
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quasi-square-shaped cadmium hydroxide nanocatalysts for electrochemical CO2 reduction with high efficiency†
Chunjun
Chen
 ab,
Xupeng
Yan
ab,
Ruizhi
Wu
ab,
Yahui
Wu
ab,
Qinggong
Zhu
a,
Minqiang
Hou
a,
Zhaofu
Zhang
a,
Honglei
Fan
a,
Jun
Ma
a,
Yuying
Huang
e,
Jingyuan
Ma
e,
Xiaofu
Sun
*ab,
Longfei
Lin
*a,
Shoujie
Liu
*cd and
Buxing
Han
*abf
ab,
Xupeng
Yan
ab,
Ruizhi
Wu
ab,
Yahui
Wu
ab,
Qinggong
Zhu
a,
Minqiang
Hou
a,
Zhaofu
Zhang
a,
Honglei
Fan
a,
Jun
Ma
a,
Yuying
Huang
e,
Jingyuan
Ma
e,
Xiaofu
Sun
*ab,
Longfei
Lin
*a,
Shoujie
Liu
*cd and
Buxing
Han
*abf
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid and Interface and Thermodynamics, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: hanbx@iccas.ac.cn; sunxiaofu@iccas.ac.cn; linlongfei@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cChemistry and Chemical Engineering of Guangdong Laboratory, Shantou 515063, China
dCollege of Chemistry and Materials Science, Anhui Normal University, Wuhu 241000, China. E-mail: jiesliu@ahnu.edu.cn
eShanghai Synchrotron Radiation Facility, Zhangjiang Laboratory (SSRF, ZJLab), Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201204, China
fShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
First published on 10th August 2021
Abstract
Powered by a renewable electricity source, electrochemical CO2 reduction reaction is a promising solution to facilitate the carbon balance. However, it is still a challenge to achieve a desired product with commercial current density and high efficiency. Herein we designed quasi-square-shaped cadmium hydroxide nanocatalysts for CO2 electroreduction to CO. It was discovered that the catalyst is very active and selective for the reaction. The current density could be as high as 200 mA cm−2 with a nearly 100% selectivity in a commonly used H-type cell using the ionic liquid-based electrolyte. In addition, the faradaic efficiency of CO could reach 90% at a very low overpotential of 100 mV. Density functional theory studies and control experiments reveal that the outstanding performance of the catalyst was attributed to its unique structure. It not only provides low Cd–O coordination, but also exposes high activity (002) facet, which requires lower energy for the formation of CO. Besides, the high concentration of CO can be achieved from the low concentration CO2via an adsorption-electrolysis device.
Introduction
The concentration of atmospheric carbon dioxide (CO2) increased dramatically with the consumption of fossil fuels, which has caused climate change and serious environmental issues.1–5 Electrochemical CO2 reduction reaction (CO2RR) is a promising solution to facilitate the carbon balance, which can not only transfer CO2 into valuable fuels and chemicals, but also provide a solution for storage of the renewable energy.6–10 CO is a versatile feedstock for producing various chemicals and fuels, which is viewed as the closest to commercialization among all the products.11–16 Although the faradaic efficiency (FE) and current density for CO have been significantly improved by using some noble-based metals (Pd, Au, Ag, etc.),17–19 non-noble based metals (Fe, Co, Ni, etc.)20–22 and metal-free carbon materials.23–26 More efficient electrocatalysts that meet the commercial purpose still remain to be developed.Recently, Cd-based materials (e.g., CdS, CdSe and metallic Cd) have been reported to convert CO2 to CO efficiently, due to the Cd sites can suppress the hydrogen evolution reaction (HER) and exhibit excellent CO anti-poisoning.27–29 The previous works showed that tuning the coverage of surface hydroxyl groups and developing special morphology of catalysts can promote CO2 reduction and inhibit the HER.30,31 As an important and conventional semiconductor, cadmium hydroxide (Cdhy) has been applied in a wide range of fields, including solar cells, sensors, and cathode electrode materials of batteries.32,33 It offers an opportunity for developing novel Cdhy catalysts, which may be favorable for improving the selectivity and activity of CO from CO2RR.
In addition, using the industrial outputs as the CO2 source is commercially viable. The concentration of CO2 in flue gas in the industrial processes (i.e. steel and cement production, power plant flue gas) is usually between 4% and 15%.34 To achieve more commercial feasibility of CO2RR, the CO2 reactant and the products separation should be considered simultaneously. Although the selectivity and activity of CO2RR have been significantly improved in the flow cell and membrane electrode assembly (MEA),35,36 the CO2 availability is low, and the product separation is also high energy consumption. In addition, the alkaline electrolyte was used, which can result in the CO2 loss, imposing a limit of carbon utilization efficiency.37 According to the previous reports,38 the low concentrations of CO2 can be directly converted to CO, however, the concentration of the CO was very low. Recently, high content of CO has been obtained via the electroreduction of the captured solution of CO2.39 However, the FE and current density were only 70% and 50 mA cm−2, which was limited to the poor electrocatalytic performance of capture solution. In our previous reports,40–42 the ionic liquid (IL) has been proven to be a good solvent for absorbing CO2, and the IL was also an excellent electrolyte for CO2RR. Thus, we can assume that combining the feature of trapping and electrocatalytic activity of IL for CO2 can promote the development of commercialization.
Herein we developed a feasible and facile strategy to synthesize quasi-square-shaped Cdhy-QS nanocatalysts for ultra-efficient CO2 electroreduction. Cdhy-QS yielded nearly 100% CO selectivity in a very wide potential range with extremely high current density (>200 mA cm−2). Moreover, the energy efficiency (EE) was also very high. These advantages made Cdhy-QS have great potential for practical application. X-ray absorption spectroscopy (XAS) and density functional theory (DFT) calculations showed that high activity facet (002) exposed on the surface and the low Cd–O coordination number resulted in the enhancement of CO2 activation. In addition, the high concentrations of CO in the gas products can be achieved from the low concentrations of CO2, when combining the Cdhy-QS and the adsorption-electrolysis device system.
Results and discussion
We used tannic acid (TA) as capping agent, which is commonly used to control synthesis of functional materials,38 to synthesize Cdhy-QS (see the experimental details in the ESI†). Firstly, X-ray diffraction (XRD) analysis was used to characterize Cdhy-QS, and the features of Cd(OH)2 can be observed (Fig. S1†). From transmission electron microscope (TEM) image of Cdhy-QS (Fig. 1A), we can observe that the Cdhy was dispersed homogeneously on the cross-linked architecture of TA. And the size of Cdhy-QS was uniform (Fig. 1B) with a quasi-square shape, which has not been reported in the literature for Cdhy. The energy dispersive X-ray elemental (EDX) mappings show that the Cd and O elements were homogenous over the entire architectures (Fig. S2†). The high-resolution TEM images reveals that Cdhy-QS had an interplanar spacing of 0.236 nm (Fig. 1C), which corresponds to the (002) plane of Cdhy. It was also identified by the corresponding fast Fourier transform (FFT) pattern (Fig. 1D). They are different with those of Cdhy nanoparticles (Cdhy-np) (see the experimental details in the ESI†), which are (100) and (110) facets (Fig. S3†). The content of Cd in Cdhy-QS and Cdhy-np was 9.5 wt% and 9.8 wt%, respectively, which were determined by inductively coupled plasma-atomic emission spectrometry (ICP-AES). Additionally, enlarged and repeated experiments for Cdhy-QS preparation were carried out (Fig. S4 and S5†), and 16 g Cdhy-QS was obtained in one experiment that only took 30 minutes, and the morphologies were similar.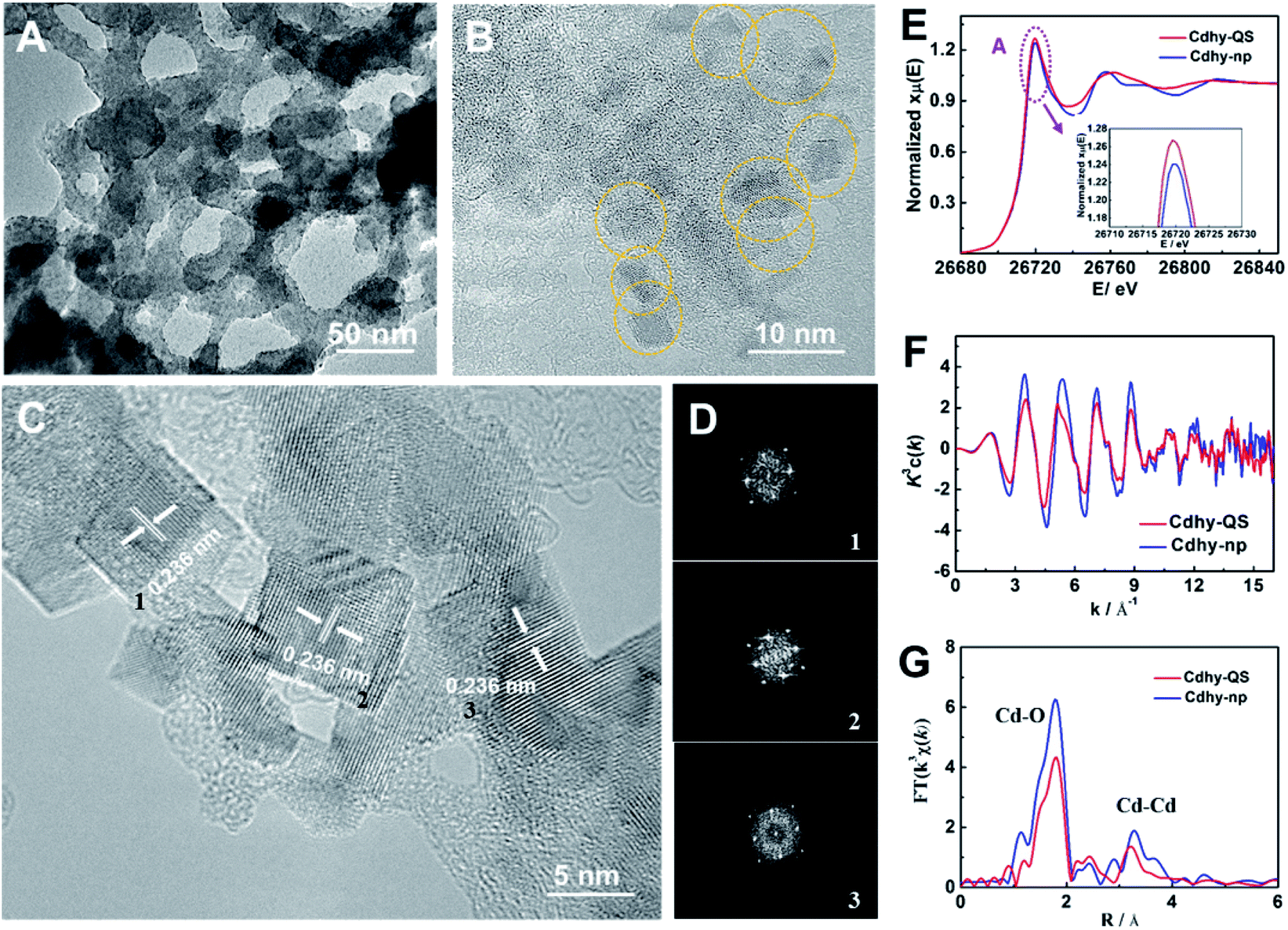 | ||
| Fig. 1 (A and B) TEM images of Cdhy-QS. (C) HR-TEM image of Cdhy-QS. (D) The FFT pattern of the corresponding HR-TEM. (E) XANES spectra at the Cd K-edge. (F) Cd K-edge extended XAFS oscillation function k3c(k). (G) The corresponding Fourier transforms FT(k3χ(k)). | ||
The local coordination environment of Cdhy-QS and Cdhy-np were characterized by synchrotron radiation X-ray absorption fine structure spectroscopy (XAFS) measurements. From the normalized Cd K-edge X-ray absorption near edge structure (XANES), we can observe that the post-edge curve for Cdhy-QS exhibited obvious differences in comparison with Cdhy-np (Fig. 1E). The white line of Cd K-edge (peak A) of Cdhy-QS is higher than that of Cdhy-np, which is corresponding to a 1s–4p transition and suggests that the electronic properties of Cd atom in Cdhy-QS are modified via changing the coordination of Cd–O. However, the threshold value of Cd K-edge for Cdhy-QS and Cdhy-np have the same spectral shape, indicating that Cdhy-QS and Cdhy-np have similar valence state. The oscillation k3χ(k) functions (Fig. 1F) showed that the k3χ(k) of Cdhy-QS was obvious different from that of Cdhy-np in all range of k region. The low k region (k = 3–8 Å−1) and high k region (k = 9–13 Å−1) can be ascribed to the Cd–O and Cd–Cd coordination, respectively. It can be seen that the amplitude of k3χ(k) for Cdhy-QS was lower than that of Cdhy-np, especially in the low k region. It suggests that the decrease of Cd–O coordination number was more significant than that of Cd–Cd. Such changes in the coordination number of Cd–O and Cd–Cd were further showed in the Fourier-transform (FT) of EXAFS in Fig. 1G. The FT curves of Cdhy showed an obvious Cd–O peak located at 1.8 Å−1, as well as Cd–Cd peak located in the range of 2.6–3.8 Å−1. For Cdhy-QS, the intensity of Cd–O coordination decreased more significantly than that of Cd–Cd coordination, indicating Cdhy-QS had less O coordination. Moreover, we used the ARTEMIS programs of IFEFFIT to fit the Cd–O and Cd–Cd coordination number for the as-prepared materials. The results are provided in Fig. S6 and Table S1.† For Cdhy-QS and Cdhy-np, the Cd–O coordination number (NCd–O) were 4.0 and 5.7, and Cd–Cd coordination number (NCd–Cd) were 1.8 and 2.2, respectively. The ratios of NCd–O/NCd–Cd for Cdhy-QS and Cdhy-np were 2.22 and 2.59, respectively, indicating that Cdhy-QS exhibited more uncoordinated Cd–O bond than Cdhy-np. Therefore, the fine structure of Cdhy-QS was distinguished from that of Cdhy-np.
To characterize the electrocatalytic activity for CO2 reduction, the as-prepared catalysts were dispersed in acetone with carbon black and poly(tetrafluoroethylene) to form a homogeneous ink. They were spread and pressed on a nickel foam substrate under 4 MPa, which served as the working electrode (Fig. S7†). Imidazolium-based ionic liquid (IL)/MeCN solution was used as the electrolyte due to the significant advantages of IL, including enhancing CO2 solubility, reducing the overpotential and suppressing the HER.43,44 The linear sweep voltammetry (LSV) was first carried out in CO2-saturated 0.5 M [Bmim]PF6/MeCN solution using a standard three-electrode configuration. Cdhy-QS showed higher current density than that of Cdhy-np (Fig. 2A). It was also significantly higher than that over Ag nanoparticles (Ag-np) and Pd nanoparticles (Pd-np), which were usually considered as efficient catalysts for CO2 electroreduction to CO. Meanwhile, much lower current density was observed in N2-saturated environment (Fig. S8†), indicating the occurrence of CO2 reduction under the applied potentials.
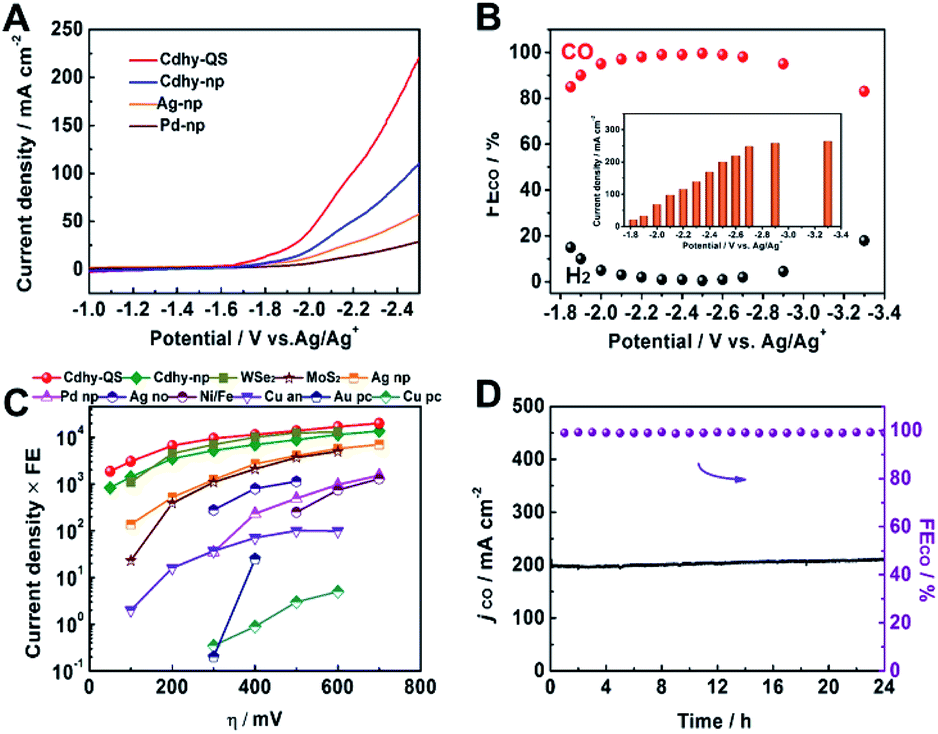 | ||
| Fig. 2 (A) LSV of different catalysts in CO2-saturated 0.5 M [Bmim]PF6/MeCN electrolyte. (B) The FE for CO and current density over Cdhy-QS in 0.5 M [Bmim]PF6/MeCN electrolyte at different potentials. (C) The dependence of current density time FE on overpotential (η) for different catalysts. The data of Cdhy-QS, Cdhy-np, Ag-np and Pd-np were obtained from chronoamperometry experiments under identical conditions. The detail data for other catalysts are listed in Table S2.† (D) Long-term stability of Cdhy-QS at −2.5 vs. Ag/Ag+ for 24 h. | ||
Controlled potential electrolysis of CO2 was performed in a typical H-type cell separated by a Nafion-117 membrane at various potentials. The gaseous and liquid products were then analyzed by gas chromatography (GC) and nuclear magnetic resonance (NMR) spectroscopy, respectively. The dominant product was CO when the [Bmim]PF6/MeCN solution was used as the electrolyte. However, only a small amount of CO was detected when the electrolysis was performed in KHCO3 or KOH aqueous solution. These results indicated that the [Bmim]PF6/MeCN solution exhibited unique advantage for CO2 electroreduction. To confirm the carbon source, the blank experiment was performed by using N2 to replace CO2 during the electrolysis, and no CO was detected. The isotope-labeled experiment was also performed by using 13CO2 to replace CO2, and only 13CO signal can be found from GC-mass spectrometry (GC-MS) spectra (Fig. S9†). These data confirmed that CO2 was the only carbon source.
It can be clearly observed that Cdhy-QS showed outstanding activity for CO2 electroreduction. The FECO was nearly 100% from −2.3 to −2.6 V vs. Ag/Ag+ (Fig. 2B). Even at a very low overpotential of 50 mV (−1.85 V vs. Ag/Ag+, Fig. S10†), the FECO can still reach 85.0%. For comparison, we also investigated the CO2 activation for Cdhy-np, Ag-np and Pd-np under the same reaction conditions (Fig. S11–S13†). The FE of Cdhy-np for CO was only 76.1%, while Ag-np and Pd-np could not reduce CO2 at such low potential. In the meantime, we also calculated the EE for the conversion of CO2 into CO. It was found that the EECO exceeded 70% at a wide potential range of −1.85 to −2.7 V vs. Ag/Ag+ for Cdhy-QS (Fig. S14†). Such high FECO and EECO in a wide range potential is unusual.
The Cdhy-QS exhibited a very high current density for CO production at all applied potentials (Fig. S15†). The current density for CO production was 18.7 mA cm−2 (normalized by geometrical surface area) at an overpotential of 50 mV, which was roughly 2.6 times higher than that of Cdhy-np. More importantly, it could reach a high value of 201.2 mA cm−2 with FE of 99.5% at −2.5 V vs. Ag/Ag+, which is outstanding in both current density and FE comparing with the data reported in the literature (Table S2†).
The catalytic activity of Cdhy-QS was compared with other reported catalysts (Fig. 2C) by multiplying current density (activity) by CO formation FE (selectivity). At an overpotential of 100 mV, the performance of Cdhy-QS exceeded that of Cdhy-np and Ag-np by a factor of about 2.1 and 22, respectively. It exceeded that of WSe2 (ref. 43) and MoS2 (ref. 45) tested under similar conditions by a factor of nearly 2.8 and 133, respectively. Additionally, the performance of Cdhy-QS was also much better than that of other commonly used metals, such as Au46 and Cu,47 by three orders of magnitude. These results indicated that Cdhy-QS had outstanding performance for CO2 reduction.
Furthermore, continuous CO2 reduction was performed at −2.5 V vs. Ag/Ag+ for 24 h to elucidate the electrode stability. As shown in Fig. 2D, there was no decay in both current density and FE for CO. We also characterized Cdhy-QS after 24 h electrolysis via TEM, X-ray photoelectron spectroscopy (XPS) and LSV (Fig. S16†). No obvious changes in morphology, structure and electrochemical characteristics were observed, further showing the excellent stability of Cdhy-QS in the reaction.
The intrinsic reason for the excellent catalytic performance of Cdhy-QS was further investigated. According to cyclic voltammograms (CV) curves under different scan rates, the electrochemical active surface areas (ECSAs) of Cdhy-QS and Cdhy-np were determined by measuring double layer capacitance (Cdl; Table S3†). The ECSAs of Cdhy-QS was slightly higher than that of Cdhy-np (Fig. 3A), suggesting that Cdhy-QS had larger amount of active sites. The total current density and partial current density for CO production were further normalized by ECSAs (Fig. S17†). Similar to the geometric current density, both of them over Cdhy-QS were significantly higher than those of Cdhy-np. Therefore, the higher activity of Cdhy-QS was mainly attributed to its special structure and morphology, rather than increasing number of active sites. Moreover, Cdhy-QS had higher CO2 adsorption capacity, verified by the CO2 adsorption isotherms in Fig. 3B. It could reach 19.6 cm3 g−1 at 1 atm, which was roughly 2.2 times higher than that of Cdhy-np. The measured Nyquist plots at an open circuit potential showed that the charge transfer resistance (Rct) between Cdhy-QS and electrolyte was smaller than that between Cdhy-np and electrolyte (Fig. 3C). This indicated more facile electron transfer from electrolyte to Cdhy-QS electrode surface, resulting in a higher overall electronic conductivity. Additionally, we also examined SO42− adsorption as a surrogate for CO2˙− by measuring single oxidative LSV scans between 1.2 V and 2.0 V (vs. RHE) at 10 mV s−1 in N2-saturated 0.1 M Na2SO4 electrolyte.48,49 The overpotential for surface SO42− adsorption on Cdhy-QS was lower than that of Cdhy-np (Fig. 3D), indicating Cdhy-QS could stabilize CO2˙− intermediates more efficiently.
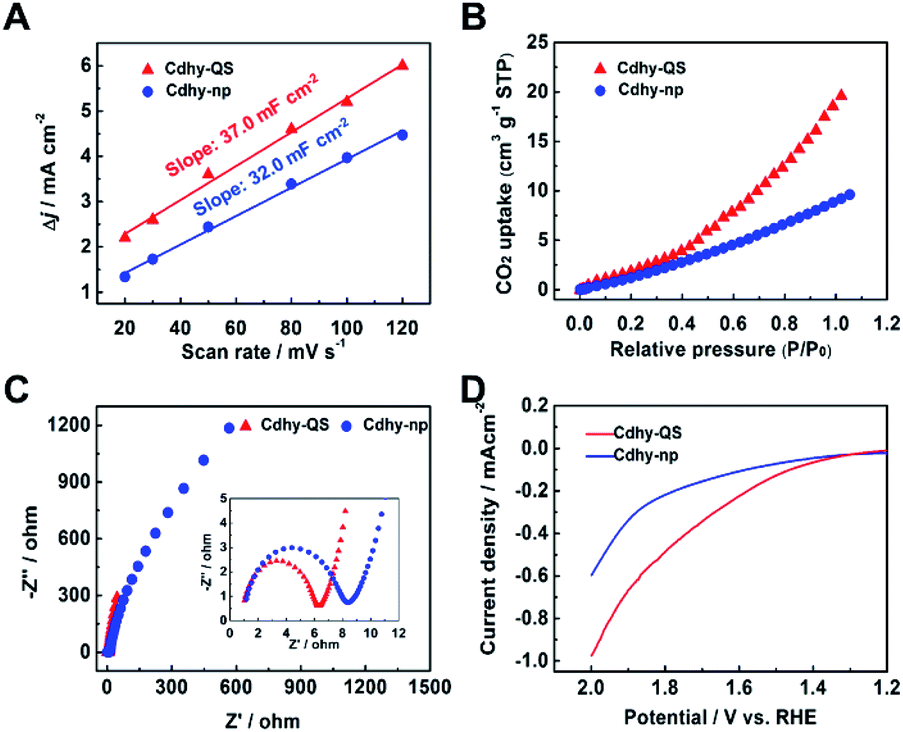 | ||
| Fig. 3 (A) Charging current density different Δj plotted against scan rates. (B) The CO2 adsorption behaviors of Cdhy-QS and Cdhy-np. (C) Nyquist plots for Cdhy-QS and Cdhy-np in CO2-saturated 0.5 M [Bmim]PF6/MeCN electrolyte. (D) Single oxidative LSV curves for SO42− adsorption in N2-saturated 0.1 M Na2SO4. | ||
Furthermore, the valence band spectra were determined to further clarify the charge-transfer process for CO2 adsorption and activation over Cdhy-QS in electrolysis. As shown in Fig. S18,† the significant change in the valence band can be observed after CO2 reduction over Cdhy-QS, which mainly results from the formation and adsorption of CO2˙− species.5 However, there is no obvious change in the valence band over Cdhy-np. These indicate that Cdhy-QS possessed higher ability for CO2 activation and CO2˙− intermediates stabilization. The desorption ability of CO is also an important factor affecting the activity of CO2 reduction to CO, and it could be probed by the electrochemical CO stripping voltammetry method.50 As shown in Fig. 4A, a sharp CO stripping profile with a dominant peak around 0.83 V vs. RHE occurred on Cdhy-QS, whereas there is a broad peak around 0.86 V vs. RHE on Cdhy-np. The negative shift indicated that Cdhy-QS had stronger CO desorption ability, leading to higher activity for CO2 reduction to CO.
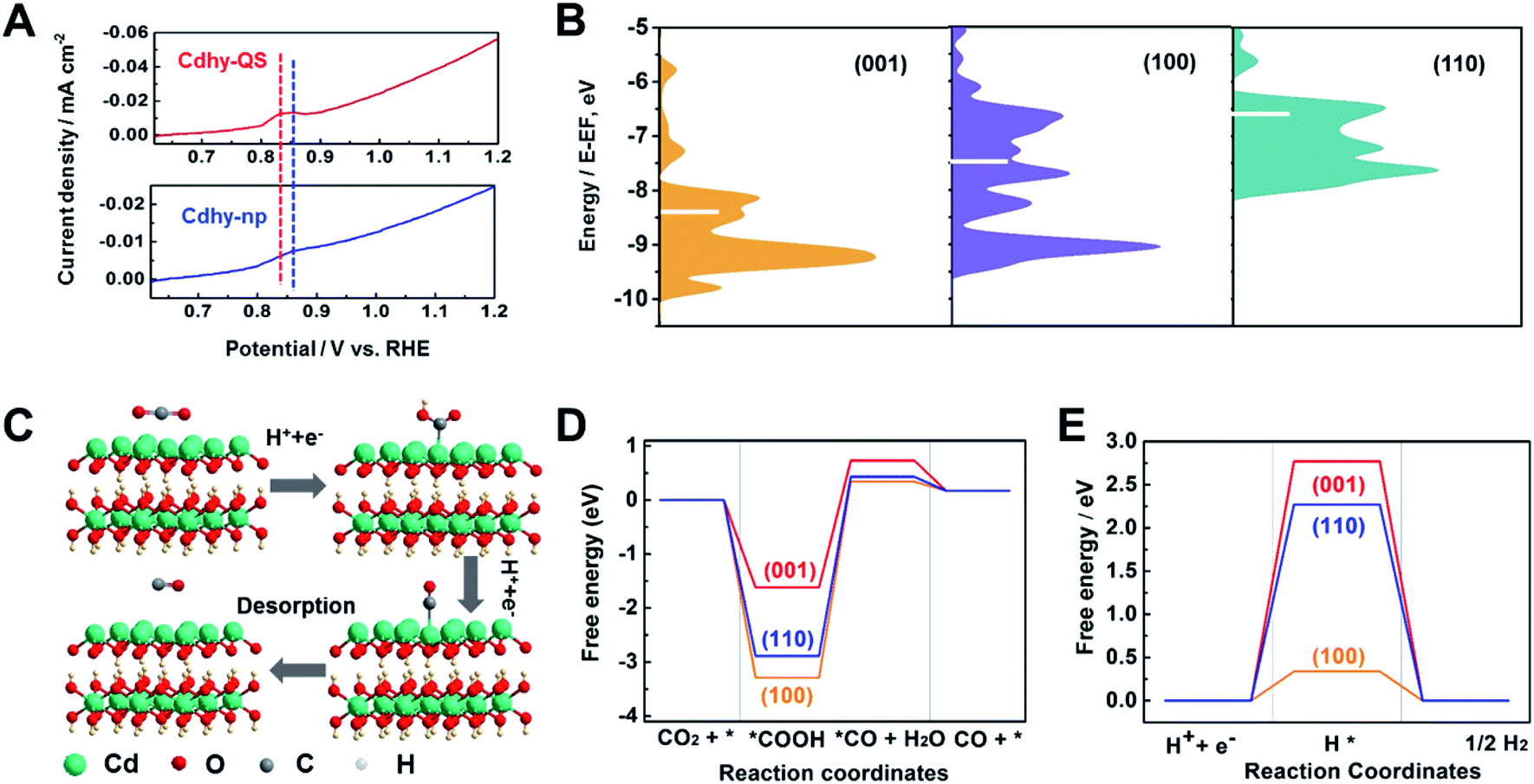 | ||
| Fig. 4 (A) The LSV curves for CO desorption from Cdhy-QS and Cdhy-np in 0.1 M Na2SO4. (B) The d-band center of gravity for different facets of Cdhy. (C) Proposed reaction paths for CO2 reduction to CO over Cdhy-QS. (D) Gibbs free energy diagrams for CO2 reduction to CO over different facets of Cdhy. (E) Gibbs free energy diagrams for HER over different facets of Cdhy. | ||
DFT calculations were then performed to gain insight into the outstanding performance of Cdhy-QS. Based on the HR-TEM images, the Cdhy-QS exposed (002) facet. In DFT calculations, the (002) facet can be represented by the basic (001) facet, because they are parallel to each other and exhibited the same atomic configuration. Thus the (001) facet can stand for Cdhy-QS, and the (110) and (100) facets represent Cdhy-np. We first optimized the three basic facets of Cdhy (Fig. S19†). For transition metal catalysts, the way that their d-band interacts with the intermediates usually determines the binding strength and the products.13 As shown in Fig. 4B, we can observe that the d-band center of (001) facet is more negative than that of other facets. It indicates that the binding strength for *COOH and *CO is weak on (001) facet, which contributes to CO production.51 Therefore, (001) facet can be considered as high activity facet of Cdhy.
In addition, the Gibbs free energy diagrams of CO2 → CO pathway were also studied (Fig. 4C, D, S20–S22 and Table S4†), *COOH formation on Cdhy surfaces is exergonic because of strong binding to the corresponding sites. The energetic ordering suggests that *COOH is less stable on (001) facet than on (110) and (100) facet, resulting in more favorable for further conversion of *COOH. The formation of *CO + H2O from *COOH is highly endergonic and acts as the rate-determining step (RDS). According to the simulation, the free energy for the RDS on (001), (110) and (100) facet are 2.30 eV, 3.32 eV and 3.64 eV, respectively. It is also kinetically favorable on (001) facet compared to (110) and (100) facet, demonstrating that the cycle of CO2 to CO is easier on (001) facet than that on (110) and (100) facet. Furthermore, the Gibbs free energy of HER was also calculated (Fig. 4E and S23†), which is a dominant competitive reaction upon CO2 reduction. It can be seen that the HER on Cdhy is endergonic. The (001) facet exhibits the highest energy barrier (2.77 eV), suggesting that it can significantly suppress the adsorption of *H. Therefore, the (001) facet can reduce the Gibbs free energy of RDS for CO2-to-CO conversion and hinder the HER simultaneously. As a result, Cdhy-QS had better performance for CO production compared with Cdhy-np.
In order to use the low concentration of CO2 and obtain high concentration of CO simultaneously, we designed an adsorption-electrolysis device. The adsorption of CO2 and the CO2 electroreduction were divided into two parts. As shown in Fig. 5A, the low concentration CO2 was bubbled into the adsorption cell first, and CO2 can be dissolved in 0.5 M [Bmim]PF6/MeCN solution, due to its high adsorption capacity of CO2. Different ratios of CO2/N2 gas were mixed to simulate the exhaust gas. Then the solution was pumped into the electrolytic cell. CO2 can be reduced to CO efficiently and high concentration CO products were obtained. After the reaction, the electrolyte can be transferred back to the adsorption cell, achieving the reuse of the electrolyte. We can see that the electroreduction of low concentration CO2 was still high-efficiency over Cdhy-QS (Fig. 5B). The FE of CO can reach 84.6%, 89.5% and 94.4%, when the concentration of CO2 was 4.9%, 10.1% and 14.8%, respectively. In addition, the concentrations of CO in the gas products were 37.1%, 49.3% and 57.0%, respectively (Fig. 5C and Table S5†). It indicated the catalyst Cdhy-QS is still high activity and the high concentration CO can be obtained when using low concentration CO2. Thus we can believe that combining the active catalysts and the adsorption-electrolysis device system will greatly promote the commercialization process of CO2RR.
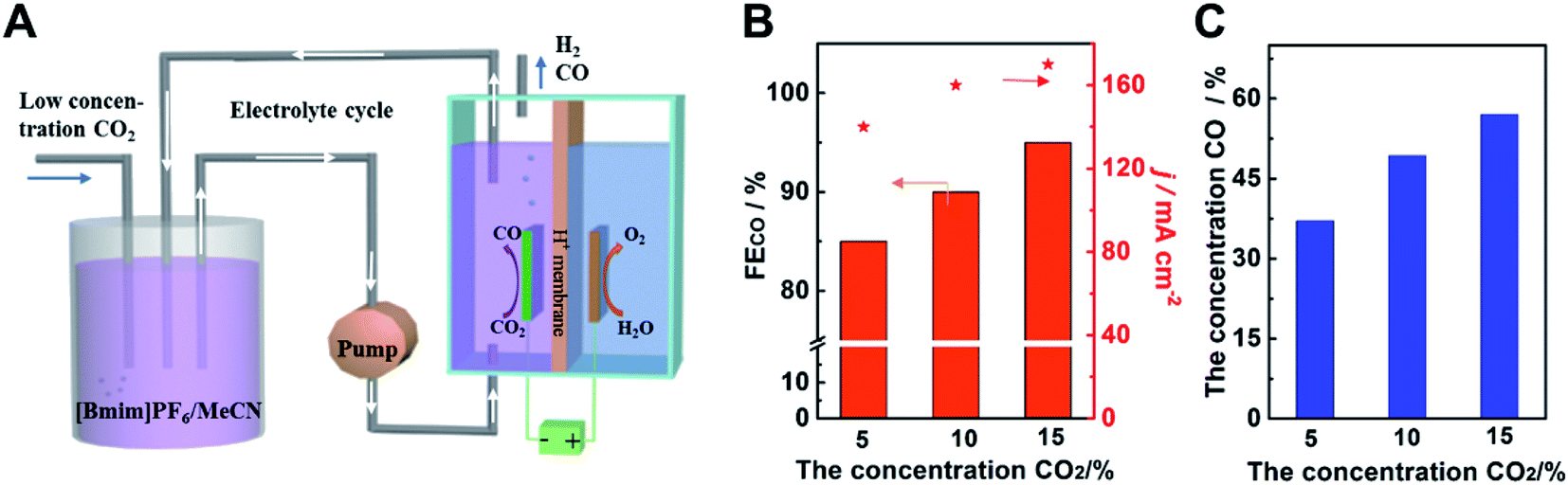 | ||
| Fig. 5 (A) The schematic diagram for the CO2 adsorption-electrolysis device system. (B) The FECO and current density for Cdhy-nc using low concentration CO2. (C) The concentration of CO in the gas products using low concentration CO2. | ||
Conclusions
In summary, we have demonstrated that Cdhy-QS has highly reactive facet on the surface with low Cd–O coordination number, resulting in very high efficiency for CO2 electroreduction to CO. The Cdhy-QS catalyst yields nearly 100% CO selectivity in a wide potential range and the current density can be higher than 200 mA cm−2 with high energy efficiency. The detailed study indicated that the outstanding catalytic performance of Cdhy-QS can be attributed to the enhanced CO2 adsorption and activation, as well as the low charge transfer resistance. We believe that the highly efficient Cdhy-QS catalyst has great potential for practical application. In addition, combining the Cdhy-QS and the adsorption-electrolysis device system can obtain high concentration of CO at low CO2 concentration. The findings of this work expand knowledge for the design of novel and efficient electrocatalysts, and also provide a guideline for promoting commercialization of CO2RR.Data availability
The data that support the findings of this study are available within the article and its ESI.†Author contributions
C. J. C., X. F.·S., L. F. L., S. J. L. and B. X. H. proposed the project, designed the experiments, and wrote the manuscript; C. J. C. performed the whole experiments; X. P.·Y., Y.·H.·W., R. Z. W., Q. G. Z., M. Q. H., Z. F. Z., H. L. F., J. M., Y.·Y.·H. and J. Y. M. performed the analysis of experimental data; X. F. S. and B. X. H. supervised the whole project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Key Research and Development Program of China (2017YFA0403003, 2017YFA0403101, 2017YFA0403102), National Natural Science Foundation of China (22002172, 21890761, 21733011), Beijing Municipal Science & Technology Commission (Z191100007219009), and Chinese Academy of Sciences (QYZDY-SSW-SLH013). The X-ray absorption experiments were conducted at Shanghai Synchrotron Radiation Facility (SSRF), China.Notes and references
- M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2013, 52, 9620–9633 CrossRef CAS PubMed.
- J. Wang, H. Li, S. Liu, Y. Hu, J. Zhang, M. Xia, Y. Hou, J. Tse, J. Zhang and Y. Zhao, Angew. Chem., Int. Ed., 2021, 60, 181–185 CrossRef CAS PubMed.
- J. Jiao, R. Lin, S. Liu, W. C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu, S. F. Hung, H. M. Chen, L. Zheng, Q. Lu, X. Yang, B. Xu, H. Xiao, J. Li, D. Wang, Q. Peng, C. Chen and Y. Li, Nat. Chem., 2019, 11, 222–228 CrossRef CAS PubMed.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H. Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- X. Sun, C. Chen, S. Liu, S. Hong, Q. Zhu, Q. Qian, B. Han, J. Zhang and L. Zheng, Angew. Chem., Int. Ed., 2019, 58, 4669–4673 CrossRef CAS PubMed.
- Y. Zhao, X. Tan, W. Yang, C. Jia, X. Chen, W. Ren, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2020, 59, 21493–21498 CrossRef CAS PubMed.
- Y. Fang and J. C. Flake, J. Am. Chem. Soc., 2017, 139, 3399–3405 CrossRef CAS PubMed.
- J. Wang, H. Yang, Q. Liu, Q. Liu, X. Li, X. Lv, T. Cheng and H. B. Wu, ACS Energy Lett., 2021, 437–444 CrossRef CAS.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- L. Han, S. Song, M. Liu, S. Yao, Z. Liang, H. Cheng, Z. Ren, W. Liu, R. Lin, G. Qi, X. Liu, Q. Wu, J. Luo and H. L. Xin, J. Am. Chem. Soc., 2020, 142, 12563–12567 CrossRef CAS PubMed.
- S. Rasul, D. H. Anjum, A. Jedidi, Y. Minenkov, L. Cavallo and K. Takanabe, Angew. Chem., Int. Ed., 2015, 54, 2146–2150 CrossRef CAS PubMed.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS PubMed.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. Kenis, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS PubMed.
- F. Li, L. Chen, G. P. Knowles, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 505–509 CrossRef CAS PubMed.
- Z. Geng, X. Kong, W. Chen, H. Su, Y. Liu, F. Cai, G. Wang and J. Zeng, Angew. Chem., Int. Ed., 2018, 57, 6054–6059 CrossRef CAS PubMed.
- D. Gao, I. T. McCrum, S. Deo, Y.-W. Choi, F. Scholten, W. Wan, J. G. Chen, M. J. Janik and B. Roldan Cuenya, ACS Catal., 2018, 8, 10012–10020 CrossRef CAS.
- R. Shi, J. Guo, X. Zhang, G. I. N. Waterhouse, Z. Han, Y. Zhao, L. Shang, C. Zhou, L. Jiang and T. Zhang, Nat. Commun., 2020, 11, 3028 CrossRef CAS PubMed.
- H. Li, Z. Cheng, Q. Zhang, A. Natan, Y. Yang, D. Cao and H. Zhu, Nano Lett., 2018, 18, 7407–7413 CrossRef CAS PubMed.
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie, F. Yang, Y. Li, G. Wang and X. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- C. Chen, X. Sun, D. Yang, L. Lu, H. Wu, L. Zheng, P. An, J. Zhang and B. Han, Chem. Sci., 2019, 10, 1659–1663 RSC.
- C. He, Y. Zhang, Y. Zhang, L. Zhao, L. P. Yuan, J. Zhang, J. Ma and J. S. Hu, Angew. Chem., Int. Ed., 2020, 59, 4914–4919 CrossRef CAS PubMed.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- C. Chen, X. Sun, X. Yan, Y. Wu, H. Liu, Q. Zhu, B. B. A. Bediako and B. Han, Angew. Chem., Int. Ed., 2020, 59, 11123–11129 CrossRef CAS PubMed.
- M. A. Ghausi, J. Xie, Q. Li, X. Wang, R. Yang, M. Wu, Y. Wang and L. Dai, Angew. Chem., Int. Ed., 2018, 57, 13135–13139 CrossRef CAS PubMed.
- J. Wu, S. Ma, J. Sun, J. I. Gold, C. Tiwary, B. Kim, L. Zhu, N. Chopra, I. Odeh, R. Vajtai, A. Z. Yu, R. Luo, J. Lou, G. Ding, P. J. A. Kenis and Pu. M. Ajayan, Nat. Commun., 2016, 7, 13869 CrossRef CAS PubMed.
- H. Jiang, J. Gu, X. Zheng, M. Liu, X. Qiu, L. Wang, W. Li, Z. Chen, X. Ji and J. Li, Energy Environ. Sci., 2019, 12, 322–333 RSC.
- Z. L. Yu, B. Qin, Z. Y. Ma, J. Huang, S. C. Li, H. Y. Zhao, H. Li, Y. B. Zhu, H. A. Wu and S. H. Yu, Adv. Mater., 2019, 31, e1900651 CrossRef PubMed.
- Z. Y. Wu, H. W. Liang, B. C. Hu and S. H. Yu, Angew. Chem., Int. Ed., 2018, 57, 15646–15662 CrossRef CAS PubMed.
- J. Shi, D. Shao, J. Zhang, D. Tan, X. Tan, B. Zhang, B. Han, F. Zhang, L. Liu and X. Cheng, Chem. Commun., 2018, 54, 5450–5453 RSC.
- P. Yang, Z. J. Zhao, X. Chang, R. Mu, S. Zha, G. Zhang and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 7724–7728 CrossRef CAS PubMed.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J.-L. Luo, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed.
- S. C. Li, B. C. Hu, Y. W. Ding, H. W. Liang, C. Li, Z. Y. Yu, Z. Y. Wu, W. S. Chen and S. H. Yu, Angew. Chem., Int. Ed., 2018, 57, 7085–7090 CrossRef CAS PubMed.
- C. Chen, J. F. Khosrowabadi Kotyk and S. W. Sheehan, Chem, 2018, 4, 2571–2586 CAS.
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2021, 4, 20–27 CrossRef CAS.
- Y. C. Tan, K. B. Lee, H. Song and J. Oh, Joule, 2020, 4, 1104–1120 CrossRef CAS.
- J. E. Huang, F. Li, A. Ozden, A. S. Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C.-T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- B. Han, X. Ou, Z. Deng, Y. Song, C. Tian, H. Deng, Y. J. Xu and Z. Lin, Angew. Chem., Int. Ed., 2018, 57, 16811–16815 CrossRef CAS PubMed.
- G. Lee, Y. C. Li, J.-Y. Kim, T. Peng, D.-H. Nam, A. Sedighian Rasouli, F. Li, M. Luo, A. H. Ip, Y.-C. Joo and E. H. Sargent, Nat. Energy, 2021, 6, 46–53 CrossRef CAS.
- Q. Zhu, D. Yang, H. Liu, X. Sun, C. Chen, J. Bi, J. Liu, H. Wu and B. Han, Angew. Chem., Int. Ed., 2020, 59, 8896–8901 CrossRef CAS PubMed.
- Q. Zhu, J. Ma, X. Kang, X. Sun, H. Liu, J. Hu, Z. Liu and B. Han, Angew. Chem., Int. Ed., 2016, 55, 9012–9016 CrossRef CAS PubMed.
- J. Hu, H. Liu and B. Han, Sci. China: Chem., 2018, 61, 1486–1493 CrossRef CAS.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS PubMed.
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato, R. Haasch, P. Zapol, B. Kumar, R. F. Klie, J. Abiade, L. A. Curtiss and A. Salehi-Khojin, Science, 2016, 353, 467–470 CrossRef CAS PubMed.
- P. Abbasi, M. Asadi, C. Liu, S. Sharifi-Asl, B. Sayahpour, A. Behranginia, P. Zapol, R. Shahbazian-Yassar, L. A. Curtiss and A. Salehi-Khojin, ACS Nano, 2017, 11, 453–460 CrossRef CAS PubMed.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS PubMed.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed.
- L. Lu, X. Sun, J. Ma, D. Yang, H. Wu, B. Zhang, J. Zhang and B. Han, Angew. Chem., Int. Ed., 2018, 57, 14149–14153 CrossRef CAS PubMed.
- A. Salehi-Khojin, H.-R. M. Jhong, B. A. Rosen, W. Zhu, S. Ma, P. J. A. Kenis and R. I. Masel, J. Phys. Chem. C, 2013, 117, 1627–1632 CrossRef CAS.
- L. Xiao, L. Zhuang, Y. Liu, J. Lu and H. D. Abruna, J. Am. Chem. Soc., 2009, 131, 602–608 CrossRef CAS PubMed.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 5948 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02328d |
| This journal is © The Royal Society of Chemistry 2021 |