
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoactive electron donor–acceptor complex platform for Ni-mediated C(sp3)–C(sp2) bond formation†
Lisa Marie
Kammer‡
,
Shorouk O.
Badir‡
,
Ren-Ming
Hu
and
Gary A.
Molander
 *
*
Roy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, Pennsylvania 19104-6323, USA
First published on 5th March 2021
Abstract
A dual photochemical/nickel-mediated decarboxylative strategy for the assembly of C(sp3)–C(sp2) linkages is disclosed. Under light irradiation at 390 nm, commercially available and inexpensive Hantzsch ester (HE) functions as a potent organic photoreductant to deliver catalytically active Ni(0) species through single-electron transfer (SET) manifolds. As part of its dual role, the Hantzsch ester effects a decarboxylative-based radical generation through electron donor–acceptor (EDA) complex activation. This homogeneous, net-reductive platform bypasses the need for exogenous photocatalysts, stoichiometric metal reductants, and additives. Under this cross-electrophile paradigm, the coupling of diverse C(sp3)-centered radical architectures (including primary, secondary, stabilized benzylic, α-oxy, and α-amino systems) with (hetero)aryl bromides has been accomplished. The protocol proceeds under mild reaction conditions in the presence of sensitive functional groups and pharmaceutically relevant cores.
Introduction
Transition metal-catalyzed cross-couplings have become indispensable tools for the rapid assembly of C(sp3)–C(sp2) linkages in medicinal chemistry settings.1 Among these platforms, net-reductive cross-electrophile couplings are particularly advantageous because they facilitate the direct integration of alkyl electrophiles,1f,j,2 bypassing the need for preformed, reactive carbon nucleophiles.3 However, the vast majority of reductive cross-coupling reactions rely on (super)stoichiometric loadings of metal powders, including manganese and zinc as chemical reductants, to restore the active catalyst.1f,j,2 In addition to safety concerns with respect to metal waste disposal, the industry's dependence on these reaction paradigms highlights the necessity for inexpensive and scalable strategies for the incorporation of abundant feedstocks in cross-coupling manifolds.1d,4 Recently, several seminal studies have demonstrated the use of organic reducing agents in cross-electrophile processes. Pioneering work from Tanaka and colleagues showcased the use of tetrakis(dimethylamino)ethylene (TDAE) as a homogeneous organic reductant to achieve the homo-coupling of aryl halides.5 Subsequently, the Weix group utilized this reductant to activate C(sp3)-hybridized electrophiles.2a,6 In 2017, Reisman demonstrated that TDAE, in place of manganese or zinc, functions as a terminal organic reductant in the enantioselective cross-coupling of alkyl-N-hydroxyphthalimide esters (redox active esters, RAEs) with alkenyl bromides (Scheme 1A).7 These protocols, however, are complicated by the air-sensitive nature of the organic super-electron-donor.8 Strategies utilizing amines and tris(trimethylsilyl)silane as non-metallic reducing agents have also been disclosed.9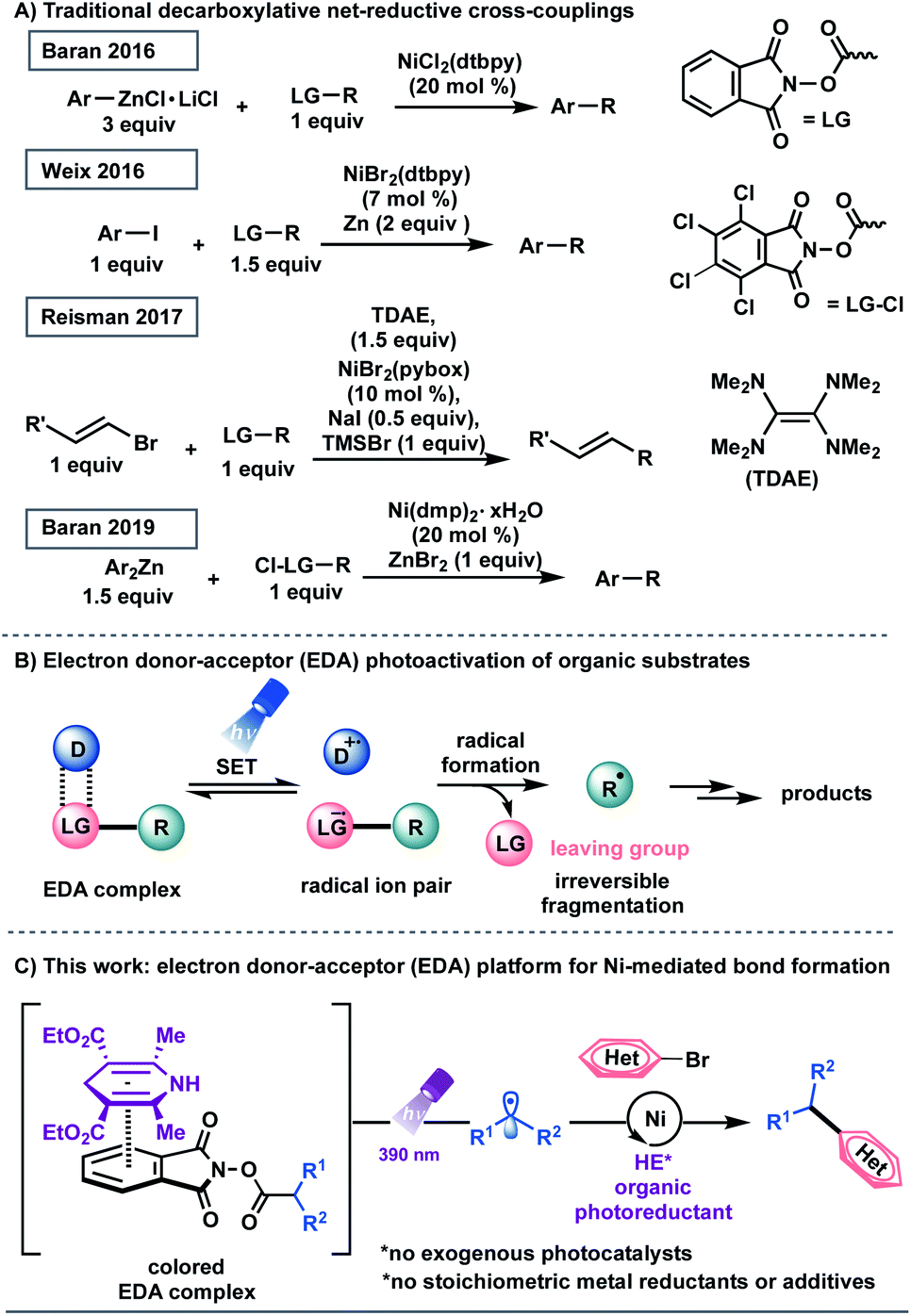 | ||
| Scheme 1 (A) Strategies toward net-reductive decarboxylative-based cross-couplings. (B) Overview of electron donor–acceptor (EDA) photoactivation. (C) Electron donor–acceptor (EDA) complex platform for Ni-mediated alkyl transfer using HE as an organic photoreductant. | ||
In recent years, photochemical methods have been enlisted to assemble challenging structural motifs using excitable catalysts under visible-light conditions. Such systems are inherently mild, efficient at room temperature, and evade the need for reactive additives (pyrophoric reagents, strong bases, harsh oxidants and reductants).10 In these dual manifolds, the reduced state of the photocatalyst has been proposed to restore the catalytically active Ni0 species through single-electron transfer (SET) events.11 However, the majority of these redox-active auxiliaries are based on precious metals including ruthenium and iridium, presenting limitations with respect to scalability and sustainability.12 These processes are further complicated by the oxidation/reduction steps of the photocatalyst. To establish a complementary reactivity mode, the Melchiorre group elegantly reported the direct photoexcitation of 4-alkyl-1,4-dihydropyridines (DHPs) to trigger the generation of C(sp3)-centered radicals in the absence of external catalysts.13,14 Although this advancement presented a milestone in its own right, the scope of the radical precursor in the reported Ni-catalyzed C(sp3)–C(sp2) cross-coupling was limited to secondary and stabilized primary systems13a owing to competitive C–H bond scission inherent to DHP feedstocks.15 In this context, the generation of heteroatom- and unactivated carbon-based radicals through direct visible-light excitation in Ni-catalyzed cross-couplings remains underdeveloped.
Recently, synthetic methods driven by the photoactivity of electron donor–acceptor (EDA) complexes (Scheme 1B) have gained considerable momentum, including borylation, thioetherification, and sulfonylation.16 Inspired by this advance, we examined the feasibility of EDA complex photoactivation as an enabling technology in Ni-mediated C(sp3)–C(sp2) cross-couplings (Scheme 1C). Under light irradiation at 390 nm, a commercially available and inexpensive electron donor, Hantzsch ester (HE, diethyl-1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylate), serves as a potent organic photoreductant to deliver diverse radical architectures from carboxylic acid feedstocks17 for further functionalization in Ni-catalyzed cross-couplings. As part of its dual role, the excited HE modulates the oxidation state of the transition-metal, delivering catalytically active Ni(0) species, thus bypassing the need for exogenous, expensive photocatalysts.
Discussion
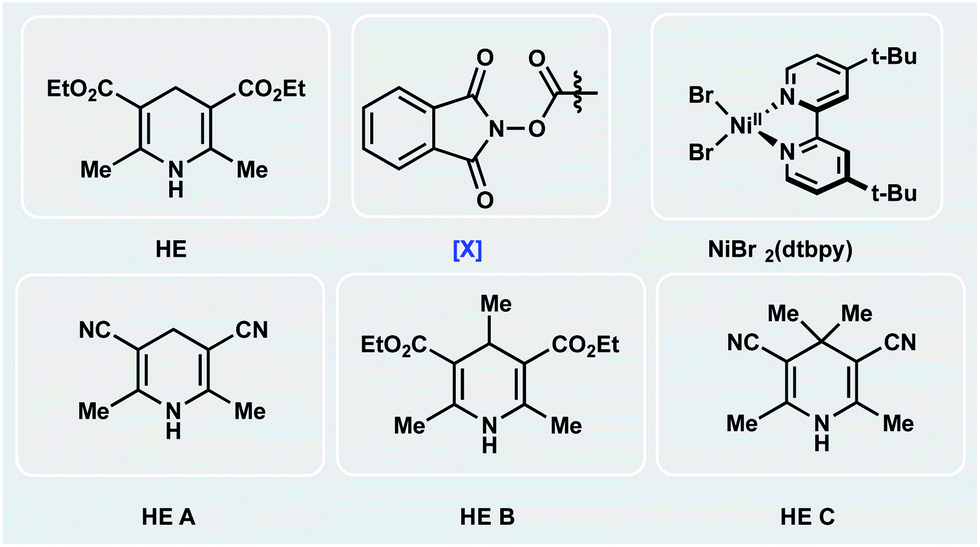
Encouraged by the potential synthetic applications of harnessing photoactive EDA complexes toward Ni-mediated bond formation, we examined the feasibility of the proposed net-reductive cross-electrophile coupling using 5-bromo-2-cyanopyridine 1 and cyclohexyl-N-hydroxyphthalimide-ester 2 as model substrates (Table 1). From the outset of our investigation, it was evident that the solvent plays a key role in this process (entries 1–4), as it heavily affects the molecular assembly and formation of EDA complexes. Once exciplex-based charge transfer occurs, the resulting radical ion pair is stabilized by interaction with solvent dipoles.18 In this vein, dimethylacetamide (DMA) proved crucial to the success of this photochemical method.|
|
|||||
|---|---|---|---|---|---|
| Entry | Deviation from std conditions | 3/IS ratiob | Entry | Deviation from std conditions | 3/IS ratiob |
| a Optimization reactions were performed using 1 (0.1 mmol), 2 (0.2 mmol), HE (0.2 mmol), and NiBr2(dtbpy) (10 mol%) in dry, degassed solvent (1.0 mL, 0.1 M) under purple Kessil irradiation for 24 h at rt. b Product to internal standard ratio (P/IS) was calculated using 1,3,5-trimethoxybenzene as internal standard using LC-MS analysis of the crude reaction mixture. c Isolated yield of 3 on 0.5 mmol scale. | |||||
| 1 | None | 1.42 (79%)c | 9 | Under air | 0.93 |
| 2 | DMSO | Traces | 10 | NiBr2(dMe-phen) | Traces |
| 3 | THF | Traces | 11 | NiBr2(bpy) | 1.32 |
| 4 | MeCN | 0.32 | 12 | NiBr2 (dMeObpy) | 1.37 |
| 5 | HE A | 1.40 | 13 | NiBr2·3H2O | 0.92 |
| 6 | HE B | 0.36 | 14 | No light | 0 |
| 7 | HE C | 0 | 15 | No Ni | 0 |
| 8 | Blue Kessil | 1.34 | 16 | No HE | Traces |
|
|||||
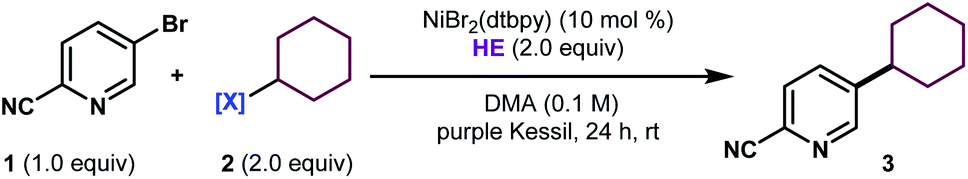
We then studied the influence of the dihydropyridine (DHP) backbone on the efficacy of the cross-coupling (Table 1, entries 5–7). To this end, we subjected four different DHP derivatives to the reaction conditions to gain a deeper understanding of their dual role in EDA complex photoactivation as well as the reduction of Ni species through SET paradigms. It was demonstrated that Hantzsch ester (HE) and cyano substitution at C3 and C5 of the DHP (HE A, entry 5) promoted the reaction most efficiently. For experimental simplicity, commercially available and inexpensive Hantzsch ester HE was adopted as the standard photoreductant. The C-4-substituted DHP (HE B, entry 6) resulted in diminished reactivity. Not surprisingly, 4,4′-dimethyl HE C (entry 7) led to no product formation. This result can be rationalized by the lack of photooxidative aromatization as an intrinsic driving force to supress a competitive back electron transfer (BET) event from the radical ion pair, restoring the ground-state EDA complex.16a,19 Finally, a ligand screen was performed (Table 1, entries 10–12), demonstrating that 4,4′-di-tert-butyl-2,2′-bipyridine (dtbpy), 2,2′-bipyridine (bpy), and electron-rich 4,4′-dimethoxy-2,2′-bipyridine (dMeObpy) function as viable ligand frameworks. Of note, modest conversion to 3 was observed using ligand-free nickel(II) bromide trihydrate.
Notably, the developed net-reductive photochemical conditions are user-friendly, employing an air-stable nickel precatalyst and a mild, homogeneous reductant (HE). Deviations from the standard reaction setup are tolerated. For example, modest product formation was observed when the reaction was carried out under air (Table 1, entry 9). Similarly, although superior reactivity was accomplished using purple Kessil irradiation (λmax = 390 nm), affording a potent photoreductant [Ered(HE*/HE˙+) = −2.28 V vs. SCE],20 comparable results were achieved under blue light (entry 8, λmax = 456 nm). Control experiments demonstrated that all reaction parameters are key to the formation of C(sp3)–C(sp2) linkages (Table 1, entries 14–16).
With suitable conditions established, we examined the scope of the decarboxylative arylation employing a broad palette of (hetero)aryl bromides (Scheme 2). In general, organic halides substituted with electron-withdrawing groups exhibited excellent reactivity, although electron-neutral and electron-donating groups also afforded the desired products in modest yields. More sterically-encumbered ortho-substituted aryl bromides (5, 9) did not hinder the cross-coupling efficacy. Furthermore, substitution at the meta position (4, 6) is tolerated. Several sensitive functional groups, including secondary sulfonamides (8, 19, 20, 31), ketones (4, 11, 18), trimethylsilylalkyne (9), and terminal alkene (22) remained intact under the developed photoredox conditions. To this end, substrates 6, 9 and 22 can be further diversified via Kumada-, sila-Sonogashira-couplings, as well as Giese-type additions, respectively.21
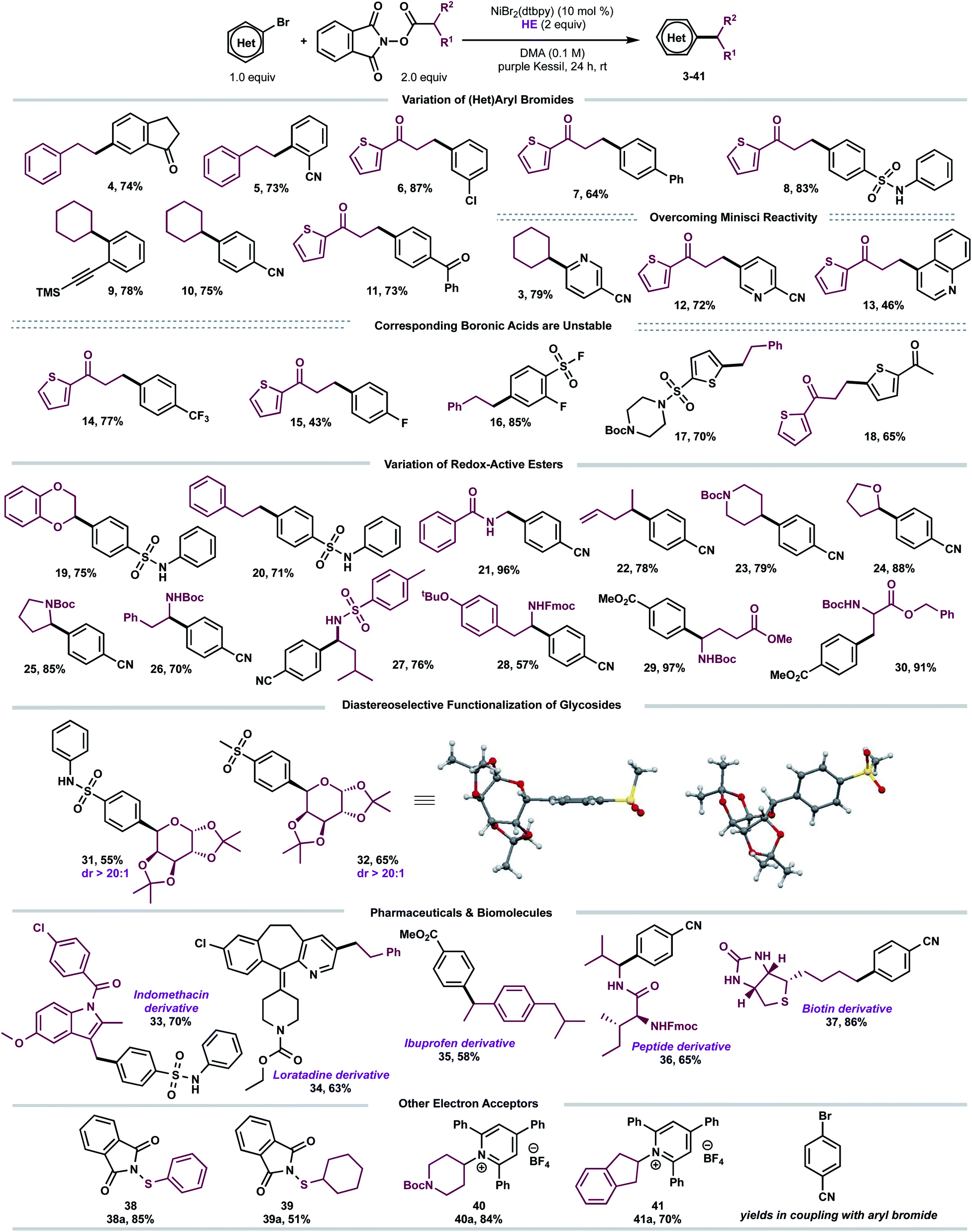 | ||
| Scheme 2 Scope of the developed C(sp3)–C(sp2) cross-coupling. All values correspond to isolated yields after purification. Reaction conditions as depicted in Table 1, entry 1 (0.5 mmol scale). | ||
Notably, several heteroarenes (electron-deficient: 3, 12, 13, 34 and electron-rich: 17, 18) were compatible structural motifs. In particular, nitrogen-containing heteroaryl bromides, including quinoline (13) and pyridine (3, 12, 34) scaffolds, reacted in a chemoselective fashion to yield C(sp3)–C(sp2) linkages, despite their propensity to undergo visible light-mediated Minisci C–H alkylation with alkyl-N-hydroxyphthalimide-esters.22 This demonstrates a complementary reactivity mode to existing Minisci protocols, delivering linchpins that drive molecular complexity.23
Next, the aliphatic photocoupling was evaluated with respect to redox-active carboxylate derivatives (Scheme 2). The reaction proceeded smoothly using a diverse array of proteinogenic and non-proteinogenic amino acids (21, 23, 25–30). Bifunctional reagents, including Boc- (17, 23, 25–26, 29–30) and Fmoc-protected (28) amines, afforded the arylated products without compromising yields. The scope was further extended to secondary, benzylic (35), and stabilized α-oxy (19, 24, 31, 32) radical architectures. Remarkably, primary alkyl systems that lack any radical stabilizing groups displayed exceptional reactivity (4–8, 11–18), providing a clear advantage in terms of scope over previously reported protocols.13a Of note, medicinally relevant structures including thiophene (6–8), piperidine (23), and pyrrolidine (25) motifs were efficiently incorporated.
To demonstrate the amenability of this cross-coupling for late-stage functionalization, including glycodiversification of drug scaffolds, photoredox-generated glycosyl radicals were successfully harnessed in this dual-catalytic manifold (Scheme 2). The desired C-aryl carbohydrates (31 and 32) were obtained in good yields and excellent diastereoselectivity (dr > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The relative configuration of the major diastereomer 32 was elucidated based on X-ray crystallography with the aryl group cis with respect to the dimethyl acetal protecting group (Scheme 2). Efficient decarboxylative arylation was observed with pharmaceutically relevant cores, displaying a high density of pendant functional groups, including indomethacin24 and loratadine25 precursors (33, 34). To evaluate the amenability toward bioactive molecules further, carboxylic acid derivatives stemming from dipeptide (36) and D-biotin (37) were subjected to the reaction conditions. The corresponding cross-coupled products were obtained in moderate to high yields (65–86%). Finally, to demonstrate the versatility of EDA paradigms toward Ni-catalyzed bond-forming processes, the cross-coupling was further extended to other electron acceptors including redox-active thiols (38a–39a)26 and pyridinium-activated amines (40a–41a)9b,16d,27 in the absence of external catalysts.
:1). The relative configuration of the major diastereomer 32 was elucidated based on X-ray crystallography with the aryl group cis with respect to the dimethyl acetal protecting group (Scheme 2). Efficient decarboxylative arylation was observed with pharmaceutically relevant cores, displaying a high density of pendant functional groups, including indomethacin24 and loratadine25 precursors (33, 34). To evaluate the amenability toward bioactive molecules further, carboxylic acid derivatives stemming from dipeptide (36) and D-biotin (37) were subjected to the reaction conditions. The corresponding cross-coupled products were obtained in moderate to high yields (65–86%). Finally, to demonstrate the versatility of EDA paradigms toward Ni-catalyzed bond-forming processes, the cross-coupling was further extended to other electron acceptors including redox-active thiols (38a–39a)26 and pyridinium-activated amines (40a–41a)9b,16d,27 in the absence of external catalysts.
To gain insight into the mechanism of this photochemical net-reductive cross-coupling, we analysed the reaction components by UV/vis absorption spectroscopy (Fig. 1). In line with seminal reports,16 although cyclohexyl-N-hydroxyphthalimide-ester shows absorption in the visible light region, mixtures of the RAE and HE in DMA at 0.2 M display a significant bathochromic shift (Fig. 1B, brick red and blue lines).
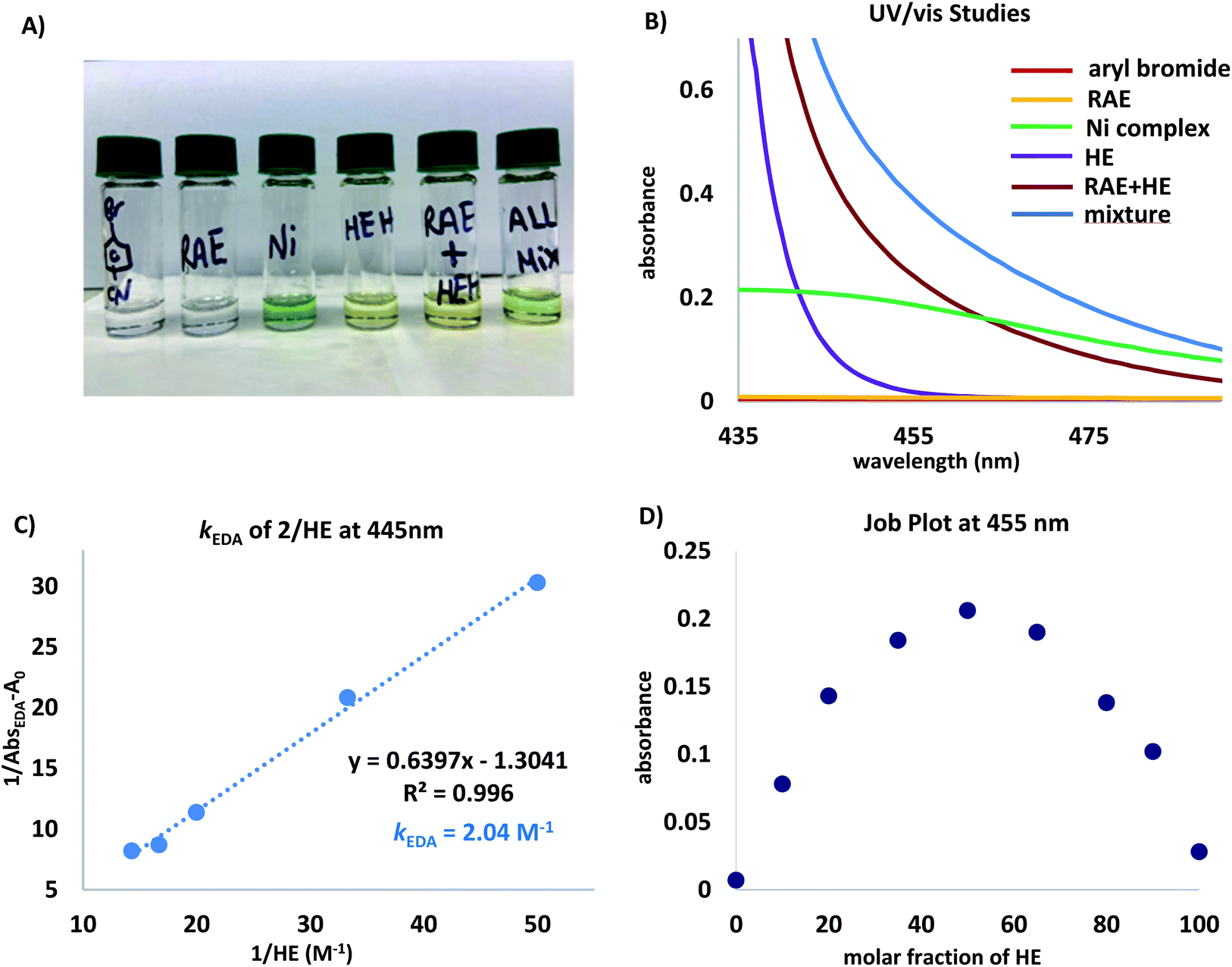 | ||
| Fig. 1 (A) Visual appearance of reaction components and mixtures thereof. (B) UV/vis absorption spectra measured in DMA (0.1 M) unless otherwise noted. Ni complex = NiBr2(dtbpy), aryl bromide = 4-bromobenzonitrile, and RAE = cyclohexyl-N-hydroxyphthalimide ester. Mixture refers to a DMA solution of all reaction components. (C) Benesi–Hildebrand plot.28 (D) Job plot29 for a mixture of N-(cyclohexyl)-hydroxyphthalimide ester (2) and HE in DMA (0.2 M). | ||
The absorption band (brick red line) stems from the formation of a new molecular aggregation, a colored EDA complex (Fig. 1A), exhibiting a wavelength band tailing to 500 nm. Preliminary studies revealed an association constant of 2.04 M−1 of HE with 2, indicating a plausible EDA complex association event prior to homolytic fragmentation (Fig. 1C). Analysis of this complex using Job's method29 revealed a 1:1 stoichiometry of the most absorbing species (Fig. 1D). Notably, concentration is a crucial parameter for effective cross-coupling. A dilute reaction mixture (10−4 M) exhibits a blue-shifted absorption band, indicating the inhibition of EDA complex formation (see ESI†). Furthermore, a DMA solution of HE was found to absorb visible light (λ > 400 nm), indicating selective photoexcitation of this species at 390 nm to generate a potent reducing agent (Fig. 1B, purple line). Under the optimized reaction conditions, near full recovery of pyridine was observed (relative to 2.0 equiv. of HE), demonstrating the role of excited HE as an organic single-electron donor in this system (Scheme 3A).
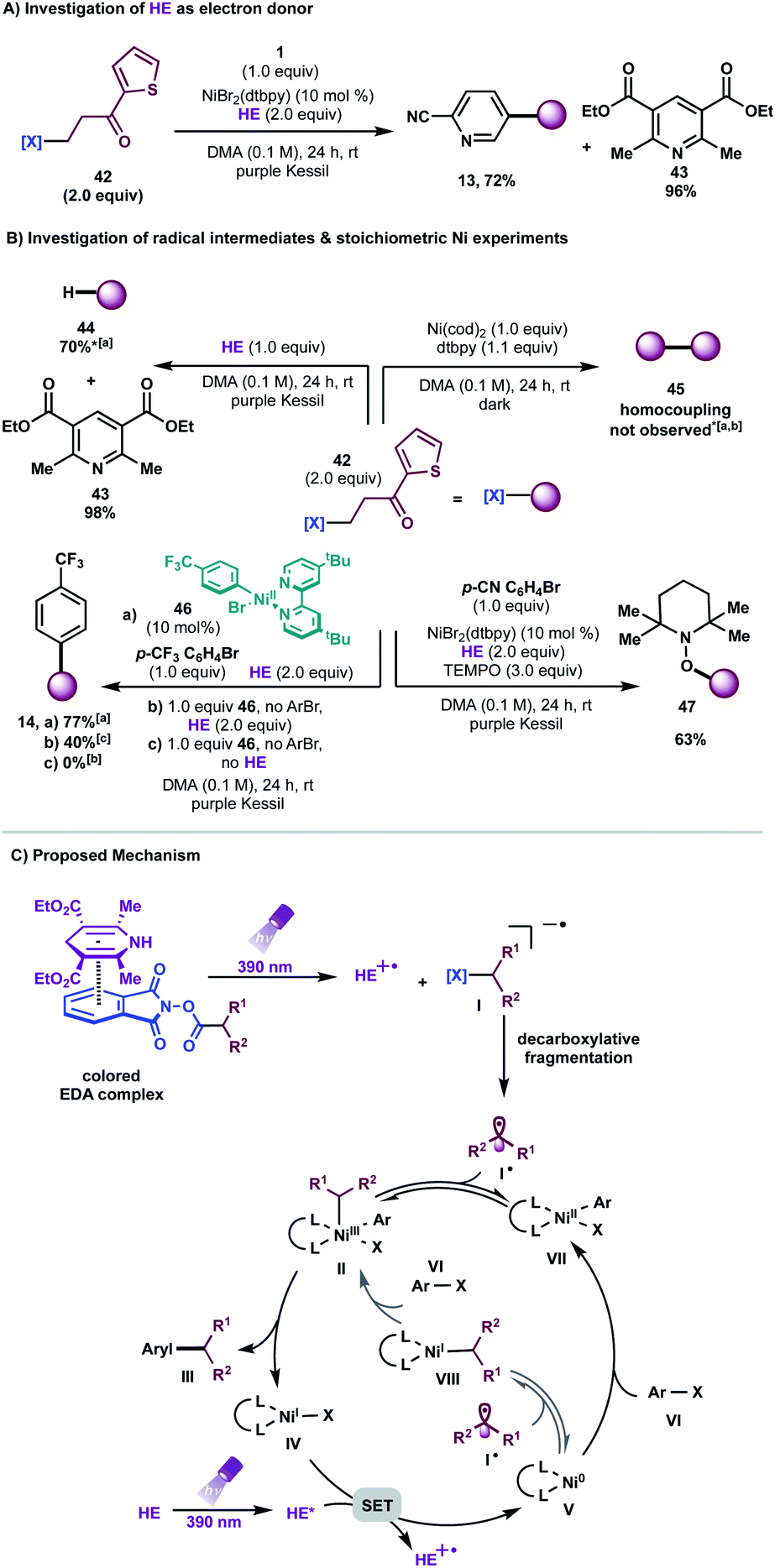 | ||
| Scheme 3 (A) Investigation of HE as electron donor. (B) Mechanistic experiments. [a]Isolated yield on 0.3 mmol scale, [b]analysed via GC-MS analysis, [c]NMR yield, *1.0 equiv. of 42. (C) Proposed mechanism. | ||
To probe the formation of alkyl radical intermediates, TEMPO trapping experiments were performed. The corresponding TEMPO-adduct was isolated in 63% yield and confirmed via HRMS analysis (Scheme 3B, bottom right). Stoichiometric experiments with Ni complex 46, synthesized through oxidative addition of 4-bromobenzotrifluoride to Ni(cod)2/dtbpy, revealed that C(sp3) alkyl transfer does not occur in the absence of HE, thus highlighting the likelihood of EDA complex activation for effective cross-coupling (Scheme 3B, bottom left). As anticipated, Ni complex 46 is catalytically active in the reaction, delivering the desired arylated product in 77% yield (Scheme 3B, bottom left).
Furthermore, negligible conversion of 42 was observed in the presence of stoichiometric amounts of Ni(COD)2/dtbpy; traces of homocoupling or alkene-side products, if any, were detected in the crude mixture, ruling out the role of Ni as a catalytic reductant toward redox-active esters (Scheme 3B, top right). Finally, it is worth noting that reduction of redox-active esters with photoredox catalysts is reported in the presence of HE as a hydrogen atom transfer (HAT) donor.30 A model reaction in the absence of Ni/dtbpy and aryl halide afforded the hydroalkylated product in 70% yield with full recovery of pyridine (Scheme 3B, top left). These findings demonstrate that the rate of alkyl radical addition to aryl-Ni(II) species VII must be faster than hydrogen atom abstraction from HE.
Based on these experiments, we propose a mechanistic scenario involving the intermediacy of an EDA complex between the electron-deficient, aliphatic redox-active esters and the electron-rich HE (Scheme 3C). Photoirradiation at 390 nm triggers an intra-complex SET event, generating a dihydropyridine radical cation and a phthalimide radical anion. The latter species undergoes decarboxylative fragmentation to yield an alkyl radical I˙. Although the absorptivity of the EDA complex is significantly greater than that of the Hantzsch ester itself, and although the entropic advantage inherent in intra-complex charge transfer provides an enormous rate enhancement over intermolecular SET, we cannot rule out the intervention of some direct electron transfer from the photoexcited Hantzsch ester to initiate radical formation from the RAE. This high-energy, C(sp3)-hybridized intermediate can suffer two potential fates. Based on previous computational studies,11 one plausible mechanistic pathway involves initial radical combination with a Ni0 species, generating a NiI intermediate that engages in oxidative addition with the aryl halides to produce high valent NiIII species II. Subsequent reductive elimination from this complex yields the desired cross-coupled product and the corresponding LnNiI species IV. At this juncture, a key SET event from the excited state HE [Ered(HE*/HE˙+) = −2.28 V vs. SCE20] to Ni [Ered(NiI/Ni0) = −1.17 V vs. SCE in THF10d] regenerates the active Ni0 catalyst. However, a process that involves initial oxidative addition of the aryl halides to Ni0 species V, affording aryl-NiII complex VII, cannot be ruled out based on stoichiometric experiments with Ni complex 46 (Scheme 3B). In this scenario, VII would engage the radical to generate NiIII complex II, which would then be carried on through the catalytic cycle.
Conclusion
In summary, we have demonstrated the implementation of an electron donor–acceptor (EDA) complex platform toward Ni-catalyzed C(sp3)–C(sp2) bond formation, circumventing the need for exogenous photocatalysts, additives, and stoichiometric metal reductants. Under the developed conditions, the generation of heteroatom- and unactivated carbon-based radicals through direct visible-light excitation of diverse electron acceptors is feasible, providing a clear advantage in terms of scope over previously reported photochemical systems.13 Upon light irradiation at 390 nm, excited HE functions as a potent organic photoreductant exhibiting a dual role: generation of reactive C(sp3)-hybridized radicals through EDA complex activation as well as restoration of the desired catalytically active Ni(0) species through SET paradigms. This decarboxylative cross-electrophile arylation is amenable to the synthesis of peptidomimetics, drug-like molecules, and the diastereoselective functionalization of carbohydrates. The commercial availability of carboxylic acids and related electron acceptor species facilitates the rapid incorporation of diverse carbon- and heteroatom-based radical architectures with high functional group tolerance. Key mechanistic and spectroscopic studies highlight the necessity for EDA photoactivation for efficient alkyl transfer and help inform the design of improved EDA-based paradigms in Ni-catalyzed cross-couplings.Author contributions
Shorouk O. Badir conceived the topic. Lisa Marie Kammer, Shorouk O. Badir, and Ren-Ming Hu completed experiments with input from Gary A. Molander. Shorouk O. Badir and Lisa Marie Kammer prepared the manuscript with input from Gary A. Molander.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support provided by NIGMS (R35 GM 131680 to G. M.). Shorouk O. Badir is supported by the Bristol-Myers Squibb Graduate Fellowship for Synthetic Organic Chemistry. The NSF Major Research Instrumentation Program (award NSF CHE-1827457), the NIH supplement awards 3R01GM118510-03S1 and 3R01GM087605-06S1, as well as the Vagelos Institute for Energy Science and Technology supported the purchase of the NMRs used in this study. We thank Dr Charles W. Ross, III (UPenn) for mass spectral data and Dr Patrick Carroll (UPenn) for X-ray crystallography data. Merck is acknowledged for donation of aryl halide informer sets. Access to a UV/vis spectrophotometer was provided by the Petersson lab (UPenn). We thank Kessil for donation of lamps.Notes and references
- For selected examples see: (a) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS; (b) P. G. Gildner and T. J. Colacot, Organometallics, 2015, 34, 5497–5508 CrossRef CAS; (c) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed; (d) L.-C. Campeau and N. Hazari, Organometallics, 2019, 38, 3–35 CrossRef CAS PubMed; (e) J. Choi and G. C. Fu, Science, 2017, 356, 7230 CrossRef PubMed; (f) J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 2, 1411–1421 RSC; (g) X. Hu, Chem. Sci., 2011, 2, 1867–1886 RSC; (h) A. Kaga and S. Chiba, ACS Catal., 2017, 7, 4697–4706 CrossRef CAS; (i) X. Ma, B. Murray and M. R. Biscoe, Nat. Rev. Chem., 2020, 4, 584–599 CrossRef CAS PubMed; (j) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767–1775 CrossRef CAS PubMed.
- For selected examples see: (a) D. A. Everson, R. Shrestha and D. J. Weix, J. Am. Chem. Soc., 2010, 132, 920–921 CrossRef CAS PubMed; (b) D. A. Everson and D. J. Weix, J. Org. Chem., 2014, 79, 4793–4798 CrossRef CAS PubMed; (c) J. Gu, C. Qiu, W. Lu, Q. Qian, K. Lin and H. Gong, Synthesis, 2017, 49, 1867–1873 CrossRef CAS; (d) X. Yu, T. Yang, S. Wang, H. Xu and H. Gong, Org. Lett., 2011, 13, 2138–2141 CrossRef CAS PubMed; (e) B. P. Woods, M. Orlandi, C.-Y. Huang, M. S. Sigman and A. G. Doyle, J. Am. Chem. Soc., 2017, 139, 5688–5691 CrossRef CAS PubMed; (f) Y. Jin, H. Yang and C. Wang, Org. Lett., 2019, 21, 7602–7608 CrossRef CAS PubMed; (g) N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2015, 137, 10480–10483 CrossRef CAS PubMed; (h) C. E. I. Knappke, S. Grupe, D. Gärtner, M. Corpet, C. Gosmini and A. Jacobi von Wangelin, Chem.–Eur. J., 2014, 20, 6828–6842 CrossRef CAS PubMed; (i) T. Moragas, A. Correa and R. Martin, Chem.–Eur. J., 2014, 20, 8242–8258 CrossRef CAS PubMed; (j) Z.-X. Tian, J.-B. Qiao, G.-L. Xu, X. Pang, L. Qi, W.-Y. Ma, Z.-Z. Zhao, J. Duan, Y.-F. Du, P. Su, X.-Y. Liu and X.-Z. Shu, J. Am. Chem. Soc., 2019, 141, 7637–7643 CrossRef CAS PubMed; (k) A. H. Cherney and S. E. Reisman, J. Am. Chem. Soc., 2014, 136, 14365–14368 CrossRef CAS PubMed; (l) K. M. M. Huihui, J. A. Caputo, Z. Melchor, A. M. Olivares, A. M. Spiewak, K. A. Johnson, T. A. DiBenedetto, S. Kim, L. K. G. Ackerman and D. J. Weix, J. Am. Chem. Soc., 2016, 138, 5016–5019 CrossRef CAS PubMed; for selected examples using electrochemical methods, see: (m) H. Li, C. P. Breen, H. Seo, T. F. Jamison, Y.-Q. Fang and M. M. Bio, Org. Lett., 2018, 20, 1338–1341 CrossRef CAS PubMed; (n) T. Koyanagi, A. Herath, A. Chong, M. Ratnikov, A. Valiere, J. Chang, V. Molteni and J. Loren, Org. Lett., 2019, 21, 816–820 CrossRef CAS PubMed; for cross-electrophile coupling in biological environments, see: (o) D. T. Flood, S. Asai, X. Zhang, J. Wang, L. Yoon, Z. C. Adams, B. C. Dillingham, B. B. Sanchez, J. C. Vantourout, M. E. Flanagan, D. W. Piotrowski, P. Richardson, S. A. Green, R. A. Shenvi, J. S. Chen, P. S. Baran and P. E. Dawson, J. Am. Chem. Soc., 2019, 141, 9998–10006 CrossRef CAS PubMed.
- For selected examples on decarboxylative arylation using carbon preformed nucleophiles, see: (a) T.-G. Chen, H. Zhang, P. K. Mykhailiuk, R. R. Merchant, C. A. Smith, T. Qin and P. S. Baran, Angew. Chem., Int. Ed., 2019, 58, 2454–2458 CrossRef CAS PubMed; (b) J. Cornella, J. T. Edwards, T. Qin, S. Kawamura, J. Wang, C.-M. Pan, R. Gianatassio, M. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 2174–2177 CrossRef CAS PubMed; (c) X.-G. Liu, C.-J. Zhou, E. Lin, X.-L. Han, S.-S. Zhang, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2018, 57, 13096–13100 CrossRef CAS PubMed; (d) F. Toriyama, J. Cornella, L. Wimmer, T.-G. Chen, D. D. Dixon, G. Creech and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 11132–11135 CrossRef CAS PubMed.
- M. J. Buskes and M.-J. Blanco, Molecules, 2020, 25, 3493 CrossRef CAS PubMed.
- (a) M. Kuroboshi, Y. Waki and H. Tanaka, Synlett, 2002, 2002, 0637–0639 CrossRef; (b) M. Kuroboshi, Y. Waki and H. Tanaka, J. Org. Chem., 2003, 68, 3938–3942 CrossRef CAS PubMed.
- (a) L. L. Anka-Lufford, K. M. M. Huihui, N. J. Gower, L. K. G. Ackerman and D. J. Weix, Chem.–Eur. J., 2016, 22, 11564–11567 CrossRef CAS PubMed; (b) D. A. Everson, B. A. Jones and D. J. Weix, J. Am. Chem. Soc., 2012, 134, 6146–6159 CrossRef CAS PubMed; for a dual catalytic system utilizing TDAE as reductant, see: (c) D. J. Charboneau, E. L. Barth, N. Hazari, M. R. Uehling and S. L. Zultanski, ACS Catal., 2020, 10, 12642–12656 CrossRef CAS PubMed.
- N. Suzuki, J. L. Hofstra, K. E. Poremba and S. E. Reisman, Org. Lett., 2017, 19, 2150–2153 CrossRef CAS PubMed.
- (a) C. Burkholder, W. R. Dolbier and M. Médebielle, J. Org. Chem., 1998, 63, 5385–5394 CrossRef CAS; (b) A. Kolomeitsev, M. Médebielle, P. Kirsch, E. Lork and G.-V. Röschenthaler, J. Chem. Soc., Perkin Trans. 1, 2000, 2183–2185 RSC; (c) F. Glaser, C. B. Larsen, C. Kerzig and O. S. Wenger, Photochem. Photobiol. Sci., 2020, 19, 1035–1041 CrossRef CAS PubMed.
- For selected examples, see: (a) K. Shimomaki, K. Murata, R. Martin and N. Iwasawa, J. Am. Chem. Soc., 2017, 139, 9467–9470 CrossRef CAS PubMed; (b) J. Yi, S. O. Badir, L. M. Kammer, M. Ribagorda and G. A. Molander, Org. Lett., 2019, 21(9), 3346–3351 CrossRef CAS PubMed; (c) T. Q. Chen and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2019, 58, 14584–14588 CrossRef CAS PubMed; (d) Q.-Y. Meng, T. E. Schirmer, K. Katou and B. König, Angew. Chem., Int. Ed., 2019, 58, 5723–5728 CrossRef CAS PubMed; (e) F. Cong, X.-Y. Lv, C. S. Day and R. Martin, J. Am. Chem. Soc., 2020, 142, 20594–20599 CrossRef CAS PubMed; (f) H. Guan, Q. Zhang, P. J. Walsh and J. Mao, Angew. Chem., Int. Ed., 2020, 59, 5172–5177 CrossRef CAS PubMed; (g) S. Patel, S. O. Badir and G. A. Molander, Trends Chem., 2021, 3, 161–175 CrossRef CAS PubMed; (h) P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087 CrossRef CAS PubMed.
- For selected examples, see: (a) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS PubMed; (b) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS; (c) D. N. Primer, I. Karakaya, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2015, 137, 2195–2198 CrossRef CAS; (d) B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed; (e) D. R. Heitz, K. Rizwan and G. A. Molander, J. Org. Chem., 2016, 81, 7308–7313 CrossRef CAS PubMed; (f) J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef CAS PubMed; (g) J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 2563–2575 CrossRef CAS PubMed; (h) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef CAS PubMed; (i) S. O. Badir and G. A. Molander, Chem, 2020, 6, 1327–1339 CrossRef CAS; (j) A. Lipp, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2021, 60, 1714–1726 CrossRef CAS PubMed.
- O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896–4899 CrossRef CAS PubMed.
- K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef CAS PubMed.
- For selected examples, see: (a) L. Buzzetti, A. Prieto, S. R. Roy and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 15039–15043 CrossRef CAS PubMed; (b) G. Goti, B. Bieszczad, A. Vega-Peñaloza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 1213–1217 CrossRef CAS PubMed; (c) T. van Leeuwen, L. Buzzetti, L. A. Perego and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 131, 5007–5011 CrossRef; (d) E. Gandolfo, X. Tang, S. Raha Roy and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 16854–16858 CrossRef CAS PubMed; (e) B. Bieszczad, L. A. Perego and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 16878–16883 CrossRef CAS PubMed; (f) N. Alandini, L. Buzzetti, G. Favi, T. Schulte, L. Candish, K. D. Collins and P. Melchiorre, Angew. Chem., Int. Ed., 2020, 59, 5248–5253 CrossRef CAS PubMed; (g) L. Cardinale, M. O. Konev and A. J. von Wangelin, Chem.–Eur. J., 2020, 26, 8239 CrossRef CAS PubMed.
- For selected example on the direct excitation of boracene-based alkylborates, see: Y. Sato, K. Nakamura, Y. Sumida, D. Hashizume, T. Hosoya and H. Ohmiya, J. Am. Chem. Soc., 2020, 142, 9938–9943 CrossRef CAS PubMed.
- (a) R. S. Varma and D. Kumar, Tetrahedron Lett., 1999, 40, 21–24 CrossRef CAS; (b) X. Bi, T. Tang, X. Meng, M. Gou, X. Liu and P. Zhao, Catal. Sci. Technol., 2020, 10, 360–371 RSC; (c) C. Zheng and S.-L. You, Chem. Soc. Rev., 2012, 41, 2498–2518 RSC; (d) G.-B. Shen, L. Xie, H.-Y. Yu, J. Liu, Y.-H. Fu and M. Yan, RSC Adv., 2020, 10, 31425–31434 RSC.
- For selected examples, see: (a) G. E. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS; (b) A. Noble, R. S. Mega, D. Pflästerer, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 2155–2159 CrossRef CAS PubMed; (c) B. Liu, C.-H. Lim and G. Miyake, J. Am. Chem. Soc., 2017, 139(39), 13616–13619 CrossRef CAS PubMed; (d) J. Wu, P. S. Grant, X. Li, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 5697–5701 CrossRef CAS PubMed; (e) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS; (f) L. Chen, J. Liang, Z.-Y. Chen, J. Chen, M. Yan and X.-j. Zhang, Adv. Synth. Catal., 2019, 361, 956–960 CAS; (g) J. Zhang, Y. Li, R. Xu and Y. Chen, Angew. Chem., Int. Ed., 2017, 129, 12793–12797 CrossRef; (h) D. Chen, L. Xu, T. Long, S. Zhu, J. Yang and L. Chu, Chem. Sci., 2018, 9, 9012–9017 RSC; (i) C. Zheng, G.-Z. Wang and R. Shang, Adv. Synth. Catal., 2019, 361, 4500–4505 CrossRef CAS.
- S. Murarka, Adv. Synth. Catal., 2018, 360, 1735–1753 CrossRef CAS.
- (a) B. Lipp and T. Opatz, in Photochemistry, RSC, 2019, vol. 46, pp. 370–394 Search PubMed; (b) G. J. Kavarnos and N. J. Turro, Chem. Rev., 1986, 86, 401–449 CrossRef CAS; (c) N. J. Turro, Pure Appl. Chem., 1981, 53, 259 CAS; (d) N. J. Turro, Modern Molecular Photochemistry, Benjamin/Cummings, Menlo Park, CA, 1978 Search PubMed; (e) N. J. Turro, V. Rammamurthy, W. Cherry and W. Farneth, Chem. Rev., 1978, 78, 125 CrossRef CAS.
- (a) R. Foster, J. Phys. Chem., 1980, 84, 2135–2141 CrossRef CAS; (b) S. V. Rosokha and J. K. Kochi, Acc. Chem. Res., 2008, 41, 641–653 CrossRef CAS PubMed.
- J. Jung, J. Kim, G. Park, Y. You and E. J. Cho, Adv. Synth. Catal., 2016, 358, 74–80 CrossRef CAS.
- (a) B. Giese, Angew. Chem., Int. Ed., 1983, 22, 753–764 CrossRef; (b) J. Huang and S. P. Nolan, J. Am. Chem. Soc., 1999, 121, 9889–9890 CrossRef CAS; (c) G. Dyker, Angew. Chem., Int. Ed., 1999, 38, 1698–1712 CrossRef.
- (a) T. C. Sherwood, N. Li, A. N. Yazdani and T. G. M. Dhar, J. Org. Chem., 2018, 83(5), 3000–3012 CrossRef CAS; (b) L. Kammer, A. Rahman and T. Opatz, Molecules, 2018, 23, 764 CrossRef PubMed; (c) R. A. Garza-Sanchez, A. Tlahuext-Aca, G. Tavakoli and F. Glorius, ACS Catal., 2017, 7, 4057–4061 CrossRef CAS; (d) M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429–1434 CrossRef CAS PubMed.
- F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575–3579 CrossRef CAS.
- G. Alván, M. Orme, L. Bertilsson, R. Ekstrand and L. Palmér, Clin. Pharmacol. Ther., 1975, 18, 364–373 CrossRef PubMed.
- R. S. Geha and E. O. Meltzer, J. Allergy Clin. Immunol., 2001, 107, 751–762 CrossRef CAS PubMed.
- (a) S. Graßl, C. Hamze, T. J. Koller and P. Knochel, Chem.–Eur. J., 2019, 25, 3752–3755 CrossRef PubMed; (b) V. P. Reddy, A. V. Kumar, K. Swapna and K. R. Rao, Org. Lett., 2009, 11, 1697–1700 CrossRef CAS; (c) B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616–13619 CrossRef CAS.
- R. Martin-Montero, V. R. Yatham, H. Yin, J. Davies and R. Martin, Org. Lett., 2019, 21(8), 2947–2951 CrossRef CAS.
- H. A. Benesi and J. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- J. S. Renny, L. L. Tomasevich, E. H. Tallmadge and D. B. Collum, Angew. Chem., Int. Ed., 2013, 52, 11998–12013 CrossRef CAS PubMed.
- For selected examples, see: G. Pratsch, G. L. Lackner and L. E. Overman, J. Org. Chem., 2015, 80, 6025–6036 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational details, as well as spectral data. CCDC 2062315. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00943e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |