DOI:
10.1039/D0SC05555G
(Review Article)
Chem. Sci., 2021,
12, 2735-2759
Recent development in transition metal-catalysed C–H olefination†
Received
8th October 2020
, Accepted 28th December 2020
First published on 29th January 2021
Abstract
Transition metal-catalysed functionalizations of inert C–H bonds to construct C–C bonds represent an ideal route in the synthesis of valuable organic molecules. Fine tuning of directing groups, catalysts and ligands has played a crucial role in selective C–H bond (sp2 or sp3) activation. Recent developments in these areas have assured a high level of regioselectivity in C–H olefination reactions. In this review, we have summarized the recent progress in the oxidative olefination of sp2 and sp3 C–H bonds with special emphasis on distal, atroposelective, non-directed sp2 and directed sp3 C–H olefination. The scope, limitation, and mechanism of various transition metal-catalysed olefination reactions have been described briefly.
 Wajid Ali | Wajid Ali was born in Uttar Pradesh, India. He received his M.Sc. degree in organic chemistry from Aligarh Muslim University and Ph.D. from the Indian Institute of Technology Guwahati in 2018 under the supervision of Prof. Bhisma K. Patel. Currently, he is working in Prof. Maiti's group at IIT Bombay as a postdoctoral fellow and his area of research interest is transition metal-catalysed distal C–H functionalization. |
 Gaurav Prakash | Gaurav Prakash was born in 1996 in Bihar, India. He completed his B.Sc. from Acharya Narendra Dev College, University of Delhi. After completing M.Sc. from Aligarh Muslim University, he joined Prof. Maiti's group as a graduate student in January 2019 at the Indian Institute of Technology Bombay, where he is currently working on transition metal-catalysed directing group assisted para-C–H functionalization. |
 Debabrata Maiti | Debabrata Maiti received his PhD from Johns Hopkins University (USA) in 2008 under the supervision of Prof. Kenneth D. Karlin. After postdoctoral studies at Massachusetts Institute of Technology (MIT) with Prof. Stephen L. Buchwald (2008–2010), he joined the Department of Chemistry at IIT Bombay in 2011, wherein he is currently an Associate Professor. His research interests focus on the development of new and sustainable catalytic methods. |
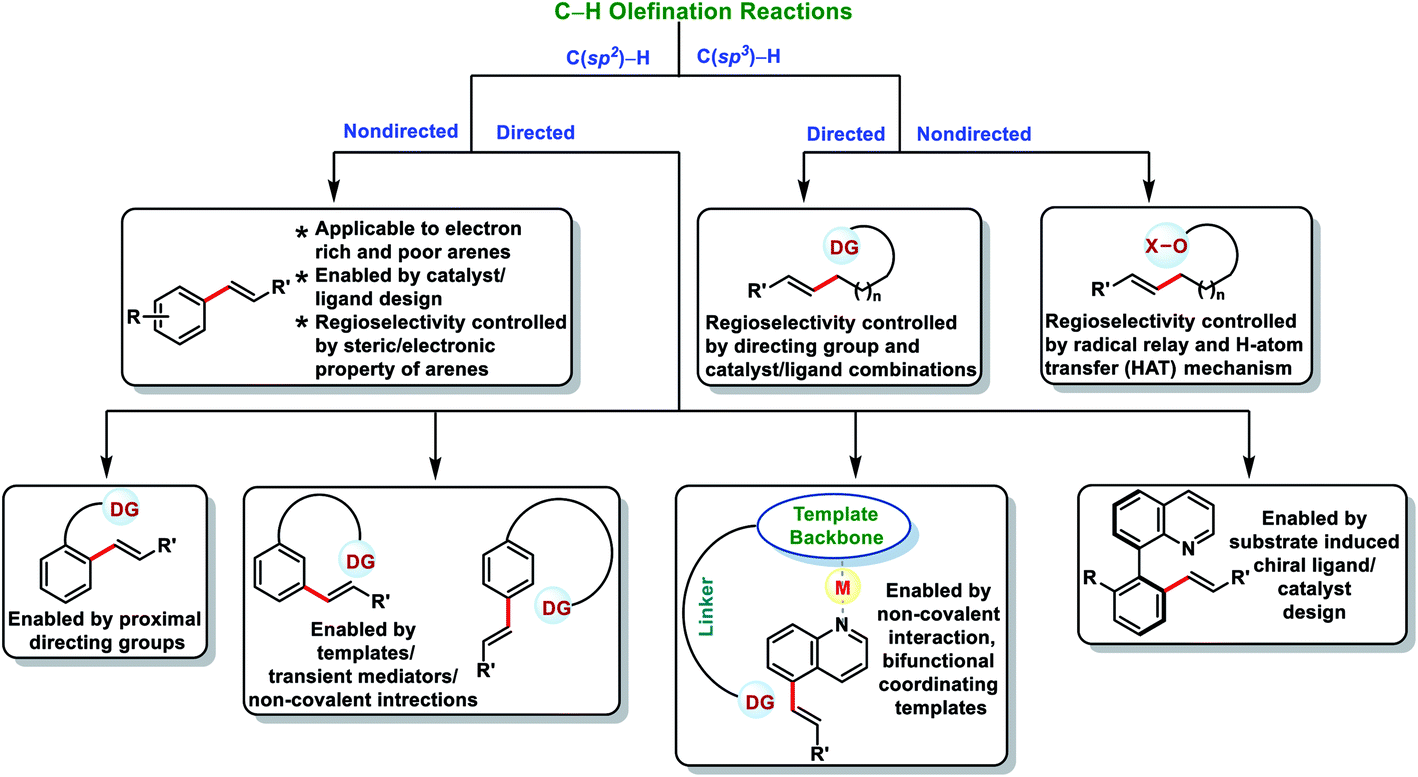
1. Introduction
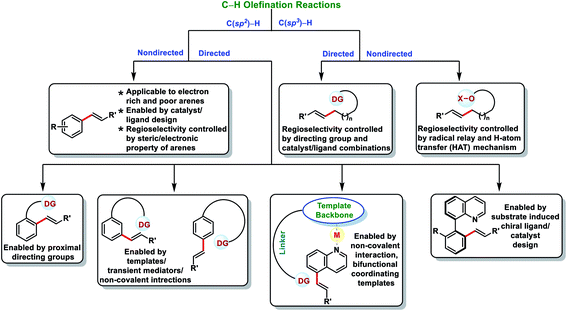
Transition metal-catalysed C–H bond activations have emerged as an important tool in synthetic organic chemistry for the construction of C–C bonds in a more economical fashion.1 In the last few decades, significant developments have been observed towards sustainable organic transformations by researchers all over the world.2 In this regard, transition metal-catalysed C–H olefination received special attention due to the versatility of olefinated compounds in many valuable products (Fig. 1). Vinyl arenes act as key intermediates in many organic transformations and are also present in numerous bioactive compounds.3–5 Pd-catalysed Mizoroki–Heck cross-coupling (Nobel Prize in 2010) of arene electrophiles with alkenes has emerged as an alternative route to the conventional olefination reactions.6 Direct oxidative C–H olefination with alkenes known as the Fujiwara–Moritani reaction represents an advanced version of Mizoroki–Heck couplings as it avoids prefunctionalized starting compounds.7 However, poor regioselectivity, low catalyst efficiency and usage of excess arenes (solvent amount) limits the Fujiwara–Moritani reaction and prompted the development of more efficient approaches. The foremost challenge in C–H activation is to selectively functionalize a particular C–H bond in the presence of several electronically equivalent C–H bonds. To overcome this issue, usage of directing groups has come up as one of the most reliable approaches. Directing groups concentrate metals in the close proximity of the desired C–H bond to be functionalized and thus improve the selectivity and reactivity. The field of directed C–H bond activations has grown tremendously and many chelating groups have been explored that can serve as the auxiliaries. Furthermore, by understanding the distal and geometrical relationships between the functional group and targeted C–H bond, U- and D-shaped templates, transient mediators and non-covalent interactions were discovered to activate meta, para and other distal C–H bonds.8
 |
| | Fig. 1 Direct C–H olefination reactions. | |
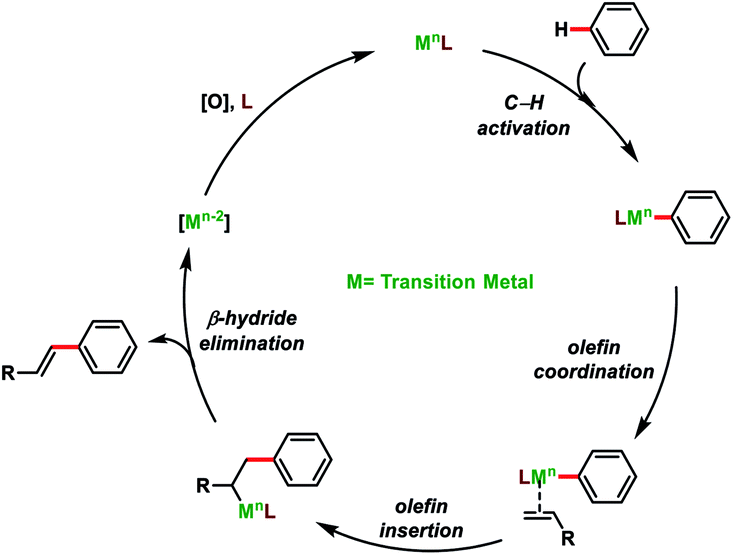
Direct olefination of the relatively unreactive C(sp3)–H bond represents one of the most fascinating and advance strategies in synthetic organic chemistry. However, in contrast to the C(sp2)–H bond, functionalization of the C(sp3)–H bond, particularly olefination, is not well investigated and limited reports are available in the literature. Selective functionalisation of the C(sp3)–H bond is much more difficult due to the lack of the assistance of the π-group, which could efficiently interact with the transition-metal centre. However, the last few decades witnessed an upsurge in distal C(sp3)–H olefination by installing the directing group as well as through the radical translocation strategy (Fig. 2).9
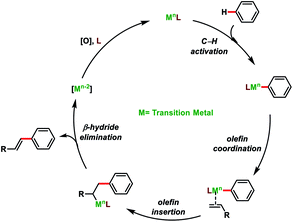 |
| | Fig. 2 General mechanism for direct C–H olefination reactions. | |
In this review, we have summarized the progress in oxidative C–H olefination reactions until summer 2020 with special emphasis on the distal, atroposelective, non-directed C(sp2)–H and directed C(sp3)–H olefinations. Also, advancements in ortho-C(sp2)–H olefination from 2017 have been discussed here.2a,b,e,f
2. Proximal C(sp2)–H olefination
2.1.
Ortho-C(sp2)–H olefination
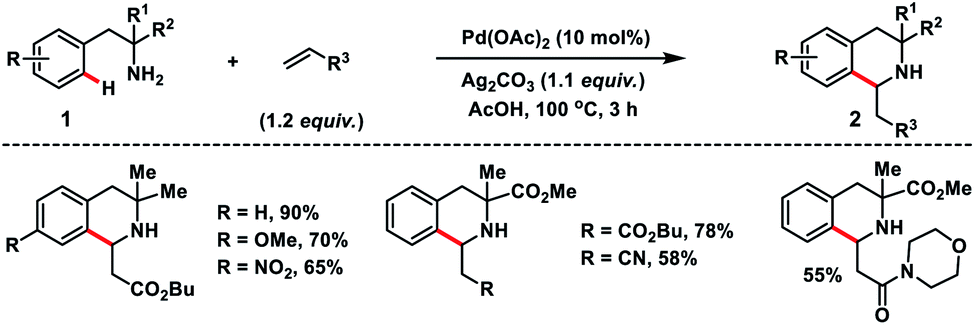
In 2017, Garcia and co-workers reported a Pd-catalysed ortho-C–H olefination of 2-phenylethylamines 1 with activated alkenes. In the reaction, a primary amine acted as the directing group which followed an intramolecular aza-Michael addition route for the construction of 3,3-disubstituted tetrahydroisoquinolines 2 (Scheme 1).10 A bidentate silver salt such as a carbonate or an acetate was found to be indispensable for the reaction. The strategy was compatible with 1,1-disubstituted-2-phenylethylamine substrates only.
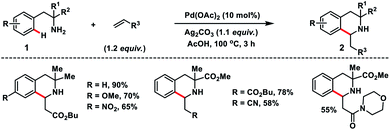 |
| | Scheme 1 Primary amine-directed ortho-C(sp2)–H olefination. | |
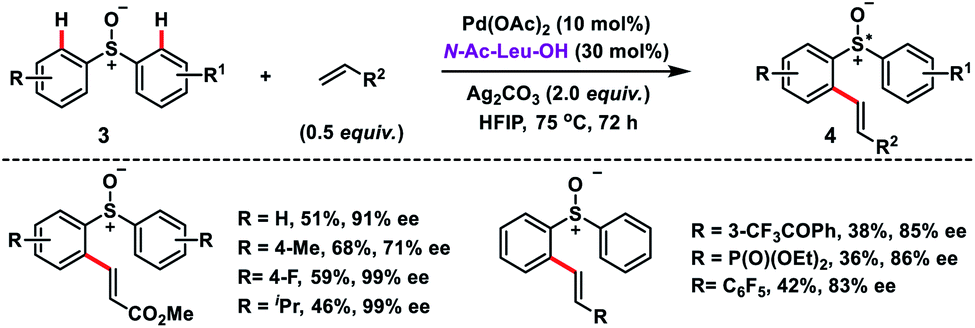
Wang's group disclosed the Pd-catalysed N-Ac-Leu-OH ligand-controlled enantioselective C–H olefination of racemic sulfoxides 3 to construct chiral diaryl sulfoxides 4 in moderate yield and excellent enantioselectivity (up to 99% ee) (Scheme 2).11 Symmetric and nonsymmetric sulfoxides were functionalized through desymmetrization and parallel kinetic resolution (PKR). Olefination of the (S)-isomer occurred at the para-substituted benzene ring while for the (R)-isomer olefination took place at the other phenyl ring with high ee. It indicated that regiodivergent parallel kinetic resolution (PKR) was involved in the reaction. The enantioselectivity in the transformation was induced during the Pd(II)/N-Ac-Leu-OH-catalysed asymmetric C–H activation step.
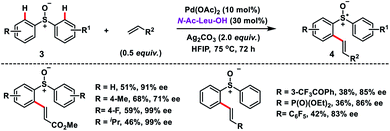 |
| | Scheme 2 Pd-catalysed enantioselective C–H olefination of diaryl sulfoxides. | |
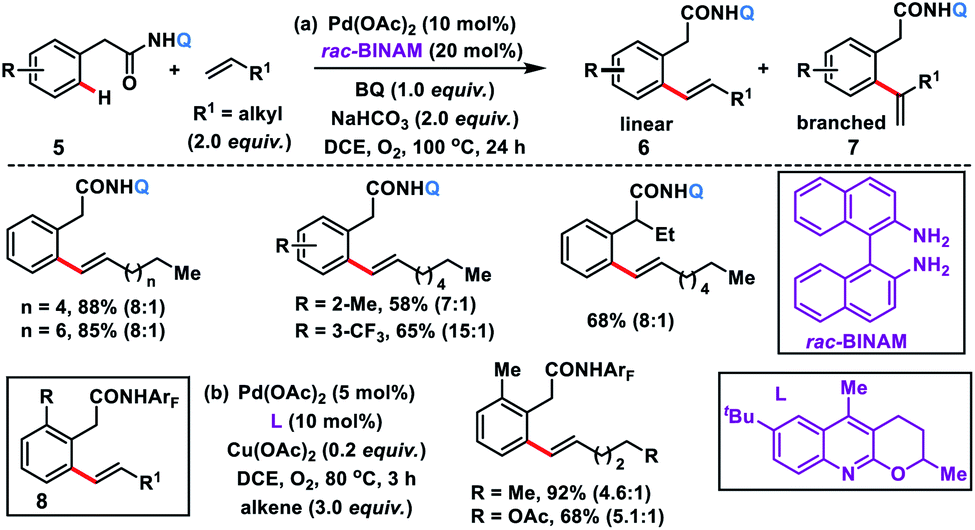
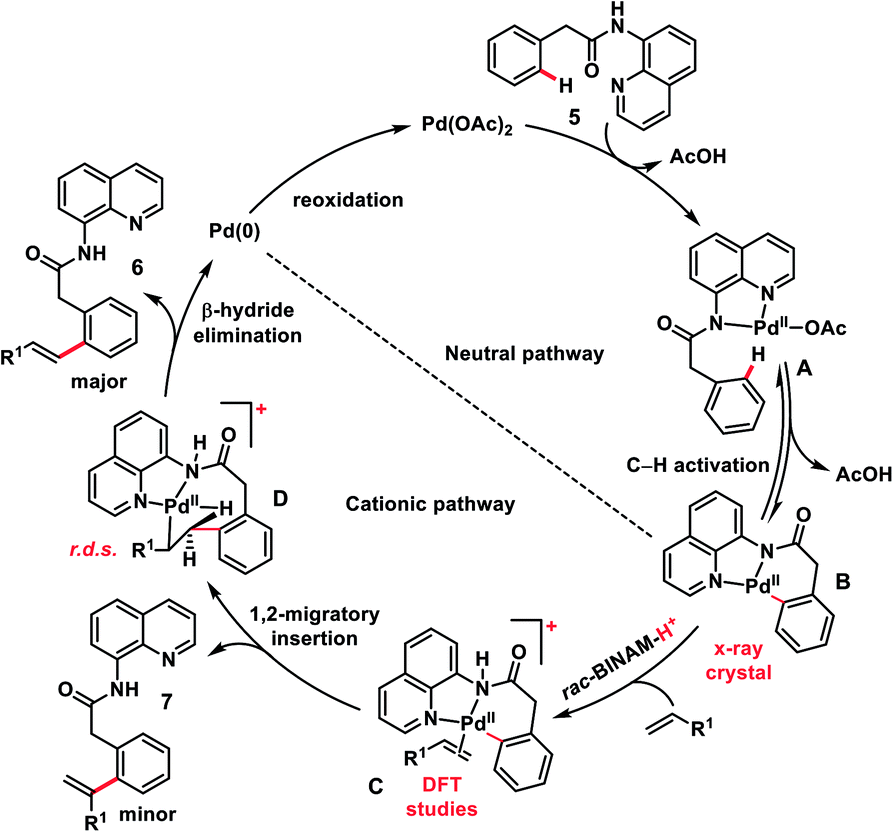
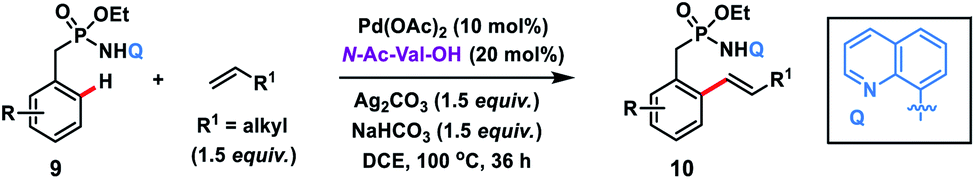
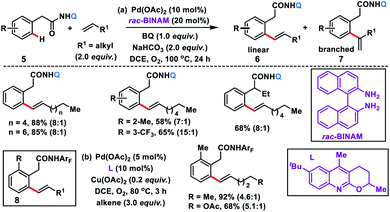
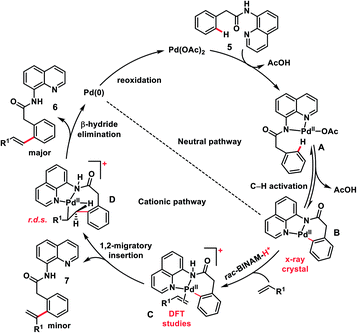
The use of unbiased aliphatic alkenes for the ortho-olefination of phenyl acetic acids 5 in high regio- and stereoselectivity was first reported by our group (Scheme 3a).12a In this Pd-catalysed olefination reaction a bidentate directing group 8-aminoquinoline was found to be the suitable auxiliary as it provided rigid coordination to Pd and formed a stable six-membered palladacycle during C–H activation. The reaction provided better yield and selectivity in the presence of the ligand rac-BINAM as it saturates the coordination site of the palladium. A library of terminal aliphatic alkenes irrespective of their chain length and functional group were well suited under the reaction conditions and provided olefinated products 6 in good yield and selectivity for linear/branched products. Several phenyl acetic acid derivatives including the commercial drug ibuprofen were olefinated efficiently. Sequential diolefination of phenyl acetic acid with two different alkenes indicated that the catalytic system used is very reactive and efficient. Similarly, Dai, Yu and co-workers also presented a Pd(II)-catalysed ortho-C(sp2)–H olefination of phenylacetic acid derivatives with unactivated aliphatic alkenes in 2018 (Scheme 3b).12b They found that a quinoline based ligand was crucial for the purpose whereas the monodentate amide coordinating group was used as a weak directing group. In the transformation, both simple and functionalized aliphatic alkenes were compatible with different acids and provided β-alkylated styrenes 8 in good yield. To make the protocol more convenient molecular oxygen was utilized as a terminal oxidant along with a catalytic copper salt as the co-oxidant. Detailed experimental and computational studies were carried out by our group to understand the origins of the selectivity.12c A series of experiments such as reaction rate and order determination, control experiments, isotopic labelling and Hammett analysis were performed to understand the reaction mechanism. NMR and kinetic studies using aryl-palladium intermediates helped to understand the arene C–H activation, olefin coordination and carbopalladation steps. KIE experiments revealed that the C–H activation step was not involved in the rate limiting step instead β-hydride elimination was controlling the overall catalytic process. The results of various control experiments were supported by DFT studies and revealed the origin of regio and stereoselectivity. The findings of experimental and computational studies suggested that the overall mechanism combines neutral and cationic pathways (Scheme 4). A neutral aryl-palladium intermediate B was formed by the fast and reversible C–H activation. Coordination of the unactivated olefin to the neutral aryl-palladium species was possible, however for efficient reactivity a positively charged intermediate C was required. Intermediate C underwent migratory insertion with an olefin to form intermediate D followed by β-hydride elimination to form the kinetically and thermodynamically favoured linear olefinated product 6. Our group further expanded the application of unbiased aliphatic alkenes for the ortho C–H olefination of benzyl phosphonamides 9 (Scheme 5).12d The strategy was well suited for various benzyl phosphonamides and aliphatic alkenes to afford linear olefinated products 10 in good yield and selectivity.
 |
| | Scheme 3 Pd-catalysed aryl C–H olefination with unbiased aliphatic alkenes. | |
 |
| | Scheme 4 Plausible mechanism for C–H olefination of phenylacetic amides. | |
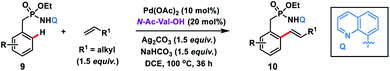 |
| | Scheme 5 Pd(II)-catalysed olefination of benzyl phosphonamide. | |
Loh and co-workers reported Pd-catalysed ortho-olefination of O-acetyl cyanohydrins aided by the synergetic directing effect of acetoxy and cyano functionalities.13 They used a mono-N-protected amino acid (MPAA), N-Ac-Gly-OH ligand, and Ag2CO3 oxidant to perform the reaction which was well suited with a variety of substrates and offered the anticipated products in good yield with high regioselectivity.
