
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multigram synthesis of an orthogonally-protected pentasaccharide for use as a glycan precursor in a Shigella flexneri 3a conjugate vaccine: application to a ready-for-conjugation decasaccharide†
Johan
Cornil‡
,
Zhaoyu
Hu‡§
,
Marion
Bouchet
and
Laurence A.
Mulard
 *
*
Unité de Chimie des Biomolécules, Institut Pasteur, UMR3523 CNRS, 28 rue du Dr Roux, 75 724 Paris Cedex 15, France. E-mail: laurence.mulard@pasteur.fr
First published on 21st July 2021
Abstract
The rapidly growing interest in carbohydrate-based bioactive molecules calls for strategies enabling the appropriate design and large-scale delivery of the glycan moiety. Herein, we describe the robust and high-yielding chemical synthesis of an orthogonally-protected pentasaccharide intended for use as a central building block in vaccine development against Shigella flexneri 3a. Elaborated from advanced crystalline intermediates and fine-tuned catalytic processes facilitating regio- and stereoselective conversions, a robust [2 + 3] strategy was designed, which avoided several tedious purifications and efficiently delivered multigram amounts of the target pentasaccharide. Conversion of this intermediate into a donor and a linker-equipped acceptor then merging then into the frame of a [5 + 5] glycosylation step furnished a decasaccharide encompassing one trichloroacetamide moiety per repeat. Chemoselective delevulination and subsequent Pd(OH)2-mediated hydrogenolysis enabling concomitant hydrodechlorination and azide reduction gave the ready-for-conjugation dimer of the repeating unit of the O-antigen from S. flexneri 3a featuring the natural stoichiometric O-acetylation. The proof-of-concept was established, opening the way to larger S. flexneri 3a oligosaccharides and fine-tuned glycoconjugates.
Introduction
Understanding the importance of carbohydrates as mediators of biological processes has substantiated major advances in oligosaccharide synthesis to overcome limitations associated with isolates from natural sources.1 Various strategies are being explored, including enzymatic, chemo-enzymatic and chemical routes. The latter feature the greatest versatility in providing access to natural, as well as non-natural oligosaccharides. Programmable, one-pot solution-phase and methods for solid-phase and HPLC-assisted automated strategies exemplify some of the major ongoing investigations to accelerate the chemical synthesis of complex glycans.2–5 Significant developments have facilitated the expedited synthesis of diverse well-defined oligosaccharides, including homopolymers of increasing chain length,6–8 and the total synthesis of the largest chemically assembled polysaccharides to date,9–11 paving the way to useful probes for further investigating poorly understood carbohydrate-mediated vital biological events. When considering large heteropolymers and highly branched targets, solution-phase iterative block synthesis has remained an attractive strategy.12–20 In particular, the successful delivery of glycans featuring several repeats strongly relies on the identification of building blocks empowering iterative homologation with high and reproducible glycosylation yields, while also obeying regio- and stereoselectivity criteria in addition to qualifying for efficient full deprotection. Another significant challenge for relevant building block design stems from the need for a synthesis enabling the large-scale production of these essential intermediates to subsequently deliver usable amounts of the extended glycan targets.21,22 Herein, we tackle this relevant issue in the context of vaccine development.Shigellae are Gram-negative bacteria and the cause of shigellosis, a major diarrheal disease responsible for a high burden, notably among children aged 1–5 years living in low- and middle-income settings.23,24Shigella is on the WHO pathogen priority list. Epidemiological data, among which the increasing antimicrobial resistance observed among field isolates, call for the development of a multivalent Shigella vaccine.23,25 Toward this goal, conjugate vaccines based on the bacterial polysaccharide antigens, or surrogates thereof, have been the subject of major interest.26 As part of the ongoing developments, we have proposed the first synthetic glycan–protein conjugate vaccine candidate against endemic shigellosis.27 While many antibacterial glycovaccine candidates use haptens corresponding to one repeating unit of the homologous natural polysaccharide antigens,28 the selected glycoconjugate prototype comprises a chemically synthesized pentadecasaccharide corresponding to a three-core repeating unit portion from the Shigella flexneri 2a O-antigen (O-Ag).29 It was produced according to good manufacturing practice and was demonstrated to be safe and immunogenic in adult volunteers in the frame of a first-in-human clinical trial.31 These achievements have provided strong support for serotype broadening. In this context, our efforts were aimed towards a vaccine candidate against S. flexneri 3a (SF3a), another prevalent Shigella serotype for which a vaccine is in high demand.24
The SF3a O-Ag is made up of a branched pentasaccharide repeat (E)ABAcCAcD (Fig. 1), featuring (1 → 2)-trans-linked L-rhamnoses (A, B, C) and an N-acetyl-D-glucosamine residue (D). Rhamnose A is 3-O-α-D-glucosylated (E). Acetylation at position 2C is stoichiometric. In contrast, position 6D is O-acetylated only to an extent of 40%.30 Epitope mapping has revealed the immunodominant 2C-O-acetyl (Ac) moiety and the importance of chain length for protective antibody recognition.32 Molecular modeling simulations were supported by NMR analysis of O-Ag segments from 12 S. flexneri serotypes featuring the same backbone, among which those relevant to S. flexneri 2a and SF3a suggested similar backbone conformational behavior.33 This study also revealed the dynamic behavior of the end-chain α-D-glucopyranosyl residue (1 → 3)-linked to rhamnose A, differing from that predicted for glucose side-chains located on internal repeats.33 Overall, convincing evidence supports the assumption that oligosaccharides achieving SF3a O-Ag functional mimicry encompass at least two repeating units. Otherwise, the role of the non-stoichiometric 6D-O-acetylation remains undisclosed.
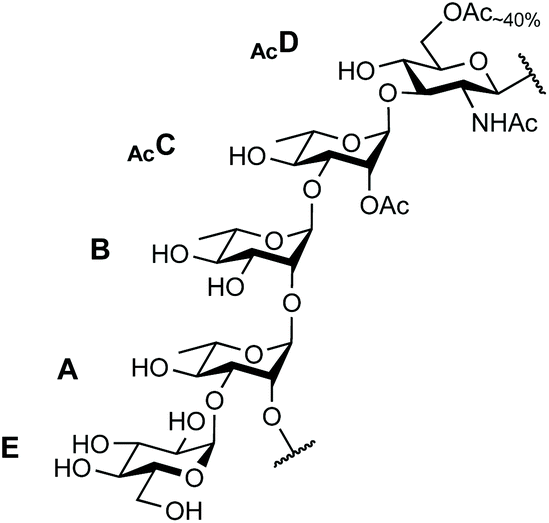 | ||
| Fig. 1 Repeating unit of the SF3a O-Ag: (E)ABAcCAcD.30 | ||
Aiming at establishing a lead hapten candidate for SF3a vaccination, we report a straightforward multi-step chemical synthesis of pentasaccharide 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 as the lead common precursor to the (E)ABAcCD and (E)ABAcCAcD modules, their combinations and oligomers thereof, as found in the native SF3a O-Ag (Scheme 1). Going beyond our previous disclosures while aiming at scalability and robustness, the orthogonally-protected pentasaccharide building block was produced in several 10-gram amounts. Emphasis was placed on the following: (i) restraining the handling of toxic and poor user-friendly reagents, in particular by circumventing the notoriously questionable tin chemistry and by avoiding concerns related to hydrazine and its derivatives, especially when involved at an advanced stage of a multi-step synthesis; (ii) limiting the repeated use of low-abundant catalysts, despite their remarkable potential as exemplified by iridium-based compounds; (iii) reducing the number of demanding purification steps involving column chromatography by promoting crystalline intermediates and the fine-tuning of reaction parameters, while (iv) achieving high-yielding conversions fulfilling regio- and stereoselectivity criteria. It is well-appreciated that concern for the latter increases when addressing glycosylation steps involved in large oligosaccharide blockwise synthesis.
17 as the lead common precursor to the (E)ABAcCD and (E)ABAcCAcD modules, their combinations and oligomers thereof, as found in the native SF3a O-Ag (Scheme 1). Going beyond our previous disclosures while aiming at scalability and robustness, the orthogonally-protected pentasaccharide building block was produced in several 10-gram amounts. Emphasis was placed on the following: (i) restraining the handling of toxic and poor user-friendly reagents, in particular by circumventing the notoriously questionable tin chemistry and by avoiding concerns related to hydrazine and its derivatives, especially when involved at an advanced stage of a multi-step synthesis; (ii) limiting the repeated use of low-abundant catalysts, despite their remarkable potential as exemplified by iridium-based compounds; (iii) reducing the number of demanding purification steps involving column chromatography by promoting crystalline intermediates and the fine-tuning of reaction parameters, while (iv) achieving high-yielding conversions fulfilling regio- and stereoselectivity criteria. It is well-appreciated that concern for the latter increases when addressing glycosylation steps involved in large oligosaccharide blockwise synthesis.
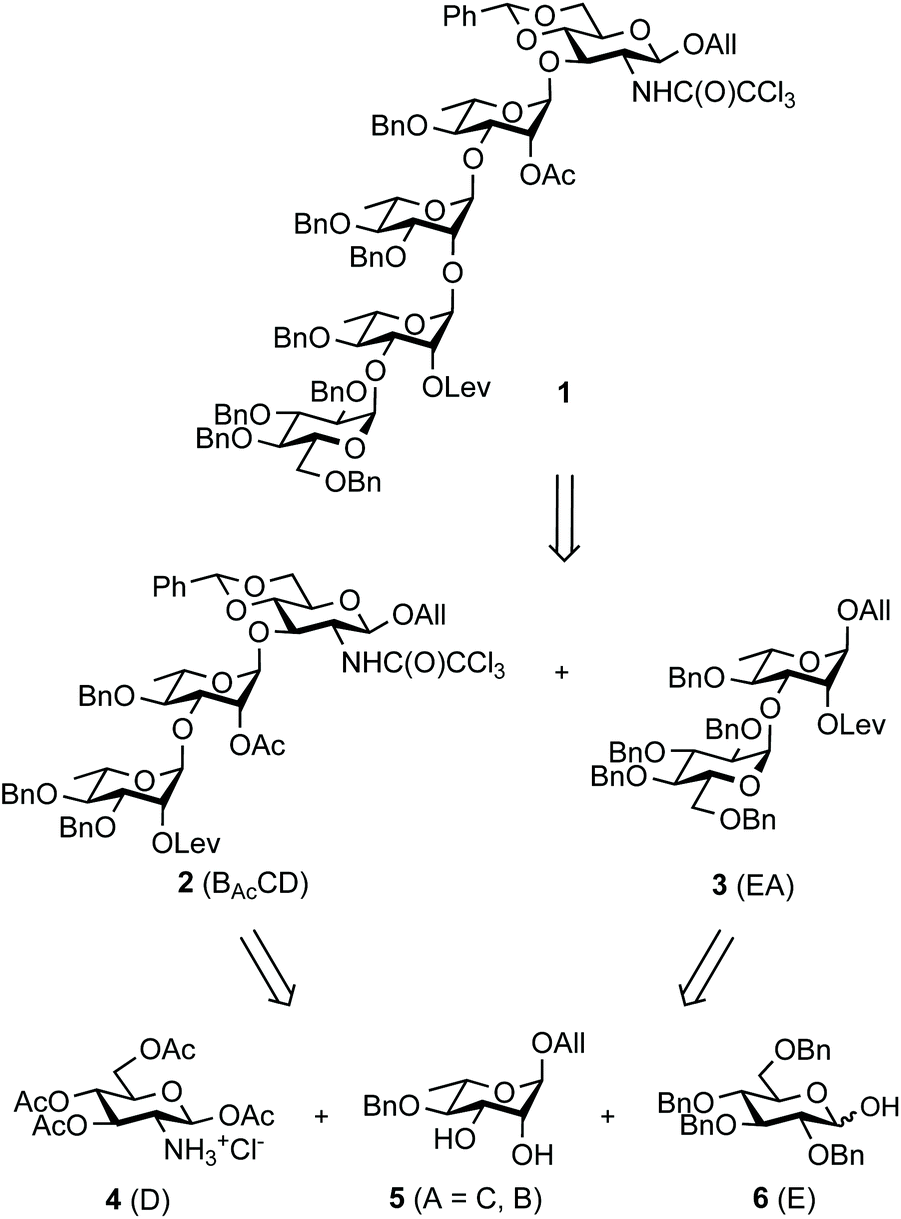 | ||
| Scheme 1 Pentasaccharide 1 and its retrosynthetic analysis. All: allyl; Lev: levulinoyl. | ||
Herein, significant inputs feature handy metal-catalyzed protecting group manipulation, advanced crystalline intermediates, fine-tuned 1,2-cis and block glycosylation steps, and a meaningful reduction of the number of columns for chromatography, the latter being known to qualify as a bottleneck when aiming at large-scale synthesis.22 Furthermore, the proof-of-concept is established since the potential of the selected pentasaccharide building block is demonstrated in the synthesis of a ready-for-conjugation linker-equipped decasaccharide corresponding to a dimer of the repeating unit of the SF3a O-Ag.
Results and discussion
Building block 1 was designed as an allyl glycoside, allowing easy conversion into a donor or an acceptor.17 It is 2C-O-acetylated as in the SF3a O-Ag. In contrast, the second site of natural O-acetylation was masked as a 4D,6D-O-benzylidene (Bzl) acetal, allowing for the chemoselective late-stage modification at OH-6D. Non-interfering hydroxyl groups are benzylated, and the site of elongation (OH-2A) features a levulinoyl ester, which fulfills the criteria for stability, anchimeric assistance and orthogonality, in particular to the 2C-acetate.34 Relying on the imidate chemistry, pentasaccharide 1 is readily accessible from the known BAcCD and EA allyl glycosides, 217 and 3,35 respectively. These key intermediates are commercially available in bulk amounts. Substantiating our previous report,36 diol 5 is routinely obtained in at least 90 g amounts in four steps and over 80% yield (Scheme 1). It performs as an exquisite common precursor to the known acceptor A/C (14) and donor B (13).35
Synthesis of the BCD trisaccharide 2
Going beyond the original tin-mediated regioselective benzylation of 1,2-cis diols37 and the inherent toxicity of tin reagents used in stoichiometric amounts, elegant procedures enabling the site-selective modification of carbohydrates have been developed.38,39 The recently reported iron(III)-based catalysts, Fe(dibm)3, offering high regioselectivity, broad scope and high reactivity,40 and its cheaper, although equally efficient, analog Fe(dipm)3,41 attracted our attention (Scheme 2). Readily obtained from the inexpensive FeCl3·6H2O, these reagents are considered non-air sensitive, non-toxic and environmentally benign.42 Gratifyingly, the Fe(dibm)3-promoted benzylation of diol 5 in the presence of base proceeded at 80 °C in acetonitrile to give the desired alcohol 7 (92%) together with its regioisomer (5%). Satisfactorily, Fe(dipm)3 performed as well. The 19:1 regioselectivity compares nicely with the 87% yield achieved using tin chemistry.35 Advantageously, purification is simpler. Next, instead of using a large excess of levulinic anhydride prepared upfront, Steglich esterification of alcohol 7 gave levulinate 8,35 which was, in turn, deallylated into hemiacetal 9.35
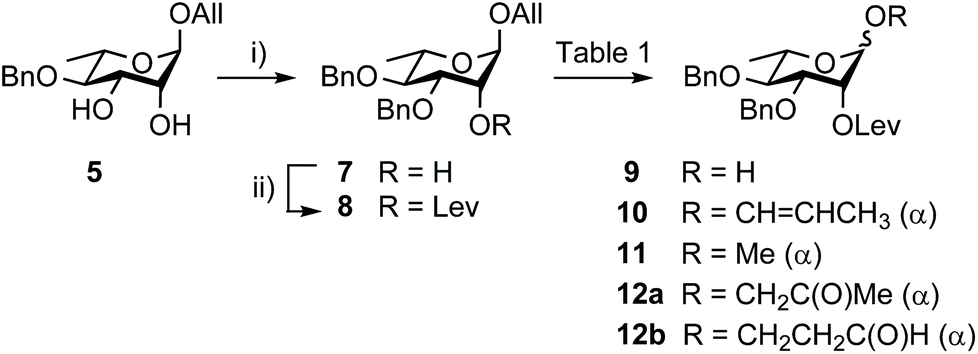 | ||
| Scheme 2 Synthesis of hemiacetal 9 from diol 5. (i) BnBr, Fe(dibm)3 or Fe(dipm)3 (5 mol%), K2CO3, MeCN, 80 °C, 92%; (ii) LevOH, EDC, DMAP, DCM, 90%. dibm: diisobutyrylmethane; dipm: dipivaloylmethane; LevOH: levulinic acid. | ||
As an attempt to avoid the previously adopted efficient, albeit expensive, [Ir(COD){PCH3(C6H5)2}2]+PF6− catalyst and its necessary hydrogen-mediated activation (Table 1, entry 1),17 we have favoured the use of more earth-abundant metal catalysts, focusing primarily on well-explored palladium derivatives (Table 1) amid numerous possible reagents,43–45 to complete the anomeric deallylation step.46 Unexpectedly, Pd(PPh3)4 used in combination with mild acids47 led at best in partial conversion to propen-1-yl 10 (entries 2 and 3). Therefore, established protocols involving Pd(II) catalysts, which are generally more stable and less expensive than Pd(0) derivatives, were considered instead. Diverging from previous observations,48 PdCl2 in buffered AcOH/AcONa was low-yielding (entry 4). Although the phenomenon was barely reported, methyl glycoside 11 was repeatedly isolated when using PdCl2 in methanol (entries 5 and 6), while the Wacker-type products43,4512a/12b were formed in DMF (entries 7 and 8). We reasoned that changing DMF to a non-polar solvent used in combination with water as the proton source would prevent side-oxidation. Indeed, conversion to propen-1-yl 10 was slow, but oxidized 12a/12b were not observed in DCM/H2O (entry 9). Otherwise, changing DMF for THF led to low conversion (entry 10). Gratifyingly, heating rhamnoside 8 to 50 °C for 2 h in DCM/H2O (3:1) containing PdCl2 (4 mol%) allowed faster completion and provided hemiacetal 9 in quantitative yield post-iodine addition (entry 11). These yet unreported easy-to-handle conditions were adopted on a large scale (entries 12 and 13).
| Entrya | Conditions | Products (yield) |
|---|---|---|
| a 60 mg scale and rt unless stated otherwise. b Post hydrolysis. c 25 g scale. d From alcohol 7 (30 g). | ||
| 1b49 |
[Ir(COD){PCH3(C6H5)2}2]+PF6− | 9 (93%) |
| 2 | Pd(PPh3)4, TsOH | 8 |
| 3 | Pd(PPh3)4, AcOH | 10 (70%) |
| 4 | PdCl2, AcOH/AcONa | 10 (58%) |
| 5 | PdCl2, MeOH | 11 |
| 6 | PdCl2, MeOH/THF | 11 |
| 7 | PdCl2, CuCl, DMF | 12a/12b (85%) |
| 8 | PdCl2, DMF/H2O | 12a/12b (74%) |
| 9 | PdCl2, DCM/H2O | 10 (74%), 9 |
| 10 | PdCl2, THF/H2O | 8, 10 |
| 11b | PdCl2, DCM/H2O, 2–4 h, 50 °C | 9 (full conversion) |
| 12b,c | PdCl2, DCM/H2O, 2–4 h, 50 °C | 9 (88%), 8 (12%) |
| 13b,d | PdCl2, DCM/H2O, 2–4 h, 50 °C | 9 (crude) |
Remarkably, trichloroacetimidate 13 is easily obtained by reacting hemiacetal 9 and trichloroacetonitrile in the presence of a base,49 and is now routinely prepared on the 40 g scale (92%) from alcohol 7 in three steps and no intermediate purification (Scheme 3). Donor 13 is stable for at least a month at −20 °C despite being isolated as a syrup.
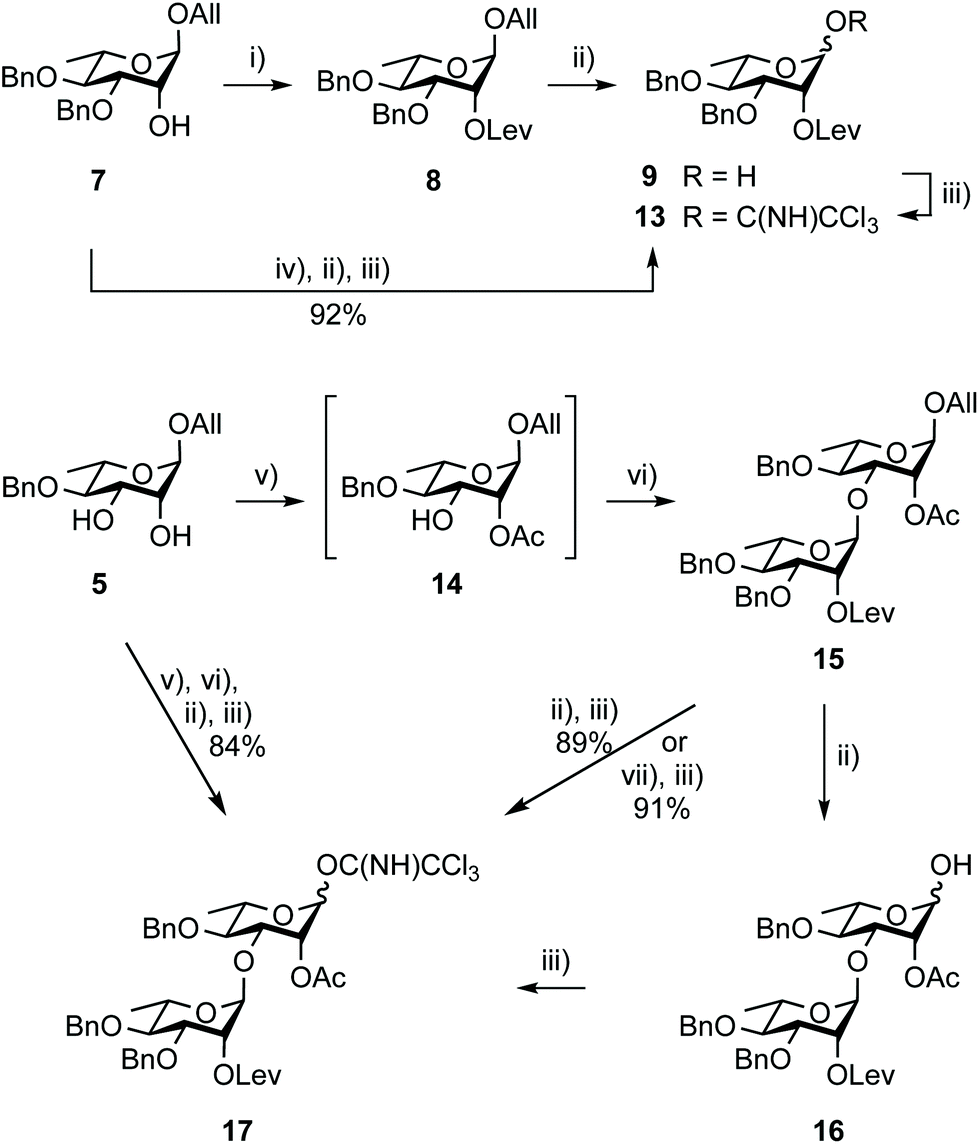 | ||
| Scheme 3 Synthesis of donor 13 from alcohol 7 (top), and donor 17 from diol 5 (bottom). (i) LevOH, EDC, DMAP, DCM, 90%. (ii) PdCl2, DCM/H2O, 50 °C then I2, THF/H2O, 88% for 9 and 90% for 16; (iii) Cl3CCN, DBU, 1,2-DCE, 87% for 13 and 84% for 17; (iv) LevOH, DCC, DMAP, DCM; (v) MeC(OMe)3, PTSA·H2O, MeCN, rt then 80% aq. AcOH, 0 °C; (vi) 13, TMSOTf, 4 Å MS, toluene, −78 °C to rt, 93%; (vii) H2-activated [Ir(COD)2{PCH3(C6H5)2}2]+PF6− (2–3 mol%), THF then I2, THF/H2O. | ||
The stepwise conversion of diol 5 into the BC donor 17 is a robust process (Scheme 3), reaching 69% over four steps on a 5–10 g scale.17 Herein, this conversion was achieved without intermediate purification reaching an overall yield of 86%, which was proven reproducible upon scaling up. Donor 17 was isolated in a 30 g amount (84%) starting from 13 g of diol 5. Noteworthy features in doing so include the reaction of acceptor 14, readily obtained from diol 5 as a 95:5 mixture of regioisomers, with a reduced excess of donor 13 (1.1 instead of 1.2 equiv.) and the use of the newly established Pd(II)-mediated anomeric deallylation protocol without any yield loss, as demonstrated for the independent conversion of rhamnobioside 15 into trichloroacetimidate 17 (91%).
Otherwise, acceptor 18 (88%) was achieved from tetraacetate 4 in four steps as described.50 In line with expectation,17 the TMSOTf-promoted [18 + 17] glycosylation proved to be highly efficient (Scheme 4). Crystalline BAcCD 2 of acceptable purity for the next step was isolated in 97% yield from 11 g of crystalline 18 and a slight excess (1.15 equiv.) of crude 17 is obtained from disaccharide 15.
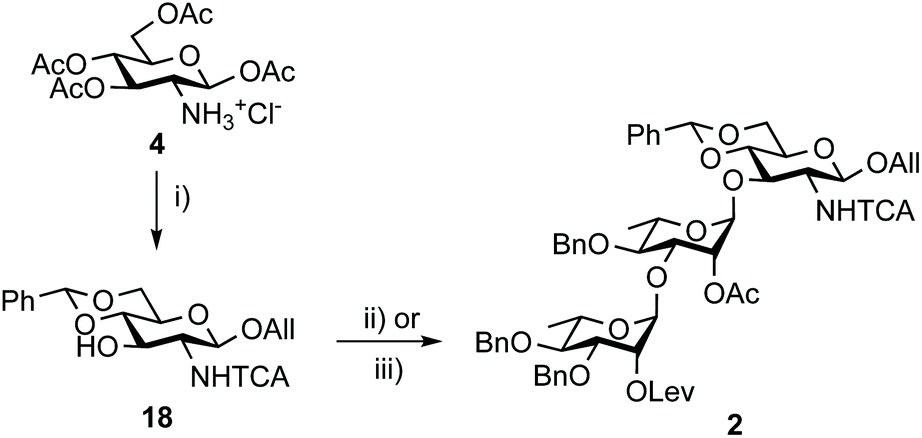 | ||
| Scheme 4 Synthesis of trisaccharide 2 from tetraacetate 4. (i) a. Cl3CC(O)Cl, pyridine, DCM; b. AllOH, TMSOTf, DCM; c. NaOMe, MeOH; d. PhCH(OMe)2, CSA, MeCN, 88%.50 (ii) 17, TMSOTf, 4 Å MS, DCM, 0 °C to rt, 86% (45 g scale). (iii) Crude 17 from 15, TMSOTf, 4 Å MS, DCM, 0 °C to rt, 97% (11 g scale). TCA: trichloroacetyl. | ||
Synthesis of the EA donors 34 and 35
The synthesis of disaccharide 3 was another opportunity for improvement (Scheme 5, Table 2). Originally, the essential 1,2-cis EA linkage was achieved from diol 5 as established in the late nineties51 to give alcohol 24 in 61% yield over three steps (entry 1).36 Since then, a better understanding of the factors affecting stereoselectivity has guided several reports on strategies addressing the challenge of anomeric control during 1,2-cis glycosylation.52,53 Therefore, going beyond original achievements,36 while considering scaling up, easy-to-implement alternatives were explored. Relying on the originally favored glucosyl donor 2154 and reinforcing previous realizations, parameters such as the promotor, its amount, and the solvent (entries 2–7) were varied without any observed meaningful improvement. While avoiding the formation of the Chapman rearrangement product 26,55 the use of the (N-phenyl)trifluoroacetimidate (PTFA) donor 2256 resulted in the loss of α/β selectivity (entry 11). In agreement with former investigations, changing donor 21 for the corresponding known thiophenyl glycoside 1957 and fluoride 2058 met no success (entries 14 and 15).
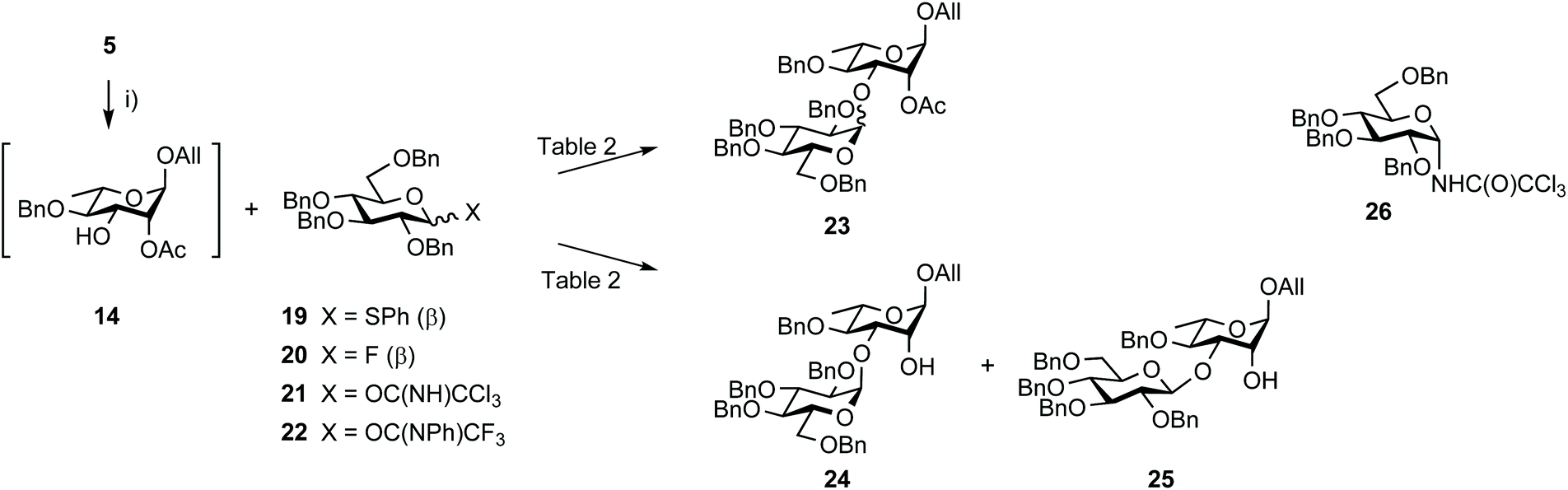 | ||
| Scheme 5 Synthesis of disaccharides EA from diol 5 and tetrabenzyl glucosyl donors. (i) MeC(OMe)3, PTSA·H2O, MeCN, rt then 80% aq. AcOH, 0 °C. | ||
63 and, in turn, the key intermediate 24 from diol 5 and donor 21, unless stated otherwise
| Entry | Conditionsa | Productsb,c (α:β ratio, yield from 5) |
|---|---|---|
| Promotor (equiv.), solvent, temperature | ||
| a Reactions were run on 70–80 mg of diol 5, using 1.3 equivalents of donor 21 and 0.3 promotor equivalent, at −78 °C unless stated otherwise. b α/β ratio based on NMR data of the crude. c Isolated yields for 23 and 24 are from diol 5, over two steps and over three steps, respectively. d 1.0 g scale. e 11 g scale. f Post transesterification. g Use of donor 22. h Use of donor 19. i Use of donor 20. | ||
| 136 | TMSOTf (0.02), toluene/DCM, −78 °C to rt |
23 (85:15), 24 (61%) |
| 2 | TMSOTf (0.3), Et2O, −78 °C |
23 (80:20, 60%) |
| 3 | TfOH (0.3), Et2O, −78 °C | 23, 14, 26 |
| 4 | Bi(OTf)3 (0.3), Et2O, −78 °C |
23 (85:15) |
| 5 | TMSOTf (0.07), Et2O, −105 °C | 23, 14, 26 |
| 6 | TMSOTf (0.3), toluene, −78 °C |
23 (80:20), 26 |
| 7 | TMSOTf (0.07), toluene, −78 °C |
23 (85:15) |
| 8 | TfOH (1.0), DMF (20), DCM, −78 °C to rt |
23 (90:10, 76%) |
| 9d,f | TfOH (1.0), DMF (20), DCM, −78 °C to rt | 24 (88%) |
| 10e,f | TfOH (1.0), DMF (20), DCM, −78 °C to rt |
24/25 (95:5, 81%) |
| 11g | TMSOTf (0.3), Et2O, −78 °C |
23 (50:50, 73%) |
| 12g | TfOH (1.0), DMF (20), DCM, rt |
23 (90:10) |
| 13d,f,g | TfOH (1.0), DMF (20), DCM, rt | 24 (79%) |
| 14h | NIS/AgOTf, DCM, −20 °C |
23 (65:35, 88%) |
| 15i | SnCl2/AgClO4, THF, −10 °C |
23 (65:35, 55%), 6 |
Next, the possible remote anchimeric assistance of the protecting groups masking the [E + A] glycosylation outcome52 was examined. In view of their orthogonal properties and easy access by means of the selective 6-O-debenzylation of hemiacetal 6 (Scheme 6),59 glucosyl donors bearing a temporary 6-O-acetyl ester or 6-O-tert-butyldiphenylsilyl ether (TBDPS), respectively, were considered. However, the enhanced α/β ratio expected from long-range 6-O-acyl-assistance using donors 2860 and 29 or from a foreseeable steric hindrance-controlled α-glucosylation by means of the silylated analogs 3161 and 32 was not observed in our hands providing the condensation products in at best a 7:3 α/β ratio (Scheme S2,† not described). Leaving aside promising albeit more demanding strategies involving specific protecting group manipulation,52 we turned to investigate the potential of exogenous nucleophiles to control stereoselectivity62 when solely considering the more readily available tetrabenzyl donors, and in particular imidates 21 and 22, as the simplest possible E precursors. We prioritized the DMF-modulated glycosylation strategy introduced by the Mong laboratory,64 a highly attractive approach as valuably demonstrated by Codée and coworkers.65 Satisfactorily, an improved α/β ratio was observed when rhamnoside 14 was reacted with trichloroacetimidate 21 in DCM containing DMF and stoichiometric TfOH (Table 2, entry 8).65 This tendency was independent of the imidate donor (entry 12). Besides the further strengthening of the potential of DMF as an external modulator of glycosylation reactions, the observed enhanced stereoselectivity was compatible with the subsequent transesterification step, therefore facilitating isolation of the product of α-glucosylation as alcohol 24 (entries 9 and 13). To our utmost satisfaction, scaling up did not interfere with stereoselectivity (entry 10). Indeed, as a clear step forward to a robust high-yielding process, these conditions delivered 25 g of the glucosylation products 24 and 25 in an excellent 95:5 α/β ratio and 81% yield over three steps from diol 5 and trichloroacetimidate 21, both of which are easily accessible crystalline materials.
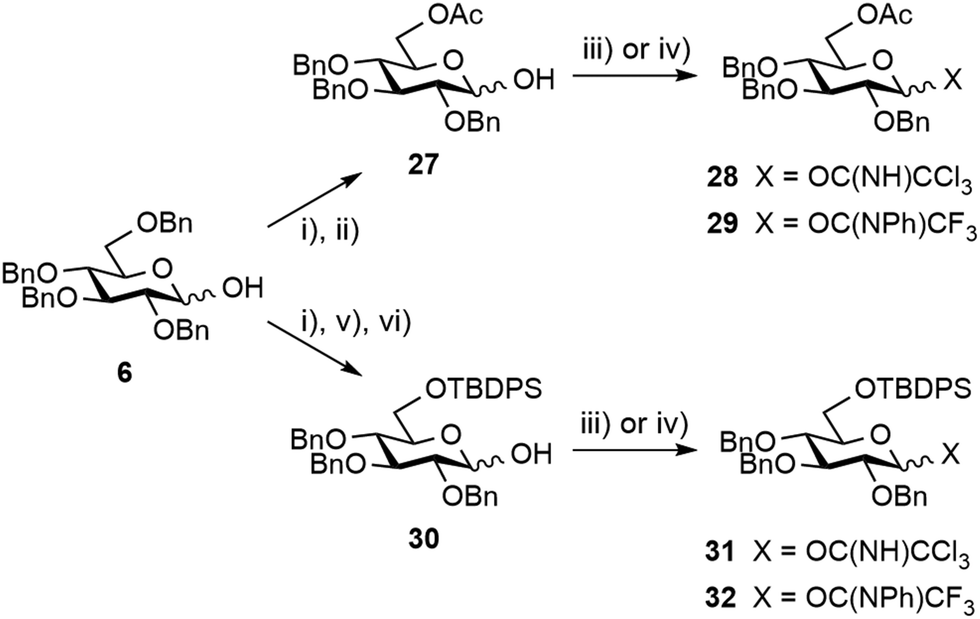 | ||
| Scheme 6 Synthesis of the 6-O-modified glucopyranosyl donors from hemiacetal 6. (i) TFA, Ac2O, 0 °C, (ii) NH2NH2·H2O, AcOH, DMF, 87% over two steps; (iii) CCl3CN, K2CO3, DCM, 85% for 28, 78% for 31; (iv) PTFACl, K2CO3 acetone, 90% for 29, 40% over 4 steps for 32; (v) MeONa, MeOH; (vi) TBDPSCl, imidazole, DMAP, DMF. | ||
Whereas Steglich levulination at OH-2A of disaccharide 24 hardly reached completion,35 full conversion into the key intermediate 3 was achieved when substituting DCC by the more reactive EDC (Scheme 7). This successful in situ activation of levulinic acid advantageously replaced the formerly adopted conditions.35 Deallylation, whether conventional35 or using the newly established aforementioned PdCl2 protocol, delivered the known hemiacetal 3335 quantitatively for direct conversion into imidates 3435 and 35. Alternatively, the fully protected 3 was evolved into those same donors without intermediate purification. Scaling up this efficient three-step conversion provided trichloroacetimidate 34 in 67% yield in combination with hemiacetal 33 (30%), post chromatography. The recovery of a meaningful amount of 33 was attributable to donor hydrolysis on the column, suggesting that careful consideration be given to the purification step for large-scale development. Satisfactorily, the more stable PTFA donor 35 was isolated in 40 g amount in an excellent 90% yield over three steps.
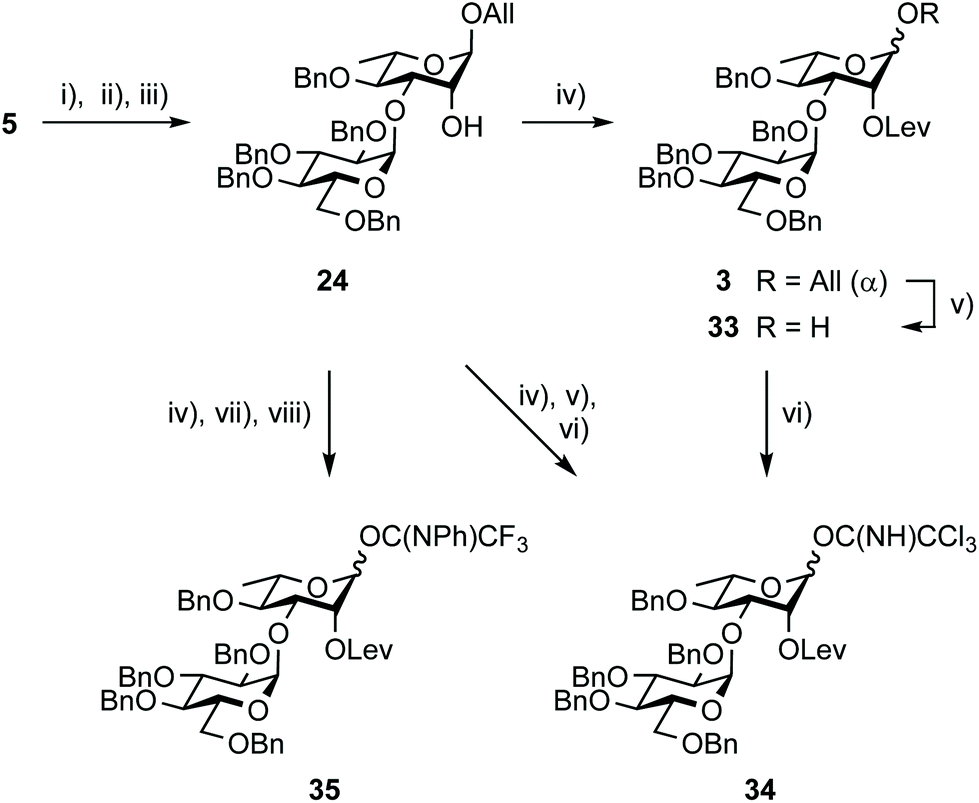 | ||
| Scheme 7 Synthesis of donors EA from diol 5 and tetrabenzyl glucosyl donors 21 or 22. (i) MeC(OMe)3, PTSA·H2O, MeCN, rt then 80% aq. AcOH, 0 °C; (iii) TfOH, DMF, 4 Å MS, DCM, −78 °C to rt; (iii) MeONa, MeOH/DCM, from 21 (1.15 equiv.), 88% (over two steps) and from 22 (1.3 equiv.), 79% (over three steps); (iv) LevOH, EDC, DMAP, DCM, 90% (30 g scale); (v) [Ir(COD){PCH3(C6H5)2}2]+PF6−, THF, then I2, THF/H2O, 85% (29 g scale); (vi) CCl3CN, DBU, DCE, −5 °C, 93% (23 g scale), also from 24, 67%, (42 g scale, 3 steps) together with 25 (30%); (vii) PdCl2, DCM/H2O then I2, THF, 50 °C; (viii) PTFACl, K2CO3, acetone, 90% (34 g scale, 3 steps). | ||
Synthesis of the EABAcCD pentasaccharide building block 1
Restraining the number of columns for chromatography, the two-step conversion of the fully protected 2 (∼25 g) into pentasaccharide 1 employed the crude acceptor 3617 (Scheme 8). Satisfactorily, the independent use of donor 34 or 35 (1.15 equiv.) ensued a good 80% yield from the fully protected 2, or rather a 88–94% corrected yield based on recovered 36. Adding to the overall improved strategy of the (E)ABAcCD building block (1), this compares favourably with original stepwise achievements.17 In particular, the proof of concept having been established, we are confident that additional fine-tuning on the two-step conversion on a large scale will contribute to further increasing the isolated yield of pentasaccharide 1. Toward this aim, we also envisioned alternatives to hydrazine acetate involving less toxic reagents for the selective delevulination at OH-2B of the 2C-O-acetyl BAcCD precursor (2). While the former remains from far the method most frequently encountered, it is not without drawback. In particular, we previously observed the partial reduction of the olefinic bond of the allyl aglycon in pentasaccharide 1 concomitant to hydrazinolysis of the 2A-O-levulinoyl ester.17 Inspiration from earlier findings66 encouraged the investigation of sulfite as a handy reagent. However, resulting at best in incomplete conversion, despite a prolonged reaction time (not described), neither the original conditions nor their modified version implemented in the context of oligonucleotide synthesis67 fulfilled our expectations. Optimization was not attempted. Instead, the implementation of user-friendly conditions enabling the high-yielding delevulination of trisaccharide 2 took advantage of a previous report from R. Adamo's group.68 Replacing hydrazine acetate by the more acceptable ethylenediamine provided alcohol 36 in a selective manner, suggesting that these conditions could be adopted in the future (Scheme 8).
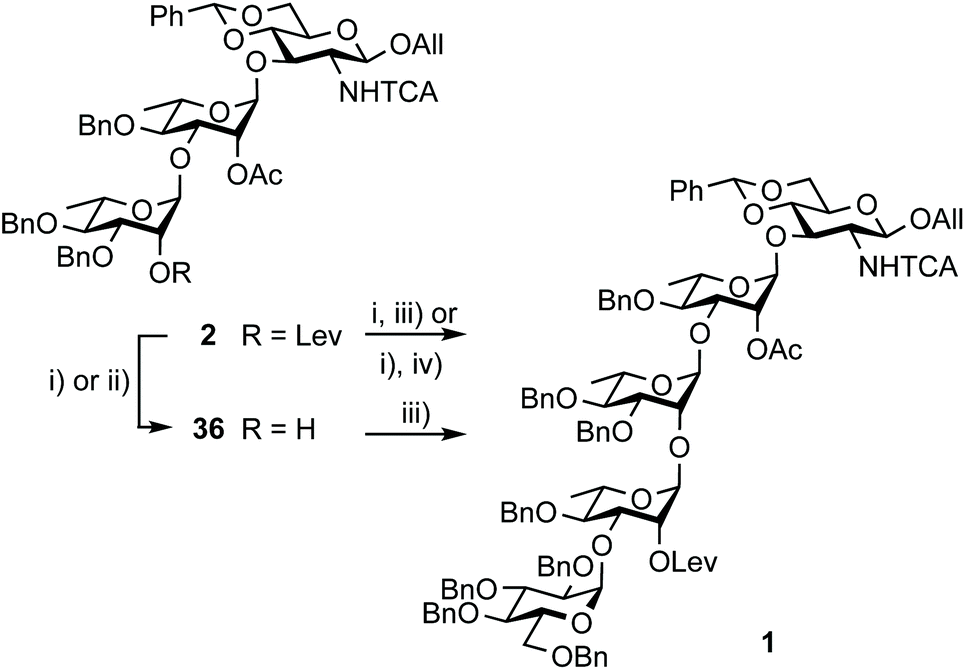 | ||
| Scheme 8 Synthesis of pentasaccharide 1 from the crystalline 2 and EA imidate donors 34 and 35. (i) NH2NH2·H2O, pyridine/AcOH (3:2), 92%; (ii) ethylenediamine, pyridine/AcOH (3:2), 74% (corrected yield: 85%); (iii) 34, TMSOTf, 4 Å MS, toluene, 94% (12 g)17 and 80% (27 g) from 2 (94% corrected based on isolated 36); (iv) 35, TMSOTf, 4 Å MS, toluene, 80% (24 g scale) from 2. | ||
Synthesis of the spacer-equipped pentasaccharide 45 and decasaccharide 46
Having achieved the improved synthesis of the fully protected 1, the next step consisted of ensuring that this key building block fulfilled expectations when evolved into a donor and a linker-equipped acceptor, respectively. Toward this aim, pentasaccharide 1 was subjected to conventional deallylation into hemiacetal 38, which was in turn converted into the corresponding PFTA donor 39 in high yield (Scheme 9). Running the two steps without any intermediate purification also resulted in an efficient conversion, repeatedly reaching over 85% yield on a multigram scale.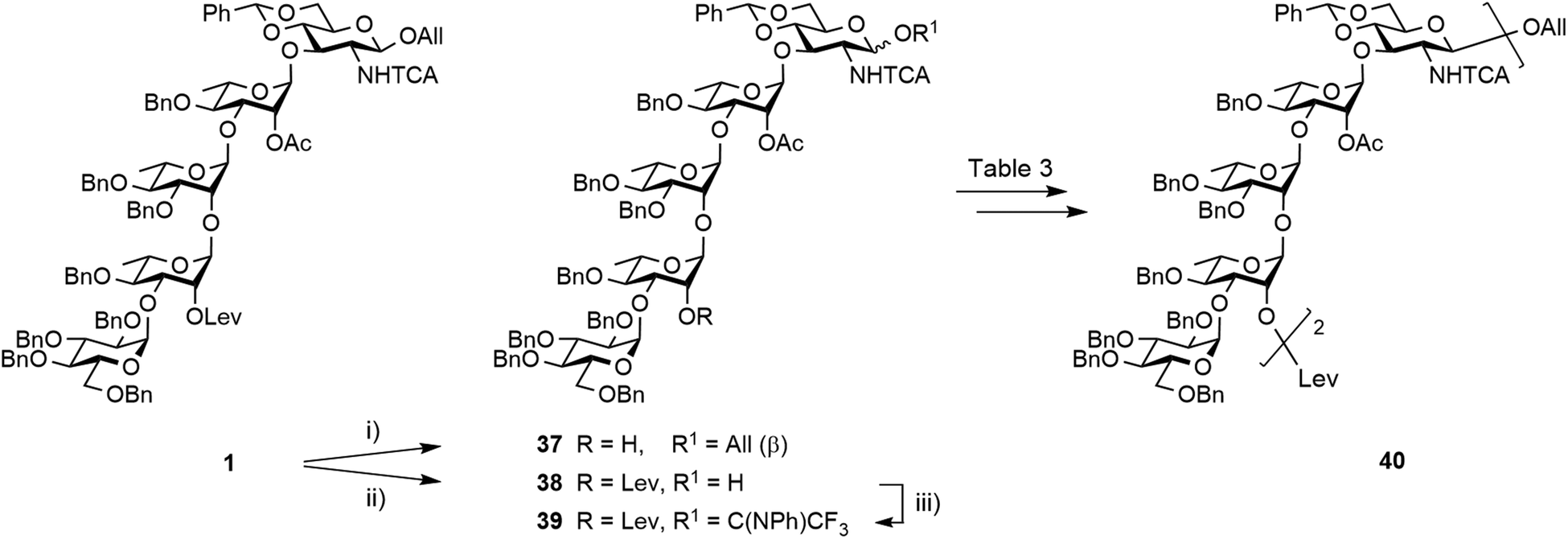 | ||
| Scheme 9 Synthesis of decasaccharide 40 from the pentasaccharide building block 1. (i) Ethylenediamine, pyridine/AcOH (3:2), 89%; (ii) [Ir(COD){PCH3(C6H5)2}2]+PF6−, THF, then I2, THF/H2O, 92%; (iii) PTFACl, K2CO3, acetone, 0 °C, 93%. | ||
Alcohol 37 was used as a model acceptor for the [5 + 5] glycosylation envisioned next (Table 3). With the promising outcome of the ethylenediamine-mediated delevulination of the BCD trisaccharide 2, it was isolated in an excellent 89% yield upon heating the fully protected 1 in the presence of excess ethylenediamine (Scheme 9). The [39 + 37] coupling proved to be high-yielding in all the conditions that were tested (Table 3). However, tendencies were revealed. In particular, some unconsumed acceptor was always observed when the 39:37 ratio was below 1.3 (entries 4–6). Glycosylation proceeded in a large range of temperature to give decasaccharide 40 in over 80% yield, but formation of an unidentified side-product was repeatedly observed at temperatures higher than −40 °C (entries 1–3). Replacing DCM with toluene while keeping the temperature at −40 °C and using 1.3 equivalents of donor had no obvious influence (entries 1 and 7). Having identified high-yielding glycosylation conditions, we turned to the synthesis of the linker-equipped decasaccharide 46.
| Entry | Conditionsa | Product (yield) | |||
|---|---|---|---|---|---|
| Acceptor | 39 equiv. | Solvent | Temperature | ||
| a Reactions were run on 100 mg of acceptor 37 using TMSOTf (0.2 equiv.) as promotor. | |||||
| 1 | 37 | 1.3 | DCM | −40 °C | 40 (90%) |
| 2 | 37 | 1.3 | DCM | rt | 40 (81%) |
| 3 | 37 | 1.3 | DCM | 0 °C | 40 (86%) |
| 4 | 37 | 1.1 | DCM | −78 °C | 40 (83%), 37 |
| 5 | 37 | 1.1 | DCM | −40 °C | 40 (85%), 37 (10%) |
| 6 | 37 | 1.2 | DCM | −40 °C | 40 (87%), 37 |
| 7 | 37 | 1.3 | Toluene | −40 °C | 40 (91%) |
Glycosylation of donor 39 with 2-azidoethanol was achieved in toluene at −40 °C to give the β-linked azidoethyl glycoside 41, which was isolated in a good 78% yield (Scheme 10). Hydrazinolysis of the 2A-O-levulinoyl ester provided acceptor 42 in a yield equivalent to that obtained for the corresponding allyl glycoside 37.17 In support of the selection of azidoethyl glycoside 42 as precursor to the larger linker-equipped SF3a oligosaccharides, its full deprotection promoted by Pd(OH)2 in an hydrogen atmosphere was uneventful, permitting the smooth concomitant hydrogenolysis and reduction of all protecting groups in place, to give the expected aminoethyl pentasaccharide 45 in a satisfactory 70% yield post-RP-HPLC chromatography.
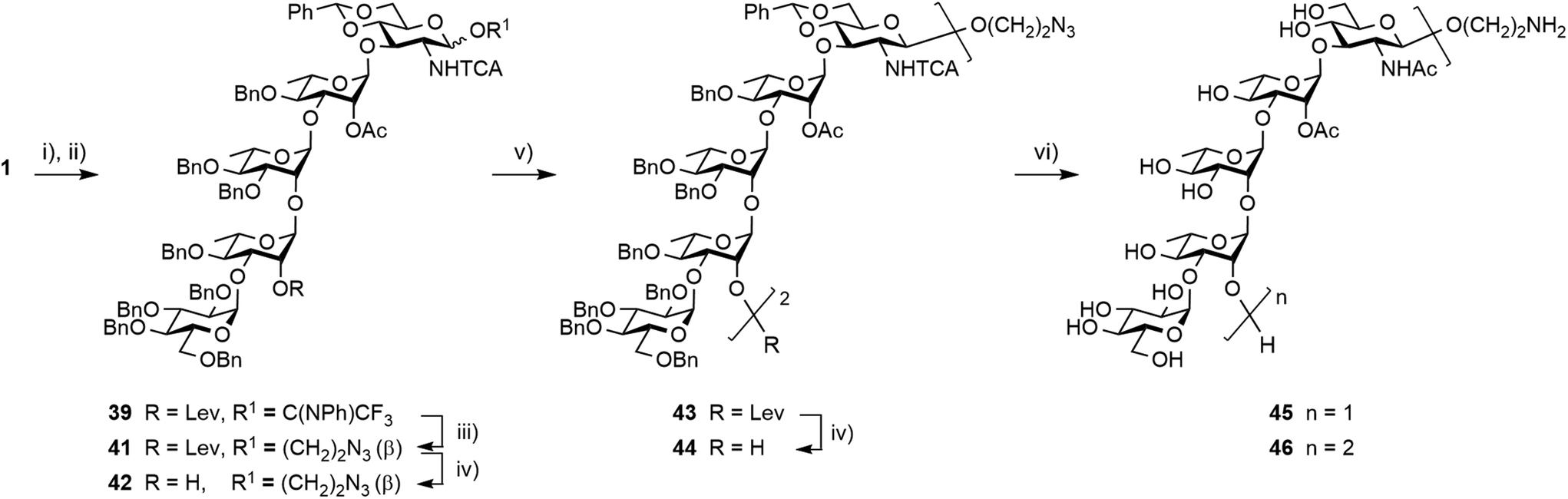 | ||
| Scheme 10 Synthesis of decasaccharide 46 from the pentasaccharide building block 1. (i) [Ir(COD){PCH3(C6H5)2}2]+PF6−, THF, then I2, THF/H2O; (ii) PTFACl, K2CO3, acetone, 0 °C, 88% (over 2 steps); (iii) 2-azidoethanol, TMSOTf, toluene, −40 °C, 78%; (iv) NH2NH2·H2O, pyridine/AcOH (3:2), 91% for 42 and 92% for 44; (v) 39 (1.3 equiv.), 4 Å MS, toluene, −40 °C, 88%; (vi) Pd(OH)2, H2, tBuOH/DCM/H2O, 70% for 45 from 42, 52% for 46 from 44. | ||
Interestingly, transferring the most promising [5 + 5] glycosylation conditions to the 2-azidoethyl-equipped acceptor revealed that the [39 + 37] glycosylation was somewhat sensitive to both solvent and temperature (not described). In agreement with original findings,17 running the condensation in non-polar toluene at −40 °C was identified as the best condition for providing decasaccharide 43 in a reproducible 90% average yield (Scheme 10). Subsequent delevulination gave alcohol 44, which was next subjected to a one-step full deprotection. While enabling the concomitant cleavage of the two 4D,6D-O-benzylidene acetals and 16 benzyl ethers in addition to the simultaneous reduction of the two 2D-trichloroacetamides and azide moiety, the Pd(OH)2-catalyzed hydrogenation/hydrogenolysis of the azidoethyl glycoside 44 in tBuOH/DCM/H2O into the aminoethyl decasaccharide 46 was more demanding than that of its counterpart 42 into pentasaccharide 45. The use of a higher Pd(OH)2 amount combined with a longer reaction time at ambient temperature and pressure furnished the conjugation-ready 46 in a good 52% yield post-RP-HPLC (Scheme 10). Nevertheless, the observed drop in the yield of the two O-Ag repeating unit segment 46versus the one repeating unit oligosaccharide 45 suggested that improvement might be needed for the full deprotection of larger oligomers featuring an aminoalkyl aglycon and a higher number of trichloroacetamide groups.
Conclusions
This study was aimed at achieving a robust process to enable the large-scale synthesis of pentasaccharide 1, and demonstrating that this orthogonally-protected building block could serve as a suitable precursor to a donor and an acceptor, whose combination would provide ready-for-conjugation oligosaccharides for use in the development of a synthetic carbohydrate-based conjugate vaccine candidate against SF3a. A robust and convenient 26-step synthesis, featuring four crystallizations and only nine columns for chromatography – mother liquors included – of pentasaccharide 1 from crystalline 1,3,4-6-tetra-O-acetyl-D-glucosamine, L-rhamnose and tetrabenzyl-D-glucose is described. The upgraded synthesis combines several independent step-specific improvements involving greener, less demanding, more stereoselective and user-friendly protocols, also promoting crystalline intermediates and multigram scale validation. Notably, relevant improvements of interest in a broader context include the implementation of the easy-to-handle PdCl2 in DCM/H2O for high-yielding anomeric deallylation and catalytic Fe(dipm)3 for the 3-O-etherification step of diol 5. Paving the way to further scale up and vaccine development against SF3a, the (E)ABAcCD pentasaccharide 1 was readily delivered in amounts over 30 g and subsequently converted into the linker-equipped hapten 45 by means of acceptor 42. Alternatively, the fully protected 1 was efficiently transformed into donor 39. Lastly, the proof-of-concept for building block selection enabling a robust [5 + 5] chain elongation strategy from pentasaccharide 1 was successfully demonstrated by delivering the ready-for-conjugation decasaccharide 46, which corresponds to a two-repeating-unit segment of the SF3a O-Ag. Aminoethyl glycosides 45 and 46 and larger SF3a O-Ag segments are ideal substrates for use as components of well-defined glycoconjugates for in vivo study, which represents the next aim.Experimental
Iron(III) dipivaloylmethane (Fe(dipm)3)41,42
To a biphasic mixture of 2,2,6,6-tetramethyl-heptane-3,5-dione (10.2 g, 55.5 mmol, 3.0 equiv.) and NaOAc (4.6 g, 55.5 mmol, 3.0 equiv.) in EtOH/H2O (1:1, 140 mL) was added FeCl3·6H2O (5.0 g, 18.5 mmol). A red slurry was formed and the mixture was heated at 60 °C for 2 h. The reaction was cooled down to rt then to 0 °C for 15 min. Filtration gave an orange powder that was washed with water and crystallized using EtOH/H2O (90:10, 70 mL). After cooling to 0 °C, crystals were filtered and rinsed using −78 °C cooled EtOH/H2O (90:10), furnishing Fe(dipm)3 as a red solid (10.9 g, 97%).
Allyl 3,4-di-O-benzyl-α-L-rhamnopyranoside (7)69
:20) indicated the total conversion of diol 5 into less polar products. Solids were filtered over a pad of Celite® and washed generously with DCM. Purification by flash column chromatography (tol/EtOAc, 100:0 to 90:10 to 80:20) gave the known 2-O-benzyl isomer70 as a yellow oil (350 mg, 5%) along with the desired 7 (6.0 g, 92%). The former had Rf = 0.75 (cHex/EtOAc, 70:30). 1H NMR (400 MHz, CDCl3) δ 7.47–7.32 (m, 10H, HAr), 5.94 (m, 1H, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif)
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.34 (dq, J = 17.2, 1.5 Hz, 1H, CHC2), 5.24 (m, 1H, CHC2), 4.98 (d, J = 11.2 Hz, 1H, HBn), 4.94 (d, J = 1.5 Hz, 1H, H-1), 4.80 (d, J = 11.8 Hz, 1H, HBn), 4.73 (d, J = 11.2 Hz, 1H, HBn), 4.68 (d, J = 11.8 Hz, 1H, HBn), 4.22 (m, 1H, C2All), 4.08 (ddapp, J = 9.0, 3.8 Hz, 1H, H-3), 4.02 (m, 1H, C2All), 3.83 (dd, J = 3.8, 1.5 Hz 1H, H-2), 3.80 (dq, J = 9.4, 6.3 Hz, 1H, H-5), 3.33 (tapp, J = 9.2 Hz, 1H, H-4), 2.53 (brs, 1H, O), 1.42 (d, J = 6.3 Hz, 3H, H-6). 13C NMR (100 MHz, CDCl3) δ 138.7 (CAr), 137.9 (CAr), 133.9 (
CH2), 5.34 (dq, J = 17.2, 1.5 Hz, 1H, CHC2), 5.24 (m, 1H, CHC2), 4.98 (d, J = 11.2 Hz, 1H, HBn), 4.94 (d, J = 1.5 Hz, 1H, H-1), 4.80 (d, J = 11.8 Hz, 1H, HBn), 4.73 (d, J = 11.2 Hz, 1H, HBn), 4.68 (d, J = 11.8 Hz, 1H, HBn), 4.22 (m, 1H, C2All), 4.08 (ddapp, J = 9.0, 3.8 Hz, 1H, H-3), 4.02 (m, 1H, C2All), 3.83 (dd, J = 3.8, 1.5 Hz 1H, H-2), 3.80 (dq, J = 9.4, 6.3 Hz, 1H, H-5), 3.33 (tapp, J = 9.2 Hz, 1H, H-4), 2.53 (brs, 1H, O), 1.42 (d, J = 6.3 Hz, 3H, H-6). 13C NMR (100 MHz, CDCl3) δ 138.7 (CAr), 137.9 (CAr), 133.9 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) HCH2), 128.6–127.7 (10CHAr), 117.2 (CHH2), 96.3 (C-1), 82.4 (C-4), 78.8 (C-2), 75.1 (H2Bn), 73.1 (H2Bn), 71.8 (C-3), 67.8 (H2All), 67.4 (C-5), 18.1 (C-6). HRMS (ESI+): m/z 407.1865 (calcd for C23H28O5Na [M + Na]+: m/z 407.1820).
:20) indicated the total conversion of diol 5 into less polar products. Solids were filtered over a pad of Celite® and washed generously with DCM. Purification by flash column chromatography (tol/EtOAc, 100:0 to 90:10 to 80:20) gave the known 3-O-benzyl isomer as a yellow oil (480 mg, 92%). The target 7 had Rf = 0.55 (cHex/EtOAc, 70:30). 1H NMR (400 MHz, CDCl3) δ 7.46–7.30 (m, 10H, HAr), 5.91 (dddd, J = 17.2, 10.4, 6.1, 5.1 Hz, 1H, CCH2), 5.30 (dq, J = 17.2, 1.6 Hz, 1H, CHC2), 5.21 (dq, J = 10.4, 1.4 Hz, 1H, CHC2), 4.92 (d, J = 10.9 Hz, 1H, HBn), 4.88 (d, J = 1.7 Hz, 1H, H-1), 4.72 (s, 2H, HBn), 4.67 (d, J = 10.9 Hz, 1H, HBn), 4.18 (ddtapp, J = 12.9, 5.1, 1.5 Hz, 1H, C2All), 4.09 (dd, J = 3.4, 1.8 Hz, 1H, H-2), 4.00 (ddtapp, J = 13.0, 6.1, 1.4 Hz, 1H, C2All), 3.90 (dd, J = 9.1, 3.4 Hz, 1H, H-3), 3.79 (dq, J = 9.5, 6.2 Hz, 1H, H-5), 3.49 (tapp, J = 9.3 Hz, 1H, H-4), 2.51 (brs, 1H, O), 1.34 (d, J = 6.3 Hz, 3H, H-6). 13C NMR (100 MHz, CDCl3) δ 138.4 (CAr), 138.0 (CAr), 133.8 (HCH2), 128.5–127.7 (10HAr), 117.4 (CHH2), 98.2 (C-1, JC,H = 168.7 Hz), 80.1 (C-3), 80.0 (C-4), 75.4 (H2Bn), 72.1 (H2Bn), 68.6 (C-2), 67.9 (H2All), 67.4 (C-5), 17.9 (C-6). HRMS (ESI+): m/z 407.1809 (calcd for C23H28O5Na [M + Na]+: m/z 407.1820).
HCH2), 128.6–127.7 (10CHAr), 117.2 (CHH2), 96.3 (C-1), 82.4 (C-4), 78.8 (C-2), 75.1 (H2Bn), 73.1 (H2Bn), 71.8 (C-3), 67.8 (H2All), 67.4 (C-5), 18.1 (C-6). HRMS (ESI+): m/z 407.1865 (calcd for C23H28O5Na [M + Na]+: m/z 407.1820).
:20) indicated the total conversion of diol 5 into less polar products. Solids were filtered over a pad of Celite® and washed generously with DCM. Purification by flash column chromatography (tol/EtOAc, 100:0 to 90:10 to 80:20) gave the known 3-O-benzyl isomer as a yellow oil (480 mg, 92%). The target 7 had Rf = 0.55 (cHex/EtOAc, 70:30). 1H NMR (400 MHz, CDCl3) δ 7.46–7.30 (m, 10H, HAr), 5.91 (dddd, J = 17.2, 10.4, 6.1, 5.1 Hz, 1H, CCH2), 5.30 (dq, J = 17.2, 1.6 Hz, 1H, CHC2), 5.21 (dq, J = 10.4, 1.4 Hz, 1H, CHC2), 4.92 (d, J = 10.9 Hz, 1H, HBn), 4.88 (d, J = 1.7 Hz, 1H, H-1), 4.72 (s, 2H, HBn), 4.67 (d, J = 10.9 Hz, 1H, HBn), 4.18 (ddtapp, J = 12.9, 5.1, 1.5 Hz, 1H, C2All), 4.09 (dd, J = 3.4, 1.8 Hz, 1H, H-2), 4.00 (ddtapp, J = 13.0, 6.1, 1.4 Hz, 1H, C2All), 3.90 (dd, J = 9.1, 3.4 Hz, 1H, H-3), 3.79 (dq, J = 9.5, 6.2 Hz, 1H, H-5), 3.49 (tapp, J = 9.3 Hz, 1H, H-4), 2.51 (brs, 1H, O), 1.34 (d, J = 6.3 Hz, 3H, H-6). 13C NMR (100 MHz, CDCl3) δ 138.4 (CAr), 138.0 (CAr), 133.8 (HCH2), 128.5–127.7 (10HAr), 117.4 (CHH2), 98.2 (C-1, JC,H = 168.7 Hz), 80.1 (C-3), 80.0 (C-4), 75.4 (H2Bn), 72.1 (H2Bn), 68.6 (C-2), 67.9 (H2All), 67.4 (C-5), 17.9 (C-6). HRMS (ESI+): m/z 407.1809 (calcd for C23H28O5Na [M + Na]+: m/z 407.1820).
3,4-Di-O-benzyl-2-O-levulinoyl-α/β-L-rhamnopyranose (9)35
PdCl2 (769 mg, 2.6 mmol, 0.05 equiv., 60% purity) was added to a solution of allyl glycoside 8 (25.1 g, 52.0 mmol) in DCM/H2O (3:1, 260 mL). After stirring for 4 h at 50 °C, a TLC control (tol/EtOAc, 80:20) showed the presence of a major less polar product in addition to some remaining 8. Nevertheless, the reaction mixture was allowed to cool to rt and I2 (26.4 g, 104.1 mmol, 2.0 equiv.) was added. After 40 min, a TLC control (tol/EtOAc, 80:20) showed the formation of two more polar products. Saturated aq. Na2S2O3 was added and the biphasic solution was filtered on a bed of Celite® and cotton. The two layers were separated and the aq. phase was extracted twice with DCM. The combined organic layers were washed with sat. aq. NaHCO3, water and brine, then dried on Na2SO4. Solids were filtered and volatiles were evaporated under reduced pressure. The crude residue was purified by flash chromatography (tol/EtOAc 90:10 then 70:30) to give, in order of elution, the unreacted 8 (3.08 g, 12%) as a brownish oil and the known hemiacetal 9 (20.19 g, α/β 85:15, 88%) as a light yellow oil. The obtained α anomer had Rf = 0.25 (tol/EtOAc, 70:30). 1H NMR (400 MHz, CDCl3) δ 7.37–7.28 (m, 10H, HAr), 5.40 (m, 1H, H-2), 5.13 (m, 1H, H-1), 4.95 (d, J = 10.9 Hz, 1H, HBn), 4.75–4.51 (m, 2H, HBn), 4.20 (d, J = 10.7 Hz, 1H, HBn), 4.03–3.98 (m, 2H, H-3, H-5), 3.43 (tapp, J = 9.4 Hz, 1H, H-4), 2.73 (m, 4H, C2Lev), 2.18 (s, 3H, C3Lev), 1.33 (d, J = 6.2 Hz, 3H, H-6). 13C NMR (100 MHz, CDCl3) δ 206.8 (OLev), 172.5 (O2Lev), 138.9 (Ar), 138.0 (Ar), 128.8–128.0 (10HAr), 92.7 (C-1, JC,H = 170.3 Hz), 80.5 (C-4), 77.9 (C-3), 75.7 (H2Bn), 72.0 (H2Bn), 70.0 (C-2), 68.1 (C-5), 38.5 (H2Lev), 30.1 (H3Lev), 28.6 (H2Lev), 18.4 (C-6). HRMS (ESI+): m/z 465.1876 (calcd for C25H30O7Na [M + Na]+: m/z 465.1889).
Prop-1-enyl 3,4-di-O-benzyl-2-O-levulinoyl-α-L-rhamnopyranoside (10)
An analytical sample of the product of allyl isomerization from the fully protected 8 had Rf = 0.7 (tol/EtOAc 70:30). 1H NMR (400 MHz, CDCl3) δ 7.44–7.30 (m, 10H, HAr), 6.18 (dq, J = 12.3 Hz, 1.8 Hz, 0.2H, CCH–CH3trans), 6.13 (dq, J = 6.2 Hz, 1.7 Hz, 0.8H, CCH–CH3cis), 5.49 (dd, J = 3.3 Hz, 1.9 Hz, 0.8H, H-2cis), 5.45 (dd, J = 3.3 Hz, 1.9 Hz, 0.8H, H-2trans), 5.14 (dq, J = 12.1 Hz, 6.8 Hz, 0.2H, CHC–CH3trans), 5.01–4.94 (m, 2H, H-1cis, H-1trans, HBn,cis, HBn,trans), 4.75 (d, J = 11.0 Hz, 0.8H, HBn,cis), 4.73 (d, J = 10.4 Hz, 0.2H, HBn,trans), 4.67 (d, J = 11.0 Hz, 0.8H, HBn,cis), 4.67 (d, J = 11.0 Hz, 0.2H, HBn,trans), 4.65–4.55 (m, 1.8H, CHC–CH3cis, HBn,cis, HBn,trans), 4.02 (dd, J = 9.4 Hz, 3.4 Hz, 0.8H, H-3cis), 4.01 (dd, J = 9.1 Hz, 3.4 Hz, 0.2H, H-3trans), 3.85–3.76 (m, 1H, H-5cis, H-5trans), 3.48 (t, J = 9.5 Hz, 0.8H, H-4cis), 3.46 (t, J = 9.3 Hz, 0.2H, H-4trans), 2.81–2.70 (m, 4H, 2CH2Lev), 2.19 (s, 2.4H, CH3Lev,cis), 2.19 (s, 0.6H, CH3Lev,trans), 1.60 (dd, J = 7.1 Hz, 1.7 Hz, 2.4H, CHCH–C3cis), 1.59 (dd, J = 5.4 Hz, 1.7 Hz, 0.6H, CHCH–C3trans), 1.36 (d, J = 6.4 Hz, 3H, H-6cis, H-6trans). 13C NMR (100 MHz, CDCl3) δ 206.2 (OLev), 172.0 (O2Lev), 142.2 (HCH–CH3trans), 141.1 (HCH–CH3cis), 138.5–137.0 (2Ar,cis, 2Ar,trans), 128.4–126.9 (10HArBn,cis, 10HArBn,trans), 104.9 (CHH–CH3trans), 104.2 (CHH–CH3cis), 97.4 (C-1cis, JC,H = 173.0 Hz), 96.9 (C-1trans, JC,H = 173.0 Hz), 79.9 (C-4trans), 79.8 (C-4cis), 77.8 (C-3trans), 77.7 (C-3cis), 75.5 (H2Bn,cis), 75.4 (H2Bn,trans), 71.8 (H2Bn,cis), 71.7 (H2Bn,trans), 68.8 (C-2cis), 68.6 (C-2trans), 68.2 (C-5cis), 68.0 (C-5trans), 38.0 (H2Lev), 29.8 (H3Lev), 28.2 (H2Lev), 18.0 (C-6), 12.4 (CHCH–H3trans), 9.3 (CHCH–H3cis).
3,4-Di-O-benzyl-2-O-levulinoyl-α-L-rhamnopyranosyl trichloroacetimidate (13)35
:10) showed that the starting material had been converted to a less polar product. DCU was filtered by passing through a pad of Celite®, and the solids were washed extensively with DCM. The organic layer was washed with water, sat. aq. NaHCO3, sat. aq. CuSO4, then twice with water and finally with brine. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. PdCl2 (920 mg, 3.0 mmol, 0.04 equiv., 60% purity) was added to a solution of crude 8 in DCM/H2O (3:1, 600 mL). The mixture was stirred for 3 h at 50 °C. TLC (cHex/EtOAc, 80:20) showed the conversion of the fully protected 8 into a less polar product. After cooling the solution to 0 °C, a solution of iodine (19.8 g, 156 mmol, 2.0 equiv.) in THF (140 mL) was added slowly and the mixture was stirred at rt for 2.5 h. TLC (cHex/EtOAc, 70:30) showed the complete disappearance of the intermediate and the presence of more polar products. Excess iodine was destroyed by adding a solution of sat. aq. Na2S2O3. The biphasic mixture was filtered on cotton and the organic phase was washed with sat. aq. NaHCO3, water and brine. The organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure. To a solution of the crude hemiacetal 9 in 1,2-DCE (260 mL) stirred under Ar at 0 °C, trichloroacetonitrile (39.1 mL, 390 mmol, 5.0 equiv.) and DBU (3.3 mL, 21.8 mmol, 0.28 equiv.) were added dropwise. The brown mixture was stirred at 0 °C for 3 h. TLC (cHex/EtOAc, 70:30 + 1% Et3N) showed the conversion of the intermediate 9 into less polar products. After incomplete concentration under reduced pressure, the mixture was purified by column chromatography on neutralized silica gel (cHex/EtOAc, 100:0 to 60:40 + 1% Et3N) to give the known donor 13 (42.6 g, 92%) as a yellow syrup along with recovered hemiacetal 9 (1.8 g, 5%).
(3,4-Di-O-benzyl-2-O-levulinoyl-α-L-rhamnopyranosyl)-(1 → 3)-2-O-acetyl-4-O-benzyl-α/β-L-rhamnopyranosyl trichloroacetimidate (17)49
:1, 575 mL) was added PdCl2 (850 mg, 2.87 mmol, 0.05 equiv., 60% purity). The mixture was stirred for 3 h at 50 °C. TLC (tol/EtOAc, 80:20) showed the disappearance of the starting 15 and the presence of a less polar product. Iodine (14.6 g, 115 mmol, 2.0 equiv.) in THF (100 mL) was added slowly at 0 °C and the mixture was stirred at rt for 2.5 h. TLC (tol/EtOAc, 70:30) showed the complete disappearance of the intermediate and the presence of a single more polar product. Excess iodine was destroyed by adding sat. aq. Na2S2O3. The biphasic mixture was filtered on cotton and the two layers were separated. The organic phase was washed with sat. aq. NaHCO3, water and brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. Trichloroacetonitrile (29 mL, 287 mmol, 5.0 equiv.) and DBU (2.5 mL, 16.1 mmol, 0.28 equiv.) were added dropwise to the obtained crude hemiacetal in anhyd. 1,2-DCE (290 mL) under Ar at 0 °C. After stirring at 0 °C for 3 h, TLC (tol/EtOAc 80:20, +1% Et3N) showed the absence of hemiacetal 16 and the presence of less polar products. The solution was concentrated to a minimum of solvent and purified by column chromatography on neutralized silica gel (tol/EtOAc 100:0 to 80:20, +1% Et3N). Donor 17 (45.0 g, 91%) was isolated as an orange syrup along with recovered 16 (3.05 g, 7%).
:20) showed orthoester consumption and the presence of more polar products. DCM was added along with water and the two layers were separated. The aq. phase was extracted with DCM and the combined organic layers were washed successively with sat. aq. NaHCO3 and brine, dried over Na2SO4, filtered and evaporated under reduced pressure to give the crude acceptor 14. TMSOTf (2.33 mL, 13.9 mmol, 0.25 equiv.) was slowly added dropwise to a solution of the latter (45.0 mmol, 1.0 equiv.) and trichloroacetimidate 13 (27.8 g, 47.3 mmol, 1.05 equiv.) in toluene (540 mL) containing activated 4 Å MS (5.0 g) at −78 °C. After stirring for 45 min at −78 °C, TLC (tol/EtOAc, 80:20) indicated acceptor consumption and the presence of a major less polar product. Et3N (3 mL) was added, the mixture was filtered over a pad of Celite® (DCM) and concentrated to dryness. Filtration over a pad of silica gel (tol/EtOAc, 100:0 to 85:15), evaporation and precipitation of TCA salts by the addition of cold toluene gave crude disaccharide 15 as an orange oil. To a solution of the latter in DCM/H2O (3:1, 430 mL) was added PdCl2 (635 mg, 2.15 mmol, 0.05 equiv., 60% purity). The mixture was stirred for 3 h at 50 °C. TLC (tol/EtOAc, 80:20) revealed a single less polar product. After cooling the mixture to 0 °C, a solution of iodine (10.9 g, 86.0 mmol, 2.0 equiv.) in THF (60 mL) was added slowly and the mixture was stirred at rt for 2.5 h. TLC (tol/EtOAc, 80:20) indicated the conversion of the intermediate into a more polar product. Excess iodine was destroyed by adding a solution of sat. aq. Na2S2O3. The mixture was filtered on cotton, and the two layers were separated. The organic phase was washed with sat. aq. NaHCO3, water and brine, dried on Na2SO4, filtered, and concentrated under reduced pressure. To a solution of crude 16 in anhyd. 1,2-DCE (215 mL) under an inert atmosphere at 0 °C, trichloroacetonitrile (21.6 mL, 215 mmol, 5.0 equiv.) and DBU (1.84 mL, 12.0 mmol, 0.28 equiv.) were added dropwise. After stirring at 0 °C for 1 h and at rt for 15 h, TLC (cHex/EtOAc 70:30, +1% Et3N) showed the conversion of hemiacetal 16 into less polar products. The solution was concentrated to a minimum of solvent and purified by flash chromatography on neutralized silica gel (cHex/EtOAc 100:0 to 80:20, +1% Et3N) to give trichloroacetimidate 17 (31.5 g, 84% over 4 steps) as an orange oil.
Allyl (2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl)-(1 → 3)-4-O-benzyl-α-L-rhamnopyranoside (24)51
:30) showed total consumption of the intermediate orthoester. DCM was added along with water and the two layers were separated. The aq. phase was extracted with DCM and the combined organic phases were washed successively with sat. aq. NaHCO3 and brine, dried over Na2SO4, filtered and concentrated to dryness to give crude acceptor 14. TfOH (300 μL, 3.4 mmol, 1.0 equiv.) was slowly added to a solution of the latter and trichloroacetimidate7121 (2.68 g, 3.91 mmol, 1.15 equiv.) in DCM (39 mL) containing DMF (5.26 mL, 68 mmol, 20 equiv.) and activated 4 Å MS (450 mg) at −78 °C. The suspension was stirred overnight while the temperature reached rt. TLC (tol/EtOAc, 70:30) indicated acceptor consumption and the presence of less polar products. Et3N (345 μL) was added and after 15 min, the suspension was filtered over a pad of Celite®. Solids were washed thoroughly with DCM and the organic phase was washed with sat. aq. NaHCO3, water and brine. The combined organic phases were dried over Na2SO4 and concentrated to dryness. The crude was solubilized in DCM/MeOH (11:8, 48 mL), 25% methanolic MeONa (1.16 mL, 5.1 mmol, 1.5 equiv.) was added, and the solution was stirred overnight. Dowex H+ resin was added to the solution under gentle stirring until neutralisation. Filtration, concentration of the filtrate to dryness, and purification of the crude by flash chromatography (tol/EtOAc, 90:10) gave the known α anomer 24 (2.45 g, 88%).
:20) indicated the total consumption of the intermediate orthoester. DCM and water were added and the two layers were separated. The aq. phase was extracted with DCM and the combined organic phases were washed successively with sat. aq. NaHCO3 and brine, dried over Na2SO4, filtered and concentrated to dryness to give the crude acceptor 14. DMF (5.28 mL, 67.9 mmol, 20 equiv.) and activated 4 Å MS (0.5 g) were added to a mix of the latter and the PTFA donor 22 (3.14 g, 4.42 mmol, 1.3 equiv.) in anhyd. DCM (44 mL) and the suspension was stirred at rt under Ar for 30 min, then at −78 °C for 15 min. TfOH (0.3 mL, 3.40 mmol, 1.0 equiv.) was added very slowly at −78 °C. The reaction mixture was then stirred for 1 h while slowly warming up to rt. TLC (cHex/EtOAc, 70:30) showed the complete disappearance of rhamnoside 14 and the presence of less polar products. The reaction mixture was neutralized with Et3N. EtOAc was added along with water and the two layers were separated. The aq. phase was extracted with EtOAc and the combined organic layers were washed successively with sat. aq. NaHCO3 and brine, dried over Na2SO4, filtered and evaporated. MeONa (25% in MeOH, 1.17 mL, 5.1 mmol, 1.5 equiv.) was added to the obtained crude in DCM/MeOH (11:8, 50 mL). After stirring at rt overnight, TLC (tol/EtOAc, 80:20) revealed that the glycosylation products had reacted and more polar products were present. DOWEX H+ resin was added and the mixture was stirred 30 minutes before filtering and washing thoroughly with MeOH. Et3N (few drops) was added and volatiles were evaporated. Purification by flash column chromatography (tol/EtOAc, 100:0 to 90:10) gave the desired α anomer 24 as a pale yellow oil (2.2 g, 79% over 3 steps) and the commercially available hemiacetal 6 (345 mg). The expected 24 had Rf = 0.45 (cHex/EtOAc, 75:25). 1H NMR (400 MHz, CDCl3) δ 7.47–7.17 (m, 25H, HAr), 5.97 (m, 1H, CCH2), 5.36 (dq, J = 17.2 Hz, 1.5 Hz, 1H, CHC2), 5.27 (dq, J = 10.4 Hz, 1.6 Hz, 1H, CHC2), 5.06–4.97 (m, 4H, H-1E, H-1A, 2HBn), 4.91 (d, J = 11.0 Hz, 1H, HBn), 4.90 (d, J = 11.6 Hz, 1H, HBn), 4.84 (d, J = 10.7 Hz, 1H, HBn), 4.78 (d, J = 11.6 Hz, 1H, HBn), 4.72 (d, J = 10.7 Hz, 1H, HBn), 4.59 (d, J = 12.2 Hz, 1H, HBn), 4.57 (d, J = 11.0 Hz, 1H, HBn), 4.38 (d, J = 12.2 Hz, 1H, HBn), 4.23 (ddt, J = 13.1 Hz, 5.5 Hz, 1.5 Hz, 1H, C2All), 4.14 (t, J = 9.3 Hz, 1H, H-3E), 4.12 (dd, J = 9.0 Hz, 3.2 Hz, 1H, H-3A), 4.08–4.00 (m, 3H, H-2A, H-5E, C2All), 3.86 (dq, J = 9.6 Hz, 6.1 Hz, 1H, H-5A), 3.81 (t, J = 9.3 Hz, 1H, H-4E), 3.69 (dd, J = 9.6 Hz, 3.7 Hz, 1H, H-2E), 3.59 (t, J = 9.3 Hz, 1H, H-4A), 3.54 (dd, J = 11.0 Hz, 2.9 Hz, 1H, H-6aE), 3.47 (brs, 1H, OH-2A), 3.43 (dd, J = 10.9 Hz, 2.0 Hz, 1H, H-6bE), 1.45 (d, J = 6.3 Hz, 3H, H-6A). 13C NMR (100 MHz, CDCl3) δ 138.7–137.6 (5Ar), 133.9 (HCH), 128.7–127.6 (25HAr), 117.3 (CHH2), 98.3 (C-1A, JC,H = 170.1 Hz), 94.0 (C-1E, JC,H = 168.4 Hz), 82.5 (C-3E), 79.4 (C-4A), 79.1 (C-2E), 77.9 (C-4E), 76.7 (C-3A), 75.6 (2H2Bn), 75.0 (H2Bn), 74.3 (H2Bn), 73.4 (H2Bn), 70.8 (C-5E), 68.0 (C-6E), 67.9 (H2All), 67.5 (C-2A), 67.3 (C-5A), 18.0 (C-6A). HRMS (ESI+): m/z 839.3749 (calcd for C50H56O10Na [M + Na]+: m/z 839.3766).
6-O-Acetyl-2,3,4-tri-O-benzyl-α/β-D-glucopyranose (27)60
Ac2O/TFA (4:1, 30 mL) was added to hemiacetal 6 (2.5 g, 5.0 mmol, 1.0 equiv.) at 0 °C and the suspension under Ar was stirred at rt for 3 h. TLC (cHex/EtOAc, 60:40) showed the conversion of the starting 6 into less polar products. Cold water (50 mL) was added at 0 °C and the mixture was stirred for 15 min at rt, then neutralized with sat. aq. NaHCO3. EtOAc was added and the two layers were separated. The aq. phase was extracted repeatedly with EtOAc and the combined organic layers were washed with sat. aq. NaHCO3, brine and dried over Na2SO4. Volatiles were evaporated and the crude was solubilized in DMF (20 mL). Hydrazine (60% in water, 0.22 mL, 7.0 mmol, 1.5 equiv.) and AcOH (0.40 mL, 7.0 mmol, 1.5 equiv.) were added at rt and the mixture was stirred at this temperature for 20 h. TLC (cHex/EtOAc, 60:40) showed the conversion of the 1,6-di-O-acetyl intermediate into more polar products. EtOAc and water were added and the two layers were separated. The aq. layer was extracted repeatedly with EtOAc and the combined organic phases were washed with sat. aq. NaHCO3, brine and dried over Na2SO4. Flash column chromatography on silica gel (cHex/EtOAc, 100:0 to 50:50) gave the known hemiacetal 27 as a white solid (α/β 65:35, 1.99 g, 87%). An analytical sample was obtained by means of a second purification. 1H NMR (400 MHz, DMSO-d6) δ 7.43–7.23 (m, 15H, HAr), 5.25 (d, J = 3.4 Hz, 0.65H, H-1α), 4.96–4.52 (m, 6.35H, HBn, H-1β), 4.30–4.10 (m, 2H, H-6), 3.96 (ddd, J = 10.1, 4.8, 2.5 Hz, 0.65H, H-5α), 3.90 (t, J = 9.2 Hz, 0.65H, H-3α), 3.72–3.59 (m, 0.7H, H-3β, H-5β), 3.49–3.38 (m, 1.65H, H-2α, H-4α, H-4β), 3.26 (dd, J = 9.2, 7.8 Hz, 0.35H, H-2β), 2.03 (s, 1.05H, Acβ), 2.00 (s, 1.95H, Acα). 13C NMR (100 MHz, DMSO-d6) δ 139.30 (CArα), 139.27 (CArβ), 139.2 (CArβ), 139.1 (CArα), 138.8 (CArα), 138.7 (CArβ), 128.7–127.8 (15HAr), 97.2 (C-1β), 90.1 (C-1α), 84.3 (C-3β), 83.5 (C-2β), 81.4 (C-3α), 80.5 (C-2α), 78.1 (C-4), 74.9–74.0 (2H2Bn), 72.4 (C-5β), 71.8 (H2Bn), 68.2 (C-5α), 63.64 (C-6β), 63.61 (C-6α), 21.1 (OH3). HRMS (ESI+): m/z 510.2479 (calcd for C29H36NO7 [M + NH4]+: m/z 510.2486).
6-O-Acetyl-2,3,4-tri-O-benzyl-α/β-D-glucopyranosyl (N-phenyl)trifluoroacetimidate (29)
K2CO3 (0.53 g, 3.86 mmol, 2.0 equiv.) and (N-phenyl)trifluoroacetimidoyl chloride (0.46 mL, 2.89 mmol, 1.5 equiv.) were added to hemiacetal 27 (950 mg, 1.93 mmol, 1.0 equiv.) in acetone (20 mL) stirred at rt. The suspension was stirred at this temperature for 2 h. TLC (cHex/EtOAc, 80:20) revealed the conversion of the starting 27 into less polar products. The suspension was filtered over a pad of Celite®, generously washed with DCM, and volatiles were evaporated. Purification of the residue by flash column chromatography on silica gel (cHex/EtOAc, 100:0 to 95:5) gave (N-phenyl)trifluoroacetimidate 29 as a 1:1 mix of α/β anomers (1.1 g, 90%). An analytical sample was obtained by means of a second purification. Donor 29 had Rf = 0.6 (cHex/EtOAc, 90:10). 1H NMR (400 MHz, CDCl3) δ 7.44–7.21 (m, 17H, HAr), 7.12 (m, 1H, HAr), 6.78–6.73 (m, J = 7.7 Hz, 2H, HAr), 6.45 (brs, 1H, H-1), 5.04 (d, J = 10.8 Hz, 1H, HBn), 4.92 (d, J = 10.8 Hz, 1H, HBn), 4.89 (d, J = 10.8 Hz, 1H, HBn), 4.81–4.75 (m, 2H, HBn), 4.62 (d, J = 10.8 Hz, 1H, HBn), 4.38–4.25 (m, 2H, H-6), 4.08 (tapp, J = 9.3 Hz, 1H, H-3), 4.02 (m, 1H, H-5), 3.73 (dd, J = 9.3, 3.4 Hz, 1H, H-2), 3.61 (tapp, J = 9.5 Hz, 1H, H-4), 2.06 (s, 3H, CH3Ac). 13C NMR (100 MHz, CDCl3) δ 170.5 (OAc), 143.5–137.6 (4Ar), 128.7–127.7 (19HAr), 124.2 (CF3), 119.4 (HAr), 81.5 (C-3), 79.3 (C-2), 77.2 (C-4), 75.8 (H2Bn), 75.3 (H2Bn), 73.4 (H2Bn), 71.5 (C-5), 62.6 (C-6), 20.8 (CH3Ac). (C-1 and CN could not be detected due to relaxation issues). HRMS (ESI+): m/z 681.2778 (calcd for C37H40F3N2O7 [M + NH4]+: m/z 681.2782).
2,3,4-Tri-O-benzyl-6-O-tert-butyldiphenylsilyl-α/β-D-glucopyranose (30)61
A mixture of Ac2O/TFA (4:1, 30 mL) was added at 0 °C to hemiacetal 6 (2.5 g, 5.0 mmol, 1.0 equiv.) under Ar and the suspension was stirred at rt for 3 h, at which time TLC (cHex/EtOAc, 60:40) indicated the conversion of the starting 6 into less polar products. Cold water (50 mL) was added at 0 °C and the mixture was stirred for 15 min at this temperature, then neutralized with 4 M aq. NaOH. EtOAc was added and the two layers were separated. The aq. layer was extracted with EtOAc and the combined organic phases were washed with brine and dried over Na2SO4. Volatiles were evaporated and MeONa (25% in MeOH, 5 mL) was added to the crude intermediate stirred in MeOH (20 mL) at rt. After stirring overnight at this temperature, TLC (cHex/EtOAc, 60:40) showed the complete disappearance of the intermediate and the presence of more polar products. Dowex H+ resin was added portion-wise while the suspension was gently stirred until neutralisation. The suspension was filtered and the volatiles were evaporated. DMAP (0.11 g, 0.93 mmol, 0.2 equiv.), imidazole (0.76 g, 11.1 mmol, 2.4 equiv.) and tert-butyldiphenylsilyl chloride (TBDPSCl, 1.44 mL, 5.55 mmol, 1.2 equiv.) were added to a solution of the crude in DMF (40 mL) at 0 °C. The mixture was stirred overnight at rt, at which time more imidazole (2.0 equiv.) and TBDPSCl (1.1 equiv.) were added. After 3 h, water and Et2O were added and the two layers were separated. The aq. phase was extracted with Et2O and the combined organic phases were washed with sat. aq. NaHCO3, brine and dried over Na2SO4. Volatiles were evaporated under reduced pressure; two successive purifications by flash column chromatography on silica gel (cHex/EtOAc, 100:0 to 95:5) gave hemiacetal 30 as a colorless oil (mostly α/β mixture, 1.7 g) contaminated with tert-butyldiphenylsilanol (10 mol%). Only the major isomer is described. 1H NMR (400 MHz, DMSO-d6) δ 7.85–7.09 (m, 25H, HAr), 5.34 (d, J = 3.4 Hz, 1H, H-1), 5.00–4.56 (m, 6H, HBn), 4.04–3.74 (m, 4H, H-3, H-5, H-6), 3.70–3.60 (m, 1H, H-4), 3.47 (dt, J = 9.6, 2.8 Hz, 1H, H-2), 1.04–0.96 (m, 9H, Si(C3)3). 13C NMR (100 MHz, DMSO-d6) δ 139.5–138.8 (5CAr), 135.8–126.9 (25HAr), 90.3 (C-1), 81.5 (C-3), 80.9 (C-2), 78.04 (C-4), 74.9–71.1 (3H2Bn), 70.8 (C-5), 63.4 (C-6), 27.2 (SiC(H3)3), 19.4 (Si(CH3)3). HRMS (ESI+): m/z 706.3548 (calcd for C43H52NO6Si [M + NH4]+: m/z 706.3558).
2,3,4-Tri-O-benzyl-6-O-tert-butyldiphenylsilyl-α/β-D-glucopyranosyl (N-phenyl)trifluoroacetimidate (32)
K2CO3 (0.32 g, 2.32 mmol, 2.0 equiv.) and (N-phenyl)trifluoroacetimidoyl chloride (PTFACl, 0.28 mL, 1.74 mmol, 1.5 equiv.) were added to hemiacetal 30 (800 mg, 1.16 mmol, 1.0 equiv.) in acetone (11.6 mL) at rt. After stirring at rt for 20 h, TLC (cHex/EtOAc, 90:10) indicated the conversion of hemiacetal 30 into less polar products. The suspension was filtered over a pad of Celite®, and the solids were generously washed with DCM. Volatiles were evaporated and the residue was purified by flash column chromatography (cHex/EtOAc, 100:0 to 90:10) to give the (N-phenyl)trifluoroacetimidate 32 as a 55:45 mix of α/β anomers (952 mg, 40% over four steps). Donor 32 had Rf = 0.65 (cHex/EtOAc, 90:10). 1H NMR (400 MHz, CDCl3) δ 7.83–7.63 (m, 4H, HAr), 7.50–7.21 (m, 23H, HAr), 7.14 (m, 1H, HAr), 6.82–6.72 (m, 2H, HAr), 6.55 (s, 0.55H, H-1a), 6.29 (s, 0.45H, H-1b), 5.12–4.89 (m, 2H, HBn), 4.85–4.63 (m, 4H, HBn), 4.33 (t, J = 9.6 Hz, 0.45H, H-4a), 4.18–3.81 (m, 5H, H-2a, H-3, H-4b, H-5, H-6), 3.76 (m, 0.55H, H-2b), 1.11 (s, 9H, SiC(C3)3). 13C NMR (100 MHz, CDCl3) δ 143.8–137.9 (4CAr), 135.9–135.6 (4HAr), 133.9–133.1 (2CAr), 129.7–124.1 (25HAr), 124.3 (CF3a), 124.1 (CF3b), 119.5 (HAr), 81.6 (C-3a), 79.7 (C-2b), 79.1 (C-3b), 77.2, 76.9, 75.9 (H2Bna), 75.8, 75.4 (H2Bn), 73.9 (C-4a), 73.4 (H2Bnb), 72.8 (H2Bna), 72.8 (H2Bnb), 62.7 (C-6a), 62.4 (C-6b), 26.9 (SiC(H3)3a), 26.8 (SiC(H3)3b), 19.4 (Si(CH3)3a), 19.4 (Si(CH3)3b) (C-1 and CN could not be detected due to relaxation). HRMS (ESI+): m/z 877.3853 (calcd for C51H56F3N2O6Si [M + NH4]+: m/z 877.3854).
(2,3,4,6-Tetra-O-benzyl-α-D-glucopyranosyl)-(1 → 3)-4-O-benzyl-2-O-levulinoyl-α-L-rhamnopyranosyl (N-phenyl)trifluoroacetimidate (35)
Levulinic acid (9.7 g, 83 mmol, 2.0 equiv.), EDC (15.5 g, 75 mmol, 1.8 equiv.) and DMAP (3.4 g, 17 mmol, 0.4 equiv.) were added to alcohol 24 (34 g, 42 mmol, 1.0 equiv.) in anhyd. DCM (210 mL). The mixture was stirred at rt for 60 h, at which time TLC (tol/EtOAc, 80:20) showed the full consumption of the starting 24 and the presence of a more polar product. The reaction mixture was diluted with water and DCM. The two layers were separated and the aq. phase was extracted with DCM repeatedly. The combined organic layers were washed successively with sat. aq. NaHCO3, water and finally brine. The organic layer was dried over Na2SO4, filtered, and volatiles were evaporated. PdCl2 (621 mg, 2.1 mmol, 0.05 equiv., 60% purity) was added to the crude 3 in DCM/H2O (3:1, 420 mL). The biphasic mixture was stirred at 50 °C for 3 h. TLC (tol/EtOAc, 80:20) showed conversion of the starting 3 into a less polar product. Iodine (10.7 g, 84 mmol, 2.0 equiv.) in THF (50 mL) was added slowly to the solution at rt. After stirring at this temperature for 2.5 h, TLC (tol/EtOAc, 8:2) showed the conversion of the intermediate into a more polar product. Sat. aq. Na2S2O3 was added and the biphasic mixture was filtered over a pad of Celite®. The organic phase was washed with sat. aq. NaHCO3, water and brine. The organic phase was dried over Na2SO4, filtered, and concentrated to dryness. PTFACl (10 mL, 63 mmol, 1.5 equiv.) and K2CO3 (11.6 g, 84 mmol, 2.0 equiv.) were added slowly to the crude hemiacetal in acetone (420 mL) under Ar, at rt. The suspension was stirred at this temperature for 16 h. TLC (cHex/EtOAc, 70:30) showed the consumption of the intermediate 33 and the presence of less polar products. After filtration over a pad of Celite®, thorough washing of the solids with DCM and concentration of the filtrate to dryness, the residue was purified by column chromatography (cHex/EtOAc, 100:0 to 80:20) to give donor 35 (39.5 g, 90% over 3 steps, 9:1 mix of anomers) as an orange oil. The PTFA donor 35 had Rf = 0.8 (tol/EtOAc, 70:30). 1H NMR (400 MHz, CDCl3) δ 7.48–7.25 (m, 23H, HAr), 7.21–7.11 (m, 5H, HAr), 6.99–6.84 (m, 2H, HAr), 6.20 (brs, 0.89H, H-1Aα), 5.95 (brs, 0.11H, H-1Aβ), 5.62 (tapp, J = 3.2 Hz, 1H, H-2A), 5.29 (d, J = 3.5 Hz, 1H, H-1E), 5.06 (d, J = 11.1 Hz, 1H, HBn), 5.03 (d, J = 10.4 Hz, 1H, HBn), 4.95 (d, J = 11.1 Hz, 1H, HBn), 4.92 (d, J = 10.9 Hz, 1H, HBn), 4.83 (d, J = 12.1 Hz, 1H, HBn), 4.77 (d, J = 12.1 Hz, 1H, HBn), 4.69 (d, J = 9.9 Hz, 1H, HBn), 4.65 (d, J = 12.0 Hz, 1H, HBn), 4.55 (d, J = 10.9 Hz, 1H, HBn), 4.44 (d, J = 12.1 Hz, 1H, HBn), 4.33 (dd, J = 9.6, 3.2 Hz, 1H, H-3A), 4.16 (t, J = 9.3 Hz, 1H, H-3E), 4.10 (m, 1H, H-5E), 3.97 (dq, J = 12.2, 6.3 Hz, 1H, H-5A), 3.84 (t, J = 9.5 Hz, 1H, H-4E), 3.69 (m, 3H, H-2E, H-4A, H-6aE), 3.59 (dd, J = 10.9, 2.1 Hz, 1H, H-6bE), 2.69–2.42 (m, 4H, C2Lev), 2.11 (s, 3H, C3Lev), 1.48 (d, J = 6.2 Hz, 3H, H-6A). 13C NMR (100 MHz, CDCl3) δ 205.9 (OLev), 171.8 (O2Lev), 143.3–137.4 (6Ar), 128.8–127.49 (29HAr), 124.5 (F3), 119.5 (HAr), 94.0 (C-1A), 93.4 (C-1E, JC,H = 170.2 Hz), 82.1 (C-3E), 79.4 (C-2E or C-4A), 79.1 (C-2E or C-4A), 77.8 (C-4E), 76.4 (H2Bn), 75.6 (H2Bn), 75.1 (H2Bn), 73.4 (H2Bn), 73.0 (H2Bn), 72.1 (C-3A), 70.7 (C-5A), 70.5 (C-5E), 68.1 (C-6E), 66.8 (C-2A), 37.8 (H2Lev), 29.7 (H3Lev), 28.0 (H2Lev), 18.0 (C-6A). HRMS (ESI+): m/z 1063.4580 (calcd for C60H66F3N2O12 [M + NH4]+: m/z 1063.4568).
Allyl (2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl)-(1 → 3)-(4-O-benzyl-2-O-levulinoyl-α-L-rhamnopyranosyl)-(1 → 2)-(3,4-di-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-(2-O-acetyl-4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-4,6-O-benzylidene-2-deoxy-2-trichloroacetamido-β-D-glucopyranoside (1)17
Hydrazine hydrate (60% in water, 5.0 mL, 103 mmol, 5.0 equiv.) was added dropwise to a solution of the fully protected BAcCD172 (23.8 g, 20.6 mmol, 1.0 equiv.) in pyridine/AcOH (3:2, 410 mL) at 0 °C. The reaction mixture was stirred at rt for 4 h. TLC (tol/EtOAc, 8:2) showed the complete disappearance of the starting material and the presence of a more polar product. The reaction mixture was diluted with water and EtOAc. The two layers were separated, the aq. phase was extracted with EtOAc and the combined organic phases were washed with sat. aq. NaHCO3, water and brine, dried over Na2SO4, filtered and evaporated to dryness. Filtration on a pad of silica (tol/EtOAc, 8:2) afforded the desired compound as a white foam. Activated 4 Å MS (5.0 g) was added to the crude acceptor 31 (20.6 mmol, 1.0 equiv.) and donor 35 (24.8 g, 23.7 mmol, 1.15 equiv.) in anhyd. toluene (410 mL) and the suspension was stirred at rt, under Ar, for 30 min. TMSOTf (750 μL, 4.12 mmol, 0.2 equiv.) was added very slowly at rt. After stirring at rt for 1 h, TLC (tol/acetone, 80:20) showed the complete disappearance of acceptor 31 and the presence of less polar products. Et3N was added and after stirring for 30 min at rt, solids were filtered over a pad of Celite® and washed generously with DCM. Successive purifications by flash column chromatography on silica gel (tol/acetone, 100:0 to 90:10 then tol/EtOAc, 100:0 to 80:20) afforded the desired pentasaccharide 1 as a beige foam (31.5 g, 80% over 2 steps).
Allyl (3,4-di-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-(2-O-acetyl-4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-4,6-O-benzylidene-2-deoxy-2-trichloroacetamido-β-D-glucopyranoside (36)17
:2 v/v, 74 mL) and the resulting solution was stirred at rt under Ar for 1 h. TLC (CH2Cl2/MeCN, 90:10) showed the complete conversion of the starting material into a closely migrating more polar compound. The mixture was partitioned between water (200 mL) and CH2Cl2 (200 mL). The aq. layer was extracted with CH2Cl2 (100 mL twice). The combined organic layers were washed with sat. aq. NaHCO3 (300 mL) and brine (300 mL), dried on Na2SO4, filtered and concentrated under reduced pressure. Flash chromatography on silica gel of the crude (CH2Cl2/MeCN, 95:5 to 85:15) afforded the alcohol 36 (3.6 g, 92%) as a white foam. The latter had Rf = 0.4 (CH2Cl2/MeCN, 90:10).
:20) to give, in order of elution, the remaining 2 (208 mg, 13%) and the acceptor 36 (1.08 g, 74%) as a white foam.
Allyl (2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl)-(1 → 3)-(4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 2)-(3,4-di-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-(2-O-acetyl-4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-4,6-O-benzylidene-2-deoxy-2-trichloroacetamido-β-D-glucopyranoside (37)
Hydrazine hydrate (50 μL, 1.0 mmol, 2 equiv.) was added to the pentasaccharide 1 (1.0 g, 0.52 mmol) in pyridine/AcOH (3:2, 10 mL) and the solution was stirred at rt under Ar for 30 min. TLC (cHex/EtOAc, 70:30) showed the complete conversion of the starting material into a less polar compound. Water (100 mL) and EtOAc (100 mL) were added and the phases were separated. The aqueous layer was extracted with EtOAc (50 mL twice) and the combined organic layers were washed with sat. aq. NaHCO3 and brine, then dried on Na2SO4, filtered, and concentrated under reduced pressure. Flash chromatography of the crude residue (cHex/EtOAc, 90:10 to 50:50) gave alcohol 37 (865 mg, 92%) as a white foam.
:20) to give pentasaccharide 37 (841 mg, 89%) as a white foam. The latter had Rf = 0.3 (tol/EtOAc, 70:30). 1H NMR (400 MHz, CDCl3) δ 7.52–7.12 (m, 45H, HAr), 7.02 (d, J = 7.4 Hz, 1H, NCO), 5.93–5.82 (m, 1H, CCH2), 5.56 (s, 1H, HBzl), 5.34–5.18 (m, 2H, CHC2), 5.16 (dd, J = 3.3, 1.9 Hz, 1H, H-2C), 5.13 (brs, 1H, H-1A), 5.12 (d, J = 8.5 Hz, 1H, H-1D), 4.99–4.81 (m, 8H, H-1B, H-1E, H-1C, 5HBn), 4.78–4.28 (m, 14H, H-3D, H-6bD, C2All, 11HBn), 4.14–4.07 (m, 1H, C2All), 4.07–3.91 (m, 7H, H-3E, H-2A, H-3A, H-2B, H-3C, H-5C, H-5E), 3.86–3.55 (m, 8H, H-5A, H-4A, H-6aD, H-4E, H-5B, H-4B, H-5D, H-2E), 3.53–3.36 (m, 7H, H-3B, H-4D, H-2D, H-6aE, H-6bE), 3.28 (tapp, J = 9.5 Hz, 1H, H-4C), 2.06 (s, 3H, OCOC3), 1.68 (bs, 1H, O), 1.26 (d, J = 6.2 Hz, 3H, H-6A), 1.25 (d, J = 6.2 Hz, 3H, H-6B), 0.73 (d, J = 6.2 Hz, 3H, H-6C). 13C NMR (100 MHz, CDCl3) δ 169.9 (OOCH3), 162.3 (ONH), 133.4 (HCH2), 138.8–137.1 (9Ar), 129.2–126.6 (45CAr), 118.5 (CHH2), 102.1 (CBzl), 101.5 (C-1B, JC,H = 170.7 Hz), 100.8 (C-1A, JC,H = 173.8 Hz), 98.3 (C-1D, JC,H = 168.9 Hz), 97.6 (C-1C, JC,H = 173.5 Hz), 94.1 (C-1E, JC,H = 170.2 Hz), 92.2 (Cl3), 82.6 (C-3E), 80.4 (C-4D), 80.3 (C-4B), 80.1 (C-4C), 79.8 (C-4A), 79.4 (C-3B), 79.0 (C-2E), 78.5 (C-3C), 77.9 (C-4E), 76.6 (C-2B), 75.8 (2C, H2Bn), 75.4 (H2Bn), 75.3 (H2Bn), 75.1 (H2Bn), 75.0 (C-3A), 74.6 (H2Bn), 74.0 (C-3D), 73.6 (H2Bn), 72.5 (H2Bn), 72.3 (C-2C), 71.1 (H2All), 70.9 (C-5E), 69.1 (C-5B), 68.9 (C-6D), 68.1 (C-5C), 68.0 (C-6E), 67.9 (C-5A), 67.4 (C-2A), 66.4 (C-5D), 60.4 (C-2D), 21.2 (OCOH3), 18.0 (2C, C-6A, C-6B), 17.4 (C-6C). HRMS (ESI+): m/z 1831.6810 (calcd for C100H110Cl3NO24NH4 [M + NH4]+: m/z 1831.6822).
(2,3,4,6-Tetra-O-benzyl-α-D-glucopyranosyl)-(1 → 3)-(4-O-benzyl-2-O-levulinoyl-α-L-rhamnopyranosyl)-(1 → 2)-(3,4-di-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-(2-O-acetyl-4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-4,6-O-benzylidene-2-deoxy-2-trichloroacetamido-α/β-D-glucopyranose (38)
1,5-Cyclooctadiene-bis(methyldiphenylphosphine)iridium hexafluorophosphate (62 mg, 73 μmol, 0.02 equiv.) was dissolved in anhyd. THF (1.5 mL) and the resulting red suspension was degassed and filled up with argon. Hydrogen was bubbled through the solution for 10 min, causing the color to change to yellow. The solution was degassed again, saturated with argon, and transferred to a solution of pentasaccharide 1 (7.02 g, 3.67 mmol, 1.0 equiv.) in anhyd. THF (18.3 mL) under argon at rt. The mixture was stirred at rt for 1 h. TLC (tol/EtOAc, 80:20, double migration) showed the complete disappearance of the starting material and the presence of a closely migrating less polar product. Iodine (2.33 g, 18.34 mmol, 5 equiv.) in THF/water (9:1, 24 mL) and solid NaHCO3 (3.08 g, 36.67 mmol, 10 equiv.) were added and the mixture was stirred vigorously at rt for 30 min. TLC (tol/EtOAc, 80:20, cHex/EtOAc, 60:40) showed the complete disappearance of the intermediate and the presence of a more polar product. Excess iodine was destroyed by adding 10% aq. Na2S2O3 until the color was stable. THF was evaporated under reduced pressure and EtOAc (300 mL) was added. The organic layer was washed with sat. NaHCO3, water, and brine, dried on Na2SO4, filtered, and concentrated under reduced pressure. Flash column chromatography of the residue (tol/EtOAc, 90:10 to 70:30) gave hemiacetal 38 (α/β: 90:10, 6.25 g, 92%) as a white foam. The latter had Rf = 0.5 (tol/EtOAc, 80:20). 1H NMR (400 MHz, CDCl3) δ 7.52–7.06 (m, 45H, HAr), 6.94 (d, J = 9.4 Hz, 1H, HNHCO), 5.56 (s, 1H, HBzl), 5.55 (brs, 1H, H-2A), 5.25 (d, J = 3.2 Hz, 1H, H-1E), 5.20 (dd, J = 7.1, 3.4 Hz, 1H, H-1Dα), 5.11 (dd, J = 3.3, 1.9 Hz, 1H, H-2C), 5.04–4.57 (m, 17H, H-1A, H-1B, H-1C, H-3D, 13HBn), 4.50 (d, J = 11.1 Hz, 1H, HBn), 4.44 (d, J = 11.5 Hz, 1H, HBn), 4.38–4.22 (m, 4H, HBn, H-6aD, H-2D, H-3A), 4.17–4.03 (m, 5H, H-5D, H-3E, H-2B, H-3C, H-5E), 4.00–3.46 (m, 14H, H-5C, H-3B, H-4E, H-5A, H-6bD, H-2E, H-4A, H-4B, H-4D, OH, H-5B, H-5D, H-6aE, H-6bE), 3.29 (tapp, J = 9.5 Hz, 1H, H-4C), 2.62–2.35 (m, 4H, C2Lev), 2.08 (s, 3H, C3Lev), 2.07 (s, 3H, OCOC3), 1.29 (d, J = 6.2 Hz, 6H, H-6A, H-6B), 0.78 (d, J = 6.2 Hz, 0.3H, H-6C), 0.74 (d, J = 6.2 Hz, 2.7H, H-6C). 13C NMR (100 MHz, CDCl3) δ 206.4 (OLev), 171.8 (O2Lev), 169.8 (OOCH3), 162.1 (ONH), 138.9–137.1 (9Ar), 129.2–126.5 (45CAr), 102.2 (CBzl), 100.6 (C-1B, JC,H = 170.7 Hz), 99.2 (C-1A, JC,H = 173.8 Hz), 98.1 (C-1C, JC,H = 173.5 Hz), 92.9 (C-1E, JC,H = 170.2 Hz), 92.4 (Cl3), 92.0 (C-1Dα, JC,H = 170.2 Hz), 82.2 (C-3E), 80.4 (2C, C-4D, C-4B), 80.3 (C-4C), 79.8 (C-4A), 79.5 (C-2E), 79.2 (C-3B), 77.8 (C-4E), 77.4 (C-3C), 76.1 (H2Bn), 75.9 (C-2B), 75.6 (H2Bn), 75.4 (2C, C-3D, H2Bn), 75.3 (H2Bn), 75.1 (H2Bn), 75.0 (H2Bn), 73.5 (H2Bn), 72.8 (H2Bn), 72.4 (C-2C), 72.3 (C-3A), 72.1 (H2Bn), 70.3 (C-5E), 69.0 (2C, C-5B, C-6D), 68.7 (C-5A), 68.4 (C-6E), 68.2 (C-2A), 68.1 (C-5C), 63.2 (C-5D), 55.6 (C-2D), 38.0 (H2Lev), 29.8 (H3Lev), 28.2 (H2Lev), 21.1 (OCOH3), 18.0 (C-6A), 17.9 (C-6B), 17.4 (C-6C). HRMS (ESI+): m/z 1891.6873 (calcd for C102H116Cl3N2O26 [M + NH4]+: m/z 1891.6886).
(α-L-Rhamnopyranosyl)-(1 → 3)-4,6-O-benzylidene-2-deoxy-2-trichloroacetamido-α/β-D-glucopyranosyl (N-phenyl)trifluoroacetimidate (39)
To a solution of hemiacetal 37 (7.04 g, 3.76 mmol) in acetone (75 mL) at rt were added sequentially K2CO3 (1.56 g, 11.27 mmol, 3 equiv.) and PTFACl (01.19 mL, 7.51 mmol, 2 equiv.). The suspension was stirred vigorously overnight at rt. TLC (tol/EtOAc, 80:20 or cHex/EtOAc, 70:30) showed the complete disappearance of the starting material and the presence of a less polar product. The suspension was filtered over a pad of Celite (DCM) and the solution was evaporated under reduced pressure. Flash column chromatography of the crude material (cHex/EtOAc, 80:20 to 50:50) gave donor 39 (7.2 g, 94%) as a white foam. The latter had Rf = 0.4 (cHex/EtOAc, 70:30). 1H NMR (400 MHz, CDCl3) δ 7.54–7.08 (m, 48H, HAr), 6.90 (d, J = 9.0 Hz, 1H, HNHCO), 6.82 (d, J = 8.0 Hz, 2H, HAr), 6.44 (brs, 1H, H-1D), 5.61 (s, 1H, HBzl), 5.55 (dd, J = 3.0, 2.0 Hz, 1H, H-2A), 5.25 (d, J = 3.2 Hz, 1H, H-1E), 5.12 (dd, J = 3.3, 1.9 Hz, 1H, H-2C), 5.04–4.57 (m, 16H, H-1A, H-1B, H-1C, 13HBn), 4.53–4.42 (m, 3H, H-2D, 2HBn), 4.39 (dd, J = 10.6, 5.0 Hz, 1H, H-6aD), 4.36 (d, J = 12.0 Hz, 1H, HBn), 4.25 (dd, J = 9.8, 3.0 Hz, 1H, H-3A), 4.18 (tapp, J = 9.5 Hz, 1H, H-3D), 4.14–3.45 (m, 18H, H-5D, H-3E, H-2B, H-3C, H-5E, H-5C, H-3B, H-4E, H-5A, H-6bD, H-2E, H-4A, H-4B, H-4D, H-5B, H-5D, H-6aE, H-6bE), 3.31 (tapp, J = 9.5 Hz, 1H, H-4C), 2.62–2.37 (m, 4H, C2Lev), 2.09 (s, 3H, OCOC3), 2.08 (s, 3H, C3Lev), 1.29 (d, J = 6.2 Hz, 3H, H-6A), 1.26 (d, J = 6.2 Hz, 3H, H-6B), 0.78 (d, J = 6.2 Hz, 3H, H-6C). 13C NMR (100 MHz, CDCl3) δ 206.3 (OLev), 171.8 (O2Lev), 170.1 (OOCH3), 162.4 (ONH), 142.9 (ONPhCF3), 138.9–136.8 (10Ar), 129.4–126.6 (49CAr), 125.0 (OCNPhF3), 119.4 (CAr), 102.4 (CBzl), 101.1 (C-1B, JC,H = 170.7 Hz), 99.3 (C-1A, JC,H = 173.8 Hz), 98.0 (C-1C, JC,H = 173.5 Hz), 93.9 (C-1D, JC,H = 170.2 Hz), 93.0 (C-1E, JC,H = 170.2 Hz), 92.1 (Cl3), 82.3 (C-3E), 80.4 (C-4B), 80.1 (C-4C), 79.9 (C-4A), 79.7 (C-4D), 79.6 (C-2E), 79.4 (C-3B), 77.9 (C-4E), 77.4 (C-3C), 76.3 (H2Bn), 75.7 (2C, C-2B, H2Bn), 75.5 (2C, H2Bn), 75.3 (H2Bn), 75.1 (2C, H2Bn), 74.9 (C-3D), 73.6 (H2Bn), 72.9 (H2Bn), 72.5 (C-2C), 72.4 (C-3A), 72.2 (H2Bn), 70.4 (C-5E), 69.1 (C-5B), 68.8 (C-5A), 68.6 (C-6D), 68.4 (C-6E), 68.5 (C-5C), 68.2 (C-2A), 65.6 (C-5D), 54.9 (C-2D), 38.1 (H2Lev), 29.9 (H3Lev), 28.3 (H2Lev), 21.2 (OCOH3), 18.1 (C-6A), 18.0 (C-6B), 17.5 (C-6C). HRMS (ESI+): m/z 2060.7185 (calcd for C110H120Cl3F3N3O26Na [M + NH4]+: m/z 2060.7178).
Allyl (2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl)-(1 → 3)-(4-O-benzyl-2-O-levulinoyl-α-L-rhamnopyranosyl)-(1 → 2)-(3,4-di-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-(2-O-acetyl-4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-(4,6-O-benzylidene-2-deoxy-2-trichloroacetamido-β-D-glucopyranosyl)-(1 → 2)-[(2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl)-(1 → 3)]-(4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 2)-(3,4-di-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-(2-O-acetyl-4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-4,6-O-benzylidene-2-deoxy-2-trichloroacetamido-β-D-glucopyranoside (40)
To a solution of the glycosyl donor 39 (146 mg, 71 μmol, 1.30 equiv.) and glycosyl acceptor1737 (100 mg, 55 μmol, 1.0 equiv.) in anhyd. toluene (0.6 mL) was added activated 4 Å MS (30 mg) and the suspension was stirred at rt under an argon atmosphere for 15 min, then for 10 min at −50 °C. TMSOTf (2 μL, 11 μmol, 0.2 equiv.) was added rapidly at −50 °C. The reaction mixture was stirred at −40 °C for 30 min, at which time a TLC (cHex/EtOAc, 70:30) follow-up indicated the consumption of the glycosyl acceptor and the presence of a major more polar product. Et3N was added at −40 °C and the suspension was stirred for another 10 min. Solids were filtered over a pad of Celite and washed generously with DCM. The combined filtrates were concentrated to dryness and the residue was purified by flash column chromatography (cHex/EtOAc, 100:0 to 50:50) to give decasaccharide 40 (184 mg, 91%) as a white foam. The latter had Rf = 0.3 (cHex/EtOAc, 70:30). 1H NMR (400 MHz, CDCl3) δ 7.50–7.02 (m, 91H, 90 HAr, HNHCO), 6.94 (d, J = 9.0 Hz, 1H, HNHCO), 5.94–5.83 (m, 1H, CCH2), 5.56 (s, 1H, HBzl), 5.53 (dd, J = 3.0, 1.9 Hz, 1H, H-2A′), 5.33–5.20 (m, 4H, HBzl, H-1E′, CHC2), 5.19–5.14 (m, 2H, H-1E, H-2C), 5.12 (d, J = 8.2 Hz, 1H, H-1D), 5.09 (brs, 1H, H-1B), 5.08–5.04 (m, 3H, H-2C′, 2HBn), 5.01 (brs, 1H, H-1A), 5.01–4.95 (m, 2H, 2HBn), 4.95 (brs, 1H, H-1B′), 4.94–4.78 (m, 9H, H-1A′, H-1C, 7HBn), 4.78–4.46 (m, 20H, H-1C′, H-1D′, H-3D, 17HBn), 4.46–4.28 (m, 6H, H-6aD, C2All, 4HBn), 4.23 (dd, J = 9.7, 2.6 Hz, 1H, H-3A′), 4.19–3.93 (m, 12H, H-3E, C2All, H-2B, H-5E, H-3B, H-3E′, H-3B′, H-5E′, H-2D′, H-3C, H-2A, H-5C), 3.93–3.40 (m, 26H, H-2B′, H-3C′, H-5C′, H-6aD′, H-5A, H-5A′, H-4E, H-4E′, H-2E, H-2E′, H-6bD, H-3A, H-5B, H-5B′, H-4D, H-5D, H-6aE, H-6bE, H-6aE′, H-6bE′, H-4A, H-4A′, H-4B, H-4B′, H-2D), 3.36 (tapp, J = 9.4 Hz, 1H, H-4D′), 3.27 (tapp, J = 9.5 Hz, 1H, H-4C), 3.25 (tapp, J = 9.5 Hz, 1H, H-4C′), 3.13 (tapp, J = 10.0 Hz, 1H, H-6bD′), 2.99–2.83 (m, 2H, H-3D′, H-5D′), 2.60–2.35 (m, 4H, CH2Lev), 2.07 (s, 3H, CH3Lev), 2.06, 2.05 (2s, 6H, HAc), 1.36–1.23 (m, 12H, H-6A, H-6A′, H-6B, H-6B′), 0.73 (d, J = 6.2 Hz, 3H, H-6C′), 0.69 (d, J = 6.2 Hz, 3H, H-6C). 13C NMR (100 MHz, CDCl3) δ 206.3 (COLev), 171.8 (CO2Lev), 170.0, 169.6 (2C, COAc), 162.3, 162.0 (2C, COCl3Ac), 139.0–137.1 (18C, CAr), 133.4 (HCH2), 129.5–126.5 (90C, HAr), 118.5 (CHH2), 102.2, 101.8 (2C, CBzl), 101.4 (C-1B′, JC,H = 171.4 Hz), 101.2 (C-1D′, JC,H = 164.6 Hz), 100.8 (C-1B, JC,H = 176.9 Hz), 100.6 (C-1A, JC,H = 170.7 Hz), 99.4 (C-1A′, JC,H = 173.8 Hz), 98.4 (C-1D, JC,H = 167.0 Hz), 97.7 (2C, C-1C′, C-1C,JC,H = 173.0 Hz), 94.7 (C-1E, JC,H = 168.2 Hz), 93.1 (2C, C-1E′, JC,H = 170.2 Hz, Cl3), 92.3 (Cl3), 83.3 (C-3E), 82.3 (C-3E′), 80.7 (C-4C′), 80.6 (C-4B), 80.4 (C-4D), 80.3 (C-4B′), 80.1 (2C, C-4A, C-4C), 80.0 (C-4D′), 79.9 (C-4A′), 79.7 (C-2E′), 79.6 (C-3A), 79.3 (C-2E), 78.8 (C-4E), 78.5 (C-3C), 77.9 (C-4E′), 77.7 (C-3D′), 77.4 (C-3C′), 76.9 (C-3B′), 76.4 (C-2B′), 76.3 (H2Bn), 76.1 (H2Bn), 75.7 (H2Bn), 75.5 (H2Bn), 75.2 (H2Bn), 75.1 (H2Bn), 75.0 (H2Bn), 74.7 (C-3B), 74.3 (C-2A), 74.0 (C-3D), 73.7 (H2Bn), 73.5 (H2Bn), 73.4(C-2B), 72.9 (H2Bn), 72.6 (C-2C′), 72.5 (C-3A′), 72.3 (C-2C), 72.2 (H2Bn), 71.0 (H2All), 70.4 (C-5E′), 70.2 (C-5E), 69.1 (C-5B′), 69.0 (C-5B), 68.9 (C-6D), 68.8 (C-5A′), 68.5 (C-6E′), 68.3(C-2A′), 68.2(C-6D′), 68.1(C-5C), 68.0 (C-6E), 67.9 (C-5C′), 66.4 (C-5D), 66.1 (C-5D′), 60.5 (C-2D), 57.7 (C-2D′), 38.1 (H2Lev), 29.9 (H3Lev), 28.3 (H2Lev), 21.2 (OCOH3), 21.1 (OCOH3), 18.1–18.0 (4C, C-6A, C-6A′, C-6B, C-6B′), 17.5 (C-6C′), 17.4 (C-6C). HRMS (ESI+): m/z 1854.1809 (calcd for C202H227Cl6N4O49 [M + 2NH4]2+: m/z 1854.1814).
2-Azidoethyl (2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl)-(1 → 3)-(4-O-benzyl-2-O-levulinoyl-α-L-rhamnopyranosyl)-(1 → 2)-(3,4-di-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-(2-O-acetyl-4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-4,6-O-benzylidene-2-deoxy-2-trichloroacetamido-β-D-glucopyranoside (41)
To a solution of 2-azidoethanol (68 μL, 897 μmol, 3.1 equiv.) and glycosyl donor 39 (600 mg, 293 μmol, 1.0 equiv.) in anhyd. toluene (2.9 mL) was added activated 4 Å MS (470 mg) and the suspension was stirred at rt under an argon atmosphere for 15 min, then for 10 min at −50 °C. TMSOTf (21 μL, 0.12 mmol, 0.2 equiv.) was added rapidly at −50 °C. The reaction mixture was stirred at −40 °C for 30 min, at which time a TLC (cHex/EtOAc, 70:30) follow-up showed the consumption of the donor. Et3N was added at this temperature. After stirring for 10 min, solids were filtered over a pad of Celite and washed generously with DCM. The combined filtrates were concentrated to dryness and the residue was purified by flash column chromatography (cHex/EtOAc, 90:10 to 50:50) to give the azidoethyl pentasaccharide 41 (461 mg, 81%) as a white foam. The latter had Rf = 0.25 (cHex/EtOAc, 70:30). 1H NMR (400 MHz, CDCl3) δ 7.52–7.06 (m, 46H, HAr, HNHCO), 5.56 (s, 1H, HBzl), 5.54 (brs, 1H, H-2A), 5.24 (d, J = 3.1 Hz, 1H, H-1E), 5.18 (d, J = 8.5 Hz, 1H, H-1D), 5.17 (dd, J = 3.3, 1.9 Hz, 1H, H-2C), 5.03–4.84 (m, 8H, H-1A, H-1B, H-1C, 5HBn), 4.78 (d, J = 13.1 Hz, 1H, HBn), 4.67 (d, J = 13.1 Hz, 1H, HBn), 4.70–4.32 (m, 11H, H-3D, H-6bD, 9HBn), 4.24 (dd, J = 9.7, 2.6 Hz, 1H, H-3A), 4.15–3.92 (m, 6H, H-2B, H-3C, OC2CH2N3, H-3E, H-5C, H-5E), 3.87–3.70 (m, 5H, H-3B, H-4E, H-5A, H-6aD, OC2CH2N3), 3.70–3.34 (m, 11H, H-2D, H-2E, H-4A, H-4B, H-4D, H-5B, H-5D, H-6aE, H-6bE, C2N3), 3.29 (tapp, J = 9.5 Hz, 1H, H-4C), 2.58–2.41 (m, 4H, C2Lev), 2.08 (s, 3H, C3Lev), 2.06 (s, 3H, OCOC3), 1.28 (d, J = 6.2 Hz, 3H, H-6A), 1.25 (d, J = 6.2 Hz, 3H, H-6B), 0.73 (d, J = 6.2 Hz, 3H, H-6C). 13C NMR (100 MHz, CDCl3) δ 206.2 (OLev), 171.8 (O2Lev), 169.9 (OOCH3), 162.3 (ONH), 138.9–137.1 (9Ar), 129.2–126.5 (45HAr), 102.1 (CBzl), 101.2 (C-1B, JC,H = 170.7 Hz), 99.1 (C-1D, JC,H = 168.9 Hz), 99.1 (C-1A, JC,H = 173.8 Hz), 97.6 (C-1C, JC,H = 173.5 Hz), 92.9 (C-1E, JC,H = 170.2 Hz), 92.2 (Cl3), 82.2 (C-3E), 80.4 (C-4D), 80.3 (C-4B), 80.0 (C-4C), 79.9 (C-4A), 79.5 (C-2E), 79.4 (C-3B), 78.5 (C-3C), 77.8 (C-4E), 76.4 (H2Bn), 75.7 (H2Bn), 75.4 (H2Bn), 75.3 (C-2B), 75.2 (H2Bn), 75.1 (H2Bn), 73.8 (C-3D), 73.5 (H2Bn), 72.9 (H2Bn), 72.3 (C-3A), 72.2 (2C, C-2C, H2Bn), 70.4 (C-5E), 69.1 (C-5B), 69.0 (OH2CH2N3), 68.7 (2C, C-6D, C-5A), 68.4 (C-6E), 68.1 (2C, C-2A, C-5C), 66.4 (C-5D), 60.4 (C-2D), 50.9 (OCH2H2N3), 38.0 (H2Lev), 29.8 (H3Lev), 28.2 (H2Lev), 21.1 (OCOH3), 18.0 (2C, C-6A, C-6B), 17.3 (C-6C). HRMS (ESI+): m/z 1963.6776 (calcd for C104H115Cl3N4O26Na [M + Na]+: m/z 1963.6763).
2-Azidoethyl (2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl)-(1 → 3)-(4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 2)-(3,4-di-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-(2-O-acetyl-4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-4,6-O-benzylidene-2-deoxy-2-trichloroacetamido-β-D-glucopyranoside (42)
Hydrazine hydrate (34 μL, 700 μmol, 2 equiv.) was added to a solution of the fully protected 41 (682 mg, 350 μmol) in pyridine–AcOH (3:2 v/v, 7 mL) and the resulting solution was stirred at rt for 30 min. TLC (cHex/EtOAc, 70:30) showed the complete conversion of the starting material into a less polar compound. Water (100 mL) and EtOAc (100 mL) were added and the phases were separated. The aq. layer was extracted with EtOAc and the combined organic layers were washed with sat. aq. NaHCO3 and brine, dried on Na2SO4, filtered, and concentrated under reduced pressure. Flash chromatography of the crude (tol/EtOAc, 90:10 to 70:30) gave alcohol 42 (589 mg, 92%) as a white foam. The latter had Rf = 0.3 (cHex/EtOAc, 70:30). 1H NMR (400 MHz, CDCl3) δ 7.52–7.06 (m, 46H, HAr, HNHCO), 5.56 (s, 1H, HBzl), 5.17 (d, J = 8.5 Hz, 1H, H-1D), 5.16 (dd, J = 3.3, 1.9 Hz, 1H, H-2C), 5.13 (brs, 1H, H-1A), 4.97 (d, J = 1.9 Hz, 1H, H-1B), 4.99–4.81 (m, 7H, H-1E, H-1C, 5HBn), 4.78–4.28 (m, 13H, H-3D, H-6bD, 11HBn), 4.09–3.91 (m, 8H, H-3E, H-2A, OC2CH2N3, H-3A, H-2B, H-3C, H-5C, H-5E), 3.87–3.56 (m, 9H, H-5A, H-4A, H-6aD, H-4E, OC2CH2N3, H-5B, H-4B, H-5D, H-2E), 3.54–3.33 (m, 7H, H-3B, H-4D, H-2D, H-6aE, H-6bE, C2N3), 3.28 (tapp, J = 9.5 Hz, 1H, H-4C), 1.68 (brs, 1H, OH), 1.26 (d, J = 6.2 Hz, 3H, H-6A), 1.25 (d, J = 6.2 Hz, 3H, H-6B), 0.73 (d, J = 6.2 Hz, 3H, H-6C). 13C NMR (100 MHz, CDCl3) δ 169.9 (OOCH3), 162.3 (ONH), 138.8–137.1 (9Ar), 129.2–127.6 (45HAr), 102.1 (CBzl), 101.4 (C-1B, JC,H = 170.7 Hz), 100.8 (C-1A, JC,H = 173.8 Hz), 99.1 (C-1D, JC,H = 168.9 Hz), 97.5 (C-1C, JC,H = 173.5 Hz), 94.0 (C-1E, JC,H = 170.2 Hz), 92.2 (Cl3), 82.5 (C-3E), 80.4 (C-4D), 80.2 (C-4B), 80.0 (C-4C), 79.7 (C-4A), 79.3 (C-3B), 79.0 (C-2E), 78.4 (C-3C), 77.8 (C-4E), 76.5 (C-2B), 75.7 (2C, H2Bn), 75.3 (H2Bn), 75.2 (H2Bn), 75.0 (2C, C-3A, H2Bn), 74.5 (H2Bn), 73.8 (C-3D), 73.5 (H2Bn), 72.4 (H2Bn), 72.2 (C-2C), 70.8 (C-5E), 69.1 (C-5B), 69.0 (OH2CH2N3), 68.7 (C-6D), 68.1 (C-5C), 68.0 (C-6E), 67.9 (C-5A), 67.4 (C-2A), 66.4 (C-5D), 60.4 (C-2D), 50.9 (OCH2H2N3), 21.1 (OCOH3), 18.0 (2C, C-6A, C-6B), 17.3 (C-6C). HRMS (ESI+): m/z 1865.6378 (calcd for C99H109Cl3N4O24Na [M + Na]+: m/z 1865.6395).
Azidoethyl (2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl)-(1 → 3)-(4-O-benzyl-2-O-levulinoyl-α-L-rhamnopyranosyl)-(1 → 2)-(3,4-di-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-(2-O-acetyl-4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-(4,6-O-benzylidene-2-deoxy-2-trichloroacetamido-β-D-glucopyranosyl)-(1 → 2)-[(2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl)-(1 → 3)]-(4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 2)-(3,4-di-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-(2-O-acetyl-4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-4,6-O-benzylidene-2-deoxy-2-trichloroacetamido-β-D-glucopyranoside (43)
To a solution of donor 39 (1.26 g, 616 μmol, 1.3 equiv.) and acceptor 42 (876 mg, 475 μmol) in anhyd. toluene (5.0 mL) was added activated 4 Å MS (282 mg). The suspension was stirred at rt under an argon atmosphere for 15 min, then for 10 min at −50 °C. TMSOTf (18 μL, 99 μmol, 0.21 equiv.) was added rapidly at −50 °C. The reaction mixture was stirred at −40 °C for 30 min, at which time a TLC (cHex/EtOAc, 70:30) follow-up showed the consumption of the acceptor. Et3N was added at −40 °C and the suspension was stirred for another 10 min. Solids were filtered over a pad of Celite and washed generously with DCM. The combined filtrates were concentrated to dryness and the residue was purified by flash column chromatography (cHex/EtOAc, 100:0 to 50:50) to deliver decasaccharide 43 (1.6 g, 91%) as a white foam. The latter had Rf = 0.3 (cHex/EtOAc, 70:30). 1H NMR (400 MHz, CDCl3) δ 7.50–7.02 (m, 91H, 90 HAr, HNHCOCl3), 6.92 (d, J = 9.0 Hz, 1H, HNHCO), 5.56 (s, 1H, HBzl), 5.53 (dd, J = 3.0, 1.9 Hz, 1H, H-2A′), 5.30 (s, 1H, HBzl), 5.22 (d, J = 3.1 Hz, 1H, H-1E′), 5.20–5.14 (m, 3H, H-1D, H-1E, H-2C), 5.11–5.04 (m, 4H, H-2C′, H-1B, 2HBn,), 5.03–4.27 (m, 38H, H-1B′, H-1A′, H-1A, H-1C, H-1C′, H-1D′, H-3D, H-6bD, 30HBn), 4.23 (dd, J = 9.7, 2.6 Hz, 1H, H-3A′), 4.19–3.93 (m, 12H, H-3E, H-2B, H-5E, H-3B, OC2CH2N3, H-3E′, H-3B′, H-5E′, H-2D′, H-3C, H-2A, H-5C), 3.93–3.33 (m, 30H, H-2B′, H-3C′, H-5C′, H-6aD′, H-5A, H-5A′, H-4E, H-4E′, H-2E, H-2E′, H-6aD, OC2CH2N3, H-3A, H-5B, H-5B′, H-4D, H-5D, H-6aE, H-6bE, H-6aE′, H-6bE′, C2N3, H-4A, H-4A′, H-4B, H-4B′, H-2D, H-4D′), 3.27 (tapp, J = 9.5 Hz, 1H, H-4C), 3.25 (tapp, J = 9.5 Hz, 1H, H-4C′), 3.13 (tapp, J = 10.0 Hz, 1H, H-6bD′), 2.99–2.83 (m, 2H, H-3D′, H-5D′), 2.60–2.35 (m, 4H, CH2Lev), 2.07 (s, 3H, CH3Lev), 2.06, 2.05 (2s, 6H, HAc), 1.36–1.23 (m, 12H, H-6A, H-6A′, H-6B, H-6B′), 0.73 (d, J = 6.2 Hz, 3H, H-6C′), 0.69 (d, J = 6.2 Hz, 3H, H-6C). 13C NMR (100 MHz, CDCl3) δ 206.3 (COLev), 171.8 (CO2Lev), 170.0, 169.6 (2C, COAc), 162.5, 162.0 (2C, COCl3Ac), 139.0–137.1 (18C, CAr), 129.5–126.5 (90C, HAr), 102.2, 101.8 (2C, CBzl), 101.5 (C-1B′, JC,H = 171.4 Hz), 101.2 (C-1D′, JC,H = 164.6 Hz), 100.8 (C-1B, JC,H = 176.9 Hz), 100.6 (C-1A, JC,H = 170.7 Hz), 99.4 (C-1A′, JC,H = 173.8 Hz), 99.2 (C-1D, JC,H = 165.0 Hz), 97.7 (2C, C-1C′, C-1C,JC,H = 173.0 Hz), 94.6 (C-1E, JC,H = 168.2 Hz), 93.1 (2C, C-1E′, JC,H = 170.2 Hz, Cl3), 92.3 (Cl3), 83.3 (C-3E), 82.3 (C-3E′), 80.7 (C-4C′), 80.6 (C-4B), 80.3 (2C, C-4D, C-4B′), 80.1 (C-4A), 80.0 (2C, C-4C, C-4D′), 79.9 (C-4A′), 79.7 (C-2E′), 79.6 (C-3A), 79.3 (C-2E), 78.8 (C-4E), 78.6 (C-3C), 77.9 (C-4E′), 77.7 (C-3D′), 77.4 (C-3C′), 76.9 (C-3B′), 76.4 (C-2B′), 76.3 (H2Bn), 76.1 (H2Bn), 75.7 (H2Bn), 75.5 (H2Bn), 75.2 (H2Bn), 75.1 (H2Bn), 75.0 (H2Bn), 74.6 (C-3B), 74.4 (C-2A), 73.9 (C-3D), 73.7 (H2Bn), 73.5 (H2Bn), 73.4(C-2B), 72.9 (H2Bn), 72.6 (C-2C′), 72.5 (C-3A′), 72.3 (C-2C), 72.2 (H2Bn), 70.3 (C-5E′), 70.2 (C-5E), 69.1 (C-5B′), 69.0 (3C, OH2CH2N3, C-6D, C-5B), 68.8 (C-5A′), 68.5 (C-6E′), 68.3 (C-2A′), 68.2 (C-6D′), 68.1 (C-5C), 68.0 (C-6E), 67.9 (C-5C′), 66.5 (C-5D), 66.1 (C-5D′), 60.5 (C-2D), 57.7 (C-2D′), 50.9 (OCH2H2N3), 38.1 (H2Lev), 29.9 (H3Lev), 28.3 (H2Lev), 21.2 (OCOH3), 21.1 (OCOH3), 18.1–18.0 (4C, C-6A, C-6A′, C-6B, C-6B′), 17.5 (C-6C′), 17.4 (C-6C). HRMS (ESI+): m/z 1868.6818 (calcd for C201H227Cl6N7O49 [M + 2NH4]2+: m/z 1868.6820).
Azidoethyl (2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl)-(1 → 3)-(4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 2)-(3,4-di-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-(2-O-acetyl-4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-(4,6-O-benzylidene-2-deoxy-2-trichloroacetamido-β-D-glucopyranosyl)-(1 → 2)-[(2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl)-(1 → 3)]-(4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 2)-(3,4-di-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-(2-O-acetyl-4-O-benzyl-α-L-rhamnopyranosyl)-(1 → 3)-4,6-O-benzylidene-2-deoxy-2-trichloroacetamido-β-D-glucopyranoside (44)
:2 v/v, 7.6 mL) and the solution was stirred at rt for 1 h. TLC (cHex/EtOAc, 70:30) showed the complete conversion of the starting material into a closely migrating less polar compound. The mixture was partitioned between water (200 mL) and EtOAc (200 mL). The aq. layer was extracted with EtOAc (100 mL twice). The combined organic layers were washed with sat. aq. NaHCO3 (300 mL) and brine (300 mL), dried on Na2SO4, filtered, and concentrated under reduced pressure. Flash column chromatography of the crude (cHex/EtOAc, 90:10 to 50:50) gave alcohol 44 (1.24 g, 91%) as a white foam. The latter had Rf = 0.3 (cHex/EtOAc, 70:30). 1H NMR (400 MHz, CDCl3) δ 7.50–7.02 (m, 91H, 90 HAr, HNHCO), 6.92 (d, J = 9.0 Hz, 1H, HNHCO), 5.55 (s, 1H, HBzl), 5.31 (s, 1H, HBzl), 5.20–5.14 (m, 3H, H-1D, H-1E, H-2C), 5.11–5.04 (m, 5H, H-1A′, H-2C′, H-1B, 2HBn,), 5.03 (brs, 1H, H-1A), 5.01–4.23 (m, 38H, H-1B′, H-1E′, H-1C, H-1C′, H-1D′, H-3D, H-6bD, H-3A′, 30HBn), 4.18–3.55 (m, 32H, H-2A′, H-3E, H-2B, H-5E, OC2CH2N3, H-3E′, H-3B′, H-5E′, H-2D′, H-3C, H-2A, H-5C, H-2B′, H-3C′, H-5C′, H-6aD′, H-5A, H-5A′, H-4E, H-4E′, H-2E, H-2E′, H-6aD, OC2CH2N3, H-3A, H-5B, H-5B′, H-4B′, H-3B), 3.52–3.32 (m, 12H, H-4D, H-5D, H-6aE, H-6bE, H-6aE′, H-6bE′, C2N3, H-4A, H-4A′, H-4B, H-2D, H-4D′), 3.27 (tapp, J = 9.5 Hz, 1H, H-4C), 3.25 (tapp, J = 9.5 Hz, 1H, H-4C′), 3.13 (tapp, J = 10.0 Hz, 1H, H-6bD′), 2.99–2.83 (m, 2H, H-3D′, H-5D′), 2.06, 2.05 (2s, 6H, HAc), 1.35 (d, J = 6.2 Hz, 3H, H-6A′), 1.29–1.23 (m, 9H, H-6A, H-6B, H-6B′), 0.72 (d, J = 6.2 Hz, 3H, H-6C′), 0.69 (d, J = 6.2 Hz, 3H, H-6C). 13C NMR (100 MHz, CDCl3) δ 170.0, 169.6 (2C, COAc), 162.5, 162.0 (2C, COCl3Ac), 139.0–137.1 (18C, CAr–C), 129.5–126.5 (90C, HAr), 102.2, 101.8 (2C, CBzl), 101.5 (C-1B′, JC,H = 170.7 Hz), 101.3 (C-1D′, JC,H = 164.6 Hz), 101.2 (C-1A′, JC,H = 173.8 Hz), 100.8 (2C, C-1B, JC,H = 176.9 Hz, C-1A, JC,H = 170.7 Hz), 99.2 (C-1D, JC,H = 165.0 Hz), 97.7 (2C, C-1C′, C-1C,JC,H = 173.0 Hz), 94.7 (C-1E, JC,H = 168.2 Hz), 94.2 (C-1E′, JC,H = 170.2 Hz), 93.1 (Cl3), 92.3 (Cl3), 83.3 (C-3E), 82.6 (C-3E′), 80.8 (C-4C′), 80.6 (C-4B), 80.3 (2C, C-4D, C-4B′), 80.1 (C-4A), 80.0 (2C, C-4C, C-4D′), 79.9 (C-4A′), 79.6 (C-2E′), 79.5 (C-3A), 79.1 (C-2E), 78.8 (C-4E), 78.6 (C-3C), 77.9 (C-4E′), 77.6 (C-3D′), 77.4 (C-3C′), 76.8 (C-3B′), 76.7 (C-2B′), 76.3 (H2Bn), 76.1 (H2Bn), 75.7 (H2Bn), 75.5 (H2Bn), 75.2 (H2Bn), 75.1 (H2Bn), 75.0 (H2Bn), 74.6 (C-3B), 74.4 (C-2A), 73.9 (C-3D), 73.7 (H2Bn), 73.5 (H2Bn), 73.4(C-2B), 72.7 (C-2C′), 72.5 (H2Bn), 72.3 (C-3A′), 72.3 (C-2C), 72.2 (H2Bn), 70.8 (C-5E′), 70.2 (C-5E), 69.1 (C-5B′), 69.0 (3C, OH2CH2N3, C-5C′, C-5B), 68.8 (C-6D), 68.3 (C-6D′), 68.2 (C-5C), 68.0 (C-6E′, C-6E), 67.9 (C-5A′), 67.6 (C-2A′), 66.5 (C-5D), 66.1 (C-5D′), 60.5 (C-2D), 57.7 (C-2D′), 50.9 (OCH2H2N3), 21.2 (OCOH3), 21.1 (OCOH3), 18.1–18.0 (4C, C-6A, C-6A′, C-6B, C-6B′), 17.5 (C-6C′), 17.4 (C-6C).
2-Aminoethyl α-D-glucopyranosyl-(1 → 3)-α-L-rhamnopyranosyl-(1 → 2)-α-L-rhamnopyranosyl-(1 → 3)-(2-O-acetyl-α-L-rhamnopyranosyl)-(1 → 3)-2-acetamido-2-deoxy-β-D-glucopyranoside (45)
A solution of alcohol 42 (255 mg, 138 μmol) in tBuOH/DCM/H2O (7:2:1, 25 mL) was degassed repeatedly. Next, 20% Pd(OH)2/C (255 mg) was added and the suspension was stirred vigorously overnight under a hydrogen atmosphere. Analytical RP-HPLC indicated the presence of the desired 45 as the only sugar detected. The suspension was centrifuged and the supernatant was passed through a PVDF membrane (0.2 μm, 25 mm). The residue was suspended in tBuOH/H2O (1:4, 5.0 mL) and centrifuged (5000 min−1). The supernatant was passed through a PVDF membrane (0.2 μm). The procedure was repeated three times. The combined filtrates were freeze-dried and the residue was filtered through a Sep-Pak C18 cartridge eluting first with 0.08% aq. TFA then with 20% MeCN in 0.08% aq. TFA. The suitable fractions were pooled, freeze-dried and the residue was purified by RP-HPLC to give pentasaccharide 45 (85 mg, 70%) as a white powder. The linker-equipped 45 had RP-HPLC (λ = 215 nm): tR = 10.46 min 1H NMR (400 MHz, D2O) δ 5.12 (brs, 1H, H-1B), 5.09 (d, J = 3.9 Hz, H-1E), 5.02–4.97 (m, 2H, H-2C, H-1A), 4.87 (brs, 1H, H-1C), 4.55 (d, J = 8.5 Hz, 1H, H-1D), 4.26 (tapp, J = 2.4 Hz, 1H, H-2A), 4.12–3.99 (m, 3H, H-5C, H-2B, OC2CH2NH2), 3.98–3.86 (m, 4H, H-5E, H-3C, H-6aD, OC2CH2NH2), 3.86–3.68 (m, 8H, H-3A, H-2D, H-3E, H-6aE, H-6bE, H-6bD, H-5A, H-3B), 3.63–3.41 (m, 9H, H-2E, H-4C, H-5B, H-4A, H-3D, H-4D, H-4E, H-4B, H-5D), 3.26–3.13 (m, 2H, C2NH2), 2.16 (s, 3H, HAc), 2.05 (s, 3H, HNAc), 1.30–1.21 (m, 9H, H-6A, H-6B, H-6C). 13C NMR (100 MHz, D2O) δ 177.4 (CONAc), 175.6 (COAc), 104.6 (C-1A, JC,H = 173.4 Hz), 103.7 (C-1B, JC,H = 172.4 Hz,), 103.1 (C-1D, JC,H = 162.7 Hz), 101.2 (C-1C, JC,H = 173.5 Hz), 98.0 (C-1E, JC,H = 170.3 Hz), 84.9 (C-3D), 80.8 (C-2B), 78.8 (C-3C), 78.6 (C-5D), 77.9 (C-3A), 75.6 (C-3E), 74.9 (C-2C), 74.5 (C-4B), 74.3 (C-2E, C-4C), 74.1 (C-5E), 72.9 (C-4A), 72.6 (C-3B), 72.1 (2C, C-5A, C-5B), 71.9 (C-4E), 71.6 (C-5C), 70.9 (C-4D), 69.3 (C-2A), 68.3 (OH2CH2NH2), 63.3 (C-6D), 63.0 (C-6E), 57.7 (C-2D), 42.1 (CH2NH2), 24.9 (CNAc), 22.9 (CAc), 19.4 (C-6A), 19.3 (C-6B), 18.9 (C-6C). HRMS (ESI+): m/z 930.3611 (calcd for C36H62N2O24Na [M + Na]+: m/z 930.3624).
2-Aminoethyl α-D-glucopyranosyl-(1 → 3)-α-L-rhamnopyranosyl-(1 → 2)-α-L-rhamnopyranosyl-(1 → 3)-(2-O-acetyl-α-L-rhamnopyranosyl)-(1 → 3)-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-(1 → 2)-[α-D-glucopyranosyl-(1 → 3)]-α-L-rhamnopyranosyl-(1 → 2)-α-L-rhamnopyranosyl-(1 → 3)-(2-O-acetyl-α-L-rhamnopyranosyl)-(1 → 3)-(2-acetamido-2-deoxy-β-D-glucopyranosyl)-(1 → 2)-[α-D-glucopyranosyl-(1 → 3)]-α-L-rhamnopyranosyl-(1 → 2)-α-L-rhamnopyranosyl-(1 → 3)-(2-O-acetyl-α-L-rhamnopyranosyl)-(1 → 3)-2-acetamido-2-deoxy-β-D-glucopyranoside (46)
Alcohol 44 (50 mg, 14 μmol) was dissolved in tBuOH/DCM/H2O (7:2:1, 5 mL) and the solution was degassed repeatedly. Then, 20 wt% Pd(OH)2/C (50 mg) was added and the suspension was stirred vigorously overnight under a hydrogen atmosphere. After 12 h, analytical RP-HPLC indicated the presence of several products corresponding to diversely N-chloroacetylated analogues of the desired 46. Et3N (4 equiv.) was added, the suspension was centrifuged and the supernatant was passed through a PVDF membrane (0.2 μm, 25 mm). The residue was suspended in tBuOH/H2O (1:4, 5.0 mL) and centrifuged (5000 min−1). The supernatant was passed through a PVDF membrane (0.2 μm). The procedure was repeated three times. The combined filtrates were freeze-dried. The residue obtained was dissolved in tBuOH/DCM/H2O (7:2:1, 5.0 mL) and the solution was degassed repeatedly. Then, 20% Pd(OH)2/C (100 mg) was added and the suspension was stirred vigorously overnight under a hydrogen atmosphere for another 2 days with analytical RP-HPLC follow-up. The suspension was centrifuged and the supernatant was passed through a PVDF membrane (0.2 μm). The residue was suspended in tBuOH/H2O (1:4, 5.0 mL) and centrifuged (5000 min−1). The supernatant was passed through a PVDF membrane (0.2 μm). This was repeated three times. The combined filtrates were concentrated by freeze-drying. The residue was dissolved in 0.5 mL H2O and passed through a Sep-Pak C18 cartridge, eluting first with 0.08% aq. TFA then with 20% CH3CN in 0.08% aq. TFA. Suitable fractions were pooled, freeze-dried and the residue was purified by RP-HPLC to give decasaccharide 46 (13 mg, 52%) as a white powder. The linker-equipped 46 had RP-HPLC (λ = 215 nm): tR = 11.73 min 1H NMR (400 MHz, D2O) δ 5.16 (d, J = 3.6 Hz, 1H, H-1E), 5.12 (brs, 1H, H-1B′), 5.11 (brs, 1H, H-1B), 5.09 (d, J = 3.9 Hz, H-1E′), 5.08 (d, J = 1.7 Hz, 1H, H-1A), 5.01–4.96 (m, 3H, H-2C, H-2C′, H-1A′), 4.88 (brs, 1H, H-1C′), 4.87 (brs, 1H, H-1C), 4.79 (d, J = 8.5 Hz, 1H, H-1D′), 4.55 (d, J = 8.5 Hz, 1H, H-1D), 4.42 (tapp, J = 2.2 Hz, 1H, H-2A), 4.26 (tapp, J = 2.4 Hz, 1H, H-2A′), 4.12–3.99 (m, 6H, H-5C, H-5C′, H-5E, H-2B, H-2B′, OC2CH2NH2), 3.98–3.75 (m, 16H, H-5E′, H-3C, H-3C′, H-3A, H-6aD, H-6aD′, OC2CH2NH2, H-3A′, H-2D, H-2D′, H-3E, H-3E′, H-6aE, H-6aE′, H-6bE, H-6bE′), 3.75–3.65 (m, 7H, H-6bD, H-6bD′, H-5A′, H-3B, H-3B′, H-5A, H-2E), 3.62–3.49 (m, 9H, H-2E′, H-4C, H-4C′, H-5B, H-5B′, H-4A′, H-3D, H-4D, H-4D′), 3.50–3.36 (m, 7H, H-4E, H-4E′, H-4B, H-4B′, H-3D′, H-5D, H-5D′), 3.33 (tapp, J = 9.8 Hz, 1H, H-4A), 3.26–3.13 (m, 2H, C2NH2), 2.16 (s, 6H, HAc), 2.09 (s, 3H, HNCO), 2.04 (s, 3H, HNAc), 1.30–1.21 (m, 18H, H-6A, H-6A′, H-6B, H-6B′, H-6C, H-6C′). 13C NMR (100 MHz, D2O) δ 177.0 (2C, CONCO), 175.7 (COAc), 175.6 (COAc), 104.6 (C-1A′, JC,H = 173.4 Hz), 104.2 (C-1D′, JC,H = 164.1 Hz), 103.8 (C-1A, JC,H = 175.2 Hz), 103.6 (2C, C-1B, C-1B′, JC,H = 174.8 Hz), 103.2 (C-1D, JC,H = 162.0 Hz), 101.2 (2C, C-1C, C-1C′, JC,H = 173.5 Hz), 98.0 (C-1E′, JC,H = 173.4 Hz), 97.0 (C-1E, JC,H = 170.7 Hz), 85.2 (C-3D′), 84.8 (C-3D), 80.9, 80.8 (2C, C-2B, C-2B′), 78.8, 78.6 (4C, C-3C, C-3C′, C-5D, C-5D′), 77.9 (C-3A′), 76.8 (C-2A), 76.1 (C-3A), 75.8 (C-3E), 75.6 (C-3E′), 74.9 (2C, C-2C, C-2C′), 74.5 (2C, C-4B, C-4B′), 74.3 (4C, C-5E′, C-2E′, C-4C, C-4C′), 74.1, 73.9 (2C, C-5E, C-2E), 73.5 (C-4A), 72.9 (C-4A′), 72.6, 72.5 (2C, C-3B, C-3B′), 72.1, 72.0 (6C, C-5A, C-5A′, C-5B, C-5B′, C-4E, C-4E′), 71.6, 71.5 (2C, C-5C, C-5C′), 71.0, 70.8 (2C, C-4D, C-4D′), 69.3 (C-2A′), 68.3 (OH2CH2NH2), 63.3 (2C, C-6D, C-6D′), 63.0 (2C, C-6E, C-6E′), 58.0, 57.7 (2C, C-2D, C-2D′) 42.1 (CH2NH2), 25.4 (CNAc), 24.9 (CNAc), 22.8 (CAc), 22.8 (2C, CAc), 19.5, 19.4, 19.2, 18.9 (6C, C-6A, C-6A′, C-6B, C-6B′, C-6C, C-6C′). HRMS (ESI+): m/z 1752.6962 (calcd for C70H117N3O47Na [M + H]+: m/z 1753.6969).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Institut Pasteur under grant agreement GPH-FLEXBIVAC (postdoctoral fellowship to J.C.) and by the European Union's seventh Framework Program for research, technological development, and demonstration under grant agreement no 261462-STOPENTERICS (postdoctoral fellowship to Z.H.). The authors would like to acknowledge Ms Catherine Guerreiro (Chemistry of Biomolecules) for her excellent advice with regard to RP-HPLC. They also thank Mr Frédéric Bonhomme (UMR3523 CNRS) for the HRMS spectra.Notes and references
- L. Krasnova and C.-H. Wong, Oligosaccharide synthesis and translational innovation, J. Am. Chem. Soc., 2019, 141, 3735–3754 CrossRef CAS PubMed.
- S. S. Kulkarni, C.-C. Wang, N. M. Sabbavarapu, A. R. Podilapu, P.-H. Liao and S.-C. Hung, “One-pot” protection, glycosylation, and protection–glycosylation strategies of carbohydrates, Chem. Rev., 2018, 118, 8025–8104 CrossRef CAS PubMed.
- M. Panza, S. G. Pistorio, K. J. Stine and A. V. Demchenko, Automated chemical oligosaccharide synthesis: Novel approach to traditional challenges, Chem. Rev., 2018, 118, 8105–8150 CrossRef CAS PubMed.
- C.-W. Cheng, Y. Zhou, W.-H. Pan, S. Dey, C.-Y. Wu, W.-L. Hsu and C.-H. Wong, Hierarchical and programmable one-pot synthesis of oligosaccharides, Nat. Commun., 2018, 9, 5202 CrossRef CAS PubMed.
- M. Guberman and P. H. Seeberger, Automated glycan assembly: A perspective, J. Am. Chem. Soc., 2019, 141, 5581–5592 CrossRef CAS PubMed.
- O. Calin, S. Eller and P. H. Seeberger, Automated polysaccharide synthesis: assembly of a 30mer mannoside, Angew. Chem., Int. Ed., 2013, 52, 5862–5865 CrossRef CAS PubMed.
- K. Naresh, F. Schumacher, H. S. Hahm and P. H. Seeberger, Pushing the limits of automated glycan assembly: synthesis of a 50mer polymannoside, Chem. Commun., 2017, 53, 9085–9088 RSC.
- Y. Zhang, H. He, Z. Chen, Y. Huang, G. Xiang, P. Li, X. Yang, G. Lu and G. Xiao, Merging reagent modulation and remote anchimeric assistance for glycosylation: highly stereoselective synthesis of α-glycans up to a 30-mer, Angew. Chem., 2021, 60, 12597–12606 CrossRef CAS PubMed.
- Y. Wu, D. C. Xiong, S. C. Chen, Y. S. Wang and X. S. Ye, Total synthesis of mycobacterial arabinogalactan containing 92 monosaccharide units, Nat. Commun., 2017, 8, 14851 CrossRef CAS PubMed.
- Q. Zhu, Z. Shen, F. Chiodo, S. Nicolardi, A. Molinaro, A. Silipo and B. Yu, Chemical synthesis of glycans up to a 128-mer relevant to the O-antigen of Bacteroides vulgatus, Nat. Commun., 2020, 11, 4142 CrossRef CAS PubMed.
- A. A. Joseph, A. Pardo-Vargas and P. H. Seeberger, Total synthesis of polysaccharides by automated glycan assembly, J. Am. Chem. Soc., 2020, 142, 8561–8564 CrossRef CAS PubMed.
- V. Pozsgay, A new strategy in oligosaccharide synthesis using lipophilic protecting groups: synthesis of a tetracosasaccharide, Tetrahedron: Asymmetry, 2000, 11, 151–172 CrossRef CAS.
- B. Fraser-Reid, J. Lu, K. N. Jayaprakash and J. C. López, Synthesis of a 28-mer oligosaccharide core of mycobacterial lipoarabinomannan (LAM) requires only two n-pentenyl orthoester progenitors, Tetrahedron: Asymmetry, 2006, 17, 2449–2463 CrossRef CAS.
- M. Joe, Y. Bai, R. C. Nacario and T. L. Lowary, Synthesis of the docosanasaccharide arabinan domain of mycobacterial arabinogalactan and a proposed octadecasaccharide biosynthetic precursor, J. Am. Chem. Soc., 2007, 129, 9885–9901 CrossRef CAS PubMed.
- A. Ishiwata and Y. Ito, Synthesis of docosasaccharide arabinan motif of mycobacterial cell wall, J. Am. Chem. Soc., 2011, 133, 2275–2291 CrossRef CAS PubMed.
- S. U. Hansen, G. J. Miller, M. J. Cliff, G. C. Jayson and J. M. Gardiner, Making the longest sugars: a chemical synthesis of heparin-related [4]n oligosaccharides from 16-mer to 40-mer, Chem. Sci., 2015, 6, 6158–6164 RSC.
- Z. Y. Hu, A. F. B. White and L. A. Mulard, Efficient iterative synthesis of O-acetylated tri- to pentadecasaccharides related to the lipopolysaccharide of Shigella flexneri type 3a through di- and trisaccharide glycosyl donors, Chem. – Asian J., 2017, 12, 419–439 CrossRef CAS PubMed.
- S. A. Thadke, B. Mishra, M. Islam, S. Pasari, S. Manmode, B. V. Rao, M. Neralkar, G. P. Shinde, G. Walke and S. Hotha, [Au]/[Ag]-Catalysed expedient synthesis of branched heneicosafuranosyl arabinogalactan motif of Mycobacterium tuberculosis cell wall, Nat. Commun., 2017, 8, 14019 CrossRef CAS PubMed.
- S. Pasari, S. Manmode, G. Walke and S. Hotha, A Versatile Synthesis of Pentacosafuranoside Subunit Reminiscent of Mycobacterial Arabinogalactan Employing One Strategic Glycosidation Protocol, Chem. – Eur. J., 2018, 24, 1128–1139 CrossRef CAS PubMed.
- J. Y. Baek, A. Geissner, D. C. K. Rathwell, D. Meierhofer, C. L. Pereira and P. H. Seeberger, A modular synthetic route to size-defined immunogenic Haemophilus influenzae b antigens is key to the identification of an octasaccharide lead vaccine candidate, Chem. Sci., 2018, 9, 1279–1288 RSC.
- V. Verez-Bencomo, V. Fernandez-Santana, E. Hardy, M. E. Toledo, M. C. Rodriguez, L. Heynngnezz, A. Rodriguez, A. Baly, L. Herrera, M. Izquierdo, A. Villar, Y. Valdes, K. Cosme, M. L. Deler, M. Montane, E. Garcia, A. Ramos, A. Aguilar, E. Medina, G. Torano, I. Sosa, I. Hernandez, R. Martinez, A. Muzachio, A. Carmenates, L. Costa, F. Cardoso, C. Campa, M. Diaz and R. Roy, A synthetic conjugate polysaccharide vaccine against Haemophilus influenzae type b, Science, 2004, 305, 522–525 CrossRef CAS PubMed.
- P. A. Driguez, P. Potier and P. Trouilleux, Synthetic oligosaccharides as active pharmaceutical ingredients: Lessons learned from the full synthesis of one heparin derivative on a large scale, Nat. Prod. Rep., 2014, 31, 980–989 RSC.
- J. Liu, J. A. Platts-Mills, J. Juma, F. Kabir, J. Nkeze, C. Okoi, D. J. Operario, J. Uddin, S. Ahmed, P. L. Alonso, M. Antonio, S. M. Becker, W. C. Blackwelder, R. F. Breiman, A. S. Faruque, B. Fields, J. Gratz, R. Haque, A. Hossain, M. J. Hossain, S. Jarju, F. Qamar, N. T. Iqbal, B. Kwambana, I. Mandomando, T. L. McMurry, C. Ochieng, J. B. Ochieng, M. Ochieng, C. Onyango, S. Panchalingam, A. Kalam, F. Aziz, S. Qureshi, T. Ramamurthy, J. H. Roberts, D. Saha, S. O. Sow, S. E. Stroup, D. Sur, B. Tamboura, M. Taniuchi, S. M. Tennant, D. Toema, Y. Wu, A. Zaidi, J. P. Nataro, K. L. Kotloff, M. M. Levine and E. R. Houpt, Use of quantitative molecular diagnostic methods to identify causes of diarrhoea in children: a reanalysis of the GEMS case-control study, Lancet, 2016, 388, 1291–1301 CrossRef CAS.
- K. L. Kotloff, M. S. Riddle, J. A. Platts-Mills, P. Pavlinac and A. K. M. Zaidi, Shigellosis, Lancet, 2018, 391, 801–812 CrossRef.
- M. M. Levine, D. Nasrin, S. Acácio, Q. Bassat, H. Powell, S. M. Tennant, S. O. Sow, D. Sur, A. K. M. Zaidi, A. S. G. Faruque, M. J. Hossain, P. L. Alonso, R. F. Breiman, C. E. O'Reilly, E. D. Mintz, R. Omore, J. B. Ochieng, J. O. Oundo, B. Tamboura, D. Sanogo, U. Onwuchekwa, B. Manna, T. Ramamurthy, S. Kanungo, S. Ahmed, S. Qureshi, F. Quadri, A. Hossain, S. K. Das, M. Antonio, D. Saha, I. Mandomando, W. C. Blackwelder, T. Farag, Y. Wu, E. R. Houpt, J. J. Verweiij, H. Sommerfelt, J. P. Nataro, R. M. Robins-Browne and K. L. Kotloff, Diarrhoeal disease and subsequent risk of death in infants and children residing in low-income and middle-income countries: analysis of the GEMS case-control study and 12-month GEMS-1A follow-on study, Lancet Glob. Health, 2020, 8, e204–e214 CrossRef PubMed.
- L. A. Barel and L. A. Mulard, Classical and novel strategies to develop a Shigella glycoconjugate vaccine: from concept to efficacy in human, Hum. Vaccines Immunother., 2019, 15, 1338–1356 CrossRef PubMed.
- A. Phalipon, M. Tanguy, C. Grandjean, C. Guerreiro, F. Belot, D. Cohen, P. J. Sansonetti and L. A. Mulard, A synthetic carbohydrate-protein conjugate vaccine candidate against Shigella flexneri, 2a infection, J. Immunol., 2009, 182, 2241–2247 CrossRef CAS PubMed.
- P. Kaplonek, N. Khan, K. Reppe, B. Schumann, M. Emmadi, M. P. Lisboa, F. F. Xu, A. D. J. Calow, S. G. Parameswarappa and M. Witzenrath, Improving vaccines against Streptococcus pneumoniae using synthetic glycans, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 13353–13358 CrossRef CAS PubMed.
- F. Belot, C. Guerreiro, F. Baleux and L. A. Mulard, Synthesis of two linear PADRE conjugates bearing a deca- or pentadecasaccharide B epitope as potential synthetic vaccines against Shigella flexneri, serotype 2a infection, Chem. – Eur. J., 2005, 11, 1625–1635 CrossRef CAS PubMed.
- A. V. Perepelov, M. E. Shekht, B. Liu, S. D. Shevelev, V. A. Ledov, S. y. N. Senchenkova, V. L. L'Vov, A. S. Shashkov, L. Feng, P. G. Aparin, L. Wang and Y. A. Knirel, Shigella flexneri, O-antigens revisited: final elucidation of the O-acetylation profiles and a survey of the O-antigen structure diversity, FEMS Immunol. Med. Microbiol., 2012, 66, 201–210 CrossRef CAS PubMed.
- D. Cohen, J. Atsmon, C. Artaud, S. Meron-Sudai, M.-L. Gougeon, A. Bialik, S. Goren, V. Asato, O. Ariel-Cohen, A. Reizis, A. Dorman, C. W. G. Hoitink, J. Westdijk, S. Ashkenazi, P. J. Sansonetti, L. A. Mulard and A. Phalipon, Safety and immunogenicity of a synthetic carbohydrate conjugate vaccine against Shigella flexneri 2a in healthy adult volunteers: a phase 1, dose escalating, single-blind, randomised, placebo-controlled study, Lancet Infect. Dis., 2020, 21, 546–558 CrossRef PubMed.
- J. Boutet, P. Blasco, C. Guerreiro, F. Thouron, S. Dartevelle, F. Nato, F. J. Canada, A. Arda, A. Phalipon, J. Jimenez-Barbero and L. A. Mulard, Detailed investigation of the immunodominant role of O-antigen stoichiometric O-acetylation as revealed by chemical synthesis, immunochemistry, solution conformation and STD-NMR spectroscopy for Shigella flexneri, 3a, Chem. – Eur. J., 2016, 22, 10892–10911 CrossRef CAS PubMed.
- F. X. Theillet, C. Simenel, C. Guerreiro, A. Phalipon, L. A. Mulard and M. Delepierre, Effects of backbone substitutions on the conformational behavior of Shigella flexneri O-antigens: implications for vaccine strategy, Glycobiology, 2011, 21, 109–121 CrossRef CAS PubMed.
- H. J. Koeners, J. Verhoeven and J. H. van Boom, Synthesis of oligosaccharides by using levulinic ester as an hydroxyl protecting group, Tetrahedron Lett., 1980, 21, 381–382 CrossRef CAS.
- J. Boutet and L. A. Mulard, Synthesis of two tetra- and four pentasaccharide fragments of Shigella flexneri serotypes 3a and X O-antigens from a common tetrasaccharide intermediate, Eur. J. Org. Chem., 2008, 5526–5542 CrossRef CAS.
- J. Boutet, C. Guerreiro and L. A. Mulard, Synthesis of branched tri- to pentasaccharides representative of fragments of Shigella flexneri serotypes 3a and/or X O-antigens, Tetrahedron, 2008, 64, 10558–10572 CrossRef CAS.
- S. David and S. Hanessian, Regioselective manipulation of hydroxyl-Groups via organotin derivatives, Tetrahedron, 1985, 41, 643–663 CrossRef CAS.
- V. Dimakos and M. S. Taylor, Site-selective functionalization of hydroxyl groups in carbohydrate derivatives, Chem. Rev., 2018, 118, 11457–11517 CrossRef CAS PubMed.
- W. Shang, B. He and D. Niu, Ligand-controlled, transition-metal catalyzed site-selective modification of glycosides, Carbohydr. Res., 2019, 474, 16–33 CrossRef CAS PubMed.
- B. Ren, O. Ramström, Q. Zhang, J. Ge and H. Dong, An Iron(III) catalyst with unusually broad substrate scope in regioselective alkylation of diols and polyols, Chem. – Eur. J., 2016, 22, 2481–2486 CrossRef CAS PubMed.
- B. Ren, N. N. Yan and L. Gan, Regioselective alkylation of carbohydrates and diols: a cheaper iron catalyst, new applications and mechanism, RSC Adv., 2017, 7, 46257–46262 RSC.
- J. C. Lo, J. Gui, Y. Yabe, C.-M. Pan and P. S. Baran, Functionalized olefin cross-coupling to construct carbon–carbon bonds, Nature, 2014, 516, 343–348 CrossRef CAS PubMed.
- J. C. Luning, U. Moller, N. Debski and P. Welzel, A new method for the cleavage of allyl glycosides, Tetrahedron Lett., 1993, 34, 5871–5874 CrossRef.
- B. Yu, J. B. Zhang, S. F. Lu and Y. Z. Hui, A novel and efficient deprotection of the allyl group at the anomeric oxygen of carbohydrates, Synlett, 1998, 29–30 CrossRef CAS.
- H. B. Mereyala and S. Guntha, A novel, mild palladium-mediated deprotection of O-allyl and prop-1-enyl ethers, Tetrahedron Lett., 1993, 34, 6929–6930 CrossRef CAS.
- W. Li and B. Yu, Temporary ether protecting groups at the anomeric center in complex carbohydrate synthesis, Adv. Carbohydr. Chem. Biochem., 2020, 77, 1–69 CrossRef PubMed.
- K. Nakayama, K. Uoto, K. Higashi, T. Soga and T. Kusama, A useful method for deprotection of the protective allyl group at the anomeric oxygen of carbohydrate moieties using tetraskis(triphenylphosphine)palladium, Chem. Pharm. Bull., 1992, 40, 1718–1720 CrossRef CAS.
- F. Segat-Dioury and L. A. Mulard, Convergent synthesis of the methyl glycosides of a tetra- and a pentasaccharide fragment of the Shigella flexneri serotype 2a O-specific polysaccharide, Tetrahedron: Asymmetry, 2002, 13, 2211–2222 CrossRef CAS.
- J. Boutet, C. Guerreiro and L. A. Mulard, Efficient synthesis of six tri- to hexasaccharide fragments of Shigella flexneri serotypes 3a and/or X O-antigen, including a study on acceptors containing N-trichloroacetylglucosamine versus N-acetylglucosamine, J. Org. Chem., 2009, 74, 2651–2670 CrossRef CAS PubMed.
- Y. Le Guen, G. Le Heiget, D. Urban, P. Chassagne and L. A. Mulard, in Carbohydrate Chemistry: Proven Synthetic Methods, ed. C. Vogel and P. Murphy, Taylor & Francis, 2017, vol. 4, ch. 39, pp. 333–342 Search PubMed.
- L. A. Mulard, M. J. Clement, F. Segat-Dioury and M. Delepierre, Synthesis and NMR study of a linear pentasaccharide fragment of the Shigella flexneri, 5a O-specific polysaccharide, Tetrahedron, 2002, 58, 2593–2604 CrossRef CAS.
- S. S. Nigudkar and A. V. Demchenko, Stereocontrolled 1,2-cis glycosylation as the driving force of progress in synthetic carbohydrate chemistry, Chem. Sci., 2015, 6, 2687 RSC.
- W. L. Leng, H. Yao, J. X. He and X. W. Liu, Venturing beyond donor-controlled glycosylation: new perspectives toward anomeric selectivity, Acc. Chem. Res., 2018, 51, 628–639 CrossRef CAS PubMed.
- R. R. Schmidt and J. Michel, O-(α-D-glucopyranosyl)trichloroacetimidate as a glucosyl donor, J. Carbohydr. Chem., 1985, 4, 141 CrossRef CAS.
- M. G. Hoffmann and R. R. Schmidt, O-Glycosyl imidates 19. Reactions of glycosyl trichloroacetimidates with silylated C-nucleophiles, Liebigs Ann. Chem., 1985, 2403–2419 CrossRef CAS.
- B. Yu and H. Tao, Glycosyl trifluoroacetimidates. Part 1: Preparation and application as new glycosyl donors, Tetrahedron Lett., 2001, 42, 2405–2407 CrossRef CAS.
- R. R. France, N. V. Rees, J. D. Wadhawan, A. J. Fairbanks and R. G. Compton, Selective activation of glycosyl donors utilising electrochemical techniques: a study of the thermodynamic oxidation potentials of a range of chalcoglycosides, Org. Biomol. Chem., 2004, 2, 2188–2194 RSC.
- G. H. Posner and S. R. Haines, A convenient, one-step, high-yield replacement of an anomeric hydroxyl group by a fluorine atom using dast. Preparation of glycosyl fluorides, Tetrahedron Lett., 1985, 26, 5–8 CrossRef CAS.
- B. La Ferla, P. Bugada and F. Nicotra, Synthesis of the dimethyl ester of 1-deoxy-L-idonojirimycin-1-methylenphosphonate: A new approach to iminosugar phosphonates, J. Carbohydr. Chem., 2006, 25, 151–162 CrossRef CAS.
- M. Hoch, E. Heinz and R. R. Schmidt, Synthesis of 6-deoxy-6-sulfo-α-D-glucopyranosyl phosphate, Carbohydr. Res., 1989, 191, 21–28 CrossRef CAS.
- W. Xu, S. A. Springfield and J. T. Koh, Highly efficient synthesis of 1-thioglycosides in solution and solid phase using iminophosphorane bases, Carbohydr. Res., 2000, 325, 169–176 CrossRef CAS PubMed.
- S. K. Mulani, W. C. Hung, A. B. Ingle, K. S. Shiau and K. K. Mong, Modulating glycosylation with exogenous nucleophiles: an overview, Org. Biomol. Chem., 2014, 12, 1184–1197 RSC.
- K. Q. Zhang, S. C. Li, J. M. Mao, H. M. Chen and M. S. Cai, Studies on Carbohydrates 25. Synthesis of disaccharide units of Serratia marcescens, O4 antigen oligosaccharide, Chem. J. Chin. Univ., 1997, 18, 1469–1473 CAS.
- S. R. Lu, Y. H. Lai, J. H. Chen, C. Y. Liu and K. K. Mong, Dimethylformamide: an unusual glycosylation modulator, Angew. Chem., Int. Ed., 2011, 50, 7315–7320 CrossRef CAS PubMed.
- L. Wang, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Reagent controlled stereoselective synthesis of α-glucans, J. Am. Chem. Soc., 2018, 140, 4632–4638 CrossRef CAS PubMed.
- M. Ono and I. Itoh, A new deprotection method for levulinyl protecting groups under neutral conditions, Chem. Lett., 1988, 585–588 CrossRef CAS.
- T. Moriguchi, M. Sekine and K. Shinozuka, Novel method of the synthesis and hybridization properties of an oligonucleotide containing non-ionic diisopropylsilyl internucleotide linkage, Bioorg. Med. Chem., 2013, 21, 8013–8018 CrossRef CAS PubMed.
- R. Adamo, M. Tontini, G. Brogioni, M. R. Romano, G. Costantini, E. Danieli, D. Proietti, F. Berti and P. Costantino, Synthesis of laminarin fragments and evaluation of a β-(1,3) glucan hexasaccaride-CRM197 conjugate as vaccine candidate against Candida albicans, J. Carbohydr. Chem., 2011, 30, 249–280 CrossRef CAS.
- P. Westerduin, P. E. de Haan, M. J. Dees and J. H. van Boom, synthesis of methyl 3-[3(2-O-α-L-rhamnopyranosyl-α-L-rhamnopyranosyloxy)decanoyloxy]decanoate, a rhamnolipid from Pseudomonas aeruginosa, Carbohydr. Res., 1988, 180, 195–205 CrossRef CAS.
- B. M. Pinto, M. M. W. Buiting and K. B. Reimer, Use of the [β-(trimethylsilyl)ethoxy]methyl (SEM) protecting group in Carbohydrate Chemistry. Fully functionalized rhamnose acceptors and donors for use in oligosaccharide synthesis, J. Org. Chem., 1990, 55, 2177–2181 CrossRef CAS.
- R. R. Schmidt and J. Michel, Direct O-glycosyl trichloroacetimidate formation, nucleophilicity of the anomeric oxygen atom, Tetrahedron Lett., 1984, 25, 821–824 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Schemes S1 and S2, general procedures, detailed experimental procedures and analytical data for compounds S1–S3, 1–3, 5, 8, 12a, 12b, 13, 15–17, 22–25, copies of the 1H and 13C NMR spectra for all new compounds. See DOI: 10.1039/d1qo00761k |
| ‡ These authors have contributed equally. |
| § Z.H.: Systems Biology Theme, Department of Biomedical Engineering, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan 430074, China |
| This journal is © the Partner Organisations 2021 |