
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHow to not build a cage: endohedral functionalization of polyoxometalate-based metal–organic polyhedra†
Ji
Guo
a,
Qing
Chang
a,
Zhiwei
Liu
a,
Yangming
Wang
a,
Chuanhong
Liu
a,
Mou
Wang
a,
Danmeng
Huang
a,
Guanying
Chen
 a,
Hongmei
Zhao
b,
Wei
Wang
*cd and
Xikui
Fang
*a
a,
Hongmei
Zhao
b,
Wei
Wang
*cd and
Xikui
Fang
*a
aMIIT Key Laboratory of Critical Materials Technology for New Energy Conversion and Storage, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, Harbin 150001, China. E-mail: xkfang@hit.edu.cn
bState Key Laboratory of Information Photonics and Optical Communications, School of Science, Beijing University of Posts and Telecommunications, Beijing 100876, China
cCAS Key Laboratory of Design and Assembly of Functional Nanostructures, Fujian Provincial Key Laboratory of Nanomaterials, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Xiamen, Fujian 361021, China. E-mail: wangwei@fjirsm.ac.cn
dXiamen Institute of Rare Earth Materials, Haixi Institutes, Chinese Academy of Sciences, Xiamen, Fujian 361021, China
First published on 3rd April 2021
Abstract
Introducing functionalities into the interior of metal–organic cage complexes can confer properties and utilities (e.g. catalysis, separation, drug delivery, and guest recognition) that are distinct from those of unfunctionalized cages. Endohedral functionalization of such cage molecules, for decades, has largely relied on modifying their organic linkers to covalently append targeted functional groups to the interior surface. We herein introduce an effective coordination method to bring in functionalities at the metal sites instead, for a set of polyhedral cages where the nodes are in situ formed polyoxovanadate clusters, [VIV6O6(OCH3)9(μ6-SO4)(COO)3]2−. Replacing the central sulfates of these hexavanadate clusters with more strongly coordinating phosphonate groups allows the installation of functionalities within the cage cavities. Organophosphonates with phenyl, biphenyl, and terphenyl tails were examined for internalization. Depending on the size/shape of the cavities, small phosphonates can fit into the molecular containers whereas larger ones inhibit or transform the framework architecture, whereby the first non-cage complex was isolated from a reaction that otherwise would lead to entropically favored regular polyhedra cages. The results highlight the complex and dynamic nature of the self-assembly process involving polyoxometalates and the scope of molecular variety accessible by the introduction of endo functional groups.
Introduction
A highly functionalized interior is a widespread attribute of biological entities, a fact well illustrated by ferritin,1–3 virus particles,4,5 and, more broadly, enzymes.6 Functional groups in the interior cavity of such macromolecular systems participate in a myriad of chemical reactions and processes, accounting for a broad spectrum of their biological functions. For more than half a century, chemists have taken inspiration from nature and developed a diverse array of organic and metal–organic cage-like molecules with well-defined cavities, i.e. “molecular containers”,7 to allow the control of reactions in ways mimicking the behavior of biological systems. However, the exploration of their endohedral functionalization so far has been rather limited.8Metal–organic polyhedra (MOP),9–16 constructed from the modular assembly of metal ions and organic linkers, are one important group of molecular containers. Although the rational design of MOPs has been widely explored and immensely successful, introducing functionalities into the cages remains challenging.8 Unlike metal–organic frameworks, many MOP cages are built with single-metal nodes,9–16 which are often coordinatively saturated and thus incapable of binding to additional functional groups. It should be noted, though, there are a handful of reports17–21 taking advantage of the open, axial binding sites of metal porphyrins to install endo functional groups or to trap guest molecules inside MOP cavities. Secondly, the construction of most MOPs utilizes the so-called “molecular paneling” approach,10 where multiple flat aromatic panels enclose a central void. This limits the probability of interior functionalization as any appended functional groups derived from a panel would have to be orthogonally positioned. Lastly, incorporating endohedral functional groups, even if successful,22–35 generally requires careful control of ligand coordination angles and lengthy organic syntheses to derivatize the parent organic linker with a functional group of choice. A notable example of such covalent functionalization has been elegantly demonstrated by Fujita22–29 for their M12L24 spherical complexes at the curvature of bis(pyridine) ligands with a bend angle of ca. 120°.
Replacing individual metal ion nodes in MOPs with metal oxide clusters such as polyoxometalates (POMs), on the other hand, could offer the opportunity for facile coordinative functionalization on the metal-cluster nodes instead, through multiple metal–ligand bonds. In recent years, Zaworotko, Su, and Wang have developed a new class of MOPs based on tritopic,36–40 tetratopic,41–46 and pentatopic (including mixed)47–50 polyoxovanadate secondary building units (SBUs) as nodes, providing us a suitable platform to test coordinative functionalization. The systems of focus here are built up from a hexavanadate SBU,36–40 [VIV6O6(OCH3)9(μ6-SO4)(COO)3]2− ({V6S}). Locked in a concave conformation, each tritopic {V6S} cluster is centered around a sulfate ion and can be connected by carboxylate linkers at its three exit points to give various platonic MOP cages with well-defined inner void spaces. Three such MOP complexes (VMOC-2,40VMOP-11,36 and VMOP-14,36) with internal cavities of different sizes and shapes (Fig. 1) were selected in this work for endohedral functionalization. First reported by Wang and Su,36,40 these MOPs were prepared from solvothermal reactions of vanadyl sulfate (from which the {V6S} SBUs are generated in situ) and their respective carboxylic acid precursors, namely 3,3′,5,5′-azobenzene tetracarboxylic acid (H4ABTC), benzene-1,4-dicarboxylic acid (H2BDC), and benzene-1,3,5-tricarboxylic acid (H3BTC).
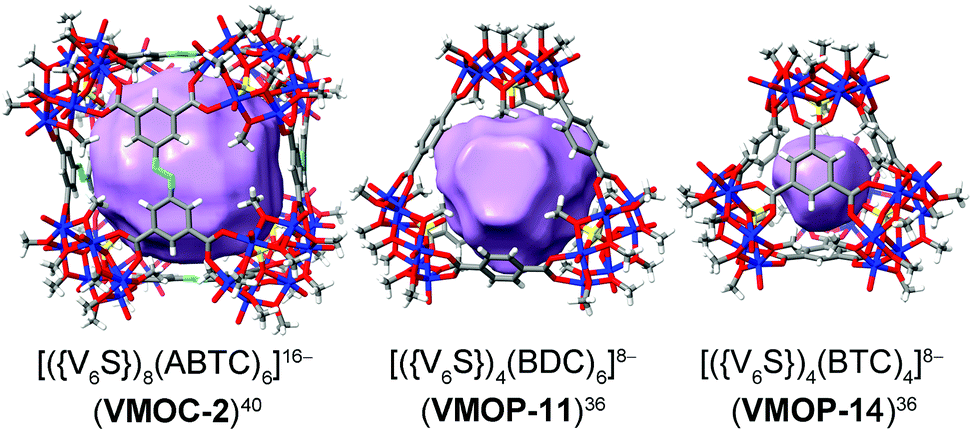 | ||
| Fig. 1 The three MOPs that are the targets of endohedral functionalization. Their interior void spaces are displayed as purple solid. Color code: V blue; S yellow; O red; C dark gray; N light blue; H white. | ||
Exploiting the above POM-based MOPs, here we present rare examples of endohedrally decorated MOPs by coordinative functionalization, as a proof-of-concept of this methodology. It was also found that the outcome hinges on the size/shape of their cavities and the functional groups; for the same cage, small functional groups can fit right into the molecular containers whereas larger ones may inhibit or alter the framework architecture, leading to unmaking of the polyhedral cages themselves.
Results and discussion
Endohedral functionalization with organophosphonates
Constructed from eight {V6S} vertices and six ABTC panels, VMOC-2 is a robust cubic complex40 with a large enclosed cavity of 1516 Å3,51 potentially allowing multiple functional groups to be appended at the inner surface of the cage. Installation of internal functionalities was accomplished by replacing the central sulfate groups of its {V6S} SBUs with organophosphonates, which have been widely used for the synthesis of hybrid polyoxovanadates.52–59 As anions of weak acids,60 organophosphonates61 are expected to form stronger bonds to the hexavanadate cluster than the sulfate ion. The ligand exchange process is also entropically favorable in that some of the enclosed solvent molecules will be released from the cavity upon complexation of endo functional groups.Phenylphosphonate was initially selected as a possible internal functional group. Thus, in a typical solvothermal experiment from which VMOC-2 was prepared, a roughly stoichiometric amount of phenylphosphonic acid was added to substitute sulfate. Its superior binding ability to the vanadium centers became so evident that 8-fold endohedral functionalization occurred even in the presence of excess sulfate ions, resulting in the endohedrally functionalized cage 1a. In IR spectra, there was a marked increase in the intensity of a band at 1068 cm−1 (asymmetric P–O stretching vibrations) that is attributed to the phosphonate functional groups. Single-crystal X-ray structure analysis (Fig. 2a) later confirmed the phosphonate groups at the interior surface of the MOP cage. The phenyl tails of all functional groups point to the center of the cavity as the void space is large enough to accommodate eight of them without unfavorable steric interactions. The appended phosphonates are also fairly ordered as a result and can be properly modelled from the electron density map.
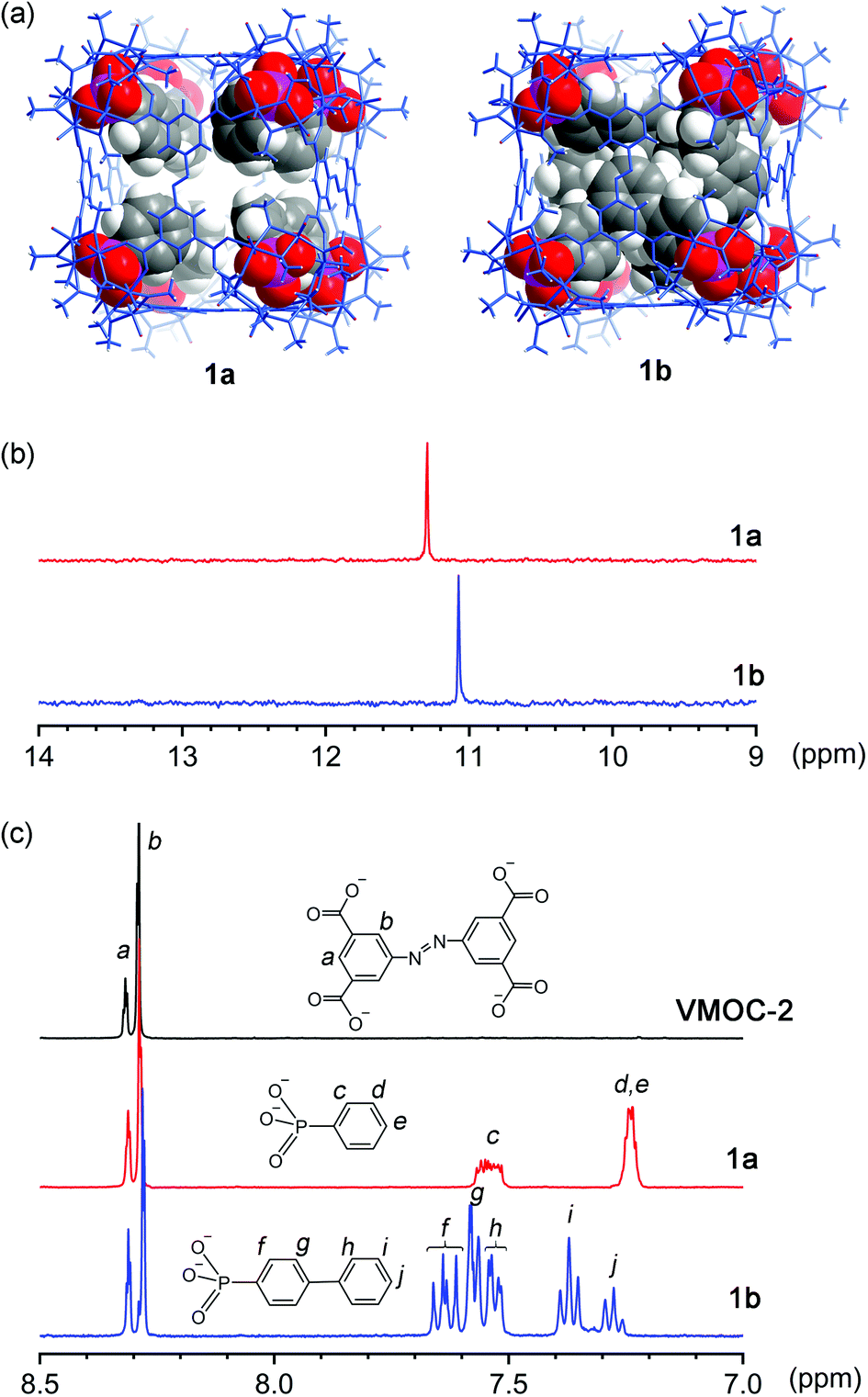 | ||
| Fig. 2 (a) X-ray crystal structure of 1a and molecular model (see text) of 1b. Biphenyl-4-phosphonate units of 1b were modeled within the outer shell, which was also determined by X-ray crystal structure analysis. The outer shells of both MOPs are in blue wire-frame representation, while the interior organophosphonates are in the space-filling model; (b) 31P NMR and (c) 1H NMR spectra of base-digested samples (400 MHz, NaOD/D2O, 293 K) of VMOC-2, 1a, and 1b. | ||
To investigate how the size of a functional group may affect the self-assembly of MOPs, we prepared two derivatives with longer rigid phenyl tails: biphenyl-4-phosphonic acid and p-terphenyl-4-phosphonic acid (see the Experimental section for details). Surprisingly, the biphenyl phosphonate groups, despite their larger size and perceived rigidity, were still able to fit in this cubic cage, leading to 1b. Like 1a, 1b crystallizes in the same monoclinic space group P21/n with similar cell parameters, suggesting that the different internal functions do not alter the framework structures or intermolecular interactions in any meaningful way. Although the shell framework of 1b could be precisely determined by X-ray structure analysis, we could not build a complete structure model of its inner phosphonate functional groups as their biphenyl moieties are severely disordered.
To elucidate the interior structure of 1b, density functional theory calculations were performed at the level of B3LYP/3-21G. The cubic framework was fixed in place, as determined by the crystal structure analysis, during the optimization. The minimum energy structure of the inner phosphonate groups (Fig. 2a) shows densely packed biphenyl moieties in the crowded interior cavity. Many of them are significantly bent and twisted to better fit into the confined space within the cage. Notably, a number of π–π stacking interactions are observed not only between interior biphenyl units but also between biphenyl phosphonates and the ABTC panel ligands on the cube faces (Fig. S13, ESI†), likely helping to stabilize this crowded assembly.
Although 1a and 1b are readily soluble in organic solvents such as DMF or DMSO, neither of them displays any 31P NMR signals from the phosphonate functional groups due to their close proximity to the paramagnetic V(IV) centers. The 1H NMR spectra of all three cubes are not very informative, either. Aromatic proton signals are significantly shifted and exceedingly broadened because of the paramagnetic effects; some are not even observable. To confirm the phosphonate functional groups, the samples were analyzed after breaking down the framework by base digestion with NaOD/D2O. The 31P NMR spectra of base-digested 1a and 1b both show a single peak at 11.30 and 11.08 ppm (Fig. 2b), respectively. Though the NMR data do not speak of the cubic structures in their entirety, the results confirmed the presence of phosphonate functional groups. Moreover, the number of functional groups, as calculated from the ratio between the resulting 1H integrals of ABTC and those of organophosphonates (Fig. 2c), was ∼8 per molecule, affirming the complete endohedral functionalization for both compounds.
Interplay of cavity size and functional groups: the steric effects
Further increasing the size of the functional groups, however, will prevent the cubic cage from being assembled, presumably because of the steric repulsion between internal groups. When p-terphenyl-4-phosphonic acid was used, no cubic MOP structures were produced. The fact that unfunctionalized VMOC-2 or partially functionalized cages (i.e. replacing some of the sulfate groups with terphenyl phosphonates) did not even form suggests that all of the hexavanadate SBUs are bound to phosphonates under the equilibrium conditions. This once again attests to the strong coordination ability of phosphonates.Besides increasing the length of functional groups, we also looked into the other side of the equation: the size of the void space of a MOP. To do this, we climbed down the MOP ladder to [(V6S)4(BDC)6]8− (VMOP-11) and [(V6S)4(BTC)4]8− (VMOP-14), which have a void space51 of 645 Å3 and 146 Å3 (Fig. 1), respectively. Both voids are significantly smaller than that of VMOC-2, but each cage only needs to accommodate four organophosphonate units.
As expected, replacement of sulfates in VMOP-11 with phenylphosphonates led to an endohedrally functionalized tetrahedral cage complex (2), shown in Fig. 3. Again, 31P and 1H NMR data supported the incorporation of four phosphonate ligands (Fig. S9 and S10, ESI†). However, we found that neither the biphenyl nor terphenyl phosphonic acid was able to generate the tetrahedral cage complex. The results thus follow the same trend seen for VMOC-2; increasingly bulky functional groups will prevent the MOP cages from forming (Scheme 1).
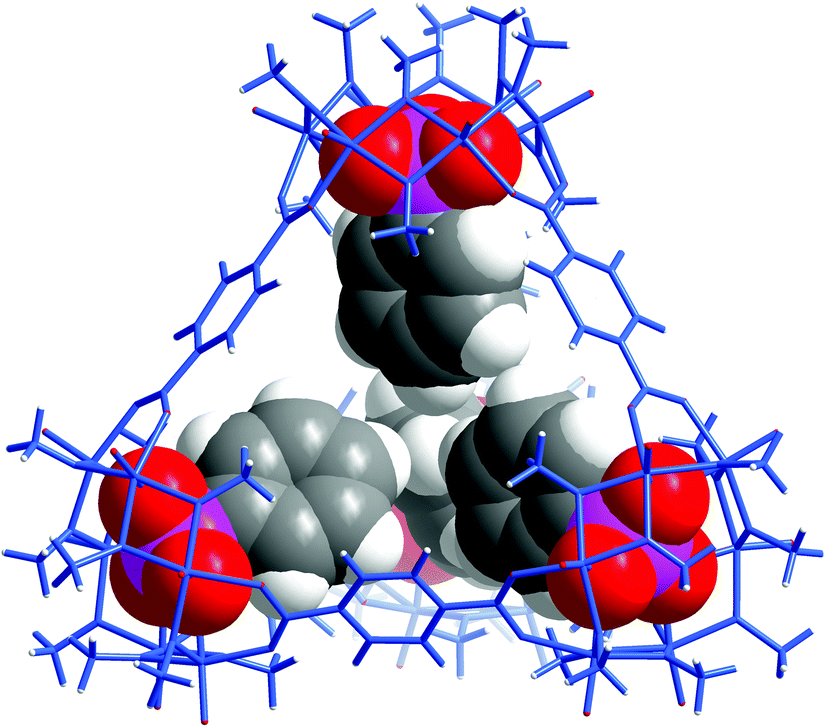 | ||
| Fig. 3 X-ray crystal structure of 2 showing the four internal phenylphosphonate functional groups. | ||
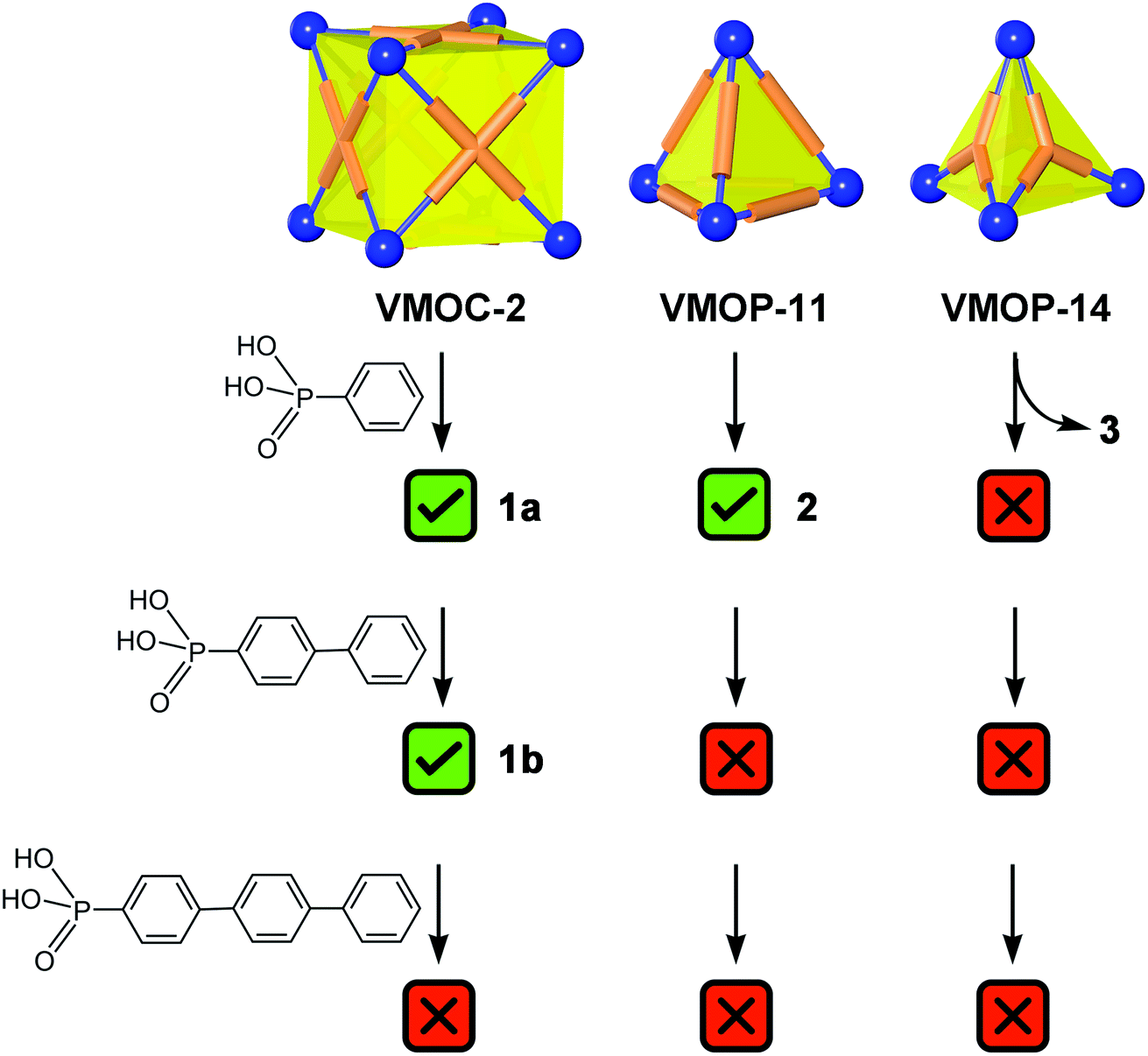 | ||
| Scheme 1 Summary of the outcome of endohedral functionalization with organophosphonic acids with increasingly lengthy phenyl tails. The vertex directing units (blue spheres) are {V6S} SBUs and the linker groups (orange tubes) represent ditopic and polytopic carboxylate ligands. | ||
Cage to non-cage transformation
Similarly, with a void space of only 146 Å3, VMOP-14 apparently is not even able to accommodate four of the smallest phenyphosphonate ligands (whose phenyl moiety itself has a van der Waals volume of ∼88 Å3), let alone the other two bulkier phosphonates. Although the incorporation of phenylphosphonates did not lead to a tetrahedral cage, it transformed the MOP framework into a non-container complex 3. This also provided insights into the unmaking of a polyhedral cage and the complex nature of the self-assembly processes involving polyoxometalates.Single crystal X-ray diffraction analysis revealed that complex 3 (Fig. 4) consists of eight hexavanadate {V6} clusters that are bridged by eight BTC ligands. On the equatorial plane of the complex sits a ring of eight vanadium atoms ({V8}), which serves as an anchor for the BTC ligands, giving rise to D4h symmetry for the whole assembly. Several features of complex 3, which make it distinct from any previously reported polyhedral cages, are worth noting.
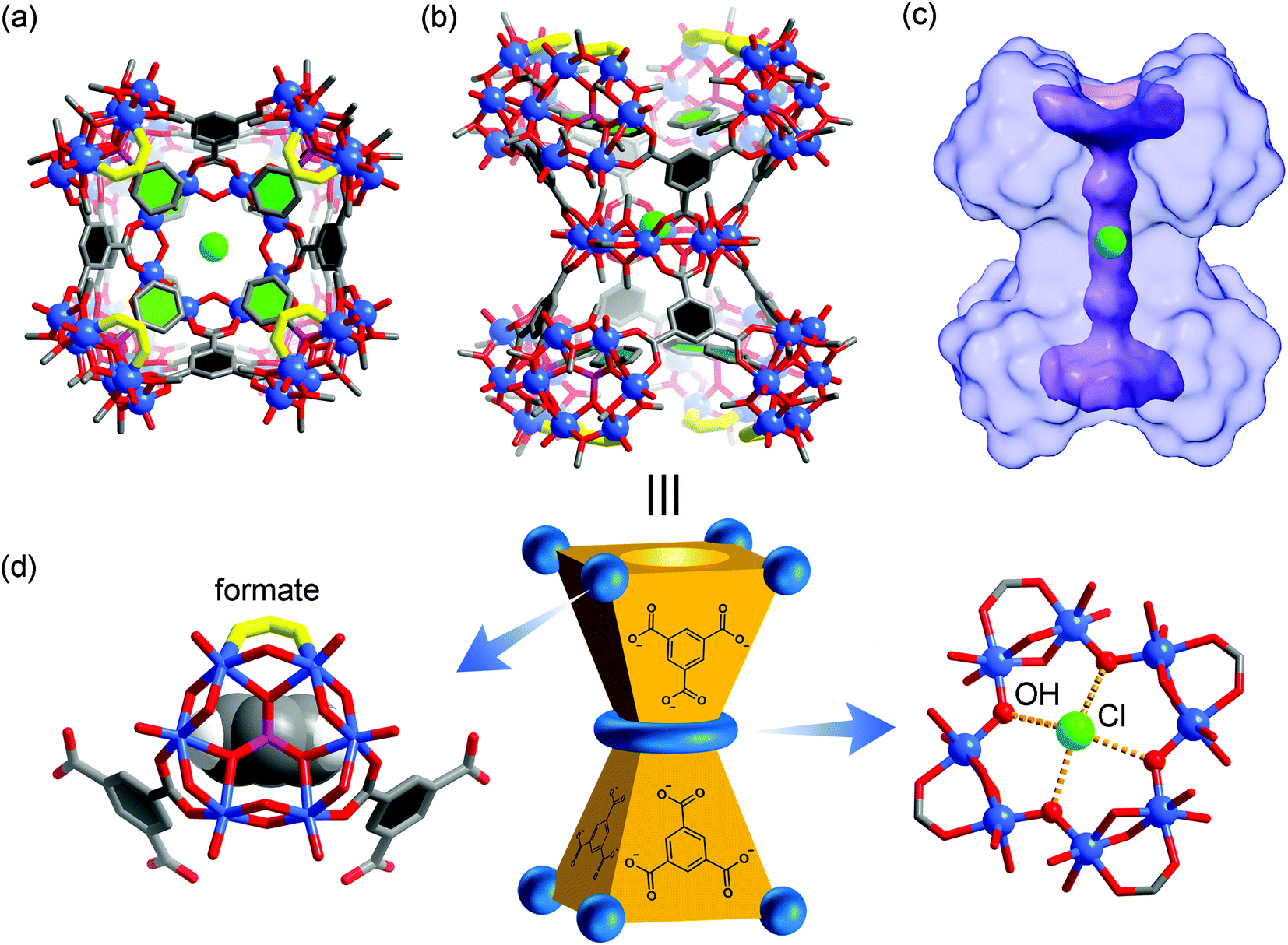 | ||
| Fig. 4 Top (a) and side (b) views of the X-ray crystal structure of 3, with hydrogen atoms omitted for clarity. The color code is the same as that of Fig. 1. The eight formate groups on the {V6} SBUs are highlighted with yellow bonds. The benzene rings in BTC and phenylphosphonates are colored black and green, respectively, for better structural visualization. The green sphere at the center is a chloride ion; (c) solvent-excluded surface of 3 with the central, ion-channel-like void space shown in purple where the Cl− guest resides; (d) schematic representation of 3 where the carboxylate binding around a {V6} SBU (methyl groups not drawn) is shown on the left and the equatorial {V8} ring on the right. | ||
First of all, the whole assembly of 3 does not enclose a discrete, interior void as for a polyhedral cage. Instead it features an open, barrel-shaped structure with a central channel (Fig. 4c) that is reminiscent of the ion channel proteins.62,63 The narrow passage, ca. 1.2 nm in length, is along the four-fold principal axis and gated by the four phenyl units from phenylphosphonates at each end of the structure. Complex 3 thus is the first non-cage structure isolated under the reaction conditions that have otherwise favored the MOP cages.
Second, the steric effect is likely the driving force for the cage transformation. In 3, the four {V6} clusters in the same half of the assembly are held together by four BTC and form a bowl-shaped motif so as to avoid steric repulsion between the phenylphosphonate groups. The average distance between adjacent {V6} SBUs, as measured from their central atoms, increases from 8.8 Å (S⋯S) in VMOP-14 to 10.6 Å (P⋯P) in 3. For each {V6} cluster, two of its extension points are guided by bridging carboxylates from BTC linkers while the third position is terminated by a formate ligand (Fig. 4d). The formate ligands, not seen in reported MOP structures,36–50 likely come from the oxidation of MeOH solvent molecules under hydrothermal conditions.64 Significantly smaller than BTC, the formates are located around the portals, leaving the central channel accessible from both ends of the structure.
Third, the observation of the equatorial {V8} ring suggests that there are likely many other equilibrating building blocks in solution aside from the tritopic, tetratopic, and pentatopic SBUs reported so far.36–50 Unlike V(IV) centers in all {V6} clusters, the eight V sites of the {V8} ring are all in the III+ oxidation state, as indicated by BVS calculations (BVS = 3.074). This oxidation state assignment was further confirmed by X-ray photoelectron spectroscopy measurements, where the integral area ratio for V4+(2p3/2) and V3+(2p3/2) is 6.14, close to their ratio (48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8) in the crystal structure of 3 (Fig. S12, ESI†). The V(III) centers, alternately sharing corners and edges through four μ-OH and eight methoxy groups, are also bridged by four peripheral formate ligands (Fig. 4d). Again, the formate motifs are a distinct feature of 3 and essential for the formation of this unique assembly. In the 1H NMR spectrum of 3 (Fig. 5, base-digested sample), the formate peak shows up at 8.23 ppm, in addition to those of the BTC (8.18 ppm) and phenylphosphonate (7.47 and 7.18 ppm) ligands.
:8) in the crystal structure of 3 (Fig. S12, ESI†). The V(III) centers, alternately sharing corners and edges through four μ-OH and eight methoxy groups, are also bridged by four peripheral formate ligands (Fig. 4d). Again, the formate motifs are a distinct feature of 3 and essential for the formation of this unique assembly. In the 1H NMR spectrum of 3 (Fig. 5, base-digested sample), the formate peak shows up at 8.23 ppm, in addition to those of the BTC (8.18 ppm) and phenylphosphonate (7.47 and 7.18 ppm) ligands.
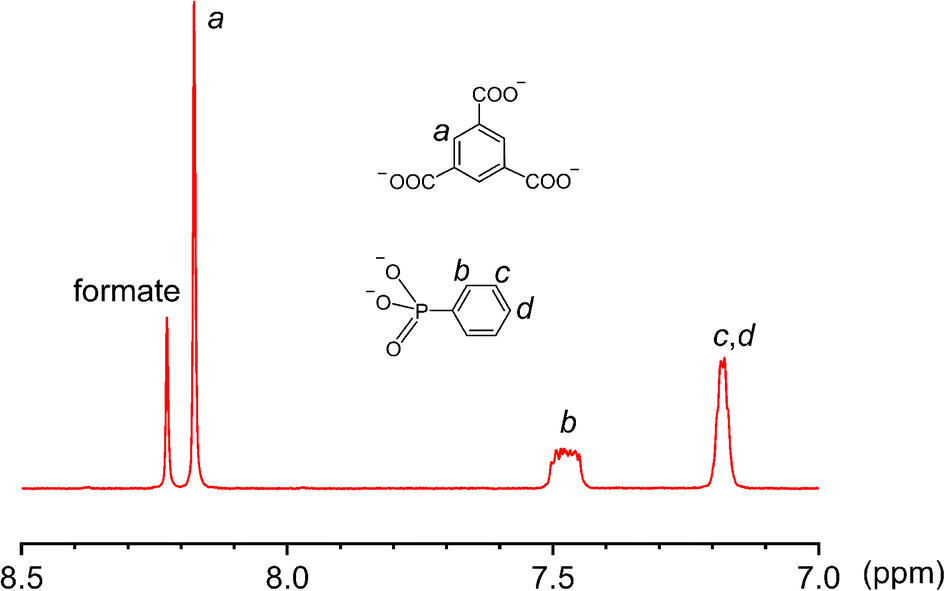 | ||
| Fig. 5 The 1H NMR spectrum of a base-digested sample of 3 (400 MHz, NaOD/D2O, 293 K). | ||
Finally, in its central channel, complex 3 is found to host a single Cl− ion, which is disordered over two equivalent positions related by a crystallographic inversion center. The Cl− ion is held in place by four O–H⋯Cl hydrogen bonds (O⋯Cl distances: 3.03–3.06 Å) with inward-pointing hydroxyl groups from the {V8} ring (Fig. 4d), and perched 0.7 Å above its equatorial plane. The inclusion of a small anion like Cl− and the hydrogen bonding interactions with its environment are similar to those observed in chloride channels,62,63 suggesting that it might be used as a synthetic ion channel,65 for which the work is ongoing.
As we expected, the bulkier biphenyl and terphenyl phosphonates were not able to fit inside a BTC-based, face-directed tetrahedron (Scheme 1). In addition, neither could generate an analogous architecture like 3, presumably also due to steric effects. Thus, to form a non-cage structure, one has to strike a right balance between the size of functional groups and the length of bridging carboxylate ligands.
Conclusions
In summary, we have introduced a facile coordination method to install organophosphonates at the interior cavity of MOPs where the metal nodes are polyoxometalate clusters. Given that the direct functionalization/post-functionalization of polyoxometalates has been well established,66–74 we anticipate that these strategies and procedures can also be applied to the functionalization of POM-based metal–organic cage complexes. This would thus allow a wider variety of functional groups beyond the current work to be incorporated and, as a result, impart unique properties and utilities (e.g. catalysis, drug delivery, guest recognition, etc.) relative to those of unfunctionalized cages. On the other hand, attempting to place oversized functional groups in the confined interior space of POM-based MOPs can disrupt the self-assembly processes, which have so far invariably given rise to MOP cages.36–50 The steric effects may prevent a MOP from being assembled, or suppress the formation of small MOPs and promote larger ones when they are all geometrically allowed. If carefully controlled, it can also allow metal–organic systems with structures/properties distinct from those of molecular cages to be constructed. In this regard, it will take effort to not build a cage.Experimental section
Materials
All reagents were from Innochem (Beijing), TCI Co., Sigma-Aldrich, Acros, and Fisher Chemical, and used without further purification. The synthesis of 3,3′,5,5′-azobenzenetetracarboxylic acid (H4ABTC) was conducted following a reported procedure.75 Biphenyl-4-phosphonic acid and p-terphenyl-4-phosphonic acid were prepared by the Michaelis–Arbuzov reaction with slight modification.76Biphenyl-4-phosphonic acid
4-Bromobiphenyl (4.66 g, 20 mmol) and 1,3-diisopropylbenzene (50 mL, 260 mmol) were added into a flask. After the suspension was degassed three times with argon, NiBr2 (0.50 g, 2.3 mmol) was added as a catalyst and the mixture was heated to 180 °C. Triethyl phosphite (5 mL, 29 mmol) was then added dropwise into the flask, and the solution was heated at this temperature for 24 h. The resulting mixture was cooled to room temperature and the faint yellow supernatant was collected. After the solvent and unreacted triethylphosphite were removed under reduced pressure, the ester was converted to phosphonic acid by refluxing in 6 M HCl (100 mL). The acid product (white solid) was washed with water and dried before use. Yield 3.2 g (68%). Elemental analysis, calcd: C, 61.5%; H, 4.7%; P, 13.2%; found: C, 60.3%; H, 4.9%; P, 12.7%. IR (2% KBr pellet, 4000–400 cm−1): 2955 (br), 2263 (br), 1602 (s), 1553 (w), 1479 (s), 1444 (w), 1389 (s), 1151 (vs), 1110 (s), 979 (vs), 976 (vs), 832 (w), 765 (vs), 724 (w), 697 (w), 665 (s), 557 (vs), 500 (vs), 457 (w), and 408 (w). 1H NMR (400 MHz, 1.0 M D2O/NaOD, δ): 7.70–7.50 (m, 6H), 7.40–7.32 (t, 2H), and 7.30–7.23 (t, 1H); 31P NMR (400 MHz, 1.0 M D2O/NaOD, δ): 11.09 (s).p-Terphenyl-4-phosphonic acid
4-Bromo-p-terphenyl (6.31 g, 20 mmol) and 1,3-diisopropylbenzene (50 mL, 260 mmol) were added into a flask. After the suspension was degassed three times with argon, NiBr2 (0.50 g, 2.3 mmol) was added as a catalyst and the mixture was heated to 180 °C. Triethyl phosphite (5 mL, 29 mmol) was then added dropwise into the flask, and the solution was heated at this temperature for 48 h. The resulting mixture was cooled to room temperature and the faint yellow supernatant was collected. After the solvent and unreacted triethylphosphite were removed under reduced pressure, the ester was converted to phosphonic acid by refluxing in 6 M HCl (100 mL). The acid product (white solid) was washed with water and dried before use. Yield 1.2 g (19%). Elemental analysis, calcd: C, 69.7%; H, 4.9%; P, 10.0%; found: C, 68.3%; H, 5.2%; P, 9.7%. IR (2% KBr pellet, 4000–400 cm−1): 2921 (br), 2344 (br), 1600 (w), 1483 (w), 1396 (w), 1149 (s), 1026 (vs), 945 (s), 821 (s), 762 (s), 729 (w), 689 (w), 649 (w), 578 (w), 538 (w), and 480 (w). 1H NMR (400 MHz, 1.0 M D2O/NaOD, δ): 7.70–7.52 (m, 10H), 7.41–7.34 (t, 2H), and 7.31–7.25 (t, 1H); 31P NMR (400 MHz, 1.0 M D2O/NaOD, δ): 11.05 (s).[NMe4]16{[(V6O6)(OMe)9(C6H5PO3)]8(ABTC)6}·12MeOH·12DMF (1a)
VOSO4·5H2O (60 mg, 0.24 mmol), H4ABTC (30 mg, 0.08 mmol), and phenylphosphonic acid (10 mg, 0.06 mmol) were suspended in a mixture of DMF:MeOH (1:2 mL); then the mixture was placed in a Parr Teflon-lined stainless steel vessel and heated at 150 °C. After 3 days, orange prismatic crystals were obtained by filtration (yield: 20 mg, 35.5% based on V). Elemental analysis, calcd: C, 35.0%; H, 5.5%; N, 4.9%; P, 2.2%; V, 21.7%; found: C, 34.5%; H, 5.2%; N, 5.0%; P, 2.1%; V, 22.3%. IR (2% KBr pellet, 4000–400 cm−1): 3443 (br), 3032 (w), 2922 (w), 2889 (w), 2814 (w), 1671 (w), 1616 (s), 1577 (s), 1488 (w), 1454 (s), 1387 (s), 1255 (w), 1228 (w), 1133 (w), 1068 (s), 945 (vs), 793 (w), 775 (sh), 724 (sh), 690 (w), 557 (s), 528 (s), and 499 (w).
[NMe4]16{[(V6O6)(OMe)9(C12H9PO3)]8(ABTC)6}·32MeOH·3DMF (1b)
VOSO4·5H2O (60 mg, 0.24 mmol), H4ABTC (30 mg, 0.08 mmol) and biphenyl-4-phosphonic acid (10 mg, 0.04 mmol) were suspended in a mixture of DMF:MeOH (1:2 mL); then the mixture was placed in a Parr Teflon-lined stainless steel vessel and heated at 150 °C. After 3 days, orange prismatic crystals were obtained by filtration (yield 22 mg, 37.1% based on V). Elemental analysis, calcd: C, 37.4%; H, 5.7%; N, 3.7%; P, 2.1%; V, 20.6%; found: C, 36.9%; H, 5.8%; N, 3.3%; P, 2.5%; V, 21.2%. IR (2% KBr pellet, 4000–400 cm−1): 3428 (br), 3028 (w), 2926 (w), 2816 (w), 1614 (s), 1573 (s), 1487 (w), 1453 (s), 1386 (s), 1253 (w), 1229 (w), 1065 (s), 946 (vs), 835 (w), 792 (w), 775 (w), 759 (w), 724 (w), 689 (w), 667 (w), 571 (s), and 545 (s).
[NMe4]8{[(V6O6)(OMe)9(C6H5PO3)]4(BDC)6}·7MeOH·3DMF (2)
VCl3 (30 mg, 0.19 mmol), terephthalic acid (H2BDC) (10 mg, 0.06 mmol) and phenylphosphonic acid (10 mg, 0.06 mmol) were suspended in a mixture of DMF:MeOH (1:2 mL); then the mixture was placed in a Parr Teflon-lined stainless steel vessel and heated at 150 °C. After 3 days, greenish yellow prismatic crystals were obtained by filtration (yield 19 mg, 44.7% based on V). Elemental analysis, calcd: C, 34.9%; H, 5.6%; N, 2.9%; P, 2.3%; V, 22.8%; found: C, 35.1%; H, 5.9%; N, 3.2%; P, 1.9%; V, 23.5%. IR (2% KBr pellet, 4000–400 cm−1): 3446 (br), 2925 (w), 2816 (w), 1670 (w), 1582 (s), 1505 (w), 1488 (s), 1436 (w), 1400 (s), 1131 (w), 1070 (s), 1003 (w), 986 (w), 948 (s), 827 (w), 746 (w), 716 (w), 699 (w), 563 (s), and 535 (s).
[NMe4]17{[(V6O6)(OMe)9(C6H5PO3)(HCOO)]8(BTC)8[V8(OH)4(OMe)8(HCOO)4]Cl}·11MeOH·6DMF (3)
VCl3 (100 mg, 0.63 mmol), 1,3,5-benzenetricarboxylic acid (H3BTC) (20 mg, 0.10 mmol) and phenylphosphonic acid (15 mg, 0.09 mmol) were suspended in a mixture of DMF:MeOH (1:2 mL); then the mixture was placed in a Parr Teflon-lined stainless steel vessel and heated at 150 °C. After 3 days, green plate crystals were obtained by filtration (yield 12 mg, 9.1% based on V). Elemental analysis, calcd: C, 31.7%; H, 5.3%; N, 2.8%; P, 2.1%; V, 24.4%; found: C, 32.3%; H, 5.4%; N, 3.1%; P, 2.4%; V, 23.9%. IR (2% KBr pellet, 4000–400 cm−1): 3442 (br), 2924 (w), 2815 (w), 1715 (w), 1618 (s), 1572 (w), 1445 (w), 1382 (s), 1137 (w), 1060 (w), 948 (vs), 753 (w),721 (w), and 562 (s).
General statement on hydrothermal reaction conditions
The reaction temperature for all hydrothermal reactions was set at 150 °C; this temperature range (130–150 °C) seems to be optimal for the formation of hexavandate-based MOPs, as reported in previous literature.36–50 The reaction temperature being too low or too high would affect the yield or result in no product formation. The volume of solution, similarly, could also affect the yield but not the nature of the products, if other parameters were kept the same. Increasing the ratio of phosphonate/carboxylate ligands from what was specified above in the Experimental section, however, did not seem to affect the product yield considerably.Author contributions
J. G. and X. F. conceived the project, designed and supervised the study. J. G., Q. C., Z. L., Y. W., C. L., M. W. and D. H. synthesized the samples, carried out the single-crystal X-ray diffraction analysis and supporting synthetic/characterization work, including thermogravimetric analysis, IR and UV. J. G., W. W. and G. C. led efforts associated with NMR and XPS measurements. H. Z. performed the DFT calculations. J. G., Q. C., W. W. and X. F. wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (grant no. 21671048, 51602310, and 51972084) and the Fundamental Research Fund for the Central Universities, China (AUGA5710094420). We thank Prof. Debin Xia for helpful discussions.Notes and references
- E. C. Theil, Annu. Rev. Biochem., 1987, 56, 289–316 CrossRef CAS PubMed.
- X. Liu and E. C. Theil, Acc. Chem. Res., 2005, 38, 167–175 CrossRef CAS PubMed.
- G. Jutz, P. van Rijn, B. S. Miranda and A. Böker, Chem. Rev., 2015, 115, 1653–1701 CrossRef CAS PubMed.
- K. M. Reinisch, M. L. Nibert and S. C. Harrison, Nature, 2000, 404, 960–967 CrossRef CAS PubMed.
- W. Chiu, R. M. Burnett and R. L. Garcea, Structural Biology of Viruses, Oxford University Press, 1997 Search PubMed.
- J. M. Berg, J. L. Tymoczko, G. J. Gatto and L. Stryer Jr, Biochemistry, W. H. Freeman and Company, 8th edn, 2015, ch. 8 Search PubMed.
- Themed issue on molecular containers: P. Ballester, M. Fujita and J. Rebek Jr, Chem. Soc. Rev., 2015, 44, 392–499 RSC.
- P. M. Bogie, T. F. Miller and R. J. Hooley, Isr. J. Chem., 2019, 59, 130–139 CrossRef CAS.
- D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS.
- M. Fujita, K. Umemoto, M. Yoshizawa, N. Fujita, T. Kusukawa and K. Biradha, Chem. Commun., 2001, 509–518 RSC.
- D. J. Tranchemontagne, Z. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136–5147 CrossRef CAS PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC.
- A. J. Gosselin, C. A. Rowland and E. D. Bloch, Chem. Rev., 2020, 120, 8987–9014 CrossRef CAS PubMed.
- S. Lee, H. Jeong, D. Nam, M. S. Lah and W. Choe, Chem. Soc. Rev., 2021, 50, 528–555 RSC.
- M. L. Merlau, M. del P. Mejia, S. T. Nguyen and J. T. Hupp, Angew. Chem., Int. Ed., 2001, 40, 4239–4242 CrossRef CAS.
- C. G. Oliveri, N. C. Gianneschi, S. T. Nguyen, C. A. Mirkin, C. L. Stern, Z. Wawrzak and M. Pink, J. Am. Chem. Soc., 2006, 128, 16286–16296 CrossRef CAS PubMed.
- C. García-Simón, R. Gramage-Doria, S. Raoufmoghaddam, T. Parella, M. Costas, X. Ribas and J. N. H. Reek, J. Am. Chem. Soc., 2015, 137, 2680–2687 CrossRef PubMed.
- S. S. Nurttila, W. Brenner, J. Mosquera, K. M. van Vliet, J. R. Nitschke and J. N. H. Reek, Chem.–Eur. J., 2019, 25, 609–620 CAS.
- F. J. Rizzuto, W. J. Ramsay and J. R. Nitschke, J. Am. Chem. Soc., 2018, 140, 11502–11509 CrossRef CAS PubMed.
- K. Suzuki, J. Iida, S. Sato, M. Kawano and M. Fujita, Angew. Chem., Int. Ed., 2008, 47, 5780–5782 CrossRef CAS PubMed.
- S. Sato, J. Iida, K. Suzuki, M. Kawano, T. Ozeki and M. Fujita, Science, 2006, 313, 1273–1276 CrossRef CAS.
- K. Suzuki, K. Takao, S. Sato and M. Fujita, J. Am. Chem. Soc., 2010, 132, 2544–2545 CrossRef CAS PubMed.
- Q.-F. Sun, S. Sato and M. Fujita, Chem. Lett., 2011, 40, 726–727 CrossRef CAS.
- D. Fujita, A. Takahashi, S. Sato and M. Fujita, J. Am. Chem. Soc., 2011, 133, 13317–13319 CrossRef CAS PubMed.
- D. Fujita, K. Suzuki, S. Sato, M. Yagi-Utsumi, Y. Yamaguchi, N. Mizuno, T. Kumasaka, M. Takata, M. Noda, S. Uchiyama, K. Kato and M. Fujita, Nat. Commun., 2012, 3, 1093–1099 CrossRef PubMed.
- K. Harris, Q.-F. Sun, S. Sato and M. Fujita, J. Am. Chem. Soc., 2013, 135, 12497–12499 CrossRef CAS PubMed.
- K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC.
- J. L. Bolliger, A. M. Belenguer and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 7958–7962 CrossRef CAS PubMed.
- M. C. Young, A. M. Johnson, A. S. Gamboa and R. J. Hooley, Chem. Commun., 2013, 49, 1627–1629 RSC.
- S. Löffler, J. Lübben, A. Wuttke, R. A. Mata, M. John, B. Dittrich and G. H. Clever, Chem. Sci., 2016, 7, 4676–4684 RSC.
- M. Krick, J. Holstein, C. Würtele and G. H. Clever, Chem. Commun., 2016, 52, 10411–10414 RSC.
- P. M. Bogie, L. R. Holloway, C. Ngai, T. F. Miller, D. K. Grewal and R. J. Hooley, Chem.–Eur. J., 2019, 25, 10232–10238 CrossRef CAS PubMed.
- X. Yan, P. Wei, Y. Liu, M. Wang, C. Chen, J. Zhao, G. Li, M. L. Saha, Z. Zhou, Z. An, X. Li and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 9673–9679 CrossRef CAS PubMed.
- Y.-T. Zhang, X.-L. Wang, S.-B. Li, Y.-R. Gong, B.-Q. Song, K.-Z. Shao and Z.-M. Su, Chem. Commun., 2016, 52, 9632–9635 RSC.
- Y.-T. Zhang, S.-B. Li, X.-L. Wang, Y.-R. Gong, K.-Z. Shao and Z.-M. Su, Dalton Trans., 2016, 45, 14898–14901 RSC.
- Y.-R. Gong, W.-C. Chen, L. Zhao, K.-Z. Shao, X.-L. Wang and Z.-M. Su, Dalton Trans., 2018, 47, 12979–12983 RSC.
- Y. Gong, Y. Zhang, C. Qin, C. Sun, X. Wang and Z. Su, Angew. Chem., Int. Ed., 2019, 58, 780–784 CrossRef CAS PubMed.
- Y. Gong, C. Qin, Y. Zhang, C. Sun, Q. Pan, X. Wang and Z. Su, Angew. Chem., Int. Ed., 2020, 59, 22034–22038 CrossRef CAS PubMed.
- Z. Zhang, L. Wojtas and M. J. Zaworotko, Chem. Sci., 2014, 5, 927–931 RSC.
- Z. Zhang, W.-Y. Gao, L. Wojtas, Z. Zhang and M. J. Zaworotko, Chem. Commun., 2015, 51, 9223–9226 RSC.
- Y.-T. Zhang, X.-L. Wang, E.-L. Zhou, X.-S. Wu, B.-Q. Song, K.-Z. Shao and Z.-M. Su, Dalton Trans., 2016, 45, 3698–3701 RSC.
- Y. Zhang, X. Wang, S. Li, B. Song, K. Shao and Z. Su, Inorg. Chem., 2016, 55, 8770–8775 CrossRef CAS PubMed.
- Y. Tao, N. Xu, X. Wang and Z. Su, Isr. J. Chem., 2019, 59, 306–310 CrossRef CAS.
- Y. Zheng, H. Gan, Y. Zhao, W. Li, Y. Wu, X. Yan, Y. Wang, J. Li, J. Li and X. Wang, Chem.–Eur. J., 2019, 25, 15326–15332 CrossRef CAS PubMed.
- Y. Zhang, H. Gan, C. Qin, X. Wang, Z. Su and M. J. Zaworotko, J. Am. Chem. Soc., 2018, 140, 17365–17368 CrossRef CAS PubMed.
- Y. Gong, Y. Tao, N. Xu, C. Sun, X. Wang and Z. Su, Chem. Commun., 2019, 55, 10701–10704 RSC.
- N. Xu, H. Gan, C. Qin, X. Wang and Z. Su, Angew. Chem., Int. Ed., 2019, 58, 4649–4653 CrossRef CAS PubMed.
- H. Gan, N. Xu, C. Qin, C. Sun, X. Wang and Z. Su, Nat. Commun., 2020, 11, 4103 CrossRef CAS PubMed.
- The size of the void space was calculated by using the 3 V web-based cavity calculator, with the radius of the small probe set at 1.4 Å (that of the water molecule) and 10 Å for that of the large probe; N. R. Voss and M. Gerstein, Nucleic Acids Res., 2010, 38, W555–W562 CrossRef CAS PubMed.
- M. I. Khan and J. Zubieta, Angew. Chem., Int. Ed., 1994, 33, 760–762 CrossRef.
- A. Müller, K. Hovemeier, E. Krickemeyer and H. Bögge, Angew. Chem., Int. Ed., 1995, 34, 779–781 CrossRef.
- Y. D. Chang, J. Salta and J. Zubieta, Angew. Chem., Int. Ed., 1994, 33, 325–327 CrossRef.
- M. I. Khan, Y. Chang, Q. Chen, H. Hope, S. Parking, D. P. Goshorn and J. Zubieta, Angew. Chem., Int. Ed., 1992, 31, 1197–1200 CrossRef.
- S. Konar and A. Clearfield, Inorg. Chem., 2008, 47, 3492–3494 CrossRef CAS PubMed.
- J. M. Breen and W. Schmitt, Angew. Chem., Int. Ed., 2008, 47, 6904–6908 CrossRef CAS PubMed.
- M. B. Mahimaidoss, S. A. Krasnikov, L. Reck, C. I. Onet, J. M. Breen, N. Zhu, B. Marzec, I. V. Shvets and W. Schmitt, Chem. Commun., 2014, 50, 2265–2267 RSC.
- A. Banerjee, B. S. Bassil, G.-V. Röschenthaler and U. Kortz, Chem. Soc. Rev., 2012, 41, 7590–7604 RSC.
- Organophosphonic acids (RPO3H2) possess two acidic protons featuring the first pKa ranging from 1.1 to 2.9 and the second pKa from 5.3 to 7.2 when R is an aromatic group, see: R. G. Franz, AAPS PharmSci, 2001, 3, 1–13 CrossRef PubMed.
- C. M. Sevrain, M. Berchel, H. Couthon and P.-A. Jaffrès, Beilstein J. Org. Chem., 2017, 13, 2186–2213 CrossRef CAS PubMed.
- R. Dutzler, E. B. Campbell, M. Cadene, B. T. Chait and R. MacKinnon, Nature, 2002, 415, 287–294 CrossRef CAS PubMed.
- R. Dutzler, FEBS Lett., 2004, 564, 229–233 CrossRef CAS PubMed.
- V. V. Kaichev, G. Y. Popova, Y. A. Chesalov, A. A. Saraev, D. Y. Zemlyanov, S. A. Beloshapkin, A. Knop-Gericke, R. Schlögl, T. V. Andrushkevich and V. I. Bukhtiyarov, J. Catal., 2014, 311, 59–70 CrossRef CAS.
- S.-P. Zheng, L.-B. Huang, Z. Sun and M. Barboiu, Angew. Chem., Int. Ed., 2021, 60, 566–597 CrossRef CAS PubMed.
- C. L. Hill, Themed issue on polyoxometalates, Chem. Rev., 1998, 98, 1 CrossRef CAS PubMed.
- A. Proust, R. Thouvenot and P. Gouzerh, Chem. Commun., 2008, 1837–1852 RSC.
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009–6048 CrossRef CAS PubMed.
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605–7622 RSC.
- M. Nyman and P. C. Burns, Chem. Soc. Rev., 2012, 41, 7354–7367 RSC.
- D.-Y. Du, J.-S. Qin, S.-L. Li, Z.-M. Su and Y.-Q. Lan, Chem. Soc. Rev., 2014, 43, 4615–4632 RSC.
- J. Zhang, Y. Huang, G. Li and Y. Wei, Coord. Chem. Rev., 2019, 378, 395–414 CrossRef CAS.
- A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382–432 RSC.
- Z.-K. Zhu, Y.-Y. Lin, H. Yu, X.-X. Li and S.-T. Zheng, Angew. Chem., Int. Ed., 2019, 58, 16864–16868 CrossRef CAS PubMed.
- S. R. Miller, E. Alvarez, L. Fradcourt, T. Devic, S. Wuttke, P. S. Wheatley, N. Steunou, C. Bonhomme, C. Gervais, D. Laurencin, R. E. Morris, A. Vimont, M. Daturi, P. Horcajada and C. Serre, Chem. Commun., 2013, 49, 7773–7775 RSC.
- R. G. Harvey and E. R. de Sombre, Top. Phosphorus Chem., 1964, 1, 57–111 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Physical characterization and additional tables and figures. The DFT-optimized interior structure of 1b (XYZ file). CCDC 2054446–2054449. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01243f |
| This journal is © The Royal Society of Chemistry 2021 |