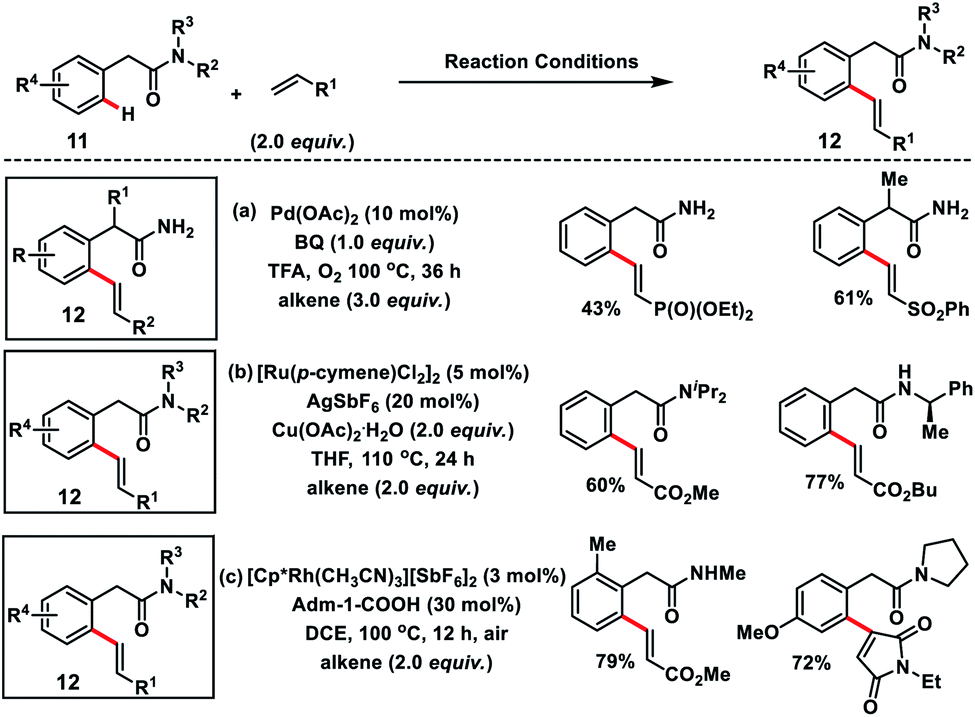
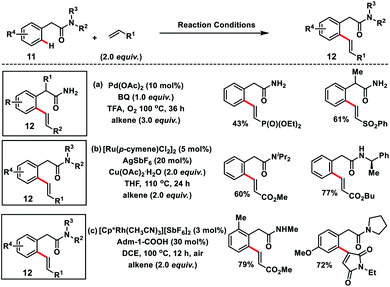
Amit and co-workers succeeded in the olefination of arylacetamides 11 using abundant and easily manipulatable primary amides as the effective directing group (Scheme 6a).14a They used benzoquinone along with oxygen as the oxidant. A series of alkenes were installed at the ortho-position of arylacetamide derivatives in moderate to good yields 12 with high regio and diastereoselectivity. Ackermann and co-workers in 2018 demonstrated the Ru(II)-catalysed ortho-C(sp2)–H olefination of weakly O-coordinating arylacetamides via a less favourable six-membered ruthenacycle (Scheme 6b).14b The strategy supports various substituted tertiary, secondary and even challenging primary amides and provided substituted olefins 12 in a chemo-, regio- and stereoselective manner. DFT studies showed that a six-membered ruthenacycle was formed via carboxylate mediated base-assisted internal electrophilic-type substitution (BIES) of the C–H bond and migratory insertion of an alkene involved in the rate-limiting step. Jeganmohan's group in 2019 utilized a Rh(III)-catalyst for the similar oxidative olefination of arylacetamides 11 with activated alkenes in good to excellent yields (Scheme 6c).14c Atmospheric oxygen served as the sole oxidant to regenerate the catalyst. A range of substituted arylacetamides underwent oxidative coupling with various acrylates and maleimides to furnish olefinated products 12 in a highly regio and diastereoselective manner. Weak coordination of acetamide oxygen with Rh directed it towards ortho-metalation via deprotonation to form a six-membered metalacyclic intermediate similar to the Ru-catalyst.
 |
| | Scheme 6 Amide-directed ortho-C(sp2)–H olefination. | |
Ir-catalysed ortho-C(sp2)–H olefination of benzylamines 13 with acrylates was reported by Fu and co-workers using pentafluoro benzoyl (PFB) as the new directing scaffold (Scheme 7).15 The reaction proceeded well with a series of substituted benzylamines in the presence of AgOAc oxidant and provided mono-olefinated products 14 in good yields. The reaction proceeded through the activation of the catalyst by AgOAc followed by the ortho-C–H bond activation via a concerted metalation–deprotonation (CMD) pathway resulting in a metallacycle. Insertion of an alkene tracked by β-hydride elimination yielded an olefinated product and Ir(I) species, which was re-oxidized to Ir(III) by AgOAc.
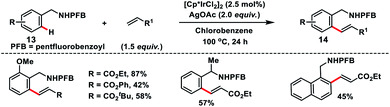 |
| | Scheme 7 Pentafluoro benzoyl-directed olefination of benzylamine. | |
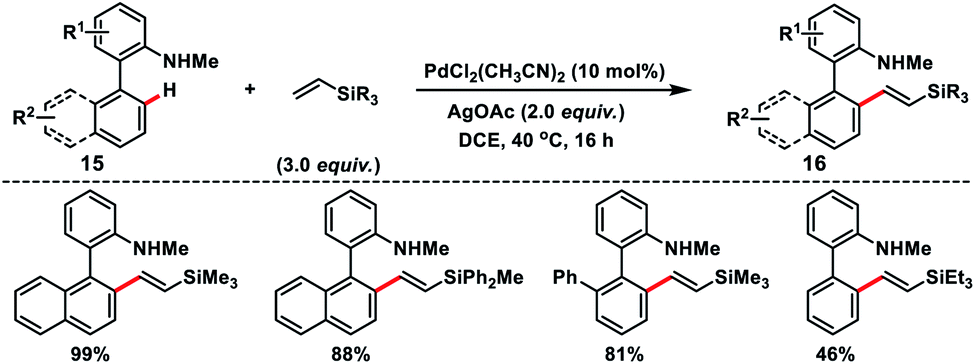
Cui and co-workers reported an interesting Pd-catalysed amino-chelation-assisted olefination of arenes 15 with an unactivated vinylsilane (Scheme 8).16 They developed a ligand-free approach to synthesize a library of arylated vinylsilanes 16 in good to excellent yield with (E)-selectivity. It was interesting to note that the geometry of the product was controlled by the directing group, since biaryl carboxylic acids under similar conditions provided olefinated products but in poor E/Z ratios.
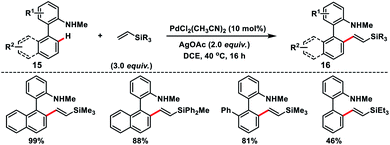 |
| | Scheme 8 C(sp2)–H olefinations of 2-amino biaryls with vinylsilanes. | |
Acid directed olefination of ferrocene carboxylic acid 17 has been developed by Wu and co-workers to synthesize 1,2-disubstituted ferrocene carboxylic acids 18 with planar chirality using the chiral MPAA ligand N-Ac-L-Phe-OH (Scheme 9).17 Diverse alkenes such as acrylates, acrylamides, vinyl phosphates, vinyl ketones and styrenes were tolerated under the given reaction conditions. Ferrocenium ion was formed in the presence of oxygen and served as the terminal oxidant to regenerate Pd(II) from Pd(0).
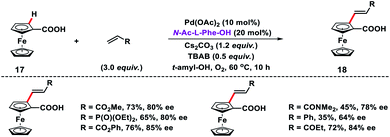 |
| | Scheme 9 Enantioselective C(sp2)–H olefinations of ferrocene carboxylic acid. | |
Mahiuddin's group further explored the directing group and used carboxylate as a weak directing group in Ru(II)-catalysed ortho-C(sp2)–H olefination of aromatic carboxylic acids with styrenes.18 The weak coordination of acid functionality serving as directing group even in the presence of the strong coordinating acetamide functional group under the reaction conditions.
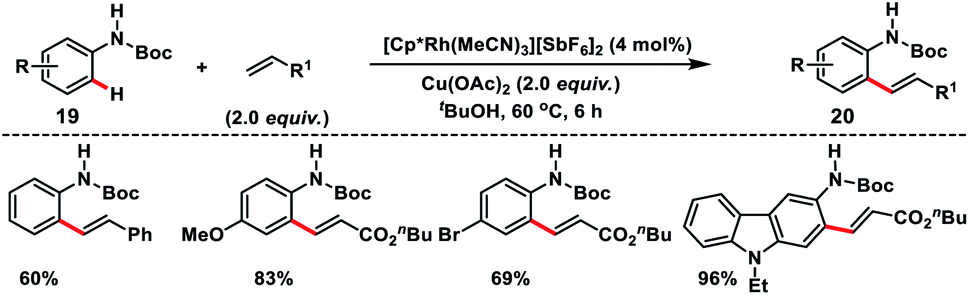
Satoh, Miura and co-workers in 2017 developed a Rh(III)-catalysed N-Boc directed ortho-olefination of anilines 19 with alkenes using a copper salt oxidant (Scheme 10).19 A series of alkenes such as acrylates, acrylamides, acrylonitrile and styrenes reacted with different anilines to afford corresponding ortho-olefinated products 20 in good yield. Styrene reacted slightly at elevated temperature and high catalyst loading. The ortho-olefinated anilines could be further derivatized into valuable nitrogen-containing heterocycles such as indoles and quinolines.
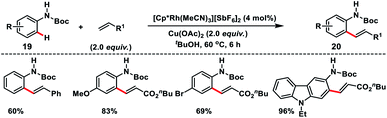 |
| | Scheme 10 Rh-catalysed N-Boc-directed ortho-olefinations of anilines. | |
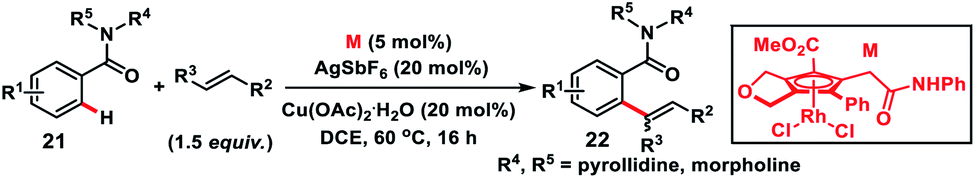
Shibata, Tanaka and co-workers in 2019 synthesized a moderately electron-deficient cyclopentadienyl Rh(III)-complex. The Rh(III)-complex bearing an ester and pendant amide moieties on the Cp ring was used for the aerobic oxidative olefination of benzamides 21 with styrene derivatives (Scheme 11).20a Diverse alkenes reacted with a multitude of benzamides to deliver corresponding products 22 in good yield. The ester moiety accelerates cleavage of the C–H bond and olefin insertion by reducing the electron density on the Rh-centre while the secondary amide eased C–H bond cleavage by intramolecular C–H extraction. Recently, Tanaka and co-workers came up with another highly active unsubstituted cyclopentadienyl rhodium(III)-complex for the oxidative ortho-olefination of sterically demanding amides.20b
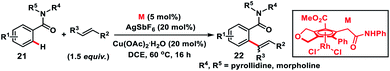 |
| | Scheme 11 Modified Rh(III)-catalysed ortho-olefination of amides. | |
Jeganmohan and co-workers established a Ru(II)-catalysed oxidative olefination followed by the intramolecular cyclisation of benzamides 23 with vinyl sulfone for the synthesis of 3-methyleneisoindolin-1-ones 24 in good yields and E/Z ratio (Scheme 12).21 Furthermore, they applied the oxidative cyclisation of benzamide for the total synthesis of aristolactam alkaloids.
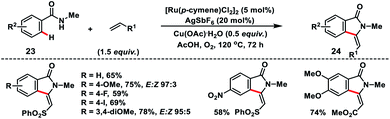 |
| | Scheme 12 Ru(II)-catalysed cyclisation of benzamides with activated alkenes. | |
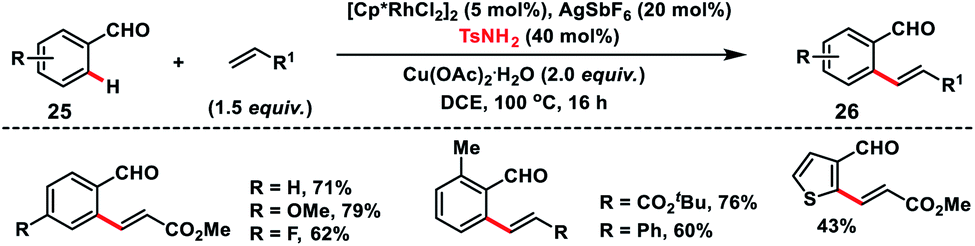
Rhodium(III)-catalysed ortho-C(sp2)–H olefination of aldehydes 25 was developed by Wang and co-workers in 2017 with the use of TsNH2 as a transient directing group (Scheme 13).22 The in situ generated imine formed via condensation of aldehyde and amine served as the directing group for the olefination of a variety of aldehydes with olefins and provided products 26 in good yield. KIE experiments suggest that cleavage of the C–H bond was involved in the rate determining step.
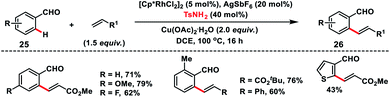 |
| | Scheme 13 Transient-directed ortho-C(sp2)–H olefinations of aldehydes. | |
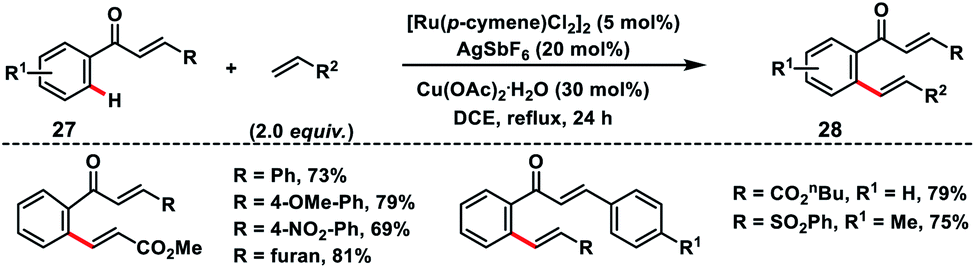
Ru(II)-catalysed enone carbonyl directed ortho-C(sp2)–H olefination of chalcones 27 was developed by Bakthadoss and co-workers in 2018 using hydrated Cu(OAc)2 as the oxidant (Scheme 14).23 A variety of activated olefins coupled with chalcone derivatives and afforded olefinated products 28 in high yield and stereoselectivity. In 2018, Ir-catalysed ortho-C(sp2)–H olefination of aromatic systems was demonstrated by Elumalai and co-workers employing diverse directing groups bearing the carbonyl functionality.24 The oxidative olefination reaction proceeded smoothly in the presence of an external oxidant Cu(OAc)2 and additive AgPF6 to provide corresponding products in good yield.
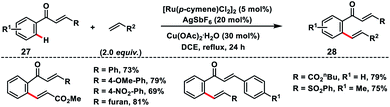 |
| | Scheme 14 Ru(II)-catalysed enone carbonyl-directed olefination of chalcones. | |
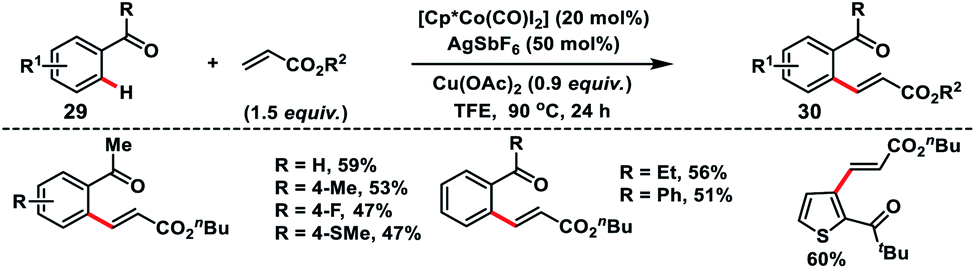
Co-catalysed olefination of aromatic ketones 29 with the assistance of the weakly coordinating carbonyl group was discussed by Maji and co-workers (Scheme 15).25 A wide range of aromatic and heteroaromatic ketones irrespective of the electronic properties coupled with activated olefins to produce olefinated products 30 in good yield and E-selectivity.
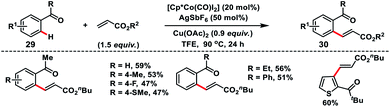 |
| | Scheme 15 Co(III)-catalysed ortho-olefination of aromatic ketones. | |
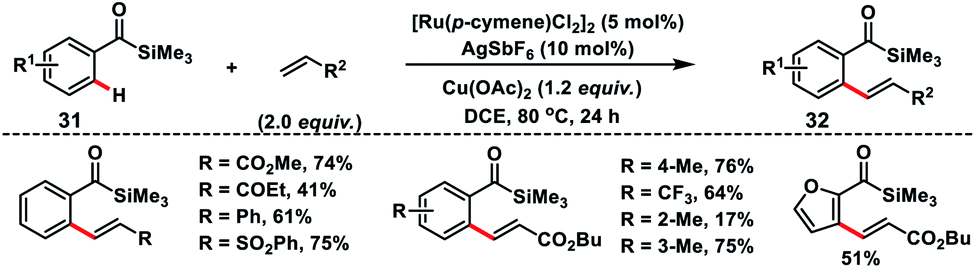
The easily manipulative acylsilane as the directing group for Ru(II)-catalysed oxidative olefination of aroylsilanes 31 with alkenes was demonstrated by Zhang and co-workers (Scheme 16).26 They utilized a copper salt as an external oxidant and the reaction was applicable to a broad range of aroylsilanes including heterocycles. Aroylsilanes coupled with a variety of alkenes and delivered ortho-olefinated products 32 in good yield with excellent regio and stereoselectivity.
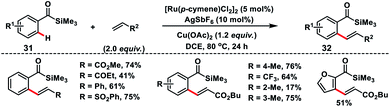 |
| | Scheme 16 Acylsilane-directed ortho-C(sp2)–H olefination. | |
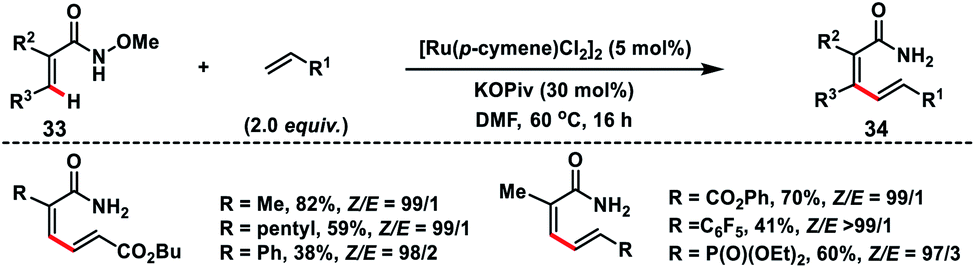
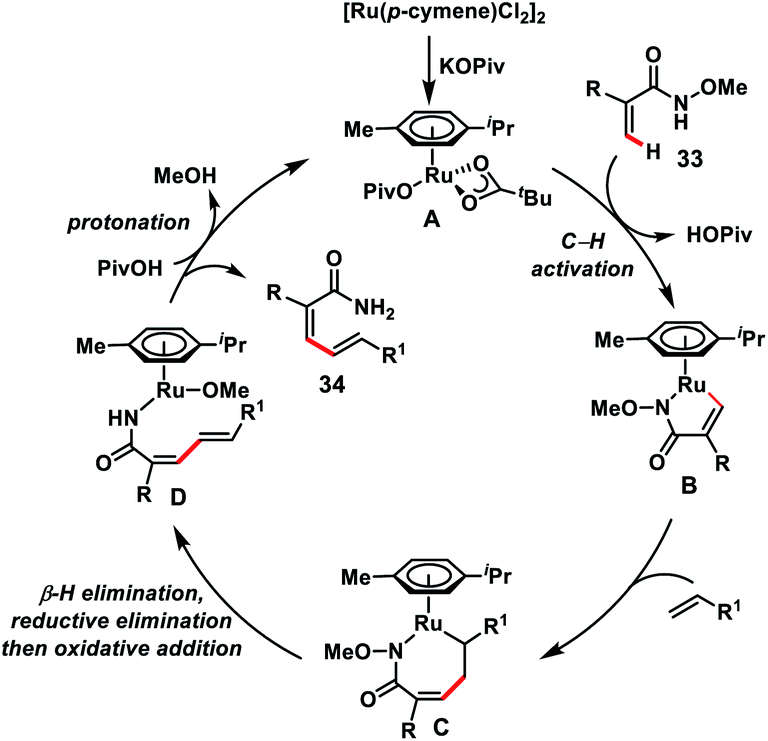
Zhong's group in 2017 demonstrated a Ru-catalysed olefination reaction at relatively difficult alkenyl C–H bonds of N-methoxy α,β-unsaturated amides 33 (Scheme 17).27a In the reaction, CONH(OMe) played a dual role of a directing group as well as an internal oxidant for the construction of synthetically important 1,3-dienamides 34 in good yield and high E-stereoselectivity. The formation of a five-membered ruthenacycle through reversible C–H activation took place followed by the alkene insertion to form a seven-membered ruthenacycle intermediate B (Scheme 18). β-Hydride elimination followed by reductive elimination generated a Ru(0) species which underwent N–O bond insertion to afford the Ru(II)-amide intermediate D. Finally, protonation with PivOH led to the formation of the desired product and Ru(II)-catalyst to continue the catalytic cycle. Later, the same group extended the olefination reaction of N-methoxy α,β-unsaturated amides with other transition metals such as Ir and Co. Ir-catalysed olefination reaction occurred in the presence of an inexpensive hydrogen acceptor chloranil and afforded the desired products in good yield and stereoselectivity.27b Similarly, construction of conjugated dienes was achieved by using [Cp*Co(CO)I2]-complex as the catalyst and AgOAc oxidant.27c
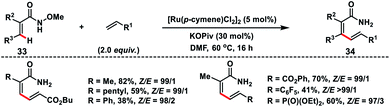 |
| | Scheme 17 Ru(II)-catalysed olefination of α,β-unsaturated amides. | |
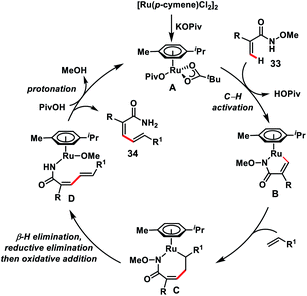 |
| | Scheme 18 Proposed mechanism for Ru-catalysed olefination of α,β-unsaturated amides. | |
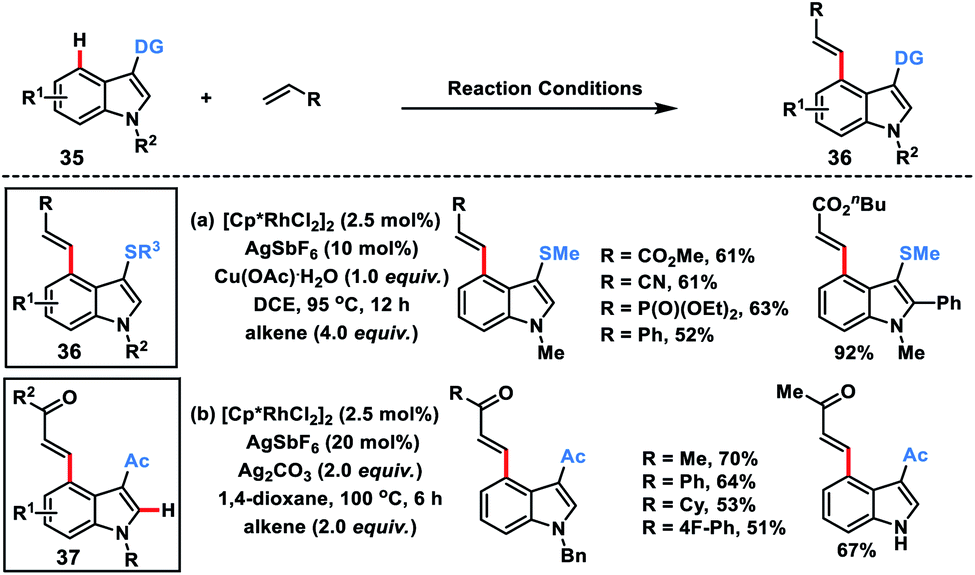
Miura's group in 2018 disclosed a Rh-catalysed thioether directed C-4 selective olefination of indoles 35 through a five-membered metallacycle intermediate (Scheme 19a).28a The transformation was applicable to a wide range of electron-deficient alkenes as well as styrene derivatives and afforded C-4 olefinated products 36 in good yield with E selectivity. The easy transformation of the thioether directing group was an added synthetic advantage to the reaction.
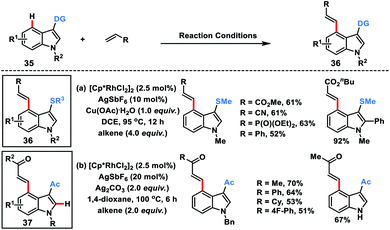 |
| | Scheme 19 Rh-catalysed thioether & acetyl-directed C-4 olefination of indoles. | |
Punniyamurthy and co-workers recently developed a Rh-catalysed C-4 alkenylation of indoles 35 with allylic alcohols employing weakly coordinating carbonyl as the directing group (Scheme 19b).28b Allyl alcohols served as either an alkenylating or alkylating agent depending on the additive used. Usage of Ag2CO3 resulted in β-hydride elimination while the presence of NaOPiv led to the alkylated products. A series of substituted indoles reacted with different allyl alcohols to afford olefinated products 37 in good yield and selectivity. Allyl alcohols in the presence of a Rh-catalyst and under basic conditions were converted into enones which served as the activated alkenes in the Rh-catalysed olefination reaction.
Indole NH directed Rh-catalysed ortho-di-olefination of 2-arylindoles was presented by Huang and co-workers in 2018.29 It is worth mentioning that when the reaction was performed in DMF instead of EtOAc, formation of 6H-isoindolo[2,1-a]indole took place through intramolecular aza-Michael reaction.
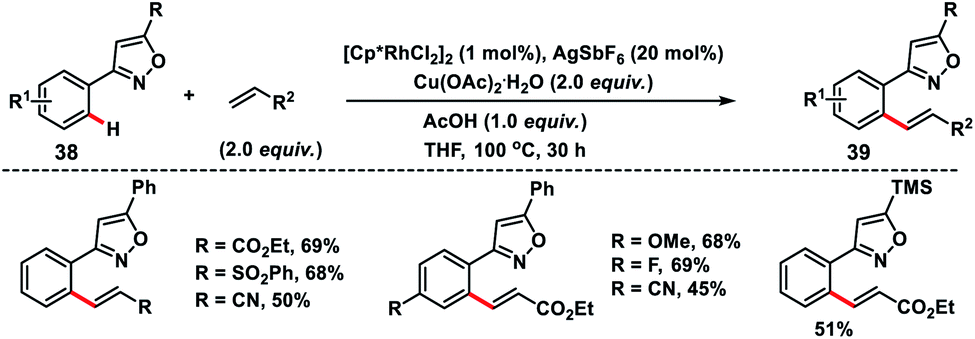
Kapur and co-workers demonstrated a precise catalyst-controlled selective C–H olefination of isoxazoles 38. When a cationic rhodium catalyst is used, the transformation was dictated by the cationic nature of the catalyst, and the strong coordination of isoxazole nitrogen led to ortho-C(sp2)–H olefination of proximal aryl rings 39 (Scheme 20),30 while a palladium-catalyst prefers electrophilic C–H activation due to the covalent nature of the catalyst and olefination took place at the distal position of the directing group. The reaction followed an interesting mechanism as KIE studies via both parallel and competitive reactions indicated the absence of the primary kinetic isotope effect highlighting that C–H activation was not the rate limiting step. The C–H activation step in the transformation most probably proceeds through a base-assisted internal electrophilic substitution (BIES) reaction and site selectivity was governed by the affinity of the Rh-catalyst for isoxazole nitrogen. Du and co-workers reported Rh(III)/[BMIM]NTf2 as a reusable catalytic system for the ortho-olefination of arenes at room temperature.31 The ionic liquid used in the transformation is chemically stable and non-volatile accounting for an environment friendly protocol.
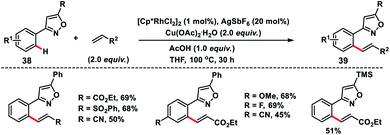 |
| | Scheme 20 Rh-catalysed isoxazole-directed olefination of proximal aryl rings. | |
2.2. Atroposelective C(sp2)–H olefination
Axially chiral biaryl scaffolds are omnipresent in a class of natural products and biologically relevant compounds, and are also used as chiral ligands or catalysts in a series of asymmetric syntheses.32 In the literature many elegant strategies are present for the synthesis of axillary chiral biaryls such as asymmetric Suzuki–Miyaura couplings, desymmetrization of prochiral biaryl compounds, atroposelective cleavage of the biaryl lactones, asymmetric oxidative homocouplings etc. Recently, asymmetric C–H activation has emerged as a potential alternative to access chiral biaryl compounds in a single step and atom economic way.
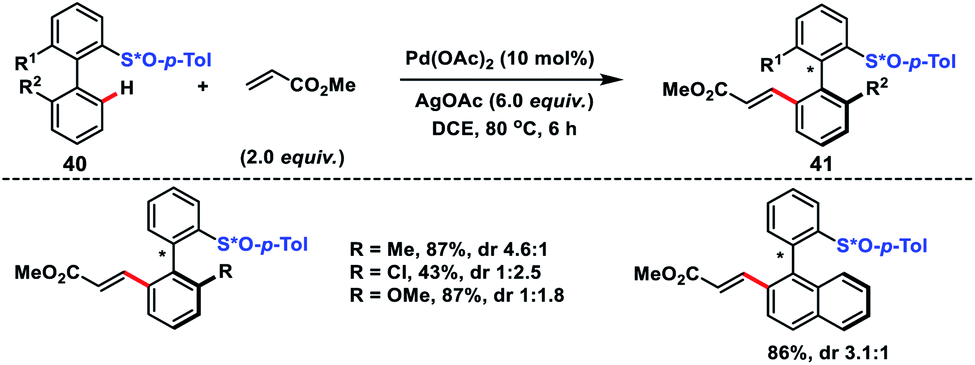
Synthesis of axillary chiral biaryls through directing group assisted C(sp2)–H olefination was first reported by Colobert and co-workers in 2013 (Scheme 21).33 The first outcome in the synthesis of axillary chiral biaryls was reported by them. They used enantiopure p-tolyl sulfoxide 40 as the directing group which induced the atropodiastereoselectivity during the olefination process. In the protocol electron-deficient acrylates served as efficient coupling partners with a variety of biaryl systems and afforded corresponding products 41 in moderate to good yield. The observed atroposelectivity during the transformation probably arose from diastereomeric discrimination during the cyclometallation step. This resulted in an atropoenriched Pd–C bond formation at the sterically less hindered side, i.e. opposite to the bulky p-tolyl group. Yang and co-workers reported a similar Pd-catalysed atroposelective C–H olefination through dynamic kinetic resolution for the synthesis of axially chiral phosphine oxide-based compounds.34 (S)-(−)-Menthyl phenylphosphinate served as the directing group as well as induced atropodiastereoselectivity during the olefination process.
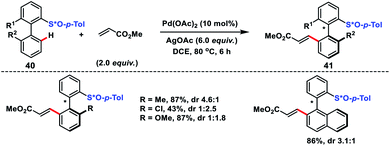 |
| | Scheme 21 Chiral auxiliary-induced atroposelective C–H olefination. | |
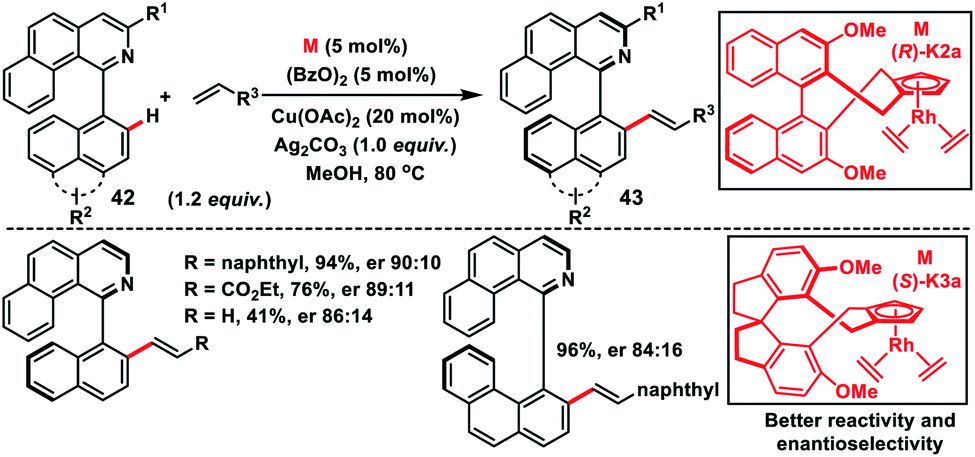
Asymmetric Rh(I)-catalysed introduction of axial chirality in biaryl systems 42 was developed by You and co-workers in 2014 (Scheme 22).35a The major challenge in the construction of axially chiral biaryls was the formation of cyclometallated species during C–H activation and it requires co-planarity of the two sterically hindered arenes which raised the energetic barrier of the reaction. For the first time chiral Cp rhodium complexes were used for the facile enantioselective synthesis of axially chiral biaryl systems in excellent yield and good enantioselectivity through the C–H activation process. Diversely substituted biaryls reacted well with a series of olefins like 2-vinylnaphthylene, styrenes and electron-deficient alkenes to grant the desired products 43. The same group in 2016 came up with another modified Rh(I)-catalyst with chiral Cp 1,1′-spiroindane ligands to synthesize axially chiral biaryls (Scheme 22).35b The modified Rh-complex (S)-K3a showed better activity and the reaction occurred even at room temperature providing products with improved enantioselectivity. The chiral biaryl products were used as suitable ligands in Rh-catalysed reactions.
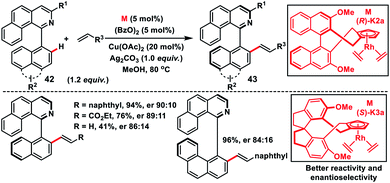 |
| | Scheme 22 Chiral Rh(I)-catalysed enantioselective C–H olefination. | |
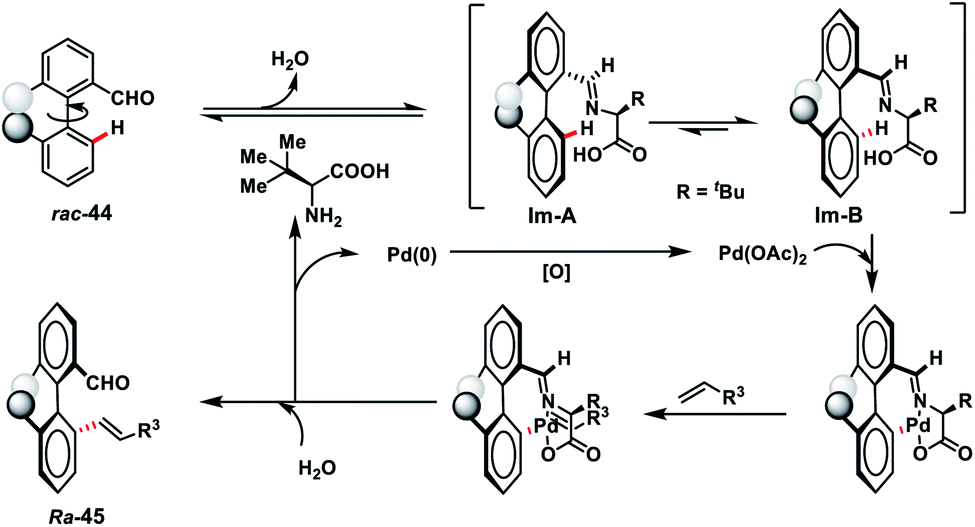
Pd-catalysed synthesis of axially chiral biaryls via C–H olefination reaction engaging tert-leucine as an inexpensive, catalytic and transient chiral auxiliary was developed by Shi and co-workers (Scheme 23).36a The reaction was compatible with diverse substituted biaryls 44 and several olefins including electron-deficient styrenes affording products 45 in good yield and excellent enantioselectivity. The reaction was proposed to progress via a reversible reaction of the chiral amino acid with 44 to form imine intermediates IM-A or IM-B. Due to the steric interaction, the C–H bond of one of the diastereomers cleaved preferentially to form an axially stereoenriched axial-biaryl palladacycle intermediate. The palladacycle intermediate underwent a typical Heck reaction followed by in situ hydrolysis of imine to yield chiral biaryls 45 (Scheme 24). Furthermore, the synthetic utility of the protocol was demonstrated by the asymmetric total synthesis of TAN-1085 in good yield and excellent enantioselectivity (>99% ee).36b The same group further elaborated an atroposelective C–H olefination reaction by the synthesis of highly enantiopure atropoisomers having pentatomic heteroaromatics (Scheme 23).36c The protocol was found to be well tolerated in terms of different five-membered biaryls containing benzothiophenes and benzofurans and provided axially chiral pentatomic biaryls in good enantioselectivity.
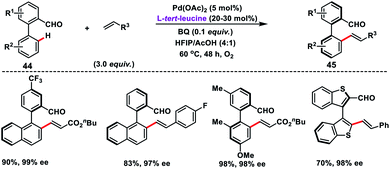 |
| | Scheme 23 Synthesis of axially chiral biaryls using a transient chiral auxiliary. | |
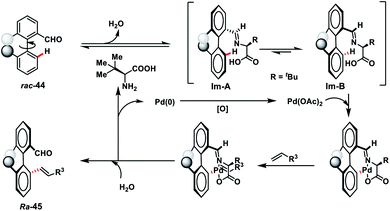 |
| | Scheme 24 Proposed path involved in the transient chiral auxiliary catalysis. | |
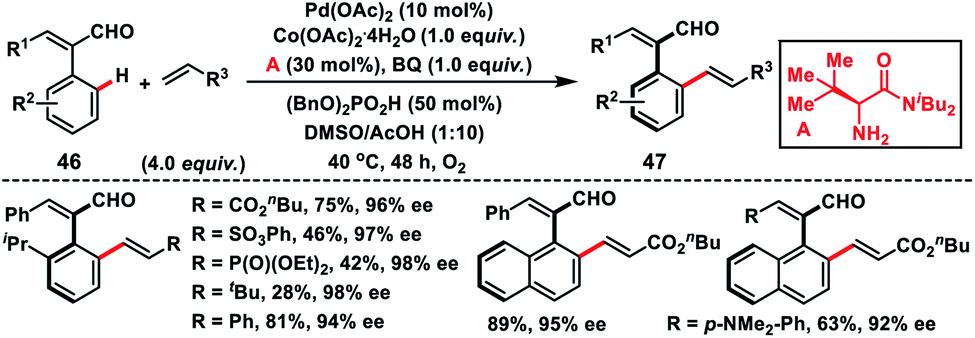
Recently, Shi's group has developed a Pd-catalysed protocol for the synthesis of challenging axially chiral styrenes 47 by introducing a bulky amino amide as the transient chiral auxiliary A (Scheme 25).37 Induction of chirality was challenging in cinnamaldehydes due to their relatively lower rotation barrier. In particular, sterically bulky substituents ortho to the chiral axis were necessary to ensure the enantioselectivity. Cinnamaldehydes 46 having a relatively large substituent at the ortho position have a higher rotational barrier compared to the smaller group. The successfully isolated palladacycle intermediate was found to have the same stereochemistry as the product.
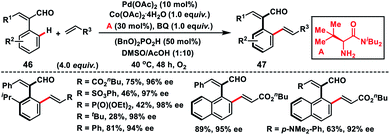 |
| | Scheme 25 Synthesis of axially chiral styrenes enabled by an amino amide transient directing group. | |
Pd-catalysed induction of atropoisomerism in biaryl systems by means of chiral ligands was first adopted by Yang and co-workers in 2017 to synthesize chiral atropoisomeric biaryl phosphine–olefins 49 (Scheme 26).38 They used MPAA Boc-L-Val-OH to execute phosphene-directed C–H olefination through dynamic kinetic resolution (DKR). Phosphene oxide 48 not only served as a directing group for C–H activation, but also controlled the outcome of the products. It was interesting to note that the electronic properties of the substituent on the biaryl system have a significant impact on the product yield and enantioselectivity.
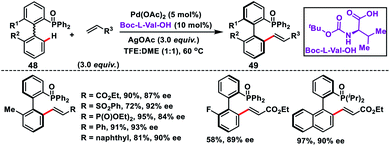 |
| | Scheme 26 Boc-L-Val-OH ligand-assisted synthesis of chiral biaryl phosphine-olefins. | |
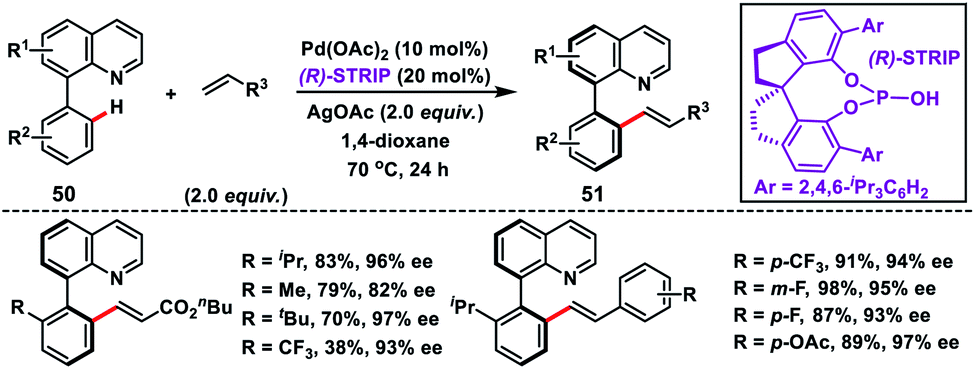
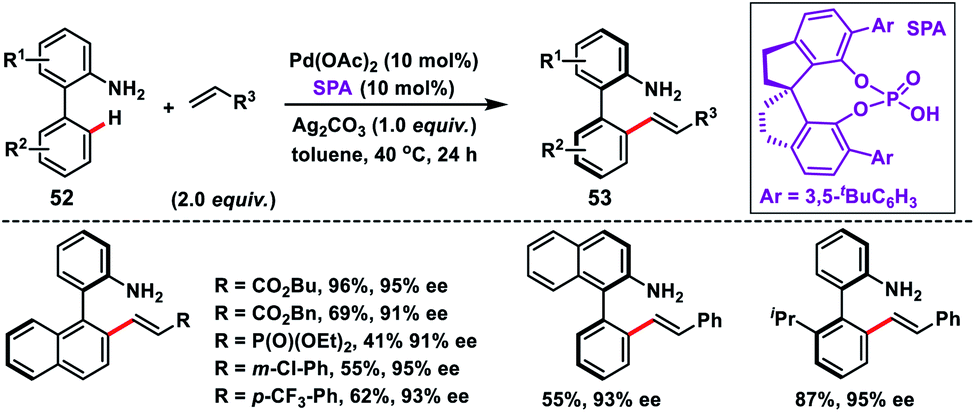
Shi and co-workers demonstrated chiral spiro phosphoric acid (R)-STRIP as an efficient chiral ligand for the Pd-catalysed synthesis of axially chiral quinoline-derived biaryls 51 in good yield and excellent enantioselectivities (99% ee) (Scheme 27).39a The better outcome of the enantioselectivity was due to the narrow and well-defined channel of SPAs which provided more rigid pockets than the BINOL-derived counterparts. This led to a healthier steric interaction with substrates 50. Density functional theory (DFT) suggested that the chiral phosphate acted as the counterion to stabilize Pd, while acetate ion served as the base in the cyclometallation deprotonation type C–H activation. Recently, a modified chiral spiro phosphoric acid (SPA) ligand was developed by the same group to execute free amine-directed C–H olefination leading to chiral biaryl-2-amines 53 in excellent yield and enantioselectivity (Scheme 28).39b The modified ligand showed an enhanced reactivity and its loading could be reduced up to 1 mol% without any alteration of enantiocontrol in gram scale synthesis.
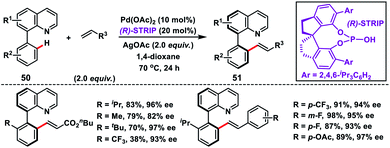 |
| | Scheme 27 (R)-STRIP as an efficient chiral ligand for the synthesis of axially chiral quinoline biaryls. | |
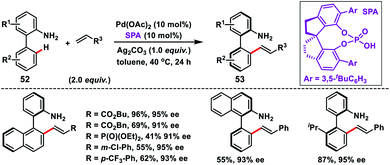 |
| | Scheme 28 Pd(II)-catalysed free-amine-directed atroposelective C–H olefination. | |
3. Distal C(sp2)–H olefination
Directing group assisted ortho-functionalization is rather facile considering the kinetic and thermodynamic stability of five, six and seven membered metallacycle transition states. However, to activate the distal C–H bond, a relatively large and energetically unfavorable 11, 12 or even higher atom containing metallacycle intermediate needs to be formed. As a result, controlling the selectivity is a major concern. Thus, it is vital to recognize the distal and geometrical relationships between the functional group and C–H bond of the substrate to overcome the entropy demand owing to the macrophane-like transition state and to achieve selective C–H activation.
3.1.
meta-C(sp2)–H olefination
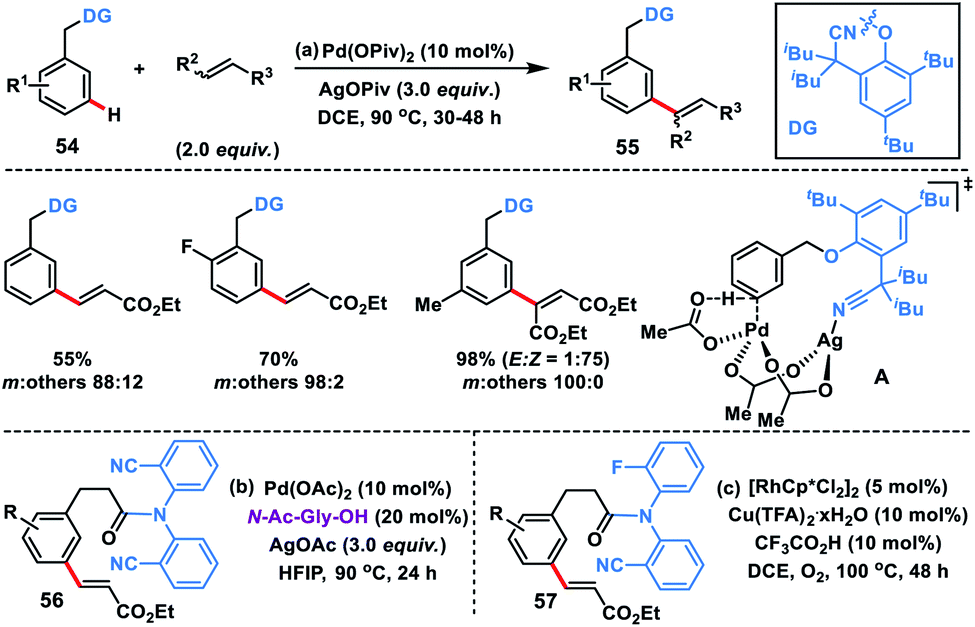
As an elementary step towards distal functionalization, Yu and co-workers in 2012 designed a U-shaped nitrile-based template. The template delivered the palladium metal to the vicinity of the meta-C–H bond of the tethered arene to execute C–H olefination with excellent selectivity overriding the electronic and steric bias (Scheme 29a).40ameta-Selective olefination (95%) of toluenes 54 was achieved in the presence of Pd(OPiv)2-catalyst and AgOPiv oxidant in DCE. The strategy was also successfully applied to the meta-olefination of hydrocinnamic acid derivatives 56 under modified reaction conditions. The reaction medium had a significant influence on the stability of the transition state as showcased by the change in the meta-selectivity on changing the solvent. DFT studies for meta-olefination of toluenes revealed that the C–H activation proceeds through a concerted metalation–deprotonation pathway and it is also the most energy demanding path. Also, C–H activation in a nitrile-based template occurred via the formation of a heterodimeric Pd–Ag transition state A having lower energy than any other form of palladium species (Scheme 29b).40b Yu and co-workers developed rhodium-catalysed meta-C–H olefination of hydrocinnamic acid derivatives 57 by a slight modification of the template used in Pd-catalysis (Scheme 29c).40c The approach was much ecofriendly as oxygen served as an external oxidant in the presence of a catalytic copper salt.
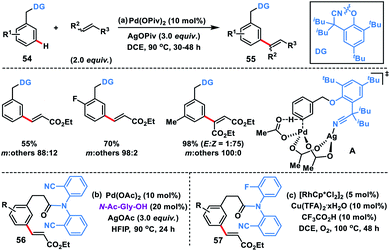 |
| | Scheme 29
meta-C–H olefination of toluenes and hydrocinnamic acids. | |
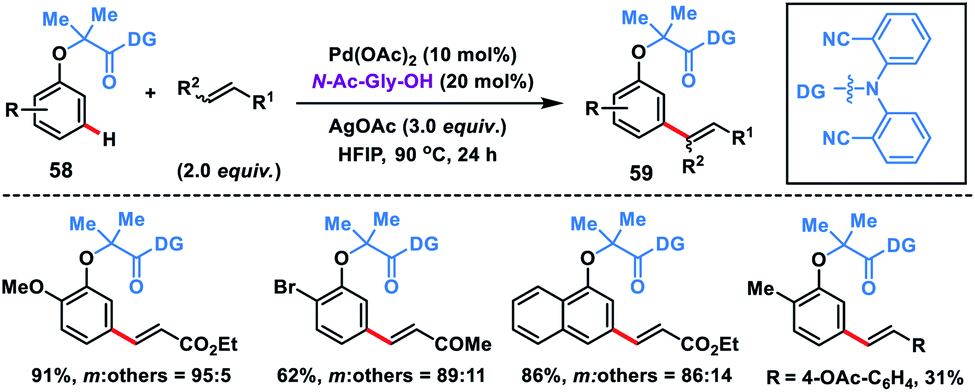
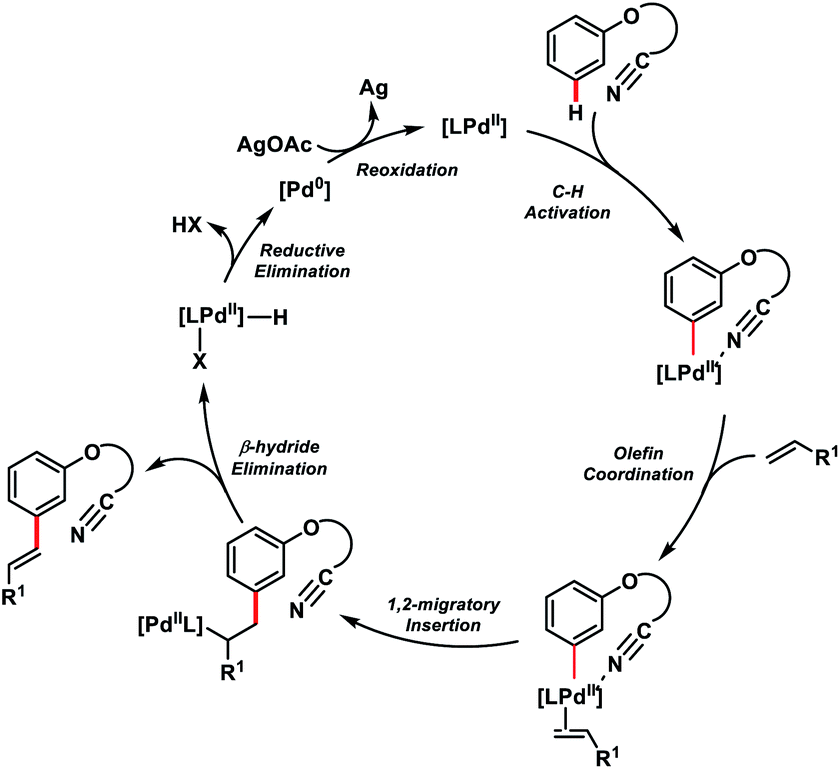
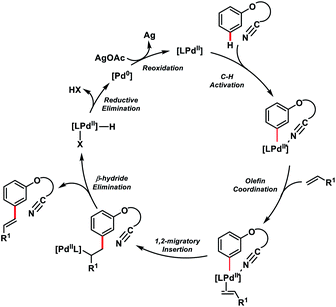
The directing group strategy for meta-C–H olefination was extended to phenol derivatives 58 by the same research group using a nitrile-based end-on template (Scheme 30).41a Selective meta-functionalization of phenol was more challenging considering that the meta position of the phenol is least active towards electrophilic attack. After rigorous optimization meta-olefinated products 59 were obtained in good yield and selectivity in the presence of Pd(OAc)2-catalyst, ligand N-Ac-Gly-OH and AgOAc oxidant in HFIP. Different phenol scaffolds including ortho-brominated substrates were well-suited under the reaction conditions and coupled with activated alkenes including electron-deficient styrenes. The mechanistic investigation41b indicated that the MPAA ligand N-Ac-Gly-OH not only acted as a dianionic bidentate ligand rather also as the internal base to abstract protons through the amidate group (Scheme 31).
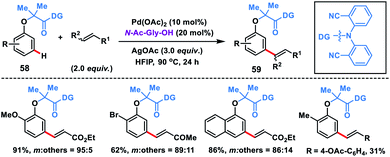 |
| | Scheme 30 Pd-catalysed meta-selective C–H olefination of phenols. | |
 |
| | Scheme 31 Catalytic cycle of meta-C–H olefination. | |
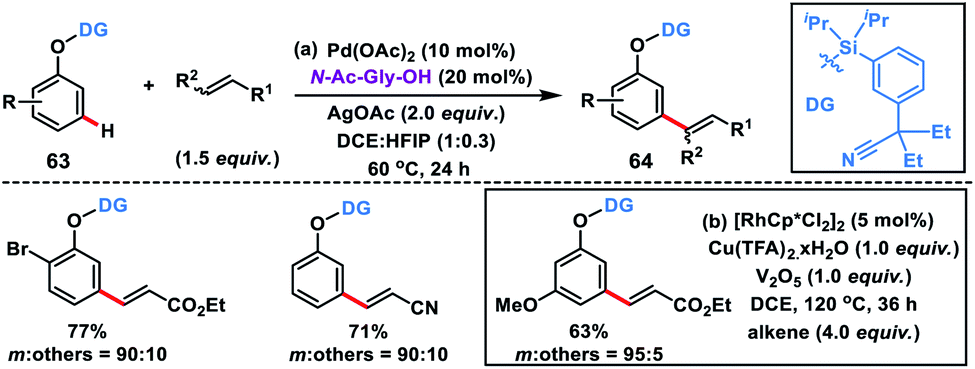
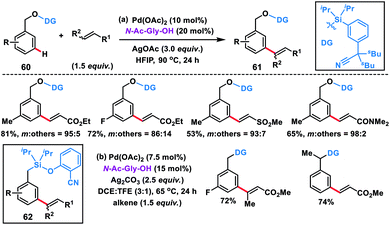
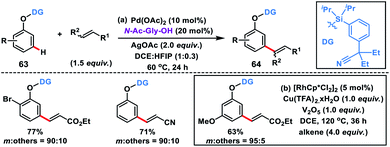
Tan's group further developed the template-based strategy for meta-selective C–H olefination of benzyl alcohols 60 using a silicon-tethered nitrile-based directing group (Scheme 32a).42a Easy incorporation of the template and its removal made the methodology more facile. meta-Selectivity up to 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 was achieved irrespective of the electronic nature of the substrate in the presence of a Pd(II)-catalyst, N-Ac-Gly-OH ligand and HFIP solvent. Extending the silyl-tethered directing group-assisted meta-olefination reactions, our group successfully olefinated benzylsilanes with very high selectivity (m:others > 20:1) to obtain 62 in good yields (Scheme 32b).42b Regioselective bis-olefination of benzylsilanes was conducted employing removable 2-hydroxy-5-methoxybenzonitrile. The silyl linker could be easily removed leading to the synthesis of olefinated toluenes, benzaldehydes and benzyl alcohols. Xu, Zhou and co-workers demonstrated meta-selective C–H olefination of phenols 63 using Pd and Rh catalysts simultaneously via incorporating a traceless organosilicon template bearing nitrile as the directing group (Scheme 33). The Pd-catalysed reaction proceeded well with AgOAc oxidant (Scheme 33a),42c while the MPAA ligand N-Ac-Gly-OH, hydrated copper salt oxidant and V2O5 co-oxidant are vital for the Rh-catalysed meta-olefination of phenols (Scheme 33b).42d The protocols were viable for a large range of olefins along with diversely substituted phenols and afforded the desired meta products 64 in good yield and excellent selectivity. meta-Olefinated phenols could be further utilized for coumarin synthesis after removal of the directing group.
:2 was achieved irrespective of the electronic nature of the substrate in the presence of a Pd(II)-catalyst, N-Ac-Gly-OH ligand and HFIP solvent. Extending the silyl-tethered directing group-assisted meta-olefination reactions, our group successfully olefinated benzylsilanes with very high selectivity (m:others > 20:1) to obtain 62 in good yields (Scheme 32b).42b Regioselective bis-olefination of benzylsilanes was conducted employing removable 2-hydroxy-5-methoxybenzonitrile. The silyl linker could be easily removed leading to the synthesis of olefinated toluenes, benzaldehydes and benzyl alcohols. Xu, Zhou and co-workers demonstrated meta-selective C–H olefination of phenols 63 using Pd and Rh catalysts simultaneously via incorporating a traceless organosilicon template bearing nitrile as the directing group (Scheme 33). The Pd-catalysed reaction proceeded well with AgOAc oxidant (Scheme 33a),42c while the MPAA ligand N-Ac-Gly-OH, hydrated copper salt oxidant and V2O5 co-oxidant are vital for the Rh-catalysed meta-olefination of phenols (Scheme 33b).42d The protocols were viable for a large range of olefins along with diversely substituted phenols and afforded the desired meta products 64 in good yield and excellent selectivity. meta-Olefinated phenols could be further utilized for coumarin synthesis after removal of the directing group.
 |
| | Scheme 32 Si-tethered nitrile-based directing group assisted meta-selective olefination of benzyl alcohol and toluene derivatives. | |
 |
| | Scheme 33
meta-Olefination of phenols using an organosilicon template. | |
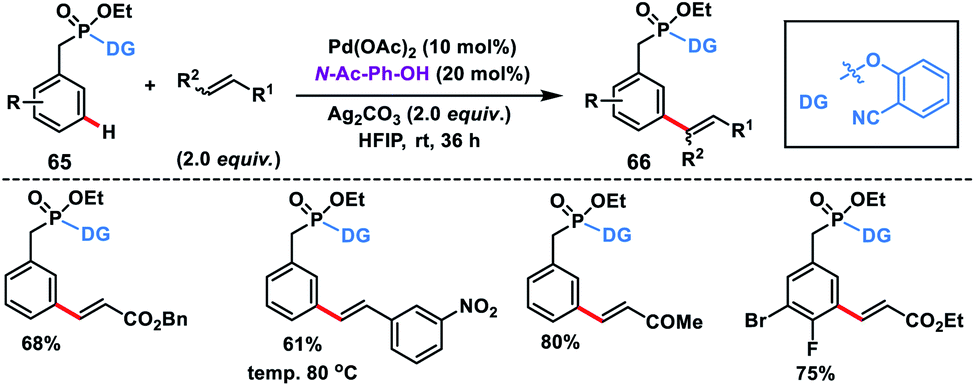
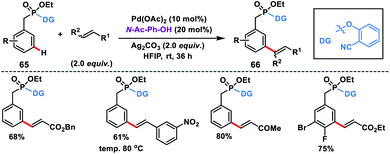
meta-C–H olefination of benzyl phosphonates 65 at room temperature was first reported by our group facilitated by a novel phosphonate-based template tethered with a 2-hydroxybenzonitrile directing group (Scheme 34).43 The reaction was performed in the presence of Pd-catalyst, N-Ac-Ph-OH ligand and silver oxidant in HFIP at room temperature to give products 66 in excellent yield and selectivity. Notably, di-olefination occurred at an elevated temperature (80 °C).
 |
| | Scheme 34
meta-Olefination of benzylic phosphonate esters. | |
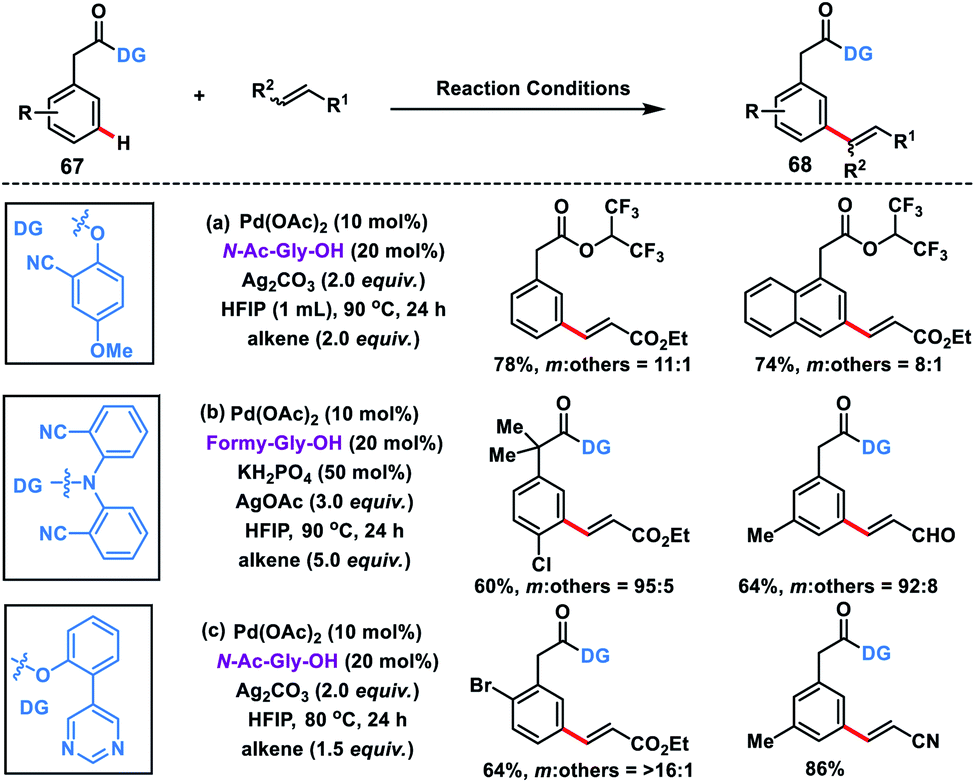
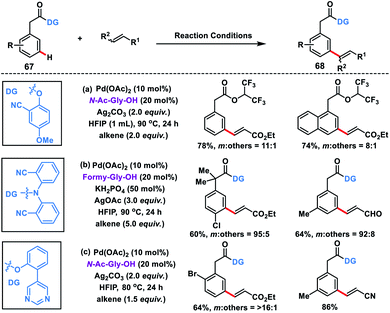
Considering the importance of arylacetic acids 67 in the pharmaceutical industry, our group developed the first meta-selective olefination of such molecules with the assistance of the 2-hydroxy-5-methoxybenzonitrile auxiliary. The reaction proceeded well in a combination of Pd-catalyst, MPAA ligand N-Ac-Gly-OH and a suitable silver oxidant, and afforded products 68 in good yield and selectivity (Scheme 35a).44a However, HFIP solvent used in the reaction led to trans-esterification thereby removal of the directing group, hence affecting the overall yield. Later on, Yu and co-workers reported meta-olefination of phenylacetic acids in the presence of modified MPAA ligand formyl-Gly-OH (Scheme 35b).44b Yu group reported a strongly coordinating pyrimidine-based template44c to execute meta-C–H olefination of arylacetic acids in order to broaden the substrate scope (Scheme 35c).
 |
| | Scheme 35
meta-Selective C–H olefination of phenylacetic acids. | |
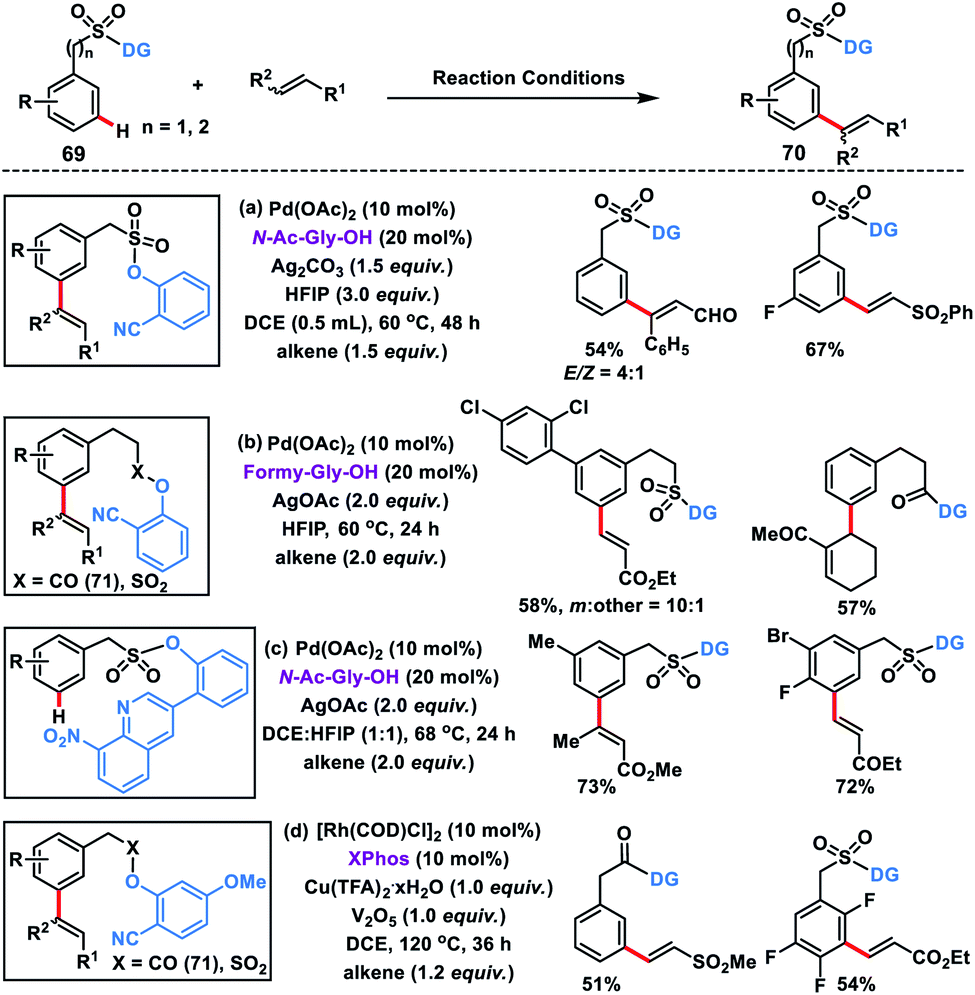
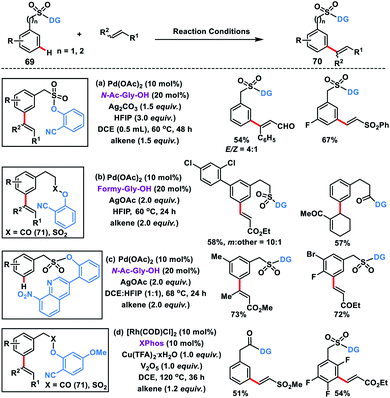
Our group developed Pd(II)-catalysed meta-C–H olefination of benzylsulfonyl ester derivatives 69 in the presence of N-Ac-Gly-OH ligand and Ag2CO3 oxidant to yield the desired products 70 in good yield and selectivity (Scheme 36a).45a Homo-diolefination and sequential hetero-diolefination in one pot with exclusive meta-selectivity and high yield were observed. Capitalizing on the same template our group extended the scope of meta-olefination for arylethane acid derivatives using sulfonyl 69 and carbonyl 71 linker (Scheme 36b).45b
 |
| | Scheme 36 Directed meta-olefination of alkylbenzene derivatives. | |
Being a weak coordinating group nitrile can be easily displaced in the presence of a strong coordinating group leading to deactivation of the catalyst. To overcome such a limitation, our group reported a strongly coordinating 8-nitroquinoline-based template tethered by a sulfonyl group for the selective meta-olefination (Scheme 36c).45c The strongly coordinating 8-nitroquinoline not only directs the metal towards the meta-position but also stabilizes the palladacycle through its high coordinating ability. Later, the scope of the meta-olefination of benzylsulfonyl esters 69 and phenylacetic acid derivatives 71 was extended by our group through Rh-catalysis in the presence of XPhos ligand (Scheme 36d).45d Unlike Pd(II), the use of Rh(I) limits the transesterification while DCE was found to be a more suitable solvent than HFIP. UV studies suggest that Rh(I) species in situ transformed into its active form Rh(III) intermediate and catalysed the reaction.
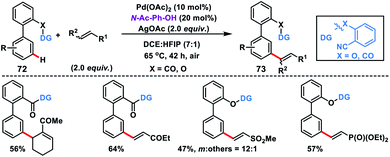
Our group in 2016 explored the distal C–H olefination of biphenyl carboxylic acids and phenols 72 with the help of a carbonyl linker and nitrile as the weak coordination group. The reaction proceeded well in the presence of a Pd-catalyst along with the MPAA ligand N-Ac-Ph-OH and AgOAc oxidant to deliver highly selective meta-olefinated products 73 (Scheme 37).46a Both substrates showed almost similar reactivity under the optimized conditions, which validated the principle of the selectivity–reactivity paradigm. Recently, Yu group developed a 2-pyridone ligand-promoted meta-selective C–H olefination of biphenyl nitrile derivatives under Pd-catalysis.46bmeta-Selective olefination in the reaction was completely controlled by the ligand without the direct involvement of the directing group. The computational study suggested that a Pd–Ag bimetallic bridge forms with the involvement of 2-pyridone and acetate molecule to lower the energy barrier, and C–H activation proceeded through a cyclometallation–deprotonation pathway.
 |
| | Scheme 37
meta-C–H-olefination of biphenyl carboxylic acids and phenols. | |
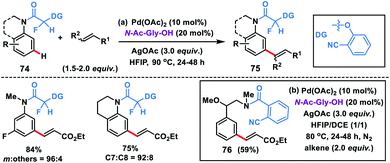
The use of a carbonyl-based template with an α-fluoro substituted nitrile auxiliary for the meta-olefination of anilines and N-heteroaromatic compounds 74 was expanded by Yu and co-workers (Scheme 38a).47a The presence of a fluorine atom α to the carbonyl group led to a conformational change and the carbonyl group drifted away from the ortho-C–H bond. The same α-fluoro substituted nitrile auxiliary was also applicable to aniline derivatives for meta-C–H olefination 75 with diverse activated alkenes. Li and co-workers in 2015 demonstrated meta-selective olefination of phenylethylamines 76 by using 2-cyanobenzoyl as the directing auxiliary (Scheme 38b).47b The same group later developed a carbamate-linked directing group by incorporating CO2 into a nitrile-based template for the Pd-catalyzed meta-olefination of anilines.47c
 |
| | Scheme 38
meta-Selective C–H olefination of amines and heteroaromatics. | |
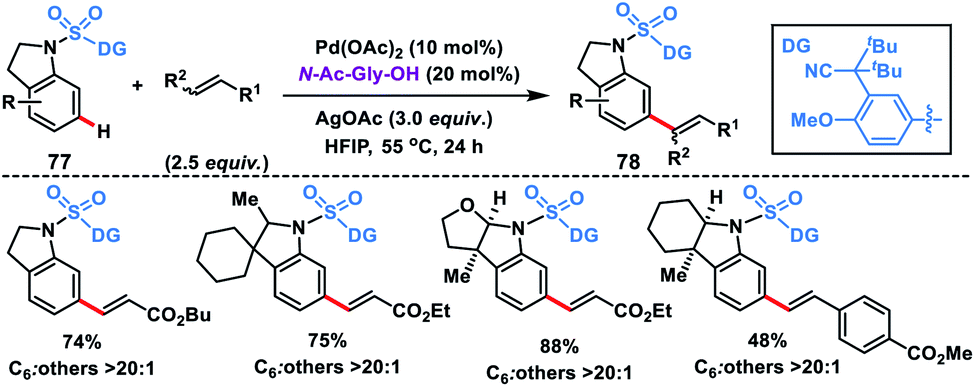
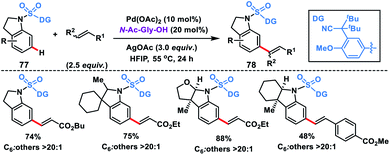
Electrophilic attack on indoline moieties is more favourable at the ortho and para positions, therefore meta-functionalizations of these substrates are extremely challenging. Yu and co-workers reported meta-selective olefination of indolines 77 by incorporating an electron-withdrawing sulfonyl bridge with an appropriate nitrile auxiliary (Scheme 39).47d The MPAA N-Ac-Gly-OH ligand-assisted meta-olefination yielding 78 proceeded through a metalation deprotonation path and provided a selectivity of >20:1 with good yields. Also, a series of N-heterocycles were olefinated at the distal position using the sulfonyl bridged template with a modified auxiliary.47e
 |
| | Scheme 39
meta-Selective olefination of indoline derivatives. | |
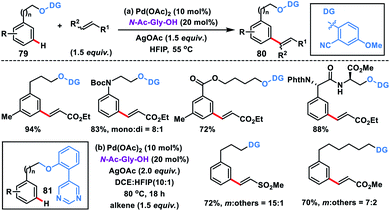
meta-Olefination of long chain arenes and tethered alcohols with very high regioselectivity was discovered by Xu, Jin and co-workers using a 2-cyanophenol based template (Scheme 40a).48a The olefination reaction proceeded well in the presence of a Pd-catalyst, MPAA ligand N-Ac-Gly-OH and a silver salt as the oxidant. A series of substrates such as 3-phenylpropanols, 2-phenoxyethanols, and 2-(phenylamino)ethanols including dipeptide-derived substrates 79 were effectively olefinated affording 80 in high yields and selectivity. In 2018, our group developed a strongly coordinating pyrimidine-based directing template for meta-functionalization of the substrate 81 which differed widely in the linker chain length (up to 20 atoms) (Scheme 40b).48b Different alkenes and arenes were tolerated under the optimized reaction conditions.
 |
| | Scheme 40
meta-C–H olefination of arenes across different linker lengths. | |
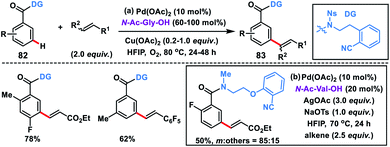
meta-Olefination of relatively electron-poor benzoic acid derivatives 82 was challenging and first reported by Li and co-workers in 2016 using an N-nosyl-substituted amide bridged template (Scheme 41a).49a A range of benzoic acid derivatives underwent meta-olefination to produce 83 in good yields and selectivity. Use of molecular oxygen as a terminal oxidant in the presence of a catalytic copper salt made the protocol more convincing. Later, Yu group designed a conformationally more flexible 2-(2-(methylamino)ethoxy)benzonitrile-based template for the meta-olefination of benzoic acids (Scheme 41b).49b They used an N-methylated amide having an additional ether linkage for the elongation of the directing group and performed the reaction with N-Ac-Val-OH as a ligand and AgOAc oxidant along with an NaOTs additive. Mechanistic investigation revealed that the reaction proceeded through a bimetallic Ag–Pd heterodimer intermediate. KIE studies suggested that the C–H activation step was the rate-limiting step.
 |
| | Scheme 41 Directing group-assisted meta-C–H olefination of benzoic acids. | |
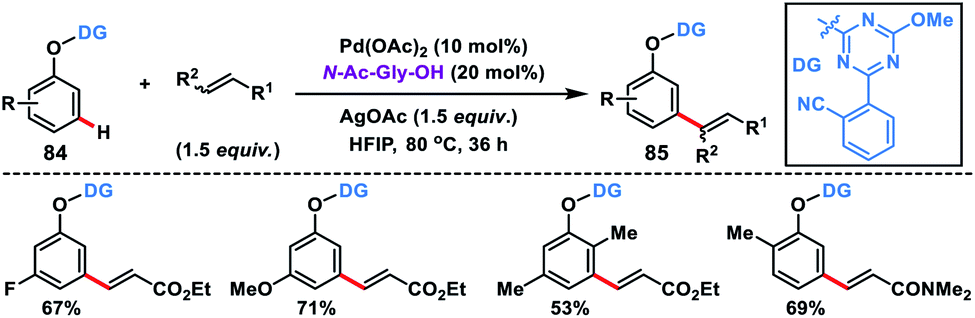
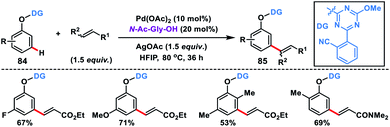
In 2019, Yu and co-workers synthesized a bifunctional template to accomplish Pd-catalysed meta-olefination of phenols 84 in the presence of N-Ac-Gly-OH ligand (Scheme 42).50 The synthesized olefinated product 85 underwent Ni-catalysed ipso coupling of the C–O bond of the phenols with aryl boronic acid to yield 1,3-disubstituted arenes. Both steps of the reaction could be performed in one pot.
 |
| | Scheme 42 Triazine linked template-assisted meta-olefination of phenols. | |
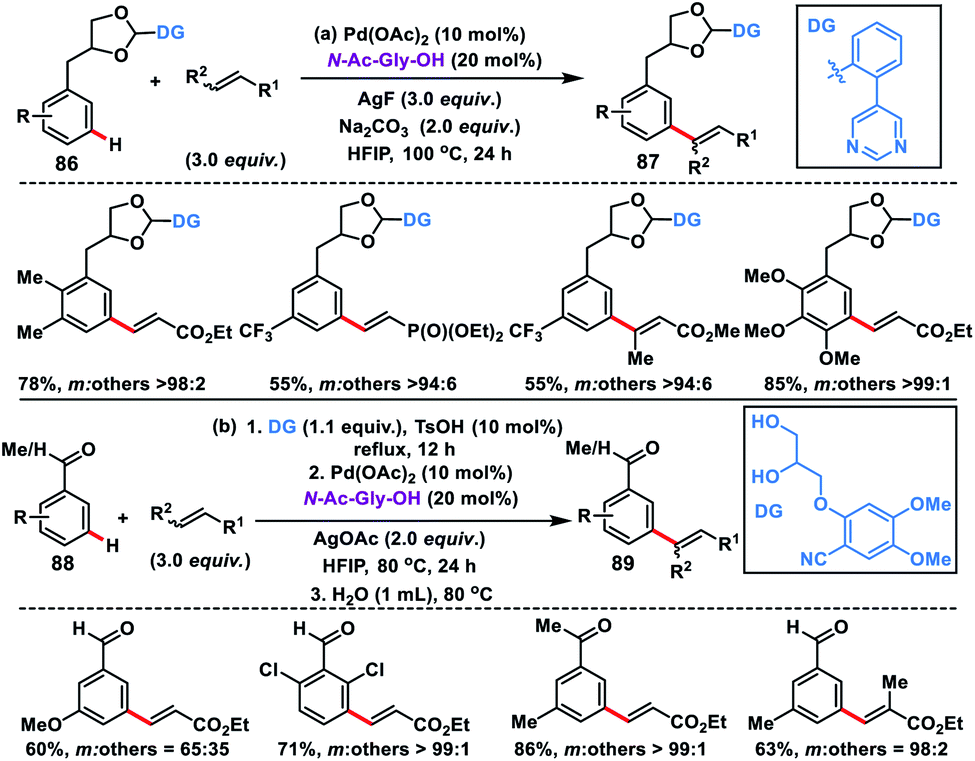
Use of an acetal or a ketal linker for the meta-olefination of 1,2-diols 86 and aldehydes or ketones 88 was developed independently by Kong and Ji group in 2019. Kong and co-workers disclosed the Pd-catalysed meta-olefination of 3-phenylpropane-1,2-diol 86 using the strongly coordinating pyrimidine directing group to yield 87 (Scheme 43a).51a The directing template cleaved up to some extent during the reaction resulting in the mixture of products. Later, Xu, Jin and co-workers used a combination of a Pd-catalyst and N-Ac-Gly-OH ligand for the meta-olefination of electron-poor aromatic aldehydes and ketones 88 using an acetal and ketal linker (Scheme 43b).51b The reaction was performed in one pot with sequential protection of carbonyl by a 1,2-diol-based nitrile template and Pd-catalysed C–H olefination reaction to yield 89 followed by removal of the template by a simple hydrolysis to access meta-olefinated aldehydes or ketones.
