
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Emergence, evidence, and effect of junction clustering in supramolecular polymer materials
Amir
Jangizehi
 ,
Mostafa
Ahmadi
* and
Sebastian
Seiffert
*
,
Mostafa
Ahmadi
* and
Sebastian
Seiffert
*
Department of Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: sebastian.seiffert@uni-mainz.de; ahmadi@uni-mainz.de
First published on 11th January 2021
Abstract
A significant fraction of biomaterials consists of supramolecular polymers and networks formed by non-covalent interactions between associative motifs. They typically contain complex structures in which on top of binary associations, phase-separation and aggregation of associative junctions occur. Such hierarchical assemblies have significant influences on the dynamics as well as the physical and mechanical properties of the materials. Similar to supramolecular biomaterials, aggregation of associative junctions has also been frequently reported to occur in synthetic supramolecular polymers and networks. Engineering of such secondary structures in a sense to create and control the extent of hierarchical assemblies can be a powerful approach not only to alter the physical and mechanical properties of these materials but also to regulate novel functions like self-healing. To implement this approach, a deep knowledge about the physical origin of clusters and the subsequent manifestation in the properties and functions of the materials are required. To this end, we review a multitude of reports on the formation of hierarchical assemblies in supramolecular polymeric materials such as networks, thermoplastic elastomers, and gels. We classify motives for phase-separation and development of clusters and the possible subsequent hierarchical assemblies, as well as their influence on the materials’ physical and mechanical properties. We summarize the specific characterization methods and present selected example applications of supramolecular materials that operate based on such hierarchical structures. This overview proposes a high potential for designing supramolecular polymeric materials with tuned properties and specified functions based on the formation of clusters.
1. Introduction
Organization of building blocks via supramolecular self-assembly driven by noncovalent interactions is ubiquitous in nature.1,2 Such organization follows a stepwise process, known as hierarchical self-assembly. This process commonly initiates by the self-assembly of building blocks via non-covalent interactions between two associative groups. In subsequent steps, phase-separation between the building blocks and binary associations followed by the aggregation of binary associations into separated domains known as clusters may occur.3,4 On top of that, ordered hierarchical assemblies may form and grow inside clusters in the following if some prerequisites are provided. In collagen, for example, three polypeptide chains fold into triple-stranded helixes, which then, in a subsequent process, self-assemble into collagen fibrils. These fibrils further aggregate to form fibers.5,6 Another well-known example of hierarchical self-assembly in nature is the tobacco mosaic virus, in which following a stepwise process, protein subunits are associated onto a single RNA chain.7,8 The assembly process includes the binding of RNA to a short helical aggregate consisting of protein coating. Likewise, amphiphilic molecules commonly show hierarchical self-assembly in biological systems.9,10 In the early steps, molecules self-assemble into micelles or bilayers, and then these units further aggregate to form larger and more complex structures.The phase-separation between building blocks and associative groups, and the subsequent aggregation of associative moieties, which are commonly found in the hierarchical structure of biomaterials, can induce specific properties and/or functions.11 In spider silk, for instance, the primary structure of proteins has alternating hydrophilic and hydrophobic blocks in their core domains. Hydrophilic blocks have numerous intra- and inter-chain hydrogen bonding, which form crystalline domains and phase separate from the hydrophobic part. Specific mechanical properties of spider silk such as high elasticity and toughness are attributed to such a hierarchical structure, where crystalline domains with β-sheet content are dispersed in tandem with a disordered protein sequence.12 In the same way, the main protein component of mussel byssus threads has a block-copolymer structure. The copolymer contains a central collagen domain, variable flanking domains, and histidine-rich domains. The latter coordinates to transition metal ions like zinc and copper. With such a multi-domain structure, the byssal threads show high stiffness and self-healing properties at the same time, which protect marine mussels in wave-swept seashore habitats.13
Inspired by nature, hierarchical self-assembly has been widely used to construct synthetic materials with well-defined associations and increasing levels of structural complexity.1,3,11,14 Hartgerink and coworkers investigated a peptide system that replicates all levels of self-assembly that are usually seen in a natural collagen: from peptide chains to triple helixes, to nanofibers, and finally to hydrogels.15 The collagen mimetic peptide consists of 36 amino acids based on lysine, aspirate, glycine, proline, and hydroxyproline. The hydrogen bonds between lysine and aspirate stabilize the triple helix structure within the nanofibers. The aggregated nanofibers self-assemble into hydrogels that can be degraded by collagenase at a similar rate than that of rat-tail collagen, suggesting a potential application as tissue-engineering scaffolds.15 Gu and coworkers utilized the self-assembly of end-functionalized linear polypeptides and peptide dendrimers to form amphiphiles. The amphiphiles then self-assemble into virosome-like nanostructures, in which peptide dendrimers aggregate. In water, these so-called peptidesome nanoparticles mimic viral capsids and have potential applications to be used as gene vectors with high gene transfection efficiency.16
In all these natural biomaterials and synthetic, bio-inspired supramolecular polymer materials, phase-separation phenomena and following aggregation of building blocks into clusters of supramolecular junctions are critical steps to organize and stabilize complex structures. In the work of Gu and coworkers, for example, the peptide dendrimer is hydrophilic generation-2 poly(L-lysine) and the linear peptide is hydrophobic poly(L-leucine) functionalized with glutamic acid. The weak hydrogen bonding between poly(L-lysine) and glutamic acid is essential for the self-assembly process, but not strong enough to form capsid-like biomimicking nanoparticles. On top of these weak interactions, the formation of such structures in water is mainly driven by the phase-separation and the subsequent aggregation of hydrophobic and hydrophilic parts. The aggregation of hydrophilic dendrimers forms a layer that protects hydrogen bonds from weakening or dissociation due to the interaction with surrounding water by the shielding effect.16 In addition, phase-separation and aggregation of hydrophilic and hydrophobic peptides in water arrange associative groups close to each other, which promotes the hierarchical assembly of associative motifs. Formation of such clusters has a significant influence on physical and mechanical properties of the complex supramolecular structures.11
In addition, the engineering of functional supramolecular materials by pushing associative groups to form cluster domains can lead to the appearance of new functions like self-healing.17,18 Chen and coworkers designed supramolecular polymer networks based on brush copolymers of poly(styrene) and poly(amide-functionalized butyl acrylate).19 They demonstrated that each chain collapses into a core–shell-like morphology with a hard poly(styrene) core and a poly(amide-functionalized butyl acrylate) shell. By self-assembly of amide groups, a two-phase nanostructure is formed, which shows self-healing features. In addition, their results revealed that the mechanical properties of the network and the self-healing efficiency can be adjusted by the ratio of the poly(styrene) and poly(amide-functionalized butyl acrylate) and with that the degree of clustering.19
In summary, the aggregation of associative junctions within the microstructure of supramolecular polymers and networks is a robust approach to tune their physical and mechanical properties and to introduce specific functions into these materials. Employing this approach, however, requires an intensive understanding of the origin and the dynamics of such clusters. The target of this review is to highlight the importance and potential applications of clusters in supramolecular polymer materials such as thermoplastic elastomers and gels by summarizing numerous studies in which aggregation of supramolecular junctions into clusters is reported. This review is structured in four sections. In the first section, the physical origin of the cluster formation in different supramolecular polymer materials is discussed. In the second section, different methods for the characterization of supramolecular clusters are summarized. The effect of such clusters on the dynamics, physical, and mechanical properties of supramolecular polymer materials is discussed in the third section. Finally, in the last section, approaches for the application of such clusters in designing functional supramolecular networks are elaborated.
2. Origin
Supramolecular polymeric materials consist of self-assembled, small-molecule functional units or oligomeric/polymeric building blocks functionalized with associative groups that can form reversible noncovalent bonds. The latter can be divided into two main types: “main-chain” and “side-chain” supramolecular systems. In the first, the polymeric backbone is constructed by noncovalent bonds between telechelic elements. In the second, the polymer backbone is covalently jointed, and supramolecular associative motifs are grafted as side-groups along that backbone.20,21 Supramolecular polymeric materials are traditionally classified based on the type of their reversible interactions. Examples of different supramolecular bonds are numerous, whereby the frequently employed ones are hydrogen bonding, π–π interaction, metal–ligand coordination, ionic interactions, host–guest interactions, and hydrophobic association. Even more, several supramolecular polymer systems are developed by integrating multiple transient bonds or even their orthogonal combinations.22Regardless of their type, non-covalently associating units in supramolecular polymeric materials follow specific reaction pathways to form large assemblies, comparable with the synthesis mechanisms of covalent polymers.20,23 The simplest growth manner is the multistep isodesmic mechanism, where the binding constants are independent of the degree of association, and so all chains grow gradually in parallel by binding to new monomers or by combining with each other. This reaction pathway is quite analogous to the classical step-growth polymerization, where long chains can only be obtained at ultimate conversions. In contrast, in many supramolecular systems that form ordered chiral assemblies, additional supramolecular interactions with dominant binding constants emerge when the association degree reaches a specific threshold. This cooperative nucleation–elongation mechanism has several analogies to the classical chain-growth polymerization mechanism, e.g., the preferential addition of monomers to the growing chains instead of forming new ones, since even if the nucleation is not thermodynamically unfavorable, it has a slower rate constant compared to the growth.23 Moreover, large association degrees can be found at all conversions, which results in faster structure formation in contrast to materials formed through the isodesmic pathway. Unlike the classical polymerization methods, these reaction lanes are not monomer specific, and depending on many factors like electronic effects, spatial limitation, or the polarity of the medium, unique types of supramolecular moieties may choose any of the growth pathways.
Irrespective of the formation pathway, aggregation of supramolecular associations provides a heterogeneous structure that exhibits different properties compared to a system of homogeneously distributed binary associations. Polymer networks, either chemical or physical, generally contain inevitable degrees of inhomogeneities on the local and global scales.24 The former consists of network defects, like unreacted dangling ends, network misconnectivities, and different types of loops, at length scales of 1–10 nm. The latter includes inhomogeneity in the spatial distribution of network junctions, which result from concentration fluctuations during the network formation, on scales of 10–100 nm.25–28 On top of that, aggregation of supramolecular associations causes another type of heterogeneity in supramolecular polymer networks, as schematized in Fig. 1. Due to the differences between the thermodynamic specificities of the supramolecular associations and the polymer precursors, often supramolecular units phase-separate into domains of irregular aggregates referred to as clusters. The irregular aggregates inside clusters can subsequently rearrange and create hierarchical assemblies by secondary forces and form clusters of ordered structures, as illustrated in Fig. 1. Therefore, these aggregates are different from the pre-shaped clusters such as polyoxometalates (POMs), fullerene (C60), or polyhedral oligomeric silsesquioxane (POSS), which are formed by the covalent bonding of inorganic building blocks, and are frequently integrated in polymer matrixes to add specific functions like catalytic, optical, and electronic activities that such clusters offer.29
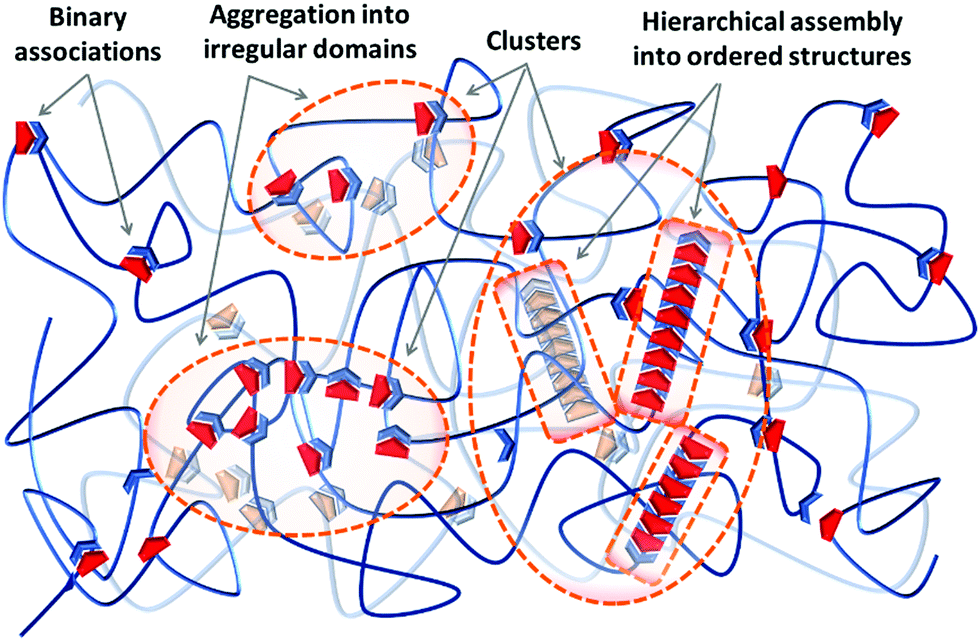 | ||
| Fig. 1 Definition of the terms used in this review: binary associations are formed by combination of two supramolecular units, either homo- or hetero-complementary. Thermodynamic specificities force binary associations to phase-separate into irregular aggregates known as clusters. The presence of secondary forces can encourage aggregates to form ordered hierarchical assemblies. | ||
In nature, biological materials integrate supramolecular bonds in such hierarchical designs as well, thereby functioning coordinately on several time and length scales. For instance, the double helix of DNA can assemble into ordered chiral structures in aqueous solutions due to the cooperation of lateral hydrogen bonding and hydrophobic interactions.23 Similarly, the hard coating of mussel byssal threads is based on irregular clusters rich in catechol–iron complexes, which exhibits a remarkable combination of hardness and extensibility.30 The emergence of alike clusters in synthetic supramolecular materials, either in ordered or irregular morphologies, can similarly change multiple essential properties that are less discussed and developed so far compared to the supramolecular polymers based on simple binary associations. Therefore, it is necessary to understand the mechanisms for the formation of such clusters either to avoid or to purposely integrate them in a tunable fashion if required. Regardless of the type of the utilized supramolecular group or the physical state of the material—either it is a gel swollen in a solvent or it is in the solid state—there are similar design concepts to create and control the extent of cluster formation in supramolecular materials, which are discussed in this section. The fundamentals of the initial phase-separation step are the same for both clusters of irregular aggregates and ordered hierarchical assemblies, as discussed in Section 2.1. The possibility of forming hierarchical assemblies aided by secondary forces is determined by additional design concepts that are discussed in Section 2.2.
2.1. Phase-separation into irregular domains
In classical block copolymers, phase-separation normally happens upon decreasing temperature, which is aided either by crystallization of a hard block or simply by thermodynamic incompatibility of blocks. Consequently, even with unentangled constituting copolymers, a robust network of interconnected phase-separated microdomains with tunable mechanical properties can be prepared. Such materials are classified as thermoplastic elastomers (TPEs) and have several industrial applications.31 These benefits of phase-separation in block copolymers have encouraged researchers to control the morphology and thereby the mechanical properties of supramolecular polymeric systems in the same way.32 The simplest analogy includes ABA-type block copolymers with hard A blocks and a soft B block, which can form a percolated network when the hard blocks phase separate into hard nanoscopic domains. Similarly, in supramolecular polymeric systems, the strength of transient interactions of kinds as discussed above is expected to be enhanced if they form phase-separated nanoscopic domains. In one of the first examples, Stadler and coworkers reported about the synthesis of polybutadiene (PBd) randomly grafted by phenyl urazole derivatives, where a thermo-reversible elastomeric network forms due to phase-segregation of the highly polar urazole groups.33 This notion has inspired many researchers to form mechanically robust thermo-reversible networks based on weakly interacting supramolecular associative groups.Boutieller and coworkers have further clarified the effect of structural design parameters on the phase-separation scope, by grafting bisurea groups through aromatic linkers on poly(dimethylsiloxane) (PDMS). The hydrogen bonding groups are either grafted along the backbone to form side-chain supramolecular polymers or as end groups on linear chains to create telechelic precursors.37 This work demonstrated that specific requirements have to be met to obtain phase-segregated TPEs. Although the side-chain systems create physical crosslinks by ordered phases of hydrogen-bonded bisureas, they are not as ordered as telechelic systems are. Moreover, too short or too long telechelic PDMS chains do not create strong TPEs, due to the lack of entanglement in the former and insufficient concentration of physical crosslinks to form a percolated network in the latter. Similarly, Goldansaz, Ahmadi and coworkers have studied diblock copolymers based on poly(n-butyl acrylate) (PnBA) and poly(hydroxyethyl acrylate) (PHEMA) segments, where the latter is functionalized by strong hydrogen bonding Ureido-pyrimidinone (UPy) motifs.38 The studied materials set up a three-dimensional network through segregation of UPy-functionalized PHEMA blocks only at a specific length of the hydrophobic PnBA segments: too short PnBA segments do not form a percolated network due to lack of entanglement, whereas too long PnBA blocks deteriorate the network percolation by lowering the segregation tendency of PHEMA blocks.
More recently, Binder and coworkers have studied the impact of phase-separation in supramolecular polymeric systems based on mono- and bifunctional poly(isobutylene) (PIB) chains grafted with weak hydrogen bonding motifs like Thy, diaminotriazole, barbituric acid, and a Hamilton wedge.17,39,40 All their studies report a similar pattern that despite the low association constants, the undirected supramolecular interactions of the homo-aggregated motifs form clusters with diverse irregular shapes and architectures, as illustrated in Fig. 2a. By contrast, formation of clusters is suppressed in blends due to the strong directional interaction between the hetero-complementary motifs. The size of micellar homo-aggregates can be reduced by introduction of hetero-complementary supramolecular chains, which results in TPEs with a percolated network due to the enhanced connectivity between clusters.40 Moreover, the phase-separated systems show stronger temperature dependences compared to the hetero-complementary hydrogen-bonded ones. This is attributed to the lower temperatures for the onset of cluster fragmentation in the former compared to the relatively high dissociation temperature of the strong binary hydrogen bonds in the latter.
