
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActinide tetra-N-heterocyclic carbene ‘sandwiches’†
Joseph F.
DeJesus
a,
Ryan W. F.
Kerr
 b,
Deborah A.
Penchoff‡
c,
Xian B.
Carroll
a,
Charles C.
Peterson
cd,
Polly L.
Arnold§¶
*b and
David M.
Jenkins
*a
b,
Deborah A.
Penchoff‡
c,
Xian B.
Carroll
a,
Charles C.
Peterson
cd,
Polly L.
Arnold§¶
*b and
David M.
Jenkins
*a
aDepartment of Chemistry, The University of Tennessee, Knoxville, Tennessee 37996, USA. E-mail: jenkins@ion.chem.utk.edu
bSchool of Chemistry, University of Edinburgh, West Mains Road, Edinburgh, EH9 3JJ, UK
cHoward H. Baker Jr. Center for Public Policy, The University of Tennessee, Knoxville, Tennessee 37996, USA
dResearch IT Services, University of North Texas, Denton, Texas 76201, USA
First published on 10th May 2021
Abstract
Highly-symmetrical, thorium and uranium octakis-carbene ‘sandwich’ complexes have been prepared by ‘sandwiching’ the An(IV) cations between two anionic macrocyclic tetra-NHC ligands, one with sixteen atoms and the other with eighteen atoms. The complexes were characterized by a range of experimental methods and DFT calculations. X-ray crystallography confirms the geometry at the metal centre can be set by the size of the macrocyclic ring, leading to either square prismatic or square anti-prismatic shapes; the geometry of the latter is retained in solution, which also undergoes reversible, electrochemical one-electron oxidation or reduction for the uranium variant. DFT calculations reveal a frontier orbital picture that is similar to thorocene and uranocene, in which the NHC ligands show almost exclusively σ-donation to the metal without π-backbonding.
Introduction
The development of N-heterocyclic carbene (NHC) ligands has transformed many fields of organometallic chemistry,1–4 but the revolutionary application of their unusually strongly σ-donating nature in transition metal chemistry has not crossed over as effectively to actinides.5–7 An electrostatic mismatch often prevents straightforward ligation of neutral NHCs to f-block complexes5 so just a few weakly bound adducts have been reported, such as the U(VI) complex (Fig. 1A).8 We (the Arnold group) first developed chelating ligands with an anionic amido group to circumvent this problem (Fig. 1B).9 This general approach was later expanded to other f-block elements,10–14 including thorium (Fig. 1C).15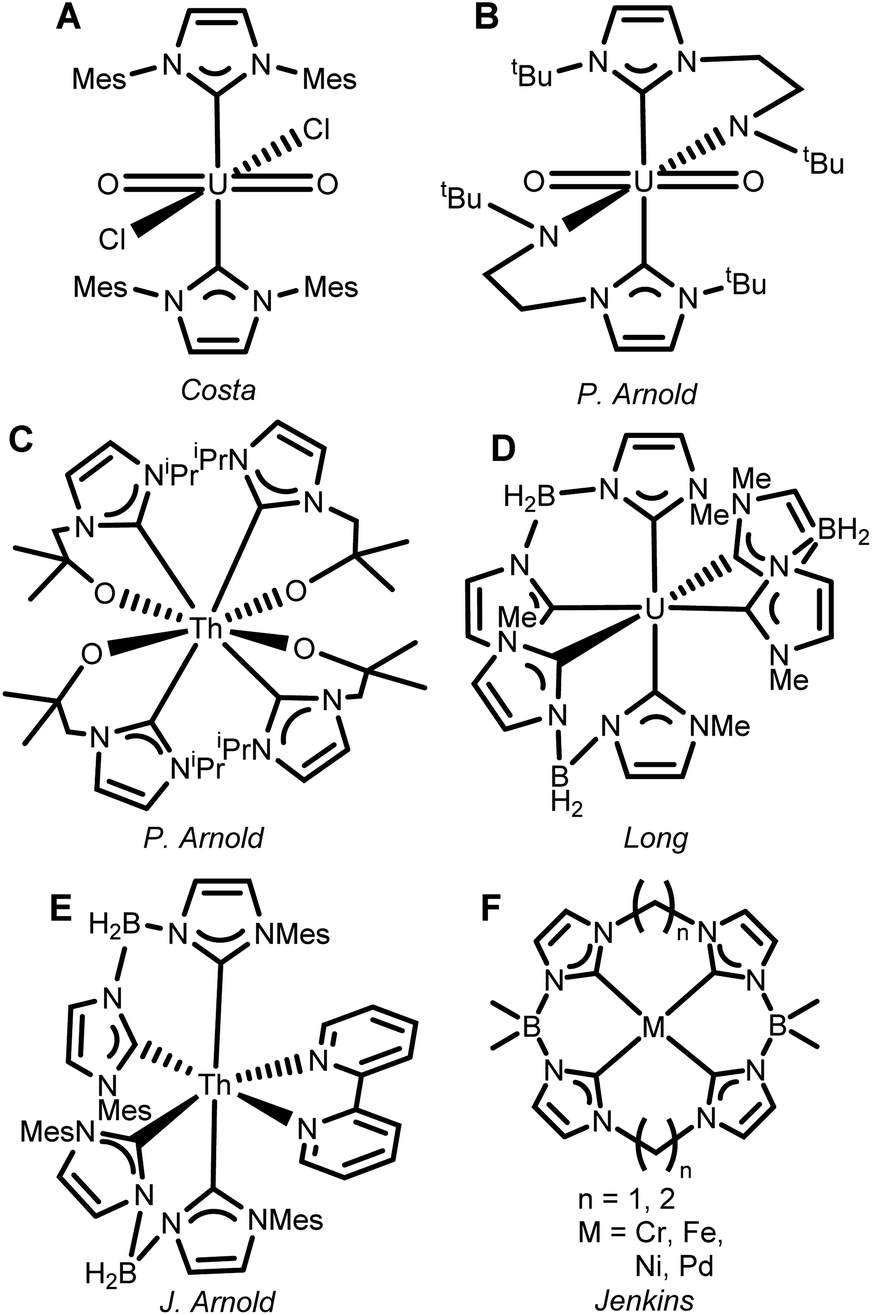 | ||
| Fig. 1 (A–E) Examples of the progression of NHC actinide complexes since 2001. (F) Dianionic macrocyclic tetracarbenes developed for transition metals by Jenkins. | ||
Incorporation of a delocalized negative charge across a polydentate ligand backbone by inclusion of quaternary boron provides an alternative strategy. Scorpionate NHCs,16 including bis- or tris-NHC borate ligands have yielded complexes with up to six coordinated NHCs per metal centre for both d- and more recently f-block cations (Fig. 1D and E).17–19 Coordinatively saturated hexa-NHC transition metal complexes (e.g. on iron) have demonstrated noteworthy electronic structure properties, spanning luminescence20,21 magnetic bistability,18 reactivity,22–26 and single molecule magnetism, often taking advantage of the impressive σ-donor strength of the NHC.27
We hypothesized that larger macrocyclic tetra-NHC ligands may be able to better stabilize complexes of the larger actinide cations.28–33 We (the Jenkins group) recently developed dianionic tetra-NHC complexes for catalysis and group transfer reactions of transition metals (Fig. 1F).34–39 We considered that the increased ionic radii of actinides versus transition metals could lead to actinide bis(NHC macrocycle) ‘sandwiches’, reminiscent of porphyrins, phthalocyanines, or even cyclooctatetraene (COT) sandwiches (e.g. (COT)2M), and, more importantly, that the strong σ-donor capacity of the NHC ligand should impart unusual electronic structures on these complexes.40–43
In this manuscript, we report the first macrocyclic NHC – f-block complexes and the first octa-NHC complexes of any metal. Isostructural square-antiprismatic thorium and uranium complexes were prepared with a 16-membered-ring macrocycle ((BMe2,MeTCH); Fig. 1F with n = 1), while an 18-membered-ring macrocycle ((BMe2,EtTCH); Fig. 1F with n = 2) yields a square prismatic geometry uranium(IV) complex. DFT calculations show that the NHCs have considerable σ-donating character which leads to significant ligand field splitting consistent with traditional COT actinide sandwich complexes.
Results and discussion
The synthesis of the octa-NHC actinide complexes is extremely challenging. For transition metal systems, we have typically deprotonated the 16-atom ring macrocyclic ligand [H4(BMe2,MeTCH)]Br2 with nBuLi at low temperature prior to adding a metal salt,34–39 but this methodology is unsuccessful with 5f salts, possibly due to lithium adduct formation.44 Other strong bases, such as NaCH2Ph in diethyl ether, led to degradation of [H4(BMe2,MeTCH)]Br2. The key breakthrough was determining that MN(SiMe3)2 (M = Li, Na or K), deprotonated the most acidic protons in the ligands in ethereal solvents. Addition of eight equivalents of NaN(SiMe3)2 to two equivalents of [H4(BMe2,MeTCH)]Br2 in DME at −35 °C followed by UI4(1,4-dioxane)2 gives an intense brown solution which is then heated to 50 °C to drive the deprotonation reaction to completion (Scheme 1). Room temperature reactions gave only very low yields of 1, which is consistent with our previous reports on iron complexes with this 16-atom macrocycle.34NMR spectra of crude reaction mixtures contain multiple sets of paramagnetic peaks as well as diamagnetic peaks attributed to degraded macrocycle, thus purification steps were critical to isolate (BMe2,MeTCH)2U (1). After the reaction was completed, the DME was removed under vacuum, and HN(SiMe3)2 was then separated from the mixture by washing it away with hexanes. The resulting burgundy residue was dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 toluene/hexanes mixture and filtered over Celite, which left the resulting salt (NaI) and other impurities behind. This dark red coloured solution was crystallized by cooling at −35 °C overnight which yielded pure (BMe2,MeTCH)2U (1) as dark purple plates in 9.3% isolated yield (Scheme 1A).
:1 toluene/hexanes mixture and filtered over Celite, which left the resulting salt (NaI) and other impurities behind. This dark red coloured solution was crystallized by cooling at −35 °C overnight which yielded pure (BMe2,MeTCH)2U (1) as dark purple plates in 9.3% isolated yield (Scheme 1A).
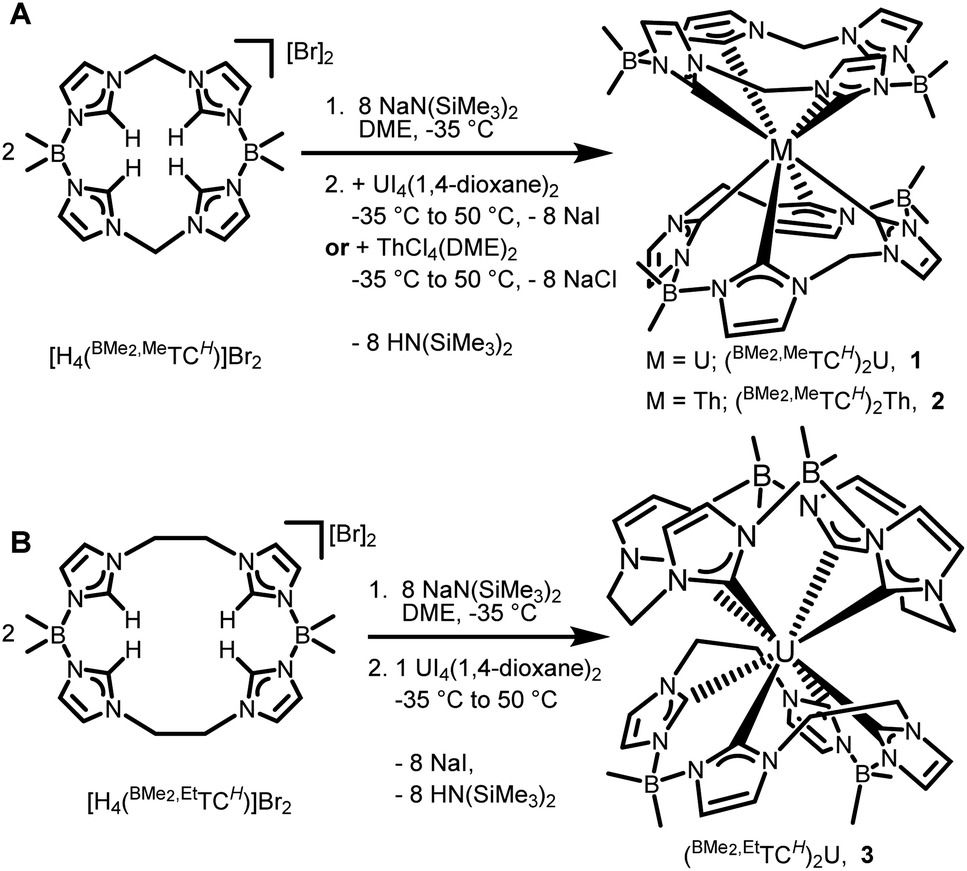 | ||
| Scheme 1 Synthesis of actinide NHC ‘sandwich’ complexes with (A) showing 16-atom macrocycle and (B) showing 18-atom macrocycle. | ||
Complex 1 is remarkably air and moisture stable and only very slowly degrades in air over months in the solid state; it appears to be indefinitely stable in an inert atmosphere at ambient temperature. The 1H NMR spectrum of a CD2Cl2 solution of complex 1 shows eight resonances (Fig. S1†) ranging from 14.8 ppm to −55.9 ppm, which demonstrates that the H atoms in the methylene bridges are diastereotopic and that there are two distinct NHC environments. In every case that we have previously reported with similar macrocycles (excluding a chiral macrocycle),39 only one NHC environment is observed by 1H NMR.34–36,38 This result suggests that the NHCs are locked into position in solution giving 1 relative D2 symmetry. Heating the solution to 50 °C in CD3CN (Fig. S10†) does not show any dynamic process or reduction in symmetry due to binding of an acetonitrile, which is consistent with the moisture stability of 1. Several reactions were carried out to target binding a ninth ligand onto 1, e.g. with diphenyldiazomethane or tolyl azide (see ESI†). However, all either showed no reaction at ambient temperatures, or decomposition of 1 at higher temperatures to decomposed ligand products and unidentified insoluble uranium decomposition products (see ESI†).
By employing similar reaction conditions to 1, with an additional separation step on C2-terminated silica gel, we were able to isolate (BMe2,MeTCH)2Th (2), albeit in 1% isolated crystalline yield (Scheme 1A). Complex 2 is colourless and stable in air. Since (BMe2,MeTCH)2Th is diamagnetic, we were able to evaluate its NMR spectra in more detail than 1. The 1H NMR spectrum in CDCl3 of 2 shows eight resonances, implying that the relative D2 symmetry is the same as 1 (Fig. S7†). Coupling (J = 13 Hz) is observed between the diastereotopic protons on the methylene bridges. In concordance with the D2 symmetry, nine carbon resonances are observed in the 13C NMR spectrum, including the two resonances for the NHC carbene carbons at 210.1 and 207.6 ppm, respectively. The 13C resonances for the NHC carbenes are consistent with the few other reported diamagnetic thorium NHC complexes.15,19
We (the Jenkins group) and others have found significant differences in structure and reactivity between 16- and 18-atom ring macrocyclic tetra-NHC ligands. For example, a chromium complex prepared with the 16-atom macrocycle, [H4(BMe2,MeTCH)]Br2, yields a relatively stable Cr(II) dimer, [(BMe2,MeTCH)Cr]2 with a chromium–chromium quadruple bond; however, switching to the 18-atom macrocycle, [H4(BMe2,EtTCH)]Br2, yields the highly unstable paramagnetic monomeric complex (BMe2,EtTCH)Cr.35,38 Similarly, iron complexes with 16-atom rings are more stable, but not catalytically active for aziridination, while 18-atom rings yield less stable complexes that perform that reaction.28,37,45,46 This trend of reduced stability for 18-atom rings translates to the actinide complexes here. Despite a wide variety of reaction conditions (see ESI†), we were only able to obtain a few single crystals of the uranium complex (BMe2,EtTCH)2U (3) (Scheme 1B). Complex 3 is highly reactive towards air and thermally unstable, precluding further characterization. We also carried out reactions to synthesize the thorium analogue of 3, but were unable to isolate any product.
The solid state structure of (BMe2,MeTCH)2U (1) reveals a square anti-prismatic geometry about the uranium centre which is in accordance with the symmetry implied by the 1H NMR (Fig. 2A). There are two separate NHC groups because one is opposite a methylene bridge and the other is opposite a BMe2 bridge. All of the U–C bonds are between 2.61 Å and 2.65 Å, which is consistent with previously characterized NHC–U bonds for tetravalent uranium.47–49 In the solid state, (BMe2,MeTCH)2Th (2) is isostructural to its uranium counterpart, giving a near perfect square anti-prismatic geometry (Fig. 2C). The Th–C bonds are between 2.69 Å and 2.73 Å which is consistent with reported NHC complexes.15,19 Notably, the M–NHC bond lengths for both 1 and 2 are similar to the M–C bonds for uranocene (2.647(4)Å) and thorocene (2.701(4)Å).43,50
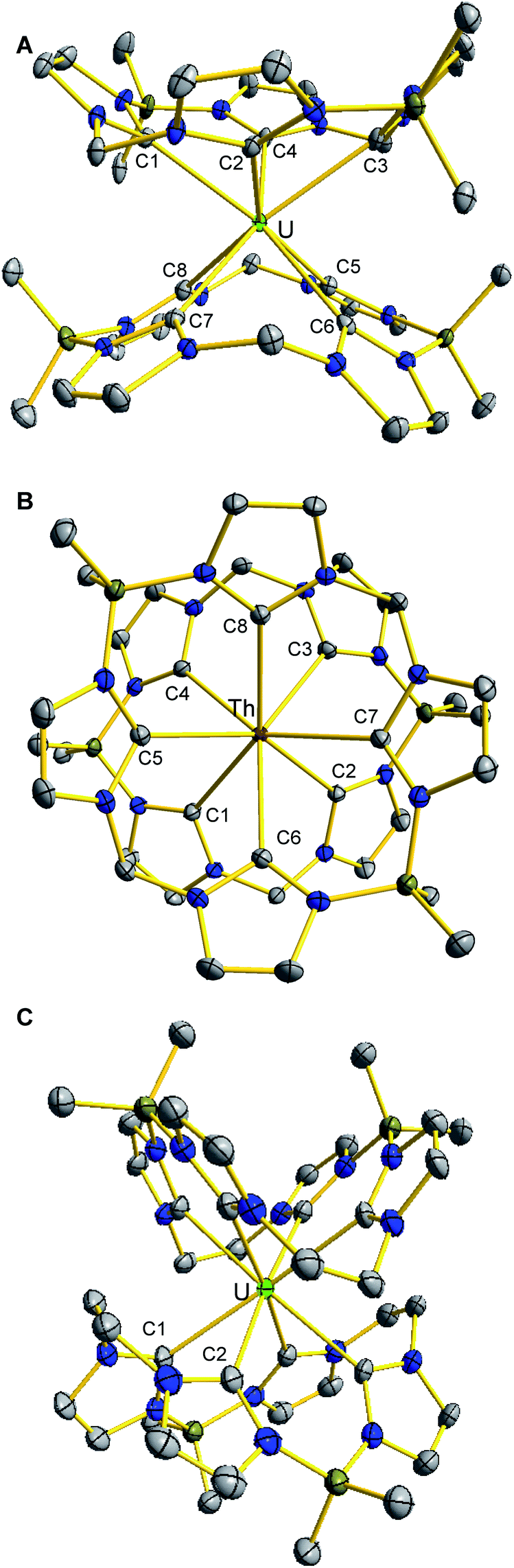 | ||
| Fig. 2 Solid state structures of (A) (BMe2,MeTCH)2U, 1, (B) (BMe2,MeTCH)2Th, 2 (shown top down), and (C) (BMe2,EtTCH)2U, 3. Green, burgundy, blue, grey, and olive ellipsoids (50% probability) represent U, Th, N, C, and B atoms, respectively. H atoms and lattice solvent molecules are omitted for clarity. | ||
As we observed with chromium complexes of (BMe2,MeTCH) and (BMe2,EtTCH), adding a single additional carbon atom to each bridge dramatically changes the structure and properties at each metal ion, so the structure of 3 was particularly noteworthy.30,35 In contrast to 1, complex 3 crystallizes in a higher symmetry space group (Fccc) and 3 has a square prismatic geometry about the uranium atom with precise D2d symmetry (Fig. 2C). In this case, the borate bridges are opposite the ethylene bridge and not offset over the opposing NHC. Presumably, the additional steric demands of the extra carbon atoms cause this change in geometry. Complex 3 only has two unique U–C bonds that are 2.615(5) Å and 2.697(4) Å for C1 and C2, respectively.
Notably, cyclic voltammetry of 1 in CH3CN with 0.1 M tetrabutylammonium hexafluorophosphate supporting electrolyte reveals two reversible one-electron redox processes centred at 0.21 V and −2.46 V versus ferrocene that we assign as oxidation and reduction of the U(IV) centre, respectively (Fig. S5 and S6†). The value of the oxidation potential is similar to other uranium(IV) complexes, but only moderately close to uranocene, at 0.06 V versus SCE (0.44 V vs. ferrocene).41,51,52 Unfortunately, reactions to oxidize 1 to uranium(V) under a variety of conditions (see ESI†) did not yet yield any isolable species.
Since these are the first homoleptic actinide NHC complexes and there are very few reports of DFT calculations on similar complexes,14,23,53–55 we were particularly interested in probing these species with theoretical investigations. Complexes 1–3 were optimized without any symmetry constraints with the double-ζ correlation consistent basis set cc-pVDZ-PP,56,57 with the multielectron fit, fully relativistic ECP60MDF58,59 pseudopotential which includes 60 core electrons for uranium and thorium. Results are obtained with the exchange-correlation hybrid B3LYP energy functional.60 No complex modes were found through vibrational frequency calculations. Geometry optimizations and harmonic vibrational frequency calculations were obtained with the NWChem 6.8 package.61 The Natural Bond Orbital 7.0 (NBO7) program62 was used to obtain NBO population analysis.63 Basis sets and effective core potentials are obtained from the Environmental Molecular Sciences Laboratory (EMSL).64 The ChemCraft package was utilized for computation visualizations.
Differences between predicted HOMO–LUMO gaps in our work and previous work53 can be attributed to the different levels of theory utilized (correlation consistent basis sets and ECP60MDF pseudopotential vs. Pople-style basis sets and Stuttgart ECP) and the application of symmetry constraints. Calculated bond lengths for the An–C bonds in all but one case were less than 0.1 Å different from the experimental crystal structures and faithfully reproduced the geometry about each actinide centre (see ESI† for details).
Complexes 1 and 3 both have HOMO and HOMO−1 (both singly occupied) orbitals that are essentially exclusively f-character (Fig. 3 and ESI S23 and S26†). For 1, the NHC bonding orbitals are lower in energy and between the HOMO−5 and HOMO−18 (Fig. S25†). Notably, in each case they show significant σ-donation which is consistent with our results14 and Liddle's account54 for NHC U(IV) complexes, but in contrast with Meyer's report on a U(III) NHC complex where he notes that π-bonding predominates.55 Despite being in a different geometry, square prismatic, complex 3 has very similar orbital depictions as 1 (Fig. 3 and S29†), underlining the ionic nature of the bonding. Finally, the HOMO orbital for 2 is ligand based, as expected for a diamagnetic complex with no electrons on the metal centre (Fig. 3). The orbitals that show NHC donation to the Th appear between HOMO−9 and HOMO−31 (Fig. S28†) and again show clear σ-bonding character to the metal centre with little d or f orbital character.
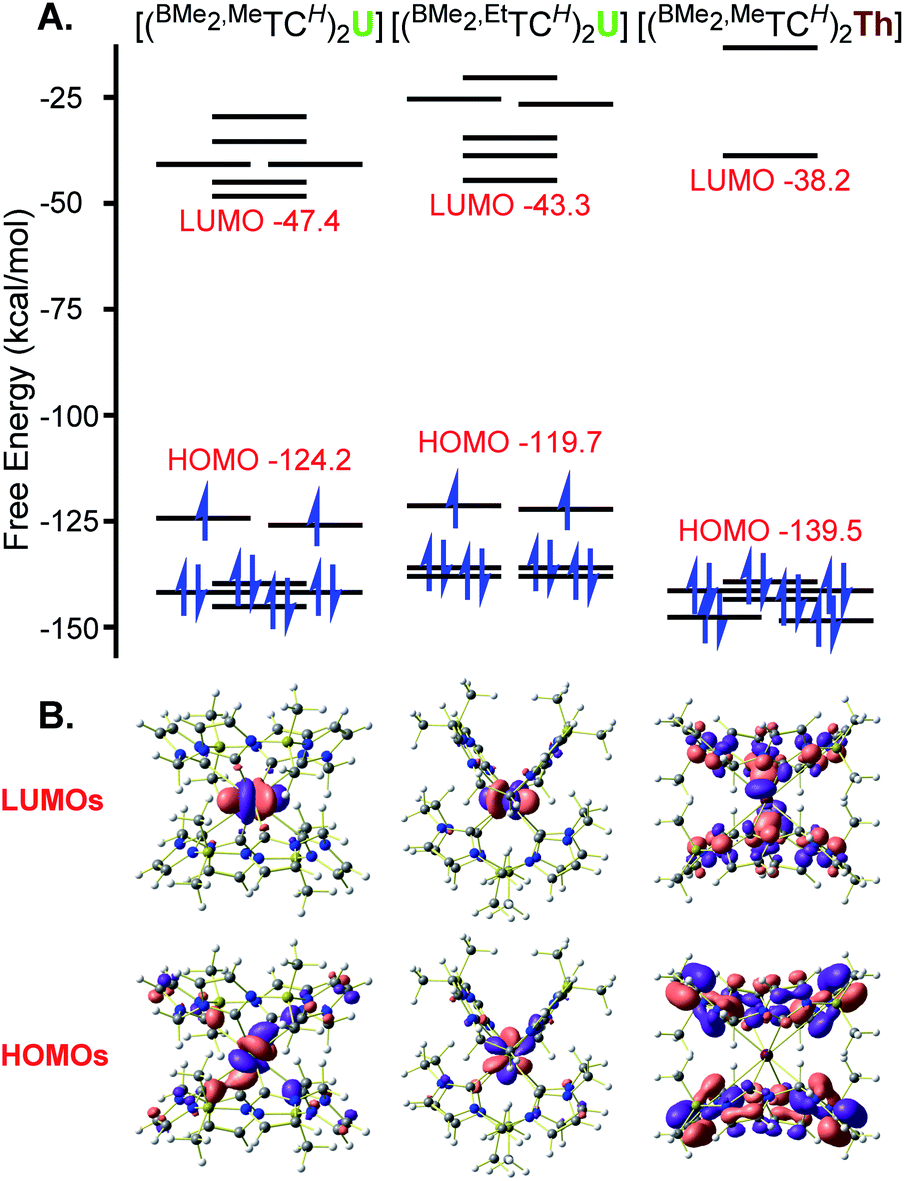 | ||
| Fig. 3 (A) Calculated frontier molecular orbital diagrams for 1–3. (B) Orbital depictions for the HOMOs and LUMOs for 1–3. HOMOs for uranium complexes 1 and 3 are singly occupied. Green, burgundy, blue, grey, olive, and white spheres represent U, Th, N, C, B, and H atoms, respectively. | ||
As complexes 1–3 represent a novel actinide ‘sandwich’, we compared their DFT calculations to the originals, uranocene ((COT)2U; COT = cyclooctatetraene) and thorocene ((COT)2Th). In particular, we wanted to determine if the strong σ-donation of the NHCs would lead to increased HOMO–LUMO gaps. For complexes 1 and 3, LUMO orbitals also maintain a high degree of f-character and the complexes each have a HOMO–LUMO gap of ∼77 kcal mol−1. A comparison with (COT)2U calculated using the same methodology as our NHC complexes shows a HOMO–LUMO gap of 88 kcal mol−1 (Table S9†), which is higher than previous calculations.53 Notably the general orbital picture is quite similar; the singly occupied HOMO orbitals, as well as the associated LUMO, LUMO+1, and LUMO+2 for 1 primarily show f-orbital character, similar to uranocene (Fig. S24 and S35†).53 Indeed, calculations for uranocene only show modest π-interactions in the HOMO−1 and HOMO−2 orbitals. Calculations for 2 show similar contours to 1, but without any populated f-electrons, the HOMO–LUMO gap expands to 101 kcal mol−1 (Fig. 3). Again, a comparison with (COT)2Th calculated using the same methodology as our NHC complexes shows a HOMO–LUMO gap of 92.1 kcal mol−1 (Table S9†), which is consistent with previous calculated HOMO–LUMO gaps for (COT)2Th.53 Complex 2 and (COT)2Th are diamagnetic complexes, which allows for more accurate energy comparisons, so these results suggest that the eight σ-donor NHC ligand set has a stronger ligand field than the two π-aromatic, cyclooctatetraenyl ligand set.
Conclusions
We have synthesized the first octa-NHC metal complexes using the large An(IV) cation. The homoleptic ‘sandwich’ organometallic complexes are either square anti-prismatic (D2 symmetry) or square prismatic (D2d symmetry) depending on the size of the macrocyclic ring. The D2 symmetry is retained in solution as evidenced by multi-nuclear NMR studies. The size of the ring greatly influences the stability despite each uranium complex having similar U–C bond distances. The 16-atom ringed species are remarkably air-stable, while the 18-atom uranium complex is incredibly frail. DFT calculations show that the NHCs donate electron density through a σ pathway and strong donation leads to very large HOMO–LUMO gaps. These experimental results (including solid state structures and electrochemistry) show similarities between these complexes and the earlier reports on uranocene and thorocene. Finally, DFT calculations suggest that although there are different bonding modes for the ligands between the different ‘sandwiches’, the ultimate ligand field strength is similar. These results set the foundation for future chemistry involving macrocyclic tetra-NHCs and actinides.Author contributions
J. F. D. and R. W. F. K. performed the synthesis and characterized the compounds. D. A. P. and C. C. P. executed the theoretical calculations. J. F. D. and X. B. C. solved the X-ray structures of the compounds. J. F. D., D. A. P., P. L. A., and D. M. J. designed the project, analyzed the results, and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Tennessee for supporting sabbatical work for J. F. D. and D. M. J. at the University of Edinburgh. The University of Tennessee also provided additional financial support for this work via the X-ray facility. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (grant agreement no 740311, P. L. A.). Additional discussion, analysis, and writing of this manuscript (P. L. A.) was supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division at the Lawrence Berkeley National Laboratory under Contract DE-AC02-05CH11231. This research used resources of the National Energy Research Scientific Computing Center (NERSC), a U.S. Department of Energy Office of Science User Facility operated under Contract No. DE-AC02-05CH11231. Computational resources were provided by the North Texas Scientific Computing, a division under the of office of the CIO for UNT and UNT System. We thank the EPSRC for funding through the Centre for Doctoral Training in Critical Resource Catalysis, CRITICAT (EP/L016419/1, R. W. F. K.).References
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- T. Scattolin and S. P. Nolan, Trends Chem., 2020, 2, 721–736 CrossRef CAS.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS PubMed.
- P. L. Arnold and I. J. Casely, Chem. Rev., 2009, 109, 3599–3611 CrossRef CAS PubMed.
- S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 8604–8641 CrossRef CAS PubMed.
- F. T. Edelmann, J. H. Farnaby, F. Jaroschik and B. Wilson, Coord. Chem. Rev., 2019, 398, 113005 CrossRef CAS.
- W. J. Oldham Jr, S. M. Oldham, W. H. Smith, D. A. Costa, B. L. Scott and K. D. Abney, Chem. Commun., 2001, 1348–1349 RSC.
- S. A. Mungur, S. T. Liddle, C. Wilson, M. J. Sarsfield and P. L. Arnold, Chem. Commun., 2004, 2738–2739 RSC.
- P. L. Arnold, R. W. F. Kerr, C. Weetman, S. R. Docherty, J. Rieb, F. L. Cruickshank, K. Wang, C. Jandl, M. W. McMullon, A. Pöthig, F. E. Kühn and A. D. Smith, Chem. Sci., 2018, 9, 8035–8045 RSC.
- P. L. Arnold, Z. R. Turner, A. I. Germeroth, I. J. Casely, G. S. Nichol, R. Bellabarba and R. P. Tooze, Dalton Trans., 2013, 42, 1333–1337 RSC.
- P. L. Arnold, Chem. Commun., 2011, 47, 9005–9010 RSC.
- Z. R. Turner, R. Bellabarba, R. P. Tooze and P. L. Arnold, J. Am. Chem. Soc., 2010, 132, 4050–4051 CrossRef CAS PubMed.
- P. L. Arnold, Z. R. Turner, N. Kaltsoyannis, P. Pelekanaki, R. M. Bellabarba and R. P. Tooze, Chem.–Eur. J., 2010, 16, 9623–9629 CrossRef CAS PubMed.
- P. L. Arnold, T. Cadenbach, I. H. Marr, A. A. Fyfe, N. L. Bell, R. Bellabarba, R. P. Tooze and J. B. Love, Dalton Trans., 2014, 43, 14346–14358 RSC.
- R. Fränkel, J. Kniczek, W. Ponikwar, H. Nöth, K. Polborn and W. P. Fehlhammer, Inorg. Chim. Acta, 2001, 312, 23–39 CrossRef.
- C. Santini, M. Marinelli and M. Pellei, Eur. J. Inorg. Chem., 2016, 2016, 2312–2331 CrossRef CAS.
- K. R. Meihaus, S. G. Minasian, W. W. Lukens, S. A. Kozimor, D. K. Shuh, T. Tyliszczak and J. R. Long, J. Am. Chem. Soc., 2014, 136, 6056–6068 CrossRef CAS PubMed.
- M. E. Garner, S. Hohloch, L. Maron and J. Arnold, Organometallics, 2016, 35, 2915–2922 CrossRef CAS.
- N. W. Rosemann, P. Chábera, O. Prakash, S. Kaufhold, K. Wärnmark, A. Yartsev and P. Persson, J. Am. Chem. Soc., 2020, 142, 8565–8569 CrossRef CAS PubMed.
- J. P. Harris, C. Reber, H. E. Colmer, T. A. Jackson, A. P. Forshaw, J. M. Smith, R. A. Kinney and J. Telser, Can. J. Chem., 2017, 95, 547–552 CrossRef.
- K. S. Kjær, N. Kaul, O. Prakash, P. Chábera, N. W. Rosemann, A. Honarfar, O. Gordivska, L. A. Fredin, K.-E. Bergquist, L. Häggström, T. Ericsson, L. Lindh, A. Yartsev, S. Styring, P. Huang, J. Uhlig, J. Bendix, D. Strand, V. Sundström, P. Persson, R. Lomoth and K. Wärnmark, Science, 2019, 363, 249 CrossRef PubMed.
- M. E. Garner, S. Hohloch, L. Maron and J. Arnold, Angew. Chem., 2016, 128, 13993–13996 CrossRef.
- M. E. Garner and J. Arnold, Organometallics, 2017, 36, 4511–4514 CrossRef CAS.
- M. E. Garner, T. D. Lohrey, S. Hohloch and J. Arnold, J. Organomet. Chem., 2018, 857, 10–15 CrossRef CAS.
- M. E. Garner, B. F. Parker, S. Hohloch, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2017, 139, 12935–12938 CrossRef CAS PubMed.
- R. Fränkel, U. Kernbach, M. Bakola-Christianopoulou, U. Plaia, M. Suter, W. Ponikwar, H. Nöth, C. Moinet and W. P. Fehlhammer, J. Organomet. Chem., 2001, 617–618, 530–545 CrossRef.
- S. A. Cramer and D. M. Jenkins, J. Am. Chem. Soc., 2011, 133, 19342–19345 CrossRef CAS PubMed.
- Z. Lu, S. A. Cramer and D. M. Jenkins, Chem. Sci., 2012, 3, 3081–3087 RSC.
- C. L. Keller, J. L. Kern, B. D. Terry, S. Roy and D. M. Jenkins, Chem. Commun., 2018, 54, 1429–1432 RSC.
- M. Ghosh, H. H. Cramer, S. Dechert, S. Demeshko, M. John, M. M. Hansmann, S. Ye and F. Meyer, Angew. Chem., Int. Ed., 2019, 58, 14349–14356 CrossRef CAS PubMed.
- Z. S. Ghavami, M. R. Anneser, F. Kaiser, P. J. Altmann, B. J. Hofmann, J. F. Schlagintweit, G. Grivani and F. E. Kühn, Chem. Sci., 2018, 9, 8307–8314 RSC.
- A. H. Mageed, J. Organomet. Chem., 2019, 902, 120965 CrossRef CAS.
- M. R. Anneser, G. R. Elpitiya, X. B. Powers and D. M. Jenkins, Organometallics, 2019, 38, 981–987 CrossRef CAS.
- M. R. Anneser, X. B. Powers, K. M. Peck, I. M. Jensen and D. M. Jenkins, Organometallics, 2019, 38, 3369–3376 CrossRef CAS PubMed.
- H. M. Bass, S. A. Cramer, A. S. McCullough, K. J. Bernstein, C. R. Murdock and D. M. Jenkins, Organometallics, 2013, 32, 2160–2167 CrossRef CAS.
- P. P. Chandrachud, H. M. Bass and D. M. Jenkins, Organometallics, 2016, 35, 1652–1657 CrossRef CAS.
- G. R. Elpitiya, B. J. Malbrecht and D. M. Jenkins, Inorg. Chem., 2017, 56, 14101–14110 CrossRef CAS PubMed.
- J. F. DeJesus and D. M. Jenkins, Chem.–Eur. J., 2020, 26, 1429–1435 CrossRef CAS PubMed.
- J. L. Sessler, P. J. Melfi and G. D. Pantos, Coord. Chem. Rev., 2006, 250, 816–843 CrossRef CAS.
- S. Hohloch, M. E. Garner, C. H. Booth, W. W. Lukens, C. A. Gould, D. J. Lussier, L. Maron and J. Arnold, Angew. Chem., Int. Ed., 2018, 57, 16136–16140 CrossRef CAS PubMed.
- B. E. Klamm, C. J. Windorff, C. Celis-Barros, M. L. Marsh, D. S. Meeker and T. E. Albrecht-Schmitt, Inorg. Chem., 2018, 57, 15389–15398 CrossRef CAS PubMed.
- A. Zalkin and K. N. Raymond, J. Am. Chem. Soc., 1969, 91, 5667–5668 CrossRef CAS.
- A. M. Tondreau, T. J. Duignan, B. W. Stein, V. E. Fleischauer, J. Autschbach, E. R. Batista, J. M. Boncella, M. G. Ferrier, S. A. Kozimor, V. Mocko, M. L. Neidig, S. K. Cary and P. Yang, Inorg. Chem., 2018, 57, 8106–8115 CrossRef CAS PubMed.
- J. W. Kück, M. R. Anneser, B. Hofmann, A. Pöthig, M. Cokoja and F. E. Kühn, ChemSusChem, 2015, 8, 4056–4063 CrossRef PubMed.
- M. R. Anneser, G. R. Elpitiya, J. Townsend, E. J. Johnson, X. B. Powers, J. F. DeJesus, K. D. Vogiatzis and D. M. Jenkins, Angew. Chem., Int. Ed., 2019, 58, 8115–8118 CrossRef CAS PubMed.
- P. L. Arnold, A. J. Blake and C. Wilson, Chem.–Eur. J., 2005, 11, 6095–6099 CrossRef CAS PubMed.
- D. Pugh, J. A. Wright, S. Freeman and A. A. Danopoulos, Dalton Trans., 2006, 775–782, 10.1039/b512133g.
- W. J. Evans, S. A. Kozimor and J. W. Ziller, Polyhedron, 2004, 23, 2689–2694 CrossRef CAS.
- A. Avdeef, K. N. Raymond, K. O. Hodgson and A. Zalkin, Inorg. Chem., 1972, 11, 1083–1088 CrossRef CAS.
- J. A. Butcher, J. Q. Chambers and R. M. Pagni, J. Am. Chem. Soc., 1978, 100, 1012–1013 CrossRef CAS.
- J. A. Butcher, R. M. Pagni and J. Q. Chambers, J. Organomet. Chem., 1980, 199, 223–227 CrossRef CAS.
- S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, D. L. Clark, S. A. Kozimor, R. L. Martin, D. K. Shuh and T. Tyliszczak, Chem. Sci., 2014, 5, 351–359 RSC.
- B. M. Gardner, J. McMaster and S. T. Liddle, Dalton Trans., 2009, 6924–6926 RSC.
- H. Nakai, X. Hu, L. N. Zakharov, A. L. Rheingold and K. Meyer, Inorg. Chem., 2004, 43, 855–857 CrossRef CAS PubMed.
- M. Vasiliu, K. A. Peterson, J. K. Gibson and D. A. Dixon, J. Phys. Chem. A, 2015, 119, 11422–11431 CrossRef CAS PubMed.
- K. A. Peterson, J. Chem. Phys., 2015, 142, 074105 CrossRef PubMed.
- M. Dolg and X. Cao, J. Phys. Chem. A, 2009, 113, 12573–12581 CrossRef CAS PubMed.
- A. Weigand, X. Cao, T. Hangele and M. Dolg, J. Phys. Chem. A, 2014, 118, 2519–2530 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. Van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. de Jong, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS.
- J. E. D. Glendening, K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2018 Search PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, J. Chem. Inf. Model., 2019, 59, 4814–4820 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details and characterization for all compounds is provided, including NMR spectra, IR spectra, UV-Vis spectra, cyclic voltammograms and X-ray diffraction data and computation data. CCDC 2047325–2047327. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01007g |
| ‡ Current address: Innovative Computing Laboratory, The University of Tennessee, Knoxville, TN 37996, USA. |
| § Current address: Department of Chemistry, University of California, Berkeley, CA 94720, USA. |
| ¶ Current address: Chemical Sciences Division, Lawrence Berkeley National Laboratory, One Cyclotron Road, Berkeley 94720, USA. E-mail: E-mail: pla@berkeley.edu. |
| This journal is © The Royal Society of Chemistry 2021 |