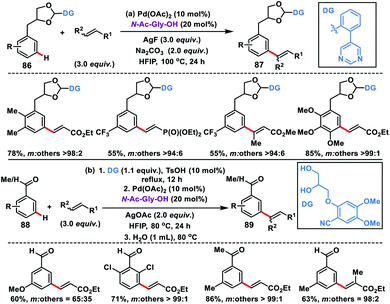 |
| | Scheme 43 Acetal or ketal linker-assisted meta-C–H olefination of arene-tethered diols and aldehydes or ketones. | |
In 2019, Li and co-workers first discovered the carboxylic acid group as the directing auxiliary for meta-olefination (Scheme 44).52 The reaction proceeded through the assistance of the carboxylic acid group, which binds with Pd possibly through k2-coordination mode instead of k1 thus overriding ortho-C–H activation. The reaction proceeded well with a series a substituted arenes 90 and different olefins including styrenes and delivered meta-olefinated products 91 in good yield and selectivity. To verify that the Pd coordinates in k1 mode in the ortho-C–H activation, a directing group without the fluorine substituent at the ortho-position was employed which afforded good remote selectivity hence supporting the hypothesis. The remote selectivity in this reaction could be rationalized as that perhaps both proximal and meta-cyclopalladation isomers were formed but the meta one was more susceptible towards functionalization due to its weaker coordination.
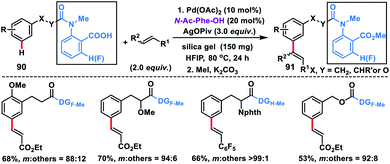 |
| | Scheme 44 Carboxy group directed meta-C–H olefination of alkylbenzenes. | |
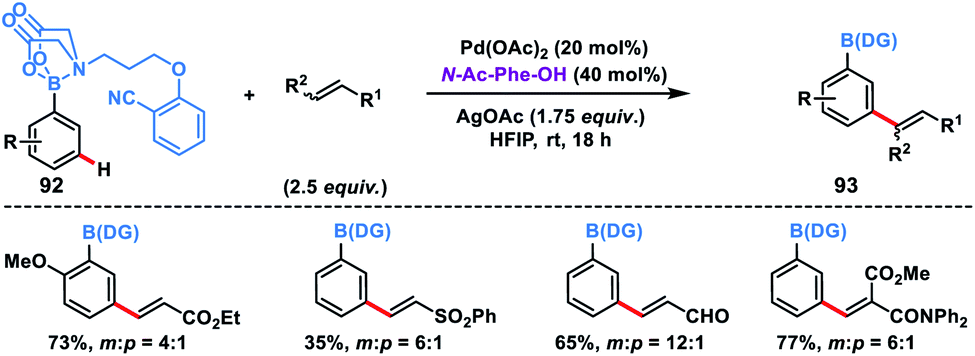
Recently, Spivey, Cordier and co-workers presented a novel N-methyl iminodiacetic acid (MIDA) boronate derivative as the protecting cum directing group for the Pd-catalysed meta-selective olefination of boronic acids 92 (Scheme 45).53 MIDA-DG boronate could be easily accessed through condensation with boronic acid and also can be removed under mild basic conditions. The reaction tolerated a series of substituted phenylboronic acids and activated olefins to give meta-olefinated products 93 in moderate yield and selectivity. The catalytic efficiency, selectivity and reaction range of this protocol needed to be further improved as it provided path for several inaccessible boronic acid derivatives.
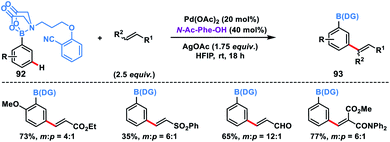 |
| | Scheme 45
meta-Olefination of aryl boronic acids directed by MIDA-derived boronate ester. | |
In recent times use of transient directing groups has emerged as a powerful and attractive strategy in chemical transformations as an alternative to covalently linked directing auxiliaries. The first study on transient directing meta-C–H olefination has been recently reported by our group, where an imine served as the linker (Scheme 46).54 We developed a modified pyrimidine-based template linked through an imine which served as the directing template for meta-olefination of synthetically important biphenyl aldehydes 94 and amines 96. In the case of biphenyl aldehyde, copper salt served as the oxidant in the presence of a catalytic silver salt while in biphenyl amine the silver salt was the sole oxidant. The MPAA ligand N-formyl-Gly-OH was essential for the reaction and the imine linkage hydrolysed in situ during acidic workup.
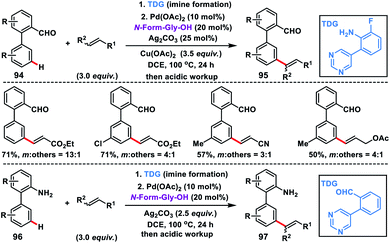 |
| | Scheme 46 Imine as a transient-directing group for meta-C–H olefination. | |
3.2.
para-C(sp2)–H olefination
Electrophilic substitution reactions in benzene at ortho and para positions are governed by the electronic nature of the substituents present. However, regioselective functionalizations at the para-position are scarce and only a few reports are available in the literature which are governed by the steric and electronic nature of the substituent. Till 2015, no reports were available which could address the issue on a striking scale for highly selective para functionalization of arenes.
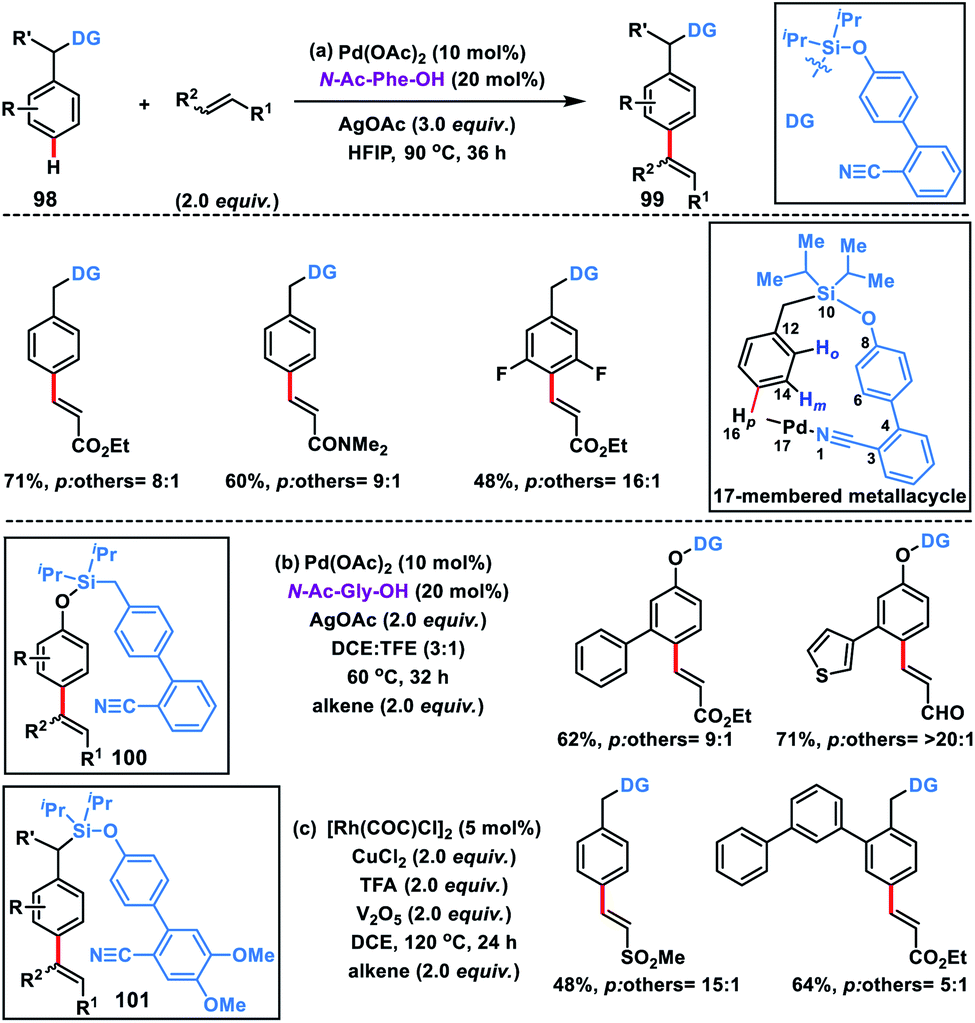
For the first time a concrete solution to address the para-selectivity was reported by our group by extending the application of template-based directing groups (Scheme 47a).55a The major apprehension in para-C–H functionalization is to deliver the metal-catalyst towards the targeted C–H bond due its distal location in the arene molecules. Also, a weakly coordinating directing group is required for the site selective interaction of metal catalysts. Additionally, the entire catalytic cycle has to proceed through a large macrocyclic cyclophane-like (16–17 membered) transition state. To overcome these obstacles, a judicially engineered directing group, linker and its length are required to reach the target C–H bond. As an initial approach to satisfy all these requirements, a biphenyl template containing the weakly coordinating cyano group was chosen. The heteroatom of the cyano group easily coordinates with the metal-catalyst, while the biphenyl system regulates the chain length of the macrocyclic assembly. After a series of optimizations, we came up with a weakly coordinating nitrile-based biphenyl template attached to the toluene ring through a silyl linker. The substituent present on the silicon atom plays a prominent role as it exerts the Thorpe–Ingold effect and pushes the directing group towards the target position. With this directing template, a series of toluene derivatives 98 were successfully olefinated regioselectively at the para position in the presence of a Pd-catalyst, N-Ac-Phe-OH ligand and silver oxidant in HFIP solvent (Scheme 47). A diverse array of toluene derivatives coupled with several activated olefins affording para-olefinated products 99 in decent yield and good selectivity by overriding all sorts of electronic and steric bias present in the substrate. A series of α,β-unsaturated esters, sulfones, amides and acrylates of vitamin E and cholesterol were used as olefin coupling partners with appreciable para-selectivity. The directing auxiliary could be easily removed by simply treating with TBAF to yield the para-olefinated toluene derivatives.
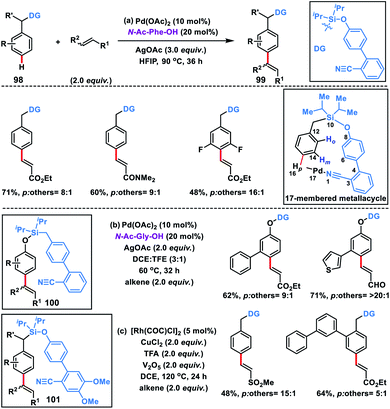 |
| | Scheme 47
para-Olefination of toluenes and phenols using a biphenyl director. | |
After the successful olefination of toluene derivatives, our group extended the application of this directing template for the para-olefination of phenol derivatives 100 by simply swapping the oxygen and methylene group on both sides of the silyl linker (Scheme 47b).55b The reaction proceeded under almost similar conditions, however the ligand N-Ac-Gly-OH was found to be optimum in the solvent combination of DCE and TFE (3:1). The methodology was applicable for a wide range of phenol derivatives and activated olefins and afforded olefinated products in 81% yield with high regioselectivity (para/others, 10:1). A wide range of functional groups like amide, ester, ketone, aldehyde and sulfonyl were well tolerated under the optimized reaction conditions. Post-synthetic application of the synthesized products was illustrated by constructing valuable natural product derivatives such as ferulic acid, the anti-inflammatory artepillin C, antimicrobial plicatin B and drupanin in good yield.
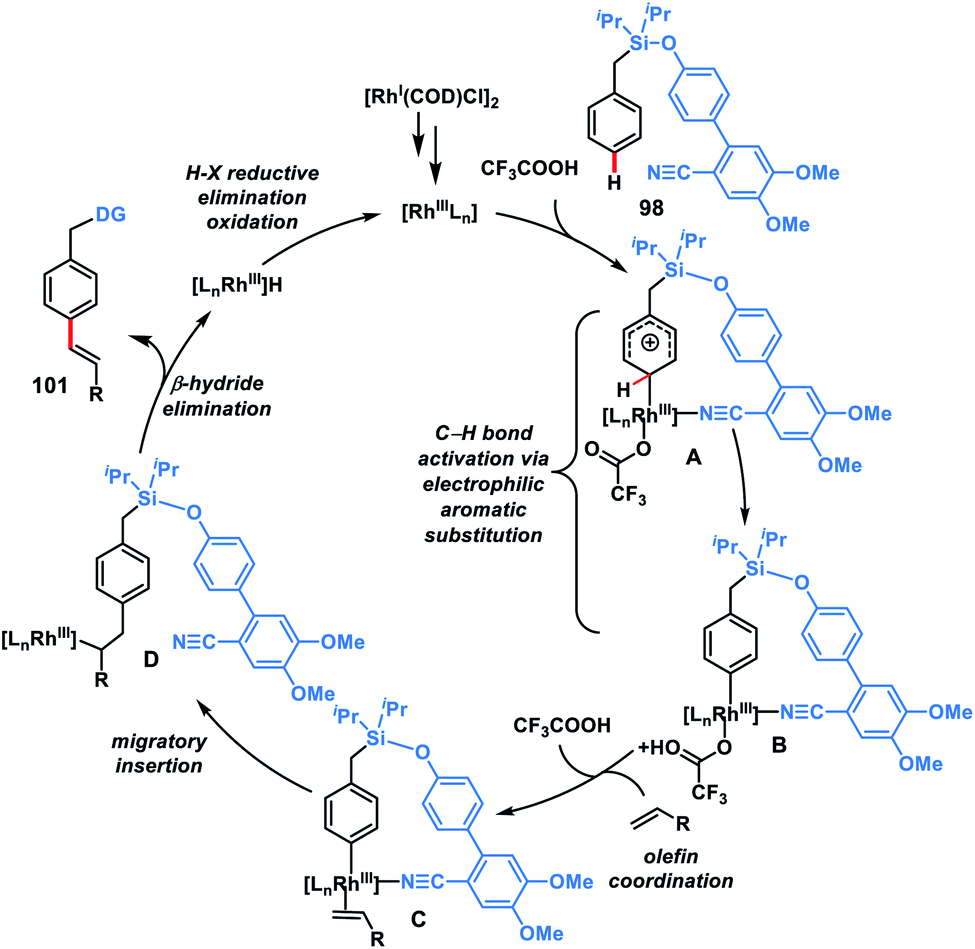
In 2019, our group modified the existing biphenyl template by introducing two OMe groups in the ring having a weakly coordinating nitrile group. This modified template has a higher electron density on the directing phenyl ring and is called the second-generation template. The second-generation template with higher electron density provided a new avenue in Rh-catalysed distal C–H functionalization. Our group utilized this second-generation template for the Rh-catalysed para-C–H-olefination of toluenes 101 under ligand and metal oxidant free conditions (Scheme 47c).55c A variety of olefins were well tolerated with different toluene derivatives along with the α-substituted one. Intermolecular isotope labelling experiments were performed and a PH/PD value of 2.9 and kH/kD value of 2.6 indicate that C–H bond activation is likely to be the rate-limiting step. Kinetics experiments performed showed that the reaction was first order with respect to the toluene substrate and zero order with respect to the olefin. Furthermore, DFT calculations suggested that C–H activation follows an electrophilic aromatic substitution path rather than a concerted metalation–deprotonation pathway (Scheme 48) and coordination of the nitrile with Rh stabilizes the C–H activation transition state. Computational studies suggested that the incorporation of two methoxy groups on the directing template activates the substrate towards C–H bond cleavage by lowering the energy barrier.
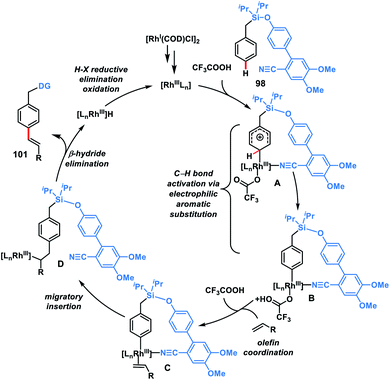 |
| | Scheme 48 Possible catalytic cycle for para C–H olefination. | |
3.3. Bifunctional-directed distal C(sp2)–H olefination
The directing group plays an important role in the site selective activation of single C–H bonds. However, the use of these directing groups is limited due to the C–H bond distance and also due to the shape of the substrates. Also, incorporation of covalently attached directing templates in molecules lacking appropriate functional groups is not feasible. Therefore, to come up with an approach not relying on covalent interaction to reach the distal position is the need of the hour since many medicinally important heterocycles lack functional groups to tether templates covalently.
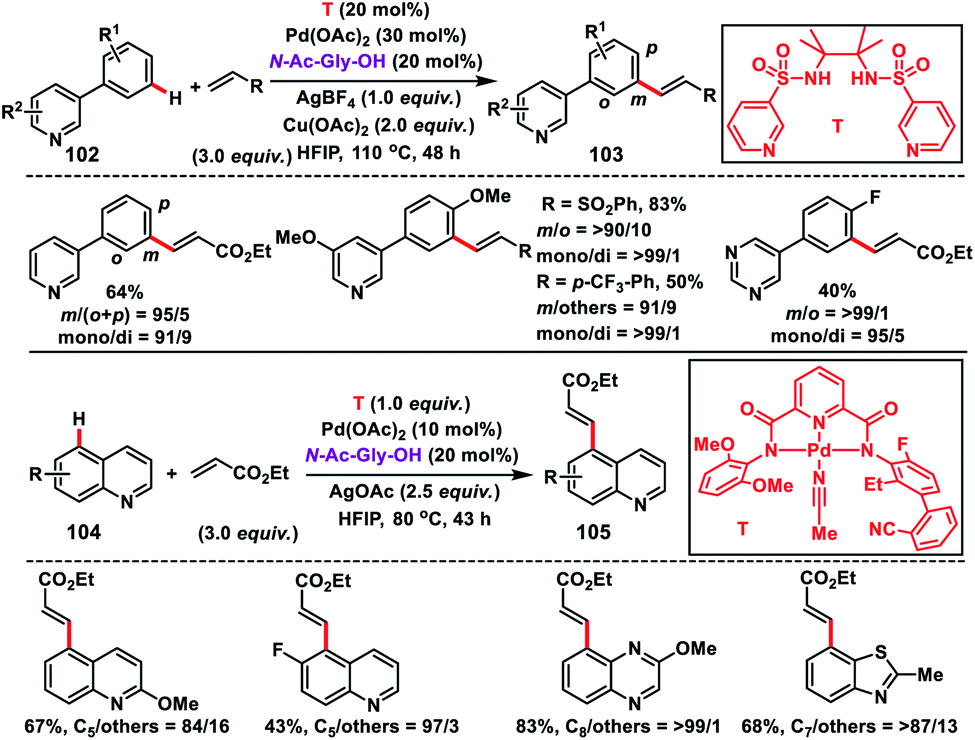
In this regard, Yu and co-workers in 2017 formulated a bifunctional template capable of coordinating with two metal centres to play two distinct roles simultaneously. For that purpose they synthesized a bis-amide based backbone-template where the sidearm holds the directing group. The bis-amide chelate with a metal centre is responsible for substrate binding and the sidearm directing group directs the Pd-catalyst towards the specific remote C–H bond. After extensive screening they found that a sulfonamide-based template (T) derived from the sterically hindered 2,3-dimethyl-2,3-butanediamine was the most effective and afforded the olefinated heterocycles 3-phenyl pyridine 103 in decent yield and selectivity (Scheme 49).56 The result obtained from the screening portrays the importance of conformational constraints of the backbone for remote C–H activation. In order to diversify the bimetallic catalysis, other heterocycles like quinolines 104 were chosen for the site-selective olefination reaction. However, the sulfonamide-based template was found to be unproductive, which demanded further optimization of templates. After screening of several covalent templates, they found a nitrile-based template (T) anchoring the first metal through tridentate coordination for the effective olefination of quinolines to give the product 105 in good yield and selectivity (Scheme 49).56 However, a quantitative amount of template is required in the presence of palladium acetate-catalyst and MPAA ligand N-Ac-Gly-OH.
