
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric synthesis of dihydro-1,3-dioxepines by Rh(II)/Sm(III) relay catalytic three-component tandem [4 + 3]-cycloaddition†
Chaoran
Xu
,
Jianglin
Qiao
,
Shunxi
Dong
 *,
Yuqiao
Zhou
,
Xiaohua
Liu
and
Xiaoming
Feng
*
*,
Yuqiao
Zhou
,
Xiaohua
Liu
and
Xiaoming
Feng
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: dongs@scu.edu.cn; xmfeng@scu.edu.cn
First published on 8th March 2021
Abstract
Heterocycles have been widely used in organic synthesis, agrochemical, pharmaceutical and materials science industries. Catalytic three-component ylide formation/cycloaddition enables the assembly of complex heterocycles from simple starting materials in a highly efficient manner. However, asymmetric versions remain a yet-unsolved task. Here, we present a new bimetallic catalytic system for tackling this challenge. A combined system of Rh(II) salt and chiral N,N′-dioxide–Sm(III) complex was established for promoting the unprecedented tandem carbonyl ylide formation/asymmetric [4 + 3]-cycloaddition of aldehydes and α-diazoacetates with β,γ-unsaturated α-ketoesters smoothly, affording various chiral 4,5-dihydro-1,3-dioxepines in up to 97% yield, with 99% ee. The utility of the current method was demonstrated by conversion of products to optically active multi-substituted tetrahydrofuran derivatives. A possible reaction mechanism was provided to elucidate the origin of chiral induction based on experimental studies and X-ray structures of catalysts and products.
Introduction
α-Diazocarbonyl compounds are widely used in organic synthesis due to their diverse reactivity profiles.1 In particular, formation of various ylides A between α-diazocarbonyl compounds and carbonyl,2a–i imine2j or thiocarbonyl2k groups, followed by subsequent cycloaddition reactions provides a concise and efficient route to heterocyclic molecules with multiple stereocenters (Scheme 1a), and thus has attracted considerable attention in the past several decades. This strategy has been successfully applied to the total synthesis of complex natural products.3 Among them, catalytic asymmetric cycloadditions4via intramolecular carbonyl ylide formation of functionalized diazo-compounds have been well-established using chiral dirhodium salts4a–c or combined catalysts.4d–f,2j On the other hand, the transformations of ylides initiated by an intermolecular reaction between an α-diazocarbonyl compound and aldehydes or imines5 have been extended to other reaction partners, including olefins,6a–c alkynes,6d–h aldehydes or ketones6i–n and imines,6o,p enabling rapid buildup of molecular complexity from simple starting materials (Scheme 1b). However, to the best of our knowledge, the highly catalytic enantioselective versions have remained unknown to date.7 The difficulty stems mainly from the following: (1) the competing formation of epoxides6h,i,k,o or dioxolanes6a,f,i,o with additional aldehydes hampers the subsequent cycloaddition with dipolarophiles (Scheme 1c); (2) rhodium catalysts could induce a background reaction, and both a Rh-associated ylide7b and a free ylide6i were proposed in the catalytic cycloaddition process, while chiral dirhodium catalysts only provided poor enantioselectivity.6i,7b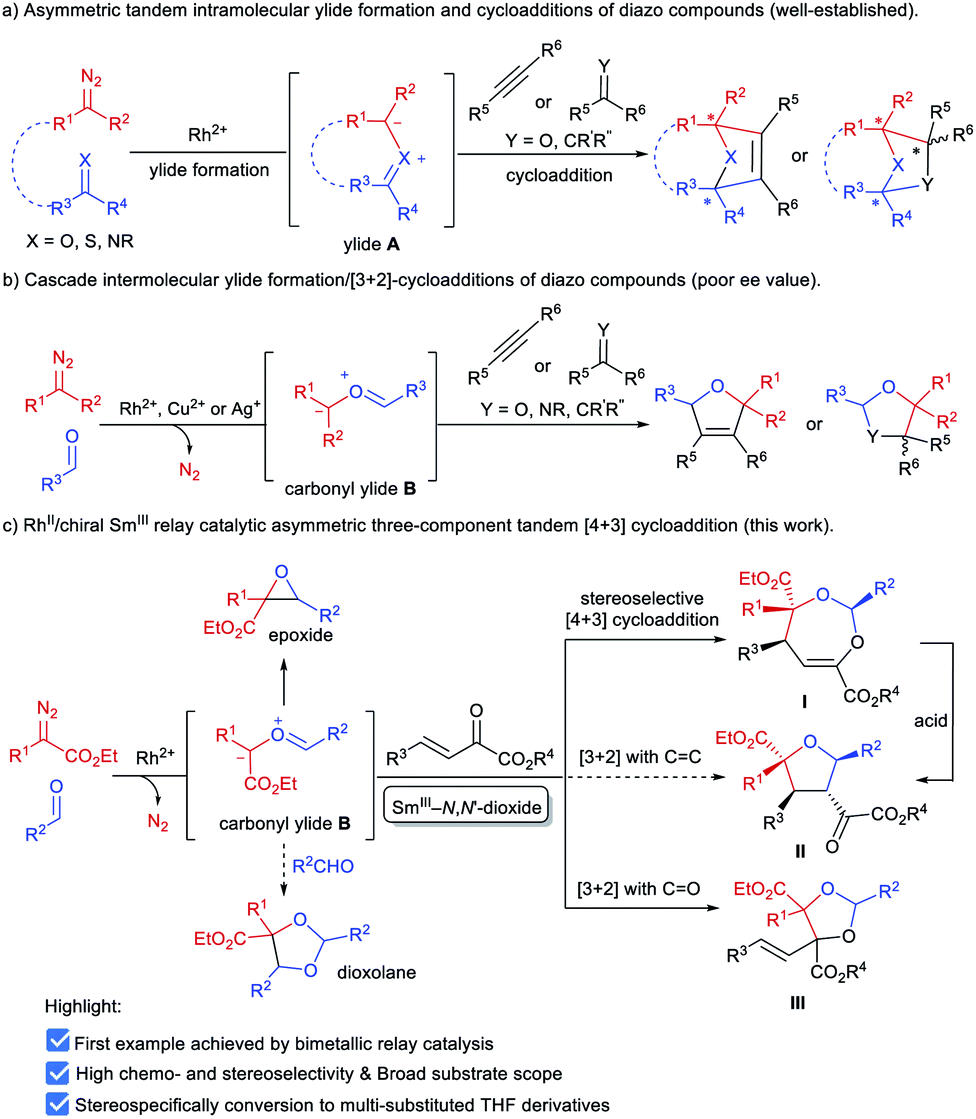 | ||
| Scheme 1 Ylide formation/cycloadditions of diazo compounds. | ||
Experiments in the literature reveal that in some cases Lewis acid catalysts are able to accelerate the cycloaddition of the in situ generated carbonyl ylide with several dipolarophiles.6a,o,p,7a Intrigued by the high performance of chiral N,N′-dioxide metal complex catalysts8 in cycloaddition reactions9 and others, we speculated that a bimetallic relay catalysis10 could potentially realize this challenging three-component tandem reaction. First, in the presence of an achiral dirhodium complex, the reaction of an aldehyde and α-diazo compound leads to carbonyl ylide B.3a Then a chiral Lewis acid catalyst preferably accelerates the subsequent stereoselective cycloaddition with a suitable dipolarophile, but not the side reactions. Herein, we wish to disclose our endeavor along this line. Rh2Piv4 and Sm(OTf)3/L4-PrPr2 combined catalysts were identified to be efficient for triggering an unprecedented enantioselective tandem ylide formation/[4 + 3]-cycloaddition reaction among β,γ-unsaturated α-ketoesters, aldehydes and α-diazoacetates. A number of chiral and densely functionalized 4,5-dihydro-1,3-dioxepines I were obtained in high yield with excellent diastereo- and enantioselectivity (Scheme 1c). It is worth noting that 4,5-dihydro-1,3-dioxepines are important intermediates in the synthesis of γ-butyrolactone derivatives,11 2-arylpropionic acids,12 and substituted tetrahydrofurans.13 Stereospecific conversion of the products to synthetically useful poly-substituted tetrahydrofuran derivatives II, the formal [3 + 2] adducts, was established as well.
Results and discussion
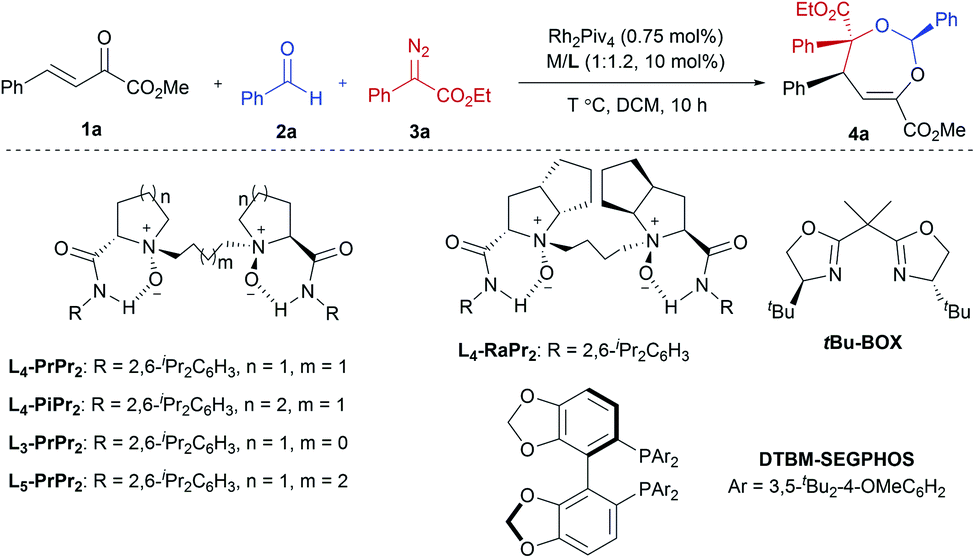
Our initial attempts involved using β,γ-unsaturated α-ketoester 1a14 as the model dipolarophile together with benzaldehyde (2a) and ethyl 2-diazo-2-phenylacetate (3a) to optimize the reaction conditions. As depicted in Scheme 1c, several competition pathways were possible including [3 + 2] of either a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond or CO bond, or [4 + 3] of CC–CO bonds. The reaction took place smoothly under the influence of Rh2Piv4 and a chiral N,N′-dioxide L4-PrPr2/Yb(OTf)3 complex at −20 °C in dichloromethane, affording 4,5-dihydro-1,3-dioxepine through a new paradigm of [4 + 3] route in 58% isolated yield with 67% ee for the major diastereomer (Table 1, entry 1). The 1H NMR spectrum of the crude mixture indicated that the other diastereomer of 1,3-dioxepine (ca. 18% yield) and [3 + 2] adduct III with the CO group of the α-ketoester (ca. 7% yield) were generated as well (for more details, see Table S6 in the ESI†). Subsequent investigation indicated that lowering the temperature from −20 °C to −78 °C resulted in a highly diastereoselective process, and the desired [4 + 3] product 4a was yielded exclusively with maintaining the ee value (entry 2, 90% yield, 66% ee). Next, several rare-earth metal salts were tested with N,N′-dioxide L4-PrPr2 as the ligand (see the ESI for details†). It was found that Sm(OTf)3 gave a higher ee value (entry 3, 84% ee). Then, screening of the backbone of the ligands suggested that L-proline derived L4-PrPr2 afforded better results than (S)-piperidine-2-carboxylic acid or L-ramipril derived ones (entries 4 and 5). The length of the carbon tether of N,N′-dioxide ligands was found to have a significant influence on the outcomes, and a four-carbon linker was superior to three- or five-carbon ones in terms of yield and enantioselectivity (entry 3 vs. entries 6 and 7). Other chiral ligands, such as tBu-Box and DTBM-SEGPHOS, gave poor results (entries 8 and 9). Without Rh2Piv4, the three-component reaction did not occur at all, even when camphor sulfonic acid15 was used as the alternative for carbene generation from diazo compounds (see Table S6 in the ESI for details†). When the reaction was carried out with only Rh(II), a mixture of at least four isomeric products (I and III) was obtained (entry 10, 4a, trace yield). The use of chiral Rh2(S-DOSP)4 furnished a racemic product (see the ESI for more details†), implying that a free carbonyl ylide might be involved. Performing the reaction with Rh(II) and Sm(OTf)3 led to the consumption of the aldehyde and diazoester and the recovery of the ketoester, but only a trace amount of the cycloaddition adduct (entry 11) was obtained. The aforementioned results clearly indicated that the chiral Lewis acid catalyst accelerated the subsequent [4 + 3]-cycloaddition and controlled the chemo- and stereoselectivity of this process. To our delight, increasing the amounts of 2a and 3a can elevate the enantioselectivity obviously, affording 4a in 92% yield and 99% ee (entry 12).
C bond or CO bond, or [4 + 3] of CC–CO bonds. The reaction took place smoothly under the influence of Rh2Piv4 and a chiral N,N′-dioxide L4-PrPr2/Yb(OTf)3 complex at −20 °C in dichloromethane, affording 4,5-dihydro-1,3-dioxepine through a new paradigm of [4 + 3] route in 58% isolated yield with 67% ee for the major diastereomer (Table 1, entry 1). The 1H NMR spectrum of the crude mixture indicated that the other diastereomer of 1,3-dioxepine (ca. 18% yield) and [3 + 2] adduct III with the CO group of the α-ketoester (ca. 7% yield) were generated as well (for more details, see Table S6 in the ESI†). Subsequent investigation indicated that lowering the temperature from −20 °C to −78 °C resulted in a highly diastereoselective process, and the desired [4 + 3] product 4a was yielded exclusively with maintaining the ee value (entry 2, 90% yield, 66% ee). Next, several rare-earth metal salts were tested with N,N′-dioxide L4-PrPr2 as the ligand (see the ESI for details†). It was found that Sm(OTf)3 gave a higher ee value (entry 3, 84% ee). Then, screening of the backbone of the ligands suggested that L-proline derived L4-PrPr2 afforded better results than (S)-piperidine-2-carboxylic acid or L-ramipril derived ones (entries 4 and 5). The length of the carbon tether of N,N′-dioxide ligands was found to have a significant influence on the outcomes, and a four-carbon linker was superior to three- or five-carbon ones in terms of yield and enantioselectivity (entry 3 vs. entries 6 and 7). Other chiral ligands, such as tBu-Box and DTBM-SEGPHOS, gave poor results (entries 8 and 9). Without Rh2Piv4, the three-component reaction did not occur at all, even when camphor sulfonic acid15 was used as the alternative for carbene generation from diazo compounds (see Table S6 in the ESI for details†). When the reaction was carried out with only Rh(II), a mixture of at least four isomeric products (I and III) was obtained (entry 10, 4a, trace yield). The use of chiral Rh2(S-DOSP)4 furnished a racemic product (see the ESI for more details†), implying that a free carbonyl ylide might be involved. Performing the reaction with Rh(II) and Sm(OTf)3 led to the consumption of the aldehyde and diazoester and the recovery of the ketoester, but only a trace amount of the cycloaddition adduct (entry 11) was obtained. The aforementioned results clearly indicated that the chiral Lewis acid catalyst accelerated the subsequent [4 + 3]-cycloaddition and controlled the chemo- and stereoselectivity of this process. To our delight, increasing the amounts of 2a and 3a can elevate the enantioselectivity obviously, affording 4a in 92% yield and 99% ee (entry 12).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Metal salt | Ligand | T (°C) | Yieldb (%) | eec (%) |
a Unless otherwise noted, the reaction was carried out with Rh2Piv4 (0.75 mol%), metal salt/ligand (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2, 10 mol%), β,γ-unsaturated α-ketoester 1a (0.10 mmol), aldehyde 2a (3.0 equiv.) and α-diazo ester 3a (3.0 equiv.) at T °C for 10 h under a N2 atmosphere.
b Isolated yield of the major diastereomer.
c Determined by HPLC analysis on a chiral stationary phase.
d
2a (4.0 equiv.) and 3a (4.0 equiv.). :1.2, 10 mol%), β,γ-unsaturated α-ketoester 1a (0.10 mmol), aldehyde 2a (3.0 equiv.) and α-diazo ester 3a (3.0 equiv.) at T °C for 10 h under a N2 atmosphere.
b Isolated yield of the major diastereomer.
c Determined by HPLC analysis on a chiral stationary phase.
d
2a (4.0 equiv.) and 3a (4.0 equiv.).
|
|||||
| 1 | Yb(OTf)3 | L4-PrPr2 | −20 | 58 | 67 |
| 2 | Yb(OTf)3 | L4-PrPr2 | −78 | 90 | 66 |
| 3 | Sm(OTf)3 | L4-PrPr2 | −78 | 90 | 84 |
| 4 | Sm(OTf)3 | L4-PiPr2 | −78 | 68 | 32 |
| 5 | Sm(OTf)3 | L4-RaPr2 | −78 | 50 | 26 |
| 6 | Sm(OTf)3 | L3-PrPr2 | −78 | 38 | 26 |
| 7 | Sm(OTf)3 | L5-PrPr2 | −78 | 85 | 54 |
| 8 | Sm(OTf)3 | tBu-Box | −78 | 56 | 11 |
| 9 | Sm(OTf)3 | DTBM-SEGPHOS | −78 | 57 | 0 |
| 10 | — | — | −78 | Trace | — |
| 11 | Sm(OTf)3 | — | −78 | Trace | — |
| 12d | Sm(OTf)3 | L4-PrPr2 | −78 | 92 | 99 |
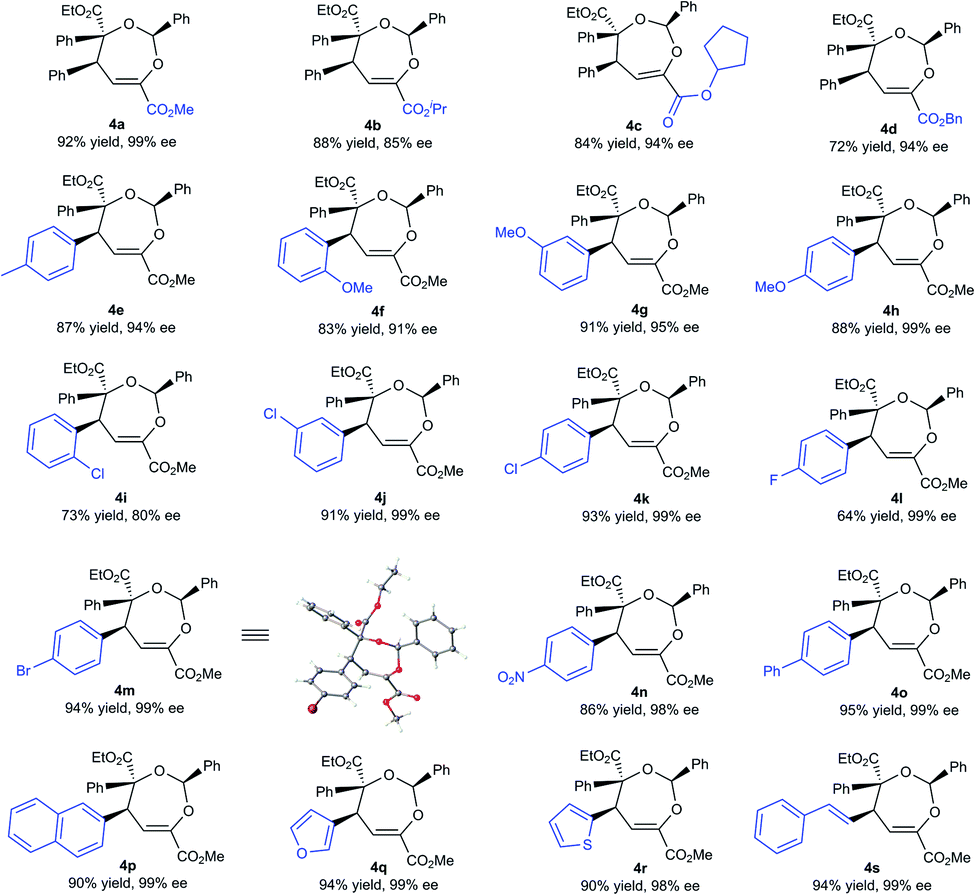
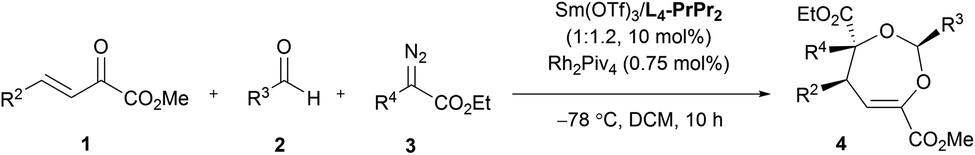
With the optimized conditions in hand, the substrate scope was next examined. As shown in Table 2, changing the ester group (R1) of the β,γ-unsaturated α-ketoester from a methyl to an isopropyl, a cyclopentyl or a benzyl group led to slightly reduced yields and ee values (4a–4d, 72–92% yield, 85–99% ee). The γ-aryl substituted β,γ-unsaturated α-ketoesters with electron-withdrawing or electron-donating substituents at 2-, 3-, and 4-positions of the phenyl group can react with benzaldehyde 2a and α-diazoester 3a smoothly to furnish the corresponding products 4e–4o in moderate to good yields (64–95% yield) and high ee values (80–99% ee). Generally, 2-substituted ones (1f and 1i) provided diminished yield and enantiomeric excess. Additionally, 2-naphthyl and heteroaromatic substrates were tolerated in this cascade reaction as well (4p–4r). Notably, β,γ,δ,ε-unsaturated α-ketoester 1s was a competent reaction partner, providing the desired [4 + 3] adduct 4s in 94% yield and 99% ee. The absolute configuration of the major enantiomer of 4m was determined to be (2S,4S,5S) by X-ray diffraction analysis, and the others were assigned by comparing their CD spectra with those of 4m.16
|
|
|---|
|
a The reaction was run with Rh2Piv4 (0.75 mol%), Sm(OTf)3/L4-PrPr2 (1:1.2, 10 mol%), β,γ-unsaturated α-ketoester 1 (0.10 mmol), aldehyde 2a (4.0 equiv.) and α-diazo ester 3a (4.0 equiv.) at −78 °C for 10 h. Yield is the isolated yield of the endo diastereoisomer. ee value was determined by HPLC analysis on a chiral stationary phase.
|
|
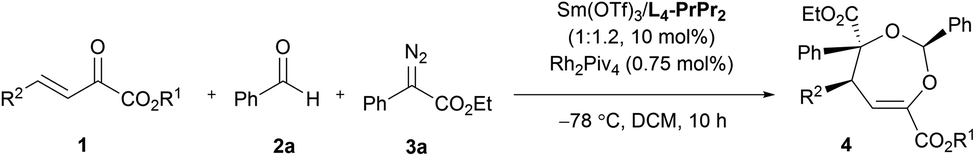
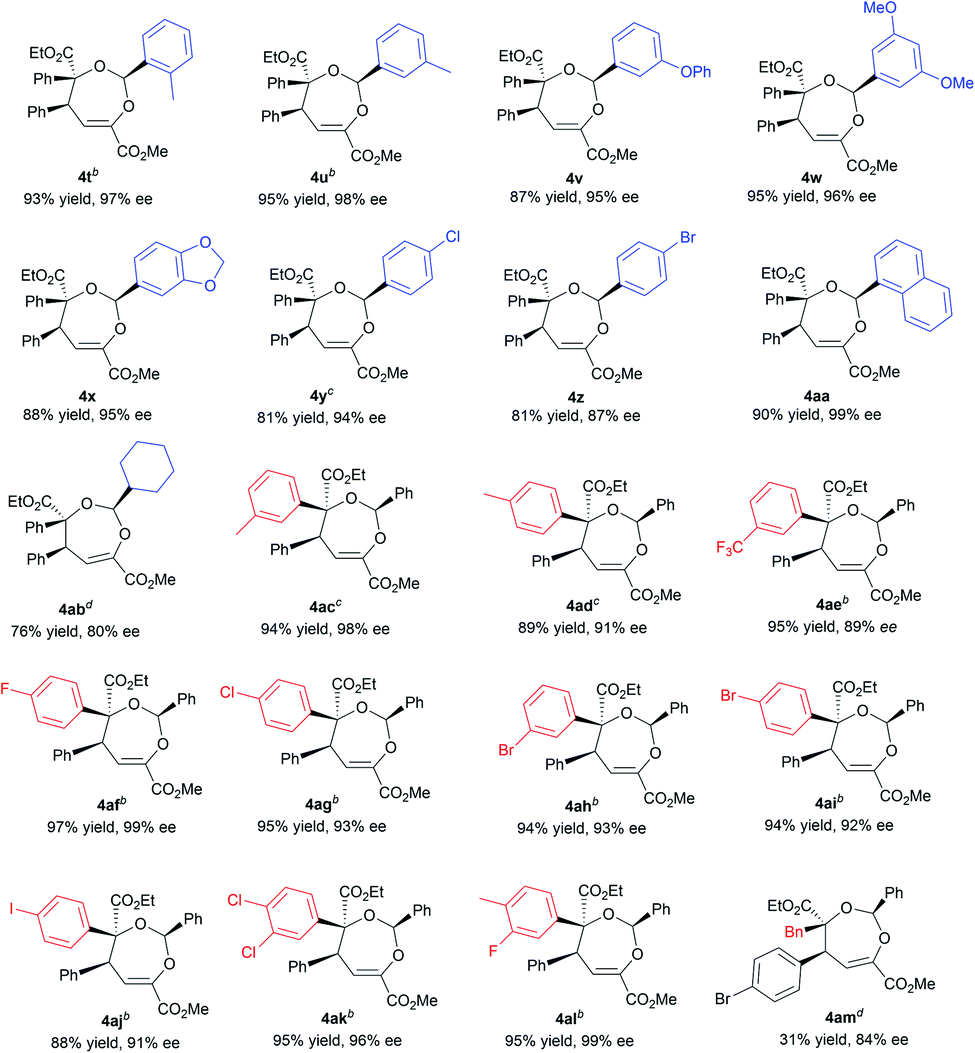
Next, various aldehydes 2 and α-diazoacetates 3 were tested (Table 3). According to the reactivity of the aldehydes, the amount of the α-diazoacetates was adjusted slightly in order to get high yield and ee. Satisfactorily, using β,γ-unsaturated α-ketoester 1a and α-phenyl diazoacetate 3a as the reaction partner, arylaldehydes 2b–2h with different electron-withdrawing or electron-donating substituents were readily converted into the corresponding products 4t–4z (81–95% yield and 87–98% ee). In this case, para-chloro- or bromo-substituted ones exhibited low reactivity and enantioselective control. 1-Naphthaldehyde 2i and cyclohexyl formaldehyde 2j were compatible in the current transformation, delivering the adducts 4aa and 4ab with good results. Furthermore, changing the phenyl group on the α-diazoacetates 3 had a limited influence on the reaction, regardless of the position and electronic effect of substituents on the phenyl group (4ac–4al, 88–97% yield, 89–99% ee). Benzyl substituted α-diazoacetate was suitable as well, affording the product 4am with 84% ee, However, low yield (31% yield) was obtained due to the elimination reaction of the α-diazoacetate.6i
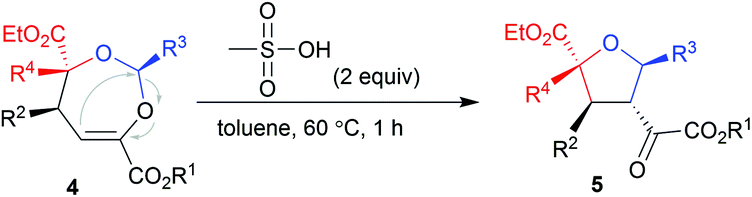
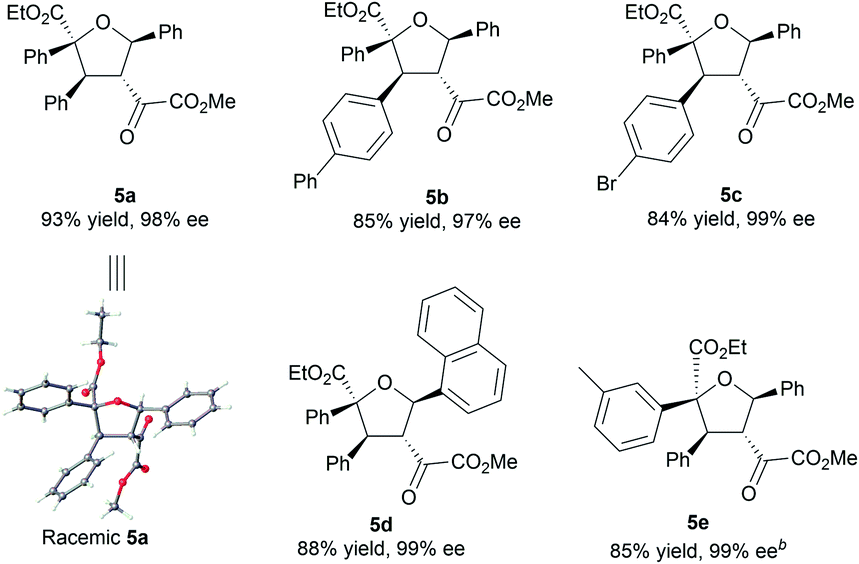
To illustrate the utility of the current methodology, further conversion of the 4,5-dihydro-1,3-dioxepines 4 was carried out. As shown in Table 4, compound 4a was stereospecifically transformed into tetrahydrofuran (THF) derivative 5a with four continuous stereogenic centers in the presence of methanesulfonic acid at 60 °C. Under such conditions, representative products 4 with different substituents rearranged to THF-derivatives 5 in high yield (84–93% yield) without loss of enantioselectivity (97–99% ee). Interestingly, compounds 5 were the formal [3 + 2]-adducts of carbonyl ylide B with the CC double bond of the β,γ-unsaturated α-ketoester (Scheme 1c, II), which could make up for the lack of the catalytic asymmetric version of the related [3 + 2] cascade reaction.17 The absolute configuration of 5a was assigned as (2S,3S,4R,5R) on the basis of the absolute configuration of 4a and the relative configuration of racemic 5a.16 The others were assigned by comparing their CD spectra with those of 5a. In addition, the alkenyl ester group in compound 4a was reduced by LiAlH4 (2.2 equiv.) to give the alcohol 6a in 76% yield with 99% ee (Scheme 2, left). Hydrogenation of 4a with Pd/C and H2 yielded the saturated 1,3-dioxepane 7a in 98% yield, 2:1 dr and 99/99% ee value (Scheme 2, right).
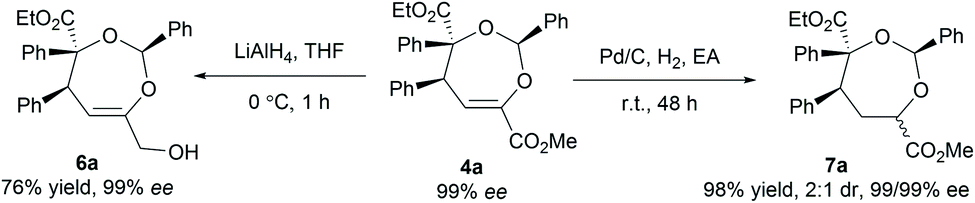 | ||
| Scheme 2 Further transformations of 4a. | ||
In order to understand the chiral induction of the N,N′-dioxide–Sm(III) Lewis acid catalyst, we got the crystals of the Sm(III) complexes of chiral N,N′-dioxides Ln-PrPr2 with different lengths of the carbon tether (n = 3, 4, or 5).16,18 The X-ray crystal structures show that each N,N′-dioxide coordinates as a tetradentate ligand to Sm(III) with two amine oxide oxygen atoms and two amide oxygen atoms, forming a square antiprismatic geometry version through the coordination with four additional species. It is interesting that L4-PrPr2 is noteworthy in view of the angles between OC–Sm–OC (narrowest, 138.5°) and ON–Sm–ON (widest, 94.4°), and the shortest bond distance of OC–Sm (2.393 and 2.423 Å) and ON–Sm (2.266 and 2.286 Å) in comparison with L3-PrPr2 and L5-PrPr2 (see the ESI for more details†). It implies that the alkyl linker between the two aminoxides affects both the geography and the electronic nature of the catalyst, delivering distinct activity and stereoselectivity.
Based on the absolute configuration of the products and control experiments, a possible reaction pathway was proposed (Scheme 3). Initially, free carbonyl ylide B is generated in situ from α-diazoacetate 3a and aldehyde 2a with the assistance of achiral Rh(II). Considering the fact that excess amounts of aldehyde and α-diazoacetate are advantageous to improve the enantioselectivity, we speculated that interaction between carbonyl ylide intermediate B and chiral SmIII–N,N′-dioxide exists, which is also supported by the operando IR experiment (see the ESI for details†). The high-coordination number of the rare-earth metal complex catalyst enabled the activation of the β,γ-unsaturated α-ketoester with Sm(III)/L4-PrPr2 in a bidentate-bonding manner at the same time. Therefore, a possible working mode was set forth to explain the stereoselectivity of the cycloaddition step. Due to the steric hindrance of the 2,6-iPr2C6H3 moiety in the chiral N,N′-dioxide ligand, the β-Re face of the unsaturated ketoester was blocked, leaving the less-hindered β-Si face available for attack by ylide B with its Re–Re face in an endo fashion.19 As a result, (2S,4S,5S)-4a was formed as the major enantiomer.
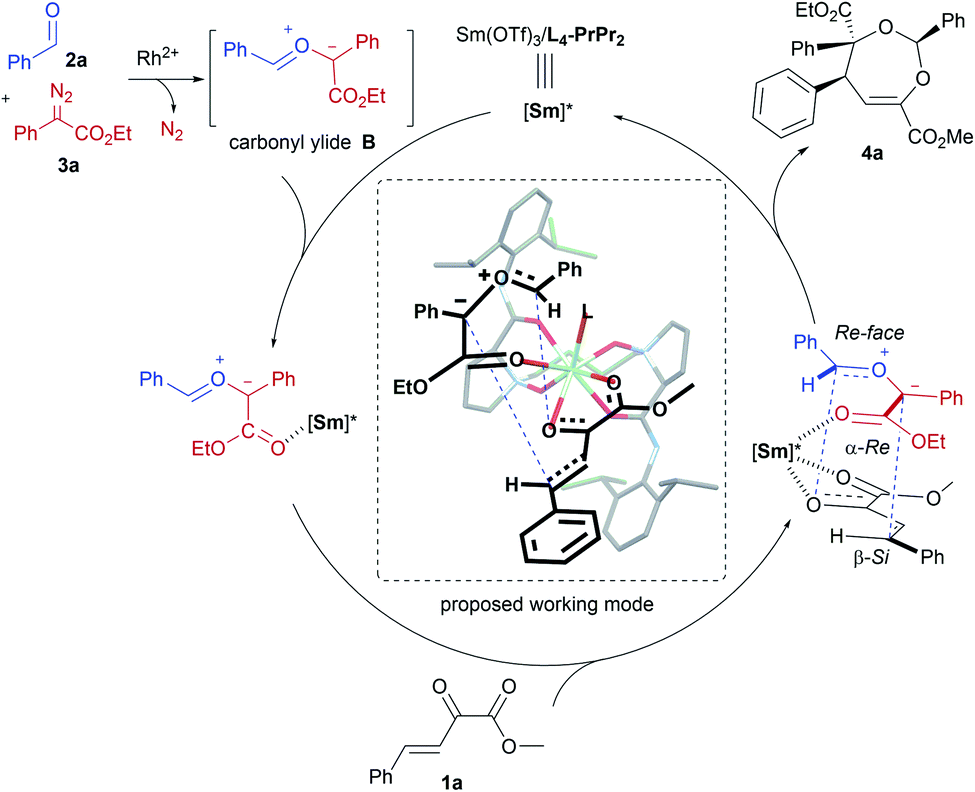 | ||
| Scheme 3 Possible reaction mechanism and working model. | ||
Conclusions
In summary, a highly efficient asymmetric cascade carbonyl ylide formation/[4 + 3]-cycloaddition reaction of β,γ-unsaturated α-ketoesters, aldehydes, and α-diazoacetates was realized by using a rhodium(II)/chiral N,N′-dioxide–Sm(III) complex bimetallic catalyst. Various chiral 4,5-dihydro-1,3-dioxepines were readily obtained in high yield with excellent ee values. Moreover, the 1,3-dioxepines can stereospecifically transform into multi-substituted tetrahydrofuran derivatives efficiently. A plausible catalytic cycle along with a working mode was proposed to explain the formation of enantioenriched products according to the experimental evidence and single crystal data. Further uses of the bimetallic relay catalysis strategy in other reactions are under investigation.Author contributions
C. R. X. performed the experiments. J. L. Q. repeated data. S. X. D. participated in structure characterization and discussion. Y. Q. Z. analyzed the X-ray diffraction crystal data. X. M. F. and X. H. L. supervised the project. X. M. F., X. H. L., S. X. D. and C. R. X. co-wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We appreciate the financial support from the National Natural Science Foundation of China (21890723 and 21772127).Notes and references
- For selected reviews on α-diazocarbonyl compounds: (a) N. R. Candeias, R. Paterna and P. M. P. Gois, Chem. Rev., 2016, 116, 2937 CrossRef CAS PubMed; (b) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS; (c) S. F. Zhu and Q. L. Zhou, Acc. Chem. Res., 2012, 45, 1365 CrossRef CAS PubMed; (d) Y. Zhang and J. B. Wang, Chem. Commun., 2009, 5350 RSC; (e) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed; (f) G. Maas, Chem. Soc. Rev., 2004, 33, 183 RSC; for selected recent examples involving α-diazoesters: (g) J. Yang, P. R. Ruan, W. Yang, X. M. Feng and X. H. Liu, Chem. Sci., 2019, 10, 10305 RSC; (h) Z. K. Xue, Y. Li and S. Z. Luo, ACS Catal., 2020, 10, 10989 CrossRef CAS; (i) R. D. C. Gallo, P. B. Momo, D. P. Day and A. C. B. Burtoloso, Org. Lett., 2020, 22, 2339 CrossRef CAS PubMed; (j) Y. Li, Y. T. Zhao, T. Zhou, M. Q. Chen, Y. P. Li, M. Y. Huang, Z. C. Xu, S. F. Zhu and Q. L. Zhou, J. Am. Chem. Soc., 2020, 142, 10557 CrossRef CAS PubMed.
- For selected examples: (a) M. E. Maier and K. Evertz, Tetrahedron Lett., 1988, 29, 1677 CrossRef CAS; (b) R. Angell, M. Fengler-Veith, H. Finch, L. M. Harwood and T. T. Tucker, Tetrahedron Lett., 1997, 38, 4517 CrossRef CAS; (c) H. Mizoguchi, H. Oguri, K. Tsuge and H. Oikawa, Org. Lett., 2009, 11, 3016 CrossRef CAS PubMed; (d) S. N. Savinov and D. J. Austin, Org. Lett., 2002, 4, 1415 CrossRef CAS PubMed; (e) S. A. Bonderoff and A. Padwa, J. Org. Chem., 2017, 82, 642 CrossRef CAS PubMed; (f) K. Inoue, H. Suga, S. Inoue, H. Sato and A. Kakehi, Synthesis, 2003, 9, 1413 Search PubMed; (g) Q. Q. Cheng, J. Yedoyan, H. Arman and M. P. Doyle, Angew. Chem., Int. Ed., 2016, 55, 5573 CrossRef CAS PubMed; (h) F. Rodier, M. Rajzmann, J. L. Parrain, G. Chouraqui and L. Commeiras, Chem.–Eur. J., 2013, 19, 2467 CrossRef CAS PubMed; (i) T. Ibata and J. Toyoda, Bull. Chem. Soc. Jpn., 1986, 59, 2489 CrossRef CAS; (j) Z. J. Jia, G. Shan, C. G. Daniliuc, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2018, 57, 14493 CrossRef CAS PubMed; (k) A. Padwa, F. R. Kinder, W. R. Nadler and L. Zhi, Heterocycles, 1993, 35, 367 CrossRef CAS.
- For selected reviews involving cycloaddition of carbonyl ylides: (a) A. Padwa and S. F. Hornbuckle, Chem. Rev., 1991, 91, 263 CrossRef CAS; (b) A. Padwa and M. D. Weingarten, Chem. Rev., 1996, 96, 223 CrossRef CAS PubMed; (c) A. Padwa, Chem. Soc. Rev., 2009, 38, 3072 RSC; (d) J. J. Medvedev and V. A. Nikolaev, Russ. Chem. Rev., 2015, 84, 737 CrossRef CAS.
- For selected examples: (a) D. M. Hodgson, P. A. Stupple, F. Y. T. M. Pierard, A. H. Labande and C. Johnstone, Chem.–Eur. J., 2001, 7, 4465 CrossRef CAS PubMed; (b) S. Murarka, Z. J. Jia, C. Merten, C. G. Daniliuc, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2015, 54, 7653 CrossRef CAS PubMed; (c) S. Kitagaki, M. Anada, O. Kataoka, K. Matsuno, C. Umeda, N. Watanabe and S. Hashimoto, J. Am. Chem. Soc., 1999, 121, 1417 CrossRef CAS; (d) C. R. Xu, K. X. Wang, D. W. Li, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2019, 58, 18438 CrossRef CAS PubMed; (e) H. Suga, Y. Hashimoto, S. Yasumura, R. Takezawa, K. Itoh and A. Kakehi, J. Org. Chem., 2013, 78, 10840 CrossRef CAS PubMed; (f) H. Suga, K. Inoue, S. Inoue and A. Kakehi, J. Am. Chem. Soc., 2002, 124, 14836 CrossRef CAS PubMed.
- Selected reviews on azomethine ylide obtained from other methods involving cycloadditions: (a) L. Wei, X. Chang and C. J. Wang, Acc. Chem. Res., 2020, 53, 1084 CrossRef CAS PubMed; (b) G. Pandey, P. Banerjee and S. R. Gadre, Chem. Rev., 2006, 106, 4484 CrossRef CAS PubMed; (c) R. Narayan, M. Potowski, Z. J. Jia, A. P. Antonchick and H. Waldmann, Acc. Chem. Res., 2014, 47, 1296 CrossRef CAS PubMed.
- For selected examples: (a) Y. Hashimoto, K. Itoh, A. Kakehi, M. Shiro and H. Suga, J. Org. Chem., 2013, 78, 6182 CrossRef CAS PubMed; (b) M. Alt and G. Maas, Tetrahedran, 1994, 50, 7435 CrossRef CAS; (c) A. J. Skaggs, E. Y. Lin and T. F. Jamison, Org. Lett., 2002, 4, 2277 CrossRef CAS PubMed; (d) S. Muthusamy, C. Gunanathan and M. Nethaji, J. Org. Chem., 2004, 69, 5631 CrossRef CAS PubMed; (e) M. P. Doyle, D. C. Forbes, M. N. Protopopova, S. A. Stanley, M. M. Vasbinder and K. R. Xavier, J. Org. Chem., 1997, 62, 7210 CrossRef CAS PubMed; (f) T. Rajasekaran, G. Karthik, B. Sridhar and B. V. S. Reddy, Org. Lett., 2013, 15, 1512 CrossRef CAS PubMed; (g) A. DeAngelis, M. T. Taylor and J. M. Fox, J. Am. Chem. Soc., 2009, 131, 1101 CrossRef CAS PubMed; (h) Y. F. Liu, Z. Wang, J. W. Shi, B. L. Chen, Z. G. Zhao and Z. L. Chen, J. Org. Chem., 2015, 80, 12733 CrossRef CAS PubMed; (i) A. DeAngelis, P. Panne, G. P. A. Yap and J. M. Fox, J. Org. Chem., 2008, 73, 1435 CrossRef CAS PubMed; (j) P. D. March and R. Huisgen, J. Am. Chem. Soc., 1982, 104, 4952 CrossRef; (k) C. D. Lu, Z. Y. Chen, H. Liu, W. H. Hu and A. Q. Mi, Org. Lett., 2004, 6, 3071 CrossRef CAS PubMed; (l) B. Jiang, X. B. Zhang and Z. H. Luo, Org. Lett., 2002, 4, 2453 CrossRef CAS PubMed; (m) V. Nair, S. Mathai, S. C. Mathew and N. P. Rath, Tetrahedron, 2005, 61, 2849 CrossRef CAS; (n) A. E. Russell, J. Brekan, L. Gronenberg and M. P. Doyle, J. Org. Chem., 2004, 69, 5269 CrossRef CAS PubMed; (o) Y. Toda, W. Kaku, M. Tsuruoka, S. Shinogaki, T. Abe, H. Kamiya, A. Kikuchi, K. Itoh and H. Suga, Org. Lett., 2018, 20, 2659 CrossRef CAS PubMed; (p) J. J. Sheng, H. Chang, Y. Qian, M. L. Ma and W. H. Hu, Tetrahedron Lett., 2018, 59, 2141 CrossRef CAS.
- For selected enantioselective examples using chiral auxiliaries: (a) X. F. Xu, X. Guo, X. C. Han, L. P. Yang and W. H. Hu, Org. Chem. Front., 2014, 1, 181 RSC; (b) S. Torssell and P. Somfai, Adv. Synth. Catal., 2006, 348, 2421 CrossRef CAS; (c) S. Torssell, M. Kienle and P. Somfai, Angew. Chem., Int. Ed., 2005, 44, 3096 CrossRef CAS PubMed.
- For selected reviews, see: (a) Z. Wang, X. H. Liu and X. M. Feng, Aldrichimica Acta, 2020, 53, 3 CAS; (b) X. H. Liu, S. X. Dong, L. L. Lin and X. M. Feng, Chin. J. Chem., 2018, 36, 791 CrossRef CAS; (c) X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2017, 50, 2621 CrossRef CAS PubMed; (d) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298 RSC; (e) M. Y. Wang and W. Li, Chin. J. Chem., 2021 DOI:10.1002/cjoc.202000508; for selected examples: (f) H. F. Zheng, Y. Wan, C. R. Xu, Q. Xiong, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2019, 58, 5327 CrossRef CAS PubMed; (g) Y. S. Chen, S. X. Dong, X. Xu, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2018, 57, 16554 CrossRef CAS PubMed.
- For selected reviews of dipolar cycloadditions, see: (a) K. V. Gothelf and K. A. Jørgensen, Chem. Rev., 1998, 98, 863 CrossRef CAS PubMed; (b) T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366 CrossRef CAS PubMed; (c) M. Kissane and A. R. Maguire, Chem. Soc. Rev., 2010, 39, 845 RSC; (d) K. E. O. Ylijoki and J. M. Stryker, Chem. Rev., 2013, 113, 2244 CrossRef CAS PubMed; (e) H. Pellissier, Adv. Synth. Catal., 2018, 360, 1551 CrossRef CAS; (f) M. Harmata, Chem. Commun., 2010, 46, 8886 RSC; (g) A. G. Lohse and R. P. Hsung, Chem.–Eur. J., 2011, 17, 3812 CrossRef CAS; (h) H. Lam and M. Lautens, Synthesis, 2020, 52, 2427 CrossRef CAS; for recent examples of [4 + 3]-cycloaddition: (i) F. Jiang, F. R. Yuan, L. W. Jin, G. J. Mei and F. Shi, ACS Catal., 2018, 8, 10234 CrossRef CAS; (j) M. Sun, C. Ma, S. J. Zhou, S. F. Lou, J. Xiao, Y. C. Jiao and F. Shi, Angew. Chem., Int. Ed., 2019, 58, 8703 CrossRef CAS PubMed; (k) P. Kumari, W. W. Liu, C. J. Wang, J. Dai, M. X. Wang, Q. Q. Yang, Y. H. Deng and Z. H. Shao, Chin. J. Chem., 2020, 38, 151 CrossRef CAS; (l) C. S. Wang, T. Z. Li, S. J. Liu, Y. C. Zhang, S. Deng, Y. C. Jiao and F. Shi, Chin. J. Chem., 2020, 38, 543 CrossRef CAS; (m) W. L. Yang, Z. S. Huang, Y. Z. Liu, X. X. Yu and W. P. Deng, Chin. J. Chem., 2020, 38, 1571 CrossRef CAS.
- For selected reviews involving relay catalysis, see: (a) Y. Wang, H. Lu and P. F. Xu, Acc. Chem. Res., 2015, 48, 1832 CrossRef CAS PubMed; (b) N. T. Patil, V. S. Shinde and B. Gajula, Org. Biomol. Chem., 2012, 10, 211 RSC; (c) D. F. Chen, Z. Y. Han, X. L. Zhou and L. Z. Gong, Acc. Chem. Res., 2014, 47, 2365 CrossRef CAS PubMed; (d) X. Guo and W. H. Hu, Acc. Chem. Res., 2013, 46, 2427 CrossRef CAS PubMed; (e) Z. T. Du and Z. H. Shao, Chem. Soc. Rev., 2013, 42, 1337 RSC.
- H. Frauenrath, D. Brethauer, S. Reim, M. Maurer and G. Raabe, Angew. Chem., Int. Ed., 2001, 40, 177 CrossRef CAS.
- T. Bando, Y. Namba and K. Shishido, Tetrahedron: Asymmetry, 1997, 8, 2159 CrossRef CAS.
- A. K. Ghosh and M. R. Belcher, J. Org. Chem., 2020, 85, 10399 CrossRef CAS PubMed.
- For selected reviews on β,γ-unsaturated α-ketoesters: (a) G. Desimoni, G. Faita and P. Quadrelli, Chem. Rev., 2013, 113, 5924 CrossRef CAS PubMed; (b) B. Eftekhari-Sis and M. Zirak, Chem. Rev., 2015, 115, 151 CrossRef CAS PubMed.
- H. Lam, Z. Qureshi, M. Wegmann and M. Lautens, Angew. Chem., Int. Ed., 2018, 57, 16185 CrossRef CAS PubMed.
- CCDC 2015654 [4m], 2046337 [5a], 2053510 [Sm(OTf)3/L3-PrPr2], 2025197 [Sm(OTf)3/L4-PrPr2], 2055726 [Sm(OTf)3/L5-PrPr2]†.
- For selected reviews on construction of tetrahydrofurans: (a) J. P. Wolfe and M. B. Hay, Tetrahedron, 2007, 63, 261 CrossRef CAS PubMed; (b) A. D. L. Torre, C. Cuyamendous, V. Bultel-Poncé, T. Durand, J. M. Galano and C. Oger, Tetrahedron, 2016, 72, 5003 CrossRef; (c) G. Jalce, X. Franck and B. Figadère, Tetrahedron: Asymmetry, 2009, 20, 2537 CrossRef CAS; for selected recent examples: (d) S. Lee, H. Y. Bae and B. List, Angew. Chem., Int. Ed., 2018, 57, 12162 CrossRef CAS PubMed; (e) P. D. Pohlhaus, S. D. Sanders, A. T. Parsons, W. Li and J. S. Johnson, J. Am. Chem. Soc., 2008, 130, 8642 CrossRef CAS PubMed; (f) S. R. Angle, G. P. Wei, Y. K. Ko and K. Kubo, J. Am. Chem. Soc., 1995, 117, 8041 CrossRef CAS.
- X-ray crystal structures indicated that the chiral pocket changed with the catalysts bearing different carbon tethers of chiral ligands (see the ESI, page S15†). Further studies are needed to illustrate the effect of the catalyst structure on diastereo- and enantioselectivity.
- Both endo and exo-selectivity were observed in previous studies (endo, 6a, f–i; exo, 6c).
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} and 19F{1H} NMR, HPLC spectra, and CD spectra (PDF). CCDC 2015654, 2046337, 2053510, 2025197 and 2055726. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01019k |
| This journal is © The Royal Society of Chemistry 2021 |