
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMXene quantum dot rivet reinforced Ni–Co LDH for boosting electrochemical activity and cycling stability†
Lili
Song‡
 a,
Shifan
Zhu‡
a,
Le
Tong
a,
Wandi
Wang
a,
Chun
Ouyang
bc,
Feng
Xu
c and
Yuqiao
Wang
*a
a,
Shifan
Zhu‡
a,
Le
Tong
a,
Wandi
Wang
a,
Chun
Ouyang
bc,
Feng
Xu
c and
Yuqiao
Wang
*a
aInstitute of Advanced Materials, School of Chemistry and Chemical Engineering, Southeast University, Nanjing, Jiangsu 211189, China. E-mail: yqwang@seu.edu.cn
bSchool of Material Science and Engineering, Jiangsu University of Science and Technology, Jiangsu 212003, China
cCETC Maritime Electronics Research Institute Co., Ltd., Ningbo Zhejiang 315000, China
First published on 2nd August 2021
Abstract
The rapid activity degradation of layered double hydroxides (LDHs) is caused by the mismatch of surface component and construction. Herein, MXene quantum dot (MQD) rivets are used to adjust the surface composition and structure of Ni–Co LDH to boost electrochemical activity and stability. The cap of MQD rivets would enrich a large number of unpaired electrons, thereby enhancing the adsorption of electrolyte ions. The surface functional groups of MQDs as rivet shafts can produce a strong interface coupling with LDH to keep stability. The coupling effect should also promote the charge transfer between LDH and MQDs and activate LDH surface adsorption. The effect can be described using the electron location function mapping, charge distribution and X-ray photoelectron spectroscopy. Ni–Co LDH@MQDs exhibits high energy storage activity (87 mA h g−1 at 1 A g−1) and durable stability (98.6% retention after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles) in a two-electrode device. This work opens up a new path for the realization of high-efficiency and long-term stable electrochemically active materials through the interface coupling between MQDs and LDH.
000 cycles) in a two-electrode device. This work opens up a new path for the realization of high-efficiency and long-term stable electrochemically active materials through the interface coupling between MQDs and LDH.
Introduction
The excellent electrochemical activity of layered double hydroxides (LDHs) depends on the two-dimensional structure, good anion exchange and selective metal cations.1–4 However, LDHs show short-term cycling stability in alkaline solution.5–7 The structure collapse may be the cause of the rapid decline in activity.8 The previous reports on a solution to the instability of LDHs are as follows: (1) controlling components,9–12 (2) introducing vacancies,13 (3) doping heteroatoms14,15 and (4) integrating composites.16–18 The reasonable design of structure and composition play a key role in achieving a durable stability at an atomic level. For instance, graphene quantum dots were used to toughen the surface structure, resulting in promoting cycling stability of LDH through bonding interaction.19 The terminal hydroxyl group can enhance the interface adhesion toughness of the interface between the electron transport layer and the halide perovskite film, thereby improving the structure reliability.20The concept of surface spatial confinement (SSC) effect was proposed to design LDH@carbon dots with excellent activity and stability in a supercapacitor.21 The SSC effect is defined as special characterization of limited spatial action caused by strong interaction between the substrate and surface modifier. The interface coupling can be expressed by the SSC effect on charge transfer and structure stability, providing an intuitive description of the interaction between different micro-nano structures. For instance, the chemical grafting of metal organic frameworks onto polypyrrole nanotubes connected by carboxyl groups can protect the inner-layer with a strong outer-surface to prolong the cycling stability of lithium-sulfur batteries.22
An abundant supply of functional groups was distributed on the surface of MXene quantum dots (MQDs).23–25 MXene is known as high metallic conductivity, good mechanical properties and complex bonding surrounding, but its major limitation is self-stacking during the electrode process.26,27 LDHs are also known as bimetallic hydroxides with excellent adsorption ability, but their swelling properties cause a collapse of the interlamellar domain.28 The bonding interaction between LDHs and MXene can be achieved through the functional groups through LDHs as the carrier and MXene as the surface modifier. The MXenes would be fixed in a specific position to avoid them gathering together. At the same time, LDHs should seem to have an added layer of protective armor to enhance the skeleton stability. It is an extremely significant challenge to express the specific effect on the surface/interface interaction in the composite structure.
Generally, it is still a difficult problem to evaluate the intensity of interface interaction.29 The electron location function (ELF) is constantly used to estimate the bond strength and electron distribution in theoretical models.30,31 The ELF mapping can be obtained by density functional theory (DFT) calculations. These theoretical methods provide potential routes for intuitively revealing the interface coupling effect on electrochemical activity and stability. Herein, we report a strategy for constructing the structure and evaluating the method. The stable structure and efficient electron transfer benefit from the MQDs being bonded to the LDH surface through their functional groups as a bridge. This occurs due to the surface space limitation, where the strong interaction between the substrate and the surface modifier is beneficial for the enhancement of the structure and activity in the special confinement space. The SSC effect can be expressed specifically as the bonding process, electron distribution and adsorption energy using the DFT calculations. The supercapacitors assembled by Ni–Co LDH@MQDs electrodes are used to reflect the actual beneficial results based on the SSC effect. The specific capacity, energy density and long-term cycling life were used to confirm the influence of MQDs on the electrochemical activity and stability of LDH.
Results and discussion
The first-principle calculations were used to describe the interfacial coupling between LDH and MQDs and predict the SSC effect on activity and stability. The calculated model was constructed by placing MQDs onto the Ni–Co LDH surface (Fig. S1a and b, ESI†). The MQD model was simplified as four carbon atoms and six titanium atoms with two hydroxyl groups, while the primary Ni–Co LDH model was composed of Ni, Co, O and H atoms. The ELF mappings were drawn to directly observe the bonding process and electron distribution. The electron distribution around the surface metal atom was redistributed in Ni–Co LDH@MQDs compared with the primary Ni–Co LDH, promoting the surface activity (Fig. 1a and Fig. S1c, ESI†). Fig. 1b shows the side view of ELF mapping in a Ni–Co LDH@MQD model. The unpaired electrons were localized at the top of the MQDs to activate the adsorption for electrolyte ions. The abundant electrons near “–Ti–O–H–O–” bonds between the surface of MQDs and LDH are beneficial for forming a strong interfacial coupling. The adhesion energy (Eadh) was calculated to estimate the interface bonding strength based on the thermodynamics. The Eadh between LDH and MQDs was −1.51 eV. The negative Eadh value should illustrate a strong bonding to strengthen the structure. The net charge of atoms was obtained to quantitatively assess the charge transfer and distribution using Mulliken charge analysis. The calculated charge was corresponding to the ELF mapping. The net charge of surface metal atoms exhibited a redistribution after MQD addition in contrast to that of Ni–Co LDH (Fig. 1c and Fig. S1d, ESI†). The Ni and Co atoms near MQDs obtained 0.01–0.06 e, and O atoms lost 0.01–0.1 e owing to the interaction. The “–Ti–O–H–O–” bonds of the MQD surface also displayed electron gains and losses (Fig. 1d), promoting the fast charge transfer and strong interface interaction. The enhanced interface charge interaction is the basic guarantee for improved reaction activity.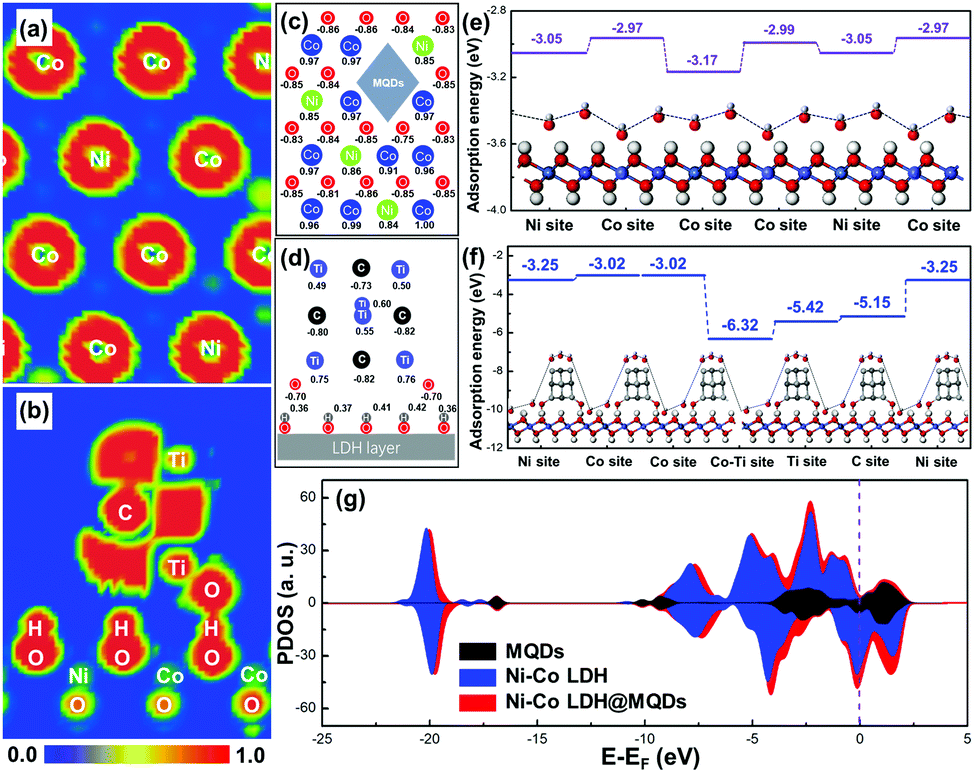 | ||
| Fig. 1 ELF mappings of (a) LDH and (b) MQDs in Ni–Co LDH@MQDs. Calculated charge of surface atoms of (c) LDH and (d) MQDs in Ni–Co LDH@MQDs. Calculated adsorption energies using hydroxide (OH−) as an example for (e) Ni–Co LDH model and (f) Ni–Co LDH@MQDs. (g) Comparative DOS plots of MQDs, Ni–Co LDH and Ni–Co LDH@MQDs. | ||
The hydroxide adsorption energy, density of states (DOS) and projected DOS (PDOS) were employed to analyse the surface activity and electron transfer after the formation of strong interaction. The adsorption energies (Ea) for hydroxides (OH−) of Ni–Co LDH@MQDs were higher than that of Ni–Co LDH (Fig. 1e and f). In particular, the Ea of surface metal sites was improved from 0.05 to 0.08 eV. The Ti and C atoms at the MQD top show higher adsorption energies than those of other sites, which is consistent with the ELF mappings and charge redistribution. MQDs can enhance the electronic conductivity of LDH in view of the high intrinsic conductivity of MQDs. This improvement is due to more electron states crossing the Fermi level after surface modification (Ef = 0, Fig. 1g). The location and shape of the 2p and 3d orbitals have undergone major changes owing to the strong interaction (Fig. S1e and f, ESI†). The results indicate that the enhanced conductivity is related to additional Ti, C and O atoms, which couples the interface of LDH and MQDs by the hydroxide adsorption. The role of MQDs can be attributed to three aspects: (1) the unpaired charges were located at the cap of MQDs to promote the hydroxide adsorption. (2) MQDs induced the charge redistributed on the LDH surface to activate the surface activity. (3) Strong coupling appeared at the interface due to surface functional groups.
Ni–Co LDH@MQDs were synthesized by a one-step hydrothermal method (Fig. 2a), indicating that MQDs were anchored on the surface of LDH as rivets (Fig. S2, ESI†). The MQD solution was prepared by separating the MXene solution using centrifugation. The pale yellow MQD solution presented blue cyan fluorescence under a UV lamp (365 nm). The MQDs with the sizes of 3.06 ± 0.78 nm were observed by TEM (Fig. 2b). The lattice fringes (d = 0.25 nm) should be assigned to the (110) crystal plane of MXene. The cleaned nickel foam was selected as the growth skeleton. The Ni–Co LDH@MQD sheets grew uniformly and densely on the nickel foam compared with LDH without MQDs in scanning electron microscopy (SEM) images (Fig. 2c and Fig. S3a, ESI†). MQDs anchoring on LDH can reduce the swelling properties to prevent a collapse of the interlamellar domain. The enhanced interlayer interaction induced by surface modification helps electron transfer.
 | ||
| Fig. 2 (a) Schematic illustration of the bonding process. (b) HRTEM images with size distribution of MQDs. (c) SEM image of Ni–Co LDH@MQDs. (d) HRTEM images of Ni–Co LDH@MQDs (Inset: Diffraction and fringe pattern). (e) The uniform distribution of Ni, Co and Ti of Ni–Co LDH@MQDs. | ||
The well-dispersed distribution of MQDs at the LDH surface was observed by high-resolution transmission electron microscopy (HRTEM) (Fig. 2d). The good dispersibility mainly depends on the strong interaction of the functional groups at specific positions on the surfaces of MQDs and LDH. MQDs were confirmed by the lattice fringes of 0.20 and 0.25 nm, referring to the (002) and (110) planes. The lattice fringes of 0.26 and 0.29 nm corresponded to the (012) and (009) plane of Ni–Co LDH. The selected area electron diffraction (SAED) patterns show the (110) plane of Ni–Co LDH and the (002) and (103) planes of MQDs. The element composition was analysed by energy-dispersive spectroscopy (EDS) (Fig. 2e and Fig. S3b, S3c, ESI†). The HADDF-STEM image and elemental mappings were tested to avoid the effect of Ni foam (Fig. S4, ESI†). The atomic ratios of Ni and Co are about 1:2 according to the element contents of Ni–Co LDH@MQDs (Fig. S5, ESI†), in agreement with the added amount and DFT calculations. The X-ray diffraction (XRD) pattern shows diffraction peaks at 11.7°, 23.6° and 39.7°, corresponding to the (003), (006), and (015) planes of LDH (JCPDS No. 15-0087) (Fig. S6, ESI†). The peak intensity of Ni–Co LDH@MQDs was lower than that of Ni–Co LDH, suggesting that MQDs covered the LDH surface. The chemical state of Ni–Co LDH@MQDs was evaluated by X-ray photoelectron spectroscopy (XPS) (Fig. 3). Co 2p and Ni 2p spectra can be deconvoluted into two spin–orbit doublets as well as two satellites due to Co2+/Ni2+ and Co3+/Ni3+ (Fig. 3a and b). The positive shift of all binding energies for Ni 2p and Co 2p indicates the electron transfer from Co and Ni to O, demonstrating surface charge redistribution on the LDH surface. The peaks of Ti–O (458.7 and 464.3 eV) and C–O (286.4 eV) indicate the strong interfacial coupling between MQDs and Ni–Co LDH through surface functional groups (Fig. 3c). The anchored state was also determined by an enhanced C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak and C–O peak shift −0.4 eV for C 1s (Fig. 3d). Fourier transform infrared (FT-IR) spectroscopy (Fig. S7, ESI†) was conducted to analyze the surface groups for Ni–Co LDH@MQDs. The broad peak at 3471 cm−1 and the peak at 1445 cm−1 should be assigned to the stretching vibration and the bending vibration of the –OH group. The peaks at 1631, 1066, 642, and 532 cm−1 correspond to the stretching vibration modes of CO, C–F, C–Ti and Ti–O groups on MQDs. This result suggested that the MQDs were successfully introduced to the surface of Ni–Co LDH. The XPS and FT-IR results provide strong experimental evidence of the formation of the SSC effect.
O peak and C–O peak shift −0.4 eV for C 1s (Fig. 3d). Fourier transform infrared (FT-IR) spectroscopy (Fig. S7, ESI†) was conducted to analyze the surface groups for Ni–Co LDH@MQDs. The broad peak at 3471 cm−1 and the peak at 1445 cm−1 should be assigned to the stretching vibration and the bending vibration of the –OH group. The peaks at 1631, 1066, 642, and 532 cm−1 correspond to the stretching vibration modes of CO, C–F, C–Ti and Ti–O groups on MQDs. This result suggested that the MQDs were successfully introduced to the surface of Ni–Co LDH. The XPS and FT-IR results provide strong experimental evidence of the formation of the SSC effect.
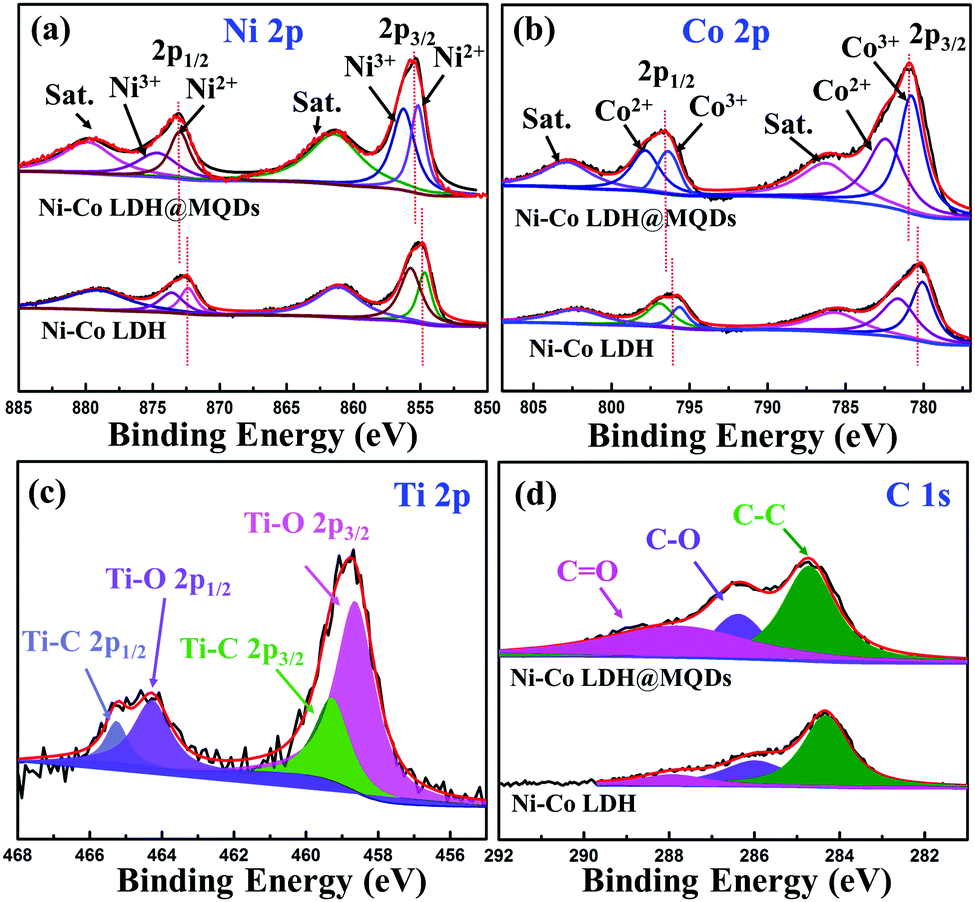 | ||
| Fig. 3 High-resolution XPS spectrum of Ni–Co LDH@MQDs: (a) Ni 2p, (b) Co 2p, (c) Ti 2p and (d) C 1s. | ||
The SSC effect on the electrochemical properties was investigated using a three-electrode cell, including cyclic voltammetry (CV), galvanostatic charge/discharge (GCD) and electrochemical impedance spectroscopy (EIS). The CV areas and discharging time indicated the improvement in energy storage after the MQD modification. (Fig. S8a–c, ESI†). The Ni–Co LDH@MQDs gained a specific capacity of 140 mA h g−1 (1260 F g−1) superior to that of Ni–Co LDH (122 mA h g−1, 1098 F g−1) (Fig. S8d, ESI†). The high conductivity helps increase specific capacitance. The capacitance retention of Ni–Co LDH@MQDs (72%) was superior to that of Ni–Co LDH (54%) (Fig. 4a). The good rate and small charge transfer resistance (Rct, 1.52 Ω) confirmed the enhanced charge transfer efficiency (Fig. 4b and Table S1, ESI†). The capacity of Ni–Co LDH sharply drops at a current density of 6 A g−1 after 5000 cycles (Fig. S8e, ESI†). For Ni–Co LDH@MQDs, the capacity retention rate increased to 61.2% over 5000 cycles. Notably, the capacity of Ni–Co LDH@MQDs sharply drops by 37.7% after the beginning 2000 cycles, while the capacity is only reduced by 1.1% after the following 3000 cycles. The surface structure of LDH@MQDs was maintained well after cycling (Fig. S8f, ESI†), demonstrating that MQD rivets can effectively reinforce the LDH surface structure, in agreement with DFT predictions.
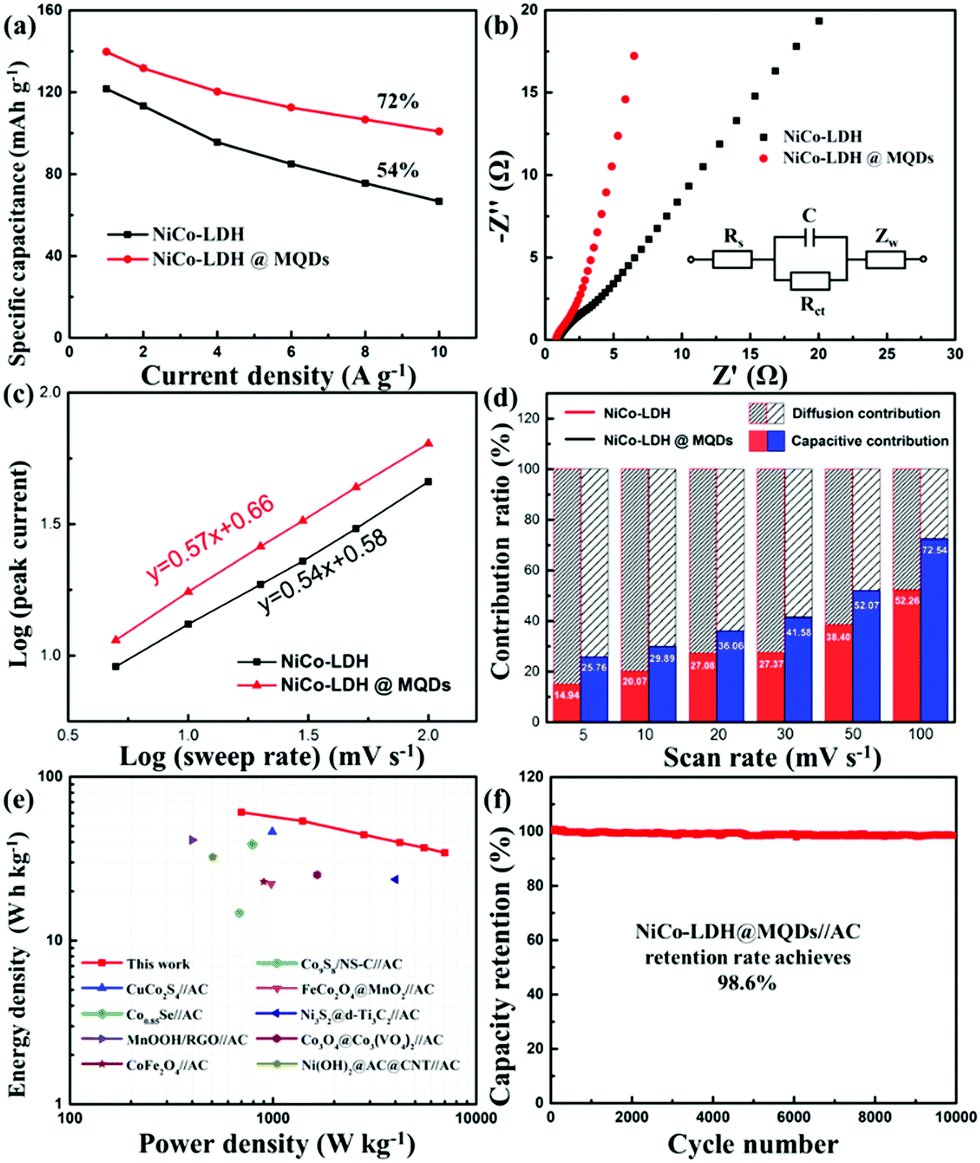 | ||
| Fig. 4 (a) Specific capacities at different current densities and (b) Nyquist plots of Ni–Co LDH and Ni–Co LDH@MQDs. (c) Plots of log(i) versus log(v) of the samples. (d) The capacitive- and diffusion-controlled processes for Ni–Co LDH@MQDs at different scan rates. (e) Ragone plots. (f) Cycling performance of Ni–Co LDH@MQDs//AC at a current density of 6 A g−1. | ||
A good linear correlation of peak current (i) versus the square root of scan rate (ν1/2) implied that the charge storage process was a diffusion-controlled process. The performance complied with the battery-type behaviour (Fig. S9, ESI†). The power law (i = avb) was further implied to explain the charge storage contribution and reaction kinetics of Ni–Co LDH@MQDs based on the CV curves. The b value is the slope of log (peak current) against log (ν) plot (Fig. 4c). The index b values were 0.54 and 0.57 (close to 0.5) corresponding to Ni–Co LDH and Ni–Co LDH@MQDs, respectively, which was in accordance with a battery-type behaviour. The surface-controlled capacitive contribution (k1v) and the diffusion-controlled contribution (k2v1/2) were discussed to quantify the ratio of contribution via the following formula as iv = k1v + k2v1/2, where k1 and k2 values can be calculated from the slope and intercept of i/v1/2versus v1/2. The CV curves of the Ni–Co LDH@MQDs were separated into surface-confined (red region) and diffusion-controlled contributions (black region) (Fig. S10, ESI†). The percent surface-confined capacitive contribution of Ni–Co LDH@MQDs (36.06%) was evidently higher than that of Ni–Co LDH (27.08%) at a scan rate of 20 mV s−1, proving the surface reconstruction of the composites by anchored MQDs. For Ni–Co LDH@MQDs, the percent capacitive contribution enhanced from 25.76% to 72.54% with the increase of scan rate from 5 to 100 mV s−1 (Fig. 4d). The excellent capacitive contribution suggested that surface redox reaction processes were dominant during charge storage at a high scan rate. This was caused by the chem-grafted MQDs with enhanced electrochemical performance.
The two-electrode device was further evaluated based on Ni–Co LDH@MQDs//AC with a voltage window of 0–1.4 V (Fig. S11a, ESI†). The CV curves suggested the excellent reversibility of the device (Fig. S11b, ESI†). The specific capacities of Ni–Co LDH@MQDs//AC were 87, 77, 63, 57, 53 and 50 mA h g−1 (224, 198, 162, 147, 136 and 129 F g−1) at current densities of 1, 2, 4, 6, 8 and 10 A g−1 (Fig. S11c and d, ESI†). The device gained an energy density of 60.7 W h kg−1 at a power density of 698 W kg−1 (Fig. 4e). The energy storage performance was better than the reports (Table S2, ESI†). The capacity remained basically unchanged as the charge/discharge process exceeded 10000 cycles (Fig. 4f). The excellent cycling stability (98.6% retention after 10000 cycles) can be attributed to the strong interfacial coupling between MQDs and Ni–Co LDH. The device performance is listed in Table S3 (ESI†).
Conclusions
The MQD-decorated LDH surface can enhance electron transfer and structure stability through mutual surface functional groups. This strong interaction at a specific surface interface can be attributed to the surface spatial confinement effect. The theoretical simulations can be calculated to deduce and quantify the surface confinement process more accurately by using the electron location function and surface adsorption energy. The actual effect is demonstrated by the performance of the assembled supercapacitor electrodes and devices. Our work finds a new way for the design and preparation of electrodes with high activity and long-term stability structures.Experimental
Model construction
All models derived from Ni(OH)2 with α = γ = 90°, β = 97.08°, a = 14.68 Å, b = 3.16 Å and c = 5.47 Å (monoclinic C2/m space group) at Materials Project database (mp-626794). The primary Ni–Co LDH cell was built by choosing the lowest layer of Ni(OH)2 (4 × 4 × 1), where the Co atom replaces the Ni position in a ratio of 3:1. A vacuum of 15 Å was used to avoid spurious interactions in a non-periodic direction (the z direction) for hybrid systems. MQDs were constructed by 4 carbon atoms and 6 titanium atoms with two hydroxyl groups. The Ni–Co LDH@MQDs model means that MQDs are placed on the primary Ni–Co LDH surface through hydroxyl groups.
DFT calculations
The first-principle calculations were carried out based on density functional theory (DFT) implemented in the CASTEP code. For the calculations, the generalized-gradient approximation of Perdew–Burke–Ernzerhof (GGA–PBE) was used for the exchange–correlation interactions.32 Spin polarization was considered in all the calculations. The energy cutoff of 400 eV and Monkhorst–Pack k-mesh of 2 × 2 × 1 were applied. Geometry optimizations have been carried out using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) minimization algorithm, with the convergence threshold of 1 × 10−5 eV for energy and 0.02 eV Å−1 for force and atomic displacements lower than 0.02 Å. The subsequent calculations of the electronic structure used the same energy cutoff and k-points described above. The adhesion energy (Eadh) was estimated as follows:| Eadh = ET − ELDH − EMQDs | (1) |
The adsorption energy (Ea) of a hydroxide ion (OH−) was computed as follows:
| Ea = Esub+OH− − Esub − EOH− | (2) |
Synthesis of MXene nanosheets
The synthesis of MXene nanosheets was based on our report.33 Typically, 1 g of LiF was first slowly added to 20 mL of a 9 M HCl solution with stirring for 5 min. Then Ti3AlC2 (purchased from 11 Technology Co. Ltd) was gradually added to the above solution to avoid overheating. The mixture was continuously stirred at 35 °C for 24 h. The resulting solution was repeatedly washed with distilled water until the pH > 6 at 3500 rpm. The above solution was sonicated for 1 h under N2 protection and further centrifuged at 3500 rpm for 1 h. The supernatant is MXene nanosheets.Synthesis of MQDs
20 mL MXene nanosheet solution was placed in a 50 mL Teflon-lined stainless steel autoclave. The pH was adjusted around 9 with ammonia. The mixture was heated at 100 °C for 6 h. The resulting solution was centrifuged at 12000 rpm for 30 minutes. The light-yellow supernatant is MXene quantum dots (MQDs) (0.5 mg L−1).
Synthesis of Ni–Co LDH@MQDs
NiCl2·6H2O (0.48 g), CoCl2·6H2O (0.95 g), hexamethylenetetramine (1.6 g) and MQDs (10 mg) were added to a Teflon-lined stainless-steel autoclave. The total volume was controlled to 60 mL with distilled water. The nickel foam with surface oxide treatment was placed in the mixed solution after continuous stirring. The autoclave was kept at 100 °C for 8 h. The treated nickel foam was washed with distilled water and absolute ethanol several times, and further dried at 60 °C in a vacuum oven. The load is about 2 mg cm−2.Characterization
The scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HRTEM) images were obtained with a Hitachi S-4800 and a TecnaiG2T20, respectively. Energy-dispersive spectroscopy (EDS) measurement was performed with SEM. The selected area electron diffraction (SAED) pattern corresponded to the corresponding TEM image. X-ray diffraction (XRD) spectra were acquired with an Ultima IV at Cu-Kα radiation. High-resolution X-ray photoelectron spectroscopy (XPS) was conducted by a Thermo Scientific K-Alpha spectrometer. The FT-IR spectrum was recorded on a PerkinElmer FT-IR spectrometer.Electrochemical measurements
All electrochemical measurements were performed with a CHI760E electrochemical workstation, including CV, GCD curves and EIS. EIS was carried out in the range of 0.01–105 Hz. The three-electrode system includes working electrode, reference electrode (saturated calomel electrode, SCE), auxiliary electrode (Pt electrode) and electrolyte (6 M KOH). The cycling stability measurement was conducted by a Land battery measurement system (LAND CT2001A). The specific capacity (Cs, mA h g−1), specific capacitances (C, F g−1), energy density (E, W h kg−1) and power density (P, W kg−1) were calculated according to the following equations:34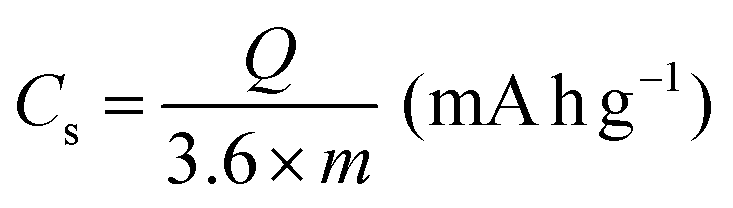 | (3) |
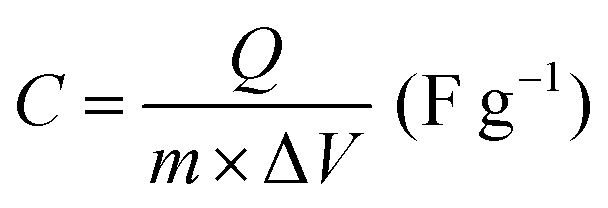 | (4) |
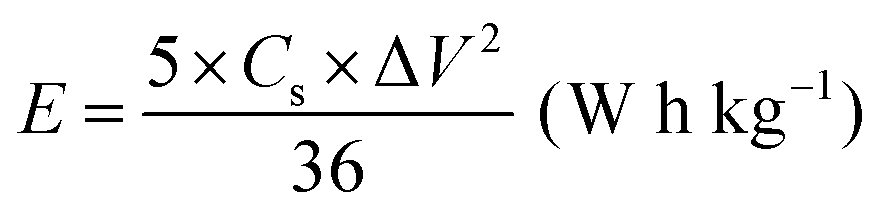 | (5) |
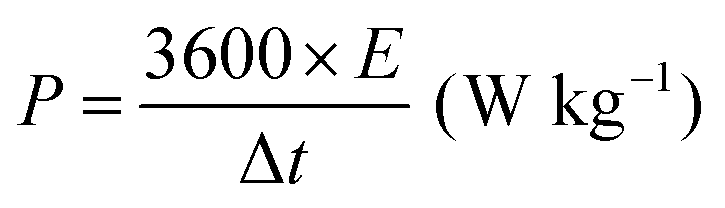 | (6) |
The two-electrode device includes positive electrode (working electrode), negative electrode (AC) and electrolyte (6 M KOH). The negative electrode (AC) was prepared by mixing AC/carbon black/poly(vinyl difluoride) (PVDF) at the weight ratio 80:10:10. The mass ratio of the positive materials (m+) and negative AC (m−) followed the equation:
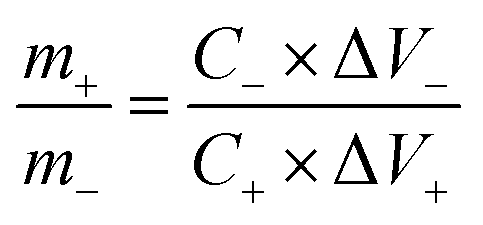 | (7) |
Author contributions
Y. W. proposed the concepts and designed the experiments. L. S., L. T. and W. W. carried out the experiments. L. S. and S. Z. conducted the DFT calculations. C. O. and F. Xu. completed the XPS measurement. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (61774033), the Natural Science Foundation of Jiangsu Province (BK20190973) and Preferential Funding of Zhejiang Post-Doctoral Research Projects (ZJ2020085). We also thank the Big Data Center of Southeast University for providing the facility support on the numerical calculations in this paper.Notes and references
- Y. Guo, X. Hong, Y. Wang, Q. Li, J. Meng, R. Dai, X. Liu, L. He and L. Mai, Adv. Funct. Mater., 2019, 29, 1809004 CrossRef.
- S. C. Sekhar, G. Nagaraju and J. S. Yu, Nano Energy, 2017, 36, 58–67 CrossRef CAS.
- Z. Yuan, S. M. Bak, P. Li, Y. Jia, L. Zheng, Y. Zhou, L. Bai, E. Hu, X. Q. Yang, Z. Cai, Y. Sun and X. Sun, ACS Energy Lett., 2019, 4, 1412–1418 CrossRef CAS.
- T. Li, X. Hao, S. Bai, Y. Zhao and Y. F. Song, Acta Phys. -Chim. Sin., 2020, 36, 1–17 Search PubMed.
- X. Gao, P. Wang, Z. Pan, J. P. Claverie and J. Wang, ChemSusChem, 2020, 13, 1226–1254 CrossRef CAS PubMed.
- H. Peng, C. Jing, J. Chen, D. Jiang, X. Liu, B. Dong, F. Dong, S. Li and Y. Zhang, CrystEngComm, 2019, 21, 470–477 RSC.
- J. Zheng, X. Pan, X. Huang, D. Xiong, Y. Shang, X. Li, N. Wang, W. M. Lau and H. Y. Yang, Chem. Eng. J., 2020, 396, 125197 CrossRef CAS.
- R. Chen, S. F. Hung, D. Zhou, J. Gao, C. Yang, H. Tao, H. Bin Yang, L. Zhang, L. Zhang, Q. Xiong, H. M. Chen and B. Liu, Adv. Mater., 2019, 31, 1903909 CrossRef CAS PubMed.
- Y. Chen, W. K. Pang, H. Bai, T. Zhou, Y. Liu, S. Li and Z. Guo, Nano Lett., 2017, 17, 429–436 CrossRef CAS PubMed.
- J. Zhang, L. Yu, Y. Chen, X. F. Lu, S. Gao and X. W. Lou, Adv. Mater., 2020, 32, 1906432 CrossRef CAS PubMed.
- Z. Zhang, Y. Du, Q. C. Wang, J. Xu, Y. N. Zhou, J. Bao, J. Shen and X. Zhou, Angew. Chem., Int. Ed., 2020, 59, 17504–17510 CrossRef CAS PubMed.
- X. F. Lu, A. L. Wang, H. Xu, X. J. He, Y. X. Tong and G. R. Li, J. Mater. Chem. A, 2015, 3, 16560–16566 RSC.
- Y. Wang, M. Qiao, Y. Li and S. Wang, Small, 2018, 14, 1800136 CrossRef PubMed.
- J. He, X. Zhou, P. Xu and J. Sun, Nano Energy, 2021, 80, 105540 CrossRef CAS.
- D. Zhou, Z. Cai, Y. Jia, X. Xiong, Q. Xie, S. Wang, Y. Zhang, W. Liu, H. Duan and X. Sun, Nanoscale Horiz., 2018, 3, 532–537 RSC.
- T. Deepalakshmi, T. T. Nguyen, N. H. Kim, K. T. Chong and J. H. Lee, J. Mater. Chem. A, 2019, 7, 24462–24476 RSC.
- X. F. Lu, J. Lin, Z. X. Huang and G. R. Li, Electrochim. Acta, 2015, 161, 236–244 CrossRef CAS.
- X. Ge, S. Liu, M. Qiao, Y. Du, Y. Li, J. Bao and X. Zhou, Angew. Chem., Int. Ed., 2019, 58, 14578–14583 CrossRef CAS PubMed.
- Y. Ma, W. Yuan, Y. Bai, H. Wu and L. Cheng, Carbon, 2019, 154, 292–300 CrossRef CAS.
- Z. Dai, S. K. Yadavalli, M. Chen, A. Abbaspourtamijani, Y. Qi and N. P. Padture, Science, 2021, 372, 618–622 CrossRef CAS PubMed.
- L. Song, C. Peng, F. Yang, L. Wang, Y. Jiang and Y. Wang, ACS Appl. Energy Mater., 2021, 4, 4654–4661 CrossRef CAS.
- Q. Li, H. Zhu, Y. Tang, P. Zhu, H. Ma, C. Ge and F. Yan, Chem. Commun., 2019, 55, 12108–12111 RSC.
- R. Tang, S. Zhou, C. Li, R. Chen, L. Zhang, Z. Zhang and L. Yin, Adv. Funct. Mater., 2020, 30, 2000637 CrossRef CAS.
- G. P. Neupane, B. Wang, M. Tebyetekerwa, H. T. Nguyen, M. Taheri, B. Liu, M. Nauman and R. Basnet, Small, 2021, 17, 2006309 CrossRef CAS.
- Z. Zeng, G. Fu, H. Bin Yang, Y. Yan, J. Chen, Z. Yu, J. Gao, L. Y. Gan, B. Liu and P. Chen, ACS Mater. Lett., 2019, 1, 432–439 CrossRef CAS.
- M. Hu, H. Zhang, T. Hu, B. Fan, X. Wang and Z. Li, Chem. Soc. Rev., 2020, 49, 6666–6693 RSC.
- K. Zhao, H. Wang, C. Zhu, S. Lin, Z. Xu and X. Zhang, Electrochim. Acta, 2019, 308, 1–8 CrossRef CAS.
- J. Zhang, Z. Li, Y. Chen, S. Gao and X. W. Lou, Angew. Chem., Int. Ed., 2018, 57, 10944–10948 CrossRef CAS PubMed.
- K. R. G. Lim, A. D. Handoko, L. R. Johnson, X. Meng, M. Lin, G. S. Subramanian, B. Anasori, Y. Gogotsi, A. Vojvodic and Z. W. Seh, ACS Nano, 2020, 14, 16140–16155 CrossRef PubMed.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- J. He, W. Sun, D. Chen, Z. Gao and C. Zhang, Langmuir, 2021, 37, 3490–3499 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- W. Wang, D. Jiang, X. Chen, K. Xie, Y. Jiang and Y. Wang, Appl. Surf. Sci., 2020, 515, 145982 CrossRef CAS.
- G. Li, X. Cui, B. Song, H. Ouyang, K. Wang, Y. Sun and Y. Wang, Chem. Eng. J., 2020, 388, 124319 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Calculated models, synthetic method and characterization, supplementary figures and tables. See DOI: 10.1039/d1ma00474c |
| ‡ L. S. and S. Z. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |