
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acetylenedicarboxylate as a linker in the engineering of coordination polymers and metal–organic frameworks: challenges and potential
Tobie J.
Matemb Ma Ntep
 *a,
Verena K.
Gramm
b,
Uwe
Ruschewitz
*b and
Christoph
Janiak
a
*a,
Verena K.
Gramm
b,
Uwe
Ruschewitz
*b and
Christoph
Janiak
a
aInstitut für Anorganische Chemie und Strukturchemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstraße 1, D-40225 Düsseldorf, Germany. E-mail: tobie.matemb.ma.ntep@uni-duesseldorf.de
bInstitut für Anorganische Chemie im Department für Chemie, Universität zu Köln, D-50939 Köln, Germany. E-mail: uwe.ruschewitz@uni-koeln.de
First published on 13th July 2022
Abstract
Despite its simplicity as a short and rod-like linear linker, acetylenedicarboxylate (ADC) has for a long time been somewhat overlooked in the engineering of coordination polymers (CPs) and especially in the construction of porous metal–organic frameworks (MOFs). This situation seems to be stemming from the thermosensitivity of the free acid (H2ADC) precursor and its dicarboxylate, which makes the synthesis of their CP- and MOF-derivatives, as well as the evacuation of guest molecules from their pores, challenging. However, an increasing number of publications dealing with the synthesis, structural characterization and properties of ADC-based CPs and MOFs, disclose ways to tackle this obstacle. In this regard, using mostly room temperature solution synthesis or mechanochemical synthesis, and very rarely solvothermal synthesis, the ADC linker has successfully been used to form one-, two-, and three-dimensional CPs with metal cations from almost all groups of the periodic table of the elements, whereby its carboxylate groups adopt mainly all types of known coordination modes. ADC-based CPs feature properties, including negative thermal expansion, formation of non-centrosymmetric networks, long-range magnetic ordering, and solid-state polymerization. The first ADC-based microporous MOFs were obtained with Ce(IV), Hf(IV) and Zr(IV), in which the presence of the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– triple-bond within their backbone results in high hydrophilicity, high CO2 adsorption capacity and enthalpy, as well as the uptake of halogen vapors. This discloses the potential of ADC-MOFs for gas storage/separation and water adsorption-based applications. Furthermore, H2ADC/ADC was discovered to undergo facile in situ hydrohalogenation to yield halogen-functionalized fumarate-based CPs/MOFs. This review surveys investigations on ADC-based coordination polymers and metal–organic frameworks, and is intended to stimulate interest on this linker in chemists working in the fields of crystal chemistry or materials science.
C– triple-bond within their backbone results in high hydrophilicity, high CO2 adsorption capacity and enthalpy, as well as the uptake of halogen vapors. This discloses the potential of ADC-MOFs for gas storage/separation and water adsorption-based applications. Furthermore, H2ADC/ADC was discovered to undergo facile in situ hydrohalogenation to yield halogen-functionalized fumarate-based CPs/MOFs. This review surveys investigations on ADC-based coordination polymers and metal–organic frameworks, and is intended to stimulate interest on this linker in chemists working in the fields of crystal chemistry or materials science.
 Tobie J. Matemb Ma Ntep | Dr Tobie Matemb Ma Ntep received his Bachelor's and Master's degrees in Cameroon at the University of Yaounde-1 and the University of Dschang, respectively. He moved to Germany in 2016 under the DAAD-doctoral scholarship and obtained his PhD in 2019 at the University of Düsseldorf, under the supervision of Prof. Christoph Janiak, working on the design of MOFs for gas adsorption and adsorption-driven heat transformation applications. Since 2021, he has spent one year in the group of Dr Christian Serre at IMAP-Paris, as a PRIME-DAAD postdoctoral fellow, where he started working on the green, large-scale synthesis and shaping of robust MOFs. |
 Verena K. Gramm | Verena K. Gramm received her PhD in the group of Uwe Ruschewitz at the University of Cologne in 2016. Her research focused on coordination polymers with acetylenedicarboxylate as the linker. In particular, structure–property relationships of anhydrous acetylenedicarboxylates as well as coordination polymers with rare earth metals were her specific research interests. Since 2017, she has been working in the chemical industry. |
 Uwe Ruschewitz | Uwe Ruschewitz received his PhD at Technical University Aachen (Germany). After postdoctoral studies with A. K. Cheetham at the UCSB in Santa Barbara he returned to Aachen, where he finished his habilitation in 2000. In the same year, he was appointed as a Professor of Inorganic Chemistry (C3) at the University of Cologne. His interests are in the field of synthetic inorganic solid-state chemistry focusing on ionic carbides, acetylenedicarboxylates, MOFs with fluorinated linkers and as hosts for photochromic dyes. His group has a strong expertise in solving and refining crystal structures from powder diffraction data. |
 Christoph Janiak | Christoph Janiak is a full professor at the Heinrich-Heine-University of Düsseldorf since 2010, with research interests in porous materials (e.g., MOFs), mixed-matrix membranes, metal nanoparticles, ionic liquids and lately electrocatalysis for water splitting. Until 2018 he was a visiting professor at Wuhan University of Technology and, currently, he is a guest professor at the Hoffmann Institute of Advanced Materials at Shenzhen Polytechnic in China. He has (co-)authored about 600 research papers and is a Fellow of the Royal Society of Chemistry (FRSC). |
1 Introduction
1.1 Linear organic linkers in coordination polymers and metal–organic frameworks
Coordination polymers (CPs) and metal–organic frameworks (MOFs) are coordination-based compounds, which are obtained by connecting metal centers or metal-containing clusters with polytopic organic ligands also known as linkers.1–3 The span of structural diversity and interesting properties featured by CPs and MOFs has made this class of materials the focus of intense research investigations for their potential application in an array of fields including hydrogen and methane storage,4–6 carbon dioxide capture,7 gas separation,8 catalysis,9 adsorption-driven heat transformation,10,11 water harvesting from air,12 and sensing.13,14 Besides the important role of the metal center in CPs and MOFs, the connectivity, geometry, size, and rigidity/flexibility of the linker also play a decisive role in defining the topology, dimensionality, and pore metrics of the obtained framework, while its functionality directs to the properties of the produced material.15,16 In this regard, linear ditopic linkers of different lengths and especially dicarboxylates have played a major role in the development of the rational design and synthesis of these materials, a concept which is also referred to as reticular chemistry.17,18 Most isoreticular MOF families including IRMOF, UiO and MIL-53 series are constructed from metal cluster nodes, the so-called secondary building units (SBUs), with linear dicarboxylate linkers such as terephthalate (benzene-1,4-dicarboxylate, BDC), fumarate (Fum), biphenyl-4,4′-dicarboxylate (BPDC), naphthalene-2,6-dicarboxylate (NDC), 2,2′-bipyridine-5,5′-dicarboxylate (BPyDC), etc. (Scheme 1).19–21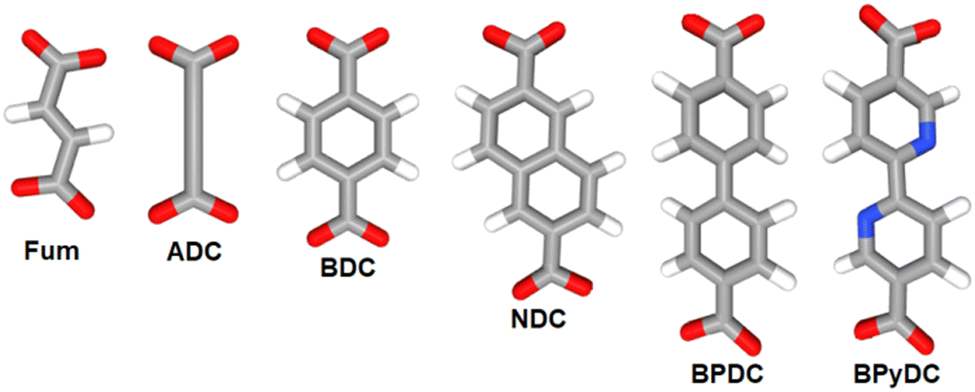 | ||
| Scheme 1 Some linear dicarboxylate linkers used in the construction of CPs and MOFs. Each of these linkers has a doubly-negative charge. Better designations would be Fum2−, ADC2−, etc. For better readability we omit the charge in the text. | ||
In this context, acetylenedicarboxylate (ADC) is the simplest and shortest straight linear alkyne-based dicarboxylate linker. However, for a long time ADC has surprisingly been used only to a comparatively small extent in the engineering of CPs and especially MOFs. A search in the MOF-subset22–24 of the Cambridge Crystallographic Database (CSD, Version 5.43 November 2021) shows 3039 entries of MOFs/CPs with the linker BDC, 652 entries with BPDC, 551 entries with Fum and only 105 entries of MOFs/CPs with ADC as the linker. The reason for the low occurrence of ADC-based CPs and MOFs might be understood from the physico-chemical properties, in particular the low thermal stability of acetylenedicarboxylic acid, which makes its use in hydro-/solvothermal syntheses difficult,25 as outlined in the next section.
1.2 Physical and chemical properties of acetylenedicarboxylic acid
Acetylenedicarboxylic acid or butynedioic acid (H2ADC) is an alkyne-based dicarboxylic acid of formula H2C4O4 or HOOC–CC–COOH. H2ADC can be partially dissociated into its monovalent anion hydrogenacetylenedicarboxylate, HC4O4− (HADC), or into the acetylenedicarboxylate dianion, C4O42− (ADC) (Scheme 2). The two dissociation constants in water are pKa1 = 0.656 and pKa2 = 2.336 at 25 °C, respectively, which classify H2ADC as a medium to strong acid (cf. pKa(HSO4−) = 1.96, pKa(H3PO4) = 2.16, and pKa(HF) = 3.18).26 H2ADC and its deprotonated forms have been reported to undergo slow decarboxylation in aqueous solutions to propiolic acid. The decarboxylation rate increases with temperature (Scheme 2) and is facilitated by the fact that the –COOH groups are bound to the unsaturated acetylenic carbon atoms. Remarkably, the mono-deprotonated hydrogenacetylenedicarboxylate monoanion decarboxylates faster than acetylenedicarboxylic acid, while the doubly deprotonated acetylenedicarboxylate dianion decarboxylates slower than the free acid.27
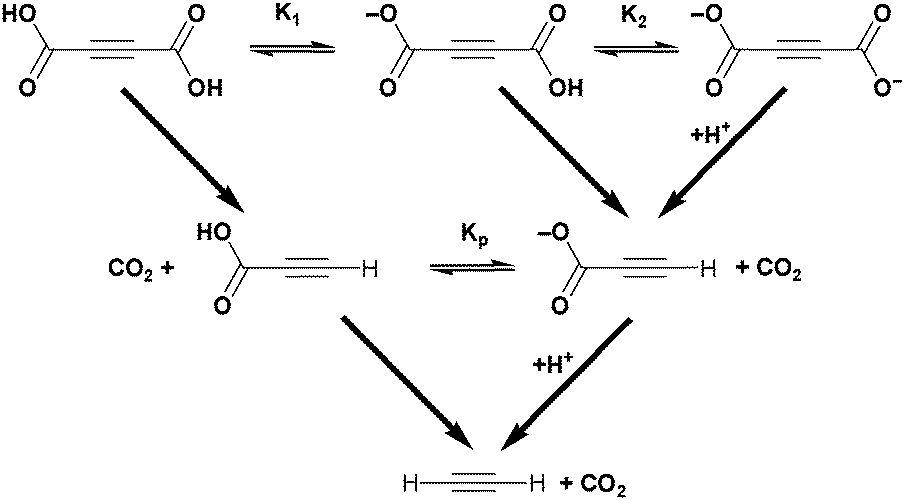 | ||
| Scheme 2 Dissociation and decarboxylation reactions of acetylenedicarboxylic acid and its ions. | ||
H2ADC can be synthesized in a two-step procedure consisting of a twofold elimination of hydrogen bromide from meso-dibromosuccinic acid, when treated with a concentrated ethanolic solution of potassium hydroxide. The obtained dipotassium acetylenedicarboxylate is then protonated by a concentrated sulfuric acid solution to yield a white to beige-colored microcrystalline powder of acetylenedicarboxylic acid (Scheme 3).28 In another method, H2ADC is obtained by reacting acetylene with carbon dioxide in the presence of a silver or cupric salt and an amine base.29
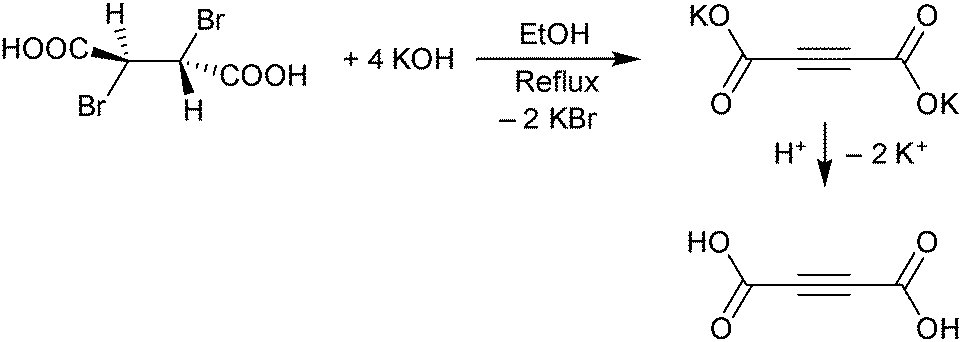 | ||
| Scheme 3 Reaction for the synthesis of H2ADC from meso-dibromosuccinic acid. | ||
H2ADC is highly soluble in water, alcohols and most organic solvents. The acid melts at about 191 °C, and then immediately starts to decompose (Fig. 1(a) and (b)).30 Its 13C NMR spectrum shows two peaks at about 156 ppm and 76 ppm, corresponding to the carbon atom of the carboxyl group and the acetylenic part, respectively (Fig. 1(c)).31 The CC bond stretching vibration is symmetry forbidden in IR spectroscopy and displays therefore no band in the IR spectrum (Fig. 1(d)).32 The triple bond is revealed by Raman spectroscopy, which displays a characteristic strong band at about 2225 cm−1 (Fig. 1(e)).33 At present, the origin of the two bands for the triple bond stretch is not fully clear. A suggestion was put forward that, since the compound is very hygroscopic, partial water absorption with hydrogen bonding to the –COOH groups will change the CC stretching frequency.34
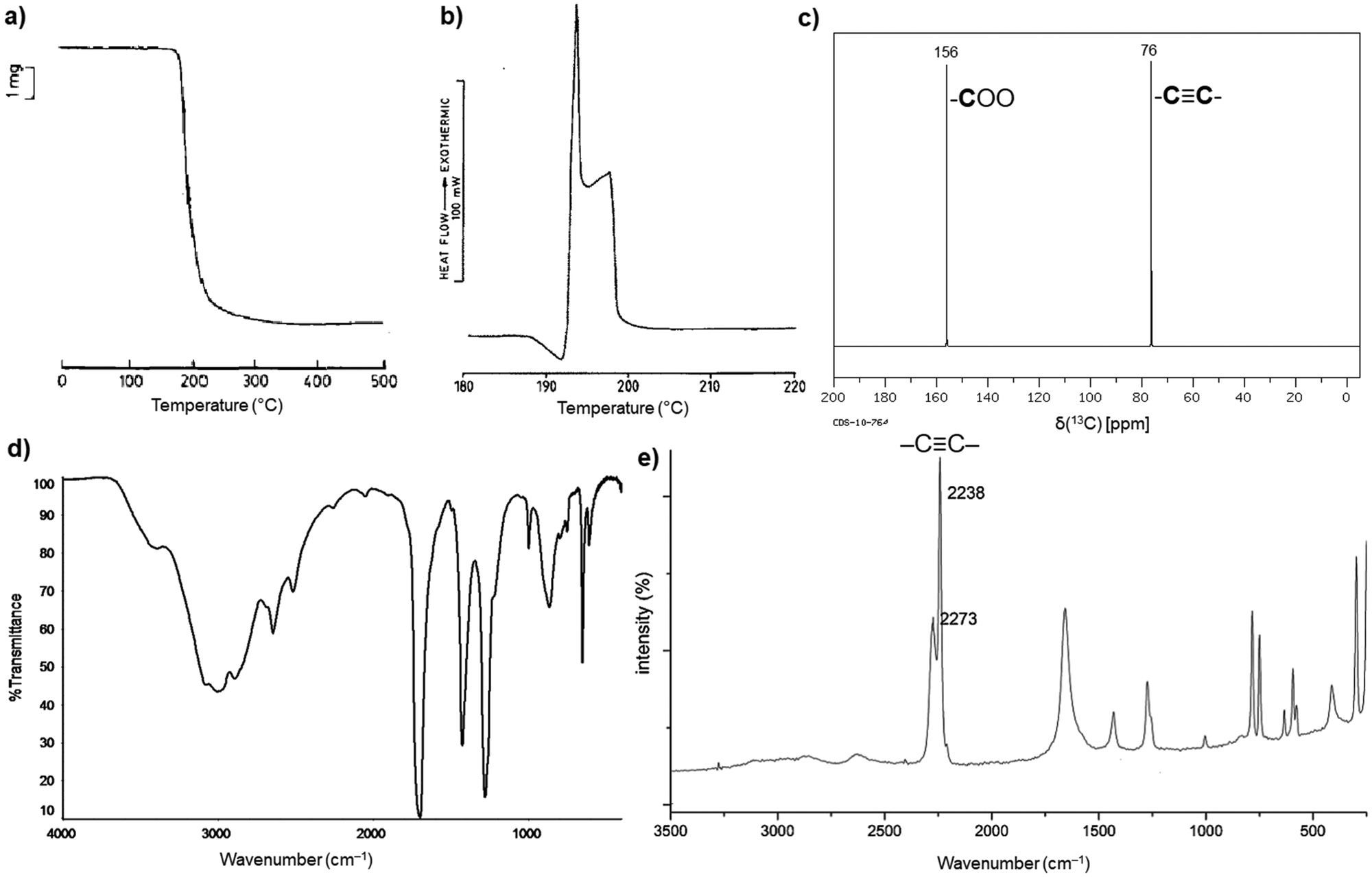 | ||
| Fig. 1 (a) Thermogravimetric curve under air, (b) differential scanning calorimetry (DSC) curve, (c) 13C NMR spectrum in D2O, (d) FT-IR spectrum and (e) Raman spectrum of H2ADC. Adapted from ref. 30–33 with permission of Elsevier, copyright 1987; copyright 2014 and permission of John Wiley & Sons, copyright 2021. | ||
With its short, rigid, linear, hydrogen-free C4 carbon backbone with d(C–C–C–C) of about 4.1 Å, acetylenedicarboxylate is among the simplest linear carboxylate-based ligands that could be used to construct CPs and MOFs. Its two carboxylate groups can adopt all relative torsion angles ranging from coplanar to perpendicular arrangements (Fig. 2(A)), while adopting various coordination fashions known for carboxylates (Fig. 2(B)).35,36 The oxalate dianion is an even simpler linker, but adopts particular coordination modes (mostly chelating, perpendicular to the C–C axis), which do not yield isoreticular MOFs.37,38
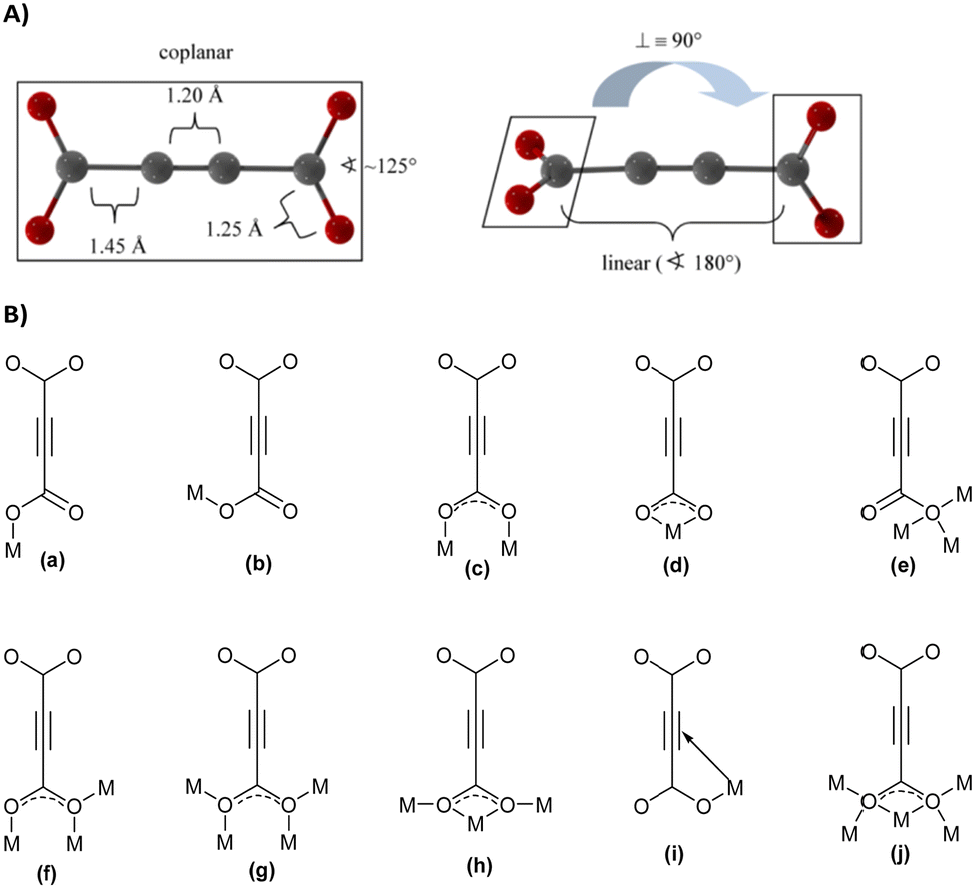 | ||
| Fig. 2 (A) Two possible conformations of the acetylenedicarboxylate linker. Reproduced from ref. 50 with permission of Wiley & Sons, copyright 2020. (B) Types of coordination modes of carboxylate groups in the acetylenedicarboxylate dianion to metal cations (Mn+) (for simplicity, only the coordination of one carboxylate is depicted; the two carboxylate groups can differ in their coordination modes): (a) monodentate syn η1:η0 mode, (b) monodendate anti η1:η0 mode, (c) bidendate syn–syn μ2-η1:η1 mode, (d) chelating η1:η1 mode, (e) monodentate μ3-η3:η0 mode (f) bridging bidentate μ3-η1:η2 mode, (g) bidentate bridging μ4-η2:η2 mode, (h) chelating bridging μ3-η2:η2 mode, (i) monodentate π-complex, and (j) chelating bridging μ5-η3:η3. | ||
1.3 Scope of this review
The present review is a survey of coordination polymers and metal–organic frameworks, based on the ADC linker. Particular attention has been given to the synthesis conditions and challenges to form and isolate such ADC-based networks, as well as structural features and distinct properties emanating from the presence of ADC in the materials. Our focus is on reports of homoleptic CPs and MOFs containing ADC as the sole organic linker. ADC-based molecular complexes, supramolecular networks, and the about 35 ADC-based networks containing additional linkers are therefore out of the scope of this work. The first part is dedicated to non-porous ADC-based coordination polymers, which we have classified according to the element group of the incorporated metal cation. The second part presents ADC-based metal–organic frameworks, first with potential porosity and second with experimentally assessed porosity. For the latter, sorption data of corresponding materials are provided. The third part presents the specific properties of ADC-based CPs and MOFs, which are derived from the presence of the ADC linker, that is, its shape and the presence of an acetylenic function within its backbone. Finally, the conclusion provides guidelines for future studies with this linker in the context of materials and structural chemistry.2 Non-porous acetylenedicarboxylate-based coordination polymers
Coordination polymers based on the ADC linker have been well documented with compounds constructed by using metals from all groups of the periodic table of the elements. The first coordination polymers (CPs) with ADC as the linker were published by Robl and Hentschel in 1990.39,40 Afterward, numerous publications on CPs with this linker were presented by one of our research groups (the Ruschewitz group), which did a systematic investigation of ADC's coordination behavior with metal cations of all elements found in the periodic table. In all these studies to which other groups also contributed, including results using monovalent, divalent, trivalent (Bi(III)) and hexavalent (U(VI)) metals, the formation of non-porous CPs was basically observed. Only for the divalent Zn(II) cation was the successful synthesis of porous MOFs reported for the first time in these early investigations (vide infra).2.1 Non-porous ADC-based CPs with alkali metal cations
The monovalent alkali metals Li+, Na+ and K+ have been used to generate ADC-based coordination polymers. The three reported ADC-based CPs with alkali metals were obtained by reacting H2ADC with LiOH, Na2CO3, and KOH, respectively, in water at room temperature, followed by slow evaporation of the solvent to yield single crystals of the compounds with formulae [Li2(ADC)(H2O)2], [Na2(ADC)(H2O)4] and [K2(ADC)(H2O)], respectively.41,42 In the crystal structure of [Li2(ADC)(H2O)2], there are two crystallographically independent Li atoms both coordinated by four oxygen atoms forming distorted LiO4 tetrahedra. One Li atom is coordinated by one water molecule and three carboxylate groups of three different ADC ligands, whereas the other Li atom is coordinated by two water molecules and two carboxylate groups of two different ADC ligands. One of these water molecules exhibits a terminating coordination, while the other bridges two Li atoms (Fig. 3(a)). Each ADC ligand bridges five Li atoms, whereby one of its carboxylates bridges two Li atoms in a syn–anti-μ2-η1:η1 mode and the other carboxylate bridges three Li atoms in a μ3-η2:η1 mode (Fig. 3(b)). Thus, four corner-sharing LiO4 tetrahedra form tetranuclear ring units (Fig. 3(a)), which are interconnected by ADC linkers into a three-dimensional framework structure (Fig. 3(c) and (d)).41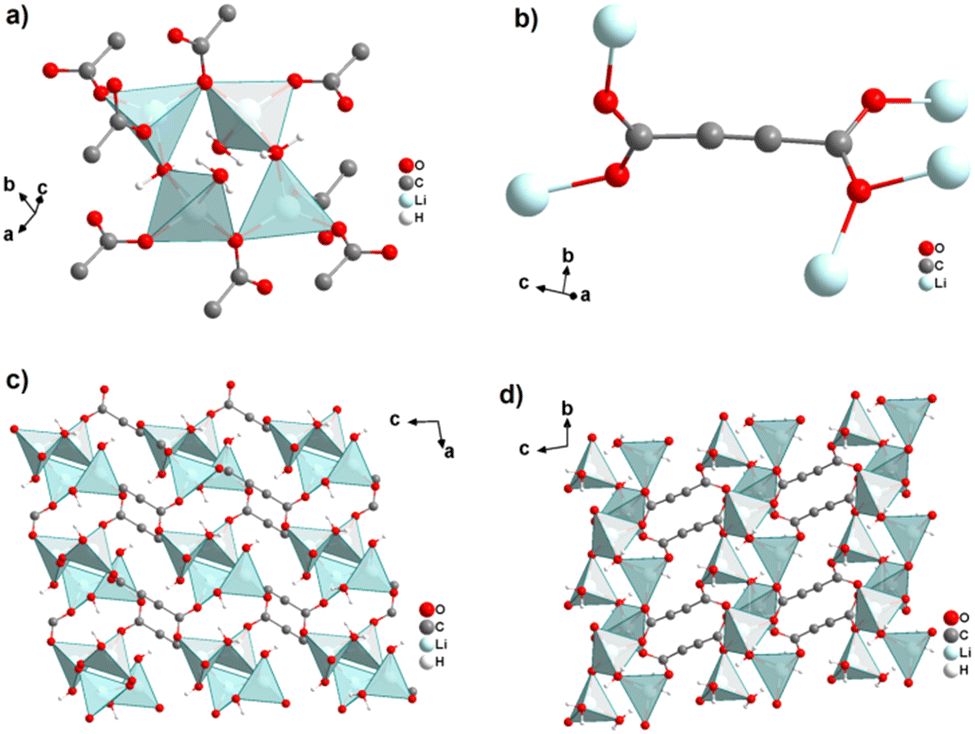 | ||
| Fig. 3 Crystal structure of hydrated lithium acetylenedicarboxylate [Li2(ADC)(H2O)2] showing: (a) the tetranuclear ring unit of corner-sharing LiO4 tetrahedra, (b) the coordination mode of the ADC linker and (c) and (d) the three-dimensional packing of the framework viewed along the [010] and [100] directions, respectively. Graphics redrawn from the cif file (CSD-Refcode ICUDOC).41 | ||
Interestingly, heating [Li2(ADC)(H2O)2] at 140 °C for 2 h yields the new coordination polymer [Li2(ADC)] as the first reported anhydrous alkali metal acetylenedicarboxylate. [Li2(ADC)] has an entirely different structure compared to [Li2(ADC)(H2O)2], with the Li atom being in this case coordinated by four oxygen atoms of four carboxylate groups from four different ADC linkers (Fig. 4(a)). Each ADC ligand bridges eight Li atoms, that is, each of its carboxylates coordinates in the μ4-η2:η2 mode. Each oxygen atom connects two Li atoms, thus forming chains of edge-sharing LiO4 tetrahedra (Fig. 4(b)). These chains are arranged in layers alternatively perpendicular to each other (Fig. 4(c)), which are connected by ADC linkers into a three-dimensional framework (Fig. 4(d)). [Li2(ADC)] is thermally stable up to 360 °C.41
 | ||
| Fig. 4 Crystal structure of anhydrous lithium acetylenedicarboxylate [Li2(ADC)] showing: (a) the coordination environment of the Li atom, (b) the chains of infinite edge-sharing LiO4 tetrahedra, (c) the coordination mode of ADC connecting the alternatively perpendicular chains and (d) the three-dimensional packing of the framework viewed along the [100] direction. Graphics redrawn from the cif file (CSD-Refcode ICUHIA).41 | ||
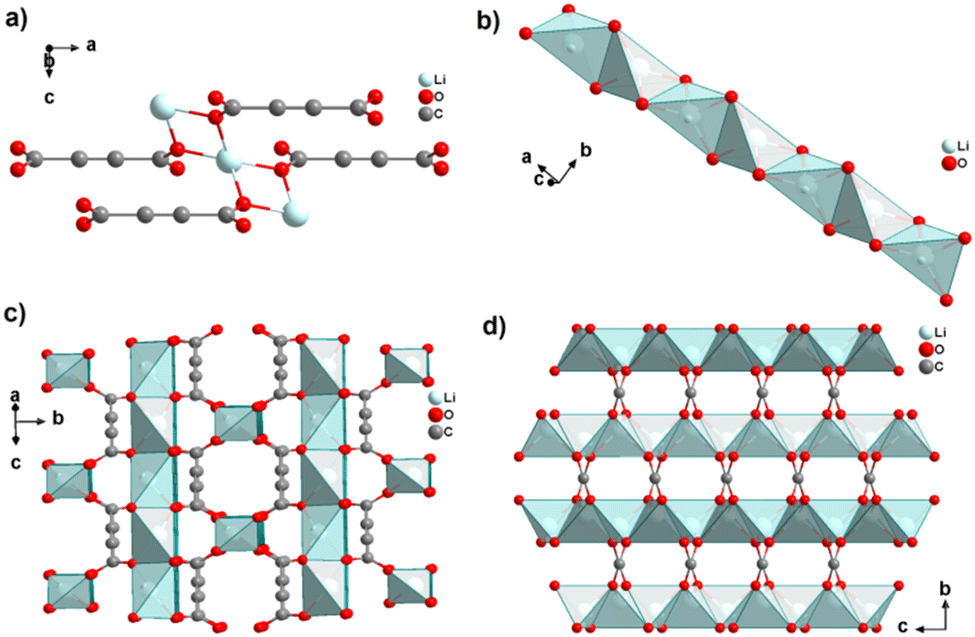
In the structure of [Na2(ADC)(H2O)4], the sodium atom is coordinated octahedrally by three water molecules (O3, O4) and three oxygen atoms (O21, O22) of carboxylate linkers from three different ADC linkers (Fig. 5(a)). Two water molecules (O3) bridge two Na atoms each, while the third water (O4) molecule exhibits a terminating coordination. One oxygen atom of the carboxylate group of ADC (O22) also bridges three Na atoms, whereas the second oxygen atom of the carboxylate group (O21) is non-coordinating. That is, ADC coordinates in a μ3-η3:η0 mode with each of its carboxylate groups (cf.Fig. 2B(e)), which results in NaO6 octahedra being interconnected into chains along the b-axis (Fig. 5(b)). These chains are linked by ADC into sheets, which are held together by hydrogen bonds (Fig. 5(c) and (d)).42
 | ||
| Fig. 5 Crystal structure of hydrated sodium acetylenedicarboxylate [Na2(ADC)(H2O)4] showing: (a) the coordination environment of the Na atom, (b) the coordination mode of ADC linkers and water oxygen atoms, and (c) and (d) sheets from chains of NaO6 octahedra connected by ADC linkers. Graphics redrawn from the cif file (CSD-Refcode XADSAX).42 | ||
In the structure of [K2(ADC)(H2O)], there are two crystallographically distinct potassium atoms, which are both heptacoordinate. One K atom is coordinated by seven oxygen atoms from a water molecule and five carboxylate groups of five different ADC ligands, of which one carboxylate coordinates in a chelating mode (Fig. 6(a)). The other K atom is coordinated by two water molecules and four carboxylate groups of four different ADC anions, of which again one carboxylate group coordinates in a chelating fashion (Fig. 6(b)). Overall, each water molecule bridges three K atoms and each ADC ligand coordinates in a μ5-η3:η3 mode with one carboxylate and in a μ4-η3:η2 mode with the other carboxylate function (Fig. 6(c)), thus yielding a complex three-dimensional framework structure (Fig. 6(d)).42
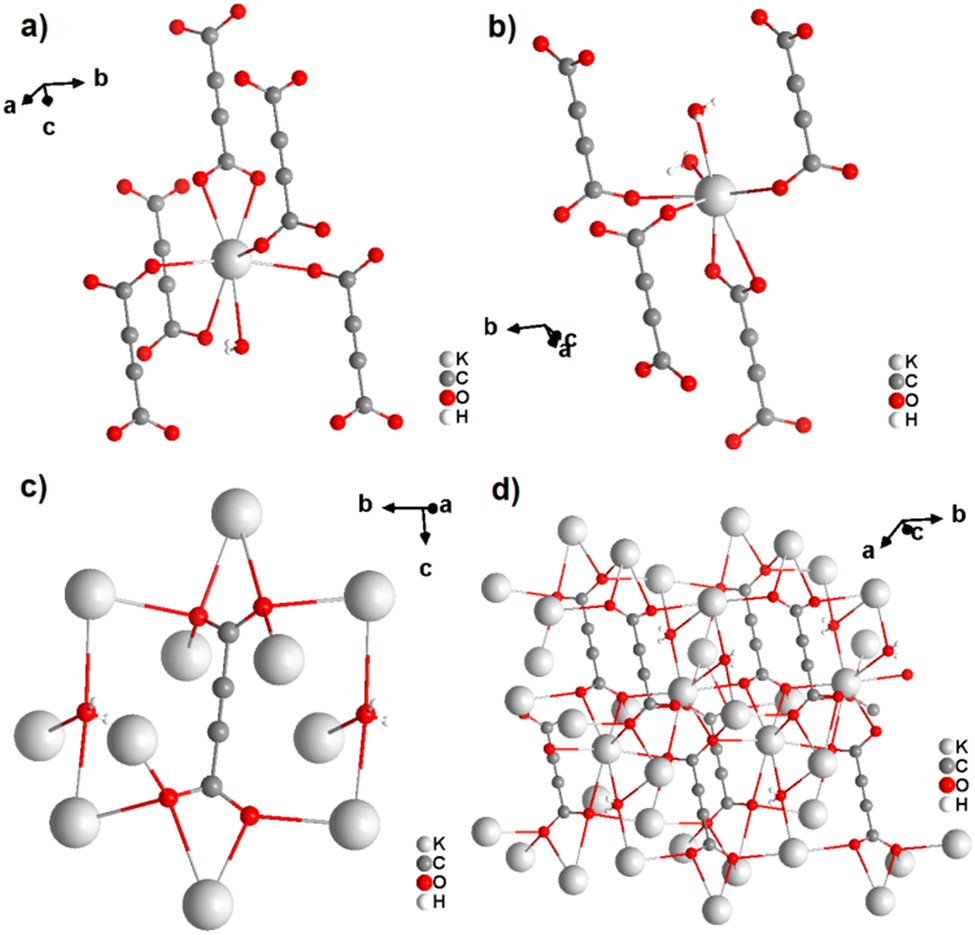 | ||
| Fig. 6 Crystal structure of hydrated potassium acetylenedicarboxylate [K2(ADC)(H2O)] showing: (a) and (b) the coordination environment of the two crystallographically distinct K atoms, (c) the coordination mode of the ADC linker and water molecules, and (d) the complex three-dimensional framework structure. Graphics redrawn from the cif file (CSD-Refcode XADRUQ).42 | ||
2.2 Non-porous ADC-based CPs with alkaline-earth metal cations
With the exception of radium, ADC-based compounds containing alkaline-earth metals have been reported for all elements in this group. Beryllium yielded a supramolecular network BeADC·4H2O in which [Be(H2O)4]2+ tetrahedra and planar ADC anions are connected by strong asymmetric hydrogen bonds. This compound will not be discussed further, as supramolecular networks are out of the scope of this review.43 Magnesium, calcium, strontium and barium yielded ADC-based CPs of respective formulae [Mg(ADC)(H2O)2], [Ca(ADC)], [Sr(ADC)] and [Ba(ADC)(H2O)].39,44,45A hydrated magnesium acetylenedicarboxylate CP [Mg(ADC)(H2O)2] was obtained via a mechanochemical approach by grinding Mg(CH3COO)2·4H2O with H2ADC in an agate mortar.45 [Mg(ADC)(H2O)2] was obtained as a microcrystalline powder so its structure was solved and refined from the powder X-ray diffraction data (PXRD). It should be noted at this point that the mechanochemical synthesis is a very straightforward method to obtain ADC-based compounds, since the possible thermally induced decarboxylation of acetylenedicarboxylic acid by heating in solution is thereby avoided.46–48
The Mg cation in [Mg(ADC)(H2O)2] is coordinated octahedrally by six oxygen atoms stemming from two water molecules and the carboxylate groups of four ADC linkers (Fig. 7(a)). The water molecules adopt a terminating coordination, while each carboxylate of the ADC linker bridges two Mg atoms in a bidentate syn–anti-μ2-η1:η1 mode (Fig. 7(b)) to yield a three-dimensional framework structure (Fig. 7(c) and (d)). The combined TGA and DTA revealed that the framework loses its crystal water at ∼150 °C and decomposition of the framework started at about 200 °C.45
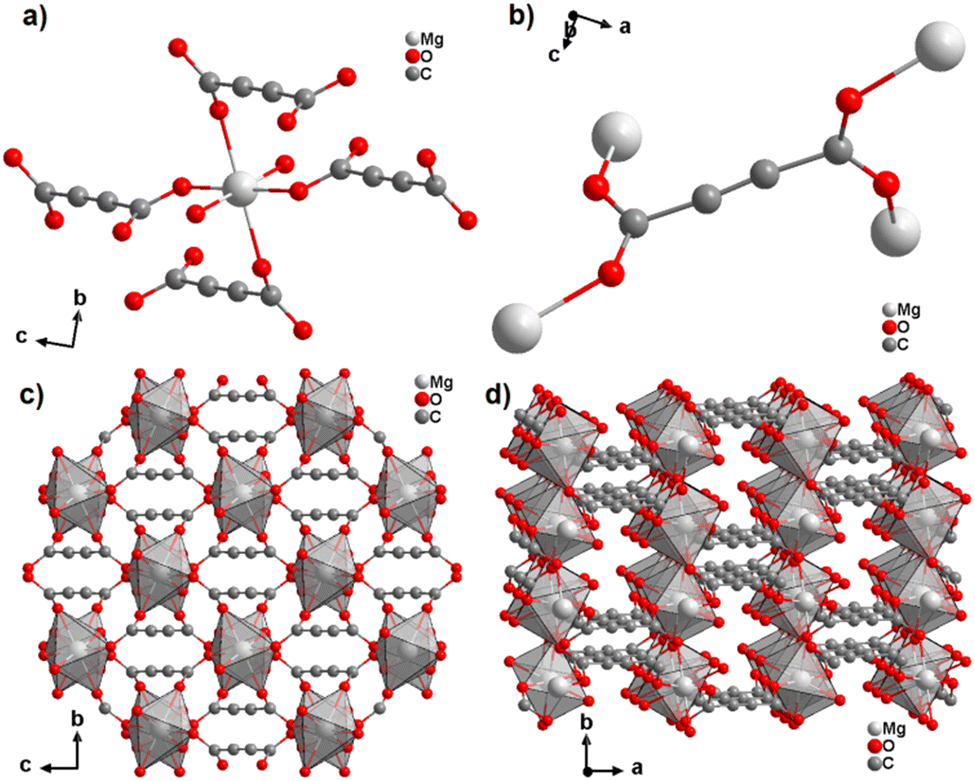 | ||
| Fig. 7 Crystal structure of magnesium acetylenedicarboxylate [Mg(ADC)(H2O)2] showing: (a) the coordination environment of the Mg atom, (b) the coordination mode of the ADC linker, and (c) and (d) the three-dimensional packing of the framework viewed along the [100] and [001] directions, respectively. Graphics redrawn from the cif file (Refcode GUWWOL).45 | ||
Anhydrous calcium acetylenedicarboxylate of formula [Ca(ADC)] was obtained via a similar mechanochemical approach by grinding Ca(CH3COO)2 with H2ADC in an agate mortar. Interestingly, [Ca(ADC)] is isotopic to [Sr(ADC)], which will be described in detail below. Their structures consist of three-dimensional frameworks, in which the Ca and Sr atoms are arranged in a diamond-like topology.45 Unlike [Sr(ADC)], [Ca(ADC)] features a very low thermal stability and starts decomposing already at ∼50 °C.
Strontium acetylenedicarboxylate of formula [Sr(ADC)] was the first reported anhydrous ADC-based CP.44 Single crystals of [Sr(ADC)] formed at the phase boundary of an aqueous silica gel containing H2ADC and an aqueous solution of SrCl2. In the crystal structure of [Sr(ADC)], the Sr atom is eightfold-coordinated by eight oxygen atoms of the carboxylate groups of six ADC linkers with two of them coordinating in a chelating-bidentate fashion (Fig. 8(a)). Each ADC linker bridges six Sr atoms with their two carboxylate functions being perpendicular to each other and coordinating in a chelating, bridging μ3–η1:η2:η1 mode (Fig. 8(b)). The resulting structure consists of a three-dimensional framework, in which all ADC anions are aligned along the [001] direction of the tetragonal unit cell. The Sr atoms are arranged in a diamond-like topology (Fig. 8(c) and (d)). Remarkably, [Sr(ADC)] displayed a very high thermal stability up to approx. 450 °C, which is very intriguing for a compound based on the thermally labile ADC ligand. No clear explanation for this observation has been given up to now.44
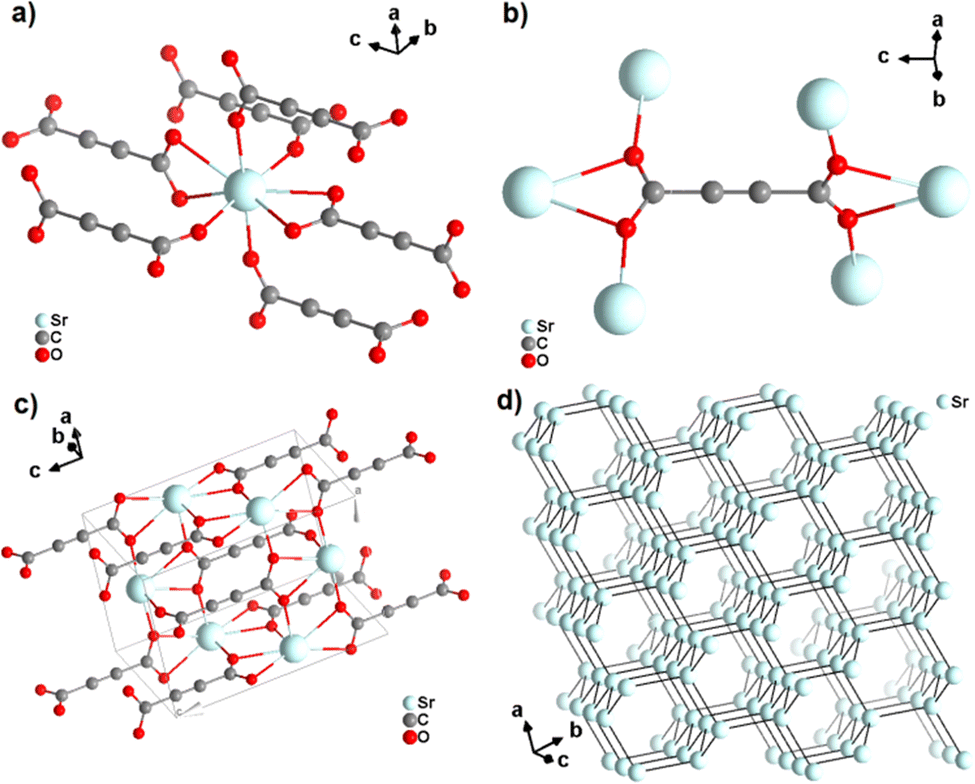 | ||
| Fig. 8 Crystal structure of anhydrous strontium acetylenedicarboxylate [Sr(ADC)] showing: (a) the coordination environment of the Sr atom, (b) the coordination mode of the ADC linker, (c) the three-dimensional packing of the framework, and (d) the diamond-like arrangement of the strontium atoms. Graphics redrawn from the cif file (CSD-Refcode XUKKET01).44 | ||
[Ba(ADC)(H2O)] was the first alkaline-earth metal acetylenedicarboxylate CP, reported by Robl et al. in 1991.39 Single crystals were obtained from slow diffusion when an aqueous solution of Ba(NO3)2 and H2ADC was overlaid with isopropanol at room temperature. Crystals grew at the phase boundary of these two solutions. In the crystal structure of [Ba(ADC)(H2O)], the Ba atom is nine-fold-coordinated with oxygen atoms from two water molecules and six carboxylate groups stemming from six different ADC linkers, of which one carboxylate group is coordinating in a chelating mode (Fig. 9(a)).
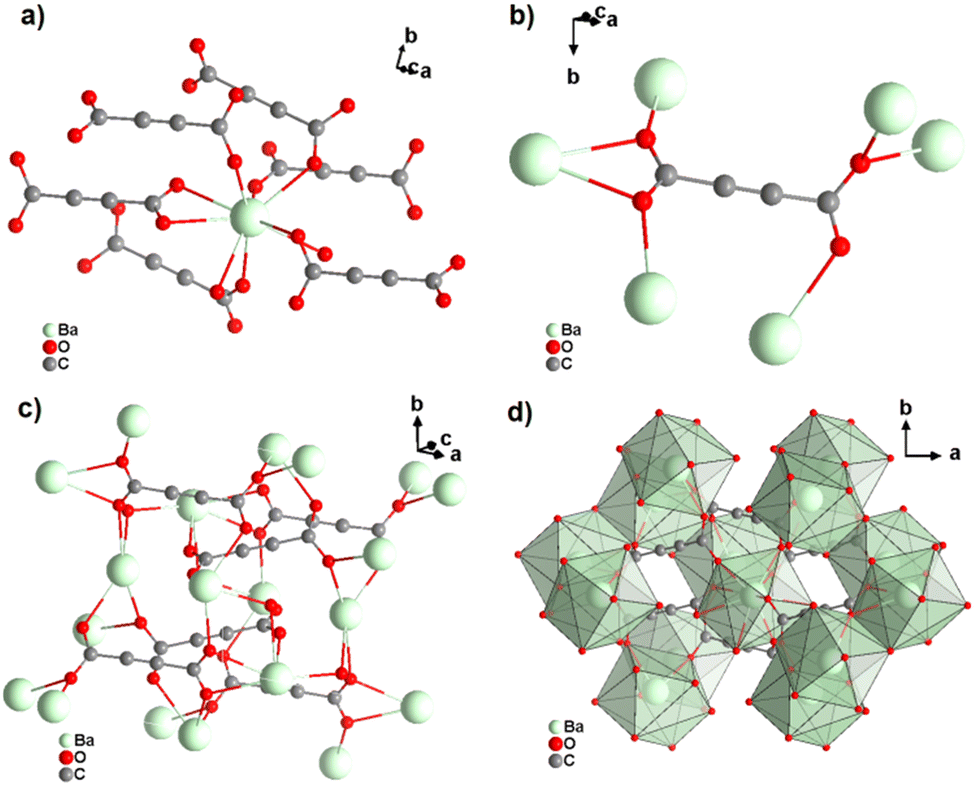 | ||
| Fig. 9 Crystal structure of hydrated barium acetylenedicarboxylate [Ba(ADC)(H2O)] showing: (a) the coordination environment of the Ba atom, (b) the coordination mode of the ADC linker and (c) and (d) the three-dimensional packing of the framework and the net of edge-sharing polyhedra, respectively. Graphics redrawn from the cif file (CSD-Refcode KIYJOR).39 | ||
Each water molecule bridges two Ba atoms, while the ADC linker bridges three Ba atoms with one of its carboxylate functions in a chelating, bridging μ3-η1:η2:η1 mode and three further Ba atoms with the other carboxylate group in a μ3-η1:η2 mode, making the ADC linker bridge six Ba atoms in total (Fig. 9(b)). This results in an intricate three-dimensional framework made up of infinite chains of edge-sharing BaO9 polyhedra connected by ADC linkers (Fig. 9(c) and (d)).
The thermal stability of [Ba(ADC)(H2O)] was not clearly stated. The authors only described the differential thermal analysis (DTA), which showed an endothermic signal starting at about 100 °C with a maximum of 160 °C. This was attributed to the release of crystal water molecules, suggesting the probable formation of an anhydrous [Ba(ADC)] CP. However, there was apparently no attempt to investigate the possible formation of such an anhydrous [Ba(ADC)] CP in this work. The authors also mentioned two successive exothermic peaks occurring at higher temperatures up to 500 °C without providing detailed thermogravimetric or DTA data. The residue after heating to 500 °C was identified as BaCO3.39 A few years later, it was shown that anhydrous [Ba(ADC)] crystallizes isotypically to [Sr(ADC)], whose crystal structure was described above.49
2.3 Non-porous ADC-based CPs with rare-earth metal and actinide cations
The only reported CP of ADC with a divalent rare-earth metal cation is anhydrous europium(II) acetylenedicarboxylate of formula [Eu(ADC)].50 [Eu(ADC)] is isostructural to [Sr(ADC)], that is, its structure consists of a three-dimensional framework, in which the EuII atoms are arranged in a diamond-like topology (vide supra). To date, all compounds with the general composition [MII(ADC)] crystallize in the SrADC-type structure, in which MIIO8 polyhedra are connected by ADC linkers with both carboxylate groups coordinating in a chelating, bridging μ3-η1:η2:η1 mode. Each MIIO8 polyhedron shares edges with four neighbouring MIIO8 polyhedra (Fig. 10(a)), while the carbon backbones of all connecting ADC linkers are aligned parallel to each other along the [001] direction of the tetragonal unit cell, yielding a 3D framework structure, whose channels are filled with the carbon atoms of the ADC linkers (Fig. 10(b)).50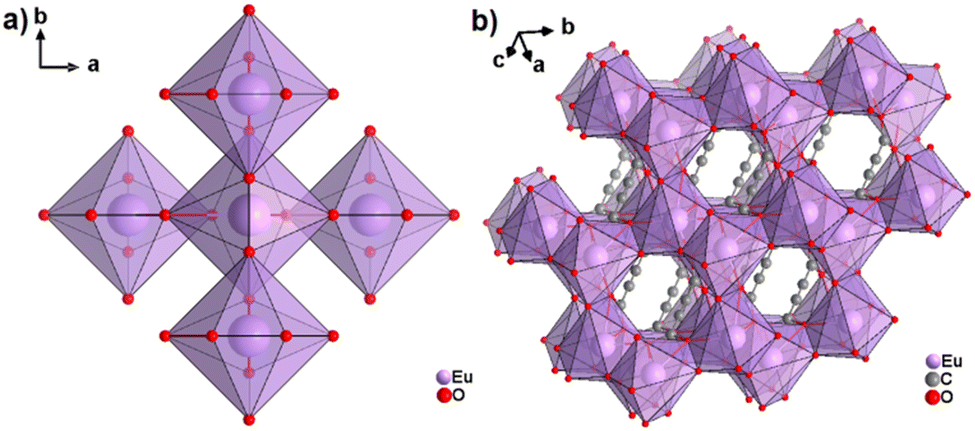 | ||
| Fig. 10 (a) MIIO8 polyhedra and four edge-sharing neighbouring analogues, and (b) the three-dimensional packing of polyhedra and linkers found in the structure of ADC-based CPs with the formula [MII(ADC)], exemplified here by [EuII(ADC)]. Graphics redrawn from the cif file (CSD-Refcode IHUCUM).50 | ||
Unlike [Sr(ADC)] and [Ca(ADC)], anhydrous [Eu(ADC)] was synthesized entirely in aqueous solutions containing EuBr2 and either K2ADC or H2ADC at room temperature. The former was obtained by grinding K(CH3COO) and H2ADC (ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in an agate mortar. Degassed water was used and the reaction was conducted under inert conditions (argon atmosphere) to prevent the oxidation of EuII to EuIII. Very interestingly, [Eu(ADC)] also displays a relatively high thermal stability like [Sr(ADC)] with a decomposition temperature of 440 °C in argon (450 °C for [Sr(ADC)]).44 This seems to be a trend of ADC-based CPs with the diamond-like arrangement of metal cations. However, the low thermal stability of [Ca(ADC)] is in sharp contrast to these findings. Furthermore, [Eu(ADC)] features a surprisingly high chemical stability for a EuII-containing compound, manifested by no degradation after exposure to humid air for several months.50 Raman spectroscopic analyses of both [Sr(ADC)] and [Eu(ADC)] were conducted to attest the presence of the ADC linker in these compounds (Fig. 11),50 and therefore ruling out a possible in situ transformation during CP formation of the ADC linker e.g. into a fumarate-like linker via hydrohalogenation (vide infra).
:1) in an agate mortar. Degassed water was used and the reaction was conducted under inert conditions (argon atmosphere) to prevent the oxidation of EuII to EuIII. Very interestingly, [Eu(ADC)] also displays a relatively high thermal stability like [Sr(ADC)] with a decomposition temperature of 440 °C in argon (450 °C for [Sr(ADC)]).44 This seems to be a trend of ADC-based CPs with the diamond-like arrangement of metal cations. However, the low thermal stability of [Ca(ADC)] is in sharp contrast to these findings. Furthermore, [Eu(ADC)] features a surprisingly high chemical stability for a EuII-containing compound, manifested by no degradation after exposure to humid air for several months.50 Raman spectroscopic analyses of both [Sr(ADC)] and [Eu(ADC)] were conducted to attest the presence of the ADC linker in these compounds (Fig. 11),50 and therefore ruling out a possible in situ transformation during CP formation of the ADC linker e.g. into a fumarate-like linker via hydrohalogenation (vide infra).
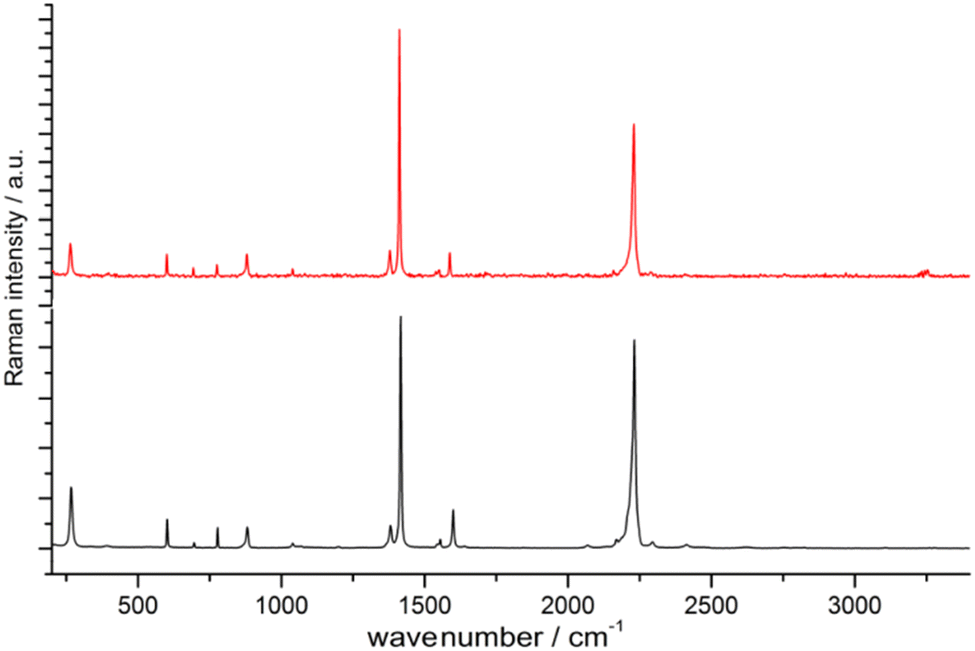 | ||
| Fig. 11 Raman spectra of [EuII(ADC)] (red) and [SrII(ADC)] (black), both displaying the strong bands of –CC–stretching vibrations of ADC at ∼2250 cm−1. Reproduced from ref. 50 with permission of Wiley & Sons, copyright 2020. | ||
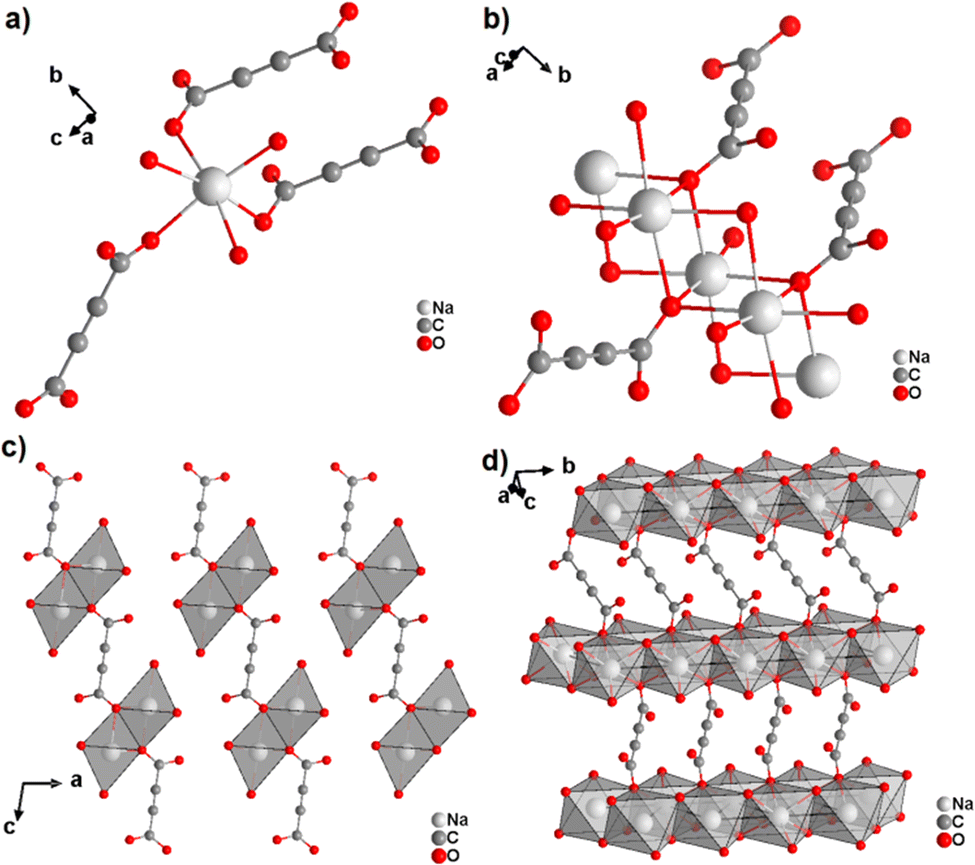
The only two reported ADC-based CPs with actinides are based on uranium(VI). K(H5O2)[UO2(ADC)2(H2O)]·2H2O and Cs2[UO2(ADC)2(H2O)]·2H2O were obtained as single crystals from aqueous solutions containing UO3, H2ADC and additionally K2CO3 and CsCO3, respectively.51 Both compounds are built of anionic [UO2(ADC)2(H2O)]2− polymeric units, in which the uranium coordination polyhedron is a pentagonal bipyramid consisting of a uranyl ion (UO22+), whose equatorial plane is formed of one oxygen atom from the water molecule and four oxygen atoms of the carboxylate groups of four ADC ligands (Fig. 12(a)).
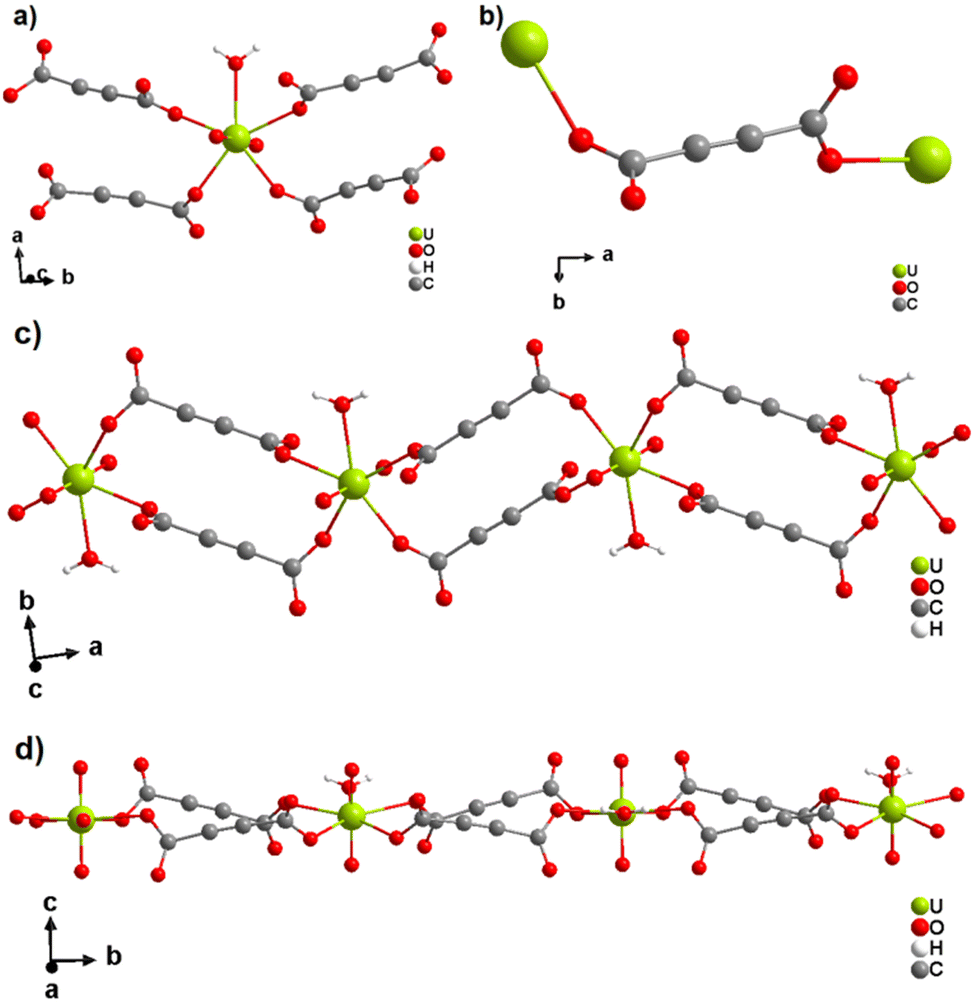 | ||
| Fig. 12 (a) Coordination environment of the U atom in the anionic [UO2(ADC)2(H2O)]2− polymeric chains, (b) coordination mode of the ADC linker; the chain arrangements in the structures of (c) K(H5O2)[UO2(ADC)2(H2O)] viewed along [001] and of (d) Cs2[UO2(ADC)2(H2O)] viewed along [100]. Graphics redrawn from the cif file which was provided by the author of ref. 51. | ||
Each carboxylate of ADC coordinates in a monodentate fashion, such that each ADC bridges two U(VI) atoms, resulting in one dimensional infinite anionic chains (Fig. 12(b) and (c)). K+ and H5O2+ are counterion species balancing the negative charge of the anionic chain in the first compound, while in the second compound, the charge of the anionic chain is balanced by two Cs+ cations.51
2.4 Non-porous ADC-based CPs with d-block metal cations
Most reported non-porous ADC-based CPs were obtained from divalent d-block metals. Manganese acetylenedicarboxylate of formula [Mn(ADC)(H2O)2] was the first reported d-block metal ADC-based CP, published in 1990.40 [Mn(ADC)(H2O)2] was formed as single crystals at the boundary of an aqueous silica gel containing H2ADC, overlaid with an aqueous solution of Mn(NO3)2. In the crystal structure of [Mn(ADC)(H2O)2], the Mn2+ cation is octahedrally coordinated by six oxygen atoms, of which four in the equatorial plan of the octahedron stem from the carboxylate groups of four ADC linkers and two oxygen atoms of the axial trans-positioned apices from connecting water molecules. [Mn(ADC)(H2O)2] is isotypic to the respective magnesium compound [Mg(ADC)(H2O)2], described earlier (Fig. 7). Each ADC linker bridges four Mn2+ cations, with each carboxylate coordinating in a bidentate bridging syn–anti μ2-η1:η1 mode, resulting in a three-dimensional framework that can be regarded as layers of MnO6 octahedra parallel to the (011) plane, which are pillared by ADC linkers. [Mn(ADC)(H2O)2] is thermally stable up to about 200 °C.40 For the isotypic magnesium CP a similar thermal stability was reported.45A cobalt acetylenedicarboxylate of formula [Co(ADC)(H2O)4]·(H2O)2 was also obtained as single crystals from a suspension of CoCO3 in an aqueous solution containing H2ADC, followed by slow evaporation of the solvent of the resulting solution.52 In the crystal structure of [Co(ADC)(H2O)4]·(H2O)2, Co2+ cations are octahedrally coordinated by four water molecules in the equatorial positions and two axial trans-positioned oxygen atoms from the carboxylate groups of two ADC linkers (Fig. 13(a)). Each carboxylate group of the ADC linker coordinates one Co2+ cation monodentately, leaving one oxygen atom of the group uncoordinated (Fig. 13(b)). Each ADC ligand therefore connects two CoO6 octahedra, resulting in chains that are held together by hydrogen bonds involving additional water molecules (Fig. 13(c) and (d)). Remarkably, already under ambient conditions [Co(ADC)(H2O)4]·(H2O)2 loses four of its six water molecules to yield a new CP of formula [Co(ADC)(H2O)2], which is isotypic to [Mn(ADC)(H2O)2] and [Mg(ADC)(H2O)2] (see Fig. 7). That is, its structure is made up of CoO6 octahedra that are connected by ADC linkers into a 3D framework. Now, a CoO6 octahedron consists of four oxygen atoms from four different ADC linkers and two oxygen atoms from two trans-positioned water molecules (vide supra). [Co(ADC)(H2O)2] is thermally stable up to 200 °C, similar to the respective manganese and magnesium CPs.52
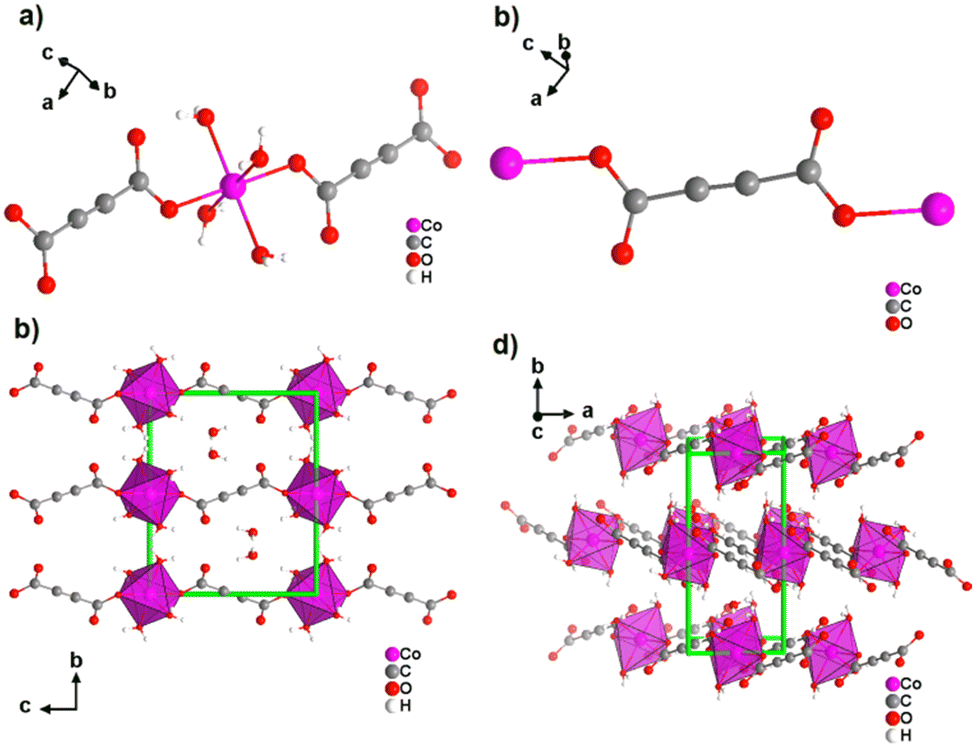 | ||
| Fig. 13 Crystal structure of cobalt acetylenedicarboxylate [Co(ADC)(H2O)4]·(H2O)2 showing: (a) the coordination environment of the Co2+ cation, (b) the coordination mode of the ADC linker, and (c) and (d) the packing of the framework viewed along [100] and [001], respectively. Graphics redrawn from the cif file (CSD-Refcode WOZKOL).52 | ||
Similar to cobalt acetylenedicarboxylate, the Ni2+ cation also yielded two ADC-based CPs with the formulae [Ni(ADC)(H2O)4]·(H2O)2 and [Ni(ADC)(H2O)2], which are isotypic to [Co(ADC)(H2O)4]·(H2O)2 and [Co(ADC)(H2O)2], respectively.53 [Ni(ADC)(H2O)4]·(H2O)2 was formed as single crystals by reacting Ni(CH3COO)2·4H2O with an aqueous solution of H2ADC, followed by slow evaporation of the solvent at room temperature. [Ni(ADC)(H2O)2] was formed as a microcrystalline powder, when heating [Ni(ADC)(H2O)4]·(H2O)2 at 100 °C under an argon atmosphere. [Ni(ADC)(H2O)2] is thermally stable up to 200 °C similar to the analogous magnesium, manganese, and cobalt CPs.53
By reacting H2ADC with Zn(NO)3·6H2O in aqueous solution, followed by slow evaporation of the solvent, single crystals of a zinc acetylenedicarboxylate CP of formula [Zn(ADC)(H2O)2] were obtained.54 Again, [Zn(ADC)(H2O)2] is isotypic to the magnesium, manganese, cobalt, and nickel CPs described above. Thus, these cations either form a hexahydrate of general formula [MII(ADC)(H2O)4]·(H2O)2 with a 1D polymeric structure or a dihydrate of formula [MII(ADC)(H2O)2] with a 3D framework structure. The hexahydrate can be transformed into the dihydrate by heating under the release of four water molecules. However, the formation of an anhydrous CP by the release of the remaining two water molecules is not possible, as decomposition of the linker occurs. As expected due to the Jahn-Teller effect, the Cu2+ compound shows a different structural behavior. The reaction of CuCl2·2H2O with H2ADC in aqueous solution, followed by slow evaporation of the solvent, yielded blue single crystals of a CP with the formula [Cu(ADC)(H2O)3]·H2O. In its structure, Cu2+ is surrounded by two monodentately coordinating carboxylate groups of two different ADC ligands in the trans position of the basal plane of a distorted square pyramid, whose remaining sites are occupied by three water molecules (Fig. 14(a)).55 This fivefold coordination of the central Cu2+ cation is noteworthy, as in all other CPs with divalent d block metals discussed so far solely octahedral sixfold coordination spheres were found. The CuO5 polyhedra are connected via the ADC linkers into linear polymeric chains parallel to the [001] direction. These chains are held together by hydrogen bonds including uncoordinated water molecules, thus leading to a three dimensional structure (Fig. 14(b)). The crystals of [Cu(ADC)(H2O)3]·H2O decomposed slowly in air, forming a black amorphous solid.55
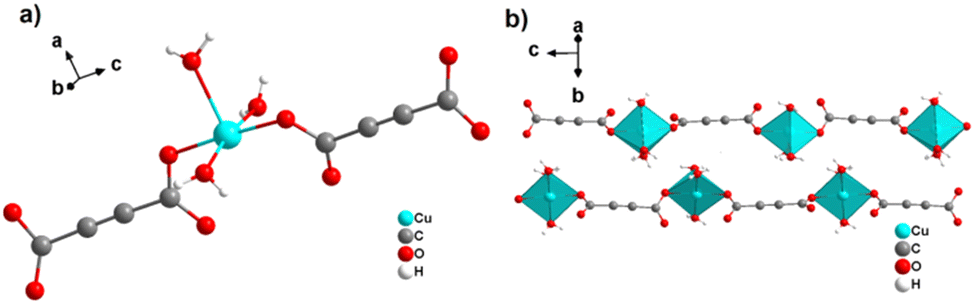 | ||
| Fig. 14 Crystal structure of copper acetylenedicarboxylate [Cu(ADC)(H2O)3]·H2O showing: (a) the coordination environment of a Cu2+ cation and (b) two one-dimensional polymeric chains, which are held together by hydrogen bonding. Graphics redrawn from the cif file (CSD-Refcode MACGAZ).55 | ||
It was demonstrated that two of the water molecules coordinating the metal cation in [MII(ADC)(H2O)4]·(H2O)2 (MII = Co, Ni) and [Cu(ADC)(H2O)3]·H2O could be replaced by other small donor ligands without changing the general topology of the underlying structure of the CP. In this regard, by allowing a slow diffusion of pyridine (Py) into an aqueous solution containing the corresponding metal cations and H2ADC, single crystals of CPs with the formulae [MII(ADC)(Py)2(H2O)2] (MII = Co, Ni) and [Cu(ADC)(Py)2(H2O)] were obtained, which are structural analogues of [MII(ADC)(H2O)4]·(H2O)2 (MII = Co, Ni) and [Cu(ADC)(H2O)3]·H2O, respectively.56 Accordingly, their crystal structures consist of MIIOx polyhedra, which are connected by ADC linkers into 1D polymeric chains. In [MII(ADC)(Py)2(H2O)2] (MII = Co, Ni), the M2+ cation is octahedrally coordinated by two axial oxygen atoms of the carboxylate groups of two trans-positioned ADC linkers, whereas in the equatorial plane two oxygen atoms of water molecules and two nitrogen atoms of pyridine ligands, which are all positioned trans to each other (Fig. 15(c)), complete the coordination sphere. In the crystal structure of [Cu(ADC)(Py)2(H2O)], the Cu2+ cation is fivefold coordinated by one axial water molecule, and in the equatorial plane, by two oxygen atoms from the carboxylate groups of two trans-positioned ADC ligands and two nitrogen atoms of two also trans coordinating pyridine molecules, thus forming a square pyramid (Fig. 16). It should be noted that the aforementioned pyridine-containing CPs do not contain any additional uncoordinated crystal water molecules, unlike their pyridine-free analogues.56 This is obviously due to the larger spatial requirements of pyridine compared to water.
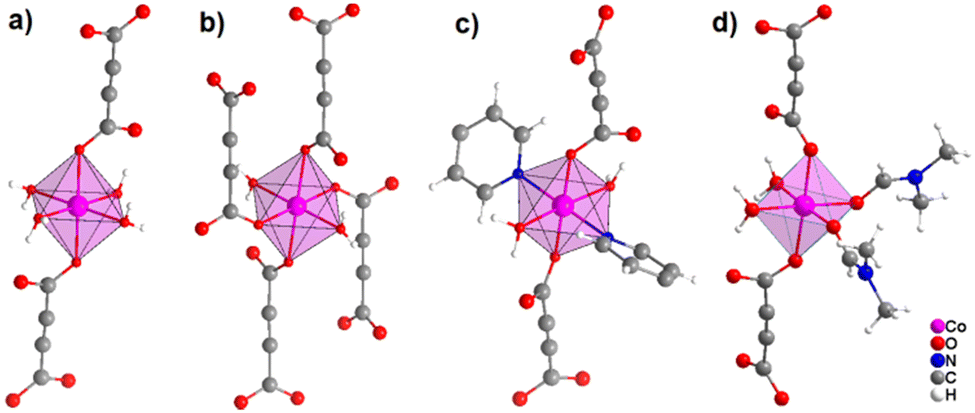 | ||
| Fig. 15 Comparison of the coordination spheres of Co2+ cations in the crystal structures of (a) [Co(ADC)(H2O)4]·(H2O)2, (b) [Co(ADC)(H2O)2], (c) [Co(ADC)(Py)2(H2O)2], and (d) [Co(ADC)(DMF)2(H2O)2]. Graphics redrawn from the cif files (CSD-Refcode WOZKOL, PAGRAU, KIFNUJ and ITICEU).40,52,56,57 | ||
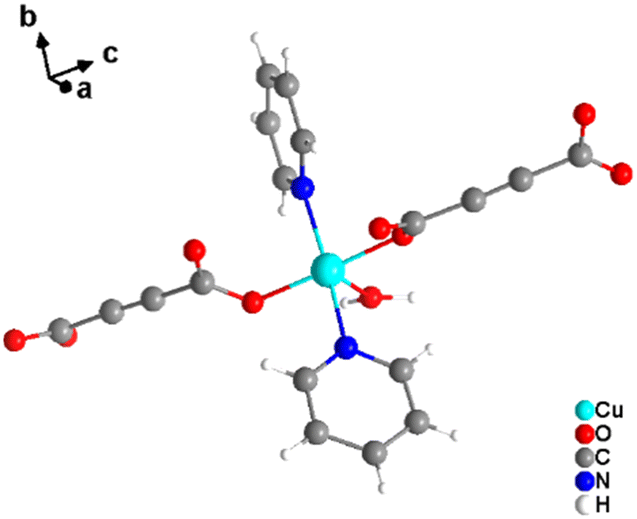 | ||
| Fig. 16 Cu2+ coordination sphere in the crystal structure of [Cu(ADC)(Py)2(H2O)]. Graphics redrawn from the cif file (CSD-Refcode KIVPEV).56 | ||
The synthetic approach described above, was also applied to the diffusion of pyridine into an aqueous solution containing Fe2+ and H2ADC, resulting in the formation of single crystals of a CP with the formula [Fe(ADC)(Py)2(H2O)2], which is isotypic to the respective CoII and NiII CPs.56 This confirms that ADC exhibits the same coordination behavior for many divalent first-row d-block metals (with the exception of copper) and forms isotypic CPs for the whole series. However, no one-dimensional FeII ADC-based CP with the specific formula [Fe(ADC)(H2O)4]·(H2O)2 or a CP with a three-dimensional framework structure of formula [Fe(ADC)(H2O)2] has yet been reported.
In subsequent studies, it was shown that when conducting the synthesis of first row d-block metal ADC-based CPs in non-aqueous solvents, the formation of CPs with similar structural units was observed. In principle, these solvent molecules were able to substitute two, but not all water molecules in the coordination spheres of the 3d metal cations found in ADC-based CPs synthesized in water. In this respect, single crystals or polycrystalline powders of CPs with formulae [MII(ADC)(DMF)2(H2O)2] (MII = Mn, Co, Ni, Zn) precipitated during slow evaporation under ambient conditions of N,N′-dimethylformamide (DMF) solutions containing the corresponding metal salts and H2ADC.57 Their structures consist of ADC linkers connecting MIIO6 octahedra into polymeric chains. In each octahedron the M2+ cation is surrounded by two trans-positioned monodentately coordinating ADC linkers, two DMF and two water molecules, which are in a cis coordination to each other (Fig. 15(d)). This structure is therefore very similar to that of [MII(ADC)(H2O)4]·(H2O)2 (MII = Co, Ni), and can simply be regarded as a substitution variant of the latter, whereby two water molecules have been exchanged by two DMF solvent molecules. The larger spatial requirement of the DMF ligand compared to water ligands also leads to the fact that no additional crystal water molecules are found in the DMF containing CPs. In this respect, they are comparable to the pyridine containing CPs mentioned earlier. But in the latter a trans coordination of the two pyridine ligands was found, whereas in the former a cis coordination of the two DMF molecules was observed (cf.Fig. 15). In summary, three different types of CPs with chain-like polymeric units were found, namely [MII(ADC)(H2O)4]·(H2O)2, [MII(ADC)(Py)2(H2O)2] and [MII(ADC)(DMF)2(H2O)2]. It should be pointed out that they have been synthesized for almost all MII 3d metals (MII = Mn, Fe, Co, Ni, Zn). For details, we would like the reader to refer to Table 1. As an example, Fig. 15 illustrates the coordination spheres around the Co2+ cations in these 1D CPs and compares them with the 3D CP [Co(ADC)(H2O)2]. Regarding the synthesis, most of the reported ADC-based CPs were synthesized in water, although there is no clear reason for this. The aforementioned results show that other organic solvents (DMF, pyridine) are also suitable for the synthesis of ADC-based CPs.
| Formula | Synthesis conditions | Crystal system | Space group | a, b, c (Å) | α, β, γ (°) | Thermal stability | Structure dimensionality | Ref. |
|---|---|---|---|---|---|---|---|---|
| a DMF = N,N′-dimethylformamide, Py = pyridine, NA = not available, RT = room temperature. | ||||||||
| [Na2(ADC)(H2O)4] | RT: slow evaporation of aqueous solution | Monoclinic | P21/n | 8.116(3) | 90(7) | NA | 2D, layers | 42 |
| 3.5771(7) | 99.21(5) | |||||||
| 15.806(5) | 90(4) | |||||||
| [K2(ADC)(H2O)] | RT: slow evaporation of aqueous solution | Triclinic |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
7.0761(14) | 90.99(3) | NA | 3D | 42 |
| 7.2388(14) | 112.02(2) | |||||||
| 8.775(2) | 117.72(2) | |||||||
| [Li2(ADC)(H2O)2] | RT: slow evaporation of aqueous solution | Triclinic |
P |
7.105(2) | 96.50(3) | Transforms to [Li2(ADC)] at 140 °C | 3D | 41 |
| 7.316(2) | 94.31(3) | |||||||
| 7.622(2) | 114.60(3) | |||||||
| [Li2(ADC)] | Dehydration of [Li2(ADC)(H2O)2] by heating at 140 °C | Orthorhombic | Fddd | 8.7017(1) | 90 | Decomposes at 360 °C | 3D | 41 |
| 16.9076(2) | 90 | |||||||
| 7.0627(1) | 90 | |||||||
| [Ba(ADC)(H2O)] | RT: diffusion at the phase boundary of an aqueous and iso-propanol solution | Monoclinic | P21/a | 7.534(2) | 90 | NA | 3D | 39 |
| 9.218(2) | 102.00(2) | |||||||
| 8.818(2) | 90 | |||||||
| [Sr(ADC)] | RT: diffusion at the phase boundary of an aqueous solution and a silica gel | Tetragonal | I41/amd | 7.2181(11) | 90 | Stable up to 450 °C | 3D | 44 |
| 7.2181(11) | 90 | |||||||
| 10.356(2) | 90 | |||||||
| [Ca(ADC)] | Mechanochemical synthesis and subsequent heating at 50 °C | Tetragonal | I41/amd | 6.8778(3) | 90 | Decomposes at 50 °C | 3D | 45 |
| 6.8778(3) | 90 | |||||||
| 10.2011(4) | 90 | |||||||
| [Mg(ADC)(H2O)2] | Mechanochemical synthesis and subsequent heating at 70 °C | Monoclinic | I2/a | 11.2022(6) | 90 | Decomposes at 200 °C | 3D | 45 |
| 7.2133(3) | 92.6674(25) | |||||||
| 7.6156(3) | 90 | |||||||
| [EuII(ADC)] | RT: precipitation from an aqueous solution | Tetragonal | I41/amd | 7.2167(1) | 90 | Stable up to 440 °C | 3D | 50 |
| 7.2167(1) | 90 | |||||||
| 10.3165(1) | 90 | |||||||
| K(H5O2)[UO2(ADC)2(H2O)]·2H2O | Crystallization from a cooled aqueous solution | Monoclinic | C2/c | 16.254(12) | 90 | NA | 1D | 51 |
| 13.508(8) | 90.91(7) | |||||||
| 7.683(6) | 90 | |||||||
| Cs2[UO2(ADC)2(H2O)]·2H2O | Crystallization from a cooled aqueous solution | Orthorhombic | Abm2 | 7.0745(10) | 90 | NA | 1D | 51 |
| 18.4246(10) | 90 | |||||||
| 13.1383(10) | 90 | |||||||
| [Mn(ADC)(H2O)2] | RT: diffusion at the phase boundary of an aqueous solution and a silica gel | Monoclinic | C2/c | 13.508(2) | 90 | Decomposes at 200 °C | 3D | 40 |
| 7.263(1) | 122.94(1) | |||||||
| 7.806(1) | 90 | |||||||
| [Co(ADC)(H2O)4]·2H2O | RT: slow evaporation of an aqueous solution | Monoclinic | P21/a | 5.1564(10) | 90 | Transforms to [Co(ADC)(H2O)2] already at RT | 1D, linear chains | 52 |
| 10.7578(15) | 96.715(15) | |||||||
| 8.982(2) | 90 | |||||||
| [Co(ADC)(H2O)2] | RT: dehydration of [Co(ADC)(H2O)4]·2H2O | Monoclinic | C2/c | 13.2355(4) | 90 | Decomposes at 200 °C | 3D | 52 |
| 7.1765(2) | 122.442(1) | |||||||
| 7.6324(2) | 90 | |||||||
| [Ni(ADC)(H2O)4]·2H2O | RT: slow evaporation of an aqueous solution | Monoclinic | P21/a | 5.132(1) | 90 | Transforms to [Ni(ADC)(H2O)2] at 100 °C | 1D, linear chains | 53 |
| 10.728(3) | 96.72(2) | |||||||
| 8.936(2) | 90 | |||||||
| [Ni(ADC)(H2O)2] | Dehydration of [Ni(ADC)(H2O)4]·H2O by heating at 100 °C | Monoclinic | C2/c | 13.1456(4) | 90 | Decomposes at 200 °C | 3D | 53 |
| 7.1786(2) | 122.241(1) | |||||||
| 7.5251(2) | 90 | |||||||
| [Zn(ADC)(H2O)2] | RT: slow evaporation of an aqueous solution | Monoclinic | C2/c | 13.245(3) | 90 | NA | 3D | 54 |
| 7.223(2) | 122.66(1) | |||||||
| 7.649(2) | 90 | |||||||
| [Cu(ADC)(H2O)3]·H2O | RT: slow evaporation of an aqueous solution | Monoclinic | P21/c | 6.5261(8) | 90 | Decomposes in air at RT | 1D, linear chains | 55 |
| 7.0683(9) | 90.418(10) | |||||||
| 18.417(2) | 90 | |||||||
| [Cd(ADC)(H2O)3]·H2O | RT: slow evaporation of an aqueous solution | Monoclinic | P21/n | 6.8195(7) | 90 | Decomposes in air at RT | 1D, zigzag chains | 59,60 |
| 7.953(1) | 99.811(8) | |||||||
| 16.387(2) | 90 | |||||||
| [MII(ADC)(DMF)2(H2O)2] MII = Mn, Co, Ni, and Zn | RT: slow evaporation of a DMF solution | Monoclinic | C2/c | 16.826(2)–17.025(4) | 90 | MII = Ni: decomposes at 146 °C; MII = Cu: decomposes at 114 °C; others: NA | 1D, linear chains | 57 |
| 9.427(3)–9.576(1) | 98.93–99.64(2) | |||||||
| 9.564(2)–9.694(2) | 90 | |||||||
| [MII(ADC)(Py)2(H2O)2] MII = Fe, Co, and Ni | RT: slow diffusion of pyridine into an aqueous solution of the reactants | Monoclinic | C2/c | 9.747(2)–9.864(1) | 90 | MII = Ni: decomposes above 160 °C; others: NA | 1D, linear chains | 56 |
| 19.270(3)–19.437(2) | 115.06–115.31(2) | |||||||
| 9.064(2)–9.155(1) | 90 | |||||||
| [Cu(ADC)(Py)2(H2O)] | RT: slow diffusion of pyridine into an aqueous solution of the reactants | Orthorhombic | P212121 | 5.653(1) | 90 | 1D, linear chains | 56 | |
| 14.340(3) | 90 | |||||||
| 18.640(2) | 90 | |||||||
| [Tl2ADC] | Slow evaporation of an aqueous solution at 1 °C | Orthorhombic | P212121 | 6.232(1) | 90 | Decomposes at 195 °C | 3D | 61,62 |
| 7.270(1) | 90 | |||||||
| 14.721(2) | 90 | |||||||
| [Co(ADC)(MeOH)4] | RT: crystallization from a methanolic solution | Monoclinic | C2/c | 12.451(5) | 90 | NA | 1D, linear chains | 58 |
| 7.082(2) | 94.07(4) | |||||||
| 14.463(5) | 90 | |||||||
| [Ni(ADC)(EtOH)4] | RT: reaction in an ethanolic solution | Triclinic |
P |
9.3478(12) | 97.40(7) | NA | 1D, linear chains | 58 |
| 9.6770(7) | 102.77(10) | |||||||
| 10.3922(10) | 103.70(8) | |||||||
| [Ni(ADC)(MeOH)(EtOH)3] | RT: reactions in ethanolic and methanolic solutions | Monoclinic | P21/n | 9.6723(5) | 90 | NA | 1D, linear chains | 58 |
| 16.1469(17) | 96.86(5) | |||||||
| 10.4128(6) | 90 | |||||||
| [Mn2(ADC)2(MeOH)5] | Crystallization from a cooled methanolic solution | Monoclinic | C2/c | 10.8875(8) | 90 | NA | 3D | 58 |
| 14.8481(14) | 102.35(7) | |||||||
| 25.7170(2) | 90 | |||||||
| [Tl2Zn(ADC)2(H2O)2] | RT: slow evaporation from an aqueous solution | Triclinic |
P |
7.289(2) | 113.18(3) | NA | 3D | 64 |
| 7.376(2) | 95.22(3) | |||||||
| 7.511(2) | 115.55(3) | |||||||
| [Pb(ADC)] | RT: diffusion at the phase boundary of an aqueous solution and a silica gel | Tetragonal | I41/amd | 7.2181(11) | 90 | Decomposes at 250 °C | 3D | 66 |
| 7.2181(11) | 90 | |||||||
| 10.356(2) | 90 | |||||||
| [Pb(ADC)(H2O)] | RT: diffusion at the phase boundary of an aqueous solution and a silica gel; mechanochemical synthesis at RT | Monoclinic | P21/c | 9.936(2) | 90 | Transforms to [Pb(ADC)] at 125 °C under vacuum | 2D, double-layers | 66 |
| 4.007(4) | 121.20(1) | |||||||
| 16.696(3) | 90 | |||||||
| [Bi(ADC)1.5(H2O)2]·2H2O | RT: slow evaporation from an aqueous solution | Triclinic |
P |
8.399(2) | 101.08(2) | Release of water molecules at 60–80 °C | 2D, double-layers | 67 |
| 8.838(2) | 111.94(2)111 | |||||||
| 9.336(2) | 11.86(2) | |||||||
Accordingly, Best-Thomson et al. showed in a very recent study that methanol and ethanol are also suitable solvents for the synthesis of ADC-based CPs with divalent 3d metals. At ambient temperature, they reacted CoCl2·4H2O with H2ADC in ethanol and Ni(NO3)2·6H2O with H2ADC in ethanol in the presence of N,N′-diisopropylethylamine as a deprotonating agent. They thus obtained two CPs of formulae [Co(ADC)(MeOH)4] and [Ni(ADC)(EtOH)4], respectively.58 The crystal structures of these two CPs show again structural units similar to those of the aforementioned [MII(ADC)(H2O)4]·(H2O)2 compounds, that is, one-dimensional chains of MIIO6 octahedra connected by ADC linkers. The MIIO6 octahedra are formed by two oxygen atoms of two axial monodentately coordinating carboxylate groups of two different ADC ligands and four equatorial oxygen atoms from four methanol or four ethanol molecules in [Co(ADC)(MeOH)4] and [Ni(ADC)(EtOH)4], respectively (Fig. 17). The following aspect of these crystal structures has to be pointed out: in contrast to the pyridine and DMF containing CPs, all water molecules are here replaced by methanol and ethanol ligands, respectively.
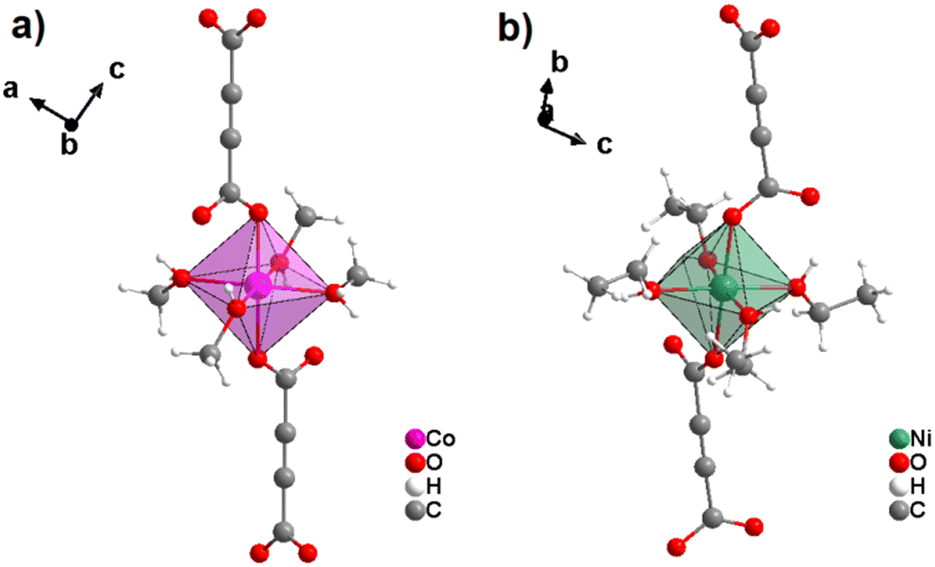 | ||
| Fig. 17 Coordination sphere of Co2+ and Ni2+ in analogous 1D chain-like CPs [Co(ADC)(MeOH)4] (left) and [Ni(ADC)(EtOH)4] (right), respectively. Their crystal structures are similar to that of [MII(ADC)(H2O)4]·(H2O)2 (MII = Co, Ni) (Fig. 13) in a way that four equatorial water molecules in the latter are substituted by methanol and ethanol molecules, respectively. Graphics redrawn from the cif files (CSD-Refcode PAGREY and PAGROI).58 | ||
Surprisingly, the reaction of Mn(NO3)2 and H2ADC in methanol at ambient temperature yielded an entirely different CP from what was obtained in water or what could be expected with respect to the aforementioned observations with Co2+ and Ni2+.58 The obtained CP of formula [Mn2(ADC)2(MeOH)5] is structurally made up of three crystallographically different Mn2+ cations and two crystallographically distinct ADC linkers. One Mn2+ cation is octahedrally coordinated by the oxygen atoms of three different ADC linkers and three methanol molecules resulting in a mer-configuration (Fig. 18(a)), while the two other Mn2+ cations are octahedrally coordinated by the oxygen atoms of four different ADC linkers and two methanol molecules, whereby the latter are arranged in a trans-position for one Mn2+ and in a cis-position for the other Mn2+ (Fig. 18(b) and (c)). In one type of ADC linker, the two carboxylate groups coordinate in a bidentate bridging syn–anti-μ2-η1:η1 mode (Fig. 18(d)), whereas in the other type of ADC linker, one carboxylate group coordinates in a bidentate syn–anti-μ2-η1:η1 mode, while the other coordinates in a monodentate syn-η1:η0 mode (Fig. 18(e)), therefore leaving one uncoordinated oxygen atom. The resulting crystal structure is a complicated non-porous three-dimensional network (Fig. 18(f)), which shows clearly that even for “simple” linkers like ADC complex and completely unexpected crystal structures can be obtained.58
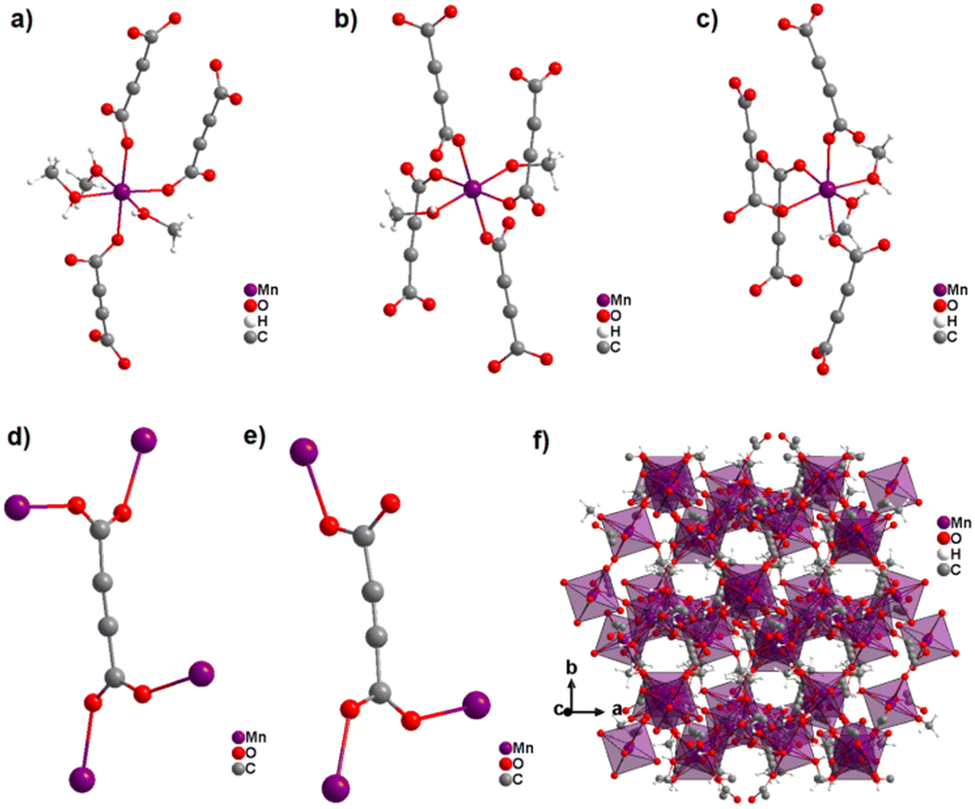 | ||
| Fig. 18 Crystal structure of the non-porous 3D CP [Mn2(ADC)2(MeOH)5]: (a–c) coordination sphere of three crystallographically distinct Mn2+ cations, (d) and (e) coordination modes of two crystallographically distinct ADC linkers, and (f) structure packing viewed along [001]. Graphics redrawn from the cif file (CSD-Refcode PAGRAU).58 | ||
The coordination geometry of the aforementioned ADC-based CPs with divalent 3d metal cations is dominated by octahedral coordination spheres. This situation changes, when going to the larger divalent cations of the second-row d-block metals. This was first observed with cadmium(II), for which a CP of formula [Cd(ADC)(H2O)3]·H2O was reported.59 In its crystal structure, the ADC linker connects two Cd2+ cations, with each of its carboxylate groups coordinating in a bidendate chelating fashion (Fig. 19(a)).
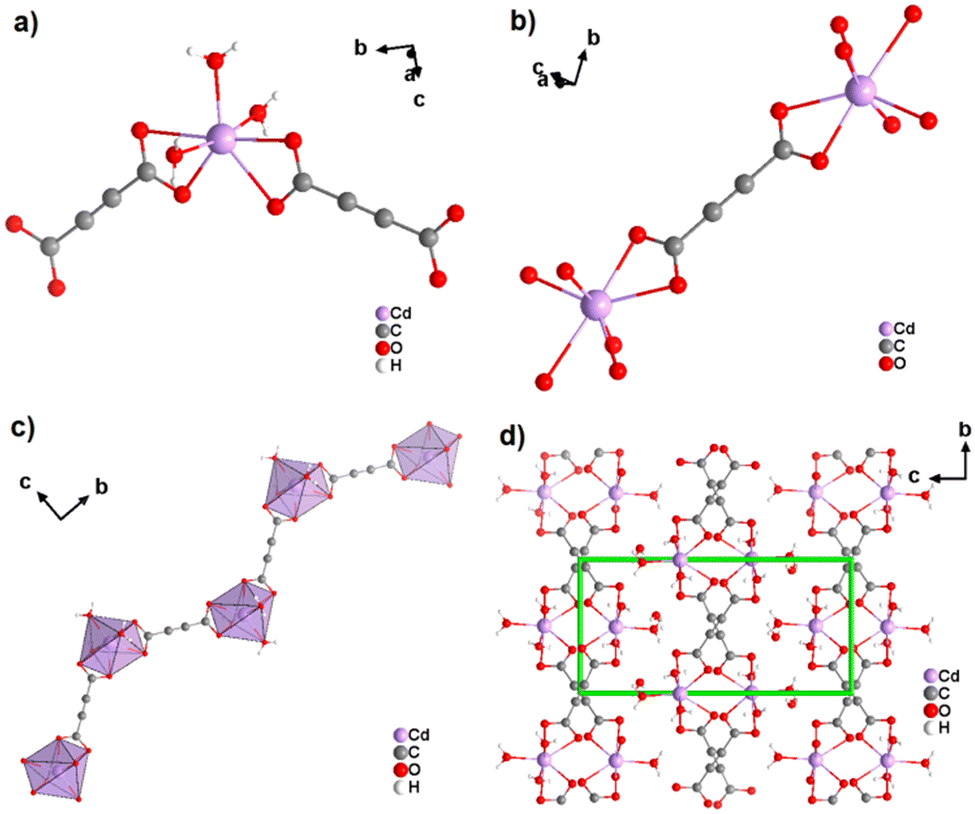 | ||
| Fig. 19 Crystal structure of cadmium acetylenedicarboxylate [Cd(ADC)(H2O)3]·H2O showing: (a) the coordination environment of the Cd2+ cation, (b) the coordination mode of the ADC linker, (c) the one-dimensional zigzag chain, and (d) the packing of the framework viewed along [100], showing chains held together by hydrogen bonds (not emphasized). Graphics redrawn from the cif file (CSD-Refcode MUGJED).59 | ||
The CdII cation is sevenfold coordinated by three water molecules and two chelating ADC ligands (Fig. 19(b)), thus forming chains (Fig. 19(c)). It is notable that the chains formed in the crystal structure of Cd-acetylenedicarboxylate are zigzag-shaped, unlike the linear-shaped chains observed in ADC-based CPs with divalent first-row d-block metals (vide supra). These zigzag chains in Cd-acetylenedicarboxylate are also interconnected by hydrogen bonds involving uncoordinated crystal water molecules to form the three dimensional crystal structure (Fig. 19(d)).
While [Cd(ADC)(H2O)3]·H2O was first synthesized by Ruschewitz et al. as single crystals by slow evaporation of an aqueous solution containing Cd(CH3COO)2·2H2O and H2ADC,59 the same compound was reported one year later by Skoulika et al., who obtained it as single crystals by slow evaporation of an aqueous solution containing Cd(NO3)2 and H2ADC.60 In both publications, there is a slight discrepancy in the reported thermal behavior of this compound. While the former team reported that [Cd(ADC)(H2O)3]·H2O decomposed already slowly in air, the latter team performed a thermogravimetric analysis in air (Fig. 20(a)), that showed that this CP loses water molecules upon heating at 90 °C to yield a microcrystalline powder of a new compound with the proposed formula [Cd(ADC)(H2O)2.3]. Although the crystal structure of the new compound could not be determined, the powder X-ray diffraction pattern showed that the two phases are not identical (Fig. 20(b) and (c)).60 [Cd(ADC)(H2O)2.3] loses further water molecules at about 150 °C to yield an amorphous anhydrous compound with the estimated formula [Cd(ADC)]·xH2O. Its decomposition starts at about 325 °C. However, the composition [Cd(ADC)]·xH2O is very unlikely, since the compound should be anhydrous and the authors observed by Raman spectroscopy that the triple bond was transformed into a double bond, probably as a result of a thermally induced polymerization or cyclisation. The formation of a C–H moiety was also concluded from the Raman spectrum.
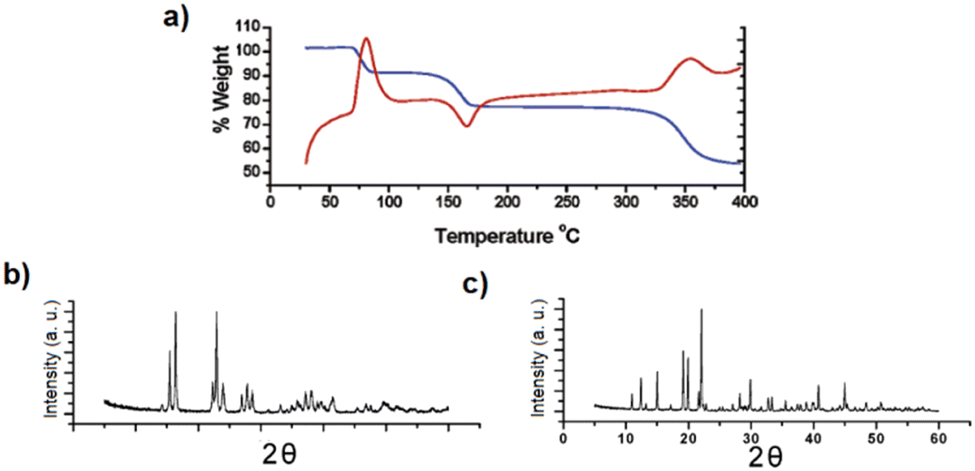 | ||
| Fig. 20 (a) Thermogravimetric (blue trace) and DTA curves (red trace) of [Cd(ADC)(H2O)3]·H2O, (b) and (c) powder X-ray diffraction patterns of [Cd(ADC)(H2O)3]·H2O and [Cd(ADC)(H2O)2.3], respectively. Reproduced from ref. 60 with permission of American Chemical Society, copyright 2003. | ||
2.5 Non-porous ADC-based CPs with p-block metal cations
The first ADC-based CP with a monovalent p-block element was reported for thallium. Single crystals of anhydrous [Tl2ADC] were formed from slow evaporation of an aqueous solution containing thallium(I)-acetate and H2ADC at room temperature.61 In another report, single crystals of [Tl2ADC] were obtained by reacting Tl(NO3) and H2ADC in methanol in the presence of KOH and a small amount of water.62 In the structure of [Tl2ADC], there are two crystallographically different TlI atoms. One Tl+ cation is sixfold-coordinated by one chelating and four monodentately coordinating ADC linkers (Fig. 21(a) and (b)). The other Tl+ cation is fivefold-coordinated by one chelating and three monodentately coordinating ADC linkers (Fig. 21(c)). The coordination spheres of both Tl+ cations are very unsymmetrical, which points to the presence of stereochemically active electron lone pairs at both Tl+ cations. All Tl+ cations are arranged in corrugated layers along the (001) plane, and the ADC linkers, aligned parallel to each other along the [001] direction, connect these layers to form a three-dimensional framework structure (Fig. 21(d)). [Tl2ADC] is thermally stable up to about 195 °C, after which it decomposes in a violent reaction releasing CO2 and forming a black residue.57 Remarkably, elemental thallium in its high (Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, Z = 2) and room temperature modifications (P63/mmc, Z = 2) were detected by X-ray powder diffraction in this residue, which is pyrophoric, as obviously a “thallium sponge” is formed.57 [Tl2ADC] crystallizes in the noncentrosymmetric space group P212121 and so investigations of its optical properties were of interest, e.g. second-harmonic generation (SHG), see Section 4.1. Indeed, very large crystals of [Tl2ADC] were grown (Fig. 22). However, in the laser beam a complete decomposition of these crystals was observed.63
m, Z = 2) and room temperature modifications (P63/mmc, Z = 2) were detected by X-ray powder diffraction in this residue, which is pyrophoric, as obviously a “thallium sponge” is formed.57 [Tl2ADC] crystallizes in the noncentrosymmetric space group P212121 and so investigations of its optical properties were of interest, e.g. second-harmonic generation (SHG), see Section 4.1. Indeed, very large crystals of [Tl2ADC] were grown (Fig. 22). However, in the laser beam a complete decomposition of these crystals was observed.63
 | ||
| Fig. 21 Crystal structure of anhydrous thallium acetylenedicarboxylate [Tl2(ADC)] showing: (a) and (b) the coordination environment of two crystallographically distinct Tl+ cations, (c) the coordination mode of the ADC linker, and (d) the three-dimensional packing of the framework. Graphics redrawn from the cif file (CSD-Refcode TILFUQ01).61 | ||
 | ||
| Fig. 22 Photograph of a crystal of [Tl2ADC].65 | ||
In a subsequent study, the same authors obtained the first ADC-based CP containing two different metal ions, namely thallium(I) and zinc(II).64 Single crystals of the CP with the formula [Tl2Zn(ADC)2(H2O)2] formed by slow evaporation of an aqueous solution containing ZnCO3, Tl(CH3COO) and H2ADC. In its crystal structure, the Zn2+ cation is octahedrally coordinated by four oxygen atoms of the carboxylate groups of four different ADC ligands and two oxygen atoms from two water molecules (Fig. 23(a), cp. Fig. 15(b)). The Tl+ cation is sixfold coordinated by six oxygen atoms of the carboxylate groups from five different ADC ligands, of which one ligand is coordinating in a chelating mode (Fig. 23(b)). The coordination sphere of Tl+ is again quite unsymmetrical, leaving one hemisphere unoccupied, which was attributed to accommodation with the stereochemically active lone pair of Tl+. One carboxylate group of the ADC linker bridges two Tl+ cations in a chelating bridging mode and one Zn2+ cation in an anti-unidentate fashion, while the other carboxylate group bridges two Tl+ cations in a bidentate anti–anti configuration and one Zn2+ cation in a syn-unidentate fashion (Fig. 23(c)). The Tl+ and Zn2+ cations are connected by ADC linkers and additional hydrogen bonds between water molecules and oxygen atoms of carboxylate groups to form a three-dimensional network (Fig. 23(d)).64
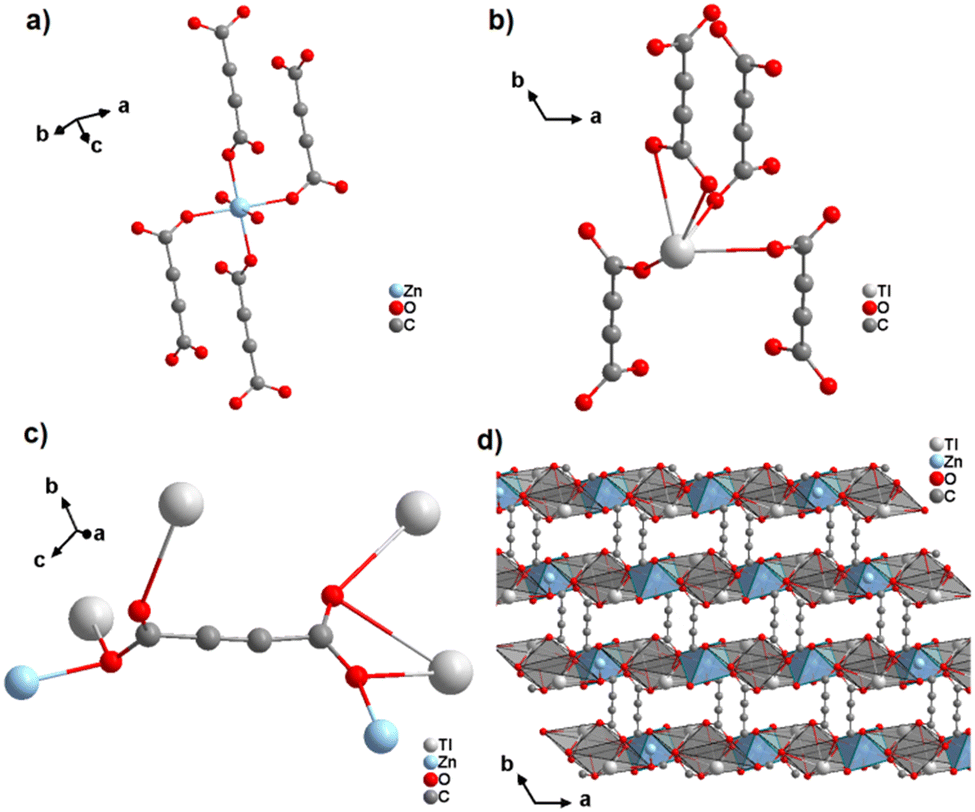 | ||
| Fig. 23 Crystal structure of mixed thallium-zinc acetylenedicarboxylate [Tl2Zn(ADC)2(H2O)2] showing: (a) and (b) the coordination environment of Zn and Tl atoms, respectively, (c) the coordination mode of ADC, and (d) the structure packing of the framework viewed along the [001] direction. Graphics redrawn from the cif file (CSD-Refcode AGURAW).64 | ||
The only reported ADC-based CP with a divalent p-block metal was obtained with lead. Single crystals of either hydrated [Pb(ADC)(H2O)] or anhydrous [Pb(ADC)] formed at the phase boundary of an aqueous silica gel containing H2ADC, overlaid with an aqueous solution of Pb(NO3)2. Noteworthy, these two CPs were obtained using almost the same procedure, whereby [Pb(ADC)] forms with a higher concentrated Pb(NO3)2 solution, while [Pb(ADC)(H2O)] forms with a solution with a lower Pb(NO3)2 concentration. [Pb(ADC)] was also obtained as a microcrystalline powder using mechanochemical methods, i.e., by grinding a solid mixture of H2ADC and Pb(CH3COO)2·3H2O in an agate mortar, or by heating [Pb(ADC)(H2O)] (vide infra).66 [Pb(ADC)] crystallizes in a structure type found for [Sr(ADC)] for the first time (Fig. 8). This seems to be a very common structure type for compounds of composition [MII(ADC)] found with MII = Ca, Sr, Ba, Pb, and Eu so far. The crystal structure consists of a three-dimensional framework, wherein eightfold coordinated Pb2+ cations are arranged in a diamond-like topology and are bridged by ADC linkers. Each carboxylate group of the ADC ligand bridges three Pb2+ cations in a μ3-η1:η2:η1 coordination mode. In the crystal structure of [Pb(ADC)(H2O)], the Pb2+ cation is ninefold coordinated by two oxygen atoms from two water molecules and seven oxygen atoms from the carboxylate groups of five different ADC linkers, of which two ADC linkers are coordinating in a chelating mode (Fig. 24(a)). Each ADC ligand connects overall five Pb2+ cations, such that one of its carboxylate groups bridges two Pb2+ cations in a μ2-η2:η1 coordination mode, while the other carboxylate group bridges three Pb2+ cations in a μ3-η3:η1 coordination mode (Fig. 24(b)). The connectivity between ADC linkers and Pb2+ cations results in the formation of double-layers that are held together by hydrogen bonds (Fig. 24(c) and (d)). [Pb(ADC)] is thermally stable up to approx. 250 °C and, most remarkably, can be converted into [Pb(ADC)(H2O)] upon suspension in water for 24 h under ambient conditions, whereas [Pb(ADC)(H2O)] is converted back into [Pb(ADC)] upon heating at 125 °C under vacuum. Such reversible topochemical transformation between hydrated and anhydrous CPs has not been reported for any other ADC-based system so far.66
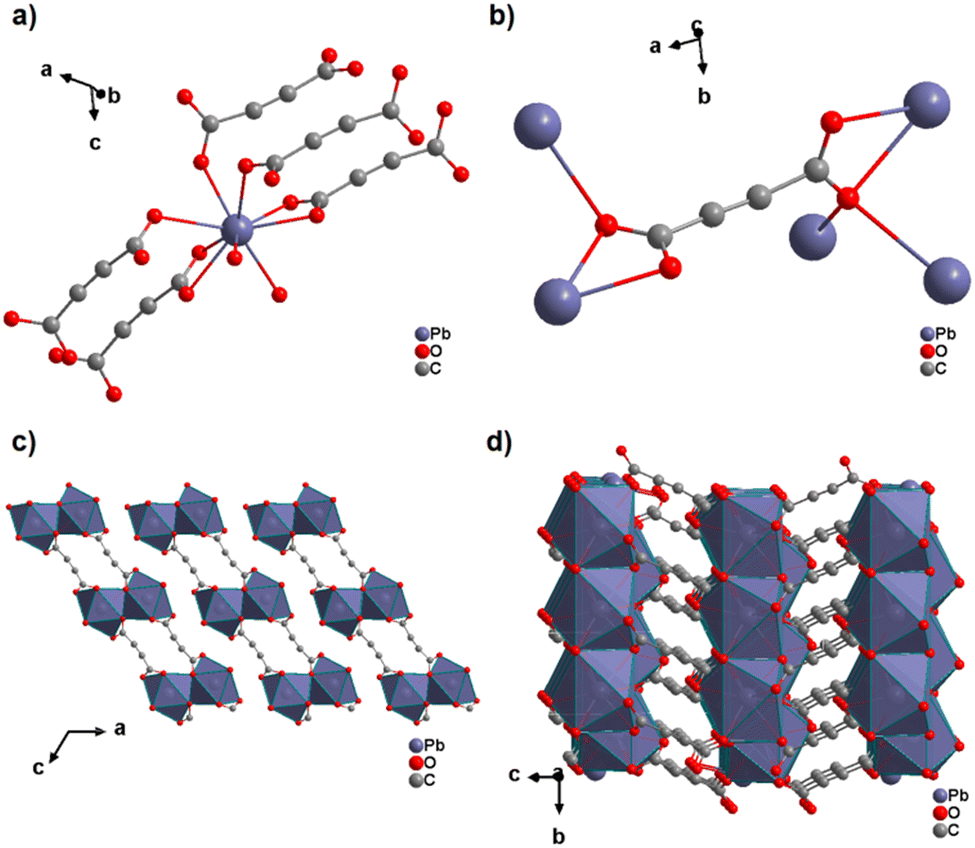 | ||
| Fig. 24 Crystal structure of lead acetylenedicarboxylate [Pb(ADC)(H2O)] showing: (a) the coordination environment of a Pb2+ cation, (b) the coordination mode of the ADC linker, and (c) and (d) the packing of the crystal structure viewed along the [010] and [100] directions, respectively. Graphics redrawn from the cif file (CSD-Refcode MUZKEY).66 | ||
An ADC-based CP with a trivalent p-block metal was obtained with bismuth, yielding single crystals of the 2D CP [Bi(ADC)1.5(H2O)2]·2H2O from a saturated solution containing Bi(NO3)3·5H2O and H2ADC at room temperature.67 In its crystal structure, Bi3+ is eightfold-coordinate with four oxygen atoms from two chelating ADC, two oxygen atoms from two ADC coordinating monodentately and two oxygen atoms from two water molecules (Fig. 25(a)). There are two crystallographically different ADC linkers which are arranged perpendicular to each other. In one ADC linker, each of the two carboxylates monodentately coordinates one Bi3+ cation in a syn η1:η0 mode (Fig. 25(b)). Meanwhile in the other ADC linker, one carboxylate is chelating and the other carboxylate bridges two Bi3+ in a chelating-bridging μ2-η2:η1 mode (Fig. 25(c)) to form dinuclear {Bi2O10(H2O)4} units made of two edge-sharing BiO8 polyhedra (Fig. 25(a)). The latter units are connected by ADC linkers into layers that are stacked in an AB fashion, resulting in a dense arrangement with no accessible porosity (Fig. 25(d)).67 It is remarkable that a 3D MOF with the same composition (vide infra, Fig. 29) is formed under very similar synthetic conditions.
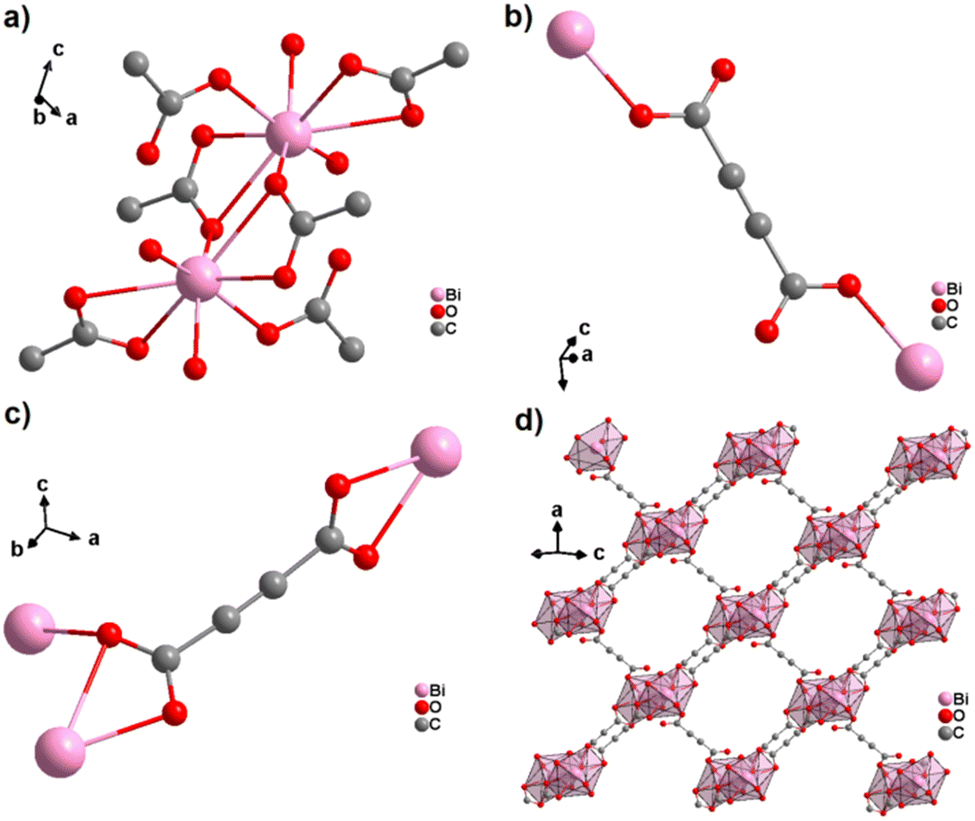 | ||
| Fig. 25 Crystal structure of bismuth acetylenedicarboxylate [Bi(ADC)1.5(H2O)2]·2H2O showing: (a) the coordination environment of two Bi3+ cations, (b) and (c) the coordination mode of two crystallographically different ADC linkers, and (d) the resulting 2D layer by connecting dinuclear {Bi2O8(H2O)6} units with ADC linkers. Graphics redrawn from the cif file (CSD-Refcode NERQOS).67 | ||
At this point we want to summarize some of the results and general aspects of the reported work on non-porous CPs based on the ADC ligand:
(i) Mainly CPs with mono- and divalent cations have been reported. [Bi(ADC)1.5(H2O)2]·2H2O with BiIII or Cs2[UO2(ADC)2(H2O)]·2H2O and K(H5O2)[UO2(ADC)2(H2O)]·2H2O with UVI are the only exceptions. For RE3+ cations (RE = rare earth metals) the formation of 3D MOFs (with no permanent porosity) has already been observed (vide infra, Fig. 30). In most CPs presented in Section 2 the ADC ligands are aligned in one direction, parallel to each other, which hinders the formation of MOFs with potential pores. In particular for transition metals (Mn, Fe, Cu, Ni, Co, Cd etc.), only CPs with single-atom metal nodes are formed and no polynuclear cluster SBUs are observed, which are typically found in many MOFs.
(ii) In most examples, these ADC-based CPs were obtained at room temperature, mainly by slow evaporation of the solvent or gel methods. Also mechanochemical syntheses were reported. Heating at moderate temperatures of 50–70 °C improved the crystallinity of the resulting CPs. Exceptions are those syntheses, where a transformation of the starting CP by the release of solvent molecules upon heating was applied.
(iii) No examples of CPs with cations of noble metals were reported due to the sensitivity of the ADC linker against oxidation.
(iv) The dimensionality of the CP increases with decreasing water/solvent content in the compound, such that all anhydrous ADC-based CPs form a three-dimensional framework structure.
(v) The carboxylate groups of the ADC ligands adopt almost all possible coordination modes in the reported CPs (Fig. 2(B)). The two carboxylate groups in one ADC linker are found to occur in all possible orientations to each other from coplanar (point group mmm) to perpendicular (point group ![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2m) and all orientations in-between (Fig. 2(A)). High symmetry orientations are mainly observed in anhydrous CPs.
2m) and all orientations in-between (Fig. 2(A)). High symmetry orientations are mainly observed in anhydrous CPs.
(vi) The ADC-based CPs presented in this Section 2 show in general a low thermal stability. Exceptions are anhydrous CPs like [Sr(ADC)], [Eu(ADC)], and [Li2(ADC)].
In Table 1 the ADC-based non-porous CPs presented in Section 2 are summarized along with the conditions of their synthesis, some structural characteristics and their thermal stabilities.
3 Acetylenedicarboxylate-based metal–organic frameworks
Unlike coordination polymers (CPs), for which ADC-based structures have been reported using metal ions from almost all groups of the periodic table of the elements, relatively few acetylenedicarboxylate-based metal–organic frameworks (MOFs) were found in the literature until 2018. The scarcity of ADC-based MOFs was explained by the low thermal stability of acetylenedicarboxylic acid, which makes the application of solvo-/hydrothermal syntheses difficult or even impossible, as higher temperatures (typically above 100 °C) are needed.68 Taking into account that the solvo-/hydrothermal approach is essential for the synthesis of almost all important MOFs, the difficulties in synthesizing ADC-based MOFs are obvious, as acetylenedicarboxylic acid is known to decompose easily in solutions at elevated temperatures.23,24 It should be noted that according to the IUPAC recommendations on terminologies, a MOF is characterized by a potential porosity, that is not required for a CP.69,70 Even more for possible applications, the guest (solvent)-filled pores of the MOF should be made guest-free without a collapse of the framework structure and the evacuated (activated) pores should be accessible, for, e.g., gases. The porosity is typically evaluated by the determination of the surface area and pore size/volume from cryogenic nitrogen or argon sorption experiments using the guest-evacuated (activated) material.71 Nevertheless, some ADC-based compounds can be considered as MOFs according to the IUPAC recommendations, as their crystal structures display the presence of pores or channels occupied by solvent molecules, although the permanent porosity was not experimentally demonstrated (vide infra). In the following, we will subdivide our survey into ADC-based MOFs with potential, but not accessible porosity (Section 3.1.) and into those with experimentally assessed porosity (Section 3.2.).3.1 ADC-based metal–organic frameworks (MOFs) with potential, but not accessible porosity
The discovery of room temperature synthesis of MOFs, including that of famous MOF-5, MOF-74, MOF-177, or MOF-199 marked also the starting point for the development of ADC-based MOFs. The very first ADC-based MOF with a divalent transition metal was a zinc-acetylenedicarboxylate, namely, MOF-31 that was reported in 2001 by Yaghi et al. MOF-31 with the formula (Et3NH)2[Zn(ADC)2] (Et3NH+ = triethylammonium cation) was synthesized at room temperature from an ethanolic mixture containing zinc nitrate Zn(NO3)2·6H2O and H2ADC under slow vapor diffusion of an ethanolic solution of triethylamine.72The crystal structure of the obtained cubic colorless crystals is constructed from tetrahedral Zn2+ cations, which are monodentately linked by four carboxylate groups from four different ADC ligands. Tetrahedral {Zn(CO2)4} SBUs are connected by ADC linkers, resulting in an anionic [Zn(ADC)2]2− network having an augmented diamond topology. The voids of the network are filled through interpenetration73–77 of a symmetry-related network. Charge-compensating triethylammonium cations fill the remaining pores with a diameter of ∼8 Å (Fig. 26(a) and (b)). It is unlikely that the triethylammonium cations may be removed from the pores, since they play an important role as counterions of the anionic framework. To the best of our knowledge, no cation exchange reactions have been attempted up to now.72
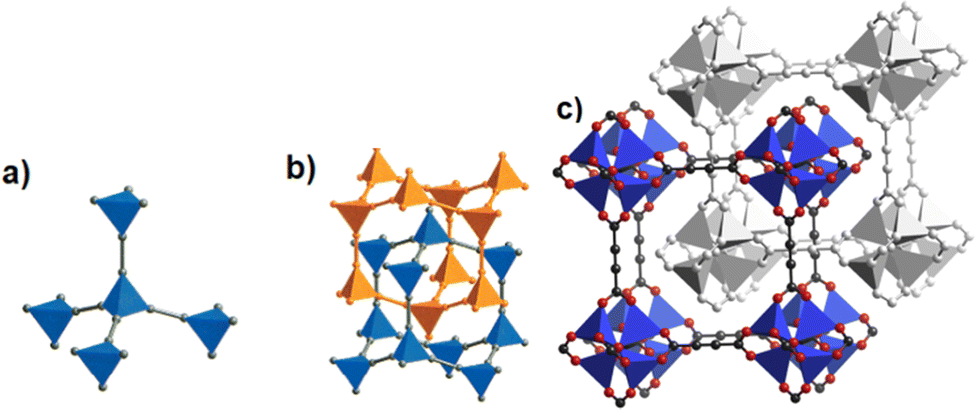 | ||
| Fig. 26 (a) Tetrahedral SBUs formed by carboxylate carbon atoms (gray spheres) connected by acetylene units (gray rods), (b) to yield a two-fold interpenetrating framework with a diamond topology in MOF-31, [Et3NH]2[Zn(ADC)2], (c) the doubly interpenetrated structure of IRMOF-0, [Zn4(μ4-O)(ADC)3] with a pcu topology modeled in Cerius2; C, black; O, red; and Zn, blue. The second interpenetrating framework is represented in gray. Adapted from ref. 72 with permission of American Chemical Society, copyright 2001, and from ref. 79 with permission of Elsevier Ltd, copyright 2008. | ||
By replacing the ethanol solvent against dimethylformamide (DMF), in the synthesis procedure, as well as Zn(NO3)2·6H2O against Zn(OOCCH3)2·2H2O, under the addition of triethylamine (Et3N) as the deprotonating agent for acetylenedicarboxylic acid (H2ADC) at room temperature, a zinc-acetylenedicarboxylate MOF was obtained, which adopts the same structural topology as MOF-5 (also known as IRMOF-1).78 It consists of tetrahedral {Zn4(μ4-O)} building blocks connected by acetylenedicarboxylate struts to form a cubic network with a pcu topology. The carboxylate groups of the ADC linker span the six edges of the {Zn4(μ4-O)} tetrahedron, coordinated to two Zn atoms, and each ADC linker bridges two {Zn4(μ4-O)} units. This material was considered as the smallest member of the IRMOF series and was thus named IRMOF-0.79 IRMOF-0 of formula [Zn4O(ADC)3]·(Et3N)6 was obtained as a microcrystalline powder and its structure was therefore elucidated from powder X-ray diffraction data and modelled using Cerius.2 Similar to MOF-31, its structure consists of doubly interpenetrated networks (Fig. 26(c)).79 Since the parent MOF-5 has reportedly been obtained both as non-interpenetrated and doubly-interpenetrated networks,80,81 it is conceivable that IRMOF-0 could also be prepared as a non-interpenetrated network.
Due to the low thermal stability of ADC, IRMOF-0 decomposes at a relatively low temperature of 120 °C, compared to the other members of the IRMOF series, which are stable up to 400 °C or above.82 It is worth noting that, although IRMOF-0 can be considered as a MOF-5 analogue, it was observed that the pore apertures were too small for any guest exchange reactions due to the double interpenetration of its frameworks. Consequently, the removal of trapped guest molecules (deduced from elemental analysis and IR spectra) was not possible and its porosity could not be accessed by gas adsorption.79
Other slight adjustments in the synthesis procedure resulted in the formation of modified structures. Best-Thompson et al. recently obtained two other 3D zinc acetylenedicarboxylates by slightly changing the synthesis approach with respect to the conditions which yielded MOF-31. By using Zn(NO3)2·6H2O and replacing trimethylamine with Hünig's base (HUN = EtiPr2N) as the deprotonating agent, two materials entitled ZnADC1 and ZnADC2 with the respective formulae (HHUN)2[Zn2(ADC)3]·(HHUN)(NO3) and (HHUN)2[Zn3(ADC)4] (HHUN+ = N,N′-diisopropylethylammonium ≡ protonated Hünigs base) were obtained as single crystals.58 ZnADC1 was obtained by a vapor diffusion technique, whereas ZnADC2 was obtained using a “layered reaction”, i.e., an ethanolic solution of the metal salt as the bottom layer was overlaid with an ethanolic solution of H2ADC and HUN as the top layer, the two solutions being separated by a layer of ethanol. ZnADC1 consists of a wine-rack type anionic 3D framework with I0O3 connectivity of formula [Zn2(ADC)3]2−, which is built on Zn2 paddle-wheel units (Fig. 27). Each ADC linker is connected such that one of its carboxylate groups bridges two Zn2+ cations in the paddle-wheel unit bidentately, while the other carboxylate group coordinates monodentately to the apical position of a Zn2+ cation in the adjacent paddle-wheel unit. This bridging mode leaves one uncoordinated oxygen atom in each ADC linker. The charge of the anionic framework [Zn2(ADC)3]2− is balanced by two HHUN+ cations per formula unit. In ZnADC1 there is an additional (HHUN)(NO3) guest pair per formula unit included in the channels.58
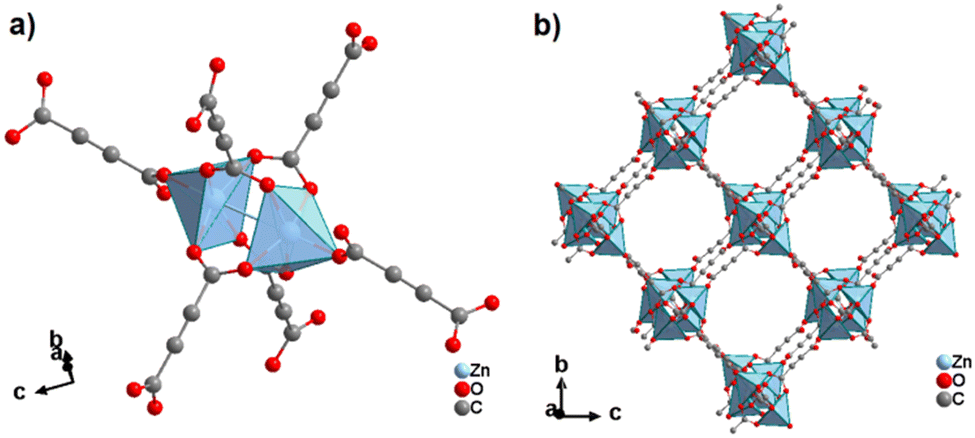 | ||
| Fig. 27 Crystal structure of ZnADC1, (HHUN)2[Zn2(ADC)3]·(HHUN)(NO3), showing: (a) the dinuclear Zn2 paddle-wheel unit connected by six ADC linkers and (b) the three-dimensional packing of the wine-rack-like framework. The HHUN+ cations and the (HHUN)(NO3) ionic pairs occupying the pores of the MOF framework are omitted for clarity. Graphics redrawn from the cif file (CSD-Refcode PAGRUO).58 | ||
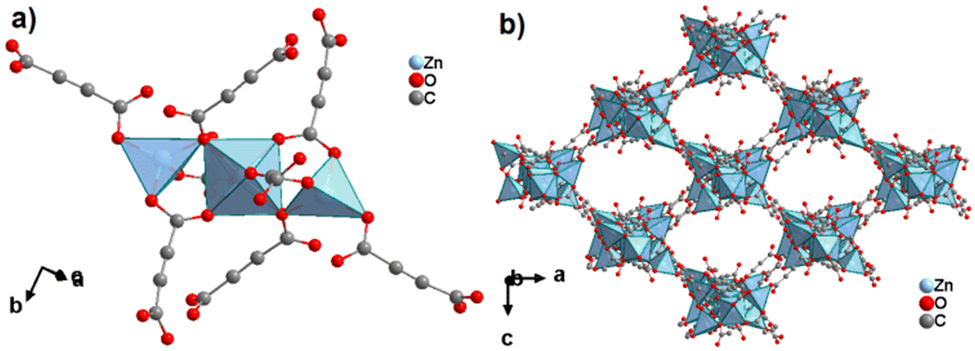
The structure of ZnADC2 consists of trinuclear Zn3 nodes made-up of a ZnO6 octahedron trans-corner connected with two ZnO4 tetrahedra. All ADC linkers – four crystallographically distinct linker anions are found in the crystal structure – coordinate in a bidentate bridging mode with one of their carboxylate groups, while the other carboxylate group coordinates monodentately, either with a single Zn2+ cation or bridging two Zn2+ cations. One oxygen atom of these carboxylate groups remains therefore uncoordinated. Each {Zn3} node is connected to others by eight ADC linkers to yield an I0O3-type 3D framework with oval-shaped channels (Fig. 28). These channels are partially occupied by HHUN+ cations, which balance the charge of the anionic [Zn3(ADC)4]2− framework. Therefore, it is unlikely that there is an accessible porosity in ZnADC1 and ZnADC2 because of the bulky (HHUN)(NO3) guests and HHUN+ counterions. In case of cation removal with charge balance, the calculation of the void space by Mercury indicates about 70% empty space for ZnADC1 and 58% for ZnACD2. One could imagine to replace HHNU+ and NO3− with smaller ions, resulting in only partly occupied pores. However, neither the stability of the frameworks of ZnADC1 and ZnADC2 in the presence of other counter ions nor the chemical stability for such an ion exchange process has been proven up to now. ZnADC1 decomposes at 130 °C.58 Such information is not available for ZnADC2, as no phase-pure sample could be synthesized.
 | ||
| Fig. 28 Crystal structure of ZnADC2, (HHUN)2[Zn3(ADC)4], showing: (a) the trinuclear Zn3 unit consisting of a ZnO6 octahedron trans-corner connected to two ZnO4 tetrahedra and (b) the three-dimensional packing of the I0O3-type framework. The HHUN+ cations occupying the pores of the MOF framework are omitted for clarity. Graphics redrawn from the cif file (CSD-Refcode PAGSAV).58 | ||
It is noticeable that 3D ADC-based coordination polymers with (potential) porosity based on low-valent metals have only been obtained with divalent Zn2+ up to now. Other mono- and divalent metals seem to have the tendency to form only non-porous coordination polymers, as in most examples the ADC ligands are aligned more or less parallel to each other (see the summary of Section 2). This tendency however changes, when moving to ADC-based compounds with high-valent M3+ metal cations, in which ADC ligands are no longer arranged parallel to each other. This is for example found in the second polymorph of bismuth acetylenedicarboxylate of formula [Bi(ADC)1.5(H2O)3]·H2O (cp. Fig. 25), which exhibits a 3D framework structure.67 This compound was obtained as single crystals by slow evaporation of an aqueous solution containing Bi(NO3)3·5H2O and H2ADC at room temperature. Remarkably, the 2D coordination polymer [Bi(ADC)1.5(H2O)2]·2H2O (see Section 2), which has actually the same composition and can be understood as a hydrate isomer of the former, is synthesized using very similar conditions with only a slightly higher concentration of H2ADC.67 The crystal structure of the 3D MOF [Bi(ADC)1.5(H2O)3]·H2O is constructed from dinuclear {Bi2O8(H2O)6} units consisting of two edge-sharing BiO8 polyhedra (Fig. 29(a)), which are connected by ADC linkers to a 3D framework structure (Fig. 29). The framework displays pores filled with non-coordinating water molecules. Each {Bi2O8(H2O)6} polyhedron consists of two symmetry-related Bi3+ cations coordinated by one carboxylate group of an ADC ligand coordinating in a chelating-bridging μ2-η2:η1 mode, and three further ADC ligands coordinating with their carboxylate groups monodentately (η1 mode). The coordination sphere of each Bi3+ is completed by three water molecules. The crystal structure contains two crystallographically different ADC ligands: one ADC ligand coordinates with each of its carboxylate groups monodentately, while the other ADC linker is connecting such that both of its carboxylate groups coordinate two Bi3+ cations in a chelating-bridging μ2-η2:η1 mode (Fig. 29(b)).67 Note that this description is slightly different from the one chosen in Section 2.5, as only Bi…O distances up to 2.75 Å are considered. As this BiIII-MOF could not be obtained in a single-phase form, no further investigations on its thermal stability and a possible release of water molecules were reported.
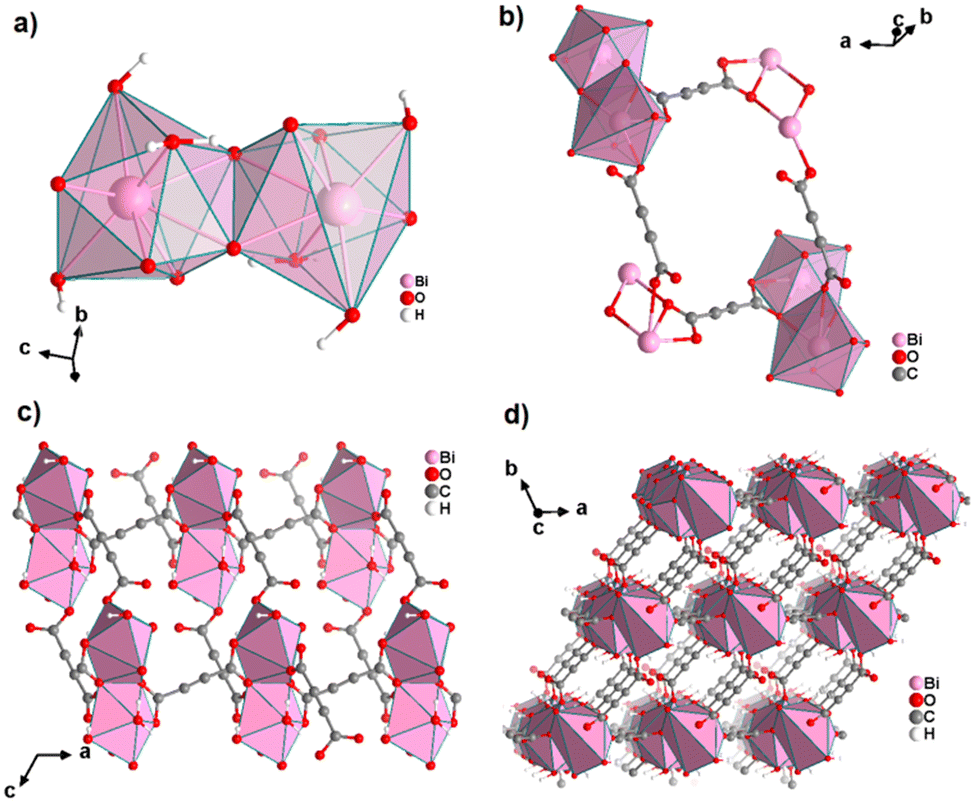 | ||
| Fig. 29 Crystal structure of [Bi(ADC)1.5(H2O)3]·H2O showing: (a) the dinuclear unit of two edge-sharing BiO8 polyhedra, (b) the connectivity of ADC linkers, (c) the connection of the dimeric units in a view along [010] and (d) the three-dimensional packing of the framework. Non-coordinating water molecules within the pores are omitted for clarity. Graphics redrawn from the cif file (CSD-Refcode NERQUY).67 | ||
The occurrence of dinuclear units consisting of two edge-sharing polyhedra was also observed in ADC-based compounds with trivalent rare earth metal cations. Again, the combination of M3+ and the ADC linker resulted in 3D coordination polymers almost across the entire 4f series (Fig. 30), which may be classified as MOFs. Independently, three different groups (Yan et al. in 1995, Michaelides et al. in 2005 and Gramm et al. in 2018) obtained MOFs of the same formula [RE2(ADC)3(H2O)6]·2H2O and crystal structure, but with different metal cations RE3+ (REIII = Gd, La, Ce, Pr, Nd, Sm, Eu, Tb, Dy, Ho, Er, and Y).83–85 Their crystal structures are built up from nine-coordinated rare earth metal cations forming monocapped square antiprisms (REO9), which are edge-shared to dinuclear units (Fig. 30(a)). The latter are nodes of an ADC-linked 3D polymer displaying one-dimensional channels filled with crystal and coordinated water molecules.
 | ||
| Fig. 30 Crystal structure of [Dy2(ADC)3(H2O)6]·2H2O showing: (a) the dinuclear unit of two edge-sharing DyO9 polyhedra, (b) the connectivity of ADC linkers, (c) the connection of the dimeric units in a view along [010] and (d) the three-dimensional packing of the framework. Non-coordinating water molecules within the pores are omitted for clarity. Graphics redrawn from the cif file (CSD-Refcode NILSUZ).85 | ||
There are two crystallographically different ADC linkers in the structure, of which one shows an inversion symmetry and acts exclusively in a chelating-bridging mode. The other ADC linker coordinates in a monodentate mode with one of its carboxylate groups and in a bridging mode with the other one (Fig. 30(b)). In general, these RE3+ MOFs were obtained at room temperature either at the phase boundary of a silica gel containing acetylenedicarboxylic acid (H2ADC) layered with an aqueous solution containing the rare earth metal nitrate or by slow evaporation of an aqueous solution containing the respective rare earth metal acetate RE(OAc)3·xH2O and H2ADC. All the aforementioned RE-ADCs are thermally only stable up to less than 200 °C.83–85 Most remarkably, Michaelides et al. demonstrated for RE = Ce that non-coordinating water molecules can be removed at 100 °C with the retention of the initial crystal structure.84 This was apparently the very first experiment that addressed the porosity of these materials.
Rare-earth metal acetylenedicarboxylates are of peculiar interest, as the very first successful synthesis of an ADC-based CP or MOF under hydrothermal conditions was obtained with europium(III). Serre et al. reported in 2005 a 3D europium(III) acetylenedicarboxylate with an open framework structure, denoted as MIL-95 and the formula [EuIII2(ADC)(H2O)2(CO3)2]·xH2O.86 The structure of MIL-95 consists of two-dimensional europium carbonate subnetworks pillared by acetylenedicarboxylate linkers, yielding a three-dimensional framework structure (Fig. 31). The framework exhibits one-dimensional channels filled with crystal water molecules. Noteworthy, MIL-95 contains the same nine-coordinated monocapped square antiprisms (EuO9) that have been found in the RE-ADC MOFs reported by Yan et al., Michaelides et al. and Gramm et al. However, in MIL-95 these polyhedra are edge-sharing along the [100] direction and corner-sharing along the [001] direction, thus forming a 2D close-packed structure of EuO9 polyhedra in the (010) plane bridged by carbonates (Fig. 31(a)).86
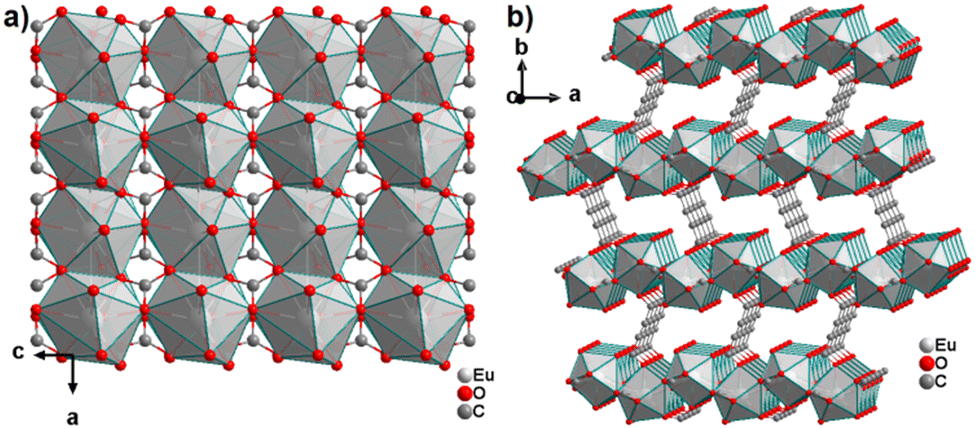 | ||
| Fig. 31 Crystal structure of MIL-95 showing: (a) the packing of edge and corner-sharing EuO9 polyhedra in the (010) plane and (b) the three-dimensional packing of the framework. The crystal water molecules in the pores are omitted for clarity. Graphics redrawn from the cif file (CSD-Refcode FIFPOA).86 | ||
All ADC linkers coordinate in a bridging mode with both carboxylate groups and thus interconnect these layers of EuO9 polyhedra (Fig. 31(b)). Interestingly, MIL-95 was synthesized under hydrothermal conditions at 120 °C from a mixture of europium(III) nitrate, acetylenedicarboxylic acid and sodium hydroxide in water.86 As the reaction time was relatively short (one night) for the hydro-/solvothermal reaction, H2ADC obviously decomposed only partly to form carbonate anions. This exemplifies clearly the difficulties in obtaining ADC-based MOFs under hydro-/solvothermal conditions. The thermal dehydration of MIL-95 at 130 °C results in an irreversible pore contraction followed by the complete collapse of the framework above 230 °C. Therefore, the porosity of MIL-95 could not be evaluated experimentally.86 However, it is not clear whether the irreversible pore contraction was solely due to water removal from the pores or stems from the ADC-inherent low thermal stability of the framework. Evacuating the pores at low temperatures (e.g. using supercritical CO2 drying) was not reported to be attempted, but might have yielded a permanently porous MIL-95.87
3.2 ADC-based metal–organic frameworks (MOFs) with experimentally assessed porosity
The introduction of tetravalent metals such as zirconium, hafnium and cerium was important in the MOF field.88 In particular, the discovery of zirconium(IV) terephthalate, the so-called UiO-66 (UiO stands for University i Oslo), has enabled the isoreticular synthesis of other (isostructural) MOFs using various linear carboxylate linkers.89 This was also a gateway for the synthesis of the first ADC-based MOF with permanent and experimentally assessed porosity by gas sorption studies with BET surface area and pore volume determination. It was only in 2018 that the first ADC-based MOF with measurable porosity was reported, namely with zirconium(IV) acetylenedicarboxylate, also denoted as Zr-HHU-1 (HHU stands for Heinrich Heine Universität/University).90 (Following the naming of this ADC-based Zr-MOF, we realized that the abbreviation HHU was also used by colleagues at Hohai University in China for their MOFs with a report on HHU-2 in 2017.91) Zr-HHU-1, whose structure is analogous to that of UiO-66, consists of octahedral [Zr6(μ3-O)4(μ3-OH)4]12+ secondary building units twelvefold connected to each other by ADC linkers yielding a microporous network with a fcu topology and the ideal formula [Zr6(μ3-O)4(μ3-OH)4(ADC)6] (Fig. 32(a) and (b)). In this structure, all ADC linkers coordinate in a bridging (μ2-η1:η1 mode) fashion with both carboxylate groups. The network possesses tetrahedral and octahedral cavities having diameters of about 5.8 Å and 9.6 Å, respectively (Fig. 32(c) and (d)). The cages are accessible through triangular windows having a diameter of about 4.4 Å.90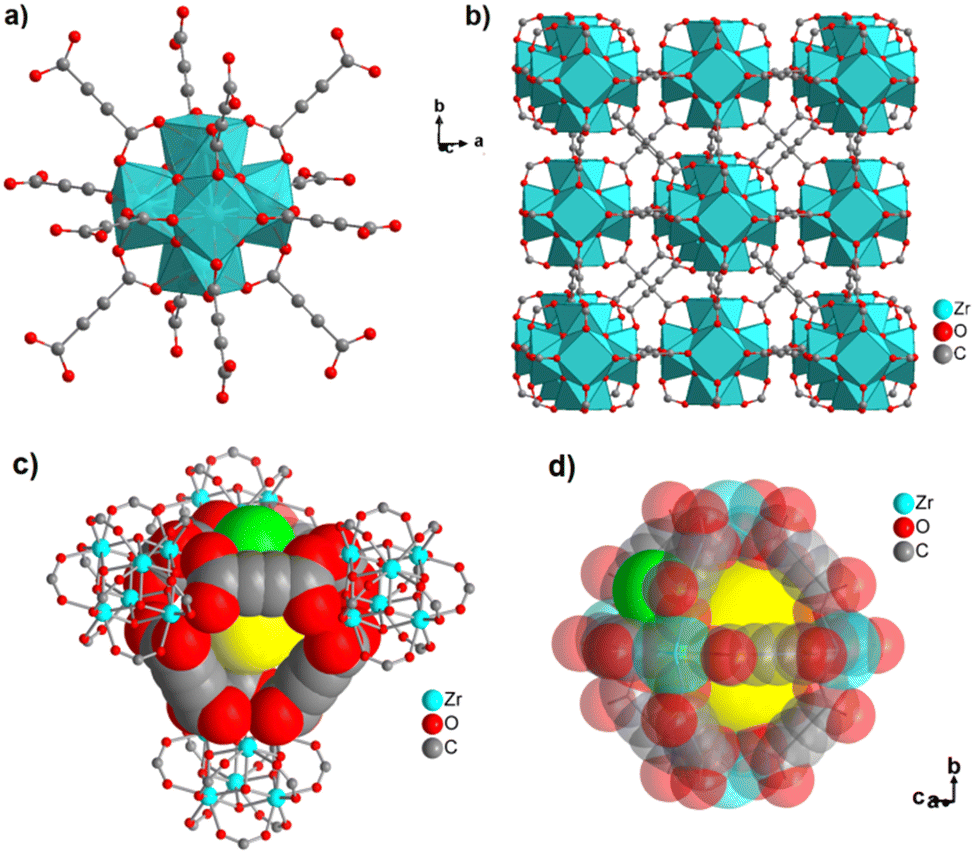 | ||
| Fig. 32 Crystal structure of Zr-HHU-1 showing: (a) the hexanuclear Zr6-cluster connected to twelve ADC linkers, (b) the three-dimensional packing of the fcu network, (c) the tetrahedral cavity, and (d) the octahedral cavity both with their available pores (yellow spheres) and triangular windows (green spheres). Graphics adapted from ref. 90 with permission of Wiley & Sons, copyright 2019. | ||
Zr-HHU-1 was synthesized by reacting zirconium oxychloride (ZrOCl2) with acetylenedicarboxylic acid in DMF at 85 °C with the addition of acetic acid as the crystallization agent (modulator). Zr-HHU-1 has like IRMOF-0 a relatively low thermal stability only up to 180 °C. However, unlike IRMOF-0, the pores of Zr-HHU-1 were successfully evacuated by supercritical CO2 and its porosity was established by the adsorption of argon, nitrogen, CO2, H2 and water vapor. N2, CO2 and H2 adsorption isotherms were all of type I according to the IUPAC classification, which attests the permanent microporous nature of Zr-HHU-1.71
After successfully synthesizing an ADC-based MOF, the next challenge in order to access its porosity was the successful evacuation of its pores without the collapse of the framework. The thermal lability of the ADC linker is usually transmitted to the ADC-based compounds, with the exception of SrADC and EuIIADC, for which an unexpectedly high thermal stability up to about 400 °C was observed, which is still not understood completely. The challenge for the successful activation of MOFs stems from the thermal activation under reduced pressure, which might be difficult for thermally labile materials. In the case of Zr-HHU-1 supercritical CO2 drying of the sample was adopted for pore evacuation, which resulted in a pore-free sample with retention of its crystallinity and permanent porosity. The Brunauer-Emmett-Teller (BET) surface area was calculated using the nitrogen sorption experiment to be 550 m2 g−1 and the micropore volume was found to be 0.19 cm3 g−1. The CO2 and H2 uptake capacities at 1 bar were 1.69 mmol g−1 (273 K) and 4.1 mmol g−1 (77 K), respectively. Zr-HHU-1 adsorbs 205 mg g−1 of water vapor at 20 °C and a relative pressure P/P0 = 0.9.
Surprisingly, Zhao et al. demonstrated some time later that Zr-HHU-1 (also denoted as UiO-66-ADC) can be synthesized at an elevated temperature of 120 °C, when the reaction mixture is made from zirconium chloride (ZrCl4), acetylenedicarboxylic acid and formic acid as the modulator (instead of acetic acid as in the initial synthesis) in DMF.92 Apparently, the composition of the reaction mixture, notably its low pH value, seems to stabilize thermally labile H2ADC in solution. This finding is of paramount importance, as it could provide a guideline on how to successfully obtain ADC-based MOFs under solvothermal conditions. Furthermore, by simply replacing DMF as a solvent with water, these authors obtained another zirconium acetylenedicarboxylate at 120 °C, namely NUS-36, whose structure is built up from eight-connected [Zr6O6(OH)2]10+ clusters and ADC linkers with cluster-capping formates, leading to an ultramicroporous framework with a bcu topology and the formula [Zr6O6(OH)2(ADC)4(HCOO)2] (Fig. 33(b)). NUS-36 is thermally stable up to about 220 °C. Its porosity was assessed by N2, CO2, H2O, O2, ethylene (C2H4) and ethane (C2H6) adsorption experiments. The pores of NUS-36 are not accessible to N2 and O2 due to their very small sizes (diameter smaller than 3.6 Å). However, NUS-36 adsorbs about 48 cm3 g−1 CO2 at 273 K and a relative pressure of P/P0 = 0.03 following a type I adsorption isotherm. NUS-36 likewise adsorbs about 150 cm3 g−1 water at 293 K and a relative pressure of P/P0 = 0.9 (Fig. 33(c) and (d)).92 The sorption behavior can be understood from the decreasing kinetic diameters in the order: N2 (3.64 Å) > O2 (3.46 Å) > CO2 (3.3 Å) > H2O (2.65 Å).
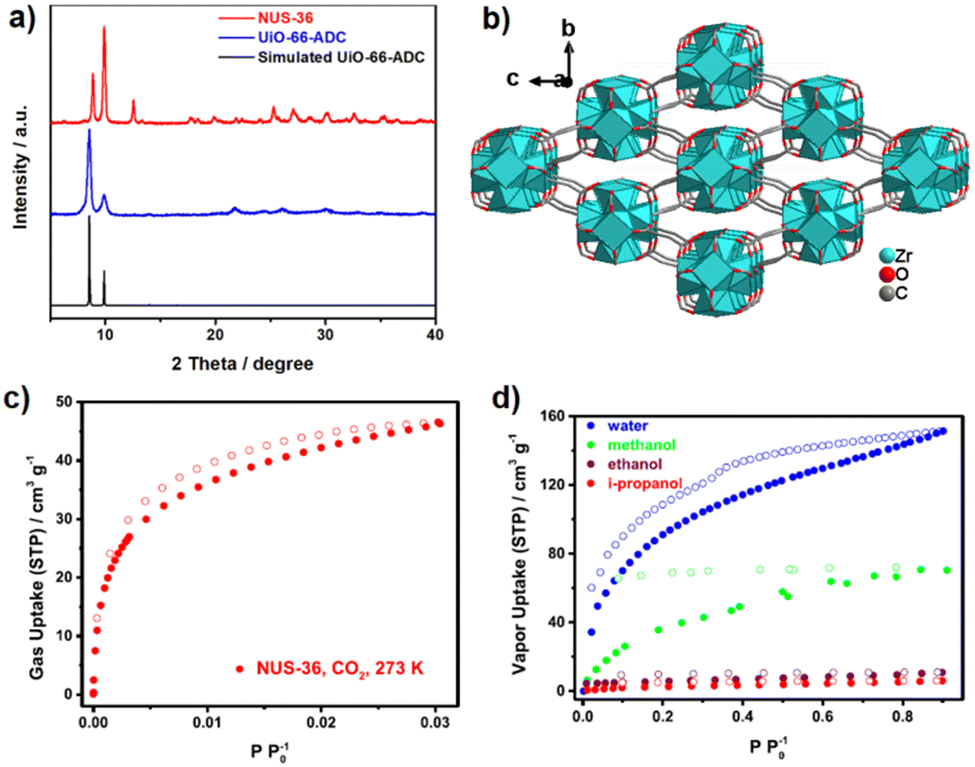 | ||
| Fig. 33 (a) Powder X-ray diffraction pattern of NUS-36 compared with that of UiO-66-ADC, (b) three-dimensional packing of the bcu network of NUS-36, (c) CO2 adsorption isotherm for NUS-36 at 273 K, and (d) adsorption isotherms of water, methanol, ethanol and i-propanol for NUS-36 at 273 K. Adapted from ref. 92 with permission of American Chemical Society, copyright 2019. | ||
The similarities in chemical properties of hafnium and zirconium enabled also to obtain an isostructural hafnium-based analogue of Zr-HHU-1, namely Hf-HHU-1 of formula [Hf6(μ3-O)4(μ3-OH)4(ADC)6]. Hf-HHU-1 was synthesized similarly to Zr-HHU-1, except that hafnium oxychloride (HfOCl2) was used as the metal salt instead of zirconium oxychloride (ZrOCl2) (Fig. 34(a)). Hf-HHU-1 is thermally stable up to about 200 °C. Its BET surface area of 476 m2 g−1 and micropore volume of 0.17 cm3 g−1 were determined from nitrogen sorption isotherms (Fig. 34(c)).34 For both Hf-HHU-1 and Zr-HHU-1, it was observed that the structures collapsed at a temperature above 100 °C even before the decomposition of the material. For this reason, the usual thermal activation under vacuum was an inappropriate means to evacuate the pores of these MOFs. As before, supercritical CO2 exchange/drying was employed to yield permanently porous Hf-HHU-1. In general, scCO2 may therefore be the method of choice for the activation of ADC-based MOFs due to their typically low thermal stability.93 However, it was determined from an elemental analysis that some residual DMF is still present in the pores of HHU-1 even after scCO2 drying. This accounts for the low BET surface area (476 m2 g−1) and micropore volume (0.17 cm3 g−1) of Hf-HHU-1 compared to the theoretically estimated BET surface area of 707 m2 g−1 and micropore volume of 0.26 cm3 g−1 obtained from the crystallographic data. This means that higher experimental surface area and pore volume of Zr-HHU-1 and Hf-HHU-1 should be reached for fully activated materials.
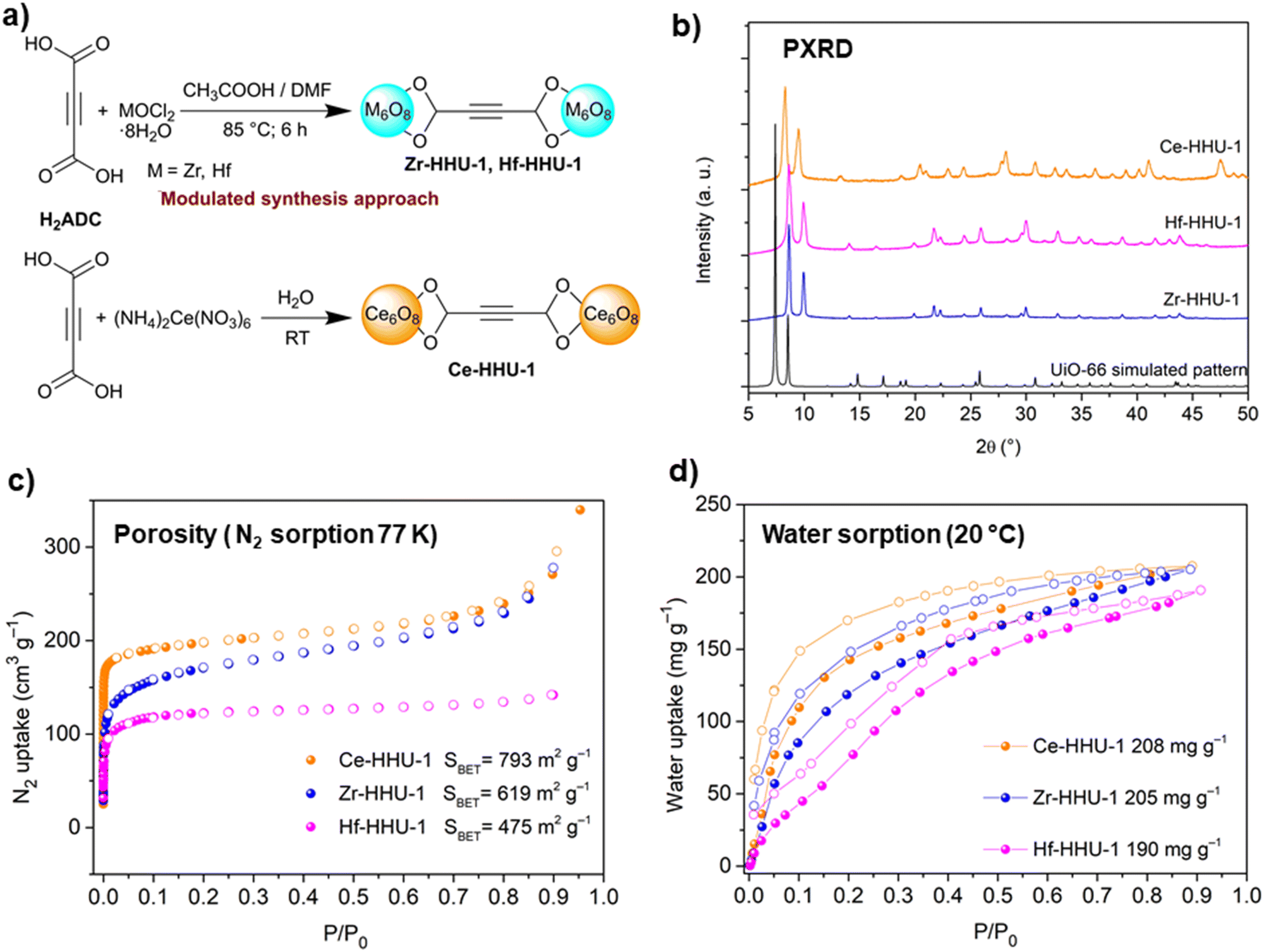 | ||
| Fig. 34 (a) Reaction schemes for the synthesis of M-HHU-1 (M = Zr, Hf, Ce), (b) powder X-ray diffraction patterns of M-HHU-1 (M = Zr, Hf, Ce), compared with the simulated pattern for UiO-66, (c) nitrogen adsorption isotherms at 77 K, and (d) water sorption isotherms of M-HHU-1 (M = Zr, Hf, Ce) at 20 °C. Graphics adapted from ref. 34, 90 and 94. | ||
The aforementioned hypothesis was confirmed by the synthesis of a cerium(IV)-based analogue of Zr-HHU-1, denoted as Ce-HHU-1 of formula [Ce6(μ3-O)4(μ3-OH)4(ADC)6].94 Ce-HHU-1 features a slightly larger cubic cell parameter a manifested in the powder X-ray diffraction pattern by a shift of the reflections to lower 2θ values compared to Zr-HHU-1 and Hf-HHU-1 (Fig. 33(b)). This is consistent with the larger ionic radius of Ce4+ (0.97 Å) in comparison to Hf4+ and Zr4+, which have almost the same ionic radius (0.84 Å),95 resulting in lattices with almost the same dimensions for the latter. Due to a heavier metal cation, Ce-HHU-1 should have a lower BET surface area and water uptake capacity per weight compared to Zr-HHU-1. However, it was observed that Ce-HHU-1 has a BET surface area of about 790 m2 g−1, a micropore volume of 0.24 cm3 g−1, and a water vapor adsorption capacity of 208 mg g−1 at 293 K and a relative pressure P/P0 = 0.9 (Fig. 34d). Table 2 gives, for comparison purposes, a summary of the porosity and water vapor uptake of Zr-HHU-1, Hf-HHU-1 and Ce-HHU-1.
| Material | S BET (m2 g−1)a | V micropore b (cm3 g−1) | H2O uptaked (mg g−1) |
|---|---|---|---|
| Zr-HHU-1 | 550 | 0.19 | 205 |
| Hf-HHU-1 | 476 | 0.17 | 190 |
| Ce-HHU-1 | 790 | 0.24 | 208 |
Ce-HHU-1 was synthesized by reacting acetylenedicarboxylic acid and ammonium cerium(IV) nitrate in water at room temperature, unlike the modulated synthesis in DMF, from which Zr-HHU-1 and Hf-HHU-1 were obtained (Fig. 34(a)). The use of water as the solvent instead of DMF, as well as the absence of monocarboxylic acid as the modulator allowed a better activation by scCO2 drying. Therefore, a higher BET surface area and water uptake were obtained (Fig. 34(c) and (d)). The CO2 uptake capacity of Ce-HHU-1 was found to be 3.2 and 2.5 mmol g−1 at 273 K and 293 K, respectively (Fig. 35(a)).94
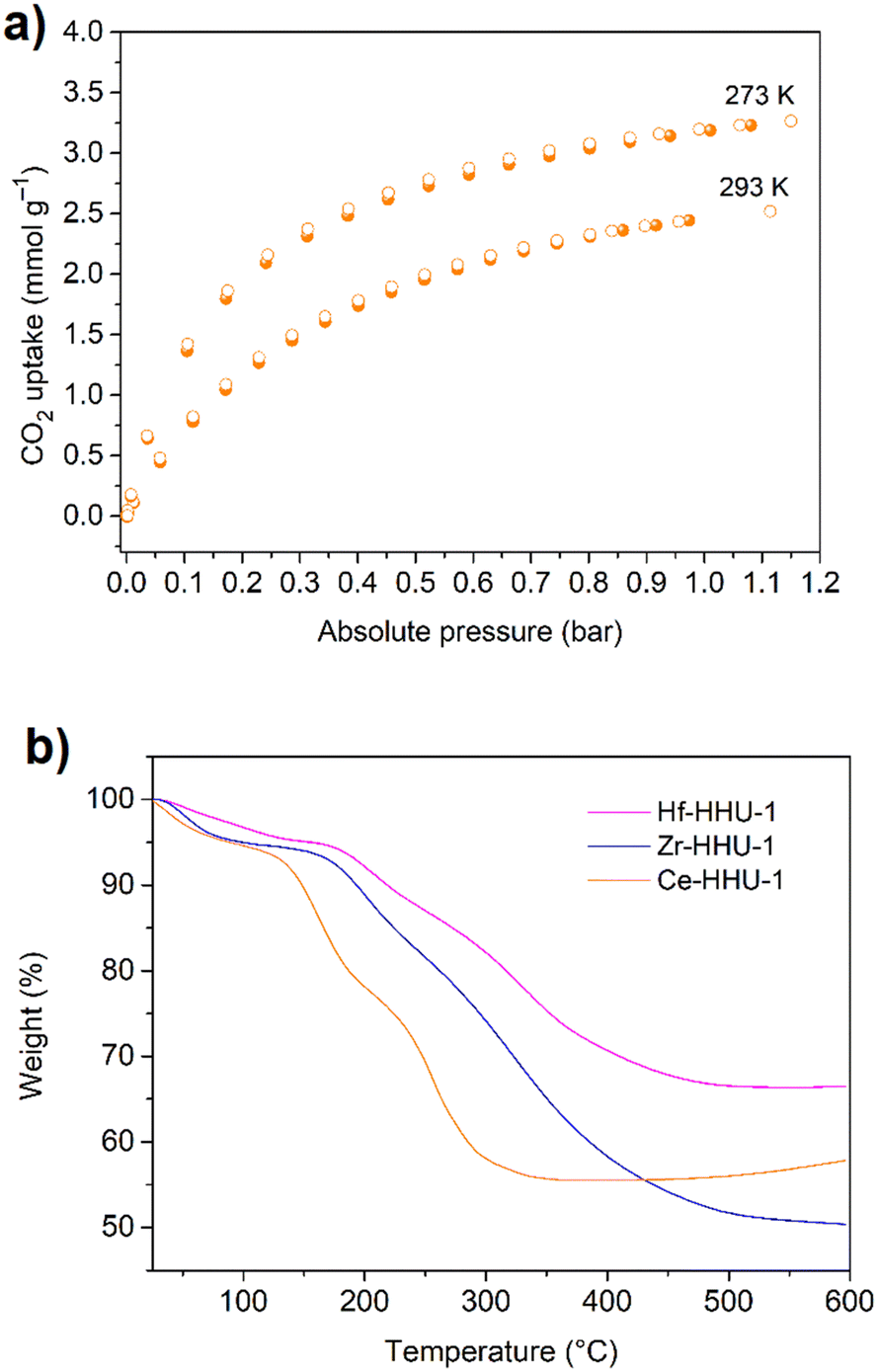 | ||
| Fig. 35 (a) CO2 adsorption isotherms of Ce-HHU-1 at 273 K and 293 K, and (b) thermogravimetric analyses of M-HHU-1 (M = Zr, Hf, and Ce). Graphics adapted from ref. 34, 90 and 94. | ||
Noteworthy, a very high yield of up to 92% was obtained in the synthesis and the absence of (missing linker or cluster)-defects in the framework of Ce-HHU-1 was deduced from thermal gravimetric analysis, using the method previously reported by Shearer et al. for the analogous UiO-66.89,96 The defect-free structure indicates that no decarboxylation reaction of H2ADC occurred during the synthesis of Ce-HHU-1 at room temperature, in contrast to the defective structure obtained for Ce-UiO-66-ADC (vide infra).
Ce-HHU-1 has a slightly lower thermal stability compared to Zr-HHU-1 and Hf-HHU-1 and decomposes already at about 120 °C (Fig. 35(b)).
Ce-HHU-1 was later also successfully synthesized by Airi et al., who denoted this material as Ce-UiO-66-ADC. In their work, Ce-UiO-66-ADC was obtained at elevated temperatures up to 100 °C in a DMF/water solvent mixture, resulting in a material with some drawbacks.97 The elevated temperature of the synthesis led to a very low yield of only 4% due to a significant decomposition of H2ADC. The yield was increased to 40% by adding a Brønsted base (triethylamine) to the reaction mixture, in order to reduce the decarboxylation of H2ADC. However, combined IR and Raman spectroscopy analyses of the obtained Ce-UiO-66-ADC revealed the presence of terminal alkyne groups (R–CC–H) in the material. This indicates that decarboxylation was not completely suppressed at a temperature of 100 °C, and therefore a very defective Ce-UiO-66-ADC sample was obtained. Raman spectroscopy revealed that the local defects in the material consist of propiolates (from the decarboxylation of H2ADC), which are connected to the Ce6O8 clusters by their carboxylate groups and remain free (uncoordinated) at their terminal alkyne ends (Fig. 36).
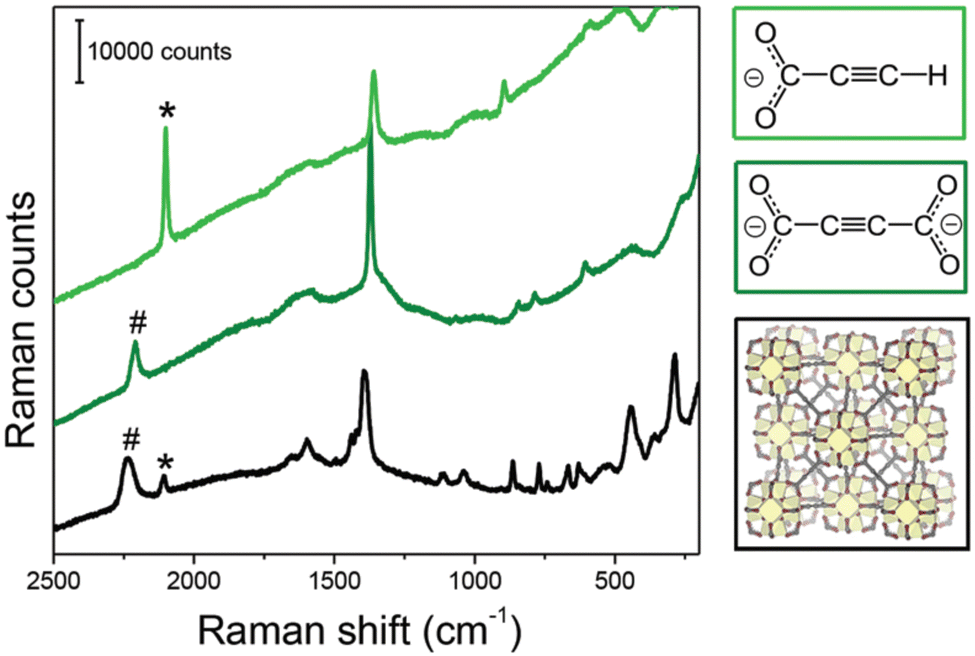 | ||
| Fig. 36 Raman spectra of Ce-UiO-66-ADC (black), an H2ADC solution (dark green) and a propiolic acid solution (light green). * and # represent the bands due to the stretching vibrations of –CC– triple bonds for H2ADC (symmetric R–CC–R) and propiolic acid (non-symmetric terminal R–CC–H), respectively. The presence of both in the spectrum of Ce-UiO-66 indicates the presence of defective sites in the framework, as the terminal alkyne cannot connect the Ce6O8 clusters. Reproduced from ref. 97 with permission of the Royal Society of Chemistry, copyright 2020. | ||
This work clearly shows the challenges that need to be overcome, when using elevated temperatures in the synthesis of ADC-based MOFs/CPs. The authors tried to activate the material thermally under vacuum, but they could not remove the DMF molecules from the pores, since the (defective) MOF structure starts collapsing as early as at 80 °C. Therefore, only a very low Langmuir surface area not exceeding 400 m2 g−1 was obtained from nitrogen sorption measurements at 77 K.97 This again highlights the importance of scCO2 drying to activate such ADC-based materials.
The metal–organic frameworks based on the acetylenedicarboxylate ligand that has been discussed in Section 3 are summarized in Table 3.
| Compound acronym | Formula | Synthesis conditions | Crystal system | Space group | a, b, c (Å) | α, β, γ (°) | Thermal stability | Porosity | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Et3N = triethylamine, Et3NH+ = triethylammonium cation, HHUN+ = EtiPr2NH+ (protonated Hünigs base), NA: not available, RT: room temperature, SBET = BET surface area, Vmicropore = micropore volume. | |||||||||
| MOF-31 | (Et3NH)2[Zn(ADC)2] | RT: vapor diffusion in EtOH in the presence of triethylamine | Cubic |
Pnm |
10.8212(13) | 90 | NA | NA, counter cations occupy pores | 72 |
| IRMOF-0 | [Zn4O(ADC)3]·(Et3N)6 | RT: in DMF in the presence of triethylamine | Cubic |
Fdc |
21.84 | 90 | Decomposes at 120 °C | NA, trapped solvent molecules in the pores | 74 |
| ZnADC1 | (HHUN)2[Zn2(ADC)3](HHUN)(NO3) | RT: vapor diffusion in EtOH in the presence of N,N′-diisopropylethylamine | Orthorhombic | Pna21 | 18.1647(3) | 90 | Decomposes at 150 °C | NA, counter cations occupy pores | 58 |
| 15.6263(2) | |||||||||
| 15.1171(2) | |||||||||
| ZnADC2 | (HHUN)2[Zn3(ADC)4] | RT: layered reaction in EtOH in the presence of N,N’-diisopropylethylamine | Orthorhombic | Pbca | 20.7992(8) | 90 | NA | NA, counter cations occupy pores | 58 |
| 15.2528(12) | |||||||||
| 27.7139(13) | |||||||||
| [Bi(ADC)1.5(H2O)3]·H2O | RT: slow evaporation of an aqueous solution | Triclinic |
P |
8.175(3) | 96.93(4) | NA | NA, pores filled with water molecules | 67 | |
| 8.643(3) | 114.33(4) | ||||||||
| 9.196(3) | 109.40(4) | ||||||||
| [Gd2(ADC)3(H2O)6]·2H2O | NA | Triclinic |
P |
8.254(2) | 95.38(2) | NA | NA | 83 | |
| 9.605(2) | 115.82(2) | ||||||||
| 9.066(2) | 110.39(2) | ||||||||
| [RE2(ADC)3(H2O)6]·2H2O REIII = La, Ce | RT: diffusion at the phase boundary of an aqueous solution and a silica gel | Triclinic |
P |
8.307, 8.304(1) | 95.63, 95.59(1) | Decomposes at 250 °C | NA, Pores filled with water molecules | 84 | |
| 8.769, 8.728(1) | 115.61, 115.70(1) | ||||||||
| 9.204, 9.177(1) | 110.26, 110.26(1) | ||||||||
| [RE2(ADC)3(H2O)6]·2H2O REIII = Pr, Nd, Sm, Eu, Tb, Dy, Ho, Er, Y | RT: slow evaporation of an aqueous solution | Triclinic |
P |
8.202–8.286(1) | 95.01–95.45(6) | Decomposes above 200 °C | NA, Pores filled with water molecules | 85 | |
| 8.514–8.648(1) | 116.15–115.94(6) | ||||||||
| 8.989–9.163(1) | 109.84–110.74(6) | ||||||||
| MIL-95 | [EuIII(ADC)(CO3)2(H2O)2]·xH2O | Hydrothermal synthesis, 120 °C in the presence of NaOH | Orthorhombic | Pba2 | 7.6762(2) | 90 | Collapses above 230 °C | NA, Pores filled with water molecules | 86 |
| 17.8448(4) | |||||||||
| 4.8105(2) | |||||||||
| Zr-HHU-1/UiO-66-ADC | [Zr6(μ3-O)4(μ3-OH)4(ADC)6] | 85 °C in DMF in the presence of acetic acid | Cubic |
Fmm |
17.925(3) | 90 | Decomposes at 180 °C | S BET = 619 m2 g−1, Vmicropore = 0.38 cm3 g−1 | 90,92 |
| Hf-HHU-1 | [Hf6(μ3-O)4(μ3-OH)4(ADC)6] | 85 °C in DMF in the presence of acetic acid | Cubic |
Fmm |
17.8529(6) | 90 | Decomposes at 200 °C | S BET = 476 m2 g−1, Vmicropore = 0.20 cm3 g−1 | 34 |
| Ce-HHU-1/Ce-UiO-66-ADC | [CeIV6(μ3-O)4(μ3-OH)4(ADC)6] | RT: in H2O | Cubic |
Fmm |
19.1733(6) | 90 | Decomposes at 120 °C | S BET = 793 m2 g−1, Vmicropore = 0.24 cm3 g−1 | 94,97 |
| NUS-36 | [Zr6O6(OH)2(ADC)4(HCOO)2] | Hydrothermal synthesis, 120 °C in the presence of formic acid | Cubic |
Pn2 |
9.9849 | 90 | Decomposes at 250 °C | S BET = 298 m2 g−1, Vmicropore = 0.12 cm3 g−1 | 92 |
4 Distinctive features and properties of ADC-based coordination polymers and MOFs
4.1 Noncentrosymmetric networks from ADC-based CPs
Noncentrosymmetric (NCS) materials, solid-state materials crystallizing in space groups without a center of symmetry, are of interest, as important properties including ferroelectricity, pyroelectricity, piezoelectricity, or nonlinear optical (NLO) behavior, e.g. second-harmonic generation (SHG), are typically associated with these compounds98,99 Despite their critical importance in many applications such as laser technology, and optical communication, the rational design of NCS materials is still a challenge, as the majority of all compounds crystallizes in centrosymmetric crystal structures. Some strategies have been reported toward the tailored design of NCS materials aiming at increasing at least the probability of synthesizing such a compound.100,101 Schuy et al. showed that CPs containing the linear (and highly symmetric) ADC ligand typically crystallize in centrosymmetric space groups (cf.Table 1), but by adding a trigonal planar anion the probability to obtain a compound that crystallizes in a noncentrosymmetric space group was significantly increased. This concept was demonstrated for the crystal structures of the ADC-based CPs [Ba2(ADC)(NO3)2]·4H2O, [Ba7(ADC)6(NO3)2]·14H2O, and [Ba3(ADC)2Cl(NO3)]·5H2O, which crystallize in the noncentrosymmetric space groups C2221, I2, and P2212, respectively.102 It was argued that the insertion of the trigonal planar nitrate ion (NO3−) is a symmetry breaking factor that results in the formation of noncentrosymmetric crystal structures with an enhanced probability. It should be pointed out that the NO3− ion is a ligand coordinated to the Ba2+ cation and not a counterion in the outer sphere of the CP (Fig. 37). This concept is confirmed by the finding that [EuIII(H2O)2(CO3)2(ADC)]·xH2O (MIL-95) also crystallizes in the noncentrosymmetric space group Pba2 (vide supra).86 Here, the trigonal planar carbonate anion (CO32−) functions as a coordinating ligand, which seems to “break” the symmetry.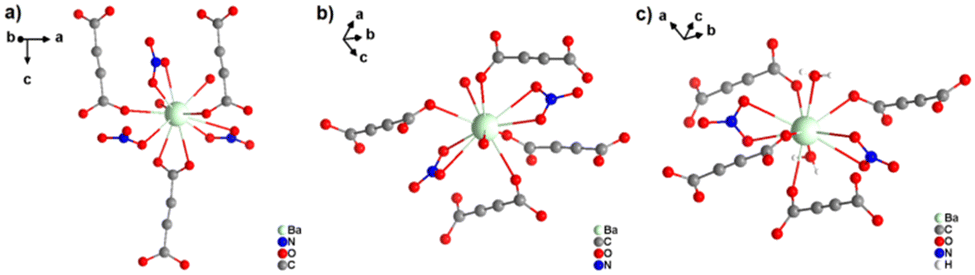 | ||
| Fig. 37 Coordination environment of Ba2+ cations in noncentrosymmetric ADC-based CPs: (a) [Ba2(ADC)(NO3)2]·4H2O, (b) [Ba7(ADC)6(NO3)2]·14H2O, and (c) [Ba3(ADC)2Cl(NO3)]·5H2O, showing also the coordination of nitrate anions in each compound. Graphics redrawn from the cif files (CSD-Refcode XAQMIN, XAQMOS and XAQMUY).102 | ||
Another concept to enhance the probability of obtaining compounds crystallizing in noncentrosymmetric space groups is the inclusion of cations with stereoactive lone pairs.100 This concept can be successfully transferred to ADC-based CPs like [Tl2(ADC)] (Fig. 21). In Section 2.5 this finding has already been discussed in more detail.
4.2 Negative thermal expansion in ADC-based CPs
Most materials have a positive coefficient of thermal expansion, that is, their volume increases upon heating or their volume decreases upon cooling, respectively. There are, however, a restricted number of materials featuring an unusual volume contraction upon heating in a much larger temperature range. In principle, this effect, which is typically called negative thermal expansion (NTE), is well-known for water in the temperature range of 0 °C to 4 °C, i.e., water exhibits a negative coefficient αv of their thermal (volume) expansion.103,104 It was quite intriguing that for all ADC-based coordination polymers with the formula [MII(ADC)] (MII = Ca,45 Sr,44 Ba,105 Pb,66 Eu50) crystallizing in the [Sr(ADC)]-type structure (Fig. 8 and 10) an NTE behavior was observed.105 It was rationalized by strong transverse MII⋯O⋯MII vibrations also known as guitar string vibrations.44,50 For [Sr(ADC)] and [EuII(ADC)] such an NTE behavior was reported for a large temperature range below room temperature (Fig. 37). The thermal volume expansion coefficients were calculated to be αv = −4.7(13) × 10−6 K−1 and αv = −9.4(12) × 10−6 K−1 for [Sr(ADC)] and [EuII(ADC)], respectively.44,50 Larger deviations from a linear behavior (dotted line in Fig. 38) are due to the fact that the tetragonal unit cell of the [Sr(ADC)] type structure is very close to a cubic metric, which leads to severe reflection overlaps in their PXRD patterns and thus hampered a very precise determination of the lattice parameters. Applying high-resolution synchrotron, powder diffraction was not always possible due to the sensitivity of some of these materials to high-intensity synchrotron beams.50 Nonetheless, a clear trend is found for the thermal volume expansion coefficient αv in these compounds: it increases with increasing atomic number. Accordingly, the largest αv value was observed for [Pb(ADC)] with αv = −18.2(1) × 10−6 K−1. However, as [Pb(ADC)] undergoes a reversible phase transition to an amorphous phase at low temperatures, this value was obtained from measurements above room temperature in the temperature range of 25 °C to 127 °C.105 These results are in good agreement with similar studies on Prussian Blue analogues, MIIPtIV(CN)6 with MII = Mn, Fe, Co, Ni, Cu, Zn, and Cd.106 | ||
| Fig. 38 Variation of unit cell volume with temperature for (a) [Sr(ADC)] and (b) [EuII(ADC)]. Reproduced from ref. 44 with permission of John Wiley & Sons, copyright 2002, and from ref. 50 with permission of John Wiley & Sons, copyright 2020. | ||
4.3 Magnetic properties of ADC-based CPs
Materials displaying magnetic interactions between their metal centers are important for many technological applications.107 Magnetism in materials occurs by coupling of individual magnetic moments through an exchange interaction. A direct magnetic exchange can take place, when the metal centers are close enough to each other for a direct spin–spin interaction through space, that is, the electronic wavefunction of the two magnetic sites overlap. Alternatively, the exchange can take place through a bridging ligand (Fig. 39(a)).108,109 For instance, there are close Mn2+⋯O2−⋯Mn2+ contacts in MnO, whereby the O2−ion mediates an antiferromagnetic (AFM) interaction between two neighbouring Mn2+ cations, the so-called super-exchange interaction (Fig. 39(b)).110 While a direct, through-space exchange between the metal atoms can only be observed in a few MOFs within the SBU like in the paddlewheel-based HKUST-1 (Fig. 39(c)),111 the metal–metal distances are typically too large in most MOFs so that a magnetic interaction can only take place via a ligand-mediated through-bond super-exchange interaction. In theoretical investigations, the acetylenedicarboxylate ligand (and extensions thereof in oligoyne-based dicarboxylates) with its linear shape and π system was identified as an ideal ligand to mediate such interactions in MOFs (Fig. 39(e) and (f)). ADC could establish a long-range magnetic ordering due to its continuous frontier orbitals. However, to achieve this ADC needs to be combined with a transition metal having a half-filled 3d shell (e.g. Mn(II), Fe(III)), while the metal–ligand–metal angle should be 180° and the two carboxylate groups of the ADC ligand should be perpendicular to each other (Fig. 2(A), right).112 In a subsequent study, the same authors computed and measured both the electronic structure and the magnetic ordering in ([Mn(ADC)(H2O)2]), which was first synthesized by Robl and Hentschel already in 1990. However, they found only a weak magnetic interaction, as the coordination of the ligand in this CP is not in a fashion as was calculated for an optimal orbital overlap, i.e., the two carboxylate groups of the ADC linker are not orthogonal to each other.113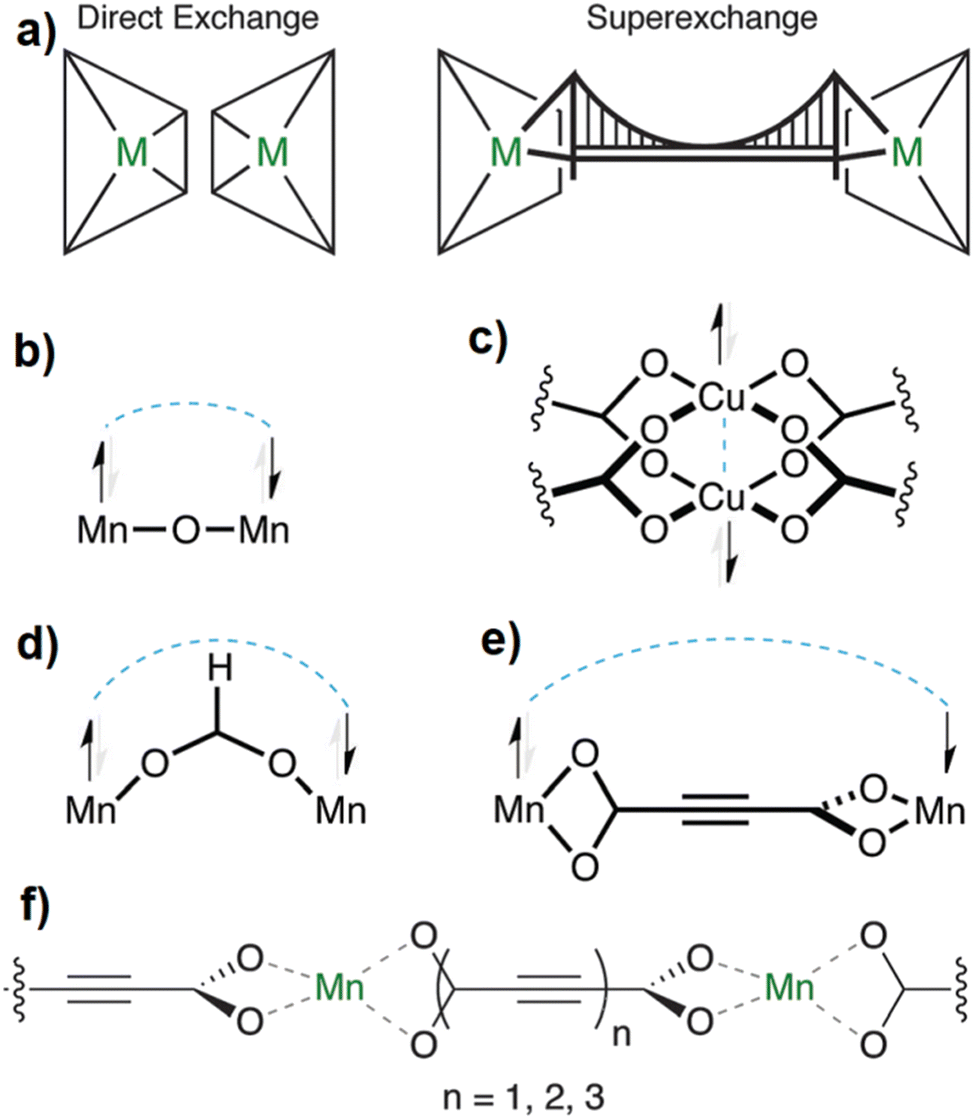 | ||
| Fig. 39 (a) Illustration of direct (left) and super-exchange magnetic interactions (right) through a bridging motif, (b) and (d) examples of magnetic super-exchange interactions in MnO and Mn(II)-formiate, (c) example of a direct exchange interaction in the paddlewheel substructure of HKUST-1, (e) and (f) illustration of a super-exchange interaction through bonds mediated by acetylenedicarboxylate and polyyne dicarboxylate ligands, respectively. Adapted from ref. 112 with permission of the Royal Society of Chemistry, copyright 2014, and from ref. 113 with permission of the Royal Society of Chemistry, copyright 2016. | ||
4.4 Adsorption properties of ADC-based MOFs
Constructing porous materials with ligands containing alkyne-based spacers has been reported by several groups, as advantages with respect to their adsorption properties were expected. Farha et al. demonstrated both experimentally and computationally, that replacing the phenyl spacer of organic linkers with a C–C triple-bond spacer has a tremendous boosting effect on the BET surface areas of the corresponding MOFs.114 Concretely, when replacing the two phenyl rings in each arm of the linker in the dicopper paddlewheel-based MOF PCN-69 (also denoted as NOTT-119) by C–C triple-bond spacers,115,116 a new isostructural MOF denoted as NU-111 was obtained (Scheme 4). This spacer substitution resulted in a remarkable increase of the BET surface area from about 4000 m2 g−1 in PCN-69 to 5000 m2 g−1 in NU-111, although the organic linker in the latter MOF is significantly shorter (Scheme 4). On the contrary, increasing the spacer's length from one phenyl ring in NOTT-112 to two phenyl rings in isostructural PCN-69, resulted only in a relatively small increase of the BET surface area from 3800 m2 g−1 to 4000 m2 g−1.114 | ||
| Scheme 4 Hexacarboxylic acid struts used to construct PCN-69 [LH6(1)], NOTT-112 [LH6(2)] and NU-111 [LH6(3)]. Reproduced from ref. 114 with permission of American Chemical Society, copyright 2012. | ||
After the computational screening of 204 hypothetical MOFs made up of octahedral [Zr6O4(OH)4]12+ SBUs and carboxylate ligands Gomez-Gualdron et al. identified NU-800 as the MOF with the highest deliverable methane capacity among all zirconium-based MOFs.117 NU-800 consists of zirconium nodes, which are connected by 1,4-benzenedipropynoate ligands (H4dbp, Scheme 5) to a network with a fcu topology. Their theoretical results were confirmed experimentally after the successful synthesis of NU-800 and measurement of its high-pressure methane adsorption, which yielded a methane deliverable capacity of 167 cm3(STP) cm−1 (0.125 g g−1) between 65 and 5.8 bar. The authors concluded from their computer simulations that alkyne groups adjacent to the inorganic zirconium nodes provide a more efficient packing around the nodes at high pressures, thereby justifying the remarkably high volumetric deliverable capacity of methane for NU-800.117
 | ||
| Scheme 5 Organic linker molecules used in the MOFs, NU-800, NOTT-101, PCN-46 and NOTT-102. | ||
Besides the positive effect of –CC– groups with respect to increasing BET surface areas and gas adsorption capacities, other reports highlight the influence of the alkyne group on the gas affinity with the framework. Zirconium acetylenedicarboxylate Zr-HHU-1 (also entitled as UiO-66-ADC) displays a very high initial heat of H2 adsorption a Q0st value of about 10 kJ mol−1. This was attributed to the synergistic interaction of hydrogen molecules with the μ3-OH groups of the Zr-oxo clusters and the –CC– triple-bond.90 In another study, the isosteric H2 adsorption enthalpy was calculated for three isoreticular dicopper paddlewheel-based MOFs, namely, NOTT-101, PCN-46 and NOTT-102, involving the respective linkers tpta4−, bdi4− and qpta4− (Scheme 5). At low coverage, the heat of adsorption reaches 7.2 kJ mol−1 for PCN-46, while this value is lower than 5 kJ mol−1 for the two other MOFs, which led the authors to the conclusion that the increased heat of adsorption in PCN-46 is attributable to the interaction between hydrogen molecules and the exposed π electrons in the polyyne units of bdi4−, which is stronger than that with the phenyl rings in tpta4− and qpta4−.118 Tranchemontagne et al. draw a similar conclusion, in which an enhanced near-zero isosteric enthalpy of H2 adsorption was attributed to the presence of C–C triple-bonds in MOFs made up of alkyne-based linkers in IRMOF-61 (ethynyldibenzoate linker) and IRMOF-62 (butadiynedibenzoate linker).119 It was also observed that the –CC– triple-bond in MOFs enhances the enthalpy of CO2 adsorption. For instance, the isosteric heat of adsorption near zero-coverage was calculated to be as high as 60 kJ mol−1 for Zr-HHU-1 and 47 kJ mol−1 for Ce-HHU-1 (also denoted as Ce-UiO-66-ADC).90,94 These values, which are much higher than those of the comparable MOFs, zirconium terephthalate UiO-66 (28 kJ mol−1) and zirconium fumarate MOF-801 (19–29 kJ mol−1),120,121 were also attributed to the effect of the –CC– triple-bonds in the ADC-based MOFs. Furthermore, Zr/Hf/Ce-HHU-1 features a high hydrophilicity displayed by a pseudo-type I water sorption isotherm (Fig. 34(d)), unlike the stepwise S-shaped isotherms displayed by UiO-66 and MOF-801,122 thereby demonstrating a higher affinity of water molecules to the alkyne units in HHU-1 MOFs, compared to the affinity of water molecules to the phenyl rings or alkene units in UiO-66 and MOF-801, respectively (Fig. 40).
 | ||
| Fig. 40 Water adsorption isotherms of isostructural UiO-66, MOF-801, and Zr-HHU-1 MOFs with a fcu topology at 293 K, showing the increased hydrophilicity induced by the ADC linker. Adapted from ref. 90 with permission of Wiley & Sons, copyright 2019. | ||
ADC-based compounds are due to the reactive C–C triple-bond in their structure a platform for chemical fixation of other reactive species via addition reactions. It was experimentally demonstrated that Ce-HHU-1 chemisorbs bromine and iodine vapours via halogenation of its alkyne units and poses therefore as an interesting material applicable for air cleaning from toxic halogen vapors.94
Finally, the short ADC linker is a good candidate to fabricate MOFs with small pores (e.g. ultramicroporous MOFs, i.e. with pore diameters of <7 Å), which are needed for CO2 capture and size-based gas separation.123–125 In this regard, the zirconium acetylenedicarboxylate MOF NUS-36 with a bcu topology has pores with a diameter of about 3.6 Å (vide supra). NUS-36 was not accessible to N2 molecules but showed a very good sorption selectivity for ethylene (C2H4) over ethane (C2H6) as indicated by an IAST (Ideal Adsorbed Solution Theory) selectivity value of up to 4.1 (Fig. 41(a)). This high selectivity was confirmed by the breakthrough experiment (Fig. 41(b)) and the finding was justified by the confined nature of the pores in NUS-36.92
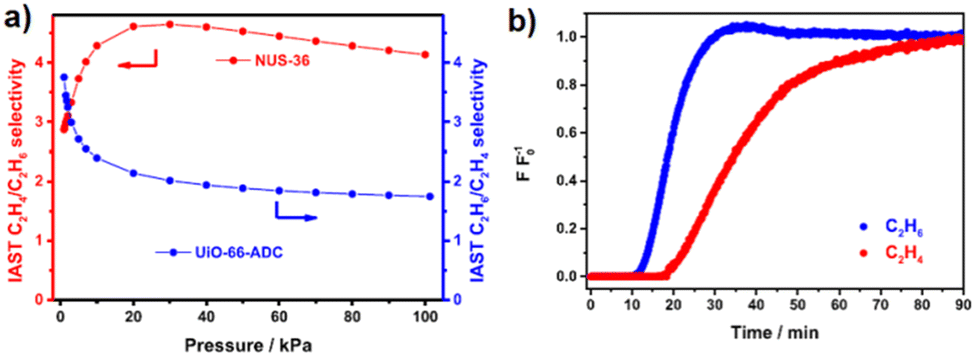 | ||
| Fig. 41 (a) Plot of the IAST selectivity of an equimolar C2H4/C2H6 mixture for UiO-66-ADC and NUS-36 at 298 K; and (b) breakthrough curves of an equimolar mixture of C2H4/C2H6 in packed columns of NUS-36 at 298 K and 1 bar. Reproduced from ref. 92 with permission of American Chemical Society, copyright 2019. | ||
4.5 In situ and post-synthetic transformations in ADC-based CPs and MOFs
The presence of the –CC– triple-bond in the backbone of the ADC ligand makes this molecule amenable to addition reactions (e.g. hydrogenation, hydration, halogenation, and hydrohalogenation.), and therefore offers the possibility to introduce functional groups in the resulting CP or MOF.126,127 Ligand functionalization in CPs and MOFs can be accomplished pre-synthetically, post-synthetically or in situ during the synthesis of the material, and the aim is to tune the functionalities of CPs and MOFs to meet or enhance specific properties.128–131
A few publications have reported the formation of halogen functionalized fumarate-based CPs and MOFs starting from acetylenedicarboxylic acid, whereby the H2ADC reactant undergoes an in situ hydrohalogenation during the formation of the CP or MOF. The first report of such a transformation was published in 2005 by Billetter et al.132
By reacting H2ADC and LiI in water at room temperature, single crystals of a CP with a layered structure and the formula [Li(HOOC–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CI–COO)(H2O)] formed. In its structure, the Li+ cation is tetrahedrally coordinated by four oxygen atoms from four iodofumarate anions and one water molecule (Fig. 42(a)). Two LiO4 tetrahedra are edge-sharing and form dinuclear units, which are connected by iodofumarate linkers to form a two-dimensional layered network (Fig. 42(b) and (c)).132 Obviously, the iodofumarate linker found in the final product originates from an in situ trans-hydroiodination of H2ADC (Scheme 6).
CI–COO)(H2O)] formed. In its structure, the Li+ cation is tetrahedrally coordinated by four oxygen atoms from four iodofumarate anions and one water molecule (Fig. 42(a)). Two LiO4 tetrahedra are edge-sharing and form dinuclear units, which are connected by iodofumarate linkers to form a two-dimensional layered network (Fig. 42(b) and (c)).132 Obviously, the iodofumarate linker found in the final product originates from an in situ trans-hydroiodination of H2ADC (Scheme 6).
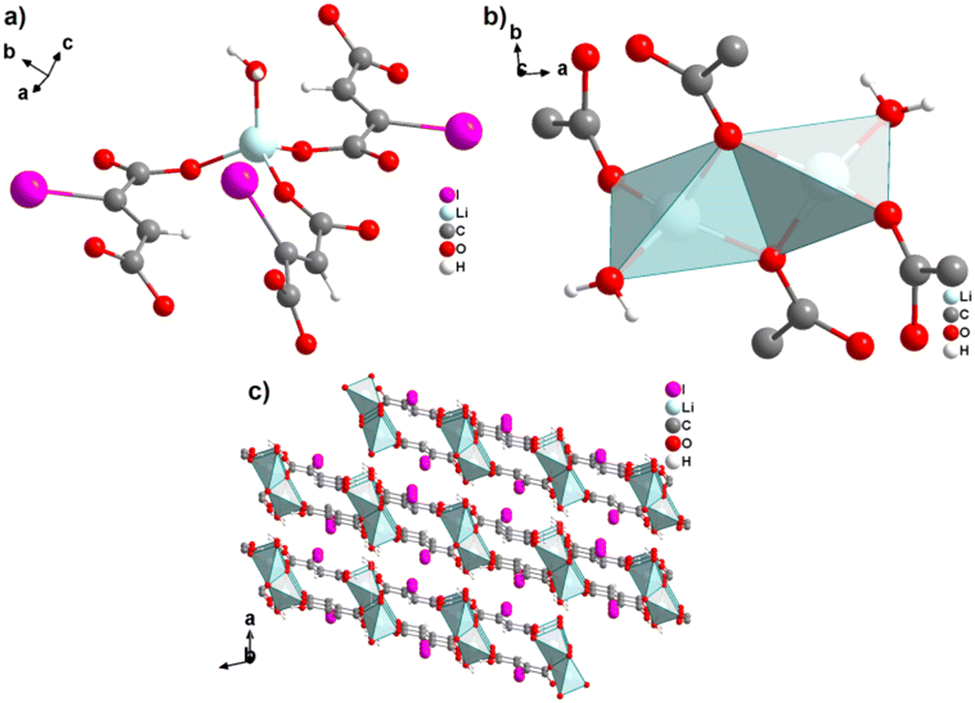 | ||
| Fig. 42 Crystal structure of [Li(HOOC–CHCI–COO)(H2O)] showing: (a) the coordination sphere of the Li+ cation, (b) the dinuclear building unit, and (c) stacked two-dimensional layers. Graphics redrawn from the cif file (CSD-Refcode WAVXEX).132 | ||
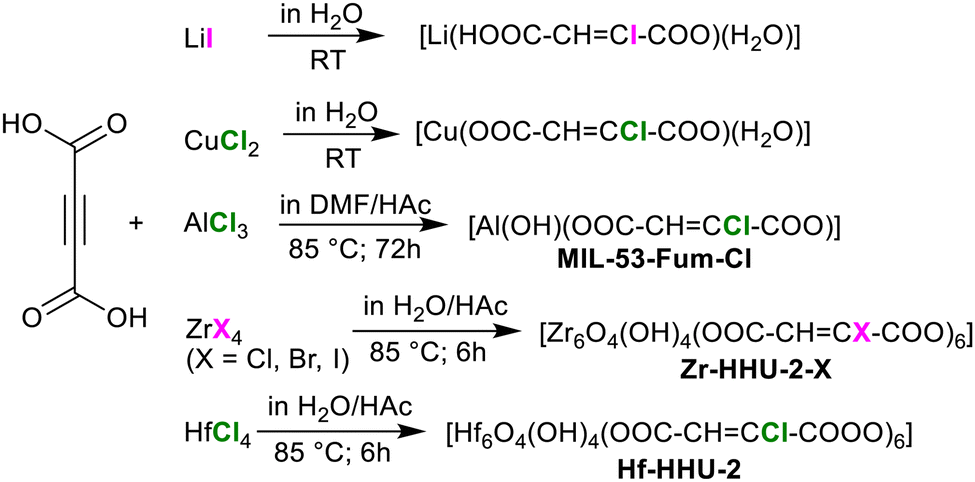 | ||
| Scheme 6 Summary of reported reactions leading to halofumarate-based CPs and MOFs from in situ hydrohalogenation of acetylenedicarboxylic acid. | ||
One year later, the same authors reported a similar observation, where single crystals of [Cu(OOC–CHCCl–COO)(H2O)2]·H2O formed upon the reaction of H2ADC with CuCl2 in water.133 In its structure, the Cu2+ cation is fivefold coordinated by three oxygen atoms from three different chlorofumarate dianions and two water molecules to form a square pyramid (Fig. 43(a)).
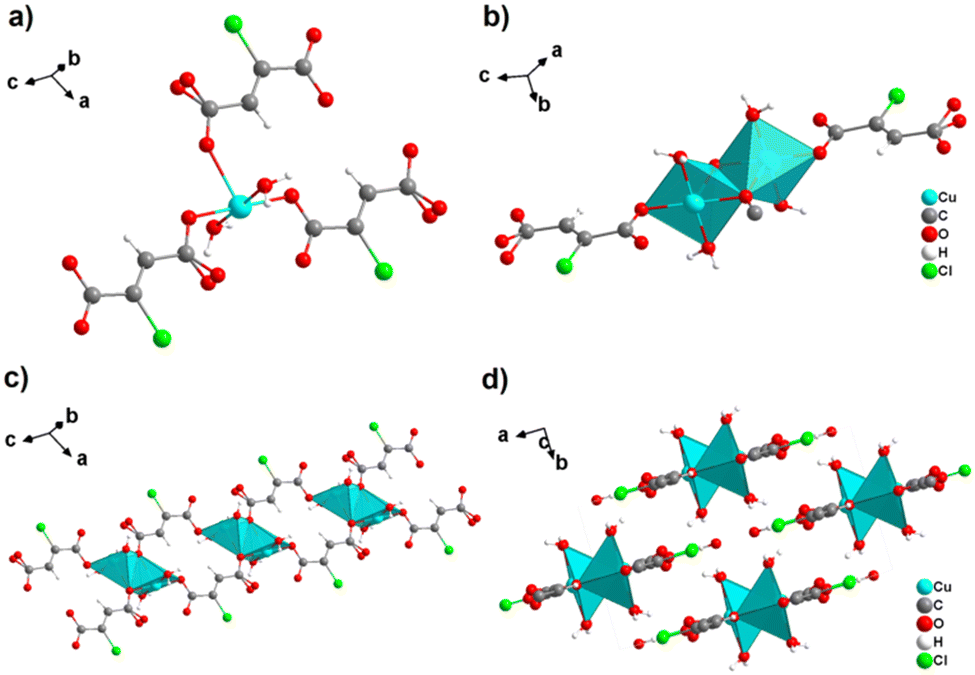 | ||
| Fig. 43 Crystal structure of [Cu(OOC–CHCCl–COO)(H2O)2]·H2O showing: (a) the coordination environment of Cu2+, (b) the dinuclear building unit with connecting chlorofumarate linkers, (c) one-dimensional ribbon extending along [001] and (d) the packing of ribbons viewed along [001]. Note: one oxygen atom of the carboxylate group neighbouring the CH moiety is disordered over two (split) positions. Graphics redrawn from the cif file (CSD-Refcode LEHJIS).133 | ||
Two such CuO5 pyramids are edge-sharing to form dinuclear units (Fig. 43(b)), which are connected by chlorofumarate linkers into one-dimensional ribbons parallel to the [001] direction (Fig. 43(c) and (d)).133 Again, the chlorofumarate linker found in the final CP originates from an unexpected in situ trans-hydrochlorination of acetylenedicarboxylic acid during the synthesis of the CP (Scheme 6).
It is worth noting that the unusual in situ hydrohalogenation of H2ADC seems to be favored when using metal halides as metal sources in aqueous solutions. In this respect, [Li2(ADC)(H2O)2] is obtained upon the reaction of H2ADC with Li(OH) in water (vide supra),41 whereas in situ hydroiodination to [Li(HOOC–CHCI–COO)(H2O)] occurs upon the reaction of H2ADC with LiI in water.132 This hypothesis is also in line with the formation of [Cu(OOC–CHCCl–COO)(H2O)2]·H2O, as H2ADC and CuCl2 as metal salts were used for its synthesis in water.133 First, the metal halide (MX) seems to react with water molecules, thereby releasing hydrogen halide (HX) into the solution. Then an addition of the in situ generated HX to the C–C triple-bond of H2ADC follows. It has not yet been demonstrated, whether the addition reaction occurs before the formation of the CP or after an intermediate ADC-based compound formed. However, there are several ADC-based CPs (that is, without the transformation of the –CC– moiety) that were obtained from metal halides. The most intriguing case is that of the reaction leading to the formation of [Cu(ADC)(H2O)3]·H2O,55 from an aqueous solution containing H2ADC and CuCl2 with the same concentrations and under very similar reaction conditions to those leading to the formation of [Cu(OOC–CHCI–COO)(H2O)2]·H2O with an in situ hydrochlorination.133 Although the authors did not address this discrepancy, the long reaction time of “one month” for the synthesis of the fumarate and the observation that “blue crystals and a green precipitate were obtained”,133 give a first hint that the formation of the latter is a slow reaction at room temperature making the transformation of the ADC-based CP with HX to the fumarate-based CP very likely.
A similar unexpected in situ hydrohalogenation of H2ADC was also observed during the reaction of ZrOCl2/ZrCl4 with H2ADC in water in the presence of acetic acid as a crystallization modulator yielding the chloro-functionalized derivative of zirconium fumarate MOF-801, which is isostructural to UiO-66 (Fig. 44).134 The chloro-functionalized MOF-801 was obtained as a microcrystalline powder and was denoted as Zr-HHU-2-Cl. Its structure consists of octahedral [Zr6O4(OH)4]12+ secondary building units, which are connected to each other by twelve chlorofumarate linkers, resulting in a microporous network with a fcu topology. It should be noted that a reaction of ZrOCl2 with H2ADC in anhydrous DMF yielded rather a UiO-type MOF (Zr-HHU-1) containing unmodified ADC as the linker (vide supra).90 This means that in situ hydrochlorination of the –CC– triple-bond in H2ADC only occurred in water.
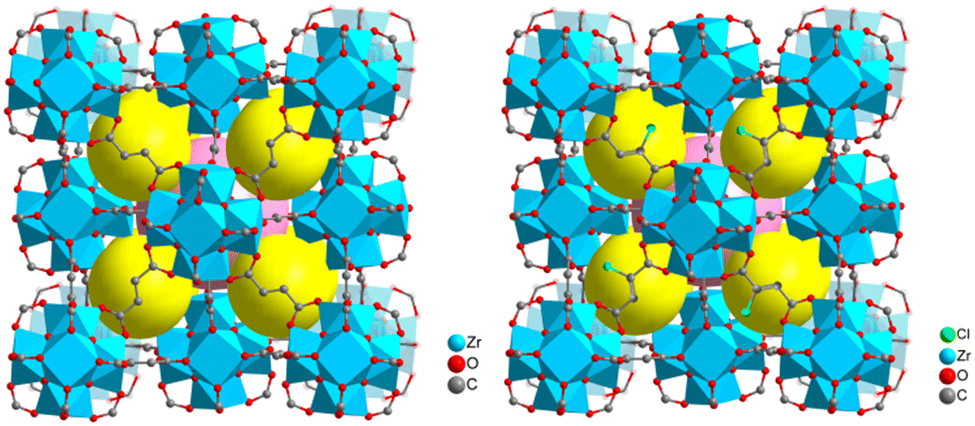 | ||
| Fig. 44 Crystal structure of the zirconium fumarate MOF-801 with a fcu toplogy (left), and its chloro-functionalized congener Zr-HHU-2-Cl (right). Graphics redrawn and adapted from the cif file (CSD-Refcode BOHJOZ).122 | ||
Using X-ray single-crystal structure analysis is straightforward to differentiate between an ADC-based MOF and its chloro-fumarate-based congener with the same topology obtained by an in situ transformation. However, discerning between these two MOFs by X-ray powder diffraction only is less straightforward and perhaps even challenging. For instance, Zr-HHU-2-Cl and Zr-HHU-1 have very similar powder X-ray diffraction (PXRD) patterns (Fig. 45(a)), which do not allow distinguishing between these two MOFs unambiguously. At this point, Raman spectroscopy is a very helpful complementary tool to follow the linker transformation, as the strong characteristic band of the –CC– stretching vibration at 2230 cm−1 in ADC-based HHU-1 completely vanishes in the Raman spectrum of Zr-HHU-2-Cl (Fig. 45(b)). Solid-state NMR spectroscopy also confirms the presence of a non-symmetrical fumarate linker in Zr-HHU-2-Cl (Fig. 45(c)). SEM-EDX elemental mapping shows the uniform distribution of Cl atoms in the material (Fig. 45(e)), while IR spectroscopy indicates the presence of the C–Cl stretching vibrations at 681 cm−1. XPS analysis reveals the chlorine in the sample to be organic by nature (C-Cl), as its peak is found at a binding energy of about 200 eV (Fig. 45(d)). All these additional methods clearly confirm the occurrence of a hydrochlorination of H2ADC leading to the final chlorofumarate linker found in Zr-HHU-2-Cl (Scheme 6).124
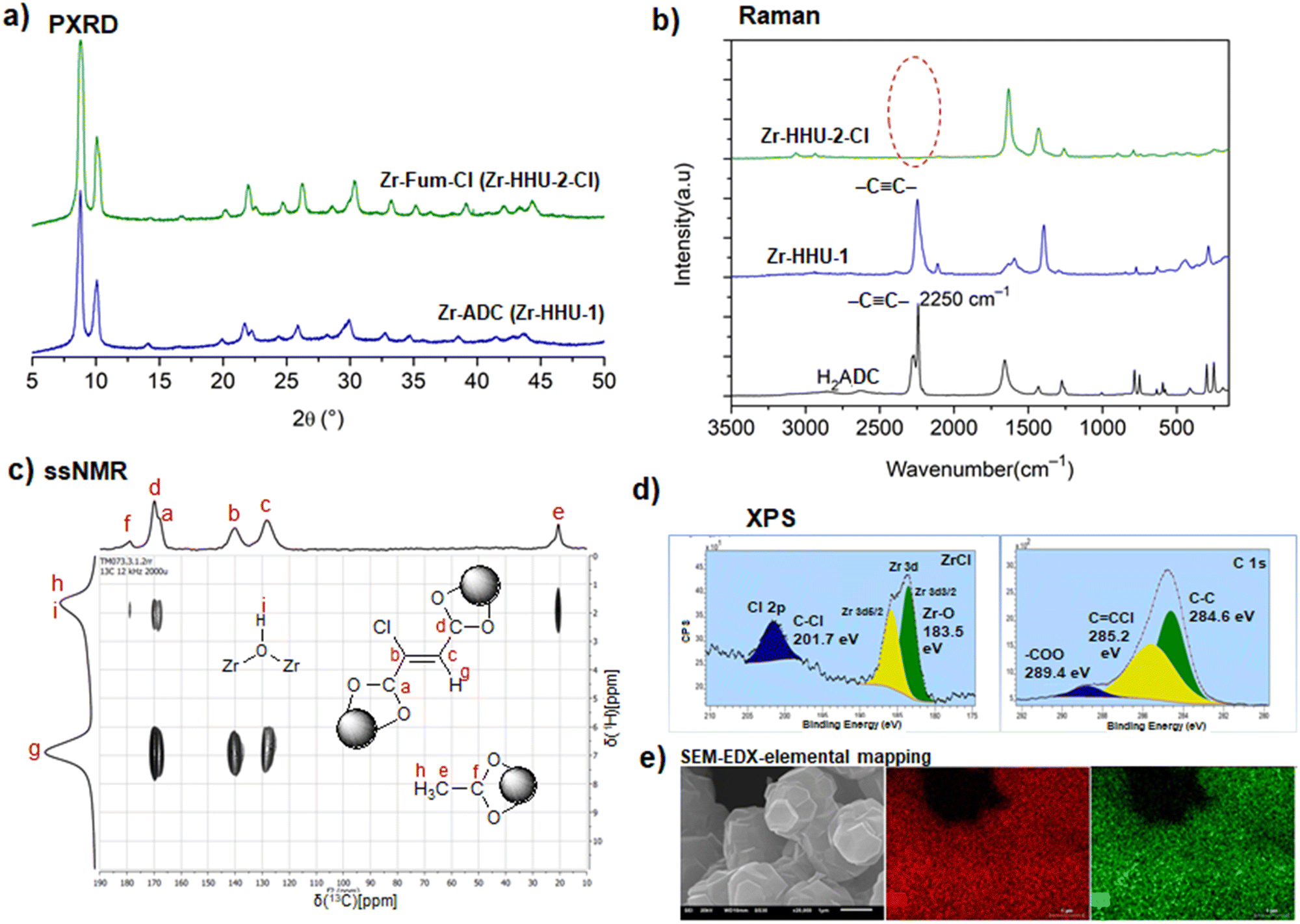 | ||
| Fig. 45 (a) Powder X-ray diffraction (PXRD) patterns of the zirconium acetylenedicarboxylate-based MOF Zr-HHU-1 and the zirconium chlorofumarate-based MOF Zr-HHU-2-Cl, showing very similar diffraction patterns; (b) Raman spectra of H2ADC, Zr-HHU-1 and Zr-HHU-2-Cl, showing the absence of the vibration of the C–C triple bond in Zr-HHU-2-Cl; (c) (ssNMR) solid state 1H–13C FSLG-HETCOR CP MAS spectrum of Zr-HHU-2-Cl showing the presence a functionalized fumarate-like linker; (d) X-ray photoelectron spectra (XPS) of Zr-HHU-2-Cl of the Cl-2p, Zr-d and C-1s regions, revealing the formation of a C–Cl bond and the presence of olefinic carbon atoms; and (e) (SEM-EDX) elemental mapping showing zirconium and chlorine at the same regions and uniformly distributed in the whole sample. Adapted from ref. 134 with permission of the American Chemical Society, copyright 2019. | ||
A similar in situ hydrochlorination of H2ADC was observed, when HfCl4 reacts with H2ADC in water in the presence of acetic acid as the crystallization modulator (Scheme 6). The resulting MOF, which was denoted as Hf-HHU-2-Cl, is the Hf-analogue of Zr-HHU-2-Cl.34 Moreover, also other halogenated (brominated and iodinated) MOFs isostructural with MOF-801, namely Zr-HHU-2-Br and Zr-HHU-2-I, were obtained by in situ hydrobromination and hydroiodination of H2ADC during its reaction with ZrBr4 and ZrI4, respectively.124
Furthermore, the reaction of H2ADC with AlCl3 yielded an aluminum chlorofumarate MOF, namely MIL-53-Fum-Cl, which exhibits the MIL-53-type structure similar to aluminum fumarate (MIL-53-Fum). Its crystal structure consists of chains of trans-corner-sharing AlO6 octahedra, which are bridged by chlorofumarate linkers to yield a network containing channels with rhombic cross-sections.135 From the aforementioned reports, it appears that any reaction mixture that produces hydrogen halides (HX, X = Cl, Br, I) in an aqueous solution is likely to induce the in situ hydrohalogenation of the –CC– triple bond in H2ADC and thereafter, the formation of a halofumarate-based MOF or CP. This transformation was observed with monovalent (Li+), divalent (Cu2+), trivalent (Al3+) and tetravalent (Hf4+ and Zr4+) metal cations and seems to occur without the need of any specific catalyst.
In addition, hydrohalogenation occurs stereoselectively yielding solely the trans-product (halofumarate-based MOF) instead of the cis-product (halomaleate-based MOF) or mixtures thereof (Scheme 7).
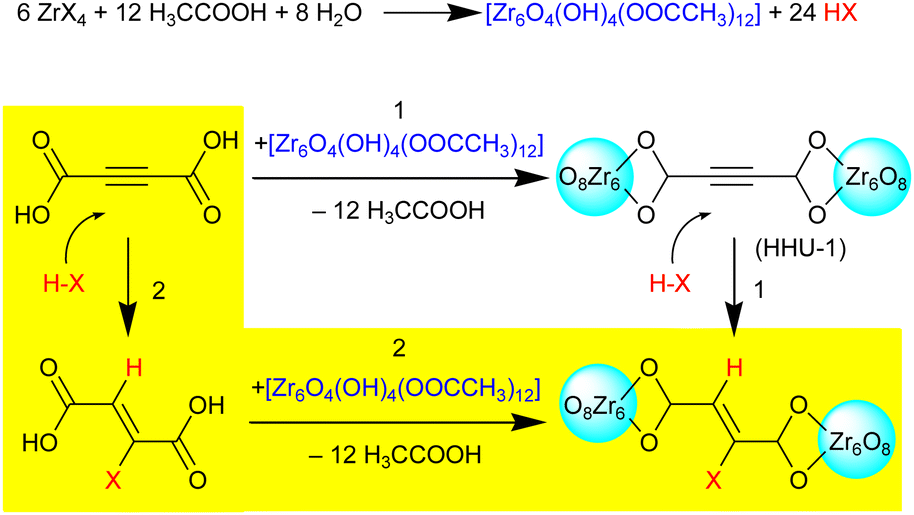 | ||
| Scheme 7 Possible reaction mechanisms for the synthesis of Zr-HHU-2-X via the formation of hydrogen halide (HX, X = Cl, Br, I) and in situ hydrohalogenation of ADC after the formation of HHU-1 (route 1) or via a prior formation of a halofumarate linker and MOF formation with this linker in a second step (route 2). Adapted from ref. 134 with permission of the American Chemical Society, copyright 2019. | ||
The stereospecific trans hydrohalogenation of electron-deficient alkynes upon reaction with lithium or sodium halides to afford the trans-adduct alkenyl has already been reported.136 It is worth noting that the in situ functionalization of MOFs is of great interest, as it reduces the number of synthesis steps, time and cost, when compared with pre-/post-synthetic modifications. Nonetheless, ADC-based CPs and MOFs should also be amenable to post-synthetic modifications via addition reactions involving the –CC– triple bond. It was demonstrated that UiO-type Zr- and Hf-based MOFs incorporating –CC– triple-bond containing ligands can undergo stereoselective halogenation or hydrobromination reactions of the unsaturated C–C bond to yield a halogenated alkene-containing Zr-/Hf-MOF, while retaining the structural features of the starting material (Scheme 8).137–139 In the case of Ce-HHU-1 (or Ce-UiO-66-ADC), it was observed that this material undergoes a solid state bromination and iodination of its C–C triple bonds to form new compounds containing dibromoethylenic or diiodoethylenic ligands, respectively, albeit with a completely different structure than that of pristine Ce-HHU-1.94
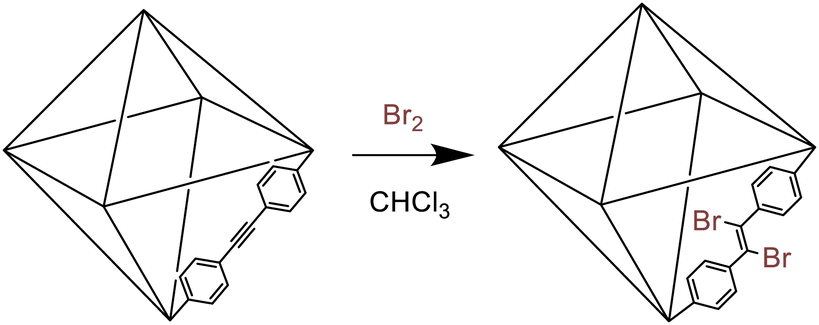 | ||
| Scheme 8 Illustration of postsynthetic bromination of a MOF with an alkyne-based linker. Reproduced from ref. 137 with permission of the Royal Society of Chemistry, copyright 2016. | ||
4.6 Solid-state polymerization in ADC-based CP and MOFs
Unsaturated molecules are amenable to solid-state polymerization (SSP) reactions, whereby the π-component of the multiple-bond opens and forms an extended chain.140 SSP can be induced photochemically,141 by high pressure,142 by ionizing radiation or thermally.143,144 The polymerization in the solid-state is favored, when the unsaturated moieties in the respective material are correctly oriented to each other with short contacts between them (<4.2 Å).145 Molecules with C–C triple bonds are especially interesting for SSP, as they would yield the formation of π-conjugated polymers, which can result in materials with interesting electrical and non-linear optical properties.146 However, the SSP of “all-organic” acetylenes is usually very limited. For instance, the SSP of acetylenedicarboxylic acid induced by 60Co γ-irradiation yielded only a conversion rate of 5% and takes place only after very long irradiation times of up to 10 days.147 Metal complexes based on acetylenecarboxylate ligands were demonstrated to be interesting starting materials for a complete solid-state polymerization. For instance, the crystal structures of dimethyl(propynoato)thallium (CH3)2Tl(O2CCCH), lanthanum propynoate La2(O2CCCH)6(H2O)4·2H2O and sodium propynoate Na(O2CCCH), contain infinite chains with short inter-chain CC⋯CC contacts in the range 3.29–3.454 Å. All these compounds undergo a solid-state polymerization upon irradiation with X-rays or 60Co γ-rays with yields up to 75%.148–150
Several ADC-based CPs were also reported to undergo SSP. Shershnev et al. obtained the SSP of [Zn(ADC)(H2O)2] in an inert atmosphere and in vacuo, upon thermal treatment at 150 °C. Alternatively, they obtained the SSP of the same compound upon 60Co γ-irradiation.151 The crystal structure of [Zn(ADC)(H2O)2] (isotypic to [Mg(ADC)(H2O)2], Fig. 7) contains infinite polymeric Zn-ADC chains with short inter-chain CC⋯CC contacts with distances of about 3.8 Å (Fig. 46(a)), which is in a good range for an SSP.54 It may therefore be expected that isotypic ADC-based CPs, that is [MII(ADC)(H2O)2] with MII = Mg, Mn, Ni, Co, should also be amenable to polymerize in the solid state.
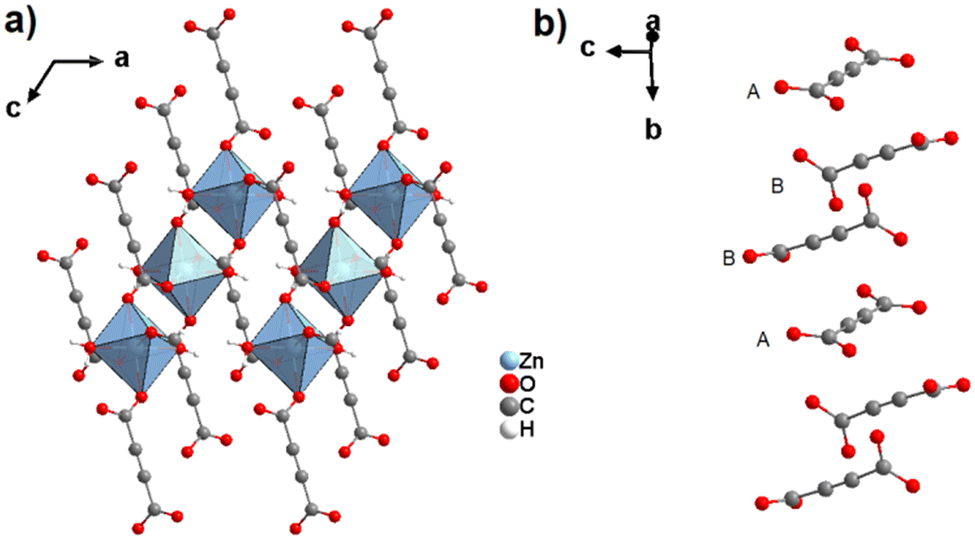 | ||
| Fig. 46 Sections of the crystal structures of [Zn(ADC)(H2O)2] (a) and [Ce2(ADC)3(H2O)6]·2H2O (b), disclosing short CC⋯CC contacts between neighbouring ADC ligands. The coordination sphere of Ce3+ is omitted in (b) for clarity. Graphics redrawn from the cif file (CSD-Refcode DAYMAS and KAPHAL).54,84 | ||
Michaelides et al. observed a thermally-induced SSP of [CeIII2(ADC)3(H2O)6]·2H2O at temperatures above 150 °C.84 The crystal structure of [CeIII2(ADC)3(H2O)6]·2H2O (isotypic to [DyIII2(ADC)3(H2O)6]·2H2O, Fig. 30) also contains infinite polymeric CeIII-ADC chains with short inter-chain CC⋯CC contacts in the sequence ⋯A⋯B⋯B⋯A⋯, where A and B represent two crystallographically different ADC ligands (Fig. 46(b)). The distance between –CC– bonds of two consecutive B ligands is 3.872 Å, while the contact of –CC– bonds for consecutive A–B ligands is at a distance of 3.432 Å, thus making the occurrence of SSP very favorable.84 Infinite polymeric Cd-ADC chains with short inter-chain CC⋯CC contacts of 3.27 Å were also found in [Cd(ADC)(H2O)3]·H2O (Fig. 19(d)). The observation of a thermally-induced SSP at about 160 °C is therefore in agreement with the results discussed before.60 It can be predicted that for many other rare earth ADC-based MOFs and CPs, e.g., those with the general formula [RE2(ADC)3(H2O)6]·2H2O (REIII = Gd, La, Pr, Nd, Sm, Eu, Tb, Dy, Ho, Er, Y), an SSP should be observable. Even more, the decomposition of anhydrous CPs [MII(ADC)] (MII = Ca, Sr, Ba, Eu, Pb) in high-intensity synchrotron beams, which goes along with a blackening of the sample (see Section 4.2), may obviously also be ascribed to SPP reactions. Finally, it is very remarkable that the SSP reactions described for ADC-based CPs/MOFs obviously proceed with a complete conversion, while for “all-organic” alkynyls or metal complexes based on alkynylcarboxylate ligands (vide supra) significantly lower conversion rates were reported.
5 Conclusions and perspectives
Acetylenedicarboxylate (ADC), as a ditopic ligand, exhibits a large variety of coordination modes that lead to the formation of both non-porous coordination polymers (1D, 2D and 3D), as well as porous metal–organic frameworks. ADC-based materials comprising all types of mono-, di-, tri-, tetra- and hexavalent metal cations from all groups across the periodic table of elements have been reported. ADC-based CPs and MOFs adopt intriguing structural features that have typically not been encountered in compounds with other linear carboxylate-based ligands, which result in unique properties including negative thermal expansion, long-range magnetic ordering, enhanced gas/vapor adsorption capacity and affinity, as well as solid-state polymerization. While the room temperature synthesis in water, DMF or alcohols and the mechanochemical synthesis have been the most employed methods to prepare ADC-based MOFs/CPs, solvothermal syntheses have also successfully been employed, although ADC starts to decompose under these conditions at (too) high temperatures on account of its thermal liability and decarboxylation in solutions. Nonetheless, some successful solvothermal syntheses indicate that this approach must not be totally excluded for the preparation of ADC-based MOFs and CPs, but the synthetic conditions have to be adjusted very carefully. Also, the occurrence of some ADC-based materials with very high thermal stabilities like strontium acetylenedicarboxylate [Sr(ADC)] contrasts the idea of a generally very low thermal stability of ADC-based compounds. Making use of acetylenedicarboxylate as the linker for constructing coordination polymers and porous metal–organic frameworks is therefore a research niche, which deserves further exploration.There has been a very recent increase in the number of publications demonstrating that many ADC-based MOFs can actually be synthesized at room temperature and probably in water.152–154 In this regard, synthesizing ADC-analogues of well-known MOFs (e.g. MIL-53-ADC, MIL-140-ADC, MIL-88-ADC) can be envisaged. Regarding the issue of porosity, a systematic determination of the porosity for already reported ADC-based CPs would be an interesting subject of investigation, as many reported ADC-based CPs show potential pores, but activation either failed or has never been attempted. In these cases, the mild activation using supercritical CO2 drying could be an interesting approach. Investigations of porous ADC-based MOFs for the storage of acetylene or the separation of acetylene-containing gas mixtures could be another interesting topic. It was demonstrated that MOFs made up of alkyne-based linkers displayed a high acetylene uptake, as well as a high isosteric heat of acetylene adsorption, which was suggestively attributed to the enhancing effect of weak π–π interactions between –CC– moieties of the adsorbate and –CC– units of the linker in the MOF adsorbent.155
The short ADC linker effectively induces small pores in the so-called ultramicroporous frameworks (with pore diameters of <7 Å). Such small pores have the potential for molecular sieving of, e.g., CO2 from N2 and for water uptake at low humidity. Small pore widths are critically needed in MOFs to achieve high quantities in low-pressure or trace gas adsorption by allowing adsorbent–adsorbate–adsorbent interactions to opposite sides of the pores with concomitant high heat of adsorption.
The postsynthetic addition of functional groups to the triple-bond (at least to a fraction of the ADC ligands), such as hydration, hydroamination, halogenation, and hydrohalogenation, should be of high potential for the ADC MOFs as the postsynthetic functionalization of MOFs is generally of great interest, due to the retention of the framework structure.
There is an interest in the thin-layer formation of MOFs as nanoporous films (named SURMOFs) which are studied for the uptake of photoactive molecules, electrical transport, exciton transport, exciton channeling, photon upconversion, the remote-controlled release of molecules, membranes with photoswitchable selectivity, ion-conduction, etc.156 While the postsynthetic functionalization at the triple-bond of the ADC linker in the bulk MOF with small pores may be admittedly a challenge; the addition of organic functional groups will be much easier in thin-layer SURMOFs where ADC-MOFs should have a high potential.
It should be noted that the versatility in properties observed with ADC-based CPs/MOFs could also be exploited in closely related fields of acetylene-/alkyne-linked covalent-organic frameworks (COFs) and covalent-organic networks (CONs).157
Finally, the typically low thermal stability of ADC-based compounds could make them good precursor candidates to produce metal oxides or metal nanoparticles from pyrolysis at moderate temperatures.158,159 We hope that this review will inspire readers with new ideas towards a better understanding of the ADC linker and stimulate the synthesis of more valuable ADC-based CP and MOF materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work leading to this publication was supported by the Postdoctoral Researchers Mobility Experience (P.R.I.M.E) program of the German Academic Exchange Service (DAAD) with funds from the German Federal Ministry of Education and Research (BMBF).References
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 2, 673–674 CrossRef PubMed.
- C. Janiak and J. K. Vieth, New J. Chem., 2010, 34, 2366–2388 RSC.
- T. Meek, J. A. Greathouse and M. D. Allendorf, Adv. Mater., 2011, 23, 249–267 CrossRef PubMed.
- J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- A. Ahmed, S. Seth, J. Purewal, A. G. Wong-Foy, M. Veenstra, A. J. Matzger and D. J. Siegel, Nat. Commun., 2019, 10, 1568 CrossRef PubMed.
- Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657–5678 RSC.
- Y. Lin, C. Kong, Q. Zhang and L. Chen, Adv. Energy Mater., 2017, 7, 1601296 CrossRef.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- L. Zhu, X.-Q. Liu, H.-L. Jiang and L.-B. Sun, Chem. Rev., 2017, 117, 8129–8176 CrossRef CAS PubMed.
- K. Henninger, H. A. Habib and C. Janiak, J. Am. Chem. Soc., 2009, 131, 2776–2777 CrossRef PubMed.
- F. de Lange, K. J. F. M. Verouden, T. J. H. Vlugt, J. Gascon and F. Kapteijn, Chem. Rev., 2015, 115, 12205–12250 CrossRef PubMed.
- J. Kalmutzki, C. S. Diercks and O. M. Yaghi, Adv. Mater., 2018, 30, 1704304 CrossRef PubMed.
- L. Liu, Y. Zhou, S. Liu and M. Xu, ChemElectroChem, 2018, 5, 6–19 CrossRef CAS.
- P. Kumar, A. Deep and K.-H. Kim, TrAC, Trends Anal. Chem., 2015, 73, 39–53 CrossRef CAS.
- A. Almeida Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tomé, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088–1110 RSC.
- T.-H. Chen, I. Popov, W. Kaveevivitchai and O. Š. Miljanić, Chem. Mater., 2014, 26, 4322–4325 CrossRef CAS.
- M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef PubMed.
- M. Yaghi, M. J. Kalmutzki and C. S. Dirercks, Introduction to reticular chemistry: metal–organic frameworks and covalent organic frameworks, Wiley-VCH, Weinheim, 2019 Search PubMed.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- F. Millange and R. I. Walton, Isr. J. Chem., 2018, 58, 1019–1035 CrossRef CAS.
- Z. Chen, S. L. Hanna, L. R. Redfern, D. Alezi, T. Islamoglu and O. K. Farha, Coord. Chem. Rev., 2019, 386, 32–49 CrossRef CAS.
- Z. Moghadam, A. Li, S. B. Wiggin, A. Tao, A. G. P. Maloney, P. A. Wood, S. C. Ward and D. Fairen-Jimenez, Chem. Mater., 2017, 29, 2618–2625 CrossRef.
- Z. Moghadam, A. Li, X.-W. Liu, R. Bueno-Perez, S.-D. Wang, S. B. Wiggin, P. A. Wood and D. Fairen-Jimenez, Chem. Sci., 2020, 11, 8373–8387 RSC.
- R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., 2016, B72, 171–179 Search PubMed.
- E. Bandrowski, Ber. Dtsch. Ges., 1979, 12, 2212–2216 CrossRef.
- M. Schwartz, R. I. Gelb and D. A. Laufer, J. Chem. Eng. Data, 1980, 25, 95–96 CrossRef.
- J. Li and T. B. Brill, J. Phys. Chem. A, 2002, 106, 9491–9498 CrossRef CAS.
- W. Abbott, R. T. Arnold and R. B. Thompson, Org. Synth., 1938, 18, 3 CrossRef.
- M. Arndt and L. J. Goossen, Germany Pat., WO2014/029689A1, 2014 Search PubMed.
- R. Allan, P. C. Beaumont, L. Macindoe, G. H. W. Milburn and A. Werninck, Thermochim. Acta, 1987, 117, 51–58 CrossRef.
- Cetylenedicarboxylic acid (142-45-0) 13C NMR-ChemicalBook, https://www.chemicalbook.com/SpectrumEN_142-45-0_13CNMR.htm (accessed on the 29th December 2021).
- D. Zidan, A. W. Allaf, A. Allahham and A. AL-Zier, Opt. Laser Technol., 2015, 68, 60–66 CrossRef.
- A. Shershnev, G. V. Shilov, G. I. Dzhardimalieva, A. D. Pomogailo, M. Izydorzak and M. Leonowicz, Macromol. Symp., 2012, 317, 180–186 CrossRef.
- J. Matemb Ma Ntep, H. Reinsch, C. Schlüsener, A. Goldman, H. Breitzke, B. Moll, L. Schmolke, G. Buntkowsky and C. Janiak, Inorg. Chem., 2019, 58, 10965–10973 CrossRef PubMed.
- B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef.
- M.-L. Hu, A. Morsali and L. Aboutorabi, Coord. Chem. Rev., 2011, 255, 2821–2859 CrossRef CAS.
- V. Krishnamurty and G. M. Harrys, Chem. Rev., 1961, 61, 213–246 CrossRef.
- M. Hernández-Molina, P. A. Lorenzo-Luis and C. Ruiz-Pérez, CrystEngComm, 2001, 3, 60–63 RSC.
- C. Robl and S. Hentschel, Z. Anorg. Allg. Chem., 1991, 596, 149–155 CrossRef CAS.
- C. Robl and S. Hentschel, Z. Anorg. Allg. Chem., 1990, 591, 188–194 CrossRef CAS.
- K. Gramm, I. Stein, E. Riesen and U. Ruschewitz, Z. Anorg. Allg. Chem., 2016, 642, 1350–1354 CrossRef.
- H. Billetter, I. Pantenburg and U. Ruschewitz, Z. Naturforsch., 2004, 59b, 903–909 CrossRef.
- C. Robl and S. Hentschel, Z. Naturforsch., 1990, 45b, 1499–1502 CrossRef.
- F. Hohn, I. Pantenburg and U. Ruschewitz, Chem. – Eur. J., 2002, 8, 4536–4541 CrossRef CAS PubMed.
- I. Stein and U. Ruschewitz, Z. Anorg. Allg. Chem., 2010, 636, 400–404 CrossRef CAS.
- A. Pichon and S. L. James, CrystEngComm, 2008, 10, 1839–1847 RSC.
- C. Chen, J. Zhao, P. Zhang and S. Dai, Polyhedron, 2019, 162, 59–64 CrossRef.
- L. Garay, A. Pichon and S. L. James, Chem. Soc. Rev., 2007, 36, 846–855 RSC.
- I. Stein, V. Gramm and U. Ruschewitz, private communication.
- K. Gramm, D. Smets, I. Grzesiak, T. Block, R. Pöttgen, M. Suta, C. Wickleder, T. Lorenz and U. Ruschewitz, Chem. – Eur. J., 2020, 26, 2726–2734 CrossRef PubMed.
- A. Charushnikova, A. M. Fedoseev, N. A. Budantseva, I. N. Polyakova and Ph Moisy, Russ. J. Coord. Chem., 2007, 33, 61–67 CrossRef.
- I. Pantenburg and U. Ruschewitz, Z. Anorg. Allg. Chem., 2002, 628, 1697–1702 CrossRef CAS.
- F. Hohn, H. Billetter, I. Pantenburg and U. Ruschewitz, Z. Naturforsch., 2002, 57b, 1375–1381 CrossRef.
- I. Stein and U. Ruschewitz, Acta Crystallogr., 2005, E61, m2680–m2682 Search PubMed.
- H. Billetter, F. Hohn, I. Pantenburg and U. Ruschewitz, Acta Crystallogr., 2003, C59, m130–m131 CAS.
- I. Stein, M. Speldrich, H. Schilder, H. Lueken and U. Ruschewitz, Z. Anorg. Allg. Chem., 2007, 633, 1382–1390 CrossRef CAS.
- D. Hermann, C. Näther and U. Ruschewitz, Solid State Sci., 2011, 13, 1096–1101 CrossRef CAS.
- L. Best-Thompson and P. J. Saines, Z. Anorg. Allg. Chem., 2020, 646, 1618–1625 CrossRef CAS.
- U. Ruschewitz and I. Pantenburg, Acta Crystallogr., 2002, C58, m483–m484 CAS.
- St Skoulika, P. Dallas, M. G. Siskos, Y. Deligiannakis and A. Michaelides, Chem. Mater., 2003, 15, 4576–4582 CrossRef CAS.
- R. Ahlers and U. Ruschewitz, Solid State Sci., 2009, 11, 1058–1064 CrossRef CAS.
- R. Askarinejad and A. Morsali, J. Coord. Chem., 2007, 60, 1903–1912 CrossRef.
- R. Ahlers, L. Bohatý and U. Ruschewitz, unpublished results.
- R. Ahlers and U. Ruschewitz, Z. Anorg. Allg. Chem., 2010, 636, 11–14 CrossRef CAS.
- R. Ahlers and U. Ruschewitz, private communication.
- A. Schuy, I. Stein and U. Ruschewitz, Z. Anorg. Allg. Chem., 2010, 636, 1026–1031 CrossRef CAS.
- S. Busch, I. Stein and U. Ruschewitz, Z. Anorg. Allg. Chem., 2012, 638, 2098–2101 CrossRef CAS.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O’Keeffe, M. P. Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef.
- K. Biradha, A. Ramanan and J. J. Vittal, Cryst. Growth Des., 2009, 9, 2969–2970 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- J. Kim, B. Chen, T. M. Reineke, H. Li, M. Eddaoudi, D. B. Moler, M. O’Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 8239–8247 CrossRef CAS PubMed.
- A. Baburin, V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, J. Solid State Chem., 2005, 178, 2452–2474 CrossRef.
- A. Baburin, V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2008, 10, 1822–1838 RSC.
- A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2004, 6, 377–395 RSC.
- L. Carlucci, G. Ciani and D. M. Proserpio, Coord. Chem. Rev., 2003, 246, 247–289 CrossRef CAS.
- Y. Alexandrov, V. A. Blatov and D. M. Proserpio, CrystEngComm, 2017, 19, 1993–2006 RSC.
- L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O’Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef PubMed.
- J. Tranchemontagne, J. R. Hunt and O. M. Yaghi, Tetrahedron, 2008, 64, 8553–8557 CrossRef.
- B. Chen, X. Wang, Q. Zhang, X. Xi, J. Cai, H. Qi, S. Shi, J. Wang, D. Yuan and M. Fang, J. Mater. Chem., 2010, 20, 3758–3767 RSC.
- H. Kim, S. Das, M. G. Kim, D. N. Dybtsev, Y. Kim and K. Kim, Inorg. Chem., 2011, 50, 3691–3696 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- Y. Xing, Z.-S. Jin, Z.-B. Duan and J.-Z. Ni, Chin. J. Struct. Chem., 1995, 14, 1 CAS.
- A. Michaelides and S. Skoulika, Cryst. Growth Des., 2005, 5, 529–533 CrossRef CAS.
- K. Gramm, A. Schuy, M. Suta, C. Wickleder, C. Sternemann and U. Ruschewitz, Z. Anorg. Allg. Chem., 2018, 644, 127–135 CrossRef.
- T. Serre, J. Marrot and G. Férey, Inorg. Chem., 2005, 44, 654–657 CrossRef PubMed.
- J. Howarth, A. W. Peters, N. A. Vermeulen, T. C. Wang, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 26–39 CrossRef.
- S. Yuan, J.-S. Qin, C. T. Lollar and H.-C. Zhou, ACS Cent. Sci., 2018, 4, 440–450 CrossRef CAS PubMed.
- H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. A. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- J. Matemb Ma Ntep, H. Reinsch, B. Moll, E. Hastürk, S. Gökpinar, H. Breitzke, C. Schlüsener, L. Schmolke, G. Buntkowsky and C. Janiak, Chem. – Eur. J., 2018, 24, 14048–14053 CrossRef PubMed.
- Z. Lu, J. Zhang, J. Duan, L. Du and C. Hang, J. Mater. Chem. A, 2017, 5, 17287–17292 RSC.
- Y. Wang, S. Yuan, Z. Hu, T. Kundu, J. Zhang, S. B. Peh, Y. Cheng, J. Dong, D. Yuan, H.-C. Zhou and D. Zhao, ACS Sustainable Chem. Eng., 2019, 7, 7118–7126 CrossRef CAS.
- E. Mondloch, O. Karagiaridi, O. K. Farha and J. T. Hupp, CrystEngComm, 2013, 15, 9258–9264 RSC.
- J. Matemb Ma Ntep, H. Reinsch, J. Liang and C. Janiak, Dalton Trans., 2019, 48, 15849–15855 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 3749–3761 CrossRef.
- A. Airi, C. Atzori, F. Bonino, A. Damin, S. Øien-Ødegaard, E. Aunan and S. Bordiga, Dalton Trans., 2020, 49, 12–16 RSC.
- S. Halasyamani and K. R. Poeppelmeier, Chem. Mater., 1998, 10, 2753–2769 CrossRef.
- M. Ok, E. O. Chi and P. S. Halasyamani, Chem. Soc. Rev., 2006, 35, 710–717 RSC.
- M. Ok, Acc. Chem. Res., 2016, 49, 2774–2785 CrossRef PubMed.
- Y. Song, J. Lindsay, Y. Zhao, A. Nairi, S.-Y. Louis, J. Ling, M. Hu and J. Hu, Comput. Mater. Sci., 2020, 183, 109792 CrossRef CAS.
- A. Schuy, H. Billetter, F. Hohn, I. Pantenburg and U. Ruschewitz, Z. Kristallogr., 2005, 220, 250–258 CAS.
- S. O. Evans, J. Chem. Soc., Dalton Trans., 1999, 3317–3326 RSC.
- W. Miller, C. W. Smith, D. S. Mackenzie and K. E. Evans, J. Mater. Sci., 2009, 44, 5441–5451 CrossRef CAS.
- V. K. Gramm, PhD thesis, University of Cologne, 2016.
- W. Chapman, P. J. Chupas and C. J. Kepert, J. Am. Chem. Soc., 2006, 128, 7009–7014 CrossRef PubMed.
- M. Krishnan, Fundamentals and Applications of Magnetic Materials, Oxford University Press, Oxford, 2016 Search PubMed.
- M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353–1379 RSC.
- M. Espallargas and E. Coronado, Chem. Soc. Rev., 2018, 47, 533–557 RSC.
- W. Anderson, Phys. Rev., 1950, 79, 350–356 CrossRef.
- H. Hendon and A. Walsh, Chem. Sci., 2015, 6, 3674–3683 RSC.
- D. Tiana, C. H. Hendon and A. Walsh, Chem. Commun., 2014, 50, 13990–13993 RSC.
- H. Hendon, F. Pradaux-Caggiano, L. E. Hatcher, W. J. Gee, C. C. Wilson, K. T. Butler, D. R. Carbery, A. Walsh and B. C. Melot, Phys. Chem. Chem. Phys., 2016, 18, 33329–33334 RSC.
- K. Farha, C. E. Wilmer, I. Eryazici, B. G. Hauser, P. A. Parilla, K. O’Neill, A. A. Sarjeant, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 9860–9863 CrossRef PubMed.
- D. J. Yuan, D. Zhao and H.-C. Zhou, Inorg. Chem., 2011, 50, 10528–10530 CrossRef CAS PubMed.
- Y. Yan, S. Yang, A. J. Blake, W. Lewis, E. Poirier, S. A. Barnett, N. R. Champness and M. Schroder, Chem. Commun., 2011, 47, 9995–9997 RSC.
- A. Gomez-Gualdron, O. V. Gutov, V. Krungleviciute, B. Borah, J. E. Mondloch, J. T. Hupp, T. Yildirim, O. K. Farha and R. Q. Snurr, Chem. Mater., 2014, 26, 5632–5639 CrossRef.
- D. Zhao, D. Yuan, A. Yakovenko and H.-C. Zhou, Chem. Commun., 2010, 46, 4196–4198 RSC.
- J. Tranchemontagne, K. S. Park, H. Furukawa, J. Eckert, C. B. Knobler and O. M. Yaghi, J. Phys. Chem. C, 2012, 116, 13143–13151 CrossRef.
- Z. Hu, Y. Peng, Z. Kang, Y. Qian and D. Zhao, Inorg. Chem., 2015, 54, 4862–4868 CrossRef CAS PubMed.
- Z. Hu, I. Castano, S. Wang, Y. Wang, Y. Peng, Y. Qian, C. Chi, X. Wang and D. Zhao, Cryst. Growth Des., 2016, 16, 2295–2301 CrossRef CAS.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- S. Shalini, S. Nandi, A. Justin, R. Maity and R. Vaidhyanathan, Chem. Commun., 2018, 54, 13472–13490 RSC.
- R.-B. Lin, S. Xiang, W. Zhou and B. Chen, Chem., 2020, 6, 337–363 CAS.
- K. Adil, Y. Belmabkhout, R. S. Pillai, A. Cadiau, P. M. Bhatt, A. H. Assen, G. Maurin and M. Eddaoudi, Chem. Soc. Rev., 2017, 46, 3402–3430 RSC.
- M. Trost and C.-J. Li, Modern alkyne chemistry: catalytic and atom-economic transformations, Wiley-VCH Verlag GmbH & Co, 2014 Search PubMed.
- M. Weiss, J. Chem. Educ., 1993, 70, 873–874 CrossRef.
- M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef PubMed.
- A. Torrisi, R. G. Bell and C. Mellot-Draznieks, Cryst. Growth Des., 2010, 10, 2839–2841 CrossRef CAS.
- X. J. Kong, T. He, Y. Zhang, X. Wu, S. Wang, M. Xu, G. Si and J. J. Li, Chem. Sci., 2019, 10, 3949–3955 RSC.
- X.-M. Zhang, Coord. Chem. Rev., 2005, 249, 1201–1219 CrossRef CAS.
- H. Billetter, I. Pantenburg and U. Ruschewitz, Acta Crystallogr., 2005, E61, m1857–m1859 Search PubMed.
- H. Billetter, I. Pantenburg and U. Ruschewitz, Acta Crystallogr., 2006, E62, m881–m883 Search PubMed.
- T. J. Matemb Ma Ntep, H. Breitzke, L. Schmolke, C. Schlüsener, B. Moll, S. Millan, N. Tannert, I. El Aita, G. Buntkowsky and C. Janiak, Chem. Mater., 2019, 31, 8629–8638 CrossRef CAS.
- J. Matemb Ma Ntep, W. Wu, H. Breitzke, C. Schlüsener, B. Moll, L. Schmolke, G. Buntkowsky and C. Janiak, Aust. J. Chem., 2019, 72, 835–841 CrossRef.
- X.-Y. Lu and S.-M. Ma, Chin. J. Chem., 1998, 16, 388–396 CAS.
- J. Marshall, T. Richards, C. L. Hobday, C. F. Murphie, C. Wilson, S. A. Moggach, T. D. Bennett and R. S. Forgan, Dalton Trans., 2016, 45, 4132–4235 RSC.
- J. Marshall, S. L. Griffin, C. Wilson and R. S. Forgan, J. Am. Chem. Soc., 2015, 137, 9527–9530 CrossRef PubMed.
- J. Marshall, S. L. Griffin, C. Wilson and R. S. Forgan, Chem. – Eur. J., 2016, 22, 4870–4877 CrossRef PubMed.
- N. Vouyiouka, E. K. Karakatsani and C. D. Papaspyrides, Progress Polym. Sci., 2005, 30, 10–37 CrossRef.
- J. Vittal and H. S. Quah, Dalton Trans., 2017, 46, 7120–7140 RSC.
- Y. Li, J. Xu, Y. Wang, H. Zheng and K. Li, Molecules, 2021, 26, 7581 CrossRef PubMed.
- J. Tsuchida, Y. Saito, S. Sato, U. Yuki, S. Inayama, Y. Tatewaki, S. Okada, A. Shindo, C. Mikura, K. Fushihara and M. Yamada, Polym. J., 2013, 45, 1007–1012 CrossRef CAS.
- M. Nishii, K. Hayashi and S. Okamura, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 891–895 CrossRef CAS.
- A. Dinca, D. G. Allis, M. D. Moskowitz, M. B. Sponsler and B. S. Hudson, Chem. Mater., 2020, 32, 1769–1783 CrossRef.
- S. Inayama, Y. Tatewaki and S. Okada, Polym. J., 2010, 42, 201–207 CrossRef CAS.
- A. Usanmaz and E. Altürk, J. Macromol. Sci., Part A: Pure Appl. Chem., 2002, A36, 379–395 CrossRef.
- J. Moloney and B. M. Foxman, Inorg. Chim. Acta, 1995, 229, 323–328 CrossRef.
- S. Brodkin and B. M. Foxman, Chem. Mater., 1996, 8, 242–247 CrossRef.
- D. Jaufmann, C. B. Case, R. B. Sandor and B. M. Foxman, J. Solid State Chem., 2000, 152, 99–104 CrossRef.
- A. Shershnev, G. I. Dzhardimalieva, D. P. Kiryukhin, V. A. Zhorin and A. D. Pomogailo, Russ. Chem. Bull., Int. Ed., 2013, 62, 1649–1658 CrossRef.
- M. Sánchez-Sánchez, N. Getachew, K. Díaz, M. Díaz-García, Y. Chebude and I. Díaz, Green Chem., 2015, 17, 1500–1509 RSC.
- S. Dai, F. Nouar, S. Zhang, A. Tissot and C. Serre, Angew. Chem., Int. Ed., 2020, 60, 4282–4288 CrossRef PubMed.
- M. Yassin, A. M. Taddesse and M. Sánchez-Sánchez, Microporous Mesoporous Mater., 2021, 324, 111303 CrossRef.
- Y. Hu, S. Xiang, W. Zhang, Z. Zhang, L. Wang, J. Bai and B. Chen, Chem. Commun., 2009, 7551–7553 RSC.
- H. Heinke and C. Wöll, Adv. Mater., 2019, 31, 1806324 CrossRef PubMed.
- L. Huang, Z. Xiang and D. Cao, J. Mater. Chem. A, 2013, 1, 3851–3855 RSC.
- S. Khullar and S. K. Mandal, RSC Adv., 2014, 4, 39204–39213 RSC.
- P. Dallas, A. B. Bourlinos, P. Komninou, M. Karakassides and D. Niarchos, Res. Lett., 2009, 4, 1358–1364 CAS.
| This journal is © The Royal Society of Chemistry 2022 |