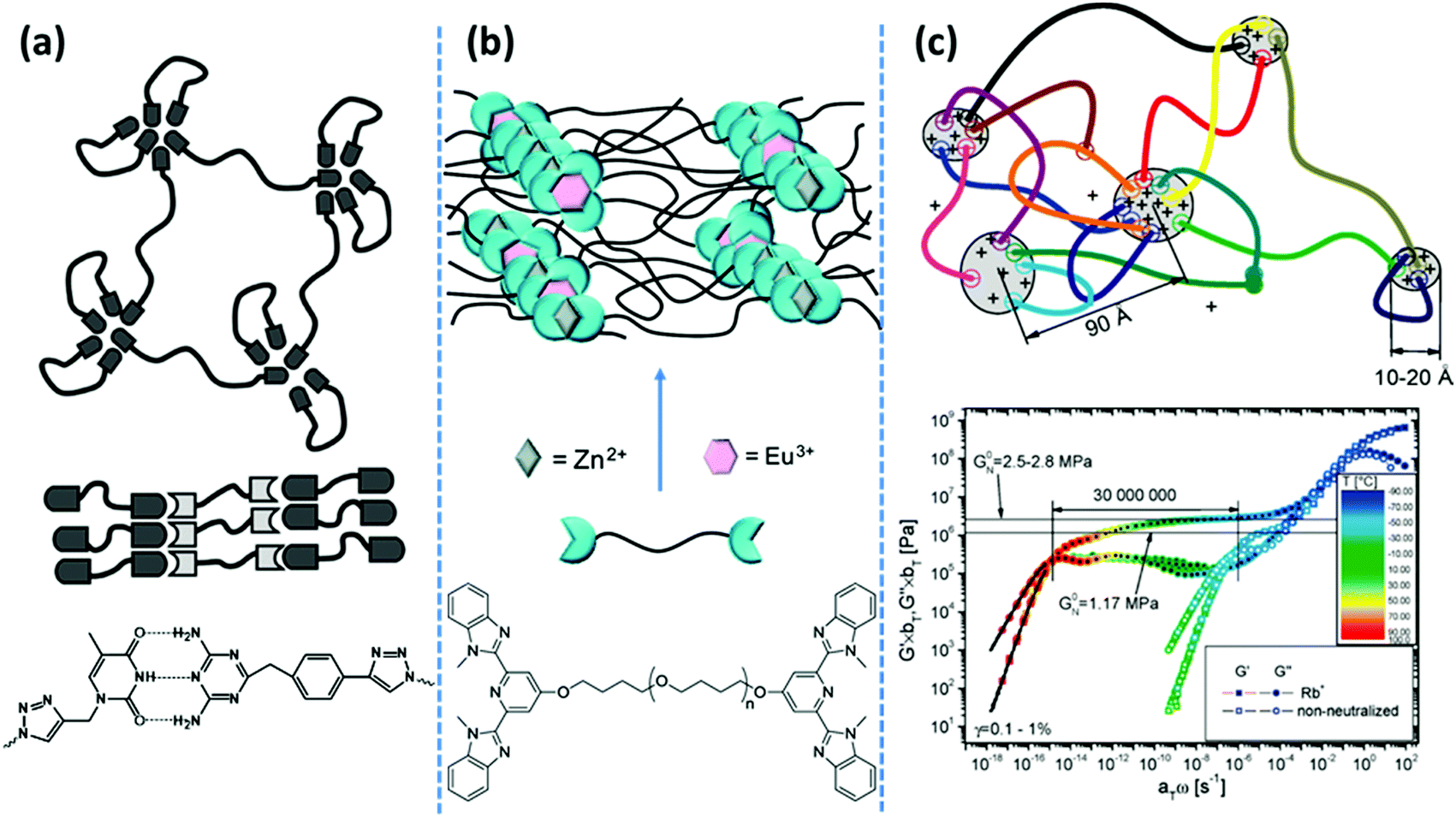 | ||
| Fig. 2 Representative examples of phase-separated supramolecular polymeric systems: (a) in hydrogen bonding-based systems, the nondirectional assembly of homo-aggregated diaminotriazine units forms segregated domains with irregular shapes, whereas its directional association with thymine suppresses phase-separation.39 (b) In metal–ligand coordination-based systems, phase-segregation of Zn2+–bis(Mebip) and Eu3+–tris(Mebip) complexes forms a 3D network from telechelic precursors.99 (c) In ionic interaction-based systems, aggregation of polar end-groups with counterions prolongs the terminal relaxation for more than seven orders of magnitude.43 Panel (a) adapted from ref. 39; copyright 2010, American Chemical Society. Panel (b) adapted from ref. 99; copyright 2012; American Chemical Society. Panel (c) taken from ref. 43; copyright 2009, American Chemical Society. | ||
Likewise, in main-chain ionomer systems based on PTHF segments that are covalently linked by sulfonated isophthalic acid units, neutralization with sodium salt results in nine orders of magnitude prolongation of the terminal relaxation. In a sharp contrast, equivalent systems with a more polar poly(ethylene glycol) (PEG) segment just show mild changes in their rheological behavior. A notable dependence of Tg on the neutralization degree in the PEG-based systems, contrary to the PTHF-based ones, demonstrates that the ionic associations reside in the polymer matrix in the former, whereas they create individual clusters in the latter.45 The aggregation of ionic groups has been also used as a tunable design parameter to form ionic hydrogels by mixing ABA-type triblock copolymers with oppositely charged end blocks. Solubility of the system in water is guaranteed by utilization of PEG as the middle block. Mixing solutions of block copolymers with strong ionic groups like sulfonate and guanidinium in the end blocks forms highly stable and resilient hydrogels even at low polymer concentrations.46 Network formation is attributed to the creation of interconnected clusters of coacervate phases. The extent and lifetime of such phases are tunable in response to external stimuli such as pH and salt concentrations. Such a design concept is similarly used in the formation of robust hydrogels by mixing oppositely charged natural polymers such as hyaluronic acid and chitosan.47
In another example, incompatible polyether ionomers extended with sulfonated isophthalic acid are blended to regulate the phase-separation.48 The ionic groups are soluble in the polar PEG matrix and thereby hold the material's electrical conductivity. In contrast, ionic groups phase-separate in the apolar PTHF domains. Consequently, a wide spectrum of combined conductivity and physicomechanical properties can be obtained by blending different fractions of PEG- and PTHF-based components and even through changing their chemical composition.48
Analogous methods are used for the formation of double network hydrogels based on polyampholytes. These hydrogels are formed by free radical polymerization of oppositely charged monomers. Aggregation of the oppositely charged ionic groups into clusters occurs along the polymerization by continuous reduction of the overall entropy due to the chain growth. Therefore, due to different formation histories, clusters with different sizes are formed throughout the reaction. The distribution of cluster size translates into different strengths and lifetimes: the stronger clusters impart elasticity whereas the reversible breakage of the weaker ones integrates toughness.49
Besides the reversible fragmentation, the colloidal diffusion of clusters is another way of stress relaxation specifically in systems based on unentangled or monofunctional precursors, which do not form a percolated network of interconnected micelles.40,44 Nevertheless, the final relaxation is not experimentally accessible in entangled systems that contain long-lasting aggregates.56,57 In those cases, a low-frequency plateau modulus is observable, which can be correlated with the fraction of chain segments that are trapped between long-lasting clusters, as shown in Fig. 3.58,59 These segments can only partially relax according to the Constraint Release Rouse (CRR) process.59 This process includes the lateral motion of the trapped segments by the Rouse mechanism, which is activated once the free neighboring chains are relaxed. In such cases, the entanglement of polymer precursors is reported to induce an upper limit for the fraction of trapped segments.56,58
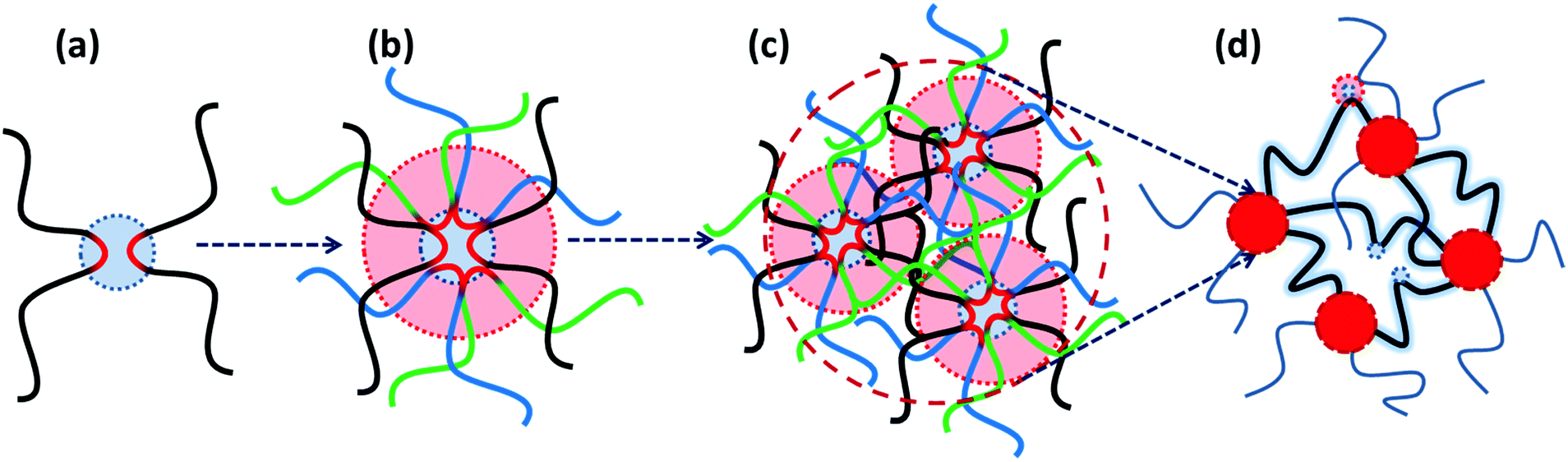 | ||
| Fig. 3 Schematic representation of the phase-separation process in supramolecular polymeric materials: (a) doublet connection; (b) aggregation of doublets into larger multiplets is limited by the necessary extension of surrounding chain segments; (c) overlapped extended segments form clusters; (d) interconnected clusters form a percolated network. The highlighted trapped segments can store elastic energy. The matrix can also contain individual supramolecular bonds, binary associations, or even multiplets. | ||
More recently, van Ruymbeke and coworkers used a combination of different techniques including dielectric spectroscopy, SAXS, and rheology to propose a general picture of the microstructure of entangled side-chain supramolecular polymeric materials.60 The material basis of their study includes PnBA bearing different fractions of weakly hydrogen bonding carboxylic acid side-groups. Quantitative agreement between the specific dielectric relaxation modes of a pure poly(acrylic acid) (PAA) and the modified PnBA systems suggests that distinct acrylic acid (AA) domains are embedded in a pure PnBA matrix. However, retardation of segmental motion found in rheology and a slight increase of Tg upon increasing the fraction of AA units suggests that the binary associations and free AA units are also present in the PnBA matrix. The temperature-dependent evolution of the structure and dynamics demonstrate a pattern similar to the former reports by Stadler and Rowan.36,43 Upon increasing the temperature, the modulus increases initially followed by a drop beyond a specific temperature. The drop of the modulus in some cases includes a two-step process. This pattern can be understood with the help of a rather historical explanation suggested by Eisenberg and coworkers for the morphology of random ionomers.61,62 This picture is quite similar to the explanation provided by Spiess and coworkers about the morphology of ethylene oxide oligomers that are tethered with imidazole heterocycles, based on the solid-state NMR studies.63 In this picture, the highly polar supramolecular associative groups tend to phase-separate from the commonly less polar polymer backbone to reach the most stable thermodynamic equilibrium. This is further encouraged once secondary interactions can form and thereby lower the Gibbs free energy even more.60 Binary associations gradually gather together to form quadruplets, sextuplets, and larger aggregates called multiplets, as shown in Fig. 3a and b.61,62,64,65 Polymer segments around multiplets have to significantly extend while matrix chains have a random coil conformation. Thus, the limited extensibility of chains sets an upper limit for the growth of multiplets. At a larger fraction of supramolecular motifs, the extended chain regions overlap and form clusters that expose their individual Tg, as illustrated in Fig. 3c. Although clusters tend to combine and thereby reduce the surface tension, this is prohibited or at least significantly slowed down in the polymeric environment due to the entanglement of neighboring chains and entrapment of polymer segments between long-lasting clusters, as highlighted in Fig. 3d.58,59 The extended polymer layer is free of supramolecular associative groups, whereas the inter-cluster region may contain free motifs, binary associations, and even individual multiplets, as noted in Fig. 3d. By increased polymeric dynamics at high temperatures, those individual entities find the chance to join the existing clusters. This results in a frequently reported initial modulus upturn upon increasing temperature.36,43 Annealing the sample at such temperatures brings the cluster closer to the most stable configuration. However, this is normally hard to be experimentally accessed, specifically in the presence of entanglements.43 By further increase in temperature, clusters finally disappear beyond a temperature threshold once the elastic energy of the extended polymer segments around multiplets compensates the gain in Gibbs free energy upon the destruction of clusters. The binary associations, however, may still exist until temperatures beyond which the association equilibrium lies on the dissociation side. This process may be reflected as a two-step relaxation process in the rheological behavior.
2.2. Hierarchical assembly into ordered structures
Examples of biological materials that employ a combination of different supramolecular motifs to function adaptively in versatile conditions are numerous. Inspired by such biomaterials, researchers have been and still are encouraged to combine multiple noncovalent forces to form nanoscopic microstructures by the hierarchical assembly of supramolecular groups.22,66,67 Such molecular designs normally lead to regular arrays of supramolecular bonds, if spatial constraints allow. Often, multiple design principles are coupled to obtain the desired microstructure. Several design factors can be extracted from the existing diverse reports by a thorough examination. In the following, we decouple such design principles.Aida and coworkers have reviewed the formation of highly-ordered shape-persistent nanostructures by the cooperative assembly of small-molecule associative units that orthogonally employ hydrogen bonding and π–π stacking.69 Such structures are incorporated in polymeric systems either as individual modifiers, like nucleating agents for promoting crystallization,70 or as covalently grafted supramolecular units. One of the associative motifs that have been widely studied for its orthogonal assembly is the quadruple hydrogen bonding UPy group. The self-complementary association of small-molecule UPy monomers in chloroform results in a significant viscosity enhancement due to the formation of extended linear chains, when bifunctional monomers are used, and the creation of a three-dimensional network in case where trifunctional monomers are utilized.71 Moreover, viscosity is reported to decrease upon addition of monofunctional stoppers, which suggests that the self-assembly of UPy monomers follows the isodesmic growth mechanism. In contrast, coupling of the same motif on π-conjugated oligo(p-phenylenevinylene) (OPV) results in unidirectional growth of polymer-like ribbons through hydrogen bonding reinforced π–π stacking, as shown in Fig. 4a.72 Here, monofunctional monomers create ordered structures, whereas bifunctional monomers, formed by chemical linking of UPy motifs with a hexamethylene spacer, provide less ordered chiral stacks.73 Similarly, mono- and bifunctional OPVs with ureidotriazine supramolecular functional groups form chiral stacks in nonpolar solvents like dodecane.73
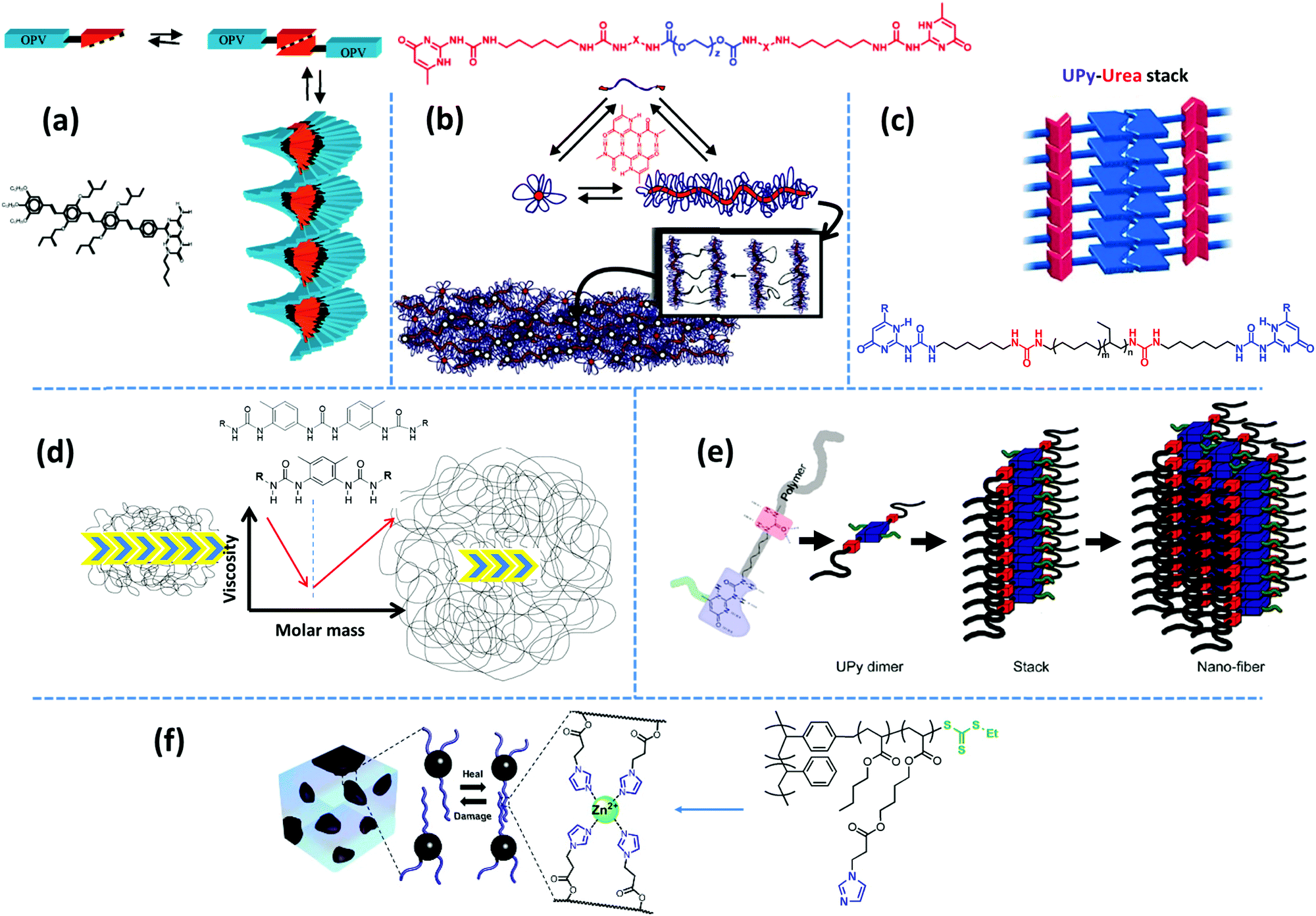 | ||
| Fig. 4 Representative examples of hierarchical supramolecular assemblies aided by secondary forces: (a) stacking of aromatic OPV units assisted by strong hydrogen bonding motifs.73 (b) Quadruple hydrogen bonding UPy groups shielded by hydrophobic alkyl linkers form nanofiber aggregates.81 (c) Fibrous aggregates form upon stacking of UPy dimers aided by lateral urea hydrogen bonds.87,89 (d) The polymer core length and compatibility affect the aggregation strength, which determines whether the mechanical behavior is dominated by supramolecular assemblies or polymeric dynamics.93,95 (e) Spatial hindrance of the substituent at the sixth position determines the inter-stack's distance.100 (f) Metal–ligand complexes are embedded in the soft matrix of polyacrylate arms in a polymer brush with a PS backbone.108 Panel (a) taken from ref. 73; copyright 2001, American Chemical Society. Panel (b) adapted from ref. 81; copyright 2012, Wiley. Panel (c) taken from ref. 89; copyright 2010, Wiley. Panel (d) taken from ref. 93; copyright 2015, Elsevier. Panel (e) taken from ref. 100; copyright 2011, American Chemical Society. Panel (f) adopted from ref. 108; copyright 2014, American Chemical Society. | ||
Benzene-1,3,5-tricarboxamide (BTA) is another famous associative motif, which forms nanorods in solution and solid by π–π stacking of benzene groups fortified by triple, rather weak, lateral hydrogen bonds.74 Introduction of bulky side groups in BTA is reported to add helicity to the columnar aggregates. Meijer and coworkers have studied the structure formation in main-chain and side-chain supramolecular polymeric systems based on PEB backbones and BTA associative motifs.75 The integration of BTA in amorphous PEB systems also results in their orthogonal assembly into phase-segregated nanorods and the consequent appearance of elastomeric properties.75,76
Following the same concept, Meijer's group has also reported on the formation of hydrogels using UPy functionalized telechelic PEG chains.80 The hydrophobic shielding of an aliphatic dodecyl spacer is proposed to be responsible for the preservation of the columnar π–π aggregates in the aqueous ambiance and thereby the formation of three-dimensional networks. In a succeeding study, the same group systematically varied the length of the alkyl spacer and the PEG backbone. The authors discovered that the network formation is enhanced by the length of the alkyl spacer whereas the length of the PEG core has an opposite destabilizing effect.81 The network structure is illustrated as interconnected bundles of nanofibers that are shielded in a hydrophobic alkyl spacer pocket, as illustrated in Fig. 4b. A first-order thermal transition is reported to happen exactly at the same temperature where rheological data show a gel–sol transition. This transition is correlated to the dissociation of bundles, which can be inverted upon cooling, but with a time delay of up to several days.81 The size of phase-separated bundles can be reduced by introducing a monofunctional supramolecular precursor. This enhances the inter-nanofiber connectivity and consequently improves the network strength and stability.82 This modification approach is quite analogous to the integration of the unidirectional hetero-complementary associating hydrogen bonds to the phase-separated network of undirected self-complementary counterparts, thereby enhancing the network connectivity with the expense of decreasing the size of supramolecular aggregates.40 Such reinforcing effect is also in accordance with the conclusion of Wang and coworkers that preventing stickers from associating into large aggregates significantly slows down the stress relaxation.52 The side-chain variant of this supramolecular design also shows phase-separated UPy clusters shielded by alkyl spacers. However, the chemical linkage between nanofibers promotes network connectivity and results in much stronger and resilient TPEs in the solid state and tough hydrogels upon immersion in water.83 In the same way, Bouteiller and coworkers have recently prepared supramolecular nanorods by the self-assembly of tris(urea) stickers connected on both sides through alkyl spacers of different lengths to short PS arms.84 They have shown that long supramolecular nanorods can be obtained when a nine carbons spacer is used, whereas five times shorter nanorods are achieved with a spacer of only five carbons, and even only spherical nanoparticles are formed in the absence of the spacer. This is attributed to the spacer's hydrophobic shielding of the hydrogen bonding associations.
Hydrophobic shielding is also reported to play an important role in stabilizing weak hydrogen bonds in aqueous media. This can be achieved by a delicate manipulation of the structure of supramolecular groups even through the addition of a small hydrophobic alkyl substituent adjacent to the associative motif. This design concept has been frequently used to form tough hybrid hydrogels.85,86 For instance, polymerization of methacrylic acid (MAAc) in aqueous media leads to the formation of polymer-rich aggregates containing multiple hydrogen bonds that are stabilized by hydrophobic interactions due to the presence of the α-methyl groups in poly(MAAc).86
Unexpectedly, a telechelic supramolecular polymeric system utilizing UPy motifs linked through urethane linkages on a polar polycaprolactone (PCL) precursor has also demonstrated the formation of fibrillar aggregates as inferred from the emergence of a low-frequency plateau modulus in the rheological spectra.88 The presence of fibrous aggregates is further confirmed by AFM imaging through the application of an amorphous PCL counterpart. In contrast, another systematic study on the aggregation of small-molecule monofunctional UPy motifs, containing a hydrophobic aromatic tail, reveals that the presence of the urea linkage is necessary for the formation of one-dimensional stacks in various solvents, whereas the urethane linked counterpart is incapable of aggregation.89 The synergistic effect of solvophobic shielding and supplementary lateral hydrogen bonds on the formation of fibrous UPy aggregates was subsequently used for the development of tough hydrogels, as discussed in the previous section.80,82
Following the original report of Boutieller and coworkers37 on the phase-separated bisurea functionalized systems, Callies, Creton, and coworkers have studied the structure and morphology of center-functionalized PnBA chains with weak bisurea and strong triurea supramolecular motifs.93–95 For either system, there is a molar mass threshold below which the assembly of supramolecular motifs into large bundles of rod-like aggregates controls the materials’ mechanical properties, as illustrated in Fig. 4d. In samples based on the strongly associating triurea motifs, this is manifested by the appearance of a low-frequency plateau modulus. Increasing the molar mass in this regime reduces the segregation tendency and consequently decreases the viscosity due to the effect of enhanced polymer dynamics.91 In contrast, above the critical molar mass, although randomly oriented small rods may appear in the strongly bonded supramolecular systems, the viscoelasticity is mainly controlled by entangled polymer chains, as depicted in Fig. 4d. Consequently, the terminal relaxation time and the viscosity increase with the molar mass in both systems.93,95 Meijer and coworkers have recently investigated the temporal structure development of UPy functionalized telechelic PEG systems in aqueous environment by using a combination of several microscopy and spectroscopy techniques.96 The authors demonstrate that the length of PEG chains destabilizes fibrillar aggregates of supramolecular units due to the steric and entropic effects. More interestingly, mixing monofunctional UPy modified PEG precursors with increasing amounts of bifunctional counterpart results in the destabilization of ordered fibers that are obtained from the sole monofunctional precursors.
Incompatibility of the polymer core is the other factor that can align supramolecular assemblies in ordered structures. For instance, the presence of the urea linkage is reported to be necessary to form fibrillar UPy aggregates in telechelic PCL and PEB systems.87,88 In contrast, aggregation of UPy groups takes place in the nonpolar PDMS system without any supplementary urea linkages.97 This is in accordance with former reports that reveal bisurea groups are capable to phase separate into microdomains with a sharp melting peak in telechelic PDMS systems.37 Along with the former discussion on the effect of chain length, the authors have shown that by increasing the molar mass of PDMS chains and the respective enhancement of the polymeric dynamics, fibrillar UPy aggregates turn into constrained spherical domains.97
In a systematic study, Meijer's group has evaluated the effect of the media's polarity on the self-assembly of small-molecule BTA motif in solution and when incorporated in telechelic polymeric systems.76 The medium polarity was gradually changed by varying the solvent composition in solution, and by replacing the nonpolar PEB polymer core with relatively more polar PTHF and PCL in the solid state. The size and stability of nanorods gradually decrease by increasing the polarity and eventually supramolecular associations disappear in most polar systems. Similarly, aggregation of monofunctional UPy monomers into one-dimensional stacks significantly diminishes upon the replacement of the hydrophobic aromatic tail with a hydrophilic triethylene glycole (TEG) group.89 Besides the lack of adequate solvophobic shielding, this is attributed to the competing hydrogen bonding of TEG groups which undermines the UPy dimerization. Likewise, in the side-chain UPy functionalized polyacrylates, it is reported that the fraction of trapped segments decreases in the presence of free HEMA units. This is interpreted as the reduction of UPy stacking due to the competitive hydrogen bonding of free hydroxyls on HEMA groups.56,57,98
In the same way, other types of supramolecular motifs demonstrate escalated phase-separation and clustering when attached to a more incompatible polymer core. For instance, telechelic metallo-supramolecular polymeric systems based on Mebip ligand show much longer relaxation times and larger storage moduli once PTHF core is replaced by an apolar PEB.41,42,99 This is also accompanied by the appearance of a higher number of Bragg peaks in the SAXS spectrum, which confirms larger microphase-separated domains with a long-range order compared to the case where PTHF core was employed.99
Along the same lines, a low-frequency plateau emerges in unentangled side-chain UPy grafted poly(lauryl methacrylate) (PLMA) systems due to the aggregation of UPy dimers, which diminishes upon introduction of the bulky adamantyl group in the sixth position.56 Surprisingly, this modification works oppositely in entangled systems. The low-frequency plateau in such systems is addressable to segments that are trapped between long-lasting clusters. Therefore, although the presence of a bulky substitute decreases the ordering of phase-separated domains, the fraction of trapped segments does not change significantly.
In another variant of the multi-phase supramolecular polymeric design, supramolecular bonds are embedded in one of the phases so that the unique specificity of the other phase can be independently employed. For instance, Guan and coworkers have designed a polymer brush with polystyrene (PS) backbone and amide functionalized PnBA arms, where the hard backbone segments phase-separates into microspheres dispersed in the matrix formed by the soft arms. The weak hydrogen bonding of amide groups provides a rapid self-healing in the virtue of the dynamic environment, whereas hard blocks strengthen the material.19 In an alternative design, as shown in Fig. 4f, an imidazole motif is embedded in poly(ethyl acrylate) arms, which can form relatively weak reversible metallo-supramolecular bonds via coordination with a metal ion like Zn2+ and provide similar self-healing properties.108
3. Structural characterization
Characterization of supramolecular polymers is usually challenging since stimuli from the surrounding environment significantly influence their molecular organization.109 This argument is valid as well for supramolecular networks. In addition, the common hierarchical assembly of supramolecular units of these networks, as discussed in Section 2, makes their characterization more difficult than conventional polymers. Structural characterization of the supramolecular polymer networks that contain aggregation of associative motifs and clusters must therefore be performed by the convergence of multiple techniques,40,110,111 as mentioned already by Stadler probably for the first time in 1998.112 To introduce the most important characterization techniques applied in such networks, this section is divided into four subsections based on the type of methods including thermal analysis, spectroscopy, scattering, and microscopy.3.1. Thermal analysis
One of the most common thermal-based analysis methods for characterization of (supramolecular) polymers is differential scanning calorimetry, DSC. This technique is generally used to determine thermal properties of materials such as heat capacity.113 In a typical DSC measurement, temperature varies with a constant rate, such that the probe sample and a reference one, with known thermal properties, follow a similar temperature profile. When the probe sample undergoes a thermal transition, a temperature lag occurs in the sample compared to the reference. To compensate this lag, the thermal power applied to the sample is changed. The difference between the initial and the new thermal powers is used to determine the heat capacity of the transition.113–115 Polymer properties like the glass transition temperature, Tg, melting point, crystallization temperature, or the degree of crystallinity are commonly characterized by DSC. These data can be even correlated to the morphology of the probed sample. As an example, DSC of a semi-interpenetrating network or a blend of two polymers may show two Tgs, due to the existence of two polymeric phases. These Tgs can be shifted in comparison to the Tg of the individual components. The origin of this shift is the hindrance of the polymeric segmental mobility of one phase due to the additional interactions with the other polymeric phase. Therefore, the magnitude of the shift is a criterion showing the degree of interpenetration or miscibility of two polymeric components and indirectly describes the phase-separation phenomenon in such systems.116DSC is widely used to characterize supramolecular polymer material as well, since this technique can indirectly provide valuable information about their microstructure. Specifically, aggregation of associative groups in these materials can be characterized by DSC if they form crystalline domains. Besides, the variation of Tg of the polymeric building blocks upon functionalization with associative groups can be studied by DSC.
Accordingly, DSC has been employed to study the clustering of PTHF with Ade and Cyc as weak hydrogen bonding associative groups.88 In a first heating run of an PTHF–Ade sample, endothermic transitions at 86 °C, 108 °C, and 135 °C were observed. The second heating exposes reduced melting peaks at 108 °C and 135 °C. The first DSC heating of PTHF–Cys sample, however, reveals only one melting transition at 88 °C.36 It has been concluded from these observations that the difference in the number of endothermic transitions between these two samples provides evidence of different aggregation types or aggregation mechanisms. Crystalline domains formed by stacking of UPy groups melt upon heating and can thereby be characterized by DSC. The enthalpy of melting is considered as a measure of the stability of stacks.88
In a library of UPy–urea functionalized telechelic polymers with an amorphous PEB backbone (Mn = 3500 g mol−1), the hierarchical self-assembly of UPy groups has been investigated by different techniques including DSC.100 While PEB is an amorphous polymer, DSC results of supramolecular associative samples demonstrate melting transitions, which confirms the stacking of UPy groups into crystalline domains. By change of the substituent at the sixth position of the UPy groups, melting points and corresponding melting enthalpies are significantly varied (81–130 °C and 9.33–0.09 J g−1, respectively). To investigate the kinetics of stacking and nanofiber formation in more detail, the authors used DSC to study the crystallization of these polymers as a function of time. Again, samples with various UPy groups with different substituents on the six position of their cytosine ring show different crystallization kinetics. They either reach the thermodynamic equilibrium in 24 h or show no crystalline domains even after several days.
Other characterization methods such as spectroscopy or scattering might be performed upon variation of temperature. In such a hybrid approach, structural characterization of supramolecular polymer materials can be achieved as a function of temperature. This strategy is discussed in the corresponding subsections.
3.2. Spectroscopy methods
Spectroscopy is a characterization technique based on the interaction between matter and electromagnetic waves. In infrared (IR) spectroscopy, data originate from the vibration of the atoms of a molecule.117 In this technique, the probe sample is irradiated by an IR beam, and the resulting interaction is characterized by the fraction of the incident radiation that gets absorbed at a particular energy. For “IR-active” molecules, this interaction leads to variation of the molecular dipoles associated with vibration and rotation of chemical bonds. Molecules with this criterion have their own specific IR spectrum including distinct characteristic peaks, each having a frequency that matches the stretching/bending vibration of a specific atomic bond. The main application of IR spectroscopy is the identification or characterization of organic molecules, polymers, and biological units.117,118In supramolecular polymers and networks, IR spectroscopy is commonly utilized to characterize hydrogen bonding between associative motifs.119 Usually, stretching of hydrogen-bonded carbonyl or amine groups shows characteristic peak at lower wave numbers (approximately 20 cm−1) compared to non-bonded ones. Variation of the intensity and/or position of the IR peaks by temperature are reliable proofs of association or dissociation of hydrogen-bonding interactions. In many examples of research on supramolecular polymeric networks, IR spectroscopy has been utilized for such temperature-dependent characterizations. One of the well-known historical examples is the work of Stadler and coworkers who studied IR spectra of PBds modified with phenylurazole groups. The authors observed that stretching of free and bonded carbonyl groups appear at 1723 cm−1 and 1701 cm−1, respectively. By increasing the temperature, the intensity of the bonded carbonyl peak decreases, while that of the free carbonyl peak increases. The authors also demonstrate that the mole fraction of bonded and free urazole groups can be calculated from the area underneath the absorption peaks.119 IR spectroscopy was also employed to study PTHF functionalized with Ade and Cyc derivatives. At 35 °C, N–H stretching of PTHF–Ade appears at 3455, 3371, and 3298 cm−1. The first and the two latter peaks correspond to the free and hydrogen-bonded N–H, respectively. At this temperature, carbonyl stretching shows three peaks at 1734, 1699, and 1661 cm−1. Similarly, the first and the two latter values are consistent with the stretching of unbound and hydrogen-bonded aromatic/aliphatic carbonyl groups. Upon heating up to 90 °C, the stretching vibration of unbounded N–H at 3455 cm−1 disappears. Moreover, the hydrogen-bonded characteristic peaks merge to a broader peak centered at 3311 cm−1 for N–H and at 1680 cm−1 for carbonyl stretching.36 This observation is attributed to the rearrangement of phase-separated domains, which includes integration of the free associative units inside existing clusters.
Temperature-variable IR spectroscopy can also be utilized to indirectly investigate the hierarchical assembly upon the aggregation of associative groups like UPy. For UPy groups linked to PEB building blocks via urea linkages, the characteristic peak of urea carbonyls at 1630 cm−1 shifts to higher wavenumbers upon heating to 140 °C, which indicates a loss of lateral urea hydrogen bonds and hence destruction of the UPy stacks. When the sample at the melt state is cooled down, vibration at 1630 cm−1 is recovered, as shown in Fig. 5a.100 The time required for this recovery depends directly on the crystallization kinetics of the UPy groups. As another example, IR spectroscopy has been used to characterize the complexation of chain-folding PI with pyrenyl end-capped PU, where a network forms via intercalation of the pyrenyl end groups into the chain-folds of PI by π–π stacking. Temperature-variable IR spectroscopy of this blend demonstrates the reduction of the intensity of bonded N–H and carbonyl peaks upon increasing temperature from 25 °C to 120 °C. In addition, the intensity of the peak correlated to the free carbonyl bond increases simultaneously.106,107
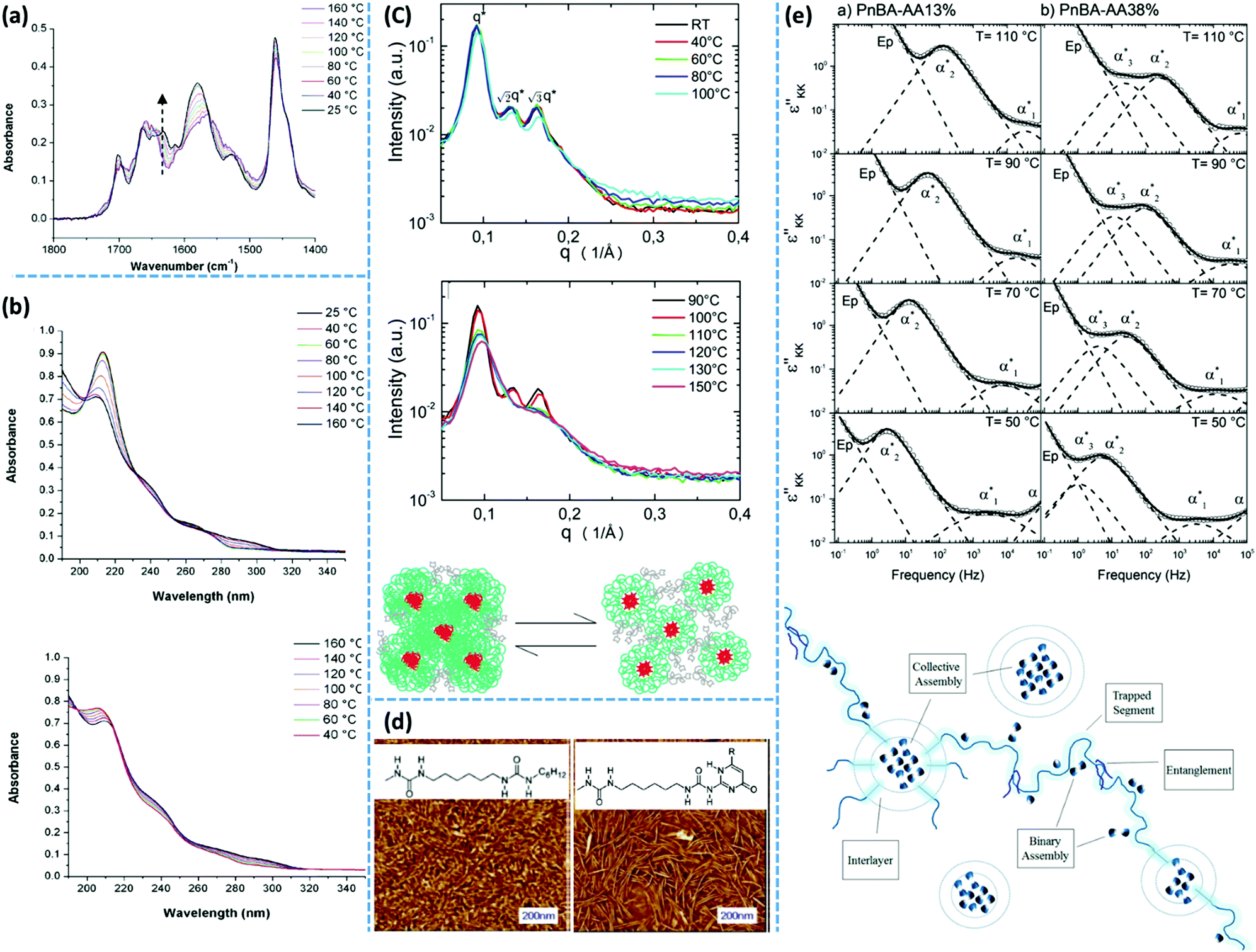 | ||
| Fig. 5 Representative applications of different characterization techniques for evaluation of supramolecular polymer networks subject to stacking of their associative groups: (a) temperature-variable infrared spectroscopy and (b) UV-vis spectroscopy of telechelic PEB functionalized with UPy groups.100 (c) SAXS spectra of monofunctional PIB with 2,6-diaminotriazine motifs and the corresponding proposed morphology.40 (d) AFM image of UPy-functionalized PEB demonstrates nanofibers while that of urea-functionalized does not.87 (e) Dielectric spectroscopy of PnBA functionalized with AA groups and the proposed general picture of the morphology.60 Panels (a and b), (c, d, and e) reprinted from ref. 100, 40, 87 and 60, respectively. Copyright 2011, 2014, 2006, and 2016, American Chemical Society. | ||
Nuclear magnetic resonance (NMR) is another spectroscopic method in which properties of certain nuclei are exploited. The nuclei are spin 1/2 isotopes such as 1H, 13C, 15N, 19F, or 31P that exhibit magnetic dipole moments.120 The diverse interactions of nuclear spins with themselves and with their surroundings including chemical shifts, dipole–dipole couplings, or J-couplings can provide information about the structure and dynamics of the probed sample.
One of the important applications of NMR is structural characterization and study of the dynamics of supramolecular materials.121,122 One dimensional NMR in solution is commonly utilized to characterize physical bonds such as hydrogen bonding,123,124 host–guest interactions,125 or metal–ligand coordination.126 As an example, Stang and coworkers have developed a hierarchical supramolecular polymerization by the combination of coordination self-assembly and hydrogen bonding. In their work, the structure of the synthesized UPy-functionalized coordination donor and coordination acceptors as well as the structure of the self-assembled unites with two different geometries (rhomboid and hexagonal) were studied by 1H NMR. Besides, the formation of linear chains upon further self-assembly of rhomboid units with two UPy end groups was confirmed by 1H NMR.127 Moreover, Stang and coworkers utilized a 2D diffusion-ordered 1H NMR approach to investigate the formation of low molar mass cyclic oligomers (instead of long linear chains) and to measure the dimensions of polydisperse aggregates.127 In this sense, more examples discussing the application of NMR in measuring the size of supramolecular polymers have been reviewed by Zhang and coworkers.109 As another example, complex multiple hydrogen bonding in the solid state can be characterized by 1H-double quantum NMR. Spiess and coworkers have illustrated that the keto and the enol tautomeric forms of the UPy group are distinguishable with this technique and the distance between four protons involved in the quadruple hydrogen bonds of this group can be measured. Moreover, the authors have studied the changes of hydrogen bonds in the solid state in response to the variation of temperature.128 In another work, Spiess, and coworkers have illustrated that non-ordered and ordered clusters (resulted from the hierarchical self-assembly) can be distinguished by NMR. By studying telechelic ethylene oxide oligomers functionalized with imidazole groups by 1H-double quantum NMR, the authors proposed the existence of three regions in the microstructure of such materials. In the first region, the imidazole groups form dimerd by strong hydrogen bonds. The subsequent hierarchical assembly of these dimers by slightly weaker hydrogen bonds leads to the formation of ordered structures. In the second region, dimers are not linked with lateral bonds, and therefore, imidazole dimers form disordered aggregates rather than ordered structures. In the third region, imidazole groups do not form any hydrogen bonding and exist as free end groups.63
Ultraviolet-visible (UV-vis) spectroscopy is another analytical spectroscopy technique based on the absorption of near-ultraviolet or visible light radiation. This radiation provides energy as required for electronic transitions. Most of these transitions involve the promotion of n or π electrons to a π* excited state, which is observed commonly in organic molecules with delocalized π electrons such as aromatic and conjugated aliphatic species. In addition, some inorganic compounds that have inherent absorption in the visible radiation region such as transition metals and their complexes can be detected by this technique.129
UV-vis spectroscopy is mainly used for quantification of organic and inorganic constituents by making a correlation between the absorbed radiation and the concentration of absorbents. This technique is also widely used in the characterization of supramolecular polymer networks containing metal–ligand complexes, π–π interactions, or other associative motifs that actively absorb radiation in this regime. As one example, UPy dimers have a characteristic absorption at 212 nm and a shoulder at 240 nm in UV spectroscopy.100 In contrast, the monomeric tautomer of UPy has a characteristic absorption at 290 nm. Therefore, this technique can be utilized to characterize dimerization of UPy groups. Meijer and coworkers have shown that after melting of PEB–UPy supramolecular polymers, and therefore, disappearance of the characteristic absorption at 212 and 240 nm, the reappearance of the original UV spectrum upon cooling, as shown in Fig. 5b, resembles the time-dependent crystallization behavior of UPy stacks.100 As another example, the visible spectrum of the blend of chain-folding PI and pyrenyl end-capped PU demonstrates an adsorption at 525 nm due to the charge transfer between the π-electron rich and π-electron deficient species.106,107 UV-vis spectroscopy is also widely used for the characterization of supramolecular polymers formed by metal–ligand complexation.130 As an example, the charge transfer between Fe2+ and terpyridine complex has a characteristic absorption peak at 558 nm.131 By replacing Fe2+ with Zn2+, this peak shifts to lower values below 350 nm.131,132
Another spectroscopic characterization technique is dielectric spectroscopy, which is based on the interaction of matter with electromagnetic waves with a frequency range of 10−6–1012 Hz.133 The data of this technique is commonly expressed by a complex dielectric function/dielectric permittivity (ε*), composed of a real part (ε′) and an imaginary part (ε″). The dielectric relaxation takes place as a consequence of reorientational motions of molecular dipoles. One of the main contributions of this technique in physics and materials science is to access the molecular dynamics in a frequency range of 10−6–1012 Hz, which exclusively separates different relaxation processes that take place on the local scale such as α or β relaxations.133,134
Dielectric spectroscopy provides a deep insight into the dynamics of supramolecular polymer networks at the molecular level.135 With this technique, independent investigation of the dynamics of individual binary association units and clusters is possible.60 Stadler and coworkers studied PBds functionalized with phenyl urazole hydrogen bonding groups by dielectric spectroscopy. The authors observed an additional relaxation process (α*) in the dielectric spectrum of supramolecular polymers that was not detected for non-associating counterparts. The relaxation strength of this additional process is correlated to the number of associated dimers.136 Similarly, Tress and coworkers observed a third relaxation process at elevated temperature in the dielectric spectrum of telechelic PDMS with carboxylic acid end groups when the end groups were grafted to the polymer chains via urethane linkages. This peak was attributed to the phase-separation of associating motifs.135,137 As another example, entangled PnBAs functionalized with different contents of AA have been studied with dielectric spectroscopy. In these samples, the α* relaxation process is composed of at least three different relaxation modes: α1*, α2*, and α3*. The obtained results demonstrate a quantitative agreement between the α* relaxations (α1* and α3*) with the α and β relaxations of a pure PAA. Accordingly, the authors concluded that in these supramolecular polymer networks, PAA domains are embedded in a PnBA-rich matrix, as illustrated in Fig. 5e.60 In another instance, Sokolov and coworkers have recently studied a series of supramolecular polymer networks with dielectric spectroscopy to characterize the segmental relaxation and lifetime of the stickers. The authors showed that one of the main advantages of this technique is to identify the timescale of the dissociation of transient bonds, even if the open stickers re-associate to their original partners. Simply put, dielectric spectroscopy can detect a process, which is invisible to the mechanical measurements like rheology as it does not contribute to the stress relaxation.138
3.3. Scattering methods
Generally, scattering is caused by the interaction of incident radiation (such as light, X-ray, and neutron) and structures within a sample.139 Scattering techniques can provide information about the average structural and dynamic properties of a material. With that, scattering techniques are particularly powerful to assess inhomogenous structures in polymer materials, including networks and gels.140X-ray scattering is a structure characterization method based on the interaction of X-ray radiation with matter.141 As described by the Bragg formula (nλ = 2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ; n is an integer multiple, λ is the radiation wavelength, d is the interplanar/lattice distance, and 2θ is the scattering angle), the length scale characterized by this method depends on the scattering angle. Wide-angle X-ray scattering, WAXS, provides structural information of the probed sample on the sub-nanometer scale (0.1–1 nm) such as atomic, crystal packing or amorphous structures, whereas small-angle X-ray scattering, SAXS, gives structural information on the length scale of 10–100 nm.141–143 In the field of polymeric materials, this length scale covers structures like crystalline lamellae or microphase-separated domains in block copolymers. With this possibility, the structure of a wide range of materials like nanomaterials, colloidal suspensions, block copolymers, polymeric gels, or polymer nanocomposites can be studied by X-ray scattering.141
sinθ; n is an integer multiple, λ is the radiation wavelength, d is the interplanar/lattice distance, and 2θ is the scattering angle), the length scale characterized by this method depends on the scattering angle. Wide-angle X-ray scattering, WAXS, provides structural information of the probed sample on the sub-nanometer scale (0.1–1 nm) such as atomic, crystal packing or amorphous structures, whereas small-angle X-ray scattering, SAXS, gives structural information on the length scale of 10–100 nm.141–143 In the field of polymeric materials, this length scale covers structures like crystalline lamellae or microphase-separated domains in block copolymers. With this possibility, the structure of a wide range of materials like nanomaterials, colloidal suspensions, block copolymers, polymeric gels, or polymer nanocomposites can be studied by X-ray scattering.141
X-ray scattering techniques are widely used to characterize supramolecular polymer materials when the self-assembly process leads to the formation of crystalline clusters or even non-ordered aggregates. The domains formed by such clusters usually cause characteristic peaks in SAXS or WAXS spectra. The position of these peaks, the ratio between the position of peaks, and the log–log slope of the SAXS pattern at very low scattering vectors, q = 4πsin(θ)/λ, are common data that gives important information about the microstructure of such networks. As an example, Herbst and coworkers studied the structure of PIB functionalized with 2,6-diaminotriazine and Thy groups by SAXS.39 In the SAXS pattern of mono-functionalized PIB with 2,6-diaminotriazine, the ratio between the observed scattering peaks is 1:√2:√3, which can be assigned to a BCC (body-centered cubic) lattice, as shown in Fig. 5c. Formation of such a BCC microstructure is typical for block copolymers made by the combination of a long and a short block. Therefore, the authors considered this sample as a block copolymer where one block is the PIB segment and the other is the 2,6-diaminotriazine moiety. SAXS patterns of the equimolar mixture of Thy and 2,6-diaminotriazine-functionalized PIB reveal excess scattering, but without a well-ordered structure. In addition, Herbst and coworkers observed that increasing the molar mass of the PIB building blocks disrupts the BCC morphology of 2,6-diaminotriazine-functionalized PIB and shifts the excess peak of an equimolar mixture of Thy- and 2,6-diaminotriazine-functionalized sample to lower q. Further SAXS investigation of samples with almost similar chemistry demonstrates that the unspecific interactions between Thy and 2,6-diaminotriazine monofunctional PIB form micelles, as illustrated in Fig. 5c.40 In the case of bifunctional telechelic PIB with these hydrogen bonding groups, the interaction between micellar aggregates leads to network formation and emergence of solid-like properties.
The complimentary method of WAXS serves for the characterization of supramolecular polymer networks as well. For example, in PTHF functionalized with Ade derivatives, the WAXS pattern shows two characteristic peaks corresponding to distances of 3.6 Å and 7.15 Å, respectively. The former is consistent with π–π stacking between nucleobases, whereas the latter is assigned to stacks of hard domains that are held together by π–π interactions and amide hydrogen bonds. This peak is not observed in the WAXS pattern of PTHF–Cys, which illustrates that this sample has no regular π–π stacking.36
Similarly, X-ray scattering techniques have been extensively used to characterize UPy-based supramolecular polymer networks. For PEB functionalized with UPy groups, temperature-dependent SAXS and WAXS were utilized to reveal the ordering of nanofibers and the order within them, respectively.100 In SAXS, the correlation peak corresponding to ∼7 nm can be attributed to the characteristic dimension of the nanofibers. Upon melting, this peak shifts to larger q (shorter distances). For samples in which the re-aggregation of UPy groups is confirmed by DSC, the correlation peak in SAXS shifts back to lower q (larger distance) upon cooling from the melt. By annealing at room temperature, this peak further shifts to lower q, which shows that objects with larger diameters develop upon annealing. Meijer and coworkers assigned this observation to the aggregation of UPy–urea end groups into nanofibers.100 The WAXS profile of the PEB–UPy supramolecular polymers is dominated by a broad halo due to the amorphous structure of the PEB backbone, but a weak reflection is observed at q = 13.8 nm−1, corresponding to 0.455 nm. This length scale correlates well with the hydrogen bonding in supramolecular bisurea polymers. Therefore, the authors correlated this peak with the urea hydrogen bonds and the inter-planar distance of two UPy dimers stacked on top of each other. In addition, a second reflection attributed to a distance of 1 nm is observed in WAXS patterns, which grows by increasing the size of the substituent at the sixth position. This length scale corresponds to the stack-to-stack distance within the aggregated nanofibers (distance between UPy cytosine alkene proton at the fifth position to the pyrimidinone carboxylate of its neighboring dimer). Because the stack-to-stack distance within nanofibers depends on the substituent type, the authors concluded that this substituent resides in between the stacks. This conclusion can explain the reduced crystallization rate of the polymers with sterically demanding substituents at the sixth position. To investigate the effect of backbone crystallinity on the clustering of UPy groups, telechelic oligocaprolactan functionalized with UPy was studied by Sijbesma and coworkers.144 Based on WAXS measurements, the authors proposed a similar microstructure as the one described by Meijer and coworkers for amorphous PEB building blocks functionalized with UPy groups.
As another example of the application of X-ray scattering techniques in the characterization of supramolecular networks, the SAXS pattern of chain-folding PI and pyrenyl end-capped PU complex shows a characteristic peak at q = 0.08 Å−1, which confirms a phase-separated morphology with a domain spacing on a 10 nm length scale. The WAXS pattern of this system represents several characteristic peaks including 5.1 and 3.4 Å−1. These peaks can be assigned to the urea–urethane hydrogen bonding and π–π stacking of the supramolecular polymers, respectively. SAXS has been also employed to characterize the microstructure of PnBAs with different molar masses (5–85 kg mol−1), center functionalized with triurea hydrogen bonding groups.94 As already mentioned, the authors of that investigation determined a molar-mass threshold for these samples (20 kg mol−1), below which SAXS patterns show an intense characteristic peak, confirming the existence of long-range order with a well-defined characteristic distance. The scattering vector and the intensity of this peak decrease by increasing the molar mass of PnBA, which demonstrates diminishing of the hierarchical supramolecular structures. In addition, samples with molar masses of 8–18 kg mol−1 show a second characteristic peak at q = √3q*, where q* is the scattering vector of the main characteristic peak. The authors attributed such a pattern to hexagonal packing of nanofibers. While the sample with the molar mass of 85 kg mol−1 does not show such a peak, the ultra SAXS, obtained at very low scattering vectors, demonstrates that for q < 0.2 nm−1 and 0.1 nm−1 < q < 1 nm−1, the scattering intensity scales with q−2/3 and q−1, respectively. Accordingly, the authors suggested that non-ordered rod-like scattering entities with characteristic dimensions in the order of several nanometers exist in the microstructure of the high molar mass samples.94
Another scattering technique is small angle neutron scattering (SANS). Some of the specific features of neutron scattering compared to X-ray scattering include a high penetration power and sensitivity to elements and isotopes. With that, by labeling samples with isotopes (like 2D), different parts of them can be selectively highlighted.139 This advantage is very useful for studying gels, as the scattering contrast can be boosted by simply preparing gels in deuterated solvents.145 Similar to X-ray scattering techniques, SANS has been widely used to study the microstructure of supramolecular polymers. Krutyeva and coworkers have studied mixtures of telechelic PEG functionalized either with DAT or with Thy groups by SANS. Analysis of the data reveals that the probability of the formation of Thy–DAT hetero-complementary association is about three times of that of DAT–DAT or Thy–Thy homo-associations.146 Sijbesma and coworkers have employed SANS to characterize the microstructure of multiblock segmented copolymers of hydrophilic PEG and hydrophobic dimer fatty acid. Phase-separation of hydrophobic and hydrophilic parts make a physically crosslinked structure, which is stable in water. Hence, such copolymers can form mechanically stable, tough hydrogels. Using SANS, the authors could determine the average radius of hydrophobic domains, the average distance between them, and the average number of hydrophobic units in these micelles.147 In earlier work, Weiss and coworkers have studied random copolymers of N-isopropylacrylamide and 2-(N-ethlylperfluorooctansulfonamido)ethyl acrylate (FOSA) as well as random copolymers of dimethyl acrylamide and FOSA. In these copolymers, hydrophobic FOSA units form clusters that act as physical crosslinks.148,149 Formation of FOSA nanodomains linked by N-isopropylacrylamide or dimethyl acrylamide have been confirmed by SAXS and SANS measurement. Details on the microstructure of the phase-separated domains, however, could only be recognized by SANS, where the contrast between different phases could be tuned using different ratios of D2O/H2O as the solvent. This approach provides the possibility to selectively investigate the microstructure of these hydrogels. As an example, for a random copolymer of dimethyl acrylamide and 9.7 mol% of FOSA, a mixture of D2O and H2O with the volume ratio of 27/73 matches the scattering length density of dimethyl acrylamide units, and therefore, FOSA nanodomains dominate the scattering pattern. By analyzing the SANS data, the authors proposed that nanodomains have a core–shell architecture, in which FOSA cores are surrounded by water-depleted acrylamide chains. Later on, the same group studied structural changes for a dimethyl acrylamide–FOSA hydrogel by in situ SANS during stress relaxation in the step-strain experiment. During the structural relaxation, the anisotropy of the scattering intensity peaks that are correlated with the size of the FOSA domains and spacing between them, have been observed, which allowed studying their relaxation process. In addition, changing the size and spacing of nanodomains during stretching and after relaxation were studied by SANS. The authors found that after relaxation, nanodomains recover their size, however, their spacing is smaller compared to the non-stretched state.150
3.4. Microscopy techniques
One of the important microscopy techniques commonly used to characterize the microstructure of supramolecular polymers is atomic force microscopy, AFM. It is a scanning probe microscopy technique in which a fine tip attached to a cantilever spring is swept over a sample surface.151 The tip-to-surface distance can be in the order of 10–100 nm (non-contact mode) or a few angstroms (contact mode). Attractive vs. repulsive interaction between the sample and tip changes the bending of the cantilever, which is commonly detected by a laser beam focused onto the end of the cantilever. Accordingly, the force between the sample and the tip can be calculated. In AFM, the topography of the surface is obtained by measuring the sample height within a sub-nanometer resolution. In addition, the bending of the cantilever can be recorded as a function of its distance from the surface in the form of a force–distance curve.152,153 AFM based single-molecule force spectroscopy is another method for characterization of supramolecular polymers. In this method, supramolecular polymer chains can form a bridge between an AFM tip and a substrate (like Au). By separation of the tip and the substrate, polymer chains are stretched. The obtained force–extension curve is utilized to determine the effective degree of polymerization and the rapture force of associative motifs like UPy groups.109,154AFM imaging of supramolecular polymer materials can directly show existence of aggregates and clusters. As one example, AFM images of PCL functionalized with UPy groups revealed that stacking of UPy groups into long fibrils is promoted via urethanes and urea hydrogen bonding linkages.88 Similarly, the AFM imaging of low-Tg oligomers (such as PEB) functionalized with UPy groups demonstrates the presence of well-defined nanofibers with a diameter of 6 to 7 nm, as demonstrated in Fig. 5d.87 Formation of this morphology depends on the microstructure of the UPy groups; if a bulky group resides on the cytosine ring, then stacking is not possible anymore, and only phase-separated UPy domains can be observed in AFM images. As another example, AFM images of PnBAs centers functionalized with a triurea hydrogen bonding group confirm the existence of nanofibers in all samples regardless of the precursor length. However, only for samples with molar masses of 5–18 kg mol−1, nanofibers are organized in bundles parallel to each other and form a hexagonally packed structure.94
Transmission electron microscopy (TEM) is another microscopy technique that is used to visualize the morphology and microstructure of self-assembled materials. Very recently, Zhu, Binder, Chen, and coworkers have studied the morphology of supramolecular particles prepared by an emulsion-solvent evaporation method using TEM. The authors synthesized comb-shaped supramolecular polymers made from a poly(styrene) randomly functionalized with barbiturate, as the backbone, and mono-functionalized poly(dimethyl siloxane) with Hamiltonian wedge end group, as side branches. TEM images from a stoichiometric mixture of these polymers in chloroform illustrate formation of irregular aggregates due to the self-assembly of barbiturate and Hamilton wedge groups. This supramolecular polymer solution can be emulsified by an aqueous solution of poly(vinyl alcohol). Upon evaporation of chloroform, the supramolecular polymer is solidified and adopts particle shapes. The morphology evolution of these particles by changing the ratio of associative groups, or by adjusting the temperature during solvent evaporation, or by the selective disassembly of Hamilton groups were investigated by TEM.155
The sample preparation of the conventional TEM, which includes drying and staining steps, may affect the microstructure and morphology of the resulting sample. In cryogenic TEM (cryo-TEM), in contrast, an aqueous solution of the sample is preserved in a frozen hydrated state via a rapid cooling process known as vitrification, and as such does not interfere with the morphology of the sample.156 Meijer, Dankers, and coworkers have utilized cryo-TEM to investigate the structure of self-assembled UPy-based macromers in water. The macromers were PEG oligomers with one or two UPy end groups connected via a hydrophobic alkyl spacer to induce the shielding effect, as explained in Section 2.2.2.80 Introduction of urea groups besides the hydrophobic alkyl spacers results in the self-assembly of macromers in water into a hierarchical structure. This is realized by the hydrogen bonding of UPy groups, phase-separation of UPy dimers from the water-soluble PEGs, which is promoted by hydrophobic spacers, and the subsequent π–π stacking of UPy dimers that is assisted by the lateral hydrogen bonding of urea groups. Comparison of the cryo-TEM images of the self-assembled structures of mono- and bifunctional macromers reveals that long fibrillar structures with diameters of 5 nm or 14 nm are created by the former, whereas, shorter structures with the diameter of 7 nm form by the latter. The authors have attributed the diameter of 14 nm to the bundle of dimers and the diameter of 7 nm or 5 nm to the single dimers.96 Very recently, Meijer, Böttcher, and coworkers utilized cryo-TEM with advanced cryo-electron tomography analysis to study details of the structure of another supramolecular polymer in water.157 In this analysis method, multiple views from different directions of the self-assembled aggregates are combined to provide three-dimensional information. The authors investigated the self-assembly of amphiphile units based on BTA core and different aliphatic tails. Using the cryo-TEM, the same research group had shown before that the self-assembly of such structures in water forms fiber structure with some periodicity.158 However, employing the cryo-electron tomography analysis they realized that these units can form micrometer-long, double helix structures with a pitch size that is tunable by the choice of the tail compositions.157
Another microscopy technique that is widely used for studying supramolecular polymer materials is fluorescence microscopy including confocal microscopy and high-resolution fluorescence microscopy such as stochastic optical reconstruction microscopy (STORM).159 Hamachi and coworkers studied the self-assembly of peptide-based and lipid-like gelators into nanofibers and further the self-sorting of these nanofibers by in situ imaging of a confocal microscope. They found that both fibers form orthogonally interpenetrating networks due to the hydrogen bonding and π–π interactions of peptide-based gelators and hydrogen bonding and hydrophobic interactions of lipid-like gelators.160
Meijer, Albertazzi, and coworkers have used the multicolor ability and the resolution enhancement of STORM to investigate the monomer exchange pathway between 1D fibers formed from the self-assembly of BTA-based monomers.161 The authors studied the structural details of the supramolecular fibers and the monomer exchange between them on a sub-aggregates scale. They mixed green and red fluorescence-labeled BTA units and probed the resulting fibers for a period of 24 h. At time zero, fibers could be observed with either red or green color. However, after 24 h, all fibers contained both monomers and no single-color fiber was observed. In addition, analyzing STORM images demonstrated that the monomer exchange along the polymer backbone is homogenous with no evidence of fragmentation–fusion or polymerization–depolymerization mechanisms.161 Similar monomer exchange process has been observed later by the same group for the supramolecular fibers formed from the hierarchical assembly of mono- and bifunctional UPy-based monomers.96 Analysis of STORM images of fibers that each monomer forms confirmed their different sizes as suggested by cryo-TEM images.
4. Manifestation of clustering in dynamics and mechanical properties
The dynamics of supramolecular polymers and networks is composed by the interplay between the reversible crosslinking and the contribution of the underlying non-associative polymeric relaxation.162 The former is typically defined by the dissociation rate of the transient bonds in the supramolecular network,163 while the latter is a hierarchical process that depends on the molecular architecture of the polymer precursor.164,165 This relaxation starts by conformational changes of short segments and proceeds over time until the length of the relaxing segments reaches the critical entanglement length. Further relaxation is prohibited once the length of the relaxing segment exceeds this threshold, since then the change in the conformation requires dragging of the neighboring entangled chains. Later on, the relaxation process is continued by a new process, once chain segments can diffuse back and forth along the axis of the confining hypothetic tube formed by entangled neighboring chains, which is known as the reptation mechanism.166,167 To precisely describe the relaxation process, other mechanisms like the thermally activated fluctuations of free ends, known as contour length fluctuation (CLF), and the loss of entanglements due to the motion of surrounding matrix chains, referred to as constraint release (CR), should be also taken into account.166–169Generally, the presence of transient crosslinks induces additional friction against all the mentioned polymeric relaxation mechanisms, which slows down the chain motion and the diffusive relaxation of polymer segments in both the melt state (i.e., in elastomers) and in the presence of solvent (i.e., in gels).53,170 Different models have been developed for main-chain and side-chain supramolecular polymers and networks to describe their relaxation based on the interplay between the kinetics of associative groups and the dynamics of polymeric constituents. In a first approach, the dynamics of entangled main-chain supramolecular polymers subjected to random scission and recombination can be assessed by application of a model by Cates that is actually devoted to wormlike micelles of surfactants.171,172 This model proposes scaling laws for dynamic parameters like the terminal relaxation time, the self-diffusion coefficient, or the viscosity as a function of the concentration of the polymeric building blocks that are functionalized with associative end-groups. Alternatively, the dynamics of side-chain supramolecular polymer networks in the melt, dilute, semi-dilute unentangled, and the semi-dilute entangled states can be described by the sticky Rouse, sticky reptation, or hindered fluctuation models developed by several authors.170,173–175 Like the Cates model, these models propose several scaling laws for the dependence of dynamic parameters on structural factors like the sticker concentration, precursor-polymer molar mass, and the polymer concentration. Such models are normally confirmed on macro- as well as microscopic scales by experiments that reveal the effects of the sticker association kinetics on the diverse multiscale dynamics of supramolecular networks.
On the macroscale, viscoelastic properties of transient networks such as their terminal relaxation time or viscosity are measured by following the networks’ responses to different types of deformation as regularly applied in oscillatory shear rheology, creep, or dynamic mechanical temperature analysis (DMTA) experiments.163,176 On the microscale, parameters such as the translational polymer-chain diffusion coefficient are measured by microscopy-based techniques like fluorescence recovery after photobleaching (FRAP) or forced Rayleigh scattering (FRS).177,178
In summary, the macroscopic properties of supramolecular polymer materials are closely tied, not only to the structure and dynamics of the polymer backbone but to that of the supramolecular bonds. However, in dilute conditions, where the entanglement of the polymer precursor is negligible and as such the polymeric dynamics is hardly traceable on experimental timescales, the macroscopic dynamics is only controlled by the supramolecular bonds. Thereby, following the dynamics of side-chain supramolecular polymer networks, transiently bonded by multiple small-molecule pincer crosslinkers, Craig and coworkers showed that the macroscopic behavior obeys a universal pattern, which is defined by the dissociation rate of the supramolecular bonds.163,179 Later on, by decoupling the association and dissociation processes in supramolecular polymer networks based on host–guest interactions, Appel and coworkers suggested that the mechanical strength is determined by the energetic barrier to dissociation of the supramolecular bonds.180,181 In the follow-up, Olsen, and coworkers highlighted that the dynamics of transient bonds in the gel environment is different from that of the small-molecule motifs in the dilute solutions and that the dynamics in the gel is the one which governs the macroscopic properties of supramolecular polymer networks.182
Clustering of the associative motifs in supramolecular polymer networks causes additional microstructural inhomogeneity that imposes extra friction against polymer relaxation. In such cases, the disengagement rate from clusters, which is commonly much longer than the dissociation rate of binary assemblies, should be considered as the main relaxation process.36 On the microscopic scale, formation of clusters can alter the translational polymer-chain diffusion due to the cooperative reduction of free volume and trapping of a fraction of polymeric segments within the long-lasting bonds.98,177 On the macroscopic scale, such clustering enhances the plateau modulus and prolongs the terminal relaxation time.88,183
4.1. Macroscopic dynamics and mechanical properties
Among a series of papers published by Stadler and coworkers about the viscoelastic properties of PBd modified with hydrogen bonding phenyl-triazoline-dione side groups,33,112,119,184,185Ref. 33 is a pioneering work that systematically addresses the effect of clusters on the dynamics and mechanical properties of model supramolecular polymer networks (Fig. 6). Stadler and coworkers observed that with the integration of hydrogen bonding side-groups, the plateau modulus enhances and extends to lower frequencies. In addition, the slope of the storage modulus versus frequency in a log–log plot decreases in the terminal flow region. Moreover, the zero-shear viscosity increases and the transition from the Newtonian to the non-Newtonian regime shifts to lower frequencies. These effects are escalated by increasing the degree of modification and eventually at high contents of associative groups no zero-shear viscosity is observed in the accessible experimental range. To investigate the effect of inter-chain hydrogen bonding on the aforesaid properties, three chemically different associative groups were utilized, as illustrated in Fig. 6a. Comparison of the thermo-rheological properties of the resulting supramolecular polymer networks revealed that the number of hydrogen bonds shared between two associative groups is a key parameter that strongly influences the dynamics and mechanical properties. The associative motif 1 can form double hydrogen bonds (see Fig. 6a) and the corresponding supramolecular network shows simple thermo-rheological properties, which means that all molecular processes contributing to the viscoelastic relaxation have the same temperature dependencies. Therefore, a unique master curve can be constructed, where shift factors obtained at different temperatures follow the Williams–Landel–Ferry (WLF) equation. When PBd is functionalized with the associative motif 3, which has an additional hydroxyl group on the phenyl ring, an extra hydrogen bond forms between the two associative groups, and each network junction is formed by triple hydrogen bonds. The thermo-rheological behavior of this network is complex, and the shift factors required for the construction of master curves cannot be explained by a single equation like WLF. For this sample, the construction of master curve of the loss modulus, G″, using experimental shift factors obtained for G′, or the other way round, fails, as demonstrated in Fig. 6d. Moreover, this network exhibits a significantly broader rubbery plateau and larger zero-shear viscosity compared to the network obtained by the associative motif 1. These observations have been attributed to the existence of extended clusters of hydrogen bonds, which form a three-dimensional network structure.33 To further confirm this hypothesis, the polymer precursor was also modified with the associative motif 2, which has a NO2 group on the phenyl ring close to the hydroxyl group. The resulting supramolecular network showed simple thermo-rheological behavior very similar to the network formed by the associative motif 1. The authors attributed this similarity to the internal hydrogen bonds between OH and NO2, which practically decreases the number of effective hydrogen bonds between the polymer chains. | ||
Fig. 6 (a) Three different hydrogen bonding phenyl-triazoline-dione associative motifs utilized in ref. 33 and (b–d) schematic representation of the thermo-rheological properties of the polymers studied in that work. Polymers functionalized with associative motifs of 1 and 2 are crosslinked via binary hydrogen bonds, whereas in the case of motif 3, triple hydrogen bonds form a three-dimensional network. (b) Storage modulus of non-associative PBd with a molar mass of 35 kg mol−1 ( ), and the functionalized polymer with 1 mol% of the associative motif 1 ( ), and the functionalized polymer with 1 mol% of the associative motif 1 ( ), the associative motif 2 ( ), the associative motif 2 ( ), and the associative motif 3 (+). (c) Viscosity of the same samples, and (d) master curve of G′ and G″ of the sample based on the associative motif 3, where horizontal shift factors of G″ are applied on G′, which highlights the thermo-rheological complexity. The original figures with the original data can be found in ref. 33. ), and the associative motif 3 (+). (c) Viscosity of the same samples, and (d) master curve of G′ and G″ of the sample based on the associative motif 3, where horizontal shift factors of G″ are applied on G′, which highlights the thermo-rheological complexity. The original figures with the original data can be found in ref. 33. | ||
As a summary, aggregation of associative groups into clusters in a supramolecular polymer network escalates the variation of dynamics and mechanical properties of polymeric precursors/building blocks upon grafting with supramolecular motifs. In addition, clusters may impose extra complexity in the thermo-rheological properties through inducing additional relaxation mechanisms. This line of thought has been pursued later in different supramolecular network elastomers and gels with either main-chain or side-chain architectures. In the following, some of these works are described.
The influence of clustering on the dynamics and mechanical properties of main-chain supramolecular polymer networks formed by strong associative groups has been also studied. For instance, when PCL building blocks are functionalized with UPy end groups through urethane linkages, the UPy groups form clusters.88 In such samples, the low-frequency terminal flow is replaced by a second plateau modulus, which extends to lower frequencies once the concentration of UPy groups is raised by decreasing the molar mass of the PCL building block. At low UPy contents, however, the second plateau is replaced with a parallel drop of moduli on a log–log scale (G′ ∼ G″ ∼ ω0.5).88 Stacking of UPy groups and the formation of long fibers were also reported to enhance the mechanical properties in UPy functionalized PEB systems.87,97,186 In diblock copolymers of poly(styrene-b-butyl acrylate) in which the acrylate block is end-functionalized with UPy groups, self-assembly of UPy groups creates a triblock supramolecular copolymer with a phase-separated microstructure. Stress–strain experiments on such a material show significantly enhanced mechanical properties in comparison to a control sample in which self-assembly is avoided through utilization of a UPy motif with a specific structure.176
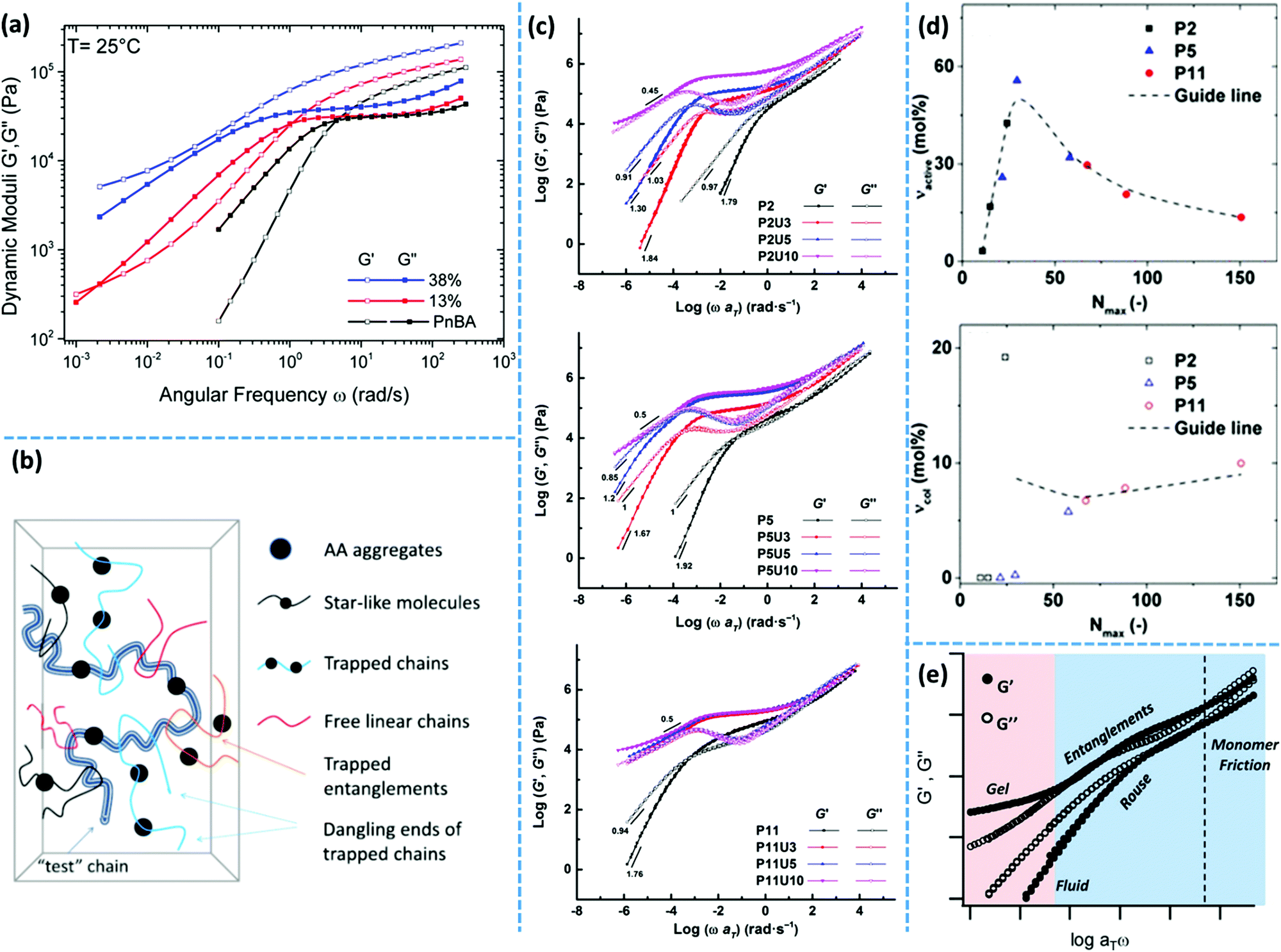 | ||
| Fig. 7 Dynamics and mechanical properties of model supramolecular networks: (a) dynamic moduli of PnBA functionalized with 13 and 38 mol% of AA associative side-groups.60,183 (b) Illustration of the test chain and different types of polymer segments that are required to theoretically describe the dynamics shown in panel (a).59 (c) Dynamic moduli of PLMA copolymers with different extent of entanglements, (2–11 denoted as P2–P11) with different UPy contents (3–10 mol% denoted as U3–U10).56 (d) Decoupling the contribution of binary associations and clusters: Vactive is the fraction of active stickers contributing to the elasticity of the network including binary associations and clusters, Vcol is the fraction of clusters among the active stickers. The abscissa, NMax, features the total number of network junctions including transient associations and chain entanglements.58 (e) Viscoelastic properties of PnBA center functionalized with triurea hydrogen bonding groups. Samples behave as viscoelastic fluid or gel depending on the possibility of clustering of associative groups, which individually depends on the length of the polymer precursor.93 Panel (a) taken from ref. 60; copyright 2016, American Chemical Society. Panel (b) adopted from ref. 59; copyright 2016, Society of Rheology. Panels (c, d, and e) reprinted from ref. 56, 58 and 95; copyright 2019, 2019, and 2015, respectively. Wiley and American Chemical Society. | ||
The parallel drop of the moduli (G′, G″ ∼ ω0.5) and the emergence of the second plateau at low frequencies have not been commonly reported for side-chain supramolecular polymer networks formed by weak associative groups like AA, in the absence of clustering. Anthamatten and coworkers have investigated the influence of the number and the association strength of sticky groups on the dynamics and mechanical properties of side-chain PnBA supramolecular networks.187 In this work, supramolecular polymers bearing weak hydrogen bonding groups like AA displayed liquid-like properties very similar to the non-sticky precursors, although at higher contents of sticky groups the crossover of the dynamic moduli shifts to lower frequencies. One of the main differences between this work and ref. 183 is the length of the precursor polymers. The absence of entanglements in Anthamatten's work187 seemingly results in the appearance of Maxwellian terminal relaxation in sharp contrast to the other works with highly entangled backbones.53,59,60,183
The interplay of chain entanglement and transient association on the dynamics of side-chain supramolecular polymer networks has also been investigated for PLMA functionalized with UPy side groups on a macroscopic scale by oscillatory shear rheology.56 The results showed that networks formed from low or medium molar mass backbones (42 and 76 kg mol−1) do not reveal terminal relaxation in the experimentally accessible frequency range if the backbone is functionalized with a high UPy content (10 mol%). A similar dynamics was also observed for the networks with a high molar mass backbone (164 kg mol−1) regardless of the UPy content. Instead of the terminal flow, these networks demonstrate a parallel drop of the moduli, and even a second plateau modulus emerges at low frequencies as shown in Fig. 7c. The emergence of the low-frequency plateau modulus is related to segments that are unable to relax the applied deformation, as they are trapped between clusters. The effective number of UPy junctions, including both individual binary associations and clusters, which contribute to the network elasticity enhances upon increase of the UPy content regardless of the backbone length. However, the ratio of the active to the available UPy associations (0.5 × UPy content determined by 1H NMR) strongly depends on the total number of network junctions, including chain entanglements and transient associations.56 This is related to the likelihood of having free UPy groups that may merge into existing clusters rather than forming new binary associations in the limit of the high molar mass precursor chains.98 Close to this comprehensive work, a TMA model was also developed to decouple the contribution of individual binary associations from those of clusters on the dynamic properties.58 This algorithm quantifies the fraction of active stickers and determines the fraction of clusters among them. The obtained master-curve of the fraction of effective UPy groups versus the total number of network junctions follows an overshoot, while the fraction of clusters reaches a plateau, in agreement with the experimental analysis of ref. 56, as shown in Fig. 7d. These results reveal how the dynamics and viscoelastic properties of side-chain supramolecular polymer networks can be controlled by tuning the formation of clusters through variation of the extent of entanglements and the sticker content.
Supramolecular polymeric networks formed by center functionalized PnBA chains including different hydrogen bonding groups expose a similar interplay between the polymeric and the supramolecular dynamics. Samples with a molar mass lower than 20 kg mol−1 resemble soft elastic solids, even at low frequencies. In contrast, those with a molar mass higher than 40 kg mol−1 behave like viscoelastic liquids demonstrating terminal flow at low frequencies, as illustrated in Fig. 7e. Considering the entanglement molar mass of PnBA, the authors attributed the elastic behavior of low molar mass samples to the molecular organization of the supramolecular groups into ordered bundles of hydrogen bonding arrays extending up to 1 µm.93 Comparison of the rheology and SAXS results demonstrated that these bundles disappear at a temperature at which the material properties shift from solid-like to liquid-like. This confirms that the aggregation of associative groups into parallel bundles, rather than their distinct binary associations, controls the mechanical properties of these materials.
The static stress–strain curves of hydrogen bonded brush copolymers constructed from a hard PS backbone and soft amide-modified PnBA brushes are similar to those of classical TPEs with an initial high stiffness followed by a large elastic deformation.19 Such multiphase supramolecular networks show enhanced mechanical strength in comparison to thermo-reversible TPE systems. By variation of the weight fraction of the hard backbone and the soft brush, the mechanical properties can be tuned such to vary the Young modulus between 9.8 and 35.7 MPa and the strain at the break between 310% and 1570%.19
In the same manner, clustering of associative groups has a significant influence on the dynamics and mechanical properties of supramolecular polymer gels. As an example, Seiffert and coworkers have studied supramolecular gels formed by transition metal-mediated linking of tetra-arm PEG end-capped with terpyridine moieties.188 The authors examined different metal ions including Mn2+, Zn2+, and Co2+ as the crosslinking agents to form supramolecular gels in different solvent mixtures. Nanoscopic structural inhomogeneity was observed in the Co2+-based network, which has the strongest association affinity among the studied sample series. In addition, the Co2+-based network does not show the onset of terminal relaxation in the experimentally accessible frequency range. The authors attributed these observations to the aggregation of the network junctions.
4.2. Microscopic dynamic properties
Similar to the macroscopic dynamic properties, as discussed in Section 4.1, the dynamics of supramolecular polymer networks on microscopic scales, which is revealed for instance by the translational diffusion coefficient, is influenced by aggregation of associative groups. The interplay of chain entanglement and the transient association on these properties has been investigated in supramolecular polymer networks based on PnBA functionalized with UPy side groups through probing the diffusion of polymeric tracers with almost identical chemical composition, molar mass, and UPy content as the network matrices by fluorescence recovery after photobleaching.98 The authors found that the diffusion of tracers is decelerated by increasing UPy content due to the enhanced transient bonding of tracers to the matrix chains. However, this deceleration gets weaker at a greater extent of chain entanglement. Consequently, the presence of chain entanglement apparently outweighs the influence of transient interactions on the dynamics of these associating networks in the melt state. This observation has been attributed to the likelihood of free UPy groups to assemble to existing clusters in the presence of chain entanglements rather than forming new ones. The authors concluded that the major consequence of an increasing the extent of entanglement is promotion of the participation in aggregates of associative groups, which therefore drastically reduces chains mobility, such that it is not observed within the timescale of the FRAP experiment.The temperature corresponding to the onset of the microscopic segmental mobility is Tg. Therefore, in addition to being a specific physical parameter, Tg can be considered as a dynamic property. The presence of transient associations within the microstructure of a supramolecular polymer is expected to increase the Tg of the precursor polymer, either by slowing down the chain dynamics or simply due to steric effects on the backbone. In copolymers of butyl acrylate and a UPy-functionalized acrylate comonomer, a linear increase in Tg with increasing UPy content has been reported by Feldman and coworkers,186 consistent with prior reports on UPy-functionalized random copolymers.189,190 Such an increase in Tg was later demonstrated for supramolecular polyacrylate systems based on weak hydrogen bonding groups like AA, aminopyridine, and carboxyl ethyl acrylate.187 In contrast, the study of PBds functionalized with hydrogen bonding phenylurazole groups or PEB functionalized with UPy have not shown such an increase of Tg.100,184 The origin of such independence of Tg from transient associations was not discussed in these works. We have observed a similar effect in polymethacrylate derivative-based side-chain supramolecular networks formed by UPy associations.56 Despite the effect of transient crosslinking on the rheological behavior was evident from the parallel drop of the moduli and/or the emergence of a low-frequency plateau, DSC results revealed a negligible change of Tg in comparison to the non-associating precursor polymers. This observation was attributed to strong phase-separation of the UPy groups from the non-polar polymer backbone, which forms a matrix enriched by non-polar polymeric segments and nanodomains of associated UPy motifs.
5. Applications
5.1. Self-healing
One of the unique properties of supramolecular polymeric materials is their self-healing ability, which is triggered by forced increase in the local dynamics of the reversible supramolecular bonds, generally by raise of temperature. Logically, there is an inherent compromise between the mechanical robustness and the self-healing ability.191–193 Therefore, specific design concepts have to be devised to preserve the required mechanical performance once dynamic self-healing ability is integrated in a system.194One of the widely used design concepts is to fortify supramolecular polymer networks based on relatively weak supramolecular bonds by the phase-separation of associative groups in microscopic domains. In this context, telechelic systems based on a low-Tg polymer core like PDMS and weak hydrogen bonds like Ade, Cys, Thy, aminotriazine that make phase-separated undirectional homo-aggregates, demonstrate exceptional low-temperature self-healing ability.17,36 Such materials reflect strongly temperature-dependent properties, in sharp contrast to their equivalent hetero-complementary blends that have much lower phase-segregation tendencies.36,39 The extent of clustering in homo-aggregated systems can be tuned by the introduction of unidirectional hetero-complementary counterparts.40 In the same manner, the supramolecular polymer blend of a chain-folding PI and a telechelic pyrenyl-functionalized PU, which are linked through nanophase-separated, weak π–π stacking interactions, demonstrates autonomous room-temperature self-healing.106 In contrast, phase-separated metallo-supramolecular polymeric systems based on a PEB core and Mebip ligand reveal self-healing features only when exposed to UV light. This is the consequence of the light-driven electronic excitement of the metal–ligand complex, which locally converts to heat.42
An alternative design concept for achieving autonomous self-healing, besides the favorable mechanical strength, is to deliberately integrate weak supramolecular bonds in one of the phases in a multi-phase supramolecular polymeric system. In the variant where the supramolecular bonds are located in soft domains, the dynamic association of reversible bonds provides room-temperature self-healing capacity, while good mechanical performance is guaranteed by the presence of non-associating hard domains. Examples include multiphase polymer brushes with PS backbone and polyacrylate derivative arms. In this design, weak transient interactions, either made by amide hydrogen bonds or the coordination of zinc ions to imidazole ligands, are embedded along the arms.19,108 The low-Tg of polyacrylate arms provides room-temperature self-healing properties. Similarly, blends of main-chain supramolecular polymers based on PEG and PTHF building blocks that are extended by ionic groups fall into the same category, since ionic groups inside PTHF domains phase-separate into strong aggregates.48
In another variant of multiphase self-healing materials, reversible bonds are inversely incorporated in hard domains.194 In this “phase-locked dynamic bonds” design, supramolecular bonds are protected inside the viscoelastic hard phase, which can be thermally unlocked to actuate the self-healing process. This concept is used to lock down the dynamic bonds in the hydrogen-bonded hard-segments of a PU elastomer. The resulting material exhibits superior transparency due to the small size of the segregated hard domains, high extensibility due to the presence of the soft matrix, and fast self-healing at 70 °C.195 In the same manner, hierarchical hydrogen bonds including urethane, urea and UPy functions are embedded as side-groups in rigid domains of a segmented PU with PTHF soft segments, as illustrated in Fig. 8a. The obtained material demonstrates high mechanical strength even after experiencing a thermally activated self-healing process.196 Such materials have shown a great potential to be applied as protective transparent coatings in a variety of fields like optics and electronics. The self-healing ability is specifically useful in rechargeable batteries since the lifetime of many of such products are limited by the formation of fractures due to the thermal expansion during multiple cycles of charging. Accordingly, Bao and coworkers have remarkably increased the lifetime of a rechargeable lithium battery by coating the silicon microparticle anodes with a thin layer of self-healing polymer based on weak amide hydrogen bonds.197
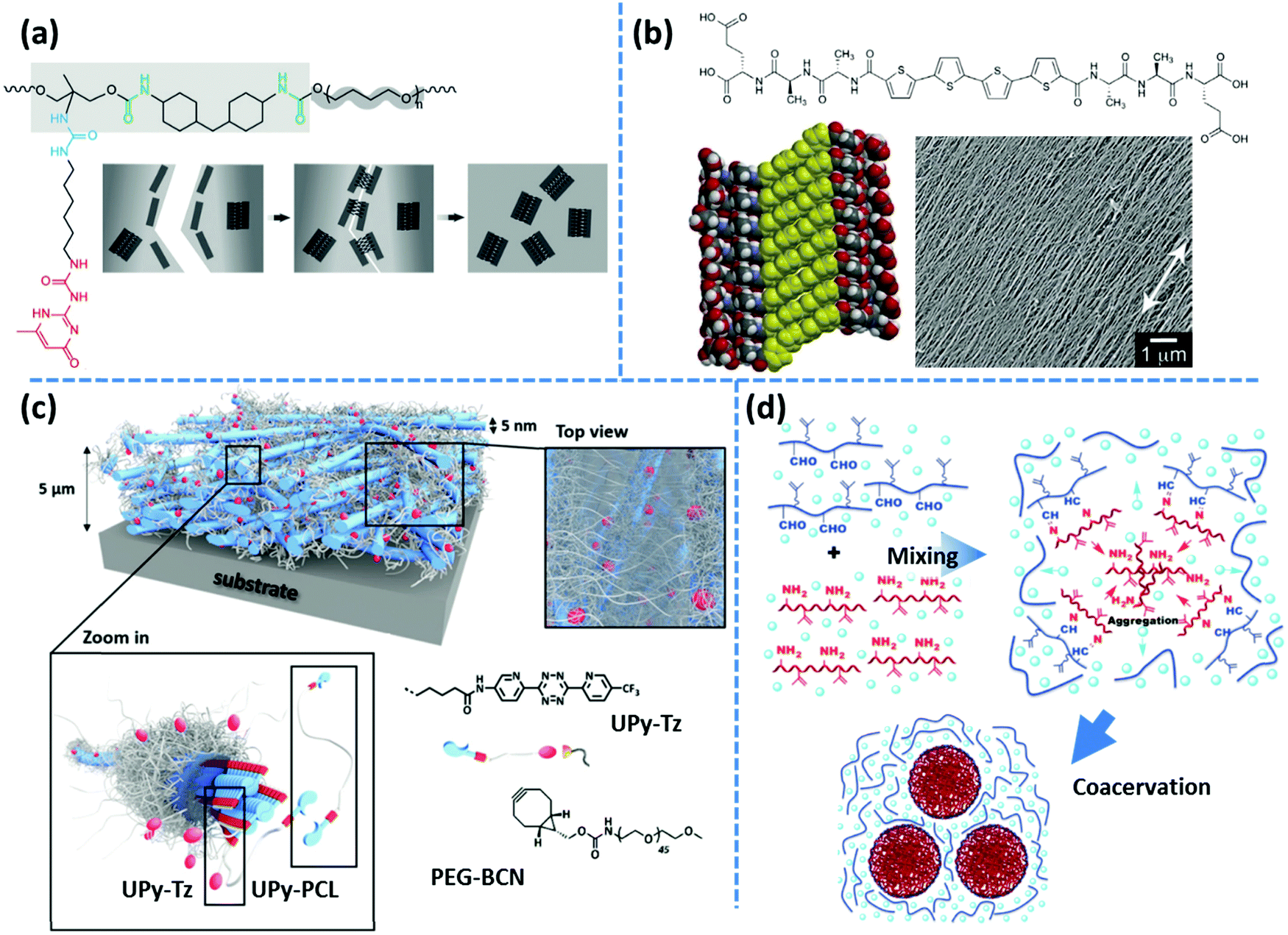 | ||
| Fig. 8 Representative applications of supramolecular polymeric systems in the presence of clusters: (a) integration of multiple hydrogen bonds in hard phases of a segmented PU imparts high-temperature self-healing.196 (b) Bifunctional thiophene derivatives with lateral hydrogen bonds self-assemble into nanowires with unique anisotropic electrical conductivity.198 (c) Immobilization of additives like the antifouling PEG–BCN reagent on the surface of fibrillar UPy aggregates through a UPy-Tz anchor incorporated inside nanofibers.205,206 (d) Phase-separated complex coacervate hydrogels that are formed by mixing oppositely charged polyelectrolytes show promise as tunable drug delivery platforms.207 Panel (a) taken from ref. 196; copyright 2018, Wiley. Panel (b) taken from ref. 198; copyright 2011, Wiley. Panel (c) taken from ref. 205 and 206; copyright 2017, Wiley and Royal Society of Chemistry, respectively. Panel (d) taken from ref. 207; copyright 2015, Wiley. | ||
5.2. Electronics
Nanostructured assemblies based on stacking of π-conjugated supramolecular units have promising applications as molecularly-designed semi-conductors in electronics.69 However, not only a perfect fit between the π-conjugated elements is required, but a decent hierarchical association into ordered clusters is crucial for achieving the desired anisotropic conductivity at the macroscopic scale. This is realized by proper coupling of π–π stacking with secondary transient forces, according to the design concepts discussed in Section 2.2, which can result in the desired spatially ordered microstructure. Widely used strategies include supplementing the π–π stacking either by lateral hydrogen bonds in the solid state or by hydrophobic shielding in aqueous media.69Following this concept, bifunctional thiophene and OPV derivatives with weakly associating amide side-groups are reported to self-assemble into nanowires with unique anisotropic electrical response, as illustrated in Fig. 8b.198 Similarly, OPV derivatives supplemented with strong quadruple hydrogen bonds have shown promises to form semiconducting nanoribbons that transport charge on long distances.72,73 Following the same concept, side-chain supramolecular polymeric materials based on a PEG polymer core and UPy motifs that are shielded by alkene spacers form tough hydrogels, which are good candidates to be used as electrolytes in lithium-ion batteries.83
5.3. Biomedicine
Supramolecular polymer hydrogels have been increasingly employed for the delivery of therapeutics and cells in drug delivery and tissue engineering applications.199,200 Traditionally, the cargo is released from the covalent hydrogel either through simple diffusion, in case of small-molecule drugs, or through degradation of the chemical network in case of large cells, growth factors, and proteins. Supramolecular polymer hydrogels employed as carriers in such designs have the advantage of releasing the load upon the dissociation of reversible crosslinks. Such reversible links either form the network or connect network segments to the load itself. The reversible dissociation of supramolecular bonds activates the diffusion of large-sized loads in the former, and assists the release of small-molecule cargos in the latter. Moreover, delivery can be site-specific by employing stimuli-responsive reversible bonds. These bonds dissociate either autonomously upon reaching the target tissue or organ, which can have a different pH, temperature, or concentration of enzymes, or through the application of external stimuli.201One of the supramolecular designs that are frequently employed as a tunable carrier in drug-delivery applications is based on the phase-separated strong hydrogen bonds in aqueous media. Dankers and coworkers have reported on the utility of main-chain and side-chain UPy functionalized PEG systems, supported by the cooperative effects of lateral urea hydrogen bonds and the hydrophobic shielding of dodecyl spacers, in intrarenal drug delivery.80 Such a design constitutes a percolated network of overlapping high-aspect ratio fibrous aggregates that forms even at low concentrations by the lateral stacking of UPy dimers upon immersion in water. Accordingly, the network erodes through dynamics of supramolecular aggregates and thereby the physically trapped cargo is released. This, however, follows different timescales for the main-chain and the side-chain design variants. The fast degrading unentangled telechelic system shows promises for short-term release purposes, whereas the entangled side-chain variant is more suitable for long-term release applications.
The same group has systematically varied structural parameters of associating units to apply the optimal design in the delivery of a thermally encapsulated protein.80 Since the decent network structure simply destructs under shear and reforms at break, it can be locally disposed in the desired organ via injection. Moreover, such hydrogels show a reversible gel–sol transition when pH is switched from neutral to slightly basic values. This unique feature has been also employed for the delivery of therapeutics by injection. As such, the solution of supramolecular polymers at basic pH values around 8.5 demonstrates in-situ gelation upon injection into physiological systems.202 After the desired time, which is conveniently tunable by varying the structural design parameters, the hydrogel cannot be further identified and the cargo is delivered along with the network erosion.
Similarly, these UPy functionalized polymers with the capacity of forming a network of nanofibrillar aggregates in water, have been widely used as films and electrospun scaffolds in tissue engineering applications.203–205 The great advantage of such systems is that different biological properties of the scaffold can be tuned simply by adding and matching UPy modified additives. For instance, UPy modified PEG derivatives are introduced for separate tuning of the cell and protein adhesion of a thin layer of UPy functionalized PCL supramolecular polymer.204 Similarly, UPy modified bioactive peptides with short oligo(ethylene glycol) (OEG) linkers reside on the surface of fibrillar UPy aggregates and tune their interaction with cells.204 In a similar fashion, UPy modified chemical anchors are used to covalently link various additives to fibrillar UPy aggregates through different types of click reactions. For instance, an anti-fouling additive is introduced in a telechelic UPy modified PCL system through a reactive UPy modified tetrazine anchor (UPy-Tz). This anchor can be modularly incorporated in fibrous aggregates from its UPy end, thereby leaving the tetrazine group on the other end available for the subsequent post-modification. Accordingly, bicyclononyne modified PEG (PEG–BCN) derivatives are selectively attached on the surface of nanofibers as anti-fouling agents via an inverse electron demand Diels–Alder cycloaddition, as shown in Fig. 8c.205,206
In a different approach, phase-separated complex coacervate hydrogels, which are formed by solution mixing of oppositely charged polyelectrolytes, are employed in tissue engineering applications. Jeon and coworkers have reported on the in situ entrapment of a growth factor inside a phase-segregated hydrogel based on two oppositely charged natural polymers, including an oxidized methacrylated alginate and a methacrylated gelatin.207 Fluorescence microscopy clearly reveals that individually segregated coacervate microdroplets rich in gelatin form upon photo-crosslinking of acrylate functions, as schematically shown in Fig. 8d. The physical entrapment of the growth factor inside segregated gelatine microdroplets significantly prolongs the release process compared to the alternative case, where it is loaded in the oxidized alginate matrix. Such tunability of the release profile provides a useful strategy to manage the fate of encapsulated cells for tissue regeneration applications.
6. Conclusions and perspective
Clustering of associative motifs in complex, hierarchical self-assembled biomaterials is an important step in the organization of structures, which introduce specific properties and/or functions. Analogously, in designing bio-inspired, artificial supramolecular polymer materials engineering such clusters is a powerful approach to adjust dynamics as well as the physical and mechanical properties. In addition, by using this strategy certain functions such as self-healing ability can be added to the synthesized networks. Considering this example, clusters of supramolecular assemblies provide the additional possibility of regulating the mechanical properties of supramolecular materials in addition to the self-healing that is provided by the reversible bonds. In this sense, understanding different mechanisms at the molecular level that can be employed to rationally motivate aggregation of associative motifs into clusters, as discussed in Section 2, is essential. For example, to synthesize a self-healing material for a specific application, the Flory interaction parameter determines the tendency and extent of the phase-separation (immiscibility) between the building blocks and associative motifs. In addition, the order of aggregates can be adjusted by rational incorporation of secondary forces, such as lateral hydrogen bonds or hydrophobic shielding, into the chemical structure of the supramolecular group. All these design concepts define how the material performs in its target application, as described by several examples in this review. The success of the selected design approach can be examined either by characterization of the clusters by the methods described in Section 3 or by studying the mechanical and dynamic properties of the designed material as discussed in Section 4. Nevertheless, clustering of associative motifs frequently impedes achieving the desired unique microstructure by inducing extra temporal and structural complexity into supramolecular polymer networks. This complexity generates many challenges to engineering functional supramolecular polymer networks. Influence of crystallinity, glass transition temperature, and polarity of polymer building blocks on the degree of clustering, the interplay of the molar mass of polymer building blocks and associative groups on the formation and dynamics of clusters and controlling the degree of trapped segments between two clusters are examples of these challenges. We hope that the highlighted examples in this review triggers new ideas and guidelines to address such challenges and to develop new supramolecular polymer networks with specific properties and functions via engineering the aggregation of associative junctions into irregular or ordered clusters.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding by the German Research Foundation (DFG) under grant number SE 1888/7-1 (Project No. 376900084) is gratefully acknowledged.References
- J. A. Elemans, A. E. Rowan and R. J. Nolte, J. Mater. Chem., 2003, 13, 2661–2670 RSC.
- I. I. Cisse, I. Izeddin, S. Z. Causse, L. Boudarene, A. Senecal, L. Muresan, C. Dugast-Darzacq, B. Hajj, M. Dahan and X. Darzacq, Science, 2013, 341, 664–667 CrossRef CAS.
- L.-J. Chen and H.-B. Yang, Acc. Chem. Res., 2018, 51, 2699–2710 CrossRef CAS.
- B. Qin, Z. Yin, X. Tang, S. Zhang, Y. Wu, J.-F. Xu and X. Zhang, Prog. Polym. Sci., 2020, 100, 101167 CrossRef CAS.
- D. J. Prockop and A. Fertala, J. Struct. Biol., 1998, 122, 111–118 CrossRef CAS.
- V. Ottani, D. Martini, M. Franchi, A. Ruggeri and M. Raspanti, Micron, 2002, 33, 587–596 CrossRef CAS.
- M. Potschka, M. Koch, M. Adams and T. Schuster, Biochemistry, 1988, 27, 8481–8491 CrossRef CAS.
- E. R. Rafikova, B. I. Kurganov, A. M. Arutyunyan, S. V. Kust, V. A. Drachev and E. N. Dobrov, Int. J. Biochem. Cell Biol., 2003, 35, 1452–1460 CrossRef CAS.
- H. Qiu, Z. M. Hudson, M. A. Winnik and I. Manners, Science, 2015, 347, 1329–1332 CrossRef CAS.
- H.-E. Jin, J. Jang, J. Chung, H. J. Lee, E. Wang, S.-W. Lee and W.-J. Chung, Nano Lett., 2015, 15, 7138–7145 CrossRef.
- S. Chen and W. H. Binder, Acc. Chem. Res., 2016, 49, 1409–1420 CrossRef CAS.
- L. Römer and T. Scheibel, Prion, 2008, 2, 154–161 CrossRef.
- M. J. Harrington, H. S. Gupta, P. Fratzl and J. H. Waite, J. Struct. Biol., 2009, 167, 47–54 CrossRef CAS.
- H. M. Keizer and R. P. Sijbesma, Chem. Soc. Rev., 2005, 34, 226–234 RSC.
- L. E. O’leary, J. A. Fallas, E. L. Bakota, M. K. Kang and J. D. Hartgerink, Nat. Chem., 2011, 3, 821–828 CrossRef.
- X. Xu, H. Yuan, J. Chang, B. He and Z. Gu, Angew. Chem., 2012, 124, 3184–3187 CrossRef.
- F. Herbst, S. Seiffert and W. H. Binder, Polym. Chem., 2012, 3, 3084–3092 RSC.
- S. Chen, N. Mahmood, M. Beiner and W. H. Binder, Angew. Chem., 2015, 127, 10326–10330 CrossRef.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS.
- J. D. Fox and S. J. Rowan, Macromolecules, 2009, 42, 6823–6835 CrossRef CAS.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS.
- H. Hofmeier and U. S. Schubert, Chem. Commun., 2005, 2423–2432 RSC.
- T. F. De Greef, M. M. Smulders, M. Wolffs, A. P. Schenning, R. P. Sijbesma and E. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS.
- M. Ahmadi, L. Löser, K. Fischer, K. Saalwächter and S. Seiffert, Macromol. Chem. Phys., 2020, 221, 1900400 CrossRef CAS.
- Y. Gu, J. Zhao and J. A. Johnson, Trends Chem., 2019, 1, 318–334 CrossRef CAS.
- Y. Gu, J. Zhao and J. A. Johnson, Angew. Chem., Int. Ed., 2020, 59, 5022–5049 CrossRef CAS.
- K. Saalwächter and S. Seiffert, Soft Matter, 2018, 14, 1976–1991 RSC.
- S. Seiffert, Polym. Chem., 2017, 8, 4472–4487 RSC.
- J. Luo, X. Sun, J.-F. Yin, P. Yin and T. Liu, Giant, 2020, 100013 CrossRef.
- M. J. Harrington, A. Masic, N. Holten-Andersen, J. H. Waite and P. Fratzl, Science, 2010, 328, 216–220 CrossRef CAS.
- J. G. Drobny, Handbook of thermoplastic elastomers, Elsevier, 2014 Search PubMed.
- J. Kim, H. Y. Jung and M. J. Park, Macromolecules, 2020, 53, 746–763 CrossRef CAS.
- L. de Lucca Freltas, J. Burgert and R. Stadler, Polym. Bull., 1987, 17, 431–438 Search PubMed.
- A. Mordvinkin, D. Döhler, W. H. Binder, R. H. Colby and K. Saalwächter, Phys. Rev. Lett., 2020, 125, 127801 CrossRef CAS.
- S. J. Rowan, P. Suwanmala and S. Sivakova, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3589–3596 CrossRef CAS.
- S. Sivakova, D. A. Bohnsack, M. E. Mackay, P. Suwanmala and S. J. Rowan, J. Am. Chem. Soc., 2005, 127, 18202–18211 CrossRef CAS.
- O. Colombani, C. Barioz, L. Bouteiller, C. Chanéac, L. Fompérie, F. Lortie and H. Montès, Macromolecules, 2005, 38, 1752–1759 CrossRef CAS.
- H. Goldansaz, A. Jangizehi, G. Nikravan, O. Verkinderen, B. Goderis, S. R. Ghaffarian, E. van Ruymbeke and M. Ahmadi, React. Funct. Polym., 2019, 137, 27–37 CrossRef CAS.
- F. Herbst, K. Schröter, I. Gunkel, S. Gröger, T. Thurn-Albrecht, J. Balbach and W. H. Binder, Macromolecules, 2010, 43, 10006–10016 Search PubMed.
- T. Yan, K. Schröter, F. Herbst, W. H. Binder and T. Thurn-Albrecht, Macromolecules, 2014, 47, 2122–2130 Search PubMed.
- W. Weng, J. B. Beck, A. M. Jamieson and S. J. Rowan, J. Am. Chem. Soc., 2006, 128, 11663–11672 CrossRef CAS.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS.
- F. J. Stadler, W. Pyckhout-Hintzen, J.-M. Schumers, C.-A. Fustin, J.-F. Gohy and C. Bailly, Macromolecules, 2009, 42, 6181–6192 CrossRef CAS.
- E. Van Ruymbeke, D. Vlassopoulos, M. Mierzwa, T. Pakula, D. Charalabidis, M. Pitsikalis and N. Hadjichristidis, Macromolecules, 2010, 43, 4401–4411 CrossRef CAS.
- Q. Chen, G. J. Tudryn and R. H. Colby, J. Rheol., 2013, 57, 1441–1462 CrossRef CAS.
- J. N. Hunt, K. E. Feldman, N. A. Lynd, J. Deek, L. M. Campos, J. M. Spruell, B. M. Hernandez, E. J. Kramer and C. J. Hawker, Adv. Mater., 2011, 23, 2327–2331 CrossRef CAS.
- A. Kayitmazer, A. Koksal and E. K. Iyilik, Soft Matter, 2015, 11, 8605–8612 RSC.
- Q. Chen, H. Masser, H.-S. Shiau, S. Liang, J. Runt, P. C. Painter and R. H. Colby, Macromolecules, 2014, 47, 3635–3644 CrossRef CAS.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS.
- T. Indei, J. Chem. Phys., 2007, 127, 144904 CrossRef.
- T. Indei, J. Chem. Phys., 2007, 127, 144905 CrossRef.
- D. Amin, A. E. Likhtman and Z. Wang, Macromolecules, 2016, 49, 7510–7524 CrossRef CAS.
- M. Ahmadi, L. G. Hawke, H. Goldansaz and E. van Ruymbeke, Macromolecules, 2015, 48, 7300–7310 CrossRef CAS.
- A. Vaccaro and G. Marrucci, J. Non-Newtonian Fluid Mech., 2000, 92, 261–273 CrossRef CAS.
- A. Semenov and M. Rubinstein, Macromolecules, 2002, 35, 4821–4837 CrossRef CAS.
- A. Jangizehi, M. Ahmadi and S. Seiffert, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 1209–1223 CrossRef CAS.
- A. Jangizehi, S. R. Ghaffarian and M. Ahmadi, Polym. Adv. Technol., 2018, 29, 726–735 CrossRef CAS.
- M. Ahmadi, A. Jangizehi, E. van Ruymbeke and S. Seiffert, Macromolecules, 2019, 52, 5255–5267 CrossRef CAS.
- L. Hawke, M. Ahmadi, H. Goldansaz and E. Van Ruymbeke, J. Rheol., 2016, 60, 297–310 CrossRef CAS.
- H. Goldansaz, C. A. Fustin, M. Wubbenhorst and E. van Ruymbeke, Macromolecules, 2016, 49, 1890–1902 CrossRef CAS.
- A. Eisenberg, Macromolecules, 1970, 3, 147–154 CrossRef CAS.
- A. Eisenberg, B. Hird and R. Moore, Macromolecules, 1990, 23, 4098–4107 CrossRef CAS.
- G. R. Goward, M. F. Schuster, D. Sebastiani, I. Schnell and H. W. Spiess, J. Phys. Chem. B, 2002, 106, 9322–9334 CrossRef CAS.
- R. K. Bose, N. Hohlbein, S. J. Garcia, A. M. Schmidt and S. van der Zwaag, Phys. Chem. Chem. Phys., 2015, 17, 1697–1704 RSC.
- J.-E. Potaufeux, J. Odent, D. Notta-Cuvier, F. Lauro and J.-M. Raquez, Polym. Chem., 2020, 11, 5914–5936 RSC.
- S.-L. Li, T. Xiao, C. Lin and L. Wang, Chem. Soc. Rev., 2012, 41, 5950–5968 RSC.
- S. Chakraborty, C. M. Berac, B. Kemper, P. Besenius and T. Speck, Macromolecules, 2019, 52, 7661–7667 CrossRef CAS.
- S. Goyal, A. Chattopadhyay, K. Kasavajhala and U. D. Priyakumar, J. Am. Chem. Soc., 2017, 139, 14931–14946 CrossRef CAS.
- T. Aida, E. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS.
- M. Blomenhofer, S. Ganzleben, D. Hanft, H.-W. Schmidt, M. Kristiansen, P. Smith, K. Stoll, D. Mäder and K. Hoffmann, Macromolecules, 2005, 38, 3688–3695 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. Folmer, J. K. Hirschberg, R. F. Lange, J. K. Lowe and E. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS.
- A. El-ghayoury, E. Peeters, A. P. Schenning and E. Meijer, Chem. Commun., 2000, 1969–1970 RSC.
- A. P. Schenning, P. Jonkheijm, E. Peeters and E. Meijer, J. Am. Chem. Soc., 2001, 123, 409–416 CrossRef CAS.
- S. Cantekin, T. F. de Greef and A. R. Palmans, Chem. Soc. Rev., 2012, 41, 6125–6137 RSC.
- J. Roosma, T. Mes, P. Leclere, A. R. Palmans and E. Meijer, J. Am. Chem. Soc., 2008, 130, 1120–1121 CrossRef CAS.
- T. Mes, M. M. Smulders, A. R. Palmans and E. Meijer, Macromolecules, 2010, 43, 1981–1991 CrossRef CAS.
- L. Brunsveld, J. Vekemans, J. Hirschberg, R. Sijbesma and E. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4977–4982 CrossRef CAS.
- J. Y. Rho, H. Cox, E. D. Mansfield, S. H. Ellacott, R. Peltier, J. C. Brendel, M. Hartlieb, T. A. Waigh and S. Perrier, Nat. Commun., 2019, 10, 1–9 CrossRef.
- J. K. Hirschberg, L. Brunsveld, A. Ramzi, J. A. Vekemans, R. P. Sijbesma and E. Meijer, Nature, 2000, 407, 167–170 CrossRef CAS.
- P. Y. Dankers, M. J. van Luyn, A. Huizinga-van der Vlag, G. M. van Gemert, A. H. Petersen, E. Meijer, H. M. Janssen, A. W. Bosman and E. R. Popa, Biomaterials, 2012, 33, 5144–5155 CrossRef CAS.
- P. Y. Dankers, T. M. Hermans, T. W. Baughman, Y. Kamikawa, R. E. Kieltyka, M. M. Bastings, H. M. Janssen, N. A. Sommerdijk, A. Larsen and M. J. Van Luyn, Adv. Mater., 2012, 24, 2703–2709 CrossRef CAS.
- R. E. Kieltyka, A. Pape, L. Albertazzi, Y. Nakano, M. M. Bastings, I. K. Voets, P. Y. Dankers and E. Meijer, J. Am. Chem. Soc., 2013, 135, 11159–11164 CrossRef CAS.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. Dankers and E. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS.
- S. Han, G. Mellot, S. Pensec, J. Rieger, F. Stoffelbach, E. Nicol, O. Colombani, J. Jestin and L. Bouteiller, Macromolecules, 2020, 53, 427–433 CrossRef CAS.
- H. J. Zhang, T. L. Sun, A. K. Zhang, Y. Ikura, T. Nakajima, T. Nonoyama, T. Kurokawa, O. Ito, H. Ishitobi and J. P. Gong, Adv. Mater., 2016, 28, 4884–4890 CrossRef CAS.
- X. Hu, M. Vatankhah-Varnoosfaderani, J. Zhou, Q. Li and S. S. Sheiko, Adv. Mater., 2015, 27, 6899–6905 CrossRef CAS.
- H. Kautz, D. Van Beek, R. P. Sijbesma and E. Meijer, Macromolecules, 2006, 39, 4265–4267 CrossRef CAS.
- D. Van Beek, A. Spiering, G. W. Peters, K. Te Nijenhuis and R. P. Sijbesma, Macromolecules, 2007, 40, 8464–8475 CrossRef CAS.
- M. M. Nieuwenhuizen, T. F. de Greef, R. L. van der Bruggen, J. M. Paulusse, W. P. Appel, M. M. Smulders, R. P. Sijbesma and E. Meijer, Chem. – Eur. J., 2010, 16, 1601–1612 CrossRef CAS.
- M. Ahmadi and S. Seiffert, J. Polym. Sci., 2020, 58, 330–342 CrossRef CAS.
- W. Schmolke, M. Ahmadi and S. Seiffert, Phys. Chem. Chem. Phys., 2019, 21, 19623–19638 RSC.
- E. R. Zubarev, M. U. Pralle, E. D. Sone and S. I. Stupp, J. Am. Chem. Soc., 2001, 123, 4105–4106 CrossRef CAS.
- X. Callies, C. Fonteneau, C. Véchambre, S. Pensec, J. M. Chenal, L. Chazeau, L. Bouteiller, G. Ducouret and C. Creton, Polymer, 2015, 69, 233–240 CrossRef CAS.
- C. Véchambre, X. Callies, C. C. Fonteneau, G. Ducouret, S. Pensec, L. Bouteiller, C. Creton, J.-M. Chenal and L. Chazeau, Macromolecules, 2015, 48, 8232–8239 CrossRef.
- X. Callies, C. Véchambre, C. Fonteneau, S. Pensec, J. M. Chenal, L. Chazeau, L. Bouteiller, G. Ducouret and C. Creton, Macromolecules, 2015, 48, 7320–7326 CrossRef CAS.
- S. I. Hendrikse, S. P. Wijnands, R. P. Lafleur, M. J. Pouderoijen, H. M. Janssen, P. Y. Dankers and E. Meijer, Chem. Commun., 2017, 53, 2279–2282 RSC.
- N. E. Botterhuis, D. Van Beek, G. M. van Gemert, A. W. Bosman and R. P. Sijbesma, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3877–3885 CrossRef CAS.
- A. Jangizehi, S. R. Ghaffarian, W. Schmolke and S. Seiffert, Macromolecules, 2018, 51, 2859–2871 CrossRef CAS.
- J. R. Kumpfer, J. J. Wie, J. P. Swanson, F. L. Beyer, M. E. Mackay and S. J. Rowan, Macromolecules, 2012, 45, 473–480 CrossRef CAS.
- W. P. Appel, G. Portale, E. Wisse, P. Y. Dankers and E. Meijer, Macromolecules, 2011, 44, 6776–6784 CrossRef CAS.
- L. Kan, P. Zhang, H. Jiang, S. Zhang, Z. Liu, X. Zhang, N. Ma, D. Qiu and H. Wei, RSC Adv., 2019, 9, 8905–8911 RSC.
- M. C. Stuparu, A. Khan and C. J. Hawker, Polym. Chem., 2012, 3, 3033–3044 RSC.
- K. E. Feldman, M. J. Kade, E. Meijer, C. J. Hawker and E. J. Kramer, Macromolecules, 2010, 43, 5121–5127 CrossRef CAS.
- L. M. Pitet, A. H. van Loon, E. J. Kramer, C. J. Hawker and E. Meijer, ACS Macro Lett., 2013, 2, 1006–1010 CrossRef CAS.
- W. H. Binder, S. Bernstorff, C. Kluger, L. Petraru and M. J. Kunz, Adv. Mater., 2005, 17, 2824–2828 CrossRef CAS.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS.
- S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. Harris, W. Hayes, M. E. Mackay and S. J. Rowan, Chem. Commun., 2009, 6717–6719 RSC.
- D. Mozhdehi, S. Ayala, O. R. Cromwell and Z. Guan, J. Am. Chem. Soc., 2014, 136, 16128–16131 CrossRef CAS.
- Y. Liu, Z. Wang and X. Zhang, Chem. Soc. Rev., 2012, 41, 5922–5932 RSC.
- S. Ge, M. Tress, K. Xing, P.-F. Cao, T. Saito and A. P. Sokolov, Soft Matter, 2020, 16, 390–401 RSC.
- T. Yan, K. Schröter, F. Herbst, W. H. Binder and T. Thurn-Albrecht, Macromolecules, 2017, 50, 2973–2985 CrossRef CAS.
- L. L. De Lucca Freitas and R. Stadler, Macromolecules, 1987, 20, 2478–2485 CrossRef CAS.
- P. Gill, T. T. Moghadam and B. Ranjbar, J. Biomol. Tech., 2010, 21, 167 Search PubMed.
- M. H. Chiu and E. J. Prenner, J. Pharm. BioAllied Sci., 2011, 3, 39 CrossRef CAS.
- R. P. D’Amelia, V. Stracuzzi and W. F. Nirode, J. Chem. Educ., 2008, 85, 109 CrossRef.
- N. Zoratto and P. Matricardi, Polymeric Gels, Elsevier, 2018, pp. 91–124 Search PubMed.
- B. Stuart, Kirk-Othmer Encyclopedia of Chemical Technology, 2000 Search PubMed.
- T. Theophanides, Infrared Spectroscopy-Materials Science, Engineering and Technology, 2012 Search PubMed.
- R. Stadler and L. de Lucca Freitas, Polym. Bull., 1986, 15, 173–179 CrossRef CAS.
- I. R. Kleckner and M. P. Foster, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 942–968 CrossRef CAS.
- H. W. Spiess, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5031–5044 CrossRef CAS.
- H. W. Spiess, Macromolecules, 2017, 50, 1761–1777 CrossRef CAS.
- J. K. Hirschberg, F. H. Beijer, H. A. van Aert, P. C. Magusin, R. P. Sijbesma and E. Meijer, Macromolecules, 1999, 32, 2696–2705 CrossRef.
- E. Kolomiets and J.-M. Lehn, Chem. Commun., 2005, 1519–1521 RSC.
- M. Miyauchi, Y. Takashima, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2005, 127, 2984–2989 CrossRef CAS.
- F. Wang, J. Zhang, X. Ding, S. Dong, M. Liu, B. Zheng, S. Li, L. Wu, Y. Yu and H. W. Gibson, Angew. Chem., Int. Ed., 2010, 49, 1090–1094 CrossRef CAS.
- X. Yan, S. Li, J. B. Pollock, T. R. Cook, J. Chen, Y. Zhang, X. Ji, Y. Yu, F. Huang and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15585–15590 CrossRef CAS.
- I. Schnell, B. Langer, S. H. Söntjens, R. P. Sijbesma, M. H. van Genderen and H. W. Spiess, Phys. Chem. Chem. Phys., 2002, 4, 3750–3758 RSC.
- P. J. Worsfold and E. A. G. Zagatto, Encyclopedia of analytical science, 2019 Search PubMed.
- M. Ahmadi and S. Seiffert, Soft Matter, 2020, 16, 2332–2341 RSC.
- H. Hofmeier, A. El-ghayoury, A. P. Schenning and U. S. Schubert, Chem. Commun., 2004, 318–319 RSC.
- R. Dobrawa, M. Lysetska, P. Ballester, M. Grüne and F. Würthner, Macromolecules, 2005, 38, 1315–1325 CrossRef CAS.
- F. Kremer and A. Schönhals, Broadband dielectric spectroscopy, Springer Science & Business Media, 2002 Search PubMed.
- F. Kremer, J. Non-Cryst. Solids, 2002, 305, 1–9 CrossRef CAS.
- M. Tress, K. Xing, S. Ge, P. Cao, T. Saito and A. Sokolov, Eur. Phys. J. E: Soft Matter Biol. Phys., 2019, 42, 133 CrossRef CAS.
- M. Müller, E. Fischer, F. Kremer, U. Seidel and R. Stadler, Colloid Polym. Sci., 1995, 273, 38–46 CrossRef.
- K. Xing, M. Tress, P. Cao, S. Cheng, T. Saito, V. N. Novikov and A. P. Sokolov, Soft Matter, 2018, 14, 1235–1246 RSC.
- B. Gold, C. Hövelmann, N. Lühmann, W. Pyckhout-Hintzen, A. Wischnewski and D. Richter, J. Rheol., 2017, 61, 1211–1226 CrossRef CAS.
- M. J. Hollamby, Phys. Chem. Chem. Phys., 2013, 15, 10566–10579 RSC.
- S. Seiffert, Prog. Polym. Sci., 2017, 66, 1–21 CrossRef CAS.
- B. Chu and B. S. Hsiao, Chem. Rev., 2001, 101, 1727–1762 CrossRef CAS.
- C. Giannini, D. Siliqi and D. Altamura, Nanocomposites: In Situ Synthesis of Polymer-Embedded Nanostructures, 2013, pp. 209–222 Search PubMed.
- Y. Nozue, Y. Shinohara and Y. Amemiya, Polym. J., 2007, 39, 1221–1237 CrossRef CAS.
- J. L. Wietor, D. J. M. van Beek, G. W. Peters, E. Mendes and R. P. Sijbesma, Macromolecules, 2011, 44, 1211–1219 CrossRef CAS.
- M. Shibayama, Polym. J., 2011, 43, 18–34 CrossRef CAS.
- M. Krutyeva, A. Brás, W. Antonius, C. Hövelmann, A. Poulos, J. Allgaier, A. Radulescu, P. Lindner, W. Pyckhout-Hintzen and A. Wischnewski, Macromolecules, 2015, 48, 8933–8946 CrossRef CAS.
- M. Mihajlovic, M. Staropoli, M.-S. Appavou, H. M. Wyss, W. Pyckhout-Hintzen and R. P. Sijbesma, Macromolecules, 2017, 50, 3333–3346 CrossRef CAS.
- J. Tian, T. A. Seery, D. L. Ho and R. Weiss, Macromolecules, 2004, 37, 10001–10008 CrossRef CAS.
- J. Tian, T. A. Seery and R. Weiss, Macromolecules, 2004, 37, 9994–10000 CrossRef CAS.
- C. G. Wiener, C. Wang, Y. Liu, R. Weiss and B. D. Vogt, Macromolecules, 2017, 50, 1672–1680 CrossRef CAS.
- D. Rugar and P. Hansma, Phys. Today, 1990, 43, 23–30 CrossRef CAS.
- E. Meyer, Prog. Surf. Sci., 1992, 41, 3–49 CrossRef CAS.
- C. S. Goldsbury, S. Scheuring and L. Kreplak, Curr. Protoc. Protein Sci., 2009, 58, 17.17.11–17.17.19 Search PubMed.
- S. Zou, H. Schönherr and G. J. Vancso, Angew. Chem., Int. Ed., 2005, 44, 956–959 CrossRef CAS.
- S. Chen, Z. Geng, X. Zheng, J. Xu, W. H. Binder and J. Zhu, Polym. Chem., 2020, 11, 4022–4028 RSC.
- Z. Kochovski, G. Chen, J. Yuan and Y. Lu, Colloid Polym. Sci., 2020, 298, 707–717 CrossRef CAS.
- R. P. Lafleur, S. Herziger, S. M. Schoenmakers, A. D. Keizer, J. Jahzerah, B. N. Thota, L. Su, P. H. Bomans, N. A. Sommerdijk and A. R. Palmans, J. Am. Chem. Soc., 2020, 142, 17644–17652 CrossRef CAS.
- C. M. Leenders, L. Albertazzi, T. Mes, M. M. Koenigs, A. R. Palmans and E. Meijer, Chem. Commun., 2013, 49, 1963–1965 RSC.
- B. Huang, M. Bates and X. Zhuang, Annu. Rev. Biochem., 2009, 78, 993–1016 CrossRef CAS.
- S. Onogi, H. Shigemitsu, T. Yoshii, T. Tanida, M. Ikeda, R. Kubota and I. Hamachi, Nat. Chem., 2016, 8, 743 CrossRef CAS.
- L. Albertazzi, D. van der Zwaag, C. M. Leenders, R. Fitzner, R. W. van der Hofstad and E. Meijer, Science, 2014, 344, 491–495 CrossRef CAS.
- S. Seiffert and J. Sprakel, Chem. Soc. Rev., 2012, 41, 909–930 RSC.
- W. C. Yount, D. M. Loveless and S. L. Craig, Angew. Chem., Int. Ed., 2005, 44, 2746–2748 CrossRef CAS.
- M. Ahmadi, C. Bailly, R. Keunings, M. Nekoomanesh, H. Arabi and E. Van Ruymbeke, Macromolecules, 2011, 44, 647–659 CrossRef CAS.
- E. Van Ruymbeke, C. Bailly, R. Keunings and D. Vlassopoulos, Macromolecules, 2006, 39, 6248–6259 CrossRef CAS.
- M. Doi and S. F. Edwards, The Theory of Polymer Dynamics, Clarendon Press, Oxford, 1986 Search PubMed.
- T. McLeish, Adv. Phys., 2002, 51, 1379–1527 CrossRef CAS.
- G. Marrucci, J. Polym. Sci., Polym. Phys. Ed., 1985, 23, 159–177 CrossRef CAS.
- M. Shivokhin, E. Van Ruymbeke, C. Bailly, D. Kouloumasis, N. Hadjichristidis and A. E. Likhtman, Macromolecules, 2014, 47, 2451–2463 CrossRef CAS.
- M. Rubinstein and A. N. Semenov, Macromolecules, 2001, 34, 1058–1068 CrossRef CAS.
- M. Cates, Macromolecules, 1987, 20, 2289–2296 CrossRef CAS.
- M. Cates and S. Candau, J. Phys.: Condens. Matter, 1990, 2, 6869–6892 CrossRef CAS.
- L. Leibler, M. Rubinstein and R. H. Colby, Macromolecules, 1991, 24, 4701–4707 CrossRef CAS.
- M. Rubinstein and A. N. Semenov, Macromolecules, 1998, 31, 1386–1397 CrossRef CAS.
- A. N. Semenov and M. Rubinstein, Macromolecules, 1998, 31, 1373–1385 CrossRef CAS.
- J. Hentschel, A. M. Kushner, J. Ziller and Z. Guan, Angew. Chem., Int. Ed., 2012, 51, 10561–10565 CrossRef CAS.
- S. Hackelbusch, T. Rossow, P. van Assenbergh and S. Seiffert, Macromolecules, 2013, 46, 6273–6286 CrossRef CAS.
- S. Tang, A. Habicht, S. Li, S. Seiffert and B. D. Olsen, Macromolecules, 2016, 49, 5599–5608 CrossRef CAS.
- W. C. Yount, D. M. Loveless and S. L. Craig, J. Am. Chem. Soc., 2005, 127, 14488–14496 CrossRef CAS.
- E. A. Appel, R. A. Forster, A. Koutsioubas, C. Toprakcioglu and O. A. Scherman, Angew. Chem., Int. Ed., 2014, 53, 10038–10043 CrossRef CAS.
- E. A. Appel, F. Biedermann, D. Hoogland, J. Del Barrio, M. D. Driscoll, S. Hay, D. J. Wales and O. A. Scherman, J. Am. Chem. Soc., 2017, 139, 12985–12993 CrossRef CAS.
- S. Tang and B. D. Olsen, Macromolecules, 2016, 49, 9163–9175 CrossRef CAS.
- A. Shabbir, H. Goldansaz, O. Hassager, E. van Ruymbeke and N. J. Alvarez, Macromolecules, 2015, 48, 5988–5996 CrossRef CAS.
- R. Stadler and L. de Lucca Freitas, Colloid Polym. Sci., 1986, 264, 773–778 CrossRef CAS.
- L. de Lucca Freitas and R. Stadler, Colloid Polym. Sci., 1988, 266, 1095–1101 CrossRef CAS.
- K. E. Feldman, M. J. Kade, E. Meijer, C. J. Hawker and E. J. Kramer, Macromolecules, 2009, 42, 9072–9081 CrossRef CAS.
- C. L. Lewis, K. Stewart and M. Anthamatten, Macromolecules, 2014, 47, 729–740 CrossRef CAS.
- T. Rossow and S. Seiffert, Polym. Chem., 2014, 5, 3018–3029 RSC.
- C. L. Elkins, T. Park, M. G. McKee and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4618–4631 CrossRef CAS.
- M. G. McKee, C. L. Elkins, T. Park and T. E. Long, Macromolecules, 2005, 38, 6015–6023 CrossRef CAS.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS.
- F. Herbst, D. Döhler, P. Michael and W. H. Binder, Macromol. Rapid Commun., 2013, 34, 203–220 CrossRef CAS.
- M. Ahmadi and M. Ghanavati, Polymer, 2020, 211, 123117 CrossRef CAS.
- Z. Wang, X. Lu, S. Sun, C. Yu and H. Xia, J. Mater. Chem. B, 2019, 7, 4876–4926 RSC.
- Y. Lai, X. Kuang, P. Zhu, M. Huang, X. Dong and D. Wang, Adv. Mater., 2018, 30, 1802556 CrossRef.
- Y. Song, Y. Liu, T. Qi and G. L. Li, Angew. Chem., Int. Ed., 2018, 57, 13838–13842 CrossRef CAS.
- C. Wang, H. Wu, Z. Chen, M. T. McDowell, Y. Cui and Z. Bao, Nat. Chem., 2013, 5, 1042 CrossRef CAS.
- B. D. Wall, S. R. Diegelmann, S. Zhang, T. J. Dawidczyk, W. L. Wilson, H. E. Katz, H. Q. Mao and J. D. Tovar, Adv. Mater., 2011, 23, 5009–5014 CrossRef CAS.
- P. Y. Dankers, M. C. Harmsen, L. A. Brouwer, M. J. Van Luyn and E. Meijer, Nat. Mater., 2005, 4, 568–574 CrossRef CAS.
- O. J. Goor, S. I. Hendrikse, P. Y. Dankers and E. Meijer, Chem. Soc. Rev., 2017, 46, 6621–6637 RSC.
- P. M. Kharkar, K. L. Kiick and A. M. Kloxin, Chem. Soc. Rev., 2013, 42, 7335–7372 RSC.
- E. A. Mol, Z. Lei, M. T. Roefs, M. H. Bakker, M. J. Goumans, P. A. Doevendans, P. Y. Dankers, P. Vader and J. P. Sluijter, Adv. Healthcare Mater., 2019, 8, 1900847 CrossRef CAS.
- B. B. Mollet, M. Comellas-Aragones, A. Spiering, S. H. Söntjens, E. Meijer and P. Y. Dankers, J. Mater. Chem. B, 2014, 2, 2483–2493 RSC.
- A. Pape, B. D. Ippel and P. Y. Dankers, Langmuir, 2017, 33, 4076–4082 CrossRef CAS.
- O. J. Goor, H. M. Keizer, A. L. Bruinen, M. G. Schmitz, R. M. Versteegen, H. M. Janssen, R. M. Heeren and P. Y. Dankers, Adv. Mater., 2017, 29, 1604652 CrossRef.
- O. J. Goor, J. E. Brouns and P. Y. Dankers, Polym. Chem., 2017, 8, 5228–5238 RSC.
- O. Jeon, D. W. Wolfson and E. Alsberg, Adv. Mater., 2015, 27, 2216–2223 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |