
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-controlled photocatalytic divergent cyclization of alkynyl aldehydes: access to cyclopentenones and dihydropyranols†‡
Haiqian
Zhu§
a,
Hanliang
Zheng§
b,
Junhua
Zhang§
a,
Jian
Feng
a,
Lichun
Kong
a,
Fang
Zhang
a,
Xiao-Song
Xue
*b and
Gangguo
Zhu
 *a
*a
aKey Laboratory of the Ministry of Education for Advanced Catalysis Materials, Department of Chemistry, Zhejiang Normal University, 688 Yingbin Road, Jinhua 321004, P. R. China. E-mail: gangguo@zjnu.cn
bState Key Laboratory of Elemento-organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: xuexs@nankai.edu.cn
First published on 26th July 2021
Abstract
Divergent synthesis is a powerful strategy for the fast assembly of different molecular scaffolds from identical starting materials. We describe here a solvent-controlled photocatalytic divergent cyclization of alkynyl aldehydes with sulfonyl chlorides for the direct construction of highly functionalized cyclopentenones and dihydropyranols that widely exist in bioactive molecules and natural products. Density functional theory calculations suggest that a unique N,N-dimethylacetamide-assisted 1,2-hydrogen transfer of alkoxy radicals is responsible for the cyclopentenone formation, whereas a C–C cleavage accounts for the selective production of dihydropyranols in acetonitrile and water at 50 °C. Given the simple and mild reaction conditions, excellent functional group compatibility, forming up to four chemical bonds, and tunable selectivity, it may find wide applications in synthetic chemistry.
Introduction
Nucleophilic addition to carbonyl groups represents one of the most classic reactions for accessing ubiquitous C–C bonds. The C–O double bonds accept the attack of nucleophiles to form an alkoxide intermediate, followed by protonation to afford alcohol products (Scheme 1a). In contrast, the radical addition to carbonyls can generate a more reactive and versatile alkoxy radical intermediate that may undergo a reduction, 1,2-hydrogen atom transfer (1,2-HAT), or β-fragmentation to produce structurally diverse products (Scheme 1b), such as alcohols,1 ketones,2,3 or aldehydes.4,5 In addition, the radical-mediated reaction usually occurs under mild, neutral conditions, thus tolerating many sensitive functional groups which are incompatible with the ionic reaction. Despite its apparent advantages, the addition of radicals to carbonyl compounds has been much less investigated due to the production of thermodynamically unfavorable alkoxy radicals.6 In this respect, we have established an aldehyde-to-ketone methodology2 with 1,2-HAT of alkoxy radicals as the key step. However, the known methods are limited to aryl or alkenyl aldehydes (R1 = Ar, alkenyl), and the transformation of alkyl aldehydes (R1 = alkyl) is still unknown owing to the lack of spin delocalization in the formed ketyl radical. Recently, Liu4 and Zhu5 reported an elegant intramolecular formyl migration reaction via the β-scission of alkoxy radicals. Key to this transformation is the release of a much more stable radical (R1˙), and therefore, it is a substrate-dependent process. So far, only α-hydroxy aldehydes work for this reaction, and the formyl shift from the more common alkyl aldehydes remains unexplored.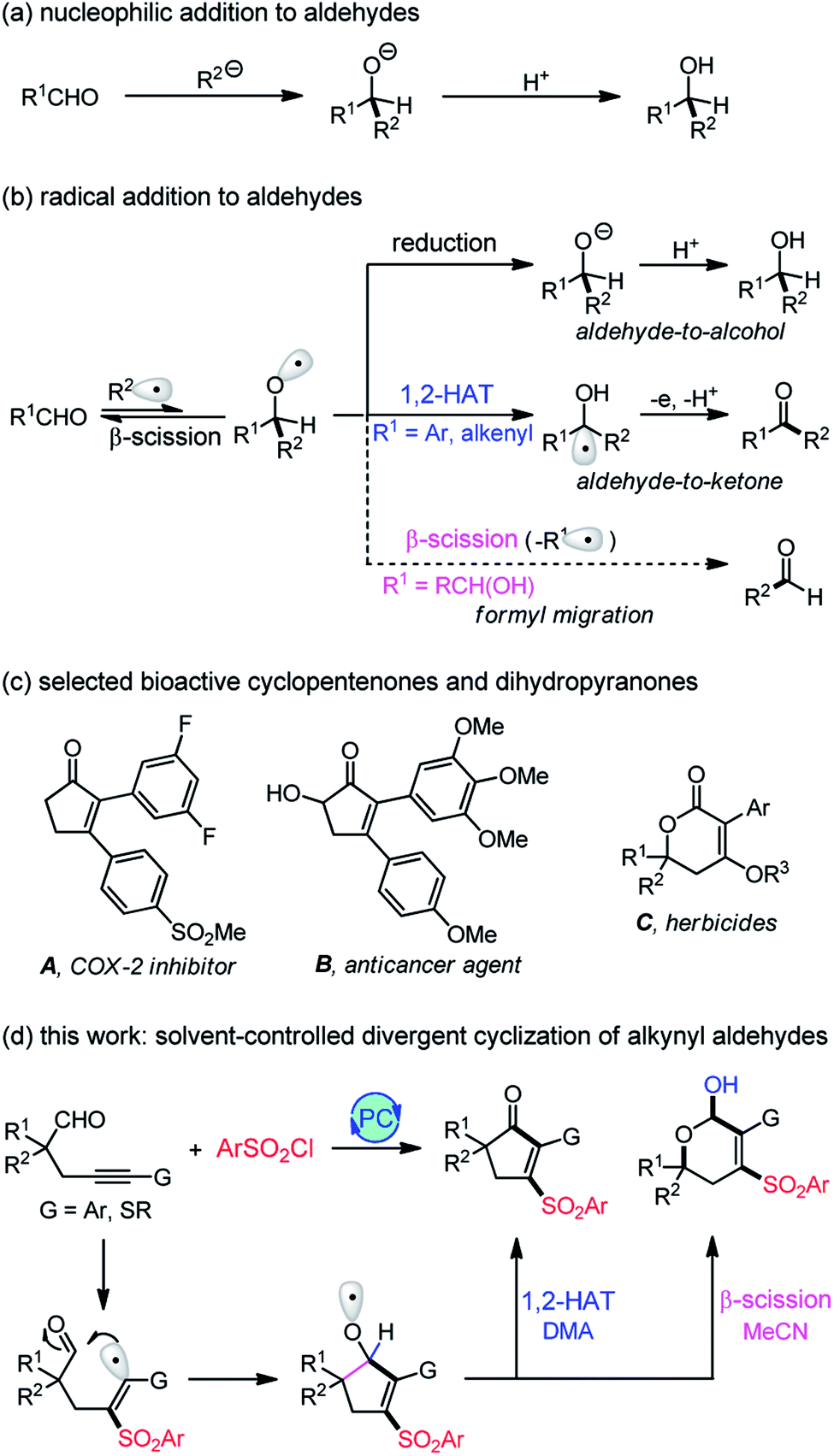 | ||
| Scheme 1 Background and summary of this work. | ||
As fundamental structural scaffolds, cyclopentenones7 and dihydropyranones8 widely exist in numerous natural products and biologically active compounds, such as the inhibitor of cyclooxygenase-2 (COX-2) A,9a potent anticancer agent B,9b and herbicides C9c (Scheme 1c). Consequently, the development of efficient methods for assembling these two motifs is highly desirable. Pursuing our recent interests in the aldehyde-to-ketone methodology,2 we envisioned that an intramolecular addition of alkenyl radicals to alkyl aldehydes would allow us to access the more reactive alkoxy radicals (Scheme 1d). If we could control the reaction pathways of the in situ generated alkoxy radicals, a divergent synthesis of cyclopentenones and dihydropyranols would be established. Herein, we disclose a visible light-induced sulfonylative cyclization of alkynyl aldehydes using commercially available sulfonyl chlorides as the sulfonylation reagent.10 The reaction offers a facile access to various structurally diverse cyclopentenones, dihydropyranols, and dihydropyranones under very mild conditions. Density Functional Theory (DFT) calculations provide insights into this unique solvent-controlled divergent transformation. Specifically, the cyclopentenone formation proceeds via a novel DMA-assisted 1,2-hydrogen transfer, while the β-scission of alkoxy radicals accounts for the selective production of dihydropyranols with MeCN as the solvent. In contrast to the existing photocatalysis divergent synthesis11–13 which is generally restricted to the substrate-11 or catalyst-controlled12 system, the solvent-controlled strategy developed here provides an operationally simple and highly efficient means for divergent synthesis.
Results and discussion
Initially, alkynyl aldehyde 1a and TsCl (2a) were chosen as model substrates for evaluating the reaction conditions. In the presence of 2 mol% of fac-Ir(ppy)3 and 2.0 equiv. of K2CO3, the 3-sulfonyl cyclopentenone 3a was isolated in 66% yield after 5 h of irradiation with 15 W of blue LEDs in DMA at 25 °C (Table 1, entry 1). Base screening revealed that Na2CO3 was the most efficient choice, delivering 3a in 76% yield (entries 2–5). Switching the solvent from DMA to MeCN led to 3a in only 5% yield, and interestingly, the 4-sulfonyl dihydropyranol 4a was isolated in 57% yield (entry 6). Encouraged by the result, we examined other bases and solvents for the dihydropyranol production, however, the reaction yields were not improved (entries 7–12). To our delight, running the reaction at 50 °C resulted in full conversion of 1a and produced 4a in 73% yield (entry 13).|
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Yield of 3ab (%) | Yield of 4ab (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.3 mmol), fac-Ir(ppy)3 (2 mol%), base (0.4 mmol), H2O (0.4 mmol), solvent, 25 °C, 5 h. b Isolated yield. c Run at 50 °C. DMA = N,N-dimethylacetamide, DCM = dichloromethane, DMSO = dimethyl sulfoxide, DMF = N,N-dimethylformamide, NR = no reaction. | ||||
| 1 | K2CO3 | DMA | 66 | Trace |
| 2 | K2HPO4 | DMA | 65 | Trace |
| 3 | K3PO4 | DMA | 66 | Trace |
| 4 | Et3N | DMA | 74 | Trace |
| 5 | Na2CO3 | DMA | 76 | Trace |
| 6 | Na2CO3 | MeCN | 5 | 57 |
| 7 | Na2HPO4 | MeCN | 30 | 30 |
| 8 | K3PO4 | MeCN | 10 | 25 |
| 9 | Na2CO3 | MeNO2 | 5 | 51 |
| 10 | Na2CO3 | DCM | Trace | 48 |
| 11 | Na2CO3 | DMSO | NR | NR |
| 12 | Na2CO3 | DMF | 31 | Trace |
| 13c | Na2CO3 | MeCN | Trace | 73 |
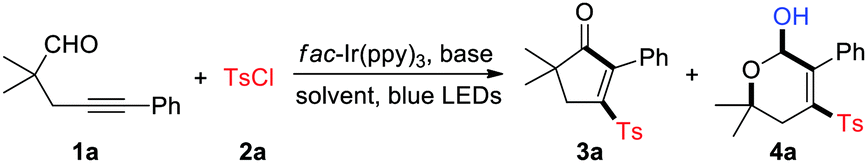
With the optimized reaction conditions in hands, we set about evaluating the scope of this cyclopentenone formation protocol with DMA as the solvent (Table 2). In general, the transformation proceeded efficiently to form densely substituted cyclopentenones in moderate to high yields (3a–3w). A broad array of functional groups, such as F, Cl, Br, CN, Ac, OMe, quinoline, pyridine, and thiofuran, are well tolerated under the reaction conditions, which may be utilized for the downtown transformations. Although both the electron-rich and -deficient substituents were accommodated on the benzene ring of 1, relatively lower yields were observed for the latter cases (3g and 3h). In addition to arylalkynyl substrates (G = Ar), the thioalkynyl aldehyde 1o reacted with 2a as well to produce the desired product 3o in 73% yield. Direct construction of spirocyclopentenones was also feasible, as exemplified by the production of 3p. The alkylalkynyl aldehyde 1q (G = Me) served as a viable substrate, while aldehydes 1r and 1s (G = SiEt3 and H) were not engaged in this reaction (3q–3s). The reaction of 1u, a dicarbonyl substrate, took place uneventfully to afford 3u in 69% yield. Substitution at the propargyl position with two methyl groups led to 3x in a low yield, which may be attributed to the increased hindrance for the radical sulfonylation of C–C triple bonds. After succeeding in synthesizing tetrasubstituted cyclopentenones, we then examined the feasibility of assembling tri- or disubstituted cyclopentenones. Starting from the α-secondary and α-primary aldehydes 1y and 1z, the 2,3,5-trisubstituted and 2,3-disubstituted cyclopentenones were successfully constructed (3y and 3z). The reaction was also amenable to the access of sulfonylated cyclohexenones, albeit in a moderate yield (3za). In contrast, construction of cyclobutenone or cycloheptenone via this method was unsuccessful at the current stage. In the meantime, sulfonyl chlorides 2 were varied with 1i as the coupling partner. Pleasingly, various arylsulfonyl chlorides proved to be efficient substrates and produced the corresponding sulfonyl cyclopentenones in medium to high yields (3zb–3zi). Unfortunately, MeSO2Cl was not engaged in this reaction (3zj). The X-ray crystallographic analysis of 3g14 clearly indicated the structure of 3-sulfonyl cyclopentenones.
| a Reaction conditions: 1 (0.2 mmol), 2 (0.3 mmol), fac-Ir(ppy)3 (2 mol%), Na2CO3 (0.4 mmol), H2O (0.4 mmol), DMA, 25 °C, 5 h. Isolated yields are given. |
|---|

|
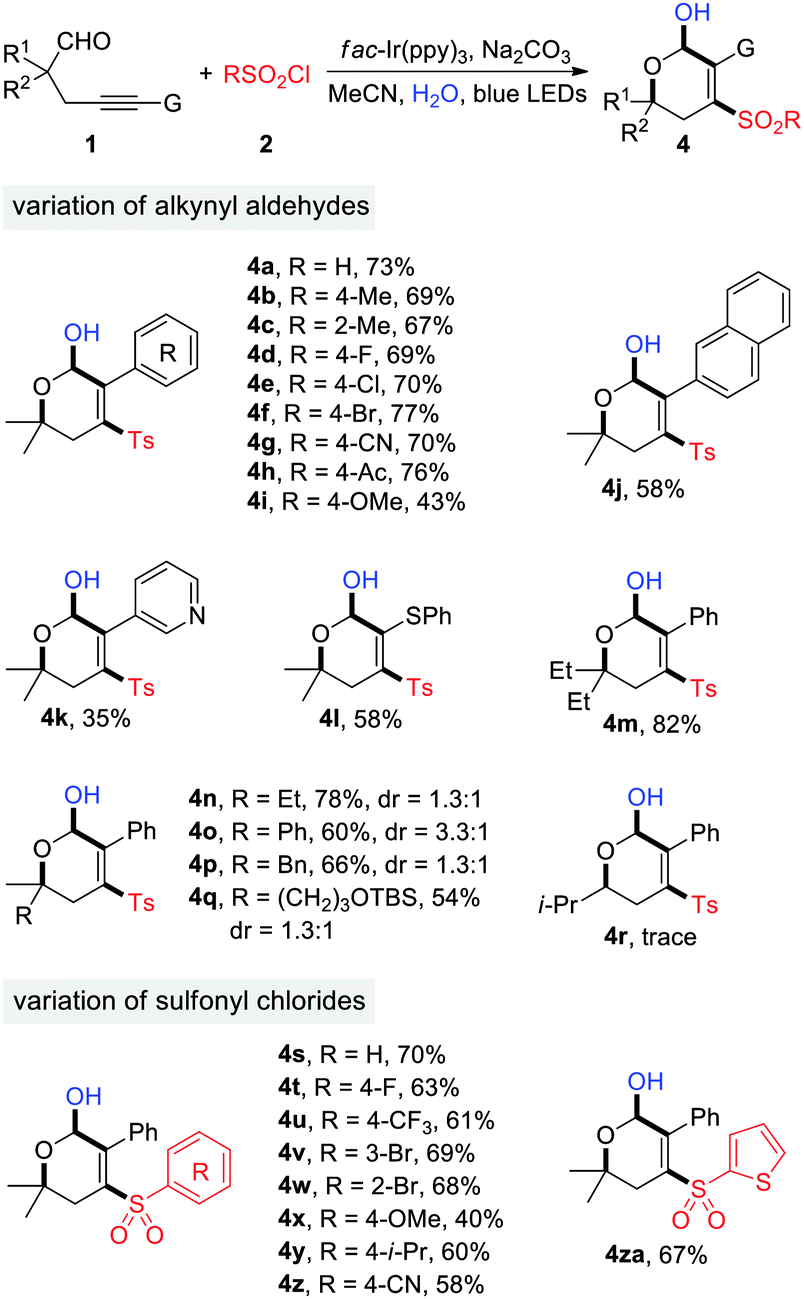
The generality of photoredox-catalyzed synthesis of dihydropyranols was then explored with MeCN as the solvent (Table 3). Alkynyl aldehydes bearing different substituents such as Me, F, Cl, Br, CN, Ac, and OMe served as competent substrates, delivering a variety of 4-sulfonyl dihydropyranols in moderate to high yields (4a–4i). The electron effects appeared to have an impact on the reaction efficiency. Specifically, the transformation of substrates 1g and 1h, having electron-withdrawing CN and Ac substituents on the benzene ring, produced the desired products 4g and 4h in 70% and 76% yield, respectively, while a moderate yield (42%) was observed in the case of 1i, bearing an electron-donating OMe group (4i). Substituents at the α-position of aldehydes were evaluated. In particular, α-tertiary aldehydes worked well for this reaction (4m–4q), whereas the α-secondary aldehyde failed to provide the corresponding product (4r), presumably due to the reduced stability of secondary alkyl radicals. Aldehydes 1q–1s were ineffective for the dihydropyranol formation. Additionally, the scope with respect to sulfonyl chlorides was investigated. A wide range of arylsulfonyl chlorides, substituted by groups such as F, CF3, Br, OMe, i-Pr, and CN, underwent the reaction smoothly to generate functionalized dihydropyranols in promising yields (4s–4z). Similarly, 2-furansulfonyl chloride served as an efficient sulfonylation reagent (4za). It's noteworthy that four new chemical bonds are concurrently created under the reaction conditions, thus highlighting the high bond-forming efficiency of this method.
| a Reaction conditions: 1 (0.2 mmol), 2 (0.3 mmol), fac-Ir(ppy)3 (2 mol%), Na2CO3 (0.4 mmol), H2O (0.4 mmol), MeCN, 50 °C, 5 h. Isolated yields are given. |
|---|
|
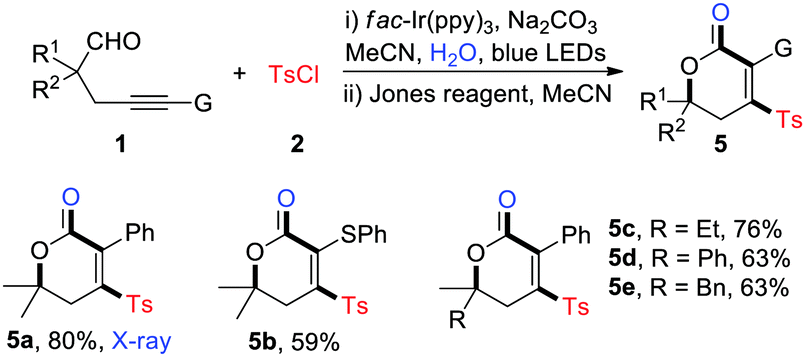
Meanwhile, the one-pot synthesis of polysubstituted dihydropyranones was explored (Table 4). As expected, the reaction furnished a set of sulfonylated dihydropyranones in medium to high yields with good functional group tolerance (5a–5e). The structure of 4-sulfonyl dihydropyranones was unambiguously identified by the X-ray crystallographic analysis of 5a.14
To demonstrate the synthetic utility of this divergent transformation of alkynyl aldehydes, we carried out the derivatization of obtained products (Scheme 2). Substitution of the Ts group of 3a by a methyl group with MeMgCl catalyzed by Ni(acac)2 afforded 6a in 54% yield.15 Michael addition of BnSH or NaOEt to 3a followed by an elimination of sulfinate formed 6b and 6c in 86% and 99% yield, respectively. Following Orita's protocol,16 photocatalytic desulfonylation of both 3a and 5a occurred readily to give 6d and 6e in high yields. Bromination at the allylic position of 5a with NBS and BPO produced 6f in a good yield. Given the significant importance of tetrasubstituted alkenes in organic synthesis, the attempts to establish stereodefined tetrasubstituted alkenes were also performed. Nucleophilic attack of MeMgCl to 5a delivered the tetrasubstituted (E)-alkene 6g in 82% yield. Furthermore, the (E)-enol 6h was selectively assembled in almost quantitative yield upon treatment of 5a with K2CO3 in MeOH at 55 °C for 10 h.
 | ||
Scheme 2 Transformation of selected products. Reaction conditions: (a) MeMgCl (1.2 equiv.), Ni(acac)2 (10 mol%), THF, 25 °C, 10 h; (b) BnSH (2.0 equiv.), DBU (2.0 equiv.), CHCl3, 25 °C, 10 h; (c) NaOEt (2.0 equiv.), THF, 25 °C, 10 h; (d) perylene (5 mol%), i-Pr2NEt (8.0 equiv.), MeCN/THF (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), blue LEDs, 25 °C, 10 h; (e) BPO (20 mol%), NBS (3.0 equiv.), CCl4, 95 °C, 24 h; (f) MeMgCl (3.0 equiv.), THF, 10 h; (g) K2CO3 (4.0 equiv.), MeOH, 55 °C, 10 h. THF = tetrahydrofuran, DBU = 1,8-diazabicyclo[5,4,0]undec-7-ene, BPO = dibenzoyl peroxide, NBS = N-bromosuccinimide. :1), blue LEDs, 25 °C, 10 h; (e) BPO (20 mol%), NBS (3.0 equiv.), CCl4, 95 °C, 24 h; (f) MeMgCl (3.0 equiv.), THF, 10 h; (g) K2CO3 (4.0 equiv.), MeOH, 55 °C, 10 h. THF = tetrahydrofuran, DBU = 1,8-diazabicyclo[5,4,0]undec-7-ene, BPO = dibenzoyl peroxide, NBS = N-bromosuccinimide. | ||
To gain insights into the reaction mechanism, some control experiments were performed, and the results are summarized in Scheme 3. In the presence of butylated hydroxytoluene (BHT, 2.0 equiv.), neither 3a nor 4a could be obtained in a noticeable yield, and instead, the sulfonyl compound 7 was isolated in 11% and 48% yield, respectively. Likewise, adding 2,2,6,6-tetramethylpiperidinooxy (TMEPO, 2.0 equiv.) to the standard conditions shut down the reaction (not shown). These results suggested a radical pathway. Additionally, the 18O isotope labeling experiments were conducted. With the addition of 5.0 equiv. of H218O, 4a-18O was isolated in 69% yield with 89% 18O incorporation. In addition to the signal of [M + Na]+ ion of 4a-18O, a signal matched with [M + Na–H218O]+ ion was observed by the HRMS analysis, thus indicating that the hydroxy group of 4a is originated from water (see ESI‡ for details). Under the standard conditions, the reaction of aldehyde 1a-D performed well to deliver 3a and 4a-D in 73% and 75% yield, respectively. Parallel intermolecular kinetic isotopic effect experiments were then conducted, and a KIE value of 1.0 was determined, implying that the aldehydic C–H cleavage may not be the rate-determing step.
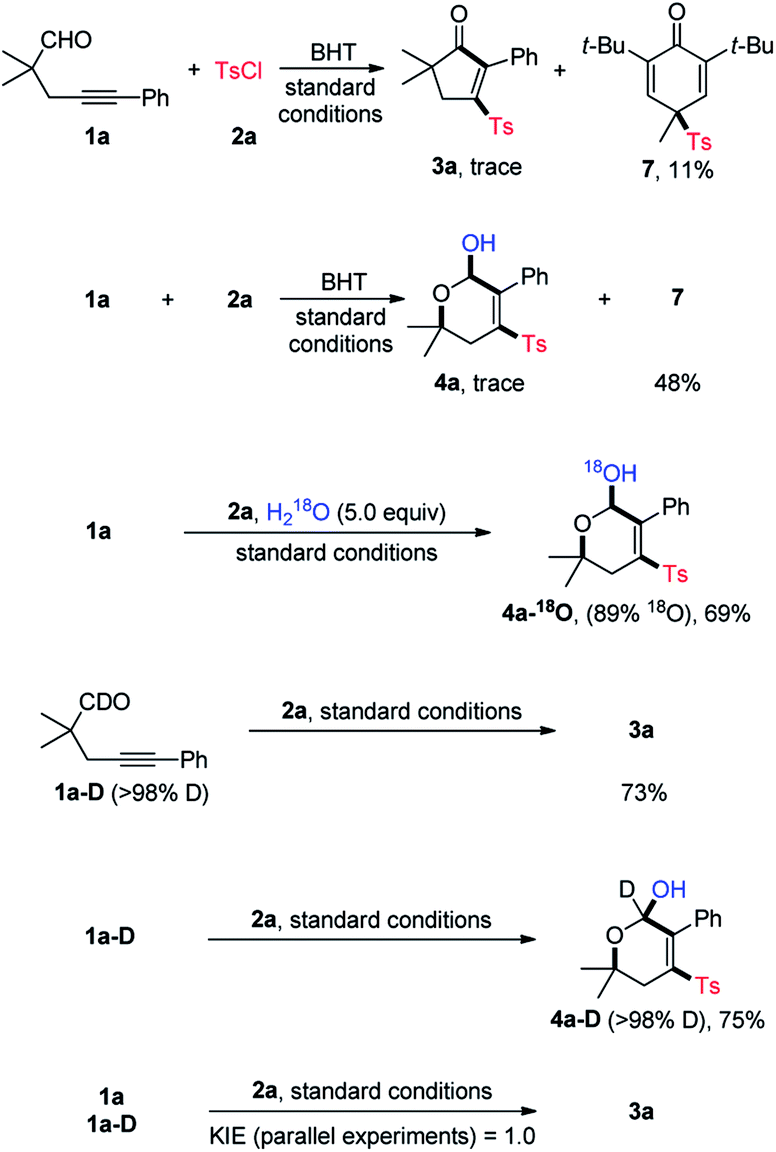 | ||
| Scheme 3 Mechanistic studies. | ||
Based on the above results and previous reports,2,4,5 a mechanistic proposal for the divergent cyclization of 1a is summarized in Scheme 4. Initially, the single electron transfer (SET) between the excited photocatalysis Ir(III)* and TsCl affords a Ir(IV) species and sulfonyl radical Ts˙.10d Radical sulfonylation of the C–C triple bond of 1a and a subsequent addition of vinyl radical I to the intramolecular CHO group produces a cyclopentenyloxy radical II. It may undergo a 1,2-HAT to deliver the neutral ketyl radical III, followed by SET with Ir(IV) and deprotonation to provide 3a (path 1a). Alternatively, the β-C-C cleavage of II17 may take place to form a tertiary alkyl radical IV (path 1b), which can be converted into the oxonium ion VIvia a 6-endo radical cyclization (path 2a)/SET oxidation sequence. Additionally, a radical oxidation to the cation VII (path 2b) followed by intramolecular nucleophilic attack may also lead to the generation of VI. Finally, the nucleophilic attack of VI by H2O generates 4a as the product.
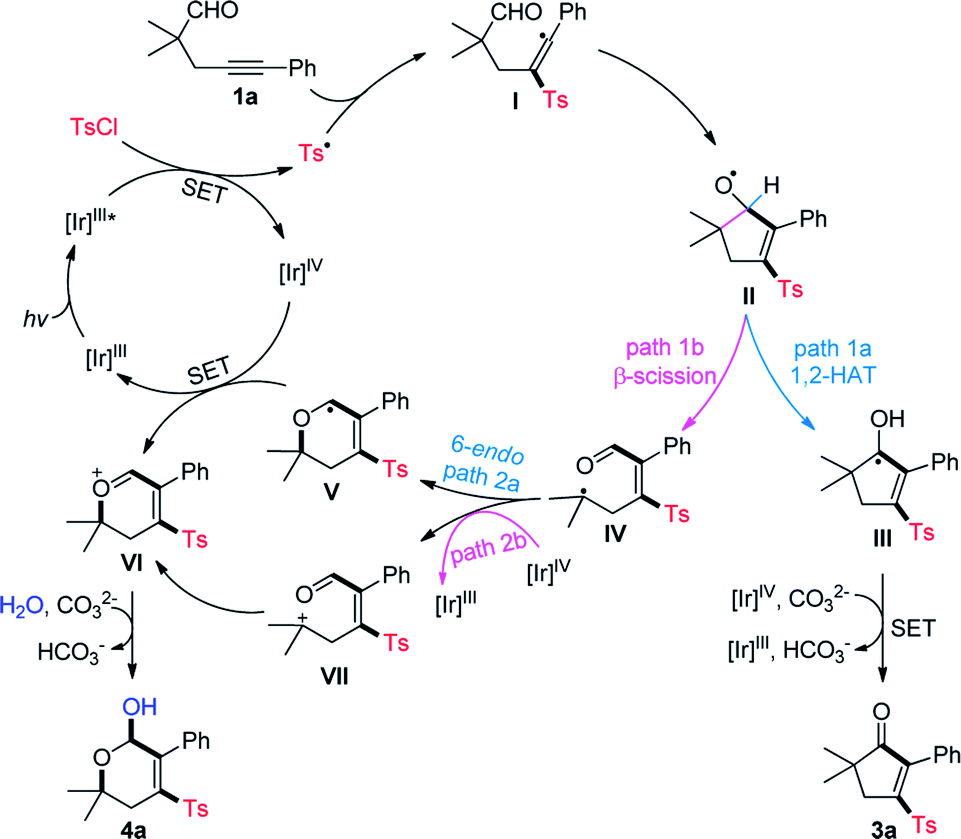 | ||
| Scheme 4 Proposed mechanism. | ||
To shed light on the unique role of solvent in tuning the reaction pathways, we carried out computational studies using DFT calculations, and the results are presented in Scheme 5. As for the reaction performed in DMA, radical sulfonylation of 1a occurring via transition state TS1 requires an activation free energy of 14.3 kcal mol−1, which is viable under the reaction conditions. Subsequently, radical addition to the intramolecular CHO group proceeds via a five-membered ring transition state TS2 to give the radical II. This step has a free energy barrier of 9.3 kcal mol−1. Starting from II, an unprecedented DMA-assisted 1,2-hydrogen transfer18 (viaTS3-DMA, with a free energy of 7.6 kcal mol−1) is favoured over the 6-endo radical cyclization (viaTS5, with a free energy of 9.4 kcal mol−1) by 1.8 kcal mol−1, thus giving rise to the ketyl radical III. Formation of III is highly exergonic by 30.7 kcal mol−1 and is therefore an irreversible process. Followed by SET oxidation and deprotonation, 3a can be constructed together with the regeneration of photocatalysis Ir(III), which is strongly exergonic by 63.2 kcal mol−1.
 | ||
| Scheme 5 The calculated relative Gibbs free energies for the intermediates and transition-states of the model reaction in DMA (in blue) and MeCN (in pink) at the wb97xd/def2tzvp-SMD//B3LYP-D3(BJ)/6-31G(d,p) level of theory. | ||
With MeCN as the solvent, the MeCN-assisted 1,2-hydrogen transfer of II (viaTS3-NCMe) has a higher energy barrier of 15.3 kcal mol−1, which is unlikely to compete with the β-C-C cleavage/6-endo cyclization sequence. Moreover, the 6-endo radical cyclization of IV requiring a free energy barrier of 4.5 kcal mol−1 is favoured over the SET oxidation, with a free energy barrier of 9.6 kcal mol−1, by 5.1 kcal mol−1. Therefore, a stable allyl radical V that lies 25.8 kcal mol−1 lower in energy than IV is selectively formed. Spin delocalization to the neighboring C–C double bond should be responsible for the increased stability of V, which provides a driving force for the C–C bond cleavage and uncommon addition of alkyl radical to the carbonyl oxygen atom19 in the 6-endo radical cyclization step. Subsequently, the radical V is oxidized by Ir(IV) to form an oxonium ion intermediate VI, followed by nucleophilic attack of water to produce 4a, which is strongly exergonic by 39.1 kcal mol−1.
Conclusions
In conclusion, a photocatalytic divergent coupling of alkynyl aldehydes with sulfonyl chlorides is developed. The reaction provides a straightforward and highly selective method for the assembly of structurally diverse cyclopentenones, dihydropyranols, and dihydropyranones that are important building blocks in organic and medicinal chemistry. Up to four new chemical bonds are concurrently constructed under mild reaction conditions, thus highlighting the high efficiency of this method. DFT calculations reveal that a DMA-assisted 1,2-HAT of alkoxy radicals is responsible for the production of cyclopentenones, whereas a β-C–C cleavage followed by 6-endo radical cyclization to formyl group accounts for the generation of dihydropyranols in MeCN. Tuneable selectivity for 1,2-HAT and β-fragmentation of alkoxy radicals is realized by change of the solvent, thus providing a novel strategy for the selective activation of inert C–H and C–C bonds.Data availability
The data supporting this article have been uploaded as part of the ESI.‡Author contributions
H. Z., J. Z., and J. F. conducted the experiments. H. Z., and X.-S. X. carried out the theoretical calculations and wrote the manuscript. L. K., and F. Z. characterized the compounds and analyzed the data. G. Z. designed the project, analyzed the results and wrote the manuscript.Conflicts of interest
There are no conficts to declare.Acknowledgements
This work is supported by the Natural Science Foundation of Zhejiang Province (LZ20B020002) and National Natural Science Foundation of China (22071218, 21672191, 21772098 and 21933004). We thank Dr Z. Zhang, Z. Yuan, and L. Zhou for their helpful discussions and Miss X. Wang for her support in the X-ray crystallographic analysis.Notes and references
- For selected reports, see: (a) T. Kawamoto, T. Fukuyama and I. Ryu, J. Am. Chem. Soc., 2012, 134, 875 CrossRef CAS PubMed; (b) L. Pitzer, F. Sandfort, F. Strieth-Kalthoff and F. Glorius, J. Am. Chem. Soc., 2017, 139, 13652 CrossRef CAS PubMed; (c) G. Wang, J. Cao, L. Gao, W. Chen, W. Huang, X. Cheng and S. Li, J. Am. Chem. Soc., 2017, 139, 3904 CrossRef CAS PubMed; (d) M. Saladrigas, C. Bosch, G. V. Saborit, J. Bonjoch and B. Bradshaw, Angew. Chem., Int. Ed., 2018, 57, 182 CrossRef CAS PubMed; (e) S. Xie, D. Li, H. Huang, F. Zhang and Y. Chen, J. Am. Chem. Soc., 2019, 141, 16237 CrossRef CAS PubMed; (f) J. Y. Kim, Y. S. Lee, Y. Choi and D. H. Ryu, ACS Catal., 2020, 10, 10585 CrossRef CAS.
- (a) C. Che, Q. Huang, H. Zheng and G. Zhu, Chem. Sci., 2016, 7, 4134 RSC; (b) Y. Zhang, D. Guo, S. Ye, Z. Liu and G. Zhu, Org. Lett., 2017, 19, 1302 CrossRef CAS PubMed; (c) D. Lu, Y. Wan, L. Kong and G. Zhu, Org. Lett., 2017, 19, 2929 CrossRef CAS PubMed; (d) Y. Chen, C. Shu, F. Luo, X. Xiao and G. Zhu, Chem. Commun., 2018, 54, 5373 RSC; (e) Y. Zhou, Z. Xiong, J. Qiu, L. Kong and G. Zhu, Org. Chem. Front., 2019, 6, 1022 RSC . For a review, see:; (f) L. Kong, Y. Zhou, F. Luo and G. Zhu, Chin. J. Org. Chem., 2018, 38, 2858 CrossRef CAS.
- For selected reports from other groups, see: (a) W.-C. Yang, P. Dai, K. Luo, Y.-G. Ji and L. Wu, Adv. Synth. Catal., 2017, 359, 2390 CrossRef CAS; (b) H. Hu, X. Chen, K. Sun, J. Wang, Y. Liu, H. Liu, L. Fan, B. Yu, Y. Sun, L. Qu and Y. Zhao, Org. Lett., 2018, 20, 6157 CrossRef CAS PubMed; (c) X.-K. He, B.-G. Cai, Q.-Q. Yang, L. Wang and J. Xuan, Chem.–Asian J., 2019, 14, 3269 CrossRef CAS PubMed; (d) Y. Mei, L. Zhao, Q. Liu, S. Ruan, L. Wang and P. Li, Green Chem., 2020, 22, 2270 RSC.
- Z.-L. Li, X.-H. Li, N. Wang, N.-Y. Yang and X.-Y. Liu, Angew. Chem., Int. Ed., 2016, 55, 15100 CrossRef CAS PubMed.
- J. Yu, D. Wang, Y. Xu, Z. Wu and C. Zhu, Adv. Synth. Catal., 2018, 360, 744 CrossRef CAS.
- S. Wilsey, P. Dowd and K. N. Houk, J. Org. Chem., 1999, 64, 8801 CrossRef CAS PubMed.
- For selected reviews, see: (a) S. P. Simeonov, J. P. M. Nunes, K. Guerra, V. B. Kurteva and C. A. M. Afonso, Chem. Rev., 2016, 116, 5744 CrossRef CAS PubMed; (b) A. J. Frontier and J. J. Hernandez, Acc. Chem. Res., 2020, 53, 1822 CrossRef CAS PubMed . For recent reports, see:; (c) K. Wang, C. Jiang, Z. Zhang, C. Han, X. Wang, Y. Li, K. Chen and J. Zhao, Chem. Commun., 2020, 56, 12817 RSC; (d) M. J. Hensinger, N. J. Dodge and M. Brewer, Org. Lett., 2020, 22, 497 CrossRef CAS PubMed; (e) J. Li, Y. Xu, X. Hu, S. Zhu and L. Liu, Org. Lett., 2020, 22, 9478 CrossRef CAS PubMed; (f) Y. H. Lee, E. H. Denton and B. Morandi, J. Am. Chem. Soc., 2020, 142, 20948 CrossRef CAS PubMed and references cited therein..
- For selected reviews, see: (a) J. A. Marco, M. Carda, J. Murga and E. Falomir, Tetrahedron, 2007, 63, 2929 CrossRef CAS; (b) X.-Y. Chen, Q. Liu, P. Chauhan and D. Enders, Angew. Chem., Int. Ed., 2018, 57, 3862 CrossRef CAS PubMed . For selected reports, see:; (c) H.-S. Yeom, J. Koo, H.-S. Park, Y. Wang, Y. Liang, Z.-X. Yu and S. Shin, J. Am. Chem. Soc., 2012, 134, 208 CrossRef CAS PubMed; (d) H. Kim, S. Y. Choi and S. Shin, Angew. Chem., Int. Ed., 2018, 57, 13130 CrossRef CAS PubMed; (e) D. D. Borisov, R. A. Novikov, A. S. Eltysheva, Y. V. Tkachev and Y. V. Tomilov, Org. Lett., 2017, 19, 3731 CrossRef CAS PubMed; (f) H.-J. Zhang and L. Yin, J. Am. Chem. Soc., 2018, 140, 12270 CrossRef CAS PubMed.
- (a) W. C. Black, C. Brideau, C.-C. Chan, S. Charleson, N. Chauret, D. Claveau, D. Ethier, R. Gordon, G. Greig, J. Guay, G. Hughes, P. Jolicoeur, Y. Leblanc, D. Nicoll-Griffith, N. Ouimet, D. Riendeau, D. Visco, Z. Wang, L. Xu and P. Prasit, J. Med. Chem., 1999, 42, 1274 CrossRef CAS PubMed; (b) M. K. Gurjar, R. D. Wakharkar, A. T. Singh, M. Jaggi, H. B. Borate, P. D. Shinde, R. Verma, P. Rajendran, S. Dutt, G. Singh, V. K. Sanna, M. K. Singh, S. K. Srivastava, V. A. Mahajan, V. H. Jadhav, K. Dutta, K. Krishnan, A. Chaudhary, S. K. Agarwal, R. Mukherjee and A. C. Burman, J. Med. Chem., 2007, 50, 1744 CrossRef CAS PubMed; (c) Y. Nakashima and Y. Jin, WO2014/084407Al, 2014.
- For selected reports on radical sulfonylation of alkynes with sulfonyl chlorides, see: (a) X. Zeng, L. Ilies and E. Nakamura, Org. Lett., 2012, 14, 954 CrossRef CAS PubMed; (b) L. Wang, H. Zhu, J. Che, Y. Yang and G. Zhu, Tetrahedron Lett., 2014, 55, 1011 CrossRef CAS; (c) A. García-Domínguez, S. Müller and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 9949 CrossRef PubMed; (d) P. Chakrasali, K. Kim, Y.-S. Jung, H. Kim and S. B. Han, Org. Lett., 2018, 20, 7509 CrossRef CAS PubMed; (e) A. Hossain, S. Engl, E. Lutsker and O. Reiser, ACS Catal., 2019, 9, 1103 CrossRef CAS.
- For selected reports, see: (a) H. Jiang, Y. Cheng, Y. Zhang and S. Yu, Org. Lett., 2013, 15, 4884 CrossRef CAS PubMed; (b) D. B. Bagal, G. Kachkovskyi, M. Knorn, T. Rawner, B. M. Bhanage and O. Reiser, Angew. Chem., Int. Ed., 2015, 54, 6999 CrossRef CAS PubMed; (c) J. Cheng, Y. Cheng, J. Xie and C. Zhu, Org. Lett., 2017, 19, 6452 CrossRef CAS PubMed; (d) Y. Liu, R.-J. Song, S. Luo and J.-H. Li, Org. Lett., 2018, 20, 212 CrossRef CAS PubMed; (e) H. Liang, G.-Q. Xu, Z.-T. Feng, Z.-Y. Wang and P.-F. Xu, J. Org. Chem., 2019, 84, 60 CrossRef CAS PubMed; (f) J. Ma, F. Schäfers, C. Daniliuc, K. Bergander, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 9639 CrossRef CAS PubMed; (g) Z.-S. Wang, Y.-B. Chen, H.-W. Zhang, Z. Sun, C. Zhu and L.-W. Ye, J. Am. Chem. Soc., 2020, 142, 3636 CrossRef CAS PubMed.
- For selected reports, see: (a) A. Singh, C. J. Fennell and J. D. Weaver, Chem. Sci., 2016, 7, 6796 RSC; (b) M. J. James, J. L. Schwarz, F. Strieth-Kalthoff, B. Wibbeling and F. Glorius, J. Am. Chem. Soc., 2018, 140, 8624 CrossRef CAS PubMed; (c) J. Li, Y. Luo, H. W. Cheo, Y. Lan and J. Wu, Chem, 2019, 5, 192 CrossRef CAS; (d) M. Mei, D. Anand and L. Zhou, Org. Lett., 2019, 21, 3548 CrossRef CAS PubMed.
- (a) S. Zhu, A. Das, L. Bui, H. Zhou, D. P. Curran and M. Rueping, J. Am. Chem. Soc., 2013, 135, 1823 CrossRef CAS PubMed; (b) C. Zhang, S. Li, F. Bureš, R. Lee, X. Ye and Z. Jiang, ACS Catal., 2016, 6, 6853 CrossRef CAS; (c) Y. Tsuchiya, R. Onai, D. Uraguchi and T. Ooi, Chem. Commun., 2020, 56, 11014 RSC.
- Crystallographic data for products 3g and 5a have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 2060837 and 2060838..
- J. L. Fabre, M. Julia and J. N. Verpeaux, Tetrahedron Lett., 1982, 23, 2469 CrossRef CAS.
- H. Watanabe, M. Takemoto, K. Adachi, Y. Okuda, A. Dakegata, T. Fukuyama, I. Ryu, K. Wakamatsu and A. Orita, Chem. Lett., 2020, 49, 409 CrossRef CAS.
- For selected reports on ring opening of unstrained cycloalkanols via β-scission of alkoxy radicals, see: (a) H. G. Yayla, H. Wang, K. T. Tarantino, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2016, 138, 10794 CrossRef CAS PubMed; (b) E. Ota, H. Wang, N. L. Frye and R. R. Knowles, J. Am. Chem. Soc., 2019, 141, 1457 CrossRef CAS PubMed; (c) J.-J. Guo, A. Hu, Y. Chen, J. Sun, H. Tang and Z. Zuo, Angew. Chem., Int. Ed., 2016, 55, 15319 CrossRef CAS PubMed; (d) A. Hu, Y. Chen, J.-J. Guo, N. Yu, Q. An and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 13580 CrossRef CAS PubMed; (e) D. Wang, J. Mao and C. Zhu, Chem. Sci., 2018, 9, 5805 RSC; (f) M. Wang, M. Li, S. Yang, X.-S. Xue, X. Wu and C. Zhu, Nat. Commun., 2020, 11, 672 CrossRef PubMed; (g) L. Huang, T. Ji and M. Rueping, J. Am. Chem. Soc., 2020, 142, 3532 CrossRef CAS PubMed . For selected reviews, see:; (h) X. Wu and C. Zhu, Chem. Commun., 2019, 55, 9747 RSC; (i) S.-H. Shi, Y. Liang and N. Jiao, Chem. Rev., 2021, 121, 485 CrossRef CAS PubMed; (j) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506 CrossRef CAS PubMed.
- For selected reports on solvent-assisted 1,2-HAT of alkoxy radicals, see: (a) K. G. Konya, T. Paul, S. Lin, J. Lusztyk and K. U. Ingold, J. Am. Chem. Soc., 2000, 122, 7518 CrossRef CAS; (b) C. Che, Z. Qian, M. Wu, Y. Zhao and G. Zhu, J. Org. Chem., 2018, 83, 5665 CrossRef CAS PubMed; (c) J. Zhang, D. Liu, S. Liu, Y. Ge, Y. Lan and Y. Chen, iScience, 2020, 23, 100755 CrossRef CAS PubMed . However, our calculations indicated that water-assisted 1,2-HAT is less likely for this work, see Fig. S1 and S2‡ of ESI for details..
- For selected reports on addition of alkyl radicals to the carbonyl oxygen atom, see: (a) D. Liu, S. Tang, H. Yi, C. Liu, X. Qi, Y. Lan and A. Lei, Chem.–Eur. J., 2014, 20, 15605 CrossRef CAS PubMed; (b) L. Lv, S. Lu, Q. Guo, B. Shen and Z. Li, J. Org. Chem., 2015, 80, 698 CrossRef CAS PubMed; (c) L.-N. Guo, S. Wang, X.-H. Duan and S.-L. Zhou, Chem. Commun., 2015, 51, 4803 RSC.
Footnotes |
| † In memory of Prof. Ei-ichi Negishi. |
| ‡ Electronic supplementary information (ESI) available: Data for all the products, experimental procedures, and computational data. CCDC 2060837 and 2060838. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03416b |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |