
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereodivergent entry to β-branched β-trifluoromethyl α-amino acid derivatives by sequential catalytic asymmetric reactions†
Vasco
Corti‡
 ,
Riccardo
Riccioli
,
Ada
Martinelli
,
Sofia
Sandri
,
Mariafrancesca
Fochi
* and
Luca
Bernardi
*
,
Riccardo
Riccioli
,
Ada
Martinelli
,
Sofia
Sandri
,
Mariafrancesca
Fochi
* and
Luca
Bernardi
*
Department of Industrial Chemistry “Toso Montanari” and INSTM RU Bologna, Alma Mater Studiorum – University of Bologna, V. Risorgimento 4, 40136 Bologna, Italy. E-mail: mariafrancesca.fochi@unibo.it; luca.bernardi2@unibo.it
First published on 29th June 2021
Abstract
Currently, conventional reductive catalytic methodologies do not guarantee general access to enantioenriched β-branched β-trifluoromethyl α-amino acid derivatives. Herein, a one-pot approach to these important α-amino acids, grounded on the reduction – ring opening of Erlenmeyer–Plöchl azlactones, is presented. The configurations of the two chirality centers of the products are established during each of the two catalytic steps, enabling a stereodivergent process.
Introduction
β-Branched α-amino acids (AAs) carrying different β-substituents – thus bearing two vicinal chirality centres, at the α- and β-carbons – are important yet challenging synthetic targets.1 The stereocontrolled preparation of these compounds has been tackled and realised with different (catalytic) methods.2–7 A non-comprehensive list of examples includes enantioselective conjugate additions and alkylations of glycinate imines,2 palladium catalysed β-C(sp3)–H alkylation and arylation of AAs,3 aziridine ring-opening,4 an engineered tryptophan synthase,5 multi-enzymatic β-methylation of AAs,6 and catalytic asymmetric hydrogenation of α,β-dehydro-amino acids (DHAAs), in its implementation with tetra-substituted substrates.7 This latter approach (Scheme 1(a)) has disclosed diastereo- and enantioselective entries to various classes of β-branched AAs, including7d less common yet relevant β-trifluoromethyl AA derivatives.8,9 Given the common stereospecificity of the hydrogenation, the E/Z geometry of the substrate dictates the relative configuration of the product (Scheme 1(a)). Thus, a diastereoisomer is obtainable only if the parent olefin isomer can be prepared. This constraint can have negative implications. For example, a straightforward synthesis of a preclinical drug candidate via asymmetric hydrogenation of the corresponding β-aryl-β-trifluoromethyl DHAA was envisioned (Scheme 1(b)).9a However, the required E-olefin isomer could not be accessed with sufficient selectivity. Furthermore, enantioselective hydrogenation of tetrasubstituted olefins can occasionally be challenging. In fact, the β-trifluoromethyl Z-DHAAs required for the target was found to be reluctant to asymmetric hydrogenation,9a preventing application of Turner's formal stereodivergent reduction of DHAAs.10 This ingenious chemo-enzymatic approach, which unlocks access to the isomer not obtainable by hydrogenation via downstream inversion of the α-amino chirality centre in the reduced DHAA, is in fact established only for β-aryl-β-methyl AAs. Eventually, to obtain the target syn-trifluoromethylated amino alcohol,§ Alimardanov et al. resorted to a longer, yet effective and scalable, route, encompassing a chiral auxiliary and introduction of the amino functionality at a late stage.9a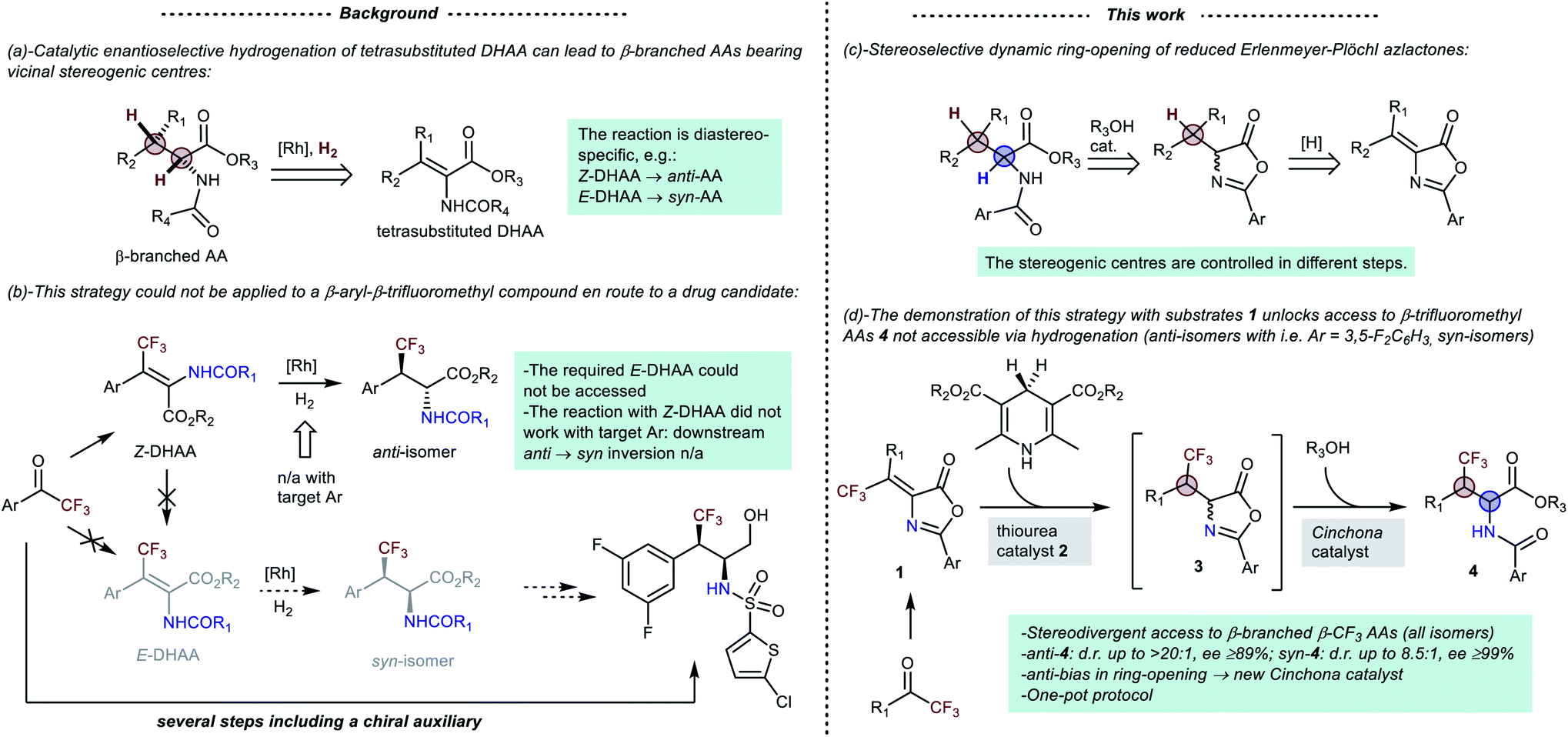 | ||
| Scheme 1 (Left) (a) catalytic asymmetric hydrogenation approach to β-branched AAs, and (b) its failure with a β-trifluoromethyl drug candidate. (Right) (c) the underlying concept of this work and (d) its demonstration with β-trifluoromethyl substrates 1. | ||
With this background in mind, we envisioned an original stereodivergent11 entry to β-branched AAs grounded on the dynamic stereoselective ring-opening of enantioselectively reduced Erlenmeyer–Plöchl azlactones (Scheme 1(c)). In contrast with catalytic hydrogenation, this formal hydrogenation of the azlactone olefin (a DHAA derivative) fixes the configurations of the two hydrogenated centres in different steps, lending itself to stereodivergency. Herein, we present the first demonstration of this strategy by its application to the one-pot preparation of β-trifluoromethyl AA derivatives 4 (Scheme 1(d)). In more detail, enantioselective transfer hydrogenation of readily available substrates 1 (ref. 9a) with Hantzsch esters12 sets the absolute configuration of the trifluoromethylated β-centre.13 Subsequent dynamic alcoholytic ring-opening14 of intermediates 3 fixes the absolute configuration of the α-carbon. In the ring-opening reaction, the substrates 3 feature a considerable bias towards the formation of anti-isomers 4. Such bias was readily leveraged with conventional Cinchona catalysts to obtain a range of anti-4 products with very high stereoselectivities (including compounds not suited for hydrogenation). Conversely, the development of the syn-selective process was less straightforward, requiring a peculiar ammonia-derived squaramide catalyst to afford the syn-4 isomers with variable diastereoselectivities.
It is worth stressing that β-branched β-trifluoromethyl AAs, and derivatives thereof, have found widespread interest in medicinal chemistry (Fig. 1). β-Trifluoromethylated analogues of natural AAs have been incorporated into peptides, wherein the trifluoromethyl group can give unique effects on stability, acidicy/basicity, folding behaviour, hydrophobicity, and ultimately biological activity.8a The β-trifluoromethyl AA framework can also be found in less canonical structures. Besides the drug candidate mentioned in Scheme 1(b), which showcases inhibition of β-amyloid production,15 another β-trifluoromethyl AA structure of medicinal interest is an analogue of thalidomide,2i which feature enhanced configurational stability compared to thalidomide. An additional example is represented by the heterocyclic compound derived from a 3-(trifluoromethyl)azetidine carboxylic acid, shown in Fig. 1. This compound is a member of a library of related heterocycles, investigated for their activity as inhibitors of phosphoinositide 3-kinases.16
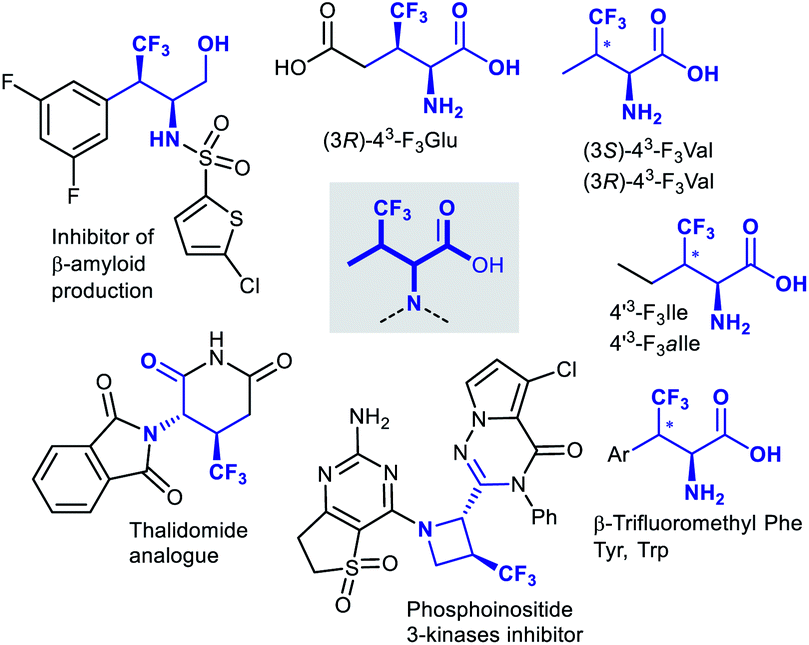 | ||
| Fig. 1 Medicinal chemistry relevant β-branched β-trifluoromethyl AAs. | ||
Results and discussion
At the outset, with transfer hydrogenation to β,β-disubstituted nitroalkenes promoted by Jacobsen-type catalysts as a lead,17 we explored different variables in the reduction of β-phenyl-β-trifluoromethyl Erlenmeyer–Plöchl substrates 1 with HEs.¶ Preliminary screening of different catalysts and HEs identified the conditions outlined in Table 1, entry 1, as promising starting point. In more detail, using 20 mol% of catalyst 2a in combination with the isobutyl HE, in dichloromethane (reagent grade) as solvent at low temperature (−30 °C), the 2-phenyl azlactone 1a could be reduced with full conversion and promising enantioselectivity (70% ee). Products 3 of the transfer hydrogenation reaction are relatively unstable. Thus, for CSP HPLC analysis they are converted to the ultimate products 4 by ring-opening with allyl alcohol using achiral tertiary amine promoters, furnishing the two diastereoisomers 4. These two isomers feature comparable ees, validating this method for the evaluation of the enantioselectivity of the reduction step. Thus, a systematic variation of the modules of this type of Jacobsen catalyst18 (AA portion, double H-bond donor, amide portion, terminal N-group), of C2 substituents of the azlactone, and of reaction conditions, was undertaken.¶ Exploration of catalysts based on double hydrogen bond donors other than thiourea (ureas and squaramides), and another AA portion (L-valine), confirmed the L-tert-leucine derived thiourea as the most efficient catalyst core. Whereas variation of solvent and/or dilution was not fruitful, investigation of different terminal N-groups in the thiourea (2a–f) indicated that the 4-(trifluoromethyl)phenyl residue (2b) could provide a distinct improvement, compared to the common 3,5-bis(trifluoromethyl)phenyl (2a) and other aryl/alkyl groups (2c–f) (entries 1–6). With catalyst 2b, three differently C2 substituted Erlenmeyer–Plöchl azlactones 1b–d were screened (entries 7–9). The 4-methoxyphenyl (PMP) derivative 1d gave better results, and was thus selected for further catalyst development, which focussed on exploring various N-substituents on the amide (entries 10–14). Amongst catalysts 2g–k, the N-benzyl-N-methyl amide derivative 2k performed best, affording the transfer hydrogenation product 3d with a respectable 85% ee, measured on the corresponding ring-opened derivative 4d.| a Conditions: substrate 1 (0.05 mmol), catalyst 2 (0.01 mmol, 20 mol%), HE (0.075 mmol), CH2Cl2 (200 μL), −30 °C, 48 h. All reactions gave >90% conversion of 1 (19F NMR). Filtration on silica, evaporation, then CH2Cl2 (0.5 mL), allyl alcohol (0.1 mmol), Et3N (1 drop), RT, 24–48 h. b Enantiomeric excess of anti-4 and syn-4, respectively, determined by CSP HPLC after chromatographic purification on silica gel. |
|---|
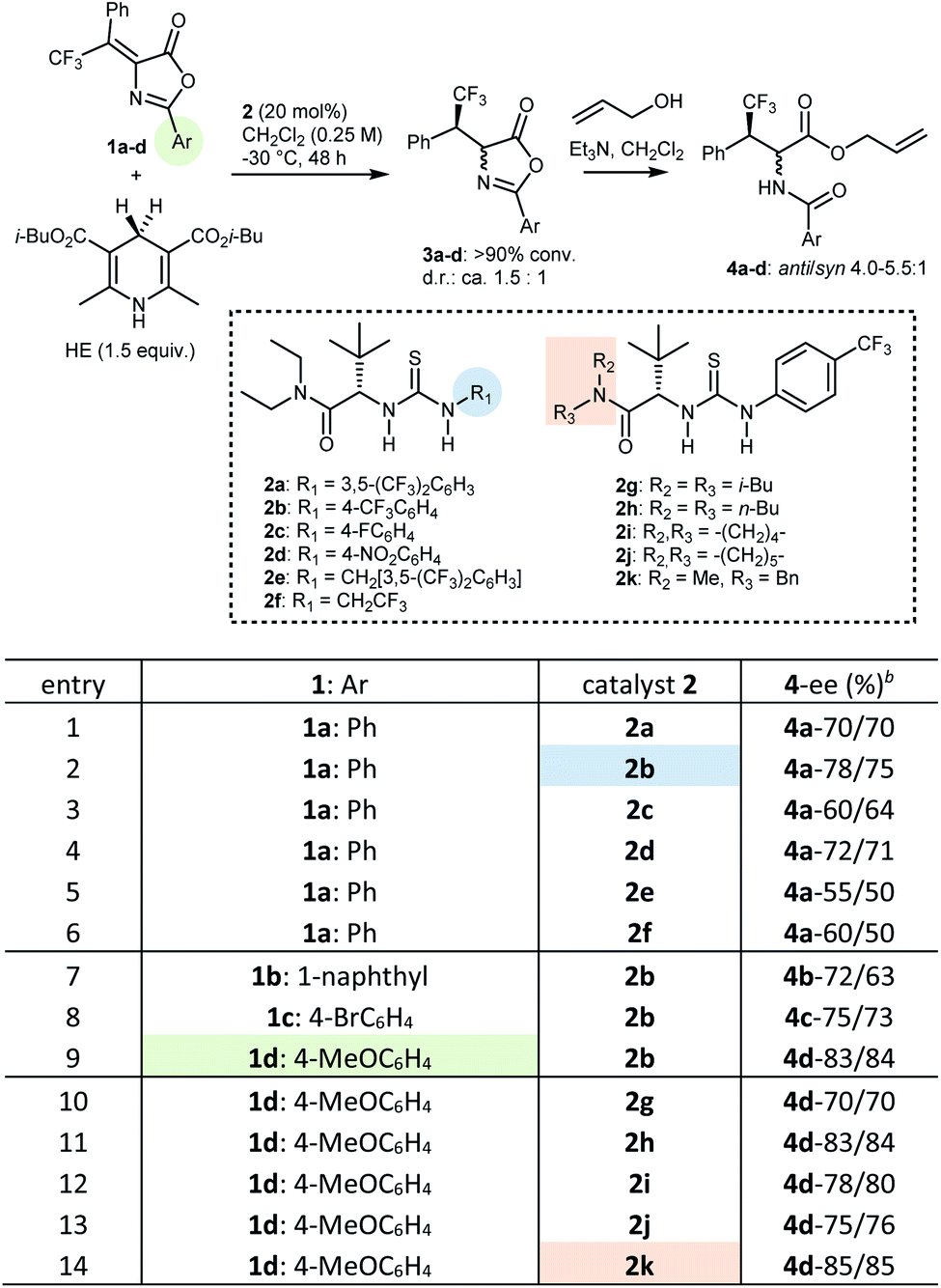
|
A closer inspection at the alcoholytic ring-opening step of compounds 3 promoted by achiral tertiary amines, indicated that the ring-opened products 4 were obtained with higher anti/syn ratios (5.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for 4d) than the parent azlactones 3 (ca. 1.5:1). Thus, the alcoholytic process was dynamic, and biased towards the anti-isomer. We initially surmised that such substrate-bias, hindering access to syn-4,19 would be circumscribed to the tertiary amine promoted alcoholytic ring-opening. Since a variety of DKRs of azlactones by ring-opening reactions, using different nucleophiles and catalytic approaches (Lewis bases, Lewis acids, Brønsted acids, enzymes), are available in the literature,14 we hoped that one of these could be subdued to our aims.
:1 for 4d) than the parent azlactones 3 (ca. 1.5:1). Thus, the alcoholytic process was dynamic, and biased towards the anti-isomer. We initially surmised that such substrate-bias, hindering access to syn-4,19 would be circumscribed to the tertiary amine promoted alcoholytic ring-opening. Since a variety of DKRs of azlactones by ring-opening reactions, using different nucleophiles and catalytic approaches (Lewis bases, Lewis acids, Brønsted acids, enzymes), are available in the literature,14 we hoped that one of these could be subdued to our aims.
However, a preliminary screening of many of these methods suggested the squaramide Cinchona catalyzed alcoholytic ring opening20 as most promising option, despite its resemblance with the biased achiral amine promoted reaction. State-of-the-art Cinchona squaramide dimeric catalysts derived from quinidine (QD-1) and quinine (QN-1) (Scheme 2) were initially employed with enantioenriched azlactone 3d under standard conditions (dichloromethane, allyl alcohol, 0 °C). While, unsurprisingly, “matched” QD-1 increased the anti/syn ratio to a high 10.0:1 value, compared to an achiral tertiary amine (ca. 5.5:1), we were pleased to observe that the corresponding “mismatched” QN-1 could reverse the selectivity of the process, forcing the ring-opening reaction towards a moderate preference (1:2.3) for syn-4d. The products 4d displayed a higher enantiomeric excess than 3d, in accordance with the Horeau effect.21 Adjusting the reaction conditions and testing additional QD and dihydroquinidine (dhQD) derived structures led to a highly anti-selective protocol. Catalyst dhQD-2 improved in fact the diastereomeric ratio of the product 4d up to 14.2:1 in favour of the anti-isomer. However, application of its quasi-enantiomeric derivative dhQN-2 did not result in the expected improvement in the syn-selectivity, providing a result similar to QN-1 (1:2.4 vs. 1:2.3). This result emphasized that the transition states leading to the anti and syn-products are “intrinsically” diastereomeric,19b due to the presence of the chiral (R)-configured β-branched chain of azlactone 3d. On these grounds, different (i.e. non quasi-enantiomeric) catalyst structures may be required for anti- and syn-selective processes. Thus, a range of (dh)QN derived squaramide catalysts and reaction conditions were examined (see also ESI†). The diastereomeric catalyst dhQN-3 (from (S)-α-methylbenzylamine instead of (R)-α-methylbenzylamine of dhQN-2) was tested first, giving however a poor result. Subsequent catalyst screening, performed at RT, suggested that the main factor affecting the stereoselectivity is the bulkiness of the squaramide portion. While catalysts QN-4, 5, 6, wherein the squaramide bears a methylene group, gave slight improvements compared to dhQN-2 (1:2.7–2.8 vs. 1:2.4), the more bulky tert-butyl substituted QN-7 provided a lower d.r. (1:1.9). The similar performances of the prototypical22 3,5-bis(trifluoromethyl)benzyl catalyst QN-4 and the simple benzyl derivative QN-5 point to a negligible influence of the electronics of this group on selectivity. Thus, aiming at reducing bulkiness, catalyst QN-8 derived from methylamine was applied, providing indeed a rewarding improvement (anti/syn = 1:3.1). A further reduction in bulkiness could be achieved only by entirely removing the N-substituent, which was finalized preparing and testing the ammonia derived catalyst QN-9. Pleasingly, this peculiar and unprecedented structure was able to afford syn-4d with a notable 1:4.6 selectivity. The very poor solubility of QN-9 in CH2Cl2 resulted however in sluggish reactivity, with only 50% conversion after 42 h at RT. Such shortcoming was overcome by switching to the more soluble dihydroquinine derivative dhQN-10, which gave 4d with >90% conversion, even by performing the reaction at 0 °C, and in a 1:5.9 diastereomeric ratio favouring the syn-isomer.23 Enantiomeric excess was found to be good (92%), as expected. At this stage, additional experiments indicated the unique requirements of the syn-selective reaction: catalyst dhQD-10, quasi-enantiomeric of dhQN-10, gave an anti-selectivity in the process comparable to an achiral tertiary amine (ca. 5:1).
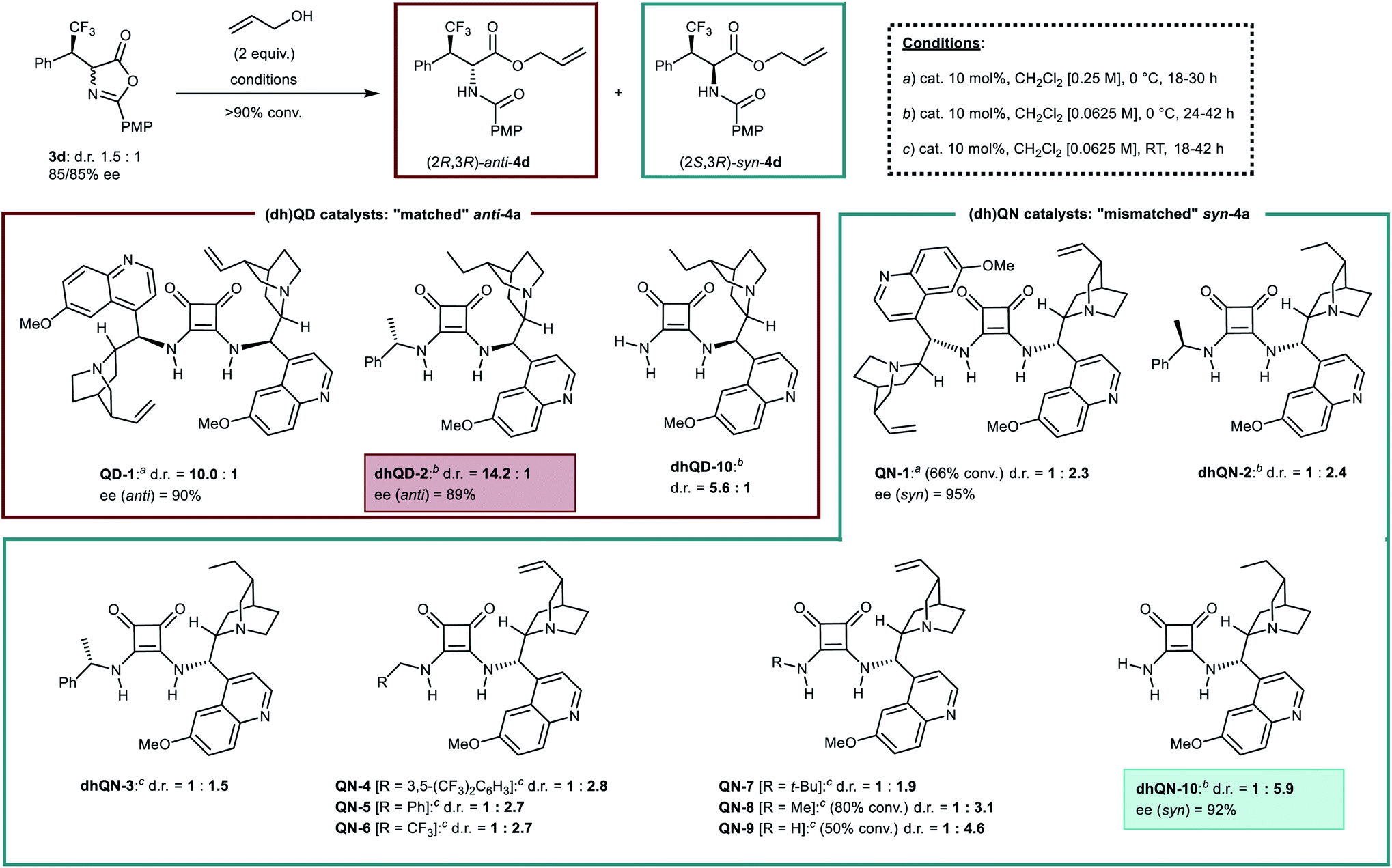 | ||
| Scheme 2 Optimization of the alcoholytic ring-opening step: selected results (d.r. values refer to anti/syn ratios). | ||
Aiming at streamlining the overall process (1d → 4d) by implementing a one pot procedure, thus circumventing the problematic purification of azlactone intermediate 3d, it was found that an excess of HE in the transfer hydrogenation reaction has to be avoided, since this species inhibits the basic squaramide catalyst used in the ring-opening step (see ESI†). In contrast, the other components of the transfer hydrogenation (thiourea catalyst 2k and pyridine co-product) do not interfere with the second step. Fortunately, it was possible to drive the transfer hydrogenation reaction to completion even by using just 1.1 equiv. of HE. Ultimately, this modification was sufficient to develop an efficient one-pot procedure.
Then, in line with the notion that enhancing the enantiopurity of 3d would result in additional improvement of syn-selectivity,19 an additional round of optimization of the catalyst used in the hydrogen transfer step was undertaken (Scheme 3). Different N-benzylic derivatives 2l–o related to 2k, and more elaborated Jacobsen catalysts bearing chiral 2-aryl pyrrolidin-1-yl amides242p–r were applied, promptly leading to improvements. Indeed, compared to N-benzyl catalyst 2k, the related 9-anthracenylmethyl derived structure 2o and the (2R)-2-phenylpyrrolidine amide 2p afforded azlactone 3a with higher enantioselectivities, with catalyst 2p providing better results (91/90% ee), even at lower catalyst loading (10 vs. 20 mol%), and in shorter reaction time (18 vs. 48 h).
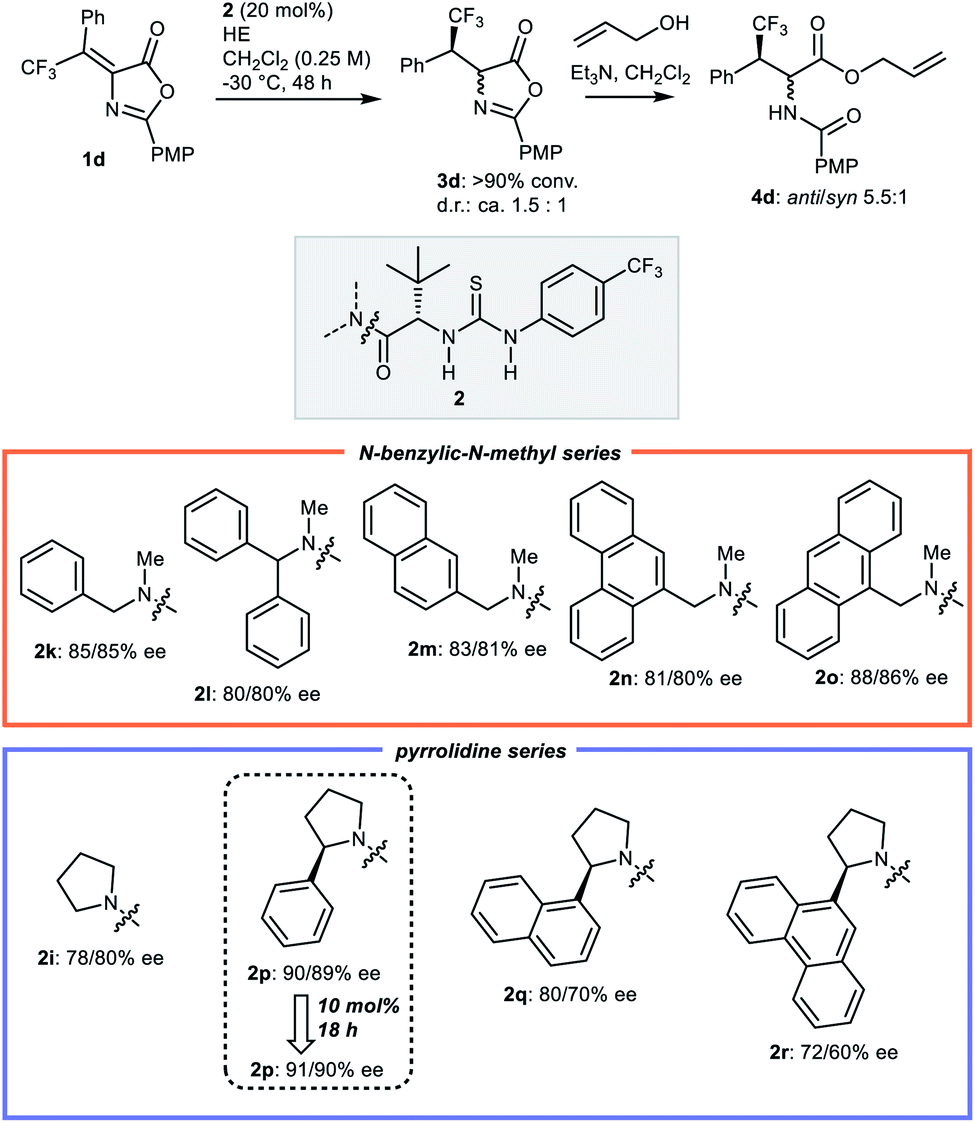 | ||
| Scheme 3 Second round of catalyst optimisation for the hydrogen transfer step: identification of optimal catalyst 2p. | ||
A tentative transition state picture can be built from a computationally validated model for the transfer hydrogenation of nitroalkenes with HEs catalysed by Jacobsen-type thiourea catalysts (2),17c complemented with recognition studies of lactones by a thiourea.25 Coordination of the acidic thiourea hydrogens to the lactone moiety, possibly assisted by its N-aryl group, and simultaneous stabilisation of the positive charge on the HE by the amide oxygen, are the key interactions between catalyst and substrates (Fig. 2). The tert-butyl group serves to “lock” the conformation of the catalyst as shown, thus leading to a match between the catalyst polar functionalities and a transition state leading to (3R)-3d. While this model does not help rationalizing the subtle effects of the amide and the thiourea aryl groups on the enantioselectivity of the reaction, it reconciles with the observed comparably high, but opposite, sense of enantioinduction exerted by catalyst 2p on the two isomeric olefins (i.e. Z-1 → (3R)-3, and E-1 → (3S)-3, see ESI†). From the experimental results shown in Scheme 3 (compare 2p with 2q and 2r), the often encountered positive relationship between the extension of the π-system of the 2-substituent of the pyrrolidine and the enantioselectivity24b is not apparent. Stabilising cation-π interactions might not be helpful to selectivity in this case.
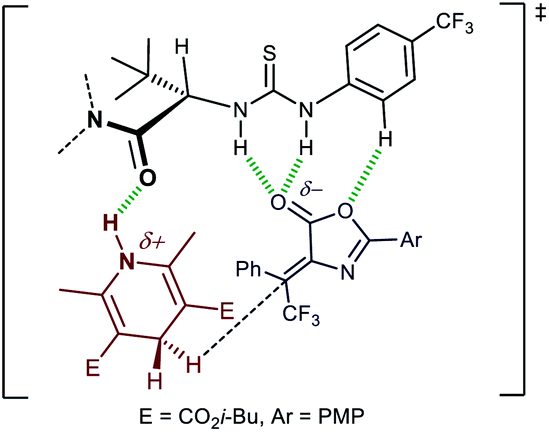 | ||
| Fig. 2 Tentative transition state model for the transfer hydrogenation reaction. | ||
The stage was thus set for the full unravelment of the stereodivergent methodology (Scheme 4). It is clear from the results reported that the improvement in enantioselectivity provided by catalyst 2p in the first step was indeed beneficial to the diastereoselectivities of the whole processes. Its combination with catalyst dhQD-2 furnished anti-4d in good yield and in essentially diastereo- and enantiopure form, while use of “mismatched” dhQN-10 in the second step afforded syn-4d in 72% yield and >99% ee, with a notable 7.5:1 diastereomeric ratio. These results are to be compared with the 14.2:1 d.r. and 89% ee for anti-4d, and the 5.9:1 d.r. and 92% ee for syn-4d obtained when catalyst 2k was used in the first step (Scheme 2). Scheme 4 shows also how the different combinations of catalysts (2p and ent-2p, dhQD-2 and dhQN-2, dhQD-10 and dhQN-10) could permit the obtainment of the full set of stereoisomeric products 4d with moderate to excellent results. Moreover, although the one pot protocol required longer reaction times (2–6 days instead of 24 h), for the ring opening step, the Cinchona loading could be lowered to 5 mol% without affecting the selectivity of the process.
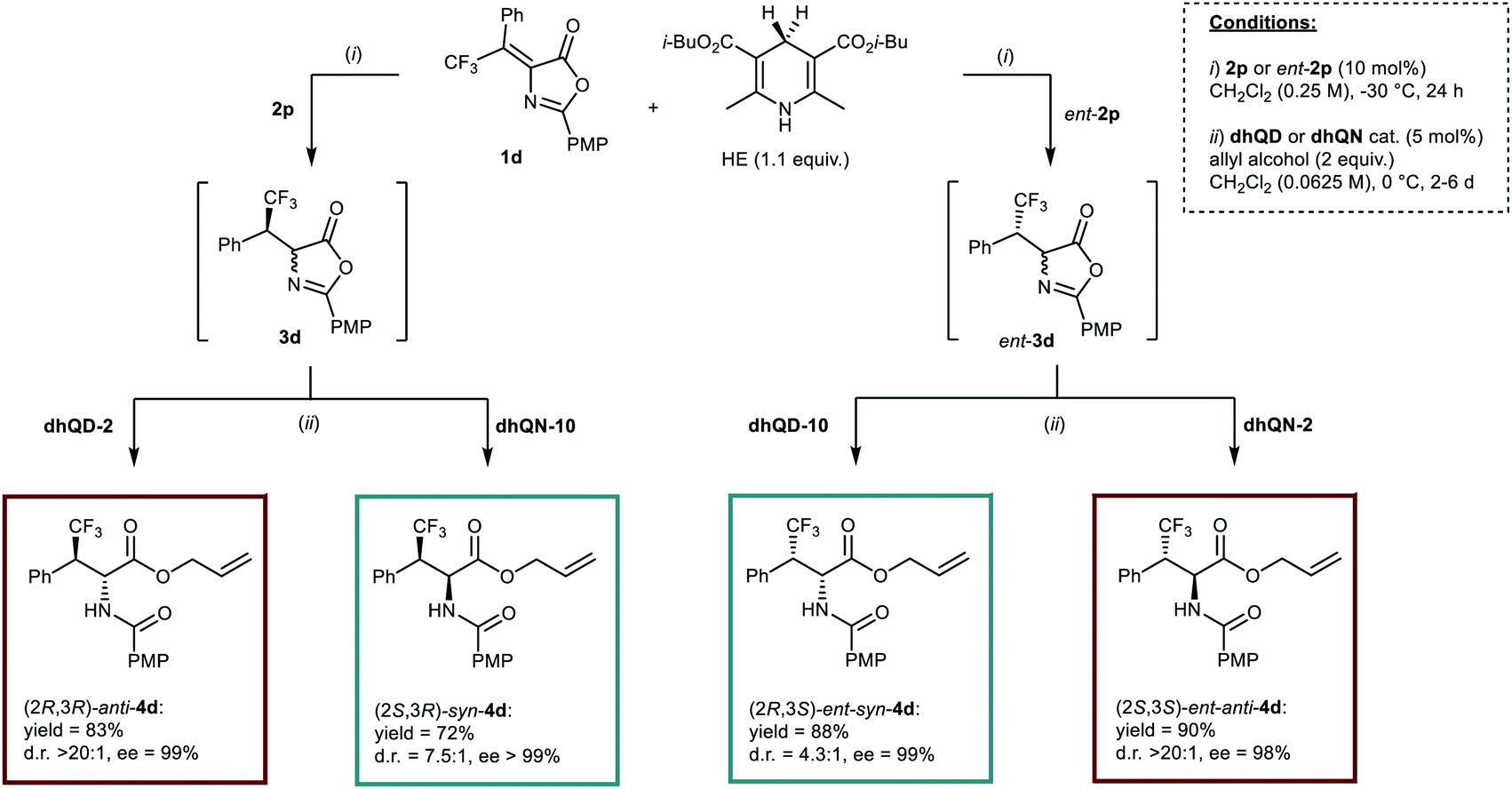 | ||
| Scheme 4 Diastereodivergent, enantioselective synthesis of the whole set of stereoisomers of 4d by applying different catalysts combinations in the one-pot process. | ||
The scope of the one-pot procedure was then studied (Table 2), by first applying a range of β-trifluoromethyl Erlenmeyer–Plöchl azlactones 1d–j, bearing electron-donating (1g, i) or electron-withdrawing (1e, f, h, j) groups at the β-aryl ring, and β-heteroaromatic substituents (1k, l). Entries 1–9 show that these substrates behaved very well in the anti-selective reaction, providing the corresponding anti-4d-l with results comparable to the parent anti-4d, that is, in good yields and outstanding diastereo- and enantioselectivities. The syn-selective processes provided variable results in terms of diastereoselectivities, ranging from a fully satisfactory 8.5:1 value for product syn-4g to less pleasing ca. 2:1 results for the β-heteroaromatic derivatives syn-4k and syn-4l. The latter results can be ascribed to a very high substrate bias towards anti-4k, l in the ring-opening process (>10:1 employing Et3N), rather than to poor catalyst dhQN-10 efficiency. Syn-4k and syn-4l were also obtained in lower yields compared to the other compounds. Nevertheless, the enantiomeric excesses of the major syn-4 isomers were found to be excellent in all cases examined (≥99% ee). Substrate 1m bearing a β-perfluoro residue rendered results similar to the β-heteroaromatic derivatives 1k and 1l, that is, excellent selectivity in the anti-4m isomer, and moderate yield and diastereoselectivity, but with >99% ee, for the syn-4m diastereoisomer (entry 10). The application of β-aliphatic substrates 1n–p required an adjustment to the conditions used in the transfer hydrogenation step, which was performed with higher (20 mol%) catalyst loading and at higher temperatures (−20 °C for the ethyl and methyl derivatives 1o and 1p, 0 °C for the more hindered cyclohexyl counterpart 1n). With these adjustments, it was possible to obtain anti-4n–p with good selectivities, while the results for syn-4n–p vary from the satisfactory level of syn-4n to the less pleasing syn-selectivity for 4p (entries 11–13). The latter result was ascribed to the deleterious combination of moderate enantioselectivity in the transfer hydrogenation step (ca. 70% ee) with high substrate bias in the ring-opening step (ca. 10:1). The last three entries 14–16 of Table 2 display the results obtained by applying alcohols other than allyl in the alcoholytic process with substrate 1d. The peculiarity of the present reaction system makes the tolerance to different primary alcohols, known for the DKR of simple azlactones,20 less than obvious, especially in the case of the syn-selective protocol. However, it was pleasing to observe that results in line with the allyl derivatives 4d were obtained for the products of methyl, benzyl and isobutyl alcohols 4q–s, although lower yields were observed in the latter case.||
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | 1: R1, Rf | R2 | Anti-4 selective processa | Syn-4 selective processa | ||||||
| Anti-4 | Yieldb (%) | Anti/sync | eed (%) | Syn-4 | Yieldb (%) | Syn/anti | eed (%) | |||
| a Conditions: 1 (0.15 mmol), HE (0.165 mmol, 1.1 equiv.), 2p (0.015 mmol, 10 mol%), CH2Cl2 (0.60 mL), −30 °C, 24–48 h, then CH2Cl2 (1.8 mL), dhQD-2 for anti-4 or dhQN-10 for syn-4 (0.0075 mmol, 5 mol%), R2OH (0.30 mmol, 2 equiv.), 0 °C, 2–8 d. b Isolated yield of combined diastereoisomers 4 after chromatography on silica gel. c Determined on the crude mixtures by 19F NMR spectroscopy. d Enantiomeric excess of major diastereoisomer, determined by CSP HPLC. e In the ring-opening step, after 2–5 d, additional catalyst dhQD-2 for anti-4 or dhQN-10 for syn-4 (0.0075 mmol) and R2OH (0.15 mmol, 1–2 equiv.), were added. f Ring-opening step warmed to RT after 2 d. g Two step reaction performed by isolating intermediate 3 by a rapid filtration on silica gel. h Reduction step performed at 0 °C. i Conditions: 1 (0.05 mmol), HE (0.055 mmol, 1.1 equiv.), 2p (0.01 mmol, 20 mol%), CH2Cl2 (0.300 mL), −20 or 0 °C, 24–48 h, then CH2Cl2 (0.60 mL), dhQD-2 for anti-4 or dhQN-10 for syn-4 (0.01 mmol, 10 mol%), allyl alcohol (0.1 mmol, 2 equiv.), 0 °C, 3–6 d. | ||||||||||
| 1 | 1d: C6H5, CF3 | Allyl | anti-4d | 83 | >20:1 |
99 | syn-4d | 72 | 7.5:1 |
>99 |
| 2 | 1e: 4-BrC6H4, CF3 | Allyl | anti-4e | 93 | >20:1 |
98 | syn-4e | 72 | 3.6:1 |
99 |
| 3 | 1f: 4-ClC6H4, CF3 | Allyl | anti-4f | 90 | >20:1 |
99 | syn-4f | 77 | 4.4:1 |
99 |
| 4 | 1g: 3-MeC6H4, CF3 | Allyl | anti-4g | 96 | >20:1 |
98 | syn-4g | 84 | 8.5:1 |
99 |
| 5 | 1h: 4-FC6H4, CF3 | Allyl | anti-4h | 88 | >20:1 |
98 | syn-4he | 78 | 4.4:1 |
99 |
| 6 | 1i: 4-MeOC6H4, CF3 | Allyl | anti-4ie,g | 90 | >20:1 |
98 | syn-4ie,f | 60 | 3.7:1 |
>99 |
| 7 | 1j: 3,5-F2C6H3, CF3 | Allyl | anti-4j | 85 | >20:1 |
99 | syn-4je | 87 | 4.0:1 |
>99 |
| 8 | 1k: 2-thienyl, CF3 | Allyl | anti-4k | 78 | >20:1 |
97 | syn-4kg | 37 | 2.1:1 |
99 |
| 9 | 1l: N-Ts-indol-3-yl, CF3 | Allyl | anti-4l | 82 | >20:1 |
96 | syn-4lg | 53 | 2.3:1 |
>99 |
| 10 | 1m: C6H5, CF3CF2CF2 | Allyl | anti-4m | 69 | >20:1 |
98 | syn-4mg,h | 50 | 2.0:1 |
>99 |
| 11i | 1n: cyclohexyl, CF3 | Allyl | anti-4ne | 98 | 16.7:1 |
99 | syn-4ne,f | 65 | 3.6:1 |
>99 |
| 12i | 1o: Et, CF3 | Allyl | anti-4o | 98 | 15.3:1 |
97 | syn-4oe | 78 | 2.6:1 |
>99 |
| 13i | 1p: Me, CF3 | Allyl | anti-4p | 97 | 10.1:1 |
89 | syn-4pe | 85 | 1.3:1 |
99 |
| 14 | 1d: C6H5, CF3 | Me | anti-4q | 91 | >20:1 |
97 | syn-4q | 77 | 5.3:1 |
>99 |
| 15 | 1d: C6H5, CF3 | Bn | anti-4r | 81 | 18:1 |
98 | syn-4re | 70 | 5.9:1 |
99 |
| 16 | 1d: C6H5, CF3 | i-Bu | anti-4se | 50 | >20:1 |
96 | syn-4se,g | 50 | 6.7:1 |
99 |

Compounds 4j were separately subjected to a two-step reduction-hydrolysis sequence (Scheme 5), delivering the corresponding aminoalcohol hydrochlorides 6, via amides 5. It is worth stressing that neither syn-6 – intermediate en route to the drug candidate (see Scheme 1(b))9a – nor anti-6 can be easily accessed by conventional asymmetric hydrogenation.7d
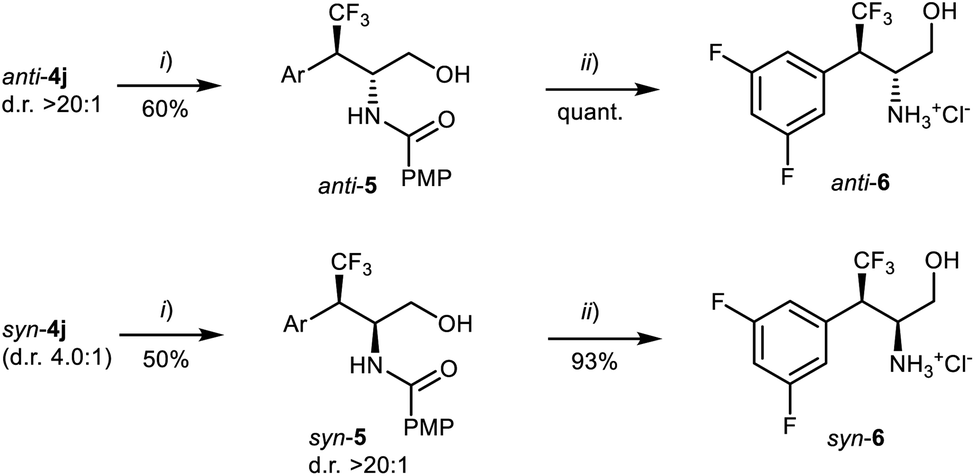 | ||
| Scheme 5 Conversion of the catalytic products 4j to the corresponding amino alcohol hydrochlorides anti-6 and syn-6. (i) NaBH4, THF/H2O, 0 °C → RT, then column chromatography on silica gel. (ii) HCl, MeOH/H2O, reflux, then work up and evaporation. | ||
Conclusions
We have proved that the conceptually new combination of two catalytic processes (transfer hydrogenation – dynamic ring-opening) on Erlenmeyer–Plöchl azlactones can provide a new stereodivergent strategy to enantioenriched β-branched AAs. The realization of this tactic with trifluoromethylated substrates has disclosed a one-pot entry to β-aryl-β-trifluoromethyl AA derivatives. Using the appropriate catalyst combination, the anti-bias of the ring-opening reaction was leveraged, giving anti-products with excellent stereoselectivities (d.r. up to >20:1, ee ≥ 89%). The scope of this reaction includes substrates reluctant to enantioselective hydrogenation. A newly designed ammonia derived squaramide catalyst afforded the syn-isomers, not obtainable by hydrogenation, with variable diastereoselectivities (d.r. up to 8.5:1) and high enantioselectivities (ee ≥ 99%).
Author contributions
Conceptualization and supervision: VC and LB. Investigation and methodology: all authors. Writing - original draft: LB. Writing - review and editing: VC, MF, LB. Funding acquisition, project administration and resources: MF and LB.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Enrico Marcantonio, Federico Curati, Riccardo Ciciriello and Giorgiana Denisa Bisag for preliminary screening and preparation of catalysts/substrates. We are grateful to Prof. Eric Jacobsen for a helpful discussion. We acknowledge financial support from the University of Bologna (RFO program), MIUR (FFABR 2017), and F.I.S. (Fabbrica Italiana Sintetici).Notes and references
- J. Michaux, G. Niel and J.-M. Campagne, Chem. Soc. Rev., 2009, 38, 2093 RSC.
- (a) B.-H. Zheng, C.-H. Ding, X.-L. Hou and L.-X. Dai, Org. Lett., 2010, 12, 1688 CrossRef CAS; (b) J. Wang, S. Zhou, D. Lin, X. Ding, H. Jiang and H. Liu, Chem. Commun., 2011, 47, 8355 RSC; (c) W.-D. Chu, L.-F. Zhang, X. Bao, X.-H. Zhao, C. Zeng, J.-Y. Du, G.-B. Zhang, F.-X. Wang, X.-Y. Ma and C.-A. Fan, Angew. Chem., Int. Ed., 2013, 52, 9229 CrossRef CAS PubMed; (d) S. Lou, G. M. McKenna, S. A. Tymonko, A. Ramirez, T. Benkovics, D. A. Conlon and F. González-Bobes, Org. Lett., 2015, 17, 5000 CrossRef CAS; (e) X.-Z. Zhang, Y.-H. Deng, X. Yan, K.-Y. Yu, F.-X. Wang, X.-Y. Ma and C.-A. Fan, J. Org. Chem., 2016, 81, 5655 CrossRef CAS PubMed; (f) F.-S. He, J.-H. Jin, Z.-T. Yang, X. Yu, J. S. Fossey and W.-P. Deng, ACS Catal., 2016, 6, 652 CrossRef CAS; (g) Y.-C. Gong, Y. Wang, E.-Q. Li, H. Cui and Z. Duan, Adv. Synth. Catal., 2019, 361, 1389 CrossRef CAS; (h) M. Winter, R. Schütz, A. Eitzinger, A. R. Ofial and M. Waser, Eur. J. Org. Chem., 2020, 3812 CrossRef CAS; (i) V. A. Soloshonok, T. Yamada, K. Sakaguchi and Y. Ohfune, Future Med. Chem., 2009, 1, 897 CrossRef CAS PubMed.
- (a) S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2013, 135, 12135 CrossRef CAS PubMed; (b) G. Chen, T. Shigenari, P. Jain, Z. Zhang, Z. Jin, J. He, S. Li, C. Mapelli, M. M. Miller, M. A. Poss, P. M. Scola, K.-S. Yeung and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 3338 CrossRef CAS PubMed , see also:; (c) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391 CrossRef CAS PubMed.
- (a) J. Legters, L. Thijs and B. Zwanenburg, Recl. Trav. Chim. Pays-Bas, 1992, 111, 16 CrossRef CAS; (b) S. C. Valdez and J. L. Leighton, J. Am. Chem. Soc., 2009, 131, 14638 CrossRef CAS PubMed; (c) A. Armstrong and A. Ferguson, Beilstein J. Org. Chem., 2012, 8, 1747 CrossRef CAS PubMed , see also:; (d) D. Wilcke, E. Herdtweck and T. Bach, Chem.–Asian J., 2012, 7, 1372 CrossRef CAS PubMed.
- C. E. Boville, R. A. Scheele, P. Koch, S. Brinkmann-Chen, A. R. Buller and F. H. Arnold, Angew. Chem., Int. Ed., 2018, 57, 14764 CrossRef CAS PubMed.
- C. Liao and F. P. Seebeck, Angew. Chem., Int. Ed., 2020, 59, 7184 CrossRef CAS PubMed.
- (a) M. J. Burk, M. F. Gross and J. P. Martinez, J. Am. Chem. Soc., 1995, 117, 9375 CrossRef CAS; (b) R. S. Haermer, D. Askin, R. P. Volante and P. J. Reider, Tetrahedron Lett., 1998, 39, 3455 CrossRef; (c) M. J. Burk, l. K. M. Bedingfield, W. F. Kiesman and J. G. Allen, Tetrahedron Lett., 1999, 40, 3093 CrossRef CAS; (d) C. Benhaim, L. Bouchard, G. Pelletier, J. Sellstedt, L. Kristofova and S. Daigneault, Org. Lett., 2010, 12, 2008 CrossRef CAS PubMed; (e) C. Molinaro, J. P. Scott, M. Shevlin, C. Wise, A. Ménard, A. Gibb, E. M. Junker and D. Lieberman, J. Am. Chem. Soc., 2015, 137, 999 CrossRef CAS PubMed.
- (a) M. Salwiczek, E. K. Nyakatura, U. I. M. Gerling, S. Ye and B. Koksch, Chem. Soc. Rev., 2012, 41, 2135 RSC; (b) J. Moschner, V. Stulberg, R. Fernandes, S. Huhmann, J. Leppkes and B. Koksch, Chem. Rev., 2019, 119, 10718 CrossRef CAS PubMed.
- For another approach to a specific β-aryl-β-trifluoromethyl AA, wherein asymmetric hydrogenation failed: (a) A. Alimardanov, A. Nikitenko, T. J. Connolly, G. Feigelson, A. W. Chan, Z. Ding, M. Ghosh, X. Shi, J. Ren, E. Hansen, R. Farr, M. MacEwan, S. Tadayon, D. M. Springer, A. F. Kreft, D. M. Ho and J. R. Potoski, Org. Process Res. Dev., 2009, 13, 1161 CrossRef CAS , for alternative approaches to β-alkyl-β-trifluoromethyl AAs, see:; (b) X. Xing, A. Fichera and K. Kumar, J. Org. Chem., 2002, 67, 1722 CrossRef CAS , correction: J. Org. Chem., 2002, 67, 8290; (c) J. A. Pigza, T. Quach and T. F. Molinski, J. Org. Chem., 2009, 74, 5510 CrossRef CAS PubMed , see also ref. 2h..
- (a) A. Enright, F. R. Alexandre, G. Roff, I. G. Fotheringham, M. J. Dawson and N. J. Turner, Chem. Commun., 2003, 2636 RSC; (b) G. J. Roff, R. C. Lloyd and N. J. Turner, J. Am. Chem. Soc., 2004, 126, 4098 CrossRef CAS PubMed , for a stereodivergent approach to β,β-diaryl AAs, see ref. 7e..
- Reviews on diastereodivergent enantioselective catalysis: (a) G. Zhan, W. Du and Y.-C. Chen, Chem. Soc. Rev., 2017, 46, 1675 RSC; (b) L. Lin and X. Feng, Chem.–Eur. J., 2017, 23, 6464 CrossRef CAS PubMed; (c) M. Bihania and J. C.-G. Zhao, Adv. Synth. Catal., 2017, 359, 534 CrossRef; (d) S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 5627 CrossRef CAS; (e) I. P. Beletskaya, C. Nájera and M. Yus, Chem. Rev., 2018, 118, 5080 CrossRef CAS PubMed , for a landmark stereodivergent olefin functionalisation by sequential reactions, see; (f) B. Simmons, A. M. Walji and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2009, 48, 4349 CrossRef CAS PubMed , for recent relevant examples, see:; (g) S.-L. Shi, Z. L. Wong and S. L. Buchwald, Nature, 2016, 532, 353 CrossRef CAS PubMed; (h) B. M. Trost, C.-I. Hung, T. Saget and E. Gnanamani, Nat. Catal., 2018, 1, 523 CrossRef CAS; (i) X.-J. Liu, S. Jin, W.-Y. Zhang, Q.-Q. Liu, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2020, 59, 2039 CrossRef CAS; (j) J. Masson-Makdissi, L. Prieto, X. Abel-Snape and M. Lautens, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202105800 , early view; (k) D.-X. Zhu, J.-G. Liu and M.-H. Xu, J. Am. Chem. Soc., 2021, 43, 8583 CrossRef PubMed.
- Recent reviews on enantioselective transfer hydrogenation with Hantzsch esters: (a) W. Dong and D. Astruc, Chem. Rev., 2015, 115, 6621 CrossRef PubMed; (b) R. P. Herrera, Top. Curr. Chem., 2016, 374, 29 CrossRef PubMed; (c) A. M. Faisca Phillips and A. J. L. Pombeiro, Org. Biomol. Chem., 2017, 15, 2307 RSC.
- For stereocontrolled formation of trifluoromethylated chirality centres, see: (a) J. Nie, H.-C. Guo, D. Cahard and J.-A. Ma, Chem. Rev., 2011, 111, 455 CrossRef CAS PubMed; (b) P. Poutrel, M. V. Ivanova, X. Pannecoucke, P. Jubault and T. Poisson, Chem.–Eur. J., 2019, 25, 15262 CrossRef CAS PubMed , and references cited therein..
- (a) A. Berkessel, F. Cleemann, S. Mukherjee, T. N. Müller and J. Lex, Angew. Chem., Int. Ed., 2005, 44, 807 CrossRef CAS , for reviews, see: ; (b) A.-N. R. Alba and R. Rios, Chem.–Asian J., 2011, 6, 720 CrossRef CAS PubMed; (c) Z. Rodríguez-Docampo and S. J. Connon, ChemCatChem, 2012, 4, 151 CrossRef; (d) P. P. de Castro, A. G. Carpanez and G. W. Amarante, Chem.–Eur. J., 2016, 22, 10294 CrossRef CAS; (e) I. F. S. Marra, P. P. de Castro and G. W. Amarante, Eur. J. Org. Chem., 2019, 5830 CrossRef CAS , for a more comprehensive list of references on DKR of azlactones by ring-opening, see ESI†..
- T. J. Caggiano, K. M. Morris, B. L. Harrison, A. F. Kreft III, D. M. Kubrak and D. M. Springer, US Pat., US20090023801A1, 2009 Search PubMed.
- W.-G. Su, G. Dai, H. Jia, Z. Zhang, J. Weng, J. D. Venable, S. D. Bembenek, W. Chai, S. P. Meduna, J. M. Keith, W. Eccles, A. D. Lebsack, W. M. Jones and R. C. Smith, WO2016119707 A1, 2016.
- (a) N. J. A. Martin, L. Ozores and B. List, J. Am. Chem. Soc., 2007, 129, 8976 CrossRef CAS PubMed; (b) E. Martinelli, A. C. Vicini, M. Mancinelli, A. Mazzanti, P. Zani, L. Bernardi and M. Fochi, Chem. Commun., 2015, 51, 658 RSC; (c) E. Massolo, M. Benaglia, M. Orlandi, S. Rossi and G. Celentano, Chem.–Eur. J., 2015, 21, 3589 CrossRef CAS PubMed; (d) T. Hostmann, J. J. Molloy, K. Bussmann and R. Gilmour, Org. Lett., 2019, 21, 10164 CrossRef CAS PubMed; (e) L. Bernardi and M. Fochi, Molecules, 2016, 21, 1000 CrossRef PubMed , and references cited therein..
- (a) A. G. Wenzel, M. P. Lalonde and E. N. Jacobsen, Synlett, 2003, 1919 CAS; (b) R. S. Klausen and E. N. Jacobsen, Org. Lett., 2009, 11, 887 CrossRef CAS PubMed; (c) S. J. Zuend, M. P. Coughlin, M. P. Lalonde and E. N. Jacobsen, Nature, 2009, 461, 968 CrossRef CAS PubMed; (d) S. J. Zuend and E. N. Jacobsen, J. Am. Chem. Soc., 2009, 131, 15358 CrossRef CAS PubMed.
- According to Masamune's multiplicity rule for double asymmetric synthesis, applied to 3a with 85% ee, even a highly selective chiral catalyst in the ring-opening of simple azlactones (20:1) would give only a moderate 2.6:1 syn/anti-4a ratio, while using enantiopure 3a the d.r. would increase to 3.6:1 (a) S. Masamune, W. Choy, J. S. Petersen and L. R. Sita, Angew. Chem., Int. Ed., 1985, 24, 1 CrossRef; (b) D. Lotter, A. Castrogiovanni, M. Neuburger and C. Sparr, ACS Cent. Sci., 2018, 4, 656 CrossRef CAS PubMed , for additional discussion, see ESI†..
- (a) J. W. Lee, T. H. Ryu, J. S. Oh, H. Y. Bae and C. E. Song, Chem. Commun., 2009, 7224 CAS; (b) J.-S. Oh, J.-W. Lee, T. H. Ryu, J. H. Lee and C. E. Song, Org. Biomol. Chem., 2012, 10, 1052 RSC; (c) S. Tallon, F. Manoni and S. J. Connon, Angew. Chem., Int. Ed., 2015, 54, 813 CrossRef CAS PubMed.
- According to the Horeau effect (or Horeau amplification), in a sequential process, like the present one, the enantiomeric excess of the major diastereomer is higher than the enantiomeric excess of the product obtained in the first step [(ee (int.)]. It can be understood by considering that the minor enantiomer produced in the first step is converted, during the second step, to the minor diastereoisomer. In simplified terms, for a reaction in which the selectivity (s) of the second step is the same on both enantiomers obtained in the first step, the ee of the major diastereoisomer can be derived from the following equation: ee = [ee (int.) + s]/[1 + s × ee(int.)] However, the reaction under study represents a more complex situation, since the second step occurs with different selectivities (s, s′) for (3R)-3d and (3S)-3d, due to substrate bias. For a rigorous treatment, based on: (a) A. M. Harned, Tetrahedron, 2008, 74, 3797 CrossRef; (b) S. E. Baba, K. Sartor, J. Poulin and H. Kagan, Bull. Soc. Chim. Fr., 1994, 131, 525 Search PubMed , see ESI†..
- J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416 CrossRef CAS PubMed.
- For reaction models built on the computational study of the alcoholytic ring-opening of an alanine derived azlactone, reported in: A. Berkessel and K. Etzenbach-Effers, in Hydrogen Bonding in Organic Synthesis, ed. P. M. Pihko, Wiley-VCH, Weinheim, 2009, ch. 3, pp. 15–42 Search PubMed, see ESI†..
- (a) S. E. Reisman, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 7198 CrossRef CAS PubMed; (b) R. R. Knowles, S. Lin and E. N. Jacobsen, J. Am. Chem. Soc., 2010, 132, 5030 CrossRef CAS.
- K. M. Lippert, K. Hof, D. Gerbig, D. Ley, H. Hausmann, S. Guenther and P. R. Schreiner, Eur. J. Org. Chem., 2012, 5919 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional optimization results, additional discussion on the ring-opening step, control experiments for the one-pot protocol, experimental section, copies of NMR spectra and HPLC traces. See DOI: 10.1039/d1sc01442k |
| ‡ Current address: Department of Chemistry, Aarhus University, 8000 Aarhus, Denmark. |
| § All through the paper, to identify the diastereomers of the β-branched β-trifluoromethyl AA derivatives, we have used Masamune's syn and anti descriptors, arbitrarily setting the β-aryl/alkyl group of these compounds in the main chain. Using CIP descriptors for the relative configuration (e.g. R*, S*), although more rigorous, would result in a less clear identification of the diastereoisomeric pairs. |
| ¶ For a more comprehensive list of screening results, see ESI.† |
| || For limitations in terms of substrate variations, see ESI.† |
| This journal is © The Royal Society of Chemistry 2021 |