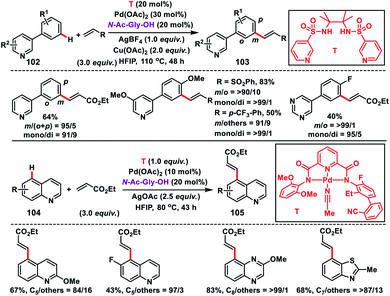 |
| | Scheme 49 Bifunctional template assisted site-selective C–H olefination. | |
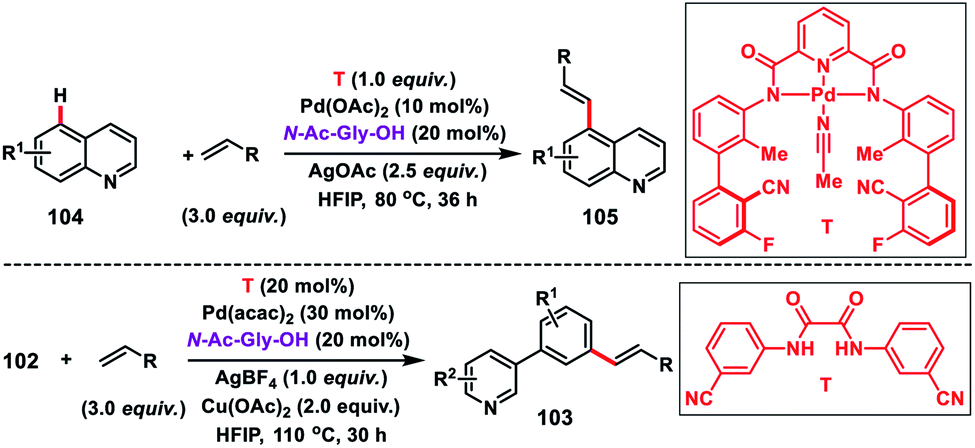
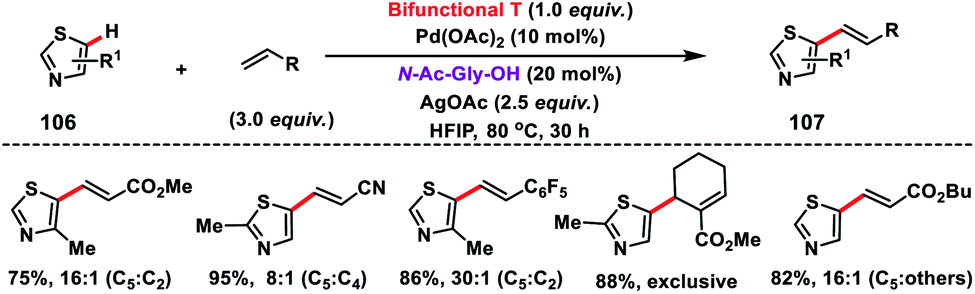
Shortly after, a similar remote C–H olefination of small heterocycles using a modified bifunctional template (T) was reported by our group (Scheme 50).57 We synthesized a symmetrical covalently attached nitrile-based tridentate template for the site-selective remote C–H olefination of heterocycles 104 in improved yield and selectivity. A variety of substituted quinolines and other heterocycles such as benzoxazole and benzothiazole were employed under the reaction conditions to afford products 105 in good yield and selectivity. We further designed a nitrile-based bifunctional amide derived from oxalic acid for the effective meta-olefination of 3-phenylpyridine derivatives 102 in synthetically useful yield and selectivity. Later, in 2019 our group utilized the same bifunctional tridentate template (T), suitable for quinoline, for the C5 selective olefination of thiazole derivatives 106 in excellent yield and selectivity (Scheme 51).58 Admirable selectivity was observed for mono-substituted or even unsubstituted thiazoles. Diverse acrylates and electron-deficient styrenes were well tolerated and provided the desired products 107 in good yield and selectivity.
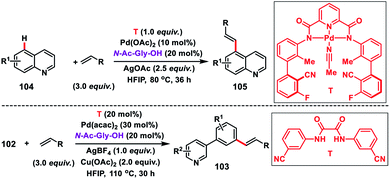 |
| | Scheme 50 C–H olefination of heterocycles with bi- and tridentate templates. | |
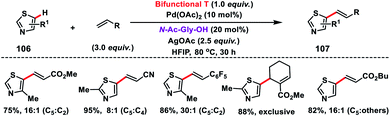 |
| | Scheme 51 C-5 selective olefination of thiazoles with a bifunctional template. | |
4. Non-directed C(sp2)–H olefination
Oxidative olefination of arenes was discovered by Fujiwara–Moritani n 1967 using a stoichiometric amount of Pd(OAc)2 and excess arenes. The major concerns in the non-directed approach are controlling the regioselectivity, use of excess arenes and harsh reaction conditions. Recently, ligand-enabled non-directed arene C–H olefination has become one of the focused areas via oxidative C–H activation to overcome the limitation of Fujiwara–Moritani reaction. The reaction mechanism involved in general in the non-directed C–H activation is the formation of a carbon–metal bond via concerted metalation–deprotonation (CMD) where a basic ligand is involved in the proton abstraction.
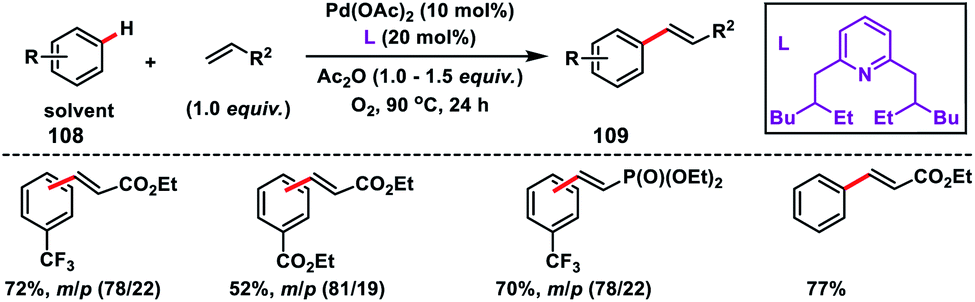
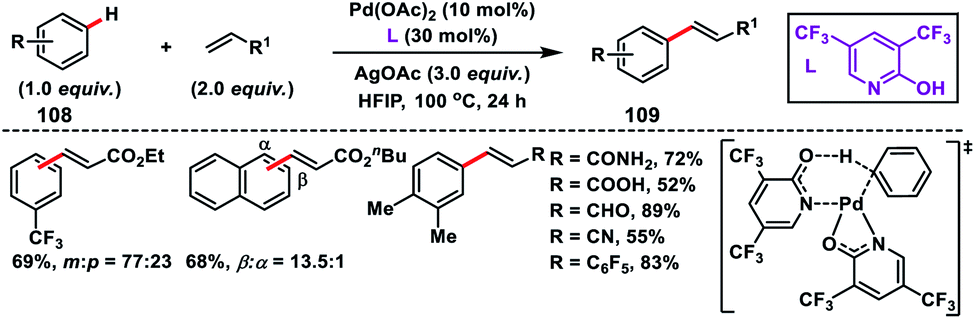
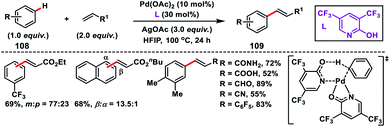
Yu and co-workers in 2009 reported the catalytic olefination of electron-deficient arenes 108 in good yield using the sterically demanding ligand 2,6-bis(2-ethylhexyl)pyridine which coordinates with palladium in a 1:1 ratio (Scheme 52).59a Oxygen was used as an oxidant to re-oxidize Pd(0) to Pd(II) with the help of the ligand. However, use of excess arenes (usually 20–30 equiv.) exerts a serious drawback especially for substituted arenes. A series of arenes with an electron-deficient group were olefinated with activated alkenes preferably at the meta position to yield 109 in good selectivity even when the substrate had an ortho directing functional group. A detailed theoretical study by Zhang on the Pd-catalysed olefination of electron-poor arenes concluded that the reaction follows a concerted metalation/deprotonation (CMD) step.59b The reaction proceeded through a six-membered metallacycle transition state and the selective meta-functionalization in the ortho-directing substrates can be rationalized by the steric repulsion caused by the ancillary pyridine type ligand as the major as well as the electronic effect.
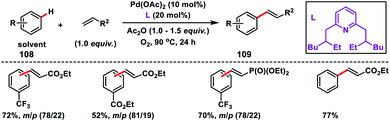 |
| | Scheme 52 Pd-catalysed olefination of arenes using 2,6-dialkylpyridine ligand. | |
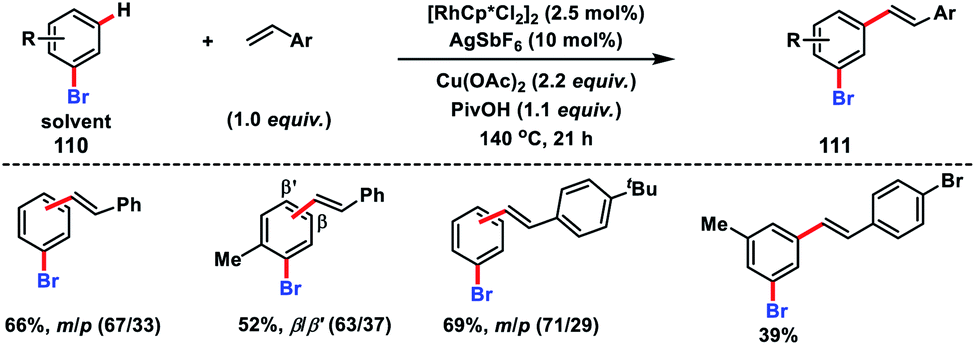
A Rh-catalysed olefination of bromoarenes 110 with substituted styrenes was demonstrated by Glorius and co-workers to synthesize meta and para products 111 (Scheme 53).60a It was demonstrated that bromoarenes were an integral part of the reaction serving as the terminal oxidant as well as a catalyst modifier. In 2012, Sanford's group established a facile oxidative olefination of arenes with a combination of a pyridine-based ligand 3,5-dichloropyridine and Pd(OAc)2 in a 1:1 ratio.60b This reaction was more efficient in terms of yield and broader substrate scope and applicable to both electron rich and deficient arenes.
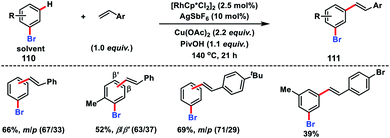 |
| | Scheme 53 Rh-catalysed olefination of bromoarenes with styrene derivatives. | |
In order to address the shortcomings of previous reports Yu and co-workers demonstrated an arene limited (reagent quantity) Fujiwara–Moritani oxidative olefination. They used a bimetallic Rh(III)-complex as the catalyst and phosphene as the ligand.61a The bimetallic Rh(III)-complex in combination with a copper salt and V2O5 was used for the mono-olefination of electron-rich and neutral arenes in good yield. A 1:1 ratio of meta/para products were obtained for monosubstituted arenes and β-selective olefination for 1,2-disubstituted arenes albeit under harsh reaction conditions (140 °C). Duan and co-workers in 2014 demonstrated a Pd-catalysed oxidative olefination of arenes using the bidentate monoanionic nitrogen ligand 2-OH-1,10-phenanthroline and catalytic Cu(OAc)2 along with oxygen as the external oxidant.61b The coordination of Pd-catalyst with the bidentate monoanionic ligand enabled the C–H activation reaction.
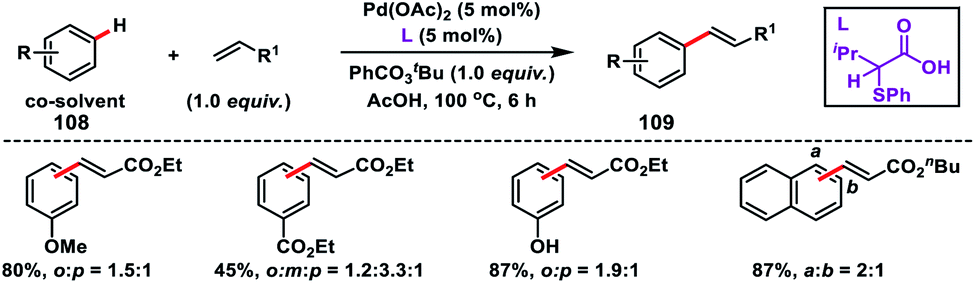
Fernández-Ibáñez and co-workers in 2017 synthesized a new class of amino acid based bidentate S,O-ligands for Pd-catalysed olefination of arenes 108 with better activity and selectivity than the Pd/pyridine combinations (Scheme 54).62a The late stage functionalization of complex molecules such as O-methylestrone and naproxen derivative was also successfully applied. An S,O-ligand was successfully utilized for the para-selective olefination of anilines under mild conditions and with high efficiency by the same group.62b The observed selectivity was solely due to the S,O-ligand.
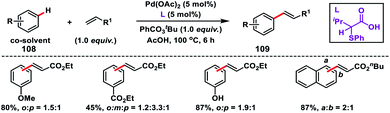 |
| | Scheme 54
S,O-Ligand-promoted Pd-catalysed olefination of arenes. | |
Yu and co-workers in 2017 demonstrated arenes as the limiting reagent in Pd-catalysed olefination of a diverse array of electron-rich and electron-poor arenes 108 including heteroarenes and biomolecules (Scheme 55).63 The electron deficient 2-pyridone ligand was used which acted as the X-type of ligand for palladium and also served as an internal base assisting the cleavage of C–H bonds via the CMD mechanism. The regioselectivity was dictated by both electronic and steric properties of the arene. Predominantly, meta and para derivatives are observed in most of the monosubstituted arenes.
 |
| | Scheme 55 2-Pyridone ligand-enabled Pd-catalysed olefination of arenes. | |
In 2018, van Gemmeren and co-workers reported Pd-catalysed arene limiting C–H olefination using two complementary ligands.64 They used N-Ac-Gly-OH in combination with a 6-methylpyridine derivative both of which combine in a 1:1 fashion with each equivalent of the Pd-catalyst and performed the olefination of a wide range of arenes with different olefins. In the dual ligand assisted olefination N-acetyl amino acid served as the internal base and also assisted in the CMD step. In 2020, Zhu and co-workers reported dual ligand enabled non-directed Pd-catalysed olefination of aryl ethers with activated alkenes as well as styrenes in good yields and moderate selectivity.65 They used a combination of N-acetyl leucine and an S,O-ligand 3-methyl-2-ipropyl-2-(phenylthio)acetic acid for better efficiency.
5. Directed C(sp3)–H olefination
The C(sp3)–H bond shows the lowest reactivity and highest thermodynamic stability, which makes its functionalization more difficult yet attractive. In the past few decades, we witnessed an incredible development in C(sp3)–H bond functionalization using a directing group strategy as a powerful tool to tackle stereo-, regio- and chemoselectivity. In this section, we shall be discussing directing group assisted transition-metal catalysed distal C(sp3)–H bond olefination reactions.
5.1. β-C(sp3)–H olefination
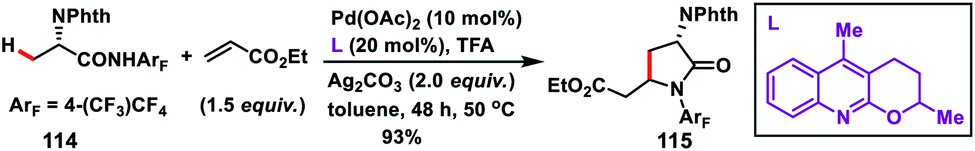
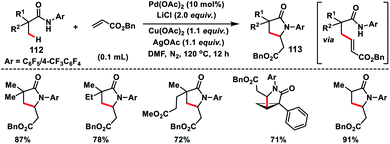
Yu and co-workers in 2010 successfully achieved β-C(sp3)–H olefination of amides 112 using a Pd-catalyst and subsequent aza-Michael addition to give the corresponding γ-lactams 113 (Scheme 56).66 Along with Pd(OAc)2, use of LiCl as an additive and combination of Cu(OAc)2 and AgOAc in 1:1 ratio as the oxidant in DMF afforded high yields. The reaction was also applied to effect olefination of cyclopropyl methylene C–H bond as well as substrates containing an α-hydrogen atom. Only cyclopropyl C–H bond was selectively olefinated in the presence of an α-methyl group. A related mechanism has been suggested where the initial amide directed C(sp3)–H insertion by Pd(II) leads to a five membered alkyl-palladium intermediate which underwent carbopalladation with olefin. Similarly, Yu and co-workers in 2014 carried out a β-C(sp3)–H olefination of protected amino acid alanine 114 with the help of a Pd-catalyst and quinoline derived ligand. The olefinated intermediate underwent intramolecular cyclisation to afford 115 (Scheme 57).67 The reaction delivered an excellent level of diastereoselectivity with respect to the α-centre. The reaction proceeded with an excellent level of diastereoselectivity with respect to the starting configuration at the α-centre. With the utilization of literature reports the lactam formed could be transformed into a β-olefinated α-amino acid with excellent enantiomeric excess (95%).
 |
| | Scheme 56 Amide-directed Pd-catalysed olefination of β-C(sp3)–H bonds. | |
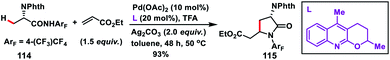 |
| | Scheme 57 Pd-catalysed C(sp3)–H olefination of the amino acid alanine. | |
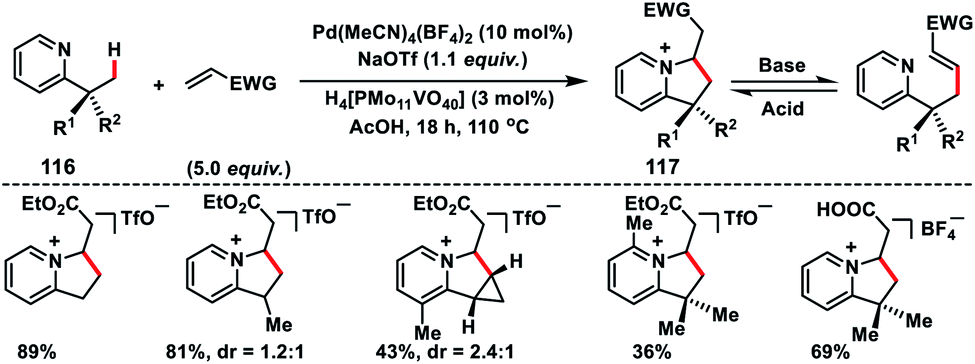
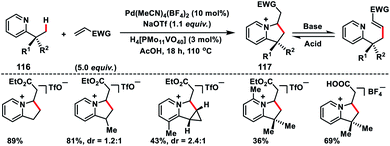
In 2011, Sanford and co-workers described a Pd-catalysed pyridine directed olefination of unactivated C(sp3)–H bonds (116) which shaped a convenient route to a 6,5-N-fused bicyclic core 117 (Scheme 58).68 After screening of several reaction conditions they found that the use of 10 mol% NaOAc, 10 mol% Pd(MeCN)4(BF4)2 and 3 mol% H4[PMo11VO40] in AcOH in air was optimal. The substrate with two types of C–H bonds, i.e. 1° and 2°, underwent C–C coupling with >20:1 selectivity for 1° over 2° C(sp3)–H bonds. Nevertheless, a 2° C(sp3)–H bond of the cyclopropane ring could also be functionalized to afford tricyclic products. A pyrazole directed C(sp3)–H olefination of the inherent alkyl group was reported by Yu and co-workers in 2016.69 For the first time, they reported the use of an MPAA ligand in a palladium-catalysed C(sp3)–H olefination reaction. The olefination reaction was compatible only with the tertiary alkyl group, however, diverse functional groups attached to the tertiary alkyl group as well as pyrazole ring were well tolerated under the given reaction conditions.
 |
| | Scheme 58 Pd-catalysed olefination of β-C(sp3)–H bonds. | |
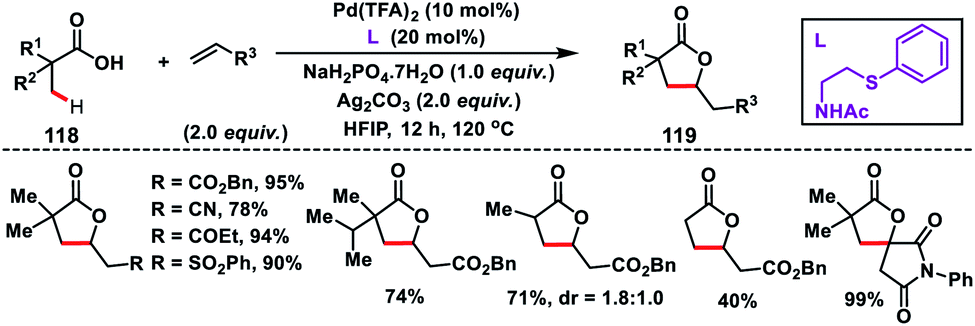
Yu group in 2018 established an inherent acid functionality as the directing group for β-C(sp3)–H olefination without an auxiliary. They synthesized an MPAA ligand, N-Ac-protected aminoethyl phenyl thioether to carry out Pd(II)-catalysed β-C(sp3)–H olefination of free carboxylic acids 118 (Scheme 59).70 Subsequent lactonization of the olefinated product with acid yielded synthetically important γ-lactones 119. It also limited the process towards being exclusively mono-selective in the presence of multiple β-C(sp3)–H bonds. Substitution of other σ-donors in place of the sulfur atom led to a drop in the yield significantly, highlighting the importance of PhS. All the substrates provided the desired γ-lactones in good to excellent yield when coupled with activated alkenes. N-Arylated or N-alkylated maleimides were also found to be suitable coupling partners and afforded spirocyclic pyrrolidines.
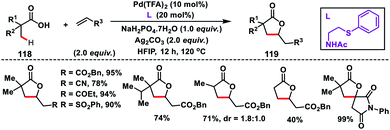 |
| | Scheme 59 Ligand-enabled β-C(sp3)–H olefination of free carboxylic acids. | |
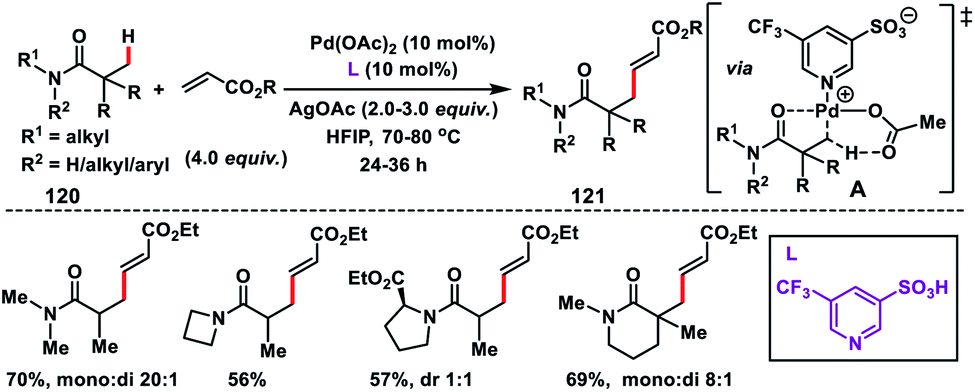
To improve the generality of the olefination reaction with a variety of substrates Yu and co-workers developed a Pd(II)-catalysed β-C(sp3)–H olefination of weakly coordinating intrinsic amides 120 (Scheme 60).71 They utilized the weakly coordinating oxygen of amides as the directing group using pyridine-3-sulfonic acid based ligands. The observed olefination of the C(sp3)–H bond was possible only in the presence of pyridine sulfonic acid ligands which indicated that the non-coordinating sulfonate group generates a highly electrophilic catalyst. For the viability of such transformations, there would be a charge separation between Pd and the ligand, which was supported by the complete inefficiency of pyridine-2-sulfonic acid. Apart from the diverse activated olefins, for the first time ethenesulfonyl fluoride, which is used in sulfur(VI) fluoride exchange (SuFEx) click reactions, was also utilized for the olefination reaction of the C(sp3)–H bond.
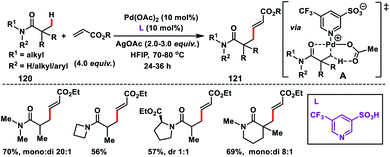 |
| | Scheme 60 Carbonyl coordination of native amides in β-C(sp3)–H olefination. | |
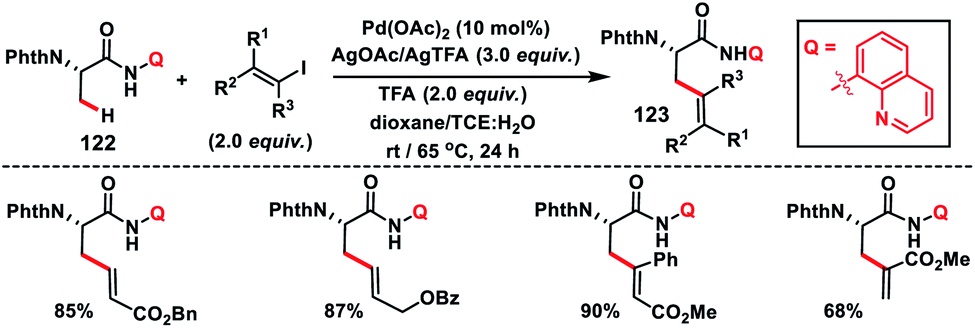
Complementary to C(sp3)–H olefination with terminal olefins under oxidative conditions, C(sp3)–H olefination could also be accomplished with vinyl halides without the requirements of any external oxidant. Such a Heck type reaction with C(sp3)–H bonds proceeds through oxidative addition followed by reductive elimination involving PdII/PdIV akin to the PdII/Pd0 intermediate for terminal olefins. In 2014, Chen and co-workers developed a stereo-retentive installation of multi-substituted olefins via N-quinolyl carboxamide-directed olefination of the β-C(sp3)–H of alanine amino acid 122 with alkenyl iodide (Scheme 61).72 This strategy provided a path for the synthesis of a wide range of exciting β-olefinated α-amino acids 123.
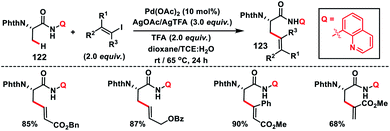 |
| | Scheme 61 Stereo-retentive olefination of the β-C(sp3)–H bond of alanine with vinyl iodides. | |
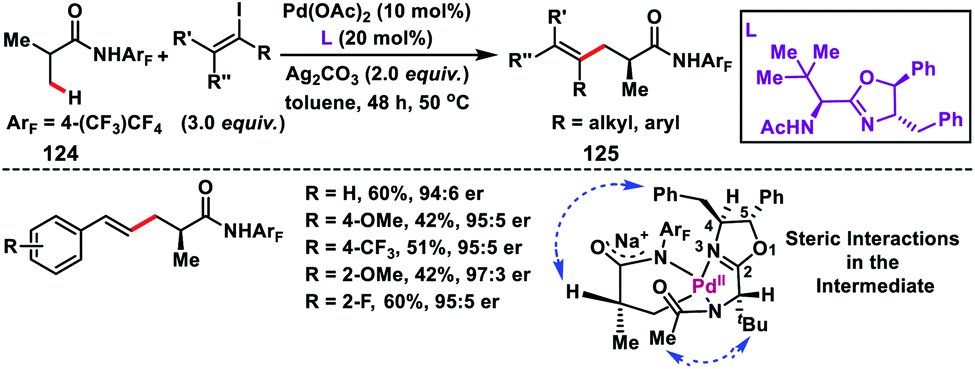
Yu and co-workers in 2017 demonstrated a Pd-catalysed enantioselective β-C(sp3)–H olefination reaction of iso-butyric acid derived substrates 124 (Scheme 62).73 The transformation provided a genuine route for the synthesis of some useful building blocks 125 with an enantioenriched α-chiral centre. Formation of a Pd–C–H intermediate was proposed, where the N-acetyl group of the coordinating nitrogen is directed towards the top face of the Pd square plane due to steric repulsion from the chiral centre on the side chain. There was a mutual relationship between the two stereocentres of the ligand as complete loss of enantioselectivity was observed when the absolute stereochemistry of one of the chiral centres was reversed. Furthermore, the 4-benzyl group of the oxazoline ring covers the top face of the intermediate. These combined steric interactions reinforced the orientation of the α-methyl group, thereby resulting in stereoselective cleavage of the C(sp3)–H bond.
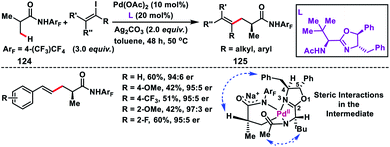 |
| | Scheme 62 Pd-catalysed enantioselective C–H alkenylation of isobutyric acid. | |
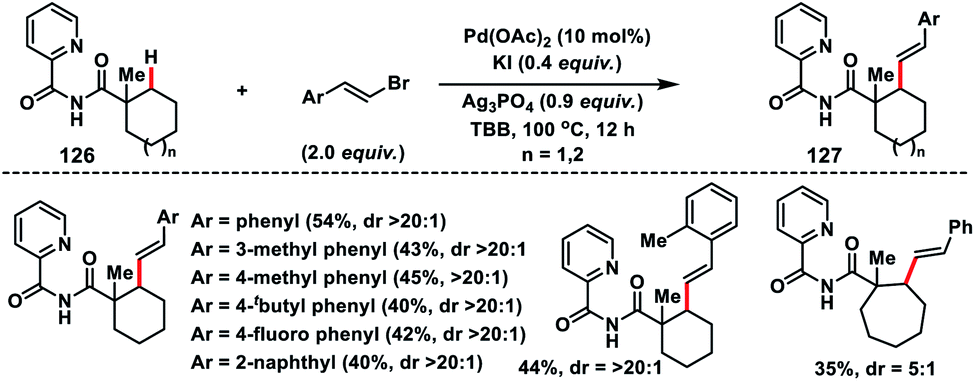
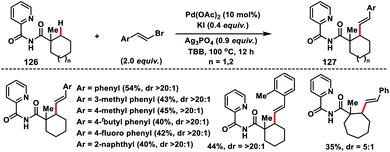
Later, Shi and co-workers developed 2-picolinamide as a new directing group for the Pd-catalysed β-C(sp3)–H alkenylation of cyclic carboxylic acids 126 with alkenyl bromides (Scheme 63).74 Interestingly, olefination reaction took place only at the secondary C(sp3)–H bond instead of primary, which was in contrast to the arylation reaction on the same substrate. A wide range of alkenyl bromides were tolerated under the reaction conditions and provided the desired 127 in good yield and excellent distereoselectivity (>20:1).
 |
| | Scheme 63 Pd-catalysed β-C(sp3)–H alkenylation with alkenyl bromides. | |
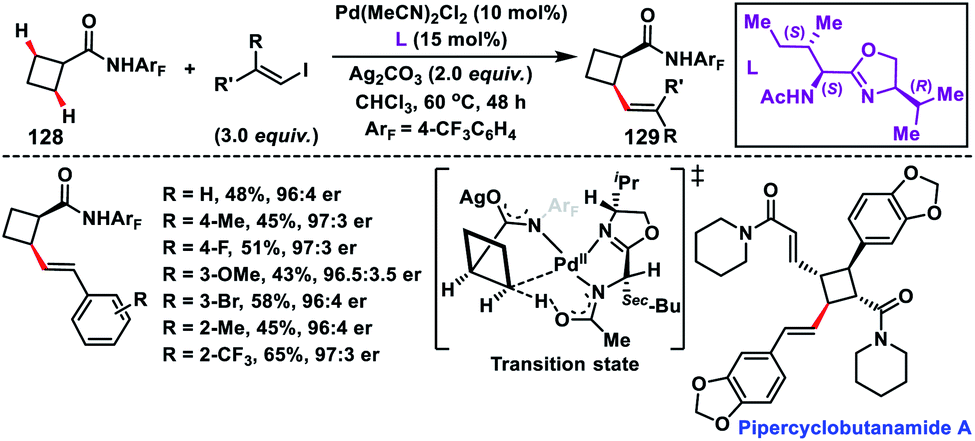
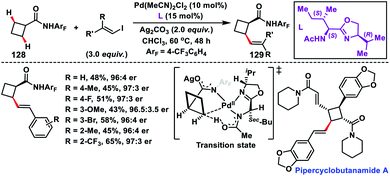
Enantioselective olefination of the C(sp3)–H bond of cyclobutyl carboxylic amides 128 was reported by Yu's group in 2018. The chiral mono-N-protected aminomethyl oxazoline (MPAO) ligand was used to obtain the desired products 129 in a moderate yield with good enantioselectivity (Scheme 64).75a The methodology was applied to synthesize diverse chiral cyclobutanes from simple monosubstituted cyclobutanes via sequential arylation and olefination. The precoordinated palladium acted as the active Pd(II) species which coordinated with amide and thus an enantio-determining C(sp3)–H metalation took place creating a chiral Pd(II) intermediate. The foremost utilization of C(sp3)–H olefination in the synthesis of the natural product pipercyclobutanamide A was reported by Baran and co-workers in 2015 (Scheme 64).75b They carried out an aminoquinoline directed Pd-catalysed alkenylation of an unactivated cyclobutane ring with alkenyl iodide. The transformation showed a glimmer of hope for the application of C(sp3)–H olefination in natural products and synthesis of their derivatives.
 |
| | Scheme 64 Enantioselective C(sp3)–H olefination of the cyclobutyl ring. | |
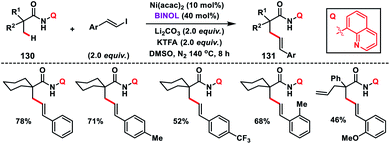
The first Ni-catalysed olefination of the β-C(sp3)–H bond with alkenyl iodide was reported by Shi and co-workers in 2015 by using aminoquinoline as the bidentate directing group (Scheme 65).76 The catalytic system employed in the transformation was an air stable Ni(acac)2-catalyst and highly efficient BINOL ligand. A wide range of alkenyl iodides and carboxamides 130 bearing an α-quaternary centre were compatible and provided products 131 in good yield and selectivity. In the case of carboxamides having a cyclic system, olefination took place only at the primary C(sp3)–H bond which was in contrast to the Pd-catalysed olefination reaction.
 |
| | Scheme 65 Ni-catalysed β-C(sp3)–H alkenylation with alkenyl iodide. | |
5.2. γ-C(sp3)–H olefination
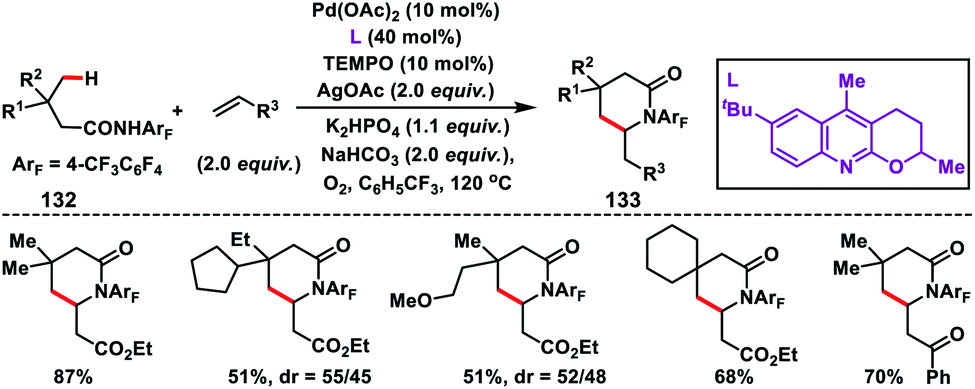
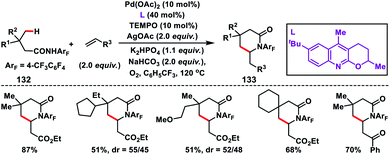
After the successful β-C(sp3)–H olefination reaction, γ-C(sp3)–H bond olefination was reported by Yu and co-workers in 2014 in order to generate a β-quaternary centre (Scheme 66).77 They achieved γ-C(sp3)–H olefination by employing a quinoline based tricyclic ligand and a weakly coordinating amide directing group 132 with a Pd-catalyst. The motive behind the use of quinoline based ligands containing a 2-alkoxy motif was that the lone pairs of oxygen constrained themselves to a plane parallel to the π-system of the quinoline ring. This resulted in higher electron density on the ligand which influenced the reactivity of the Pd(II)-catalyst. After extensive optimization, use of 10 mol% TEMPO as a co-oxidant in oxygen enhanced the yield of the product 133 significantly.
 |
| | Scheme 66 Ligand-enabled construction of β-quaternary carbon centres. | |
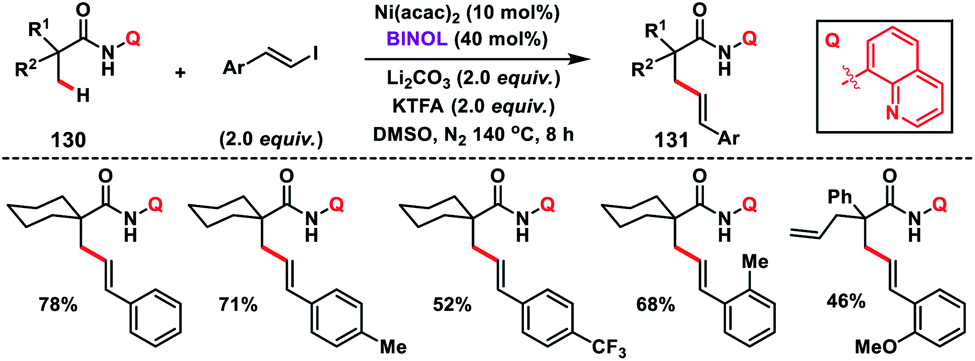
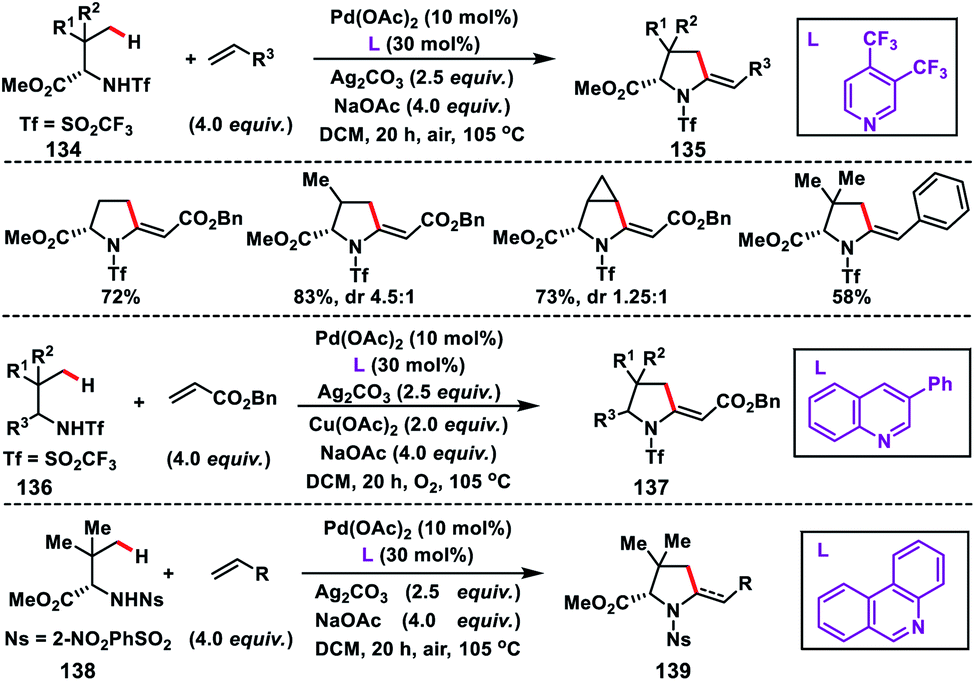
In 2016, Yu and co-workers developed another excellent approach for Pd(II)-catalysed γ-C(sp3)–H olefination of Tf- and Ns-protected amines. The olefinated intermediate underwent subsequent aza-Wacker oxidative cyclisation or conjugate addition to harvest a variety of C2-alkylated pyrrolidines.78 They figured out three different classes of pyridine and quinoline based ligands to match the requirements for C(sp3)–H olefination of different classes of amines depicting a sporadic example of ligand enabled C(sp3)–H olefination. Interestingly, amino acids 134 containing both γ-methyl and γ-methylene C(sp3)–H bond, preferentially γ-methyl C(sp3)–H bond, were olefinated to 135 (Scheme 67). For α-amino acids the electron-deficient ligand 3,4-bis(trifluoromethyl)pyridine was effective to achieve the transformation. 3-Phenylquinoline was found suitable for the Tf-protected alkyl amines 136 including β-amino alcohols and afforded the desired products 137 in good yield (Scheme 67). The applicability of the strategy was further enhanced by the use of a common protecting group 2-nitrobenzenesulfonyl (Ns) in 138 instead of Tf with a different ligand phenanthridine (Scheme 67).
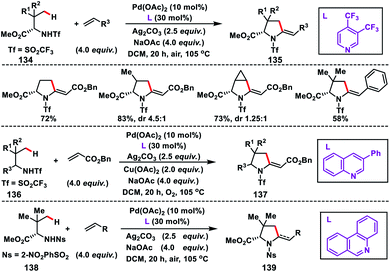 |
| | Scheme 67 γ-C(sp3)–h olefination of Tf and Ns-protected α-amino acids and alkyl amine. | |
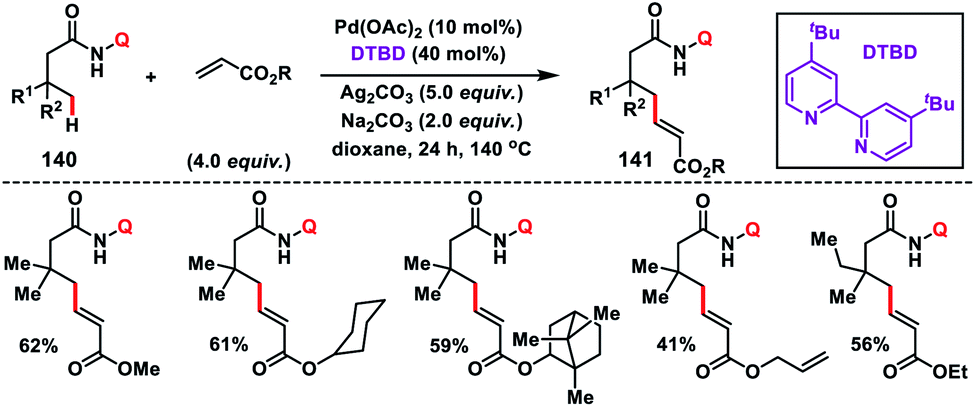
In 2017, our group developed a protocol for the Pd-catalysed γ-C(sp3)–H olefination of aliphatic acids 140 with activated olefins using 8-aminoquinoline as the directing group (Scheme 68).79 The γ-C(sp3)–H olefination with acrylates occurred in the presence of a quinoline based ligand 4,4′-di-tert-butyl-2,2′-bipyridine (DTBD) along with Ag salt oxidant and Na2CO3 base. A variety of acrylates and acid derivatives provided the desired products 141 in good yield. Vinyl iodides were also equally compatible under the reaction conditions for the γ-C(sp3)–H olefination of protected amino acids, however no ligands and external oxidants were required. This transformation occurred through the aminoquinoline-directed formation of a six-membered palladacycle intermediate. The olefin coordination followed by the migratory insertion of activated olefin and β-hydride elimination resulted in the final product and Pd(0), which was re-oxidized to Pd(II) with Ag(I) salt (Scheme 69).
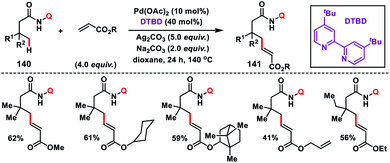 |
| | Scheme 68 Pd-catalysed γ-C(sp3)–H olefination with activated alkenes. | |
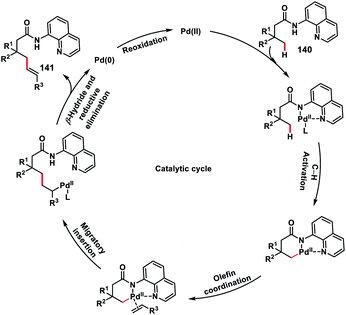 |
| | Scheme 69 Proposed mechanism for Pd-catalysed γ-C(sp3)–H olefination. | |
In 2015, Gaunt and co-workers reported a Pd-catalysed γ-C(sp3)–H olefination of amino alcohols 142 followed by intramolecular aza-Michael addition to afford pyrrolidine moieties 143 in good yields (Scheme 70).80 This transformation was achieved by the temporary conversion of the catalytically incompetent primary amino alcohol to a hindered secondary amine. The sterically hindered secondary amine was capable of promoting Pd-catalysed C–H activation reaction. The γ-C(sp3)–H olefination reaction became feasible due to H-bonding between the catalyst and amine. This led to the intensive interaction of substrates around Pd and orientation of the aliphatic amine substituent in a perfect geometry needed for C–H activation. Later, Gaunt's group achieved a ligand assisted Pd-catalysed γ-C(sp3)–H olefination of aliphatic amines 144 guided by the free (NH)-amine (Scheme 71).81 A cyclopalladated five-membered ring formed, which allowed olefin insertion and aza-Michael cyclisation leading to pyrrolidine moieties 145 in good yields and excellent regio-and diastereoselectivity. They observed that the amino acid derived ligand strongly influences the C–H activation step for aliphatic amines containing the competitive sites of reactivity. By making the cyclopalladation step reversible, the ligand enables a productive five-membered cyclopalladation pathway.
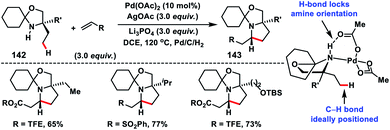 |
| | Scheme 70 Pd-catalysed γ-C(sp3)–H olefination of primary amino alcohols. | |
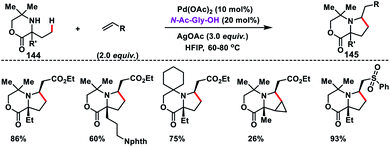 |
| | Scheme 71 Ligand induced γ-C(sp3)–H olefination of aliphatic amines. | |
Gemmeren's group reported ligand assisted Pd-catalysed γ-C(sp3)–H olefination of free carboxylic acids 146 without incorporating any exogenous directing group. The alkene intermediate followed an in situ intramolecular Michael addition to provide δ-lactones 147 in good yields (Scheme 72).82a Simultaneously, our group also demonstrated γ-C(sp3)–H olefination of free aliphatic acids 146 under similar reaction conditions except the use of N-Ac-L-val-OH ligand instead of N-Ac-β-alanine (Scheme 72).82b Both δ-lactones and ε-lactones 147 were formed depending upon the olefin coupling partners. Six membered δ-lactones were formed when coupled with acrylates, ketones, nitriles and sulfones while ε-lactones resulted when coupled with N-substituted maleimides. It was worth noting that acids containing only a quaternary centre at the β-position were compatible which signify the Thorpe–Ingold effect. Mechanistic investigations suggested that the γ-C(sp3)–H bond cleavage was the rate-determining step in the transformation.
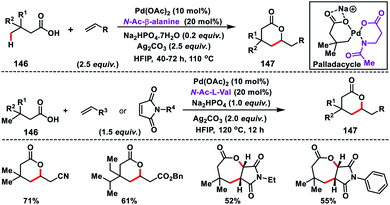 |
| | Scheme 72 Ligand-enabled γ-C(sp3)–H olefination of free carboxylic acids. | |
Recently, Pd-catalysed γ-C(sp3)–H olefination of weakly coordinating ketone derivatives 148 was reported by Yu and co-workers. They used a combination of mono-N-protected amino acid, electron-deficient 2-pyridone ligand and modified imino-amide L, X-type directing group (DG) for the purpose (Scheme 73).83 The protocol has a wide scope of applicability and substituents on both ketones and olefins were well tolerated providing products 149 in moderate to good yield.
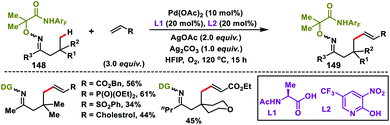 |
| | Scheme 73 L,X-type directing group-assisted C–H olefination of ketones. | |
6. Miscellaneous
In 2019, Gevorgyan reported the unusual Pd-catalysed site-selective β-, γ- and δ-C(sp3)–H olefination of alcohols 150via radical relay Heck reaction under visible light at room temperature (Scheme 74).84 The transformation took place at the sterically demanding site resulting in the formation of a quaternary carbon centre in the presence of Xantphos ligand. The strategy was compatible with a variety of alcohols and olefin substrates including styrenes and afforded the corresponding alkenylated products 151 in moderate to good yield. In substrates containing the competitive C(sp3)–H site (i.e. β- vs. γ- and γ- vs. δ-), γ-C(sp3)–H olefination took place preferentially due to the higher preference of 1,6-HAT (hydrogen atom transfer) for the Si auxiliary.
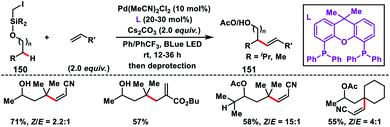 |
| | Scheme 74 Pd-catalysed radical relay Heck reaction. | |
7. Conclusion and outlook
The Fujiwara–Moritani reaction opened a new avenue in the synthesis of olefinated compounds with excellent efficiency, economy and environmental benignity by the direct coupling of C–H bonds. As discussed in the review, the last few decades of research demonstrated that irrespective of the nature of the C–H bond, transition-metal catalysts such as Pd, Rh, Ru, Ni, Co, and Ir made olefination reaction efficient and regioselective. Regioselectivity in the olefination reaction (sp2 & sp3) can be achieved by designing strongly as well as weakly coordinating directing groups along with the judiciary usage of a suitable ligand. Also, selective access of meta- and para-C–H bond was achieved by anchoring the covalently linked directing group which could form a cyclophane-like macrocyclic transition state. Very recently, researchers devised catalytic systems that enable nondirected C–H olefination reaction under arene limited conditions with broad substrate scope and improved selectivity. Site specific olefination in many medicinally important small heterocycles devoid of functional group to tether a template covalently can be achieved by designing bifunctional templates coordinated with two metal centres. Recent advancement in the asymmetric C–H activation resulted in an enantioselective C–H olefination reaction to access axially chiral biaryl compounds in a single step and atom economic way.
Despite these advances, oxidative C–H olefination reactions still face a number of challenges. For example, low regioselectivity is one of the major concerns. In some cases, this issue was addressed by the installation of the directing group. The poor selectivity in non-directed and distal C–H olefination limits its use in the bulk. Considering the practical importance of atom and step economical C–H bond olefination there are still many research areas left to be explored for the effective C–H olefination. Development of new traceless directing groups for the utilization of base metals such as Co, Ni, Cu and Fe under milder reaction conditions is one of the areas to be explored in the near future. New directions, such as use of frustrated Lewis ion pairs or acid–base pairs, are still unknown for remote sp2 & sp3 C–H bond olefination reaction. Often C–H olefination reaction requires high temperature which resulted in poor regio-selection leading to multiple site functionalization. Therefore, merger of metal/photoredox catalysis generally enabled the reaction to take place at room temperature ensuring higher regioselectivity. Another aspect of photoredox catalysis is that it avoids use of super-stoichiometric metal salts making the process more economical and environmentally benign. There is plenty of scope in photoredox C–H olefination reactions which remains to be explored especially for distal sp2 & sp3 C–H bonds. Recent development in the artificial metalloenzyme catalyzed asymmetric C–H functionalization opens a new avenue to be investigated for asymmetric C–H olefination reactions. Success in artificial metalloenzyme catalyzed asymmetric C–H olefination reactions will help to mimic the natural metalloenzyme and in vivo C–H olefination could be utilized for targeted drug delivery. Also, C–H olefination reaction of DNA encoding can lead to the formation of a DNA encoded library. Therefore, to find more applications in natural product synthesis, drug discovery and agrochemicals, further improvement in the C–H bond olefination is expected. To address these inadequacies, development of effective templates, catalysts, ligands and new approaches are required with the detailed understanding of the olefination mechanism.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank SERB India (CRG/2018/003915) for financial support. Fellowship from Indian Institute of Technology Bombay (WA), CSIR New Delhi, India (GP) is greatly acknowledged.
Notes and references
-
(a) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC
 ;
(b) J. C. Fox, R. E. Gilligan, A. K. Pitts, H. R. Bennett and M. J. Gaunt, Chem. Sci., 2016, 7, 2706–2710 RSC ;
(c) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS PubMed ;
(d) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC ;
(e) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed .
;
(b) J. C. Fox, R. E. Gilligan, A. K. Pitts, H. R. Bennett and M. J. Gaunt, Chem. Sci., 2016, 7, 2706–2710 RSC ;
(c) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS PubMed ;
(d) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC ;
(e) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed .
-
(a) W. Ma, P. Gandeepan, J. Lid and L. Ackermann, Org. Chem. Front., 2017, 4, 1435–1467 RSC ;
(b) S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886–896 RSC ;
(c) P. Wedi and M. van Gemmeren, Angew. Chem., Int. Ed., 2018, 57, 13016–13027 CrossRef CAS PubMed ;
(d) A. Dey, S. K. Sinha, T. K. Achar and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 10820–10843 CrossRef CAS PubMed ;
(e) A. Deb and D. Maiti, Eur. J. Org. Chem., 2017, 1239–1252 CrossRef CAS ;
(f) S. Bag and D. Maiti, Synthesis, 2016, 48, 804–815 CrossRef CAS ;
(g) R. Manikandana and M. Jeganmohan, Chem. Commun., 2017, 53, 8931–8947 RSC ;
(h) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS PubMed .
-
(a) P.-H. Nguyen, J.-L. Yang, M. N. Uddin, S.-L. Park, S.-I. Lim, D.-W. Jung, D. R. Williams and W.-K. Oh, J. Nat. Prod., 2013, 76, 2080–2087 CrossRef CAS PubMed ;
(b) J. Cheel, C. Theoduloz, J. Rodríguez, G. Saud, P. D. S. Caligari and G. Schmeda-Hirschmann, J. Agric. Food Chem., 2005, 53, 8512–8518 CrossRef CAS PubMed .
-
(a) R. S. P. Singh, D. Michel, U. Das, J. R. Dimmock and J. Alcorn, Bioorg. Med. Chem. Lett., 2014, 24, 5199–5202 CrossRef CAS PubMed ;
(b) T. Chuprajob, C. Changtam, R. Chokchaisiri, W. Chunglok, N. Sornkaew and A. Suksamrarn, Bioorg. Med. Chem. Lett., 2014, 24, 2839–2844 CrossRef CAS PubMed .
-
(a) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed ;
(b) A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed ;
(c) M. M. Silva Paula, C. V. Franco, M. C. Baldin, L. Rodrigues, T. Barichello, G. D. Savi, L. F. Bellato, M. A. Fiori and L. Silva, Mater. Sci. Eng., C, 2009, 29, 647–650 CrossRef .
-
(a) J. Ruan and J. Xiao, Acc. Chem. Res., 2011, 44, 614–626 CrossRef CAS PubMed ;
(b) D. Mc Cartney and P. J. Guiry, Chem. Soc. Rev., 2011, 40, 5122–5150 RSC ;
(c) R. F. Heck, Acc. Chem. Res., 1979, 12, 146–151 CrossRef CAS .
-
(a) I. Moritani and Y. Fujiwara, Tetrahedron Lett., 1967, 8, 1119–1122 CrossRef ;
(b) Y. Fujiwara, I. Moritani, M. Matsuda and S. Teranishi, Tetrahedron Lett., 1968, 9, 3863–3865 CrossRef ;
(c) Y. Fujiwara, I. Noritani, S. Danno, R. Asano and S. Teranishi, J. Am. Chem. Soc., 1969, 91, 7166–7169 CrossRef CAS PubMed .
-
(a) H. J. Davis and R. J. Phipps, Chem. Sci., 2017, 8, 864–877 RSC ;
(b) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed ;
(c) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CrossRef CAS ;
(d) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed ;
(e) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed ;
(f)
J. Li, S. de Sarkar and L. Ackermann, in Topics in Organometallic Chemistry, ed. P. H. Dixneuf and H. Doucet, Springer International Publishing, Cham, 2016 Search PubMed ;
(g) A. Dey, S. Agasti and D. Maiti, Org. Biomol. Chem., 2016, 14, 5440–5453 RSC ;
(h) B. Niu, K. Yang, B. Lawrence and H. Ge, ChemSusChem, 2019, 12, 2955–2969 CrossRef CAS PubMed .
-
(a) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed ;
(b) R.-Y. Zhu, M. E. Farmer, Y.-Q. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578–10599 CrossRef CAS PubMed ;
(c) G. He, B. Wang, W. A. Nack and G. Chen, Acc. Chem. Res., 2016, 49, 635–645 CrossRef CAS PubMed ;
(d) Y. Xu and G. Dong, Chem. Sci., 2018, 9, 1424–1432 RSC ;
(e) R. R. Karimov and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 4234–4241 CrossRef CAS PubMed ;
(f) H. Sterckx, B. Morel and B. U. W. Maes, Angew. Chem., Int. Ed., 2019, 58, 7946–7970 CrossRef CAS PubMed .
- A. Mancinelli, C. Alamillo, J. Albert, X. Ariza, H. Etxabe, J. Farràs, J. Garcia, J. Granell and F. J. Quijada, Organometallics, 2017, 36, 911–919 CrossRef CAS .
- Y.-C. Zhu, Y. Li, B.-C. Zhang, F.-X. Zhang, Y.-N. Yang and X.-S. Wang, Angew. Chem., Int. Ed., 2018, 57, 5129–5133 CrossRef CAS PubMed .
-
(a) A. Deb, S. Bag, R. Kancherla and D. Maiti, J. Am. Chem. Soc., 2014, 136, 13602–13605 CrossRef CAS PubMed ;
(b) M.-Z. Lu, X.-R. Chen, H. Xu, H.-X. Dai and J.-Q. Yu, Chem. Sci., 2018, 9, 1311–1316 RSC ;
(c) A. Deb, A. Hazra, Q. Peng, R. S. Paton and D. Maiti, J. Am. Chem. Soc., 2017, 139, 763–775 CrossRef CAS PubMed ;
(d) K. Seth, M. Bera, M. Brochetta, S. Agasti, A. Das, A. Gandini, A. Porta, G. Zanoni and D. Maiti, ACS Catal., 2017, 7, 7732–7736 CrossRef CAS .
- Q.-J. Liang, B. Jiang, Y.-H. Xu and T.-P. Loh, J. Org. Chem., 2018, 83, 8265–8271 CrossRef CAS PubMed .
-
(a) Y. Jaiswal, Y. Kumar and A. Kumar, J. Org. Chem., 2018, 83, 1223–1231 CrossRef CAS PubMed ;
(b) Q. Bu, T. Rogge, V. Kotek and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 765–768 CrossRef CAS PubMed ;
(c) S. Jambu, R. Sivasakthikumaran and M. Jeganmohan, Org. Lett., 2019, 21, 1320–1324 CrossRef CAS PubMed .
- X. Yang, R. Sun, C. Zhang, X. Zheng, M. Yuan, H. Fu, R. Li and H. Chen, Org. Lett., 2019, 21, 1002–1006 CrossRef CAS PubMed .
- Q.-Y. Sun, Z. Li, Z. Xu, Z.-J. Zheng, J. Cao, K.-F. Yang, Y.-M. Cui and L.-W. Xu, Chem. Commun., 2019, 55, 6229–6232 RSC .
- Y. Huang, C. Pi, X. Cui and Y. Wu, Adv. Synth. Catal., 2020, 362, 1385–1390 CrossRef CAS .
- S. Dana, A. Mandal, H. Sahoo, S. Mallik, G. S. Grandhi and M. Baidya, Org. Lett., 2018, 20, 716–719 CrossRef CAS PubMed .
- T. Morita, T. Satoh and M. Miura, Org. Lett., 2017, 19, 1800–1803 CrossRef CAS PubMed .
-
(a) R. Yoshimura, Y. Shibata, T. Yamada and K. Tanaka, J. Org. Chem., 2019, 84, 2501–2511 CrossRef CAS PubMed ;
(b) R. Yoshimura and K. Tanaka, Chem.–Eur. J., 2020, 26, 4969–4973 CrossRef CAS PubMed .
- M. C. Reddy and M. Jeganmohan, Chem. Sci., 2017, 8, 4130–4135 RSC .
- X. Liu, Z. Wang, Q. Chen, M.-y. He and L. Wang, Appl. Organomet. Chem., 2017, e4039 Search PubMed .
- M. Bakthadossa and P. V. Kumara, Adv. Synth. Catal., 2018, 360, 2650–2658 CrossRef .
- K. Elumalai and W. K. Leong, Tetrahedron Lett., 2018, 59, 113–116 CrossRef CAS .
- M. Raja Sk, S. S. Bera and M. S. Maji, Adv. Synth. Catal., 2019, 361, 585–590 CrossRef .
- X. Lu, C. Shen, K. Meng, L. Zhao, T. Li, Y. Sun, J. Zhang and G. Zhong, Chem. Commun., 2019, 55, 826–829 RSC .
-
(a) C. Yu, F. Li, J. Zhang and G. Zhong, Chem. Commun., 2017, 53, 533–536 RSC ;
(b) K. Meng, Y. Sun, J. Zhang, K. Zhang, X. Ji, L. Ding and G. Zhong, Org. Lett., 2019, 21, 8219–8224 CrossRef CAS PubMed ;
(c) T. Li, C. Shen, Y. Sun, J. Zhang, P. Xiang, X. Lu and G. Zhong, Org. Lett., 2019, 21, 7772–7777 CrossRef CAS PubMed .
-
(a) C. N. Kona, Y. Nishii and M. Miura, Org. Lett., 2018, 20, 4898–4901 CrossRef CAS PubMed ;
(b) S. Pradhan, M. Mishra, P. B. De, S. Banerjee and T. Punniyamurthy, Org. Lett., 2020, 22, 1720–1725 CrossRef CAS PubMed .
- Q. Han, X. Guo, Z. Tang, L. Su, Z. Yao, X. Zhang, S. Lin, S. Xiang and Q. Huang, Adv. Synth. Catal., 2018, 360, 972–984 CrossRef CAS .
- P. Kumar and M. Kapur, Org. Lett., 2019, 21, 2134–2138 CrossRef CAS PubMed .
- T. Yao and K. Du, ACS Sustainable Chem. Eng., 2019, 7, 6068–6077 CrossRef CAS .
-
(a) J. E. Smyth, N. M. Butler and P. A. Keller, Nat. Prod. Rep., 2015, 32, 1562–1583 RSC ;
(b) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed ;
(c) M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193–3207 RSC .
- T. Wesch, F. R. Leroux and F. Colobert, Adv. Synth. Catal., 2013, 355, 2139–2144 CrossRef CAS .
- Y.-N. Ma, H.-Y. Zhang and S.-D. Yang, Org. Lett., 2015, 17, 2034–2037 CrossRef CAS PubMed .
-
(a) J. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2014, 53, 13244–13247 CrossRef CAS PubMed ;
(b) J. Zheng, W.-J. Cui, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2016, 138, 5242–5245 CrossRef CAS PubMed .
-
(a) Q.-J. Yao, S. Zhang, B.-B. Zhan and B.-F. Shi, Angew. Chem., Int. Ed., 2017, 56, 6617–6621 CrossRef CAS PubMed ;
(b) J. Fan, Q.-J. Yao, Y.-H. Liu, G. Liao, S. Zhang and B.-F. Shi, Org. Lett., 2019, 21, 3352–3356 CrossRef CAS PubMed ;
(c) H.-M. Chen, S. Zhang, G. Liao, Q.-J. Yao, X.-T. Xu, K. Zhang and B.-F. Shi, Organometallics, 2019, 38, 4022–4028 CrossRef CAS .
- H. Song, Y. Li, Q.-J. Yao, L. Jin, L. Liu, Y.-H. Liu and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 6576–6580 CrossRef CAS PubMed .
- S.-X. Li, Y.-N. Ma and S.-D. Yang, Org. Lett., 2017, 19, 1842–1845 CrossRef CAS PubMed .
-
(a) J. Luo, T. Zhang, L. Wang, G. Liao, Q.-J. Yao, Y.-J. Wu, B.-B. Zhan, Y. Lan, X.-F. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2019, 58, 6708–6712 CrossRef CAS PubMed ;
(b) B.-B. Zhan, L. Wang, J. Luo, X.-F. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 3568–3572 CrossRef CAS PubMed .
-
(a) D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 CrossRef CAS PubMed ;
(b) Y.-F. Yang, G.-J. Cheng, P. Liu, D. Leow, T.-Y. Sun, P. Chen, X. Zhang, J.-Q. Yu, Y.-D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 344–355 CrossRef CAS PubMed ;
(c) H.-J. Xu, Y. Lu, M. E. Farmer, H.-W. Wang, D. Zhao, Y.-S. Kang, W.-Y. Sun and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 2200–2203 CrossRef CAS PubMed .
-
(a) H.-X. Dai, G. Li, X.-G. Zhang, A. F. Stepan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 7567–7571 CrossRef CAS ;
(b) G.-J. Cheng, Y.-F. Yang, P. Liu, P. Chen, T.-Y. Sun, G. Li, X. Zhang, K. N. Houk, J.-Q. Yu and Y.-D. Wu, J. Am. Chem. Soc., 2014, 136, 894–897 CrossRef CAS PubMed .
-
(a) S. Lee, H. Lee and K. L. Tan, J. Am. Chem. Soc., 2013, 135, 18778–18781 CrossRef CAS PubMed ;
(b) T. Patra, R. Watile, S. Agasti, T. Naveen and D. Maiti, Chem. Commun., 2016, 52, 2027–2030 RSC ;
(c) R.-J. Mi, J. Sun, F. E. Kühn, M.-D. Zhou and Z. Xu, Chem. Commun., 2017, 53, 13209–13212 RSC ;
(d) R.-J. Mi, Y.-Z. Sun, J.-Y. Wang, J. Sun, Z. Xu and M.-D. Zhou, Org. Lett., 2018, 20, 5126–5129 CrossRef CAS PubMed .
- M. Bera, S. K. Sahoo and D. Maiti, ACS Catal., 2016, 6, 3575–3579 CrossRef CAS .
-
(a) M. Bera, A. Modak, T. Patra, A. Maji and D. Maiti, Org. Lett., 2014, 16, 5760–5763 CrossRef CAS PubMed ;
(b) Y. Deng and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 888–891 CrossRef CAS PubMed ;
(c) Z. Jin, L. Chu, Y.-Q. Chen and J.-Q. Yu, Org. Lett., 2018, 20, 425–428 CrossRef CAS PubMed .
-
(a) M. Bera, A. Maji, S. K. Sahoo and D. Maiti, Angew. Chem., Int. Ed., 2015, 54, 8515–8519 CrossRef CAS PubMed ;
(b) A. Modak, A. Mondal, R. Watile, S. Mukherjee and D. Maiti, Chem. Commun., 2016, 52, 13916–13919 RSC ;
(c) U. Dutta, A. Modak, B. Bhaskararao, M. Bera, S. Bag, A. Mondal, D. W. Lupton, R. B. Sunoj and D. Maiti, ACS Catal., 2017, 7, 3162–3168 CrossRef CAS ;
(d) M. Bera, S. Agasti, R. Chowdhury, R. Mondal, D. Pal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 5272–5276 CrossRef CAS PubMed .
-
(a) S. Maity, E. Hoque, U. Dhawa and D. Maiti, Chem. Commun., 2016, 52, 14003–14006 RSC ;
(b) Z. Fan, K. L. Bay, X. Chen, Z. Zhuang, H. S. Park, K.-S. Yeung, K. N. Houk and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 4770–4777 CrossRef CAS PubMed .
-
(a) R.-Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215–220 CrossRef CAS PubMed ;
(b) S. Li, H. Ji, L. Cai and G. Li, Chem. Sci., 2015, 6, 5595–5600 RSC ;
(c) L. Yang, L. Fu and G. Li, Adv. Synth. Catal., 2017, 359, 2235–2240 CrossRef CAS ;
(d) G. Yang, P. Lindovska, D. Zhu, J. Kim, P. Wang, R.-Y. Tang, M. Movassaghi and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 10807–10813 CrossRef CAS PubMed ;
(e) G. Yang, D. Zhu, P. Wang, R.-Y. Tang and J.-Q. Yu, Chem.–Eur. J., 2018, 24, 3434–3438 CrossRef CAS PubMed .
-
(a) L. Zhang, C. Zhao, Y. Liu, J. Xu, X. Xu and Z. Jin, Angew. Chem., Int. Ed., 2017, 56, 12245–12249 CrossRef CAS PubMed ;
(b) R. Jayarajan, J. Das, S. Bag, R. Chowdhury and D. Maiti, Angew. Chem., Int. Ed., 2018, 57, 7659–7663 CrossRef CAS PubMed .
-
(a) S. Li, L. Cai, H. Ji, L. Yang and G. Li, Nat. Commun., 2016, 7, 10443 CrossRef PubMed ;
(b) L. Fang, T. G. Saint-Denis, B. L. H. Taylor, S. Ahlquist, K. Hong, S. S. Liu, L. L. Han, K. N. Houk and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 10702–10714 CrossRef CAS PubMed .
- J. Xu, J. Chen, F. Gao, S. Xie, X. Xu, Z. Jin and J.-Q. Yu, J. Am. Chem. Soc., 2019, 141, 1903–1907 CrossRef CAS PubMed .
-
(a) S. Fang, X. Wang, F. Yin, P. Cai, H. Yang and L. Kong, Org. Lett., 2019, 21, 1841–1844 CrossRef CAS PubMed ;
(b) S. Xie, S. Li, W. Ma, X. Xu and Z. Jin, Chem. Commun., 2019, 55, 12408–12411 RSC .
- S. Li, H. Wang, Y. Weng and G. Li, Angew. Chem., Int. Ed., 2019, 58, 18502–18507 CrossRef CAS PubMed .
- A. F. Williams, A. J. P. White, A. C. Spivey and C. J. Cordier, Chem. Sci., 2020, 11, 3301–3306 RSC .
- S. Bag, S. Jana, S. Pradhan, S. Bhowmick, N. Goswami, S. K. Sinha and D. Maiti, ChemRxiv. DOI:10.26434/chemrxiv.11521827.v1 .
-
(a) S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137(37), 11888–11891 CrossRef CAS PubMed ;
(b) T. Patra, S. Bag, R. Kancherla, A. Mondal, A. Dey, S. Pimparkar, S. Agasti, A. Modak and D. Maiti, Angew. Chem., Int. Ed., 2016, 55, 7751–7755 CrossRef CAS PubMed ;
(c) U. Dutta, S. Maiti, S. Pimparkar, S. Maiti, L. R. Gahan, E. H. Krenske, D. W. Lupton and D. Maiti, Chem. Sci., 2019, 10, 7426–7432 RSC .
- Z. Zhang, K. Tanaka and J.-Q. Yu, Nature, 2017, 543, 538–542 CrossRef CAS PubMed .
- T. K. Achar, K. Ramakrishna, T. Pal, S. Porey, P. Dolui, J. P. Biswas and D. Maiti, Chem.–Eur. J., 2018, 24, 17906–17910 CrossRef CAS PubMed .
- T. K. Achar, J. P. Biswas, S. Porey, T. Pal, K. Ramakrishna, S. Maiti and D. Maiti, J. Org. Chem., 2019, 84, 8315–8321 CrossRef CAS PubMed .
-
(a) Y.-H. Zhang, B.-F. Shi and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131(14), 5072–5074 CrossRef CAS PubMed ;
(b) S. Zhang, L. Shi and Y. Ding, J. Am. Chem. Soc., 2011, 133, 20218–20229 CrossRef CAS PubMed .
-
(a) F. W. Patureau, C. Nimphius and F. Glorius, Org. Lett., 2011, 13, 6346–6349 CrossRef CAS ;
(b) A. Kubota, M. H. Emmert and M. S. Sanford, Org. Lett., 2012, 14, 1760–1763 CrossRef CAS PubMed .
-
(a) H. U. Vora, A. P. Silvestri, C. J. Engelin and J.-Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 2683–2686 CrossRef CAS PubMed ;
(b) C.-H. Ying, S.-B. Yan and W.-L. Duan, Org. Lett., 2014, 16, 500–503 CrossRef CAS PubMed .
-
(a) K. Naksomboon, C. Valderas, M. Gómez-Martínez, Y. Álvarez-Casao and M. A. Fernández-Ibáñez, ACS Catal., 2017, 7, 6342–6346 CrossRef CAS PubMed ;
(b) K. Naksomboon, J. Poater, F. M. Bickelhaupt and M. A. Fernández-Ibáñez, J. Am. Chem. Soc., 2019, 141, 6719–6725 CrossRef CAS PubMed .
- P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K.-S. Yeung and J.-Q. Yu, Nature, 2017, 551, 489–493 CrossRef CAS PubMed .
- H. Chen, P. Wedi, T. Meyer, G. Tavakoli and M. van Gemmeren, Angew. Chem., Int. Ed., 2018, 57, 2497–2501 CrossRef CAS PubMed .
- B. Yin, M. Fu, L. Wang, J. Liu and Q. Zhu, Chem. Commun., 2020, 56, 3293–3296 RSC .
- M. Wasa, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132(11), 3680–3681 CrossRef CAS PubMed .
- J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216–1220 CrossRef CAS PubMed .
- K. J. Stowers, K. C. Fortner and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 6541–6544 CrossRef CAS .
- W. Yang, S. Ye, Y. Schmidt, D. Stamos and J.-Q. Yu, Chem.–Eur. J., 2016, 22, 7059–7062 CrossRef CAS PubMed .
- Z. Zhuang, C.-B. Yu, G. Chen, Q.-F. Wu, Y. Hsiao, C. L. Joe, J. X. Qiao, M. A. Poss and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 10363–10367 CrossRef CAS PubMed .
- H. Park, Y. Li and J.-Q. Yu, Angew. Chem., Int. Ed., 2019, 58, 11424–11428 CrossRef CAS PubMed .
- B. Wang, C. Lu, S.-Y. Zhang, G. He, W. A. Nack and G. Chen, Org. Lett., 2014, 16, 6260–6263 CrossRef CAS PubMed .
- Q.-F. Wu, P.-X. Shen, J. He, X.-B. Wang, F. Zhang, Q. Shao, R.-Y. Zhu, C. Mapelli, J. X. Qiao, M. A. Poss and J.-Q. Yu, Science, 2017, 355, 499–503 CrossRef CAS PubMed .
- Y.-F. Zhang, H.-W. Zhao, H. Wang, J.-B. Wei and Z.-J. Shi, Angew. Chem., Int. Ed., 2015, 54, 13686–13690 CrossRef CAS PubMed .
-
(a) Q.-F. Wu, X.-B. Wang, P.-X. Shen and J.-Q. Yu, ACS Catal., 2018, 8, 2577–2581 CrossRef CAS PubMed ;
(b) W. R. Gutekunst, R. Gianatassio and P. S. Baran, Angew. Chem., Int. Ed., 2012, 51, 7507–7510 CrossRef CAS PubMed .
- Y.-J. Liu, Z.-Z. Zhang, S.-Y. Yan, Y.-H. Liu and B.-F. Shi, Chem. Commun., 2015, 51, 7899–7902 RSC .
- S. Li, G. Chen, C.-G. Feng, W. Gong and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 5267–5270 CrossRef CAS PubMed .
- H. Jiang, J. He, T. Liu and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 2055–2059 CrossRef CAS PubMed .
- N. Thrimurtulu, S. Khan, S. Maity, C. M. R. Volla and D. Maiti, Chem. Commun., 2017, 53, 12457–12460 RSC .
- J. Calleja, D. Pla, T. W. Gorman, V. Domingo, B. Haffemayer and M. J. Gaunt, Nat. Chem., 2015, 7, 1009–1016 CrossRef CAS PubMed .
- C. He and M. J. Gaunt, Chem. Sci., 2017, 8, 3586–3592 RSC .
-
(a) K. K. Ghosh, A. Uttry, A. Mondal, F. Ghiringhelli, P. Wedi and M. van Gemmeren, Angew. Chem., Int. Ed., 2020, 59, 12848–12852 CrossRef CAS PubMed ;
(b) J. Das, P. Dolui, W. Ali, J. P. Biswas, H. B. Chandrashekar, G. Prakash and D. Maiti, Chem. Sci., 2020, 11, 9697–9702 RSC .
- H. S. Park, Z. Fan, R.-Y. Zhu and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 12853–12859 CrossRef CAS PubMed .
- P. Chuentragoo, D. Yadagiri, T. Morita, S. Sarkar, M. Parasram, Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 1794–1798 CrossRef PubMed .
Footnotes |
| † Dedicated to Prof. Wolfgang Kaim on the occasion of his 70th birthday. |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2021 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab