, 16, 8363–8374 RSC.
- J. Bella, M. Eaton, B. Brodsky and H. M. Berman, Science, 1994, 266, 75–81 CrossRef CAS PubMed.
- N. Panasik Jr, E. S. Eberhardt, A. S. Edison, D. R. Powell and R. T. Raines, Int. J. Pept. Protein Res., 1994, 44, 262–269 CrossRef CAS PubMed.
- J. A. Hodges and R. T. Raines, Org. Lett., 2006, 8, 4695–4697 CrossRef CAS PubMed.
- E. S. Eberhardt, N. Panasik and R. T. Raines, J. Am. Chem. Soc., 1996, 118, 12261–12266 CrossRef CAS PubMed.
- N. A. Wenzell, H. K. Ganguly, A. K. Pandey, M. R. Bhatt, G. P. A. Yap and N. J. Zondlo, ChemBioChem, 2019, 20, 963–967 CrossRef CAS PubMed.
- L. E. Bretscher, C. L. Jenkins, K. M. Taylor, M. L. DeRider and R. T. Raines, J. Am. Chem. Soc., 2001, 123, 777–778 CrossRef CAS PubMed.
- F. W. Kotch, I. A. Guzei and R. T. Raines, J. Am. Chem. Soc., 2008, 130, 2952–2953 CrossRef CAS PubMed.
- A. Choudhary, D. Gandla, G. R. Krow and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 7244–7246 CrossRef CAS PubMed.
- R. W. Newberry, B. VanVeller, I. A. Guzei and R. T. Raines, J. Am. Chem. Soc., 2013, 135, 7843–7846 CrossRef CAS PubMed.
- M. Kuemin, Y. A. Nagel, S. Schweizer, F. W. Monnard, C. Ochsenfeld and H. Wennemers, Angew. Chem., 2010, 122, 6468–6471 CrossRef.
- J. K. R. Deka, B. Sahariah, S. S. Sakpal, A. K. Bar, S. Bagchi and B. K. Sarma, Org. Lett., 2021, 23, 7003–7007 CrossRef CAS PubMed.
- R. F. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- J. R. Lane, J. Contreras-Garcia, J. P. Piquemal, B. J. Miller and H. G. Kjaergaard, J. Chem. Theory Comput., 2013, 9, 3263–3266 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. T. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. T. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef CAS PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
- L. Monterrosas-Pérez, J. Sandoval-Lira, M. Amador-Ramírez, H. Flores-Segura, J. M. Hernández-Pérez and J. Solano-Altamirano, Struct. Chem., 2020, 31, 305–317 CrossRef.
- M. E. Sanz, A. Lesarri, M. I. Pena, V. Vaquero, V. Cortijo, J. C. Lopez and J. L. Alonso, J. Am. Chem. Soc., 2006, 128, 3812–3817 CrossRef CAS PubMed.
- S. Blanco, J. C. López, S. Mata and J. L. Alonso, Angew. Chem., Int. Ed., 2010, 49, 9187–9192 CrossRef CAS PubMed.
- A. Lesarri, E. J. Cocinero, J. C. Lopez and J. L. Alonso, J. Am. Chem. Soc., 2005, 127, 2572–2579 CrossRef CAS PubMed.
- I. León, E. Alonso, C. Cabezas, S. Mata and J. Alonso, Commun. Chem., 2019, 2, 1–8 CrossRef.
- R. G. Bird, V. Vaquero-Vara, D. P. Zaleski, B. H. Pate and D. W. Pratt, J. Mol. Spectrosc., 2012, 280, 42–46 CrossRef CAS.
- C. Pérez, J. L. Neill, M. T. Muckle, D. P. Zaleski, I. Peña, J. C. Lopez, J. L. Alonso and B. H. Pate, Angew. Chem., 2015, 127, 993–996 CrossRef.
- S. Blanco and J. C. Lopez, J. Phys. Chem. Lett., 2018, 9, 4632–4637 CrossRef CAS PubMed.
- S. Blanco, A. Macario and J. C. Lopez, Phys. Chem. Chem. Phys., 2019, 21, 20566–20570 RSC.
- S. K. Singh, J. K. Vaishnav and A. Das, J. Chem. Phys., 2016, 145, 104302 CrossRef PubMed.
- P. Deb, G. Y. Jin, S. K. Singh, J. Moon, H. Kwon, A. Das, S. Bagchi and Y. S. Kim, J. Phys. Chem. Lett., 2018, 9, 5425–5429 CrossRef CAS PubMed.
- H. Ye, Y. Hai, Y. Ren and L. You, Chem. – Eur. J., 2017, 23, 3804–3809 CrossRef CAS PubMed.
- A. G. Santana, E. Jimenez-Moreno, A. M. Gomez, F. Corzana, C. Gonzalez, G. Jimenez-Oses, J. Jimenez-Barbero and J. L. Asensio, J. Am. Chem. Soc., 2013, 135, 3347–3350 CrossRef CAS PubMed.
- H. Zheng, H. B. Ye, X. X. Yu and L. You, J. Am. Chem. Soc., 2019, 141, 8825–8833 CrossRef CAS PubMed.
- I. W. Windsor, B. Gold and R. T. Raines, ACS Catal., 2019, 9, 1464–1471 CrossRef CAS PubMed.
- E. C. Vik, P. Li, P. J. Pellechia and K. D. Shimizu, J. Am. Chem. Soc., 2019, 141, 16579–16583 CrossRef CAS PubMed.
- A. Mazzanti, L. Lunazzi, M. Minzoni and J. E. Anderson, J. Org. Chem., 2006, 71, 5474–5481 CrossRef CAS PubMed.
- A. Mazzanti, L. Lunazzi, R. Ruzziconi, S. Spizzichino and M. Schlosser, Chem. – Eur. J., 2010, 16, 9186–9192 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2022 |