
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrometer-scale transient ion transport for real-time pH assay in living rat brains†
Kailin
Zhang
ac,
Huan
Wei
ac,
Tianyi
Xiong
ac,
Yanan
Jiang
a,
Wenjie
Ma
a,
Fei
Wu
a,
Ping
Yu
 *ac and
Lanqun
Mao
abc
*ac and
Lanqun
Mao
abc
aBeijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Analytical Chemistry for Living Biosystems, Institute of Chemistry, The Chinese Academy of Sciences (CAS), CAS Research/Education Center for Excellence in Molecular Science, Beijing 100190, China. E-mail: yuping@iccas.ac.cn
bCollege of Chemistry, Beijing Normal University, Beijing 100875, China
cCollege of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
First published on 19th April 2021
Abstract
Ion transport has been widely used for various applications such as sensing, desalination and energy conversion; however, nearly all applications are based on steady-state ion transport. Herein, we for the first time demonstrate the capability of transient ion transport for in vivo sensing with both high spatial (∼μm) and temporal (∼ms) resolution by using pH as the model target. Transient ion transport behavior (i.e., time-dependent ion current change) was observed by applying high-frequency pulse potential. Importantly, we proposed the ion distribution transient model for this time-dependent ion transport behavior. With this model, the temporal resolution of the as-developed pH microsensor based on ion current was improved to the ms level, thus satisfying the requirement of neurochemical recording. Moreover, our microsensor features good reproducibility, selectivity, and reversibility, and can thus real-time monitor the pH change in living rat brains. This study demonstrates the first example of in vivo sensing based on ion transport, opening a new way to neurochemical monitoring with ultrahigh spatiotemporal resolution. This study is also helpful to understand the transient process of asymmetric ion transport.
Introduction
Development of new sensing principles and techniques for in vivo monitoring of molecules in the central nervous system has attracted ever increasing interest because of the important but unrevealed roles of molecular events in brain activity.1–3 However, the complexity of the brain, such as the diversity and spatial distribution as well as the dynamics of chemical species, poses great challenges to conventional methods for in vivo monitoring of neurochemicals.4,5 In this case, the methods should simultaneously satisfy the requirements of selectivity, sensitivity, temporal resolution (∼ms, the rapid release of neurotransmitters) and spatial resolution (∼μm, the precise measurement of the extracellular environment).6 So far, microprobe-based methods employing optical fibers and carbon fiber electrodes have led to rapidly increasing activity in this research area.7–10 For example, the introduction of carbon fiber electrodes into brain chemistry has boosted the research on real-time monitoring of neurochemicals since 1970s due to their high spatiotemporal resolution and good designability.11,12 However, monitoring of electroinactive species remains a great challenge because the poor electroactivity of the neurochemicals essentially makes them very difficult to oxidize or reduce at carbon fiber electrodes. In this case, the introduction of new sensing tools with high spatiotemporal resolution is highly desirable for understanding chemistry-related physiological and pathological processes.Recently, ion transport at the micro/nanoscale has been of great interest in both fundamental and applied research areas.13–21 Previous attempts have revealed that sensors based on ion transport in confined nanochannels can bring new opportunities for sensing, especially for sensing electroinactive molecules, by virtue of the resistance-pulse technique or ion current rectification behavior.22–26 For example, Pourmand’s group pioneered in the biosensor application of nanopipette technology, which exhibits great potential for in situ intracellular chemical sensing.27–33 Intracellular H2O2 sensing has also been reported by using a DNAzyme-modified nanopipette.34 However, these principles and methods cannot be used for in vivo analysis due to the limitation in spatial and temporal resolution. This is because the nanoscale tip of a pipette is not suitable for extracellular neurochemical analysis in vivo, even though it easily penetrates into the cells for intracellular chemical analysis. Moreover, the principles for most pipette-based sensors reported so far are based on steady-state ion transport (i.e., ion current rectification), bearing poor temporal resolution, which makes it difficult to apply these sensors in in vivo monitoring of the fast dynamics of neurochemicals in the brain.
Recently, we have successfully observed micrometer-scale asymmetric ion transport (i.e., micrometer-scale ion current rectification, MICR) in a polyelectrolyte brush-modified micropipette, which lays the foundation to develop microsensors based on asymmetric ion transport on the microscale.15,35,36 Moreover, the micropipette bears a robust tip for tissue-implantable sensing of extracellular species due to its size matching with that of neurons. Herein we demonstrated the applicability of micrometer-scale transient ion transport in in vivo analysis with high spatiotemporal resolution by using pH as the model target (Scheme 1). To sense pH, polyimidazole brushes were first attached onto the inner wall of micropipettes by the atom transfer radical polymerization reaction. The association/dissociation of protons on the imidazole with pH variation would increase/decrease the surface charge density, consequently resulting in ion current change for pH assay. Different from traditional methods based on ion current rectification, high-frequency square-wave pulse potential was used to controllably tune the transient ion transport behavior, which could largely increase the temporal resolution and stability of the microsensor (Fig. 1). The high temporal and spatial resolution as well as the good selectivity based on the association/dissociation of protons enable the microsensor as a new platform for in vivo pH sensing. To the best of our knowledge, this is the first report of an in vivo microsensor based on ion transport, which opens a new door for fabrication of various in vivo microsensors by rationally designing the surface chemistry of the inner wall. This study is also helpful to understand the transient process of asymmetric ion transport.
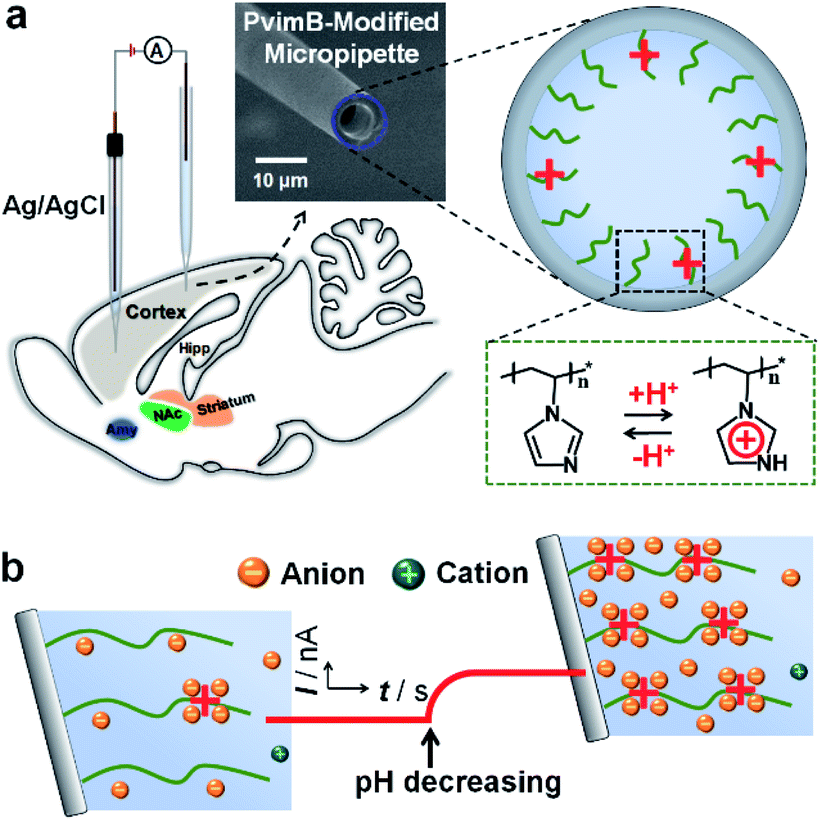 | ||
| Scheme 1 Schematic illustration of the setup (a) and the principle (b) for in vivo pH monitoring using a functionalized micropipette based on transient ion transport. | ||
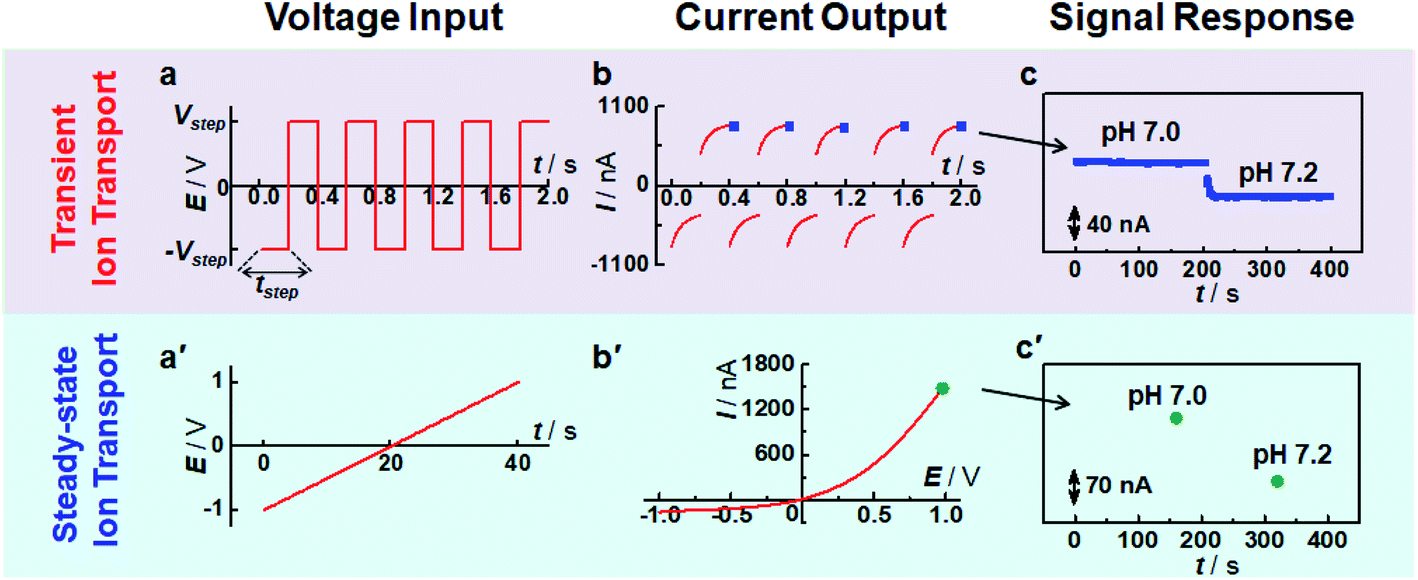 | ||
| Fig. 1 Comparison of sensing response time based on transient ion transport and steady-state ion transport. (a and a′) The waveform of the applied potential pulse. (b and b′) The recorded current trace. (c and c′) The output signal with the change of solution pH. | ||
Results and discussion
Fabrication and characterization of a poly(N-vinylimidazole) brush-modified micropipette
To demonstrate the possibility of using transient ion transport for in vivo analysis, poly(N-vinylimidazole) brushes (PvimB) were attached onto the inner surface of a micropipette by surface-initiated atom transfer radical polymerization (Fig. S1†) since imidazole bears excellent ability of protonation/deprotonation in the physiological pH range (Fig. S2†). Moreover, the recognition between imidazole and protons was fast, reversible and selective, which are all beneficial for constructing a high-performance in vivo microsensor.37 The successful modification with PvimB was confirmed by comparing the rectified current–voltage (I–V) curve of the PvimB-modified micropipette with that of a bare micropipette (Fig. S3†) in 50 mM phosphate buffer (pH 5.8) containing 100 mM NaCl. The result was consistent with our previous report on polyelectrolyte brushes since polyimidazole was positively charged in a solution of pH 5.8.35 Moreover, the rectification ratio increased with the decrease of solution pH (Fig. S4†), indicating that the PvimB-modified micropipette could be used for pH sensing based on ion current change.Current response at steady and transient states
To validate the use of transient ion transport in sensing applications, the current responses at transient and steady states were investigated. As shown in Fig. 1, compared with steady-state ion transport (i.e., the ion current is independent of time at a specific potential), high-frequency pulse potential results in a time-dependent ion current response. Moreover, this time-dependent ion current is strongly related to pulse frequency (Fig. S5†). Specifically, the ratio of end current at the positive potential and that at the negative potential decreases with the increase of pulse frequency. When an ultralow frequency pulse potential was applied (e.g., tstep = 20 s), the current response tends to be stable at the end of each potential pulse (Fig. S6a†). Moreover, the current values at the end point (i.e., 20 s and 40 s) are almost the same as those at the same potential recorded with the I–V curve (i.e., +1 V and −1 V in Fig. S6b†), indicating that a steady current response was obtained. This phenomenon was similar to the previous observation of scan-rate dependent ion current rectification.38,39 For a low-frequency potential pulse, there was enough time for the redistribution of freely moving ions, consequently achieving a “steady-state” response. In contrast, when a high-frequency potential pulse was applied, it was difficult for the current to reach a stable response since the ionic redistribution was relatively sluggish under a high-frequency voltage perturbation, resulting in a “transient” response. By using pulse potential as the polarization potential, the processes of ion accumulation and depletion could be obtained in real-time, which is not only beneficial to study the transient process of ion transport but also suitable for sensing devices, as demonstrated below.Fig. 1c and c′ show the current change at different pH values. Compared with the traditional sensing principle based on steady-state ion transport, the present strategy shows much higher time-resolution for target response. Moreover, the end current at each step potential decreased with the increase of solution pH because of the decrease of surface charge density. The current change shows a strong and sensitive dependence on solution pH (Fig. 1c). And the current response was stable at a specific pH, suggesting that this transient technique could be used for pH assay. To rule out the influence of solution conductivity, a poly(1-vinyl-3-butylimidazolium) brush (PvbimB)-modified micropipette was prepared (Fig. S7a†), and no current change was observed even when the pH changed from 5.8 to 7.0 (Fig. S7b,† black curve), further demonstrating that the current response originated from the surface charge density by the dissociation/association of imidazole protons rather than the conductivity variation of the bath solution with different pH values. These results essentially demonstrated that transient ion transport is expected to result in high sensing performance with high temporal resolution.
Model for transient ion transport
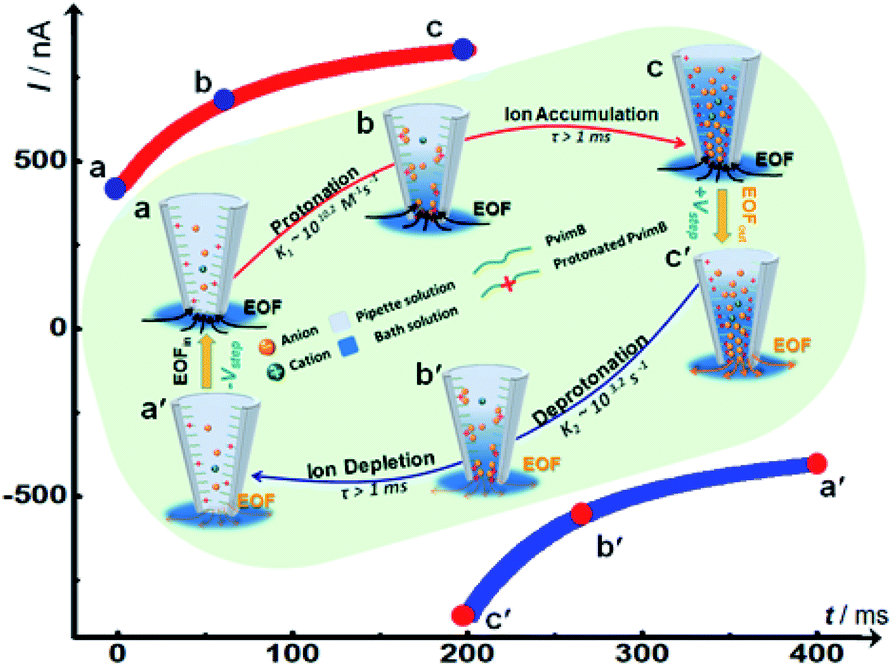
To better understand the time-dependent nature of the ion transport behavior observed in this study, a schematic transient model based on ion distribution is given in Scheme 2 to demonstrate the transient processes of ion transport under one potential pulse cycle. When a potential was applied across the micropipette, mainly three interacting factors influenced the ion distribution at the tip of the micropipette, that is, the rate of ion redistribution, the surface charge density, and the direction and velocity of the electroosmotic flow (EOF). As shown in Scheme 2a–c, when a positive potential was applied, the direction of the EOF was inward. In this case, the outer solution with low pH would instantaneously enter the tip of the micropipette, which would consequently increase the surface charge density by the protonation of imidazole moieties, resulting in the accumulation of anions, and vice versa (Scheme 2c′–a′). This is a feedback process between the surface charge density-modulated electrostatic field and the electrostatic field-modulated EOF, in which the surface charge density and EOF are interdependent on each other. In other words, the increase of surface charge density due to the protonation of polyimidazole would result in the increase of the inward EOF. Meanwhile, the increase of the EOF can generate higher surface charge density, which is a positive feedback process for the accumulation of anions at the tip of the micropipette. In this process, the rate of proton association (∼1010.2 s−1 M−1)/dissociation (∼103.2 s−1) is very fast, which would not become the rate-limiting step.40 The velocity of the EOF could be estimated using the Helmholtz–Smoluchowski equation:where Ex is the electric field through the micropipette and parallel to the surface, η is the viscosity of the fluid, and ε0 and εr are the permittivity of vacuum and dielectric constant of water, respectively. The zeta potentials (ζ) of PvimB-modified silica nanoparticles (SiNPs) at different pH values were used to estimate the zeta potentials of the micropipette surface (Fig. S2†). Ex adopted the value from finite element simulations at the point of z = 0, x = 0 (Fig. S8 and S9†). The velocity of the EOF was calculated to be 21.16 μm s−1 at 1.0 V bias potential and 8.78 μm s−1 at −0.4 V bias potential in pH 5.8 electrolyte. Moreover, the velocity of the EOF was also experimentally determined by using a home-made setup (Fig. S10†). The determined EOF velocity is shown in Table S1,† which is at the same level as the estimated value based on the Helmholtz–Smoluchowski equation.41 This result demonstrates the correlation between surface charge density and EOF velocity. Another important rate-limiting step is the ion redistribution, which is related to the mobility of ions. To estimate this rate, we conducted linear scan experiments at different scan rates (Fig. S11†). The rectification totally disappeared when the scan rate was increased to 2000 V s−1, indicating that the time for ion redistribution is longer than 1 ms (herein, the potential scan range was 2 V). All these results demonstrate that the surface charge density, the rate of ion redistribution as well as the direction and velocity of the EOF contribute to the final ion distribution at the tip of the micropipette, and consequently the ion current intensity. These processes are interdependent and time-dependent, resulting in a time-dependent current response as shown in Fig. 1b. When a low-frequency pulse potential was applied, all the processes had enough time to reach an equilibrium state, consequently resulting in a steady-state ion current. In contrast, when a high-frequency pulse potential was applied, the ion redistribution was insufficient due to the relatively slow mobility of ions, giving rise to a time-dependent current response. With this model, this time-dependent transient current response could be used for stable and reproducible in vivo analysis by rationally controlling the parameters of pulse potential, as demonstrated below.
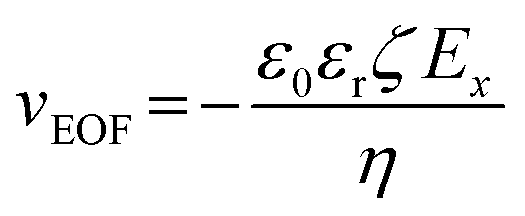
 | ||
| Scheme 2 Schematic illustration of the transient process and time-dependent ion current change based on the ion distribution model under high frequency pulse potential. | ||
Stability and sensitivity
We then investigated the stability of the signal output since stable sensing through the transient process is intuitively difficult. However, our pH microsensor showed a stable output over one hour, and the typical current fluctuation was less than 3 nA h−1 (Fig. 2a, red curve). We think that this stable response is due to to the renewed step induced by the negative potential as demonstrated in Scheme 2. The polarization under negative potential would result in the inner solution returning back to the initial state (Scheme 2c′–a′), essentially contributing to the stable output. In contrast, when the pipette was polarized at a constant positive potential, that is, without the polarization of a negative potential, the output signal shows a sluggish increase even for the same pH solution (Fig. 2a, black curve). This is reasonable because the protons continuously move into (or out of) the pipette tip in a single direction under a constant potential, making it difficult to form a stable ion distribution at the tip of the micropipette within a short time. This phenomenon would become more obvious when the tip size of the pipette is changed from the nanoscale to microscale. Moreover, the stability was strongly related to the step potential applied (Fig. S12†). When a larger negative step potential was applied, it was difficult to obtain a stable current response (Fig. S12a†). In contrast, a stable current response was obtained regardless of the pH of the backfilled solution when a smaller negative step potential was applied (Fig. S12b†). This is understandable as the EOF rectification occurs at the opposite potential as demonstrated above (Table S1†).41 When a larger negative potential was applied, the larger outward EOF would drive the inner solution to occupy the tip of the micropipette. In this case, the outer solution cannot totally enter the tip of the micropipette under the polarization of a smaller positive potential, consequently resulting in an unstable response. In contrast, when a smaller negative potential is applied, the larger inner EOF would drive the outer solution into the pipette, consequently resulting in a stable and sensitive response. This result suggests that our pH microsensor features good stability while maintaining high temporal resolution as a result of rationally tuning the parameters of pulse potential.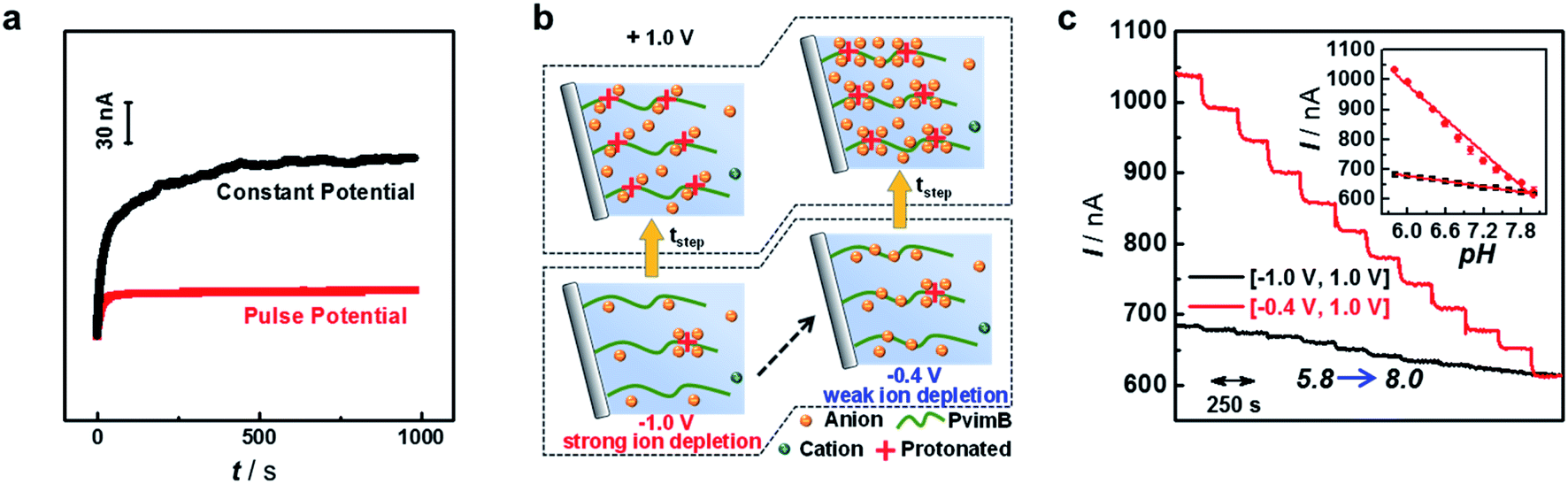 | ||
| Fig. 2 (a) Long-time response of the microsensor polarized by a constant potential (black curve) and pulse potential (red curve). (b) Schematic illustration of ion concentration changes under different pulse potentials. (c) Current traces and calibration curves (error bars were obtained for the same micropipette from three independent measurements) obtained for the PvimB-modified micropipette towards the successive pH variation from 5.8 to 8.0 with different −Vstep. The electrolyte was 100 mM NaCl in 50 mM phosphate buffer (pH 5.8). | ||
More interestingly, we found that the current response at the same positive step potential was different when the applied negative step potential was changed (Fig. S13†). Fig. 2c shows the current response towards successive pH change (pH 5.8 to 8.0) at +1 V when the applied negative step potential was −1 V (black curve) and −0.4 V (red curve). To well understand this phenomenon, the ion concentration distribution under different polarization potentials is given in Fig. 2b. As shown, a smaller negative potential (e.g., −0.4 V) would result in weak-ion depletion, which would result in strong-ion accumulation in the positive potential. In contrast, a larger negative potential (e.g., −1.0 V) features strong-ion depletion, further giving rise to weak-ion accumulation. These results essentially demonstrated that the sensitivity could also be increased without the loss of temporal resolution. All these attempts revealed that with the proposed ion distribution transient model, the sensing performance could be rationally modulated according to the requirement of specific targets, which lays the principal foundation for developing various microsensors based on transient ion transport. For in vivo sensing of pH in the present study, a pulse frequency of 2.5 Hz, negative step potential of −0.4 V and positive step potential of +1 V were used to simultaneously satisfy the requirements of in vivo analysis in terms of temporal resolution, sensitivity and stability.
In vivo pH assay
We next investigated the sensing performance of the as-prepared microsensor in a real-time pH assay. As shown in Fig. 3a, the microsensor shows good reproducibility, which was obtained with the same micropipette from three independent measurements. Some bioactive molecules and inorganic salt ions would concomitantly change during brain pH variation.42 Therefore, potential interferents were added to investigate the selectivity of the microsensor. As shown in Fig. 3b, the microsensor shows good selectivity towards the coexisting biological molecules. Moreover, the response of the microsensor toward pH in the range from 5.8 to 8.0 was reversible as shown in Fig. 3c. The as-prepared microsensor was well responsive to pH changes, and the current response change (Is) showed good linearity with pH ranging from 5.8 to 8.0 (Is (nA) = −326.9 pH + 3140.3, r2 = 0.978) (Fig. 3d). Moreover, we compared the performance of our microsensor to that of previously reported pH microsensors for in vivo analysis (Table S2†). As shown, our microsensor features high spatiotemporal resolution, good reversibility, good stability and high selectivity, substantially enabling its use in real-time monitoring of pH change in living rat brains.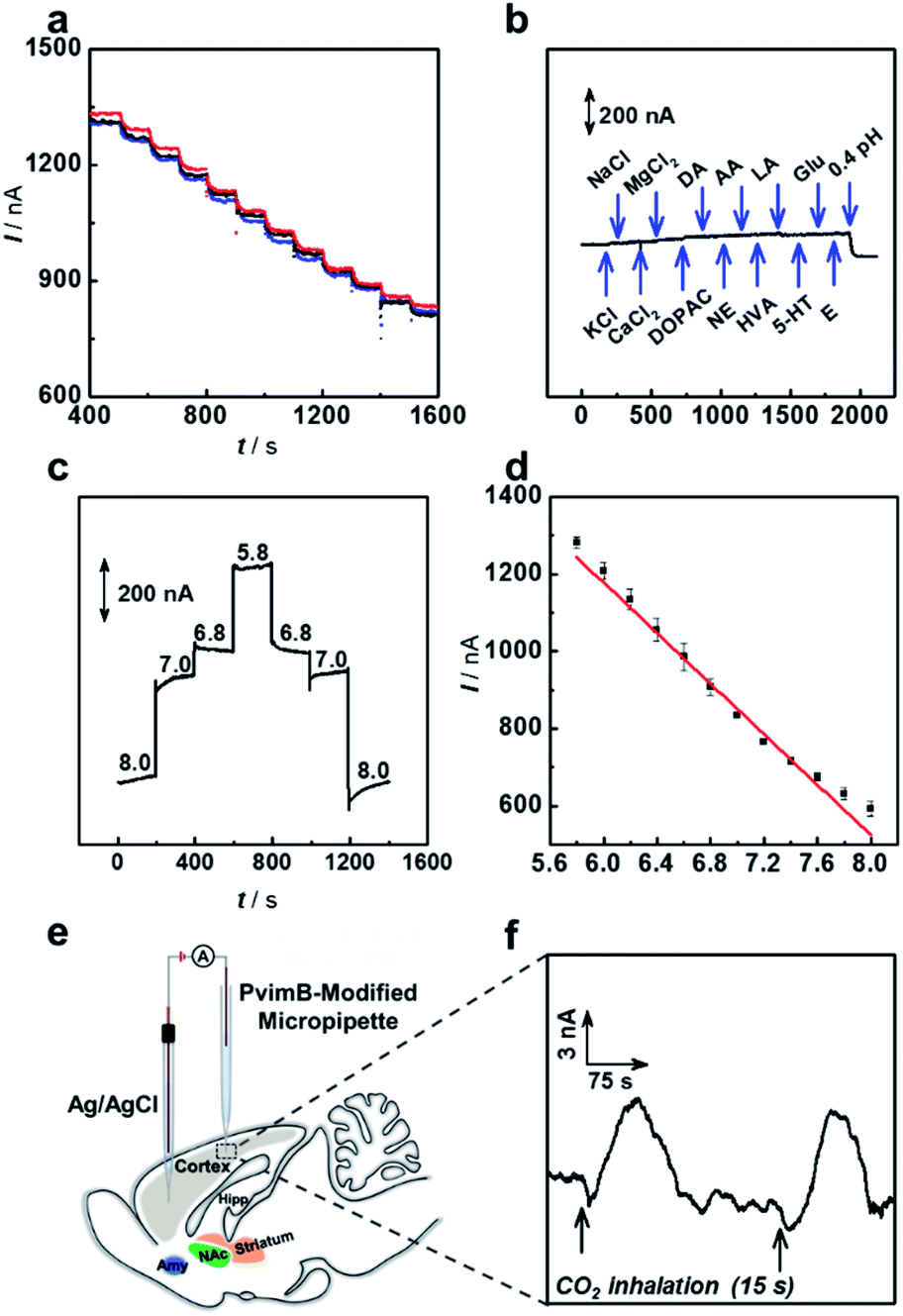 | ||
| Fig. 3 Performance of the as-prepared pH microsensor based on transient ion transport: (a) reproducibility (currents were obtained with the same micropipette from three independent measurements); (b) selectivity (1 mM NaCl, 1 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 50 μM DOPAC, 10 μM DA, 10 μM NE, 200 μM AA, 30 μM HVA, 1 mM LA, 30 μM 5-HT, 10 mM Glu and 10 μM E); (c) reversibility and (d) linearity (error bars were obtained for the same micropipette from three independent measurements). (e) Schematic illustration of the as-prepared pH microsensor implanted in the cortex of a rat brain. (f) Real-time monitoring of pH dynamics in the cortex by using the as-prepared pH microsensor under the stimulation of CO2 inhalation (black arrows). The electrolyte that back-filled into the PvbimB-modified micropipette was aCSF (pH 7.4). | ||
As a proof of concept, the in vivo sensing performance of the pH microsensor based on transient ion transport was validated by using a CO2 inhalation model in Sprague–Dawley rat brains.43,44 As illustrated in Fig. 3e, the PvimB-modified micropipette was implanted into the brain cortex to monitor pH changes during acid–base disturbance upon breathing in pure CO2 gas. As shown in Fig. 3f, before the stimulation, the recorded current was relatively stable. When rats were subjected to pure CO2 inhalation for 15 s (Fig. 3f, black arrows), a current increase was observed, and it then went back to the baseline for 115. To determine the amount of this pH change, calibration after use is adopted because the electrodes lose some of the sensitivity upon implantation in brain tissue. According to the post-calibration (Fig. S14†), breathing in pure CO2 gas rapidly decreased the pH level by about 0.10 ± 0.05 (n = 5 independent measurements for 3 rats), which is consistent with our previous observation during CO2 inhalation.44 These results essentially evaluated the applicability of the microsensor based on transient ion transport in in vivo analysis with high spatiotemporal resolution.
Conclusions
In summary, we have for the first time demonstrated that micrometer-scale transient ion transport could be used for highly spatiotemporal in vivo sensing by using pH as the model target. The introduction of the micrometer-scale asymmetric ion transport successfully meets the requirement of temporal and spatial resolution for real-time neurochemical analysis. The transient model was proposed to understand the time-dependent ion transport process. The as-prepared pH microsensor shows a high temporal resolution (∼ms), sensitivity, selectivity, repeatability, reversibility, and stability, which could be used for real-time monitoring the change of pH in rat brains. This study not only provides a novel method for pH assay in rat brains but also establishes a universal platform for other kinds of neurochemicals by rationally designing the surface chemistry of a micropipette. Moreover, the present study is also helpful to understand the transient process of asymmetric ion transport.Experimental section
Reagents and solutions
1-Vinylimidazole (Vim), copper(I) bromide (CuBr, 98%), N,N,N′,N′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%), 3-aminopropyl-triethoxysilane (APS), 1-chlorobutane and 2-bromoisobutyryl bromide (BIBB) were purchased from J&K Chemical (Tianjin, China). Dopamine (DA), noradrenaline (NE), adrenaline (E), sodium ascorbate (AA), homovanillic acid (HVA), sodium lactate (LA), serotonin (5-HT), glucose (Glu) and inorganic salts (i.e., NaCl, KCl, CaCl2 and MgCl2) were purchased from Sigma-Aldrich. Artificial cerebrospinal fluid (aCSF) was prepared by mixing NaCl (126 mM), KCl (2.4 mM), KH2PO4 (0.5 mM), MgCl2 (0.85 mM), NaHCO3 (27.5 mM), Na2SO4 (0.5 mM), and CaCl2 (1.1 mM) in aqueous solution, and pH values were adjusted to 7.4. The chemicals were all of analytical grade and all solutions were prepared with ultrapurified water (18 MΩ cm) from a Milli-Q purification system.Apparatus and characterization
Current–voltage (I–V) curves were recorded on a CHI 660E electrochemical workstation (Shanghai Chenhua Instrument Co. Ltd, China). To apply the potential and measure the ion current, an Ag/AgCl wire (0.6 mm diameter) was inserted into a glass micropipette and used as a working electrode, and another Ag/AgCl electrode with the same size was used as a counter/reference electrode and immersed in bath solution. In order to satisfy the requirement of in vivo electrochemical measurements, a microsized Ag/AgCl electrode was prepared as described previously.44 It could be implanted into brain tissue and used as a counter/reference electrode. All the potentials given herein were internal vs. external. For the electrochemical experiment in fundamental section (i.e., Fig. 1 and 2), the pipette was filled with a solution of 100 mM NaCl in 50 mM phosphate buffer (pH 5.8), and the bath solution was the same solution. Different volumes of 2 M NaOH solution were added to regulate solution pH accurately from 5.8 to 8.0. For the electrochemical experiment in the in vivo section (i.e., Fig. 3), the filled solution was aCSF solution (artificial cerebrospinal fluid) to mimic the brain environment.Scanning electron microscopy (SEM) imaging was performed using an S4800 (Hitachi, Japan). The zeta potential of silica nanoparticles (SiNPs) was measured using a Zetasizer Nano ZS ZEN 3600 (Malvern Instruments Ltd, UK). For the EOF experiment, a glass capillary (I.D. 100 μm) was purchased from Ningke (Xinpeng, Shanghai). The moving distance of the liquid column within the glass capillary was observed using a metalloscope (9XF-PC, Shanghai Optical Instrument Co. Ltd, China).
Preparation of PvimB-modified micropipettes
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7) for 12 h to remove organic contaminants. Then, the borosilicate capillaries were washed with cleaning solution (30% H2O2/NH4OH/H2O = 1:1:5) and deionized water. The washed capillaries were dried at 60 °C overnight and stored in a clean and cool place. A clean bare nanopipette was fabricated using a CO2-laser-based pipette puller (P-2000, Sutter Instrument) from borosilicate glass capillaries (0.86 mm (I.D.) and 1.50 mm (O.D.), Sutter Instrument). Pulling parameters were as follows: heat = 325, fil = 4, vel = 15, del = 128, pul = 50. The micropipettes (∼10 μm) were obtained by polishing the as-prepared nanopipette with a micro forge (MF-900, Narishige Instrument).
:7) for 12 h to remove organic contaminants. Then, the borosilicate capillaries were washed with cleaning solution (30% H2O2/NH4OH/H2O = 1:1:5) and deionized water. The washed capillaries were dried at 60 °C overnight and stored in a clean and cool place. A clean bare nanopipette was fabricated using a CO2-laser-based pipette puller (P-2000, Sutter Instrument) from borosilicate glass capillaries (0.86 mm (I.D.) and 1.50 mm (O.D.), Sutter Instrument). Pulling parameters were as follows: heat = 325, fil = 4, vel = 15, del = 128, pul = 50. The micropipettes (∼10 μm) were obtained by polishing the as-prepared nanopipette with a micro forge (MF-900, Narishige Instrument).
In vivo experiments
Adult female Sprague-Dawley rats (weighing 300–350 g) purchased from Health Science Center, Peking University, were housed on a 12:12 h light–dark schedule with food and water ad libitum. All procedures were approved by the Beijing Association on Laboratory Animal Care and the Association for Assessment and Accreditation of Laboratory Animal Care and performed according to their guidelines. Animal experiments were performed with a method described in our earlier work.44 The animals were anaesthetized with chloral hydrate (345 mg kg−1, i.p.) and positioned onto a stereotaxic frame by using ear rods. The PvimB-modified micropipette was implanted into the cerebral cortex using standard stereotaxic procedures (AP = 2.0 mm, L = 2.0 mm from bregma, V = −1.5 mm from dura). A home-made microsized Ag/AgCl electrode was implanted into the dura of the brain (Fig. 3e). For carrying out CO2 inhalation, one end of rubber tubing was linked to the reducing valve of a CO2 bottle, and the other end was placed under the nose of the rat, and brain acidosis was induced by exposing the animal for a short time (ca. 15 s) to CO2.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
We acknowledge the financial support from the Natural Science Foundation of Beijing (JQ19009), the National Natural Science Foundation of China (22074149, 21775151 for P. Y., 21790390, 21790391 for L. M.), and the National Key Research and Development Project (2018YFE0200800).Notes and references
- S.-K. Kang, R. K. J. Murphy, S.-W. Hwang, S. M. Lee, D. V. Harburg, N. A. Krueger, J. Shin, P. Gamble, H. Cheng, S. Yu, Z. Liu, J. G. McCall, M. Stephen, H. Ying, J. Kim, G. Park, R. C. Webb, C. H. Lee, S. Chung, D. S. Wie, A. D. Gujar, B. Vemulapalli, A. H. Kim, K.-M. Lee, J. Cheng, Y. Huang, S. H. Lee, P. V. Braun, W. Z. Ray and J. A. Rogers, Nature, 2016, 530, 71–76 CrossRef CAS PubMed.
- T. Someya, Z. Bao and G. G. Malliaras, Nature, 2016, 540, 379–385 CrossRef CAS PubMed.
- G. Shan, X. Li and W. Huang, The Innovation, 2020, 1, 100031 CrossRef.
- P. Yu, X. He and L. Mao, Chem. Soc. Rev., 2015, 44, 5959–5968 RSC.
- D. L. Robinson, A. Hermans, A. T. Seipel and R. M. Wightman, Chem. Rev., 2008, 108, 2554–2584 CrossRef CAS PubMed.
- T. Xiao, F. Wu, J. Hao, M. Zhang, P. Yu and L. Mao, Anal. Chem., 2017, 89, 300–313 CrossRef CAS PubMed.
- M. L. Huffman and B. J. Venton, Analyst, 2009, 134, 18–24 RSC.
- T. Xiao, Y. Wang, H. Wei, P. Yu, Y. Jiang and L. Mao, Angew. Chem., Int. Ed., 2019, 58, 6616–6619 CrossRef CAS PubMed.
- X. Liu, T. Xiao, F. Wu, M. Y. Shen, M. Zhang, H. h. Yu and L. Mao, Angew. Chem., Int. Ed., 2017, 56, 11802–11806 CrossRef CAS PubMed.
- F. Zhao, Y. Liu, H. Dong, S. Feng, G. Shi, L. Lin and Y. Tian, Angew. Chem., Int. Ed., 2020, 59, 10426–10430 CrossRef CAS PubMed.
- R. N. Adams, Anal. Chem., 1976, 48, 1126A–1138A CrossRef CAS PubMed.
- Y. T. Li, S. H. Zhang, L. Wang, R. R. Xiao, W. Liu, X. W. Zhang, Z. Zhou, C. Amatore and W. H. Huang, Angew. Chem., Int. Ed., 2014, 53, 12456–12460 CAS.
- R. J. Yu, Y. L. Ying, R. Gao and Y. T. Long, Angew. Chem., Int. Ed., 2019, 58, 3706–3714 CrossRef CAS PubMed.
- C. Dekker, Nat. Nanotechnol., 2007, 2, 209–215 CrossRef CAS PubMed.
- T. Xiong, K. Zhang, Y. Jiang, P. Yu and L. Mao, Sci. China: Chem., 2019, 62, 1346–1359 CrossRef CAS.
- X. He, K. Zhang, Y. Liu, F. Wu, P. Yu and L. Mao, Angew. Chem., Int. Ed., 2018, 57, 4590–4593 CrossRef CAS PubMed.
- D. Wang, M. Kvetny, J. Liu, W. Brown, Y. Li and G. Wang, J. Am. Chem. Soc., 2012, 134, 3651–3654 CrossRef CAS PubMed.
- C.-Y. Lin, C. Combs, Y.-S. Su, L.-H. Yeh and Z. S. Siwy, J. Am. Chem. Soc., 2019, 141, 3691–3698 CrossRef CAS PubMed.
- Z.-Y. Jiang, H.-L. Liu, S. A. Ahmed, S. Hanif, S.-B. Ren, J.-J. Xu, H.-Y. Chen, X.-H. Xia and K. Wang, Angew. Chem., Int. Ed., 2017, 56, 4767–4771 CrossRef CAS PubMed.
- C. Wang, F. F. Liu, Z. Tan, Y. M. Chen, W. C. Hu and X. H. Xia, Adv. Funct. Mater., 2020, 30, 1908804 CrossRef CAS.
- Y. Li, D. Wang, M. M. Kvetny, W. Brown, J. Liu and G. Wang, Chem. Sci., 2015, 6, 588–595 RSC.
- W. J. Lan, D. A. Holden, B. Zhang and H. S. White, Anal. Chem., 2011, 83, 3840–3847 CrossRef CAS PubMed.
- T. Li, X. He, K. Zhang, K. Wang, P. Yu and L. Mao, Chem. Sci., 2016, 7, 6365–6368 RSC.
- N. Sa, Y. Fu and L. A. Baker, Anal. Chem., 2010, 82, 9963–9966 CrossRef CAS PubMed.
- S. Howorka and Z. Siwy, Chem. Soc. Rev., 2009, 38, 2360–2384 RSC.
- Y. Sun, F. Zhang, J. Quan, F. Zhu, W. Hong, J. Ma, H. Pang, Y. Sun, D. Tian and H. Li, Nat. Commun., 2018, 9, 1–7 CrossRef PubMed.
- S. Umehara, M. Karhanek, R. W. Davis and N. Pourmand, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4611–4616 CrossRef CAS PubMed.
- R. E. Ozel, A. Lohith, W. H. Mak and N. Pourmand, RSC Adv., 2015, 5, 52436–52443 RSC.
- R. A. Nascimento, R. E. Özel, W. H. Mak, M. Mulato, B. Singaram and N. Pourmand, Nano Lett., 2016, 16, 1194–2200 CrossRef CAS PubMed.
- R. E. Ozel, G. Bulbul, J. Perez and N. Pourmand, ACS Sens., 2018, 3, 1316–1321 CrossRef CAS PubMed.
- H. Ouldali, K. Sarthak, T. Ensslen, F. Piguet, P. Manivet, J. Pelta, J. C. Behrends, A. Aksimentiev and A. Oukhaled, Nat. Biotechnol., 2020, 38, 176–181 CrossRef CAS PubMed.
- G. Baaken, I. Halimeh, L. Bacri, J. Pelta, A. Oukhaled and J. C. Behrends, ACS Nano, 2015, 9, 6443–6449 CrossRef CAS PubMed.
- C. B. Rosen, D. Rodriguez-Larrea and H. Bayley, Nat. Biotechnol., 2014, 32, 179–181 CrossRef CAS PubMed.
- J. Song, C.-H. Xu, S.-Z. Huang, W. Lei, Y.-F. Ruan, H.-J. Lu, W. Zhao, J.-J. Xu and H.-Y. Chen, Angew. Chem., Int. Ed., 2018, 57, 13226–13230 CrossRef CAS PubMed.
- X. He, K. Zhang, T. Li, Y. Jiang, P. Yu and L. Mao, J. Am. Chem. Soc., 2017, 139, 1396–1399 CrossRef CAS PubMed.
- K. Zhang, X. He, Y. Liu, P. Yu, J. Fei and L. Mao, Anal. Chem., 2017, 89, 6794–6799 CrossRef CAS PubMed.
- R. J. Sundberg and R. B. Martin, Chem. Rev., 1974, 74, 471–517 CrossRef CAS.
- J. P. Guerrette and B. Zhang, J. Am. Chem. Soc., 2010, 132, 17088–17091 CrossRef CAS PubMed.
- D. Momotenko and H. H. Girault, J. Am. Chem. Soc., 2011, 133, 14496–14499 CrossRef CAS PubMed.
- M. Figen, G. G. Hammes and K. Kustin, J. Am. Chem. Soc., 1960, 82, 3482–3483 CrossRef.
- P. Jin, H. Mukaibo, L. P. Horne, G. W. Bishop and C. R. Martin, J. Am. Chem. Soc., 2010, 132, 2118–2119 CrossRef CAS PubMed.
- M. Chesler, Physiol. Rev., 2003, 83, 1183–1221 CrossRef CAS PubMed.
- V. A. Magnotta, H.-Y. Heo, B. J. Dlouhy, N. S. Dahdaleh, R. L. Follmer, D. R. Thedens, M. J. Welsh and J. A. Wemmie, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8270–8273 CrossRef CAS PubMed.
- J. Hao, T. Xiao, F. Wu, P. Yu and L. Mao, Anal. Chem., 2016, 88, 11238–11243 CrossRef CAS PubMed.
- D. Janssen, R. D. Palma, S. Verlaak, P. Heremans and W. Dehaen, Thin Solid Films, 2006, 515, 1433–1438 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00061f |
| This journal is © The Royal Society of Chemistry 2021 |