
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic asymmetric synthesis of quaternary trifluoromethyl α- to ε-amino acid derivatives via umpolung allylation/2-aza-Cope rearrangement†
Xi-Shang
Sun
a,
Xing-Heng
Wang
a,
Hai-Yan
Tao
a,
Liang
Wei
*a and
Chun-Jiang
Wang
 *ab
*ab
aEngineering Research Center of Organosilicon Compounds & Materials, Ministry of Education, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, 430072, China. E-mail: cjwang@whu.edu.cn
bState Key Laboratory of Elemento-organic Chemistry, Nankai University, Tianjin 300071, China
First published on 17th September 2020
Abstract
In this study, we developed an efficient Ir-catalyzed cascade umpolung allylation/2-aza-Cope rearrangement of tertiary α-trifluoromethyl α-amino acid derivatives for the preparation of a variety of quaternary α-trifluoromethyl α-amino acids in high yields with excellent enantioselectivities. The umpolung reactivity empowered by the activation of the key isatin-ketoimine moiety obviates the intractable enantioselectivity control in Pd-catalyzed asymmetric linear α-allylation. In combination with quasi parallel kinetic resolution or kinetic resolution, the generality of this method is further demonstrated by the first preparation of enantioenriched quaternary trifluoromethyl β-, γ-, δ- and ε-amino acid derivatives.
Introduction
Enantioenriched fluorinated amino acids (AAs) and their biopolymers have received great attention in the past decades due to their potential in preparing bioactive molecules with unique characteristics.1 The introduction of the trifluoromethyl group into amino acids often exerts increased environmental and metabolic stability with enhanced biological activity,2 thus the products have found widespread bio-organic and medical applications as, for example, biological tracers, mechanistic probes, and enzyme inhibitors.3 Accordingly, the development of innovative synthetic protocols to construct chiral trifluoro-methylated amino acids with yet unknown chemical and biological attributes is in extremely high demand. Although some achievement has been made in this field, the progress in developing reliable synthetic methodologies to synthesize diverse quaternary trifluoromethyl amino acids is far from being fully explored. For example, the catalytic asymmetric synthesis of trifluoromethylated α-amino acids has been well established with many addition methods, such as catalytic asymmetric Strecker-type reaction, nucleophilic addition and amination.4,5 In sharp contrast, the asymmetric construction of enantioenriched trifluoromethylated AAs other than α-amino acids, such as β-, γ-, δ- and ε-AAs, has been scarcely reported and in most of the methods developed so far chiral auxiliaries or enzymes were employed.6 In addition, the asymmetric construction of quaternary trifluoromethyl amino acids, which are particularly useful in the design of peptides and proteins with enhanced properties,7 represents another synthetic challenge in this area because of the disfavored steric hindrance.4cIn our continuous interest in the asymmetric synthesis of unnatural amino acids,8 we recently disclosed an Ir-catalyzed asymmetric α-allylation of an α-CF3 aldimino ester (Scheme 1a, n = 0).9 Quasi-kinetic resolution of the formed diastereomeric allylation intermediates would produce enantioenriched quaternary α-trifluoromethyl α-amino acids (α-Tfm α-AAs) and homoallylic amines simultaneously. However, further attempts to extend this protocol for the preparation of more challenging quaternary trifluoromethyl β-, γ-, δ- and ε-amino acids through elongating the length of the ester carbon chain all failed due to the reduced nucleophilicity of the carbon connected to CF3 in the corresponding aldimino ester. Inspired by previous investigation on the 2-azaallyl anion,10,11 we surmised that an appropriate imino activating group connected to the N-terminus of a tertiary trifluoromethylated amino ester should be capable of regulating the reactivity of the in situ-formed 2-azaallyl carbanion and allowing an Ir-catalyzed asymmetric branched-selective allylation12 exclusively at the α′-position in an umpolung manner. The generated allylation intermediate containing two adjacent multi-substituted stereogenic centers would readily undergo subsequent 2-aza-Cope rearrangement11a,b,13 to release the steric congestion and produce linear allyl substituted α-Tfm α-AAs in high enantioselectivity control. Most importantly, the umpolung strategy not only obviates the intractable Pd-catalyzed linear α-allylation but might tolerate broader α-ester carbon chains, and therefore enable precise construction of diverse types of quaternary trifluoromethylated amino acids. Only one report of palladium-catalyzed linear allylation of an α-CF3 aldimino ester has been disclosed so far with a single example of enantioenriched quaternary α-Tfm α-AAs with 50% ee (Scheme 1b, left side).14 To realize this design, several challenging issues need to be considered: (1) regioselectivity: the competitive nucleophilicity of the α and α′ positions of the 2-azaallyl anion in the first allylation step; (2) stereoselectivity: it is a formidable task to achieve high diastereo-/enantioselectivity control in the allylation/2-aza-Cope rearrangement with the bulky CF3-containing 2-azaallyl anion as a prochiral nucleophile; (3) chemoselectivity: it is well-known that a CF3 group attached to a carbon atom bearing a hydrogen atom easily undergoes β-fluorine elimination.15 Herein, we report the development, stereochemical modulation, substrate generality and synthetic applications of Ir-catalyzed umpolung allylation/2-aza-Cope rearrangement for the preparation of chiral quaternary trifluoromethyl α-, β-, γ-, δ- and ε-amino acid derivatives that are not readily accessed by other methodologies.
 | ||
| Scheme 1 Catalytic asymmetric synthesis of quaternary trifluoromethyl α-AAs (previous work) and α- to ε-AAs (this work). | ||
Results and discussion
Our initial trials were conducted between methyl cinnamyl carbonate 2a and different potential 2-azaallyl anion precursors 1a–1c using [Ir(cod)Cl]2/(S,S,S)-L1 (ref. 16 and 17) as the catalyst and Cs2CO3 as the base in 1,2-dichloroethane (DCE) at room temperature. No reaction occurred with ketoimine esters derived from benzophenone or fluorenone (Table 1, entries 1 and 2). To our delight, with isatin-activated ketoimine ester 1c, the desired quaternary trifluoromethyl α-amino acid derivative 3a was isolated in 99% yield with 96% ee. Considering the significant effect of the isatin-derived imino moiety on the reactivity and the predicted pKa values (in DMSO: 17.38 (1a), 12.66 (1b), 12.44 (1c)),18,19 we believed that both the steric effect and the electronic effect affect the reactivity of the tested ketoimine esters. Encouraged by these promising results, further screening of other parameters was performed. No desired product was observed in the absent of a base or using a weak organic base Et3N, indicating that an appropriate base with sufficient basicity is essential for the formation of 2-azaallyl carbanion (entries 4 and 5). When DBU was employed, 3a was obtained in 80% yield along with a small amount of β-fluorine elimination byproduct (entry 6). Performing the reaction in other solvents including dichloromethane, benzene, tetrahydrofuran, and toluene all led to decreased yield although the enantioselectivities remain excellent (entries 7–10).|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | Base | Solvent | Yieldb (%) | eec (%) |
| a All reactions were carried out with 0.2 mmol 1, 0.22 mmol 2a and 0.2 mmol base in 2 mL of solvent. b Isolated yield. c Determined by HPLC analysis. | |||||
| 1 | 1a | Cs2CO3 | (CH2Cl)2 | NR | — |
| 2 | 1b | Cs2CO3 | (CH2Cl)2 | NR | — |
| 3 | 1c | Cs2CO3 | (CH2Cl)2 | 99 | 96 |
| 4 | 1c | — | (CH2Cl)2 | NR | — |
| 5 | 1c | NEt3 | (CH2Cl)2 | NR | — |
| 6 | 1c | DBU | (CH2Cl)2 | 80 | 95 |
| 7 | 1c | Cs2CO3 | CH2Cl2 | 90 | 94 |
| 8 | 1c | Cs2CO3 | Benzene | 85 | 95 |
| 9 | 1c | Cs2CO3 | THF | 37 | 96 |
| 10 | 1c | Cs2CO3 | Toluene | 80 | 93 |
With optimized reaction conditions in hand, we set out to evaluate the generality of this cascade reaction. Firstly, a variety of substituted allyl carbonates were investigated. As summarized in Table 2, para-, meta-substituted and 3,4-disubstituted cinnamyl carbonates reacted smoothly with 1a, giving α-Tfm α-AAs 3b–3i in 87–99% yield with 90–96% ee (Table 2, entries 2–9). The current transformation shows good tolerance towards the electronic effect of the substituents; both electron-withdrawing and electron-donating groups were well-tolerated. In line with previous Ir-catalyzed asymmetric allylation with Feringa-type ligands, ortho-methyl substituted cinnamyl carbonate 2j was not a viable π-allyl precursor in this catalytic system. Fortunately, the corresponding product 3j could be obtained in 82% yield with 64% ee when using You's ligand (R,Ra)-L2 (ref. 20) (entry 10). Fused 2-naphthyl, heteroaromatic 2-thienyl and 2-furyl substituted carbonates 2k–m worked well, affording the corresponding 3k–m in good yields with high to excellent enantioselectivities (entries 11–13). In addition, crotyl carbonate 2n was also proven to be a suitable reaction partner, delivering the desired quaternary CF3-containing α-amino acids 3n in 95% yield with 86% ee (entry 14).
|
|
||||
|---|---|---|---|---|
| Entry | R | 3 | Yieldb (%) | eec (%) |
| a All reactions were carried out with 0.2 mmol 1c, 0.22 mmol 2, Cs2CO3 (0.2 mmol) in 2 mL of DCE. b Isolated yield. c Ee determined by HPLC analysis. d (R,Ra)-L2 was used. e X-ray structure of hydrolyzed (R)-3k′ was obtained (Fig. 1). | ||||
| 1 | Ph(2a) | 3a | 99 | 96 |
| 2 | p-MeC6H4 (2b) | 3b | 91 | 94 |
| 3 | p-MeOC6H4 (2c) | 3c | 89 | 96 |
| 4 | p-ClC6H4 (2d) | 3d | 99 | 94 |
| 5 | p-BrC6H4 (2e) | 3e | 87 | 94 |
| 6 | 3,4-Cl2-C6H3 (2f) | 3f | 99 | 90 |
| 7 | m-MeOC6H4 (2g) | 3g | 99 | 94 |
| 8e | m-MeC6H4 (2h) | 3h | 92 | 94 |
| 9 | Piperonyl (2i) | 3i | 98 | 94 |
| 10d | o-MeC6H4 (2j) | 3j | 82 | 63 |
| 11e | 2-Naphthyl (2k) | 3k | 86 | 94 |
| 12 | 2-Thienyl (2l) | 3l | 90 | 94 |
| 13 | 2-Furyl (2m) | 3m | 92 | 86 |
| 14 | Me (2n) | 3n | 95 | 86 |

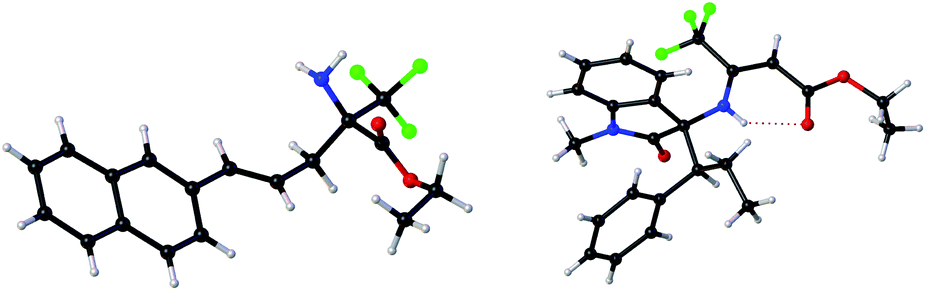 | ||
| Fig. 1 X-ray of (R)-3k′ and (S,S)-5a.21 | ||
Having established the asymmetric construction of quaternary trifluoromethyl α-amino acids, we then extended the current methodology for the preparation of more challenging trifluoromethyl-containing β-amino acids using isatin-activated ketoimine ester 1d as the 2-azaallyl carbanion precursor. The reaction between 1d and 2a under identical conditions except that DBU was used as the base instead of Cs2CO3 affords quaternary β-CF3 β-amino acid (β-Tfm β-AA) 4a in 37% yield along with 54% yield of isomerized allylation product 5a with excellent enantioselectivities (Table 3, entry 1). It is believed that a quasi-parallel kinetic resolution (PKR) process occurred with the two diastereomeric branched allylation intermediates formed. Although no better improvement in the ratio of the two products could be achieved via further reaction optimization, compounds 4a and 5a could be readily separated through silica gel column purification due to the distinct polar difference, which is ascribed to the intramolecular hydrogen bond interaction existing in the isomerized allylation product 5a (Fig. 1). The isolated compound 5a (98% ee) could be further converted to ent-4a with 64% ee (opposite configuration) upon heating in dichloroethane or toluene (see the ESI† for more details). In view of the fact that both enantioenriched β-Tfm β-AAs and 3-amino oxindole derivatives22 are useful building blocks in organic synthesis, developing a general method to prepare the two chiral compounds in one-pot is of particular interest. Thus, the generality of this transformation was then evaluated using 1d as the 2-azaallyl anion precursor. As shown in Table 3, meta- and para-substituted cinnamyl carbonates are all compatible reaction partners, giving the desired 4b–j and 5b–j in high overall yields with excellent enantioselectivities (Table 3, entries 2–10). No reaction occurred with ortho-substituted cinnamyl carbonate due to the disfavored steric hindrance at the nucleophilic 2-azaallyl anion and electronic Ir–π-allyl intermediate. 2-Naphthyl substituted 2k reacted smoothly with 1d, producing 4k and 5k in 35% and 45% yield, respectively, with excellent enantioselectivities (entry 11). To our surprise, when using 2-furyl and 2-thienyl allyl carbonates as the π-allyl precursors, β-Tfm β-AA derivatives 4l and 4m were observed as the major products in high yields and good enantioselectivity along with a trace amount of 5 (entries 12 and 13). The varying ratios of products 4 and 5 were attributed to the different diastereoselectivities in the first allylation step. Furthermore, when crotyl carbonate 2n was tested in this reaction, only allylation product 5n was isolated in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr without further rearrangement (entry 14).
:1 dr without further rearrangement (entry 14).
|
|
|||
|---|---|---|---|
| Entry | R | Yieldb,c (ee) (%) | |
| 4 | 5 | ||
| a All reactions were carried out with 0.40 mmol 1d, 0.44 mmol 2, base (0.4 mmol) in 2 mL of DCE. b Isolated yield. c Determined by HPLC analysis. d X-ray structure of (S,S)-5a was obtained. e 89% ee (major) and 81% ee (minor). | |||
| 1 | Ph (2a) | 4a, 37(91) | 5a, 54(98)d |
| 2 | p-MeC6H4 (2b) | 4b, 35(90) | 5b, 45(98) |
| 3 | p-MeOC6H4 (2c) | 4c, 34(85) | 5c, 57(96) |
| 4 | p-ClC6H4 (2d) | 4d, 38(90) | 5d, 52(98) |
| 5 | p-BrC6H4 (2e) | 4e, 35(90) | 5e, 55(97) |
| 6 | 3,4-Cl2C6H3 (2f) | 4f, 34(92) | 5f, 48(87) |
| 7 | m-MeOC6H4 (2g) | 4g, 32(89) | 5g, 47(91) |
| 8 | m-MeC6H4 (2h) | 4h, 39(94) | 5h, 57(97) |
| 9 | Piperonyl (2i) | 4i, 41(85) | 5i, 53(95) |
| 10 | m-ClC6H4 (2j) | 4j, 43(93) | 5j, 56(97) |
| 11 | 2-Naphthyl (2k) | 4k, 35(90) | 5k, 45(97) |
| 12 | 2-Thienyl (2l) | 4l, 90(94) | 5l, trace |
| 13 | 2-Furyl (2m) | 4m, 81(85) | 5m, trace |
| 14 | Me (2n) | — | 5n, 88(89)e |
Remarkably, the current method is also capable of constructing quaternary trifluoromethyl γ-, δ- and ε-amino acids through further elongating the length of the ester carbon chain. As shown in Table 4, the reaction between isatin-activated γ-CF3 γ-amino ester 1e and 2a undergoes an umpolung Ir-catalyzed allylation followed by quasi-kinetic resolution via 2-aza-Cope rearrangement, delivering the desired product 6a (44% yield, 90% ee) and less reactive allylation product 7a (35% yield, 94% ee). Similarly, using isatin-activated δ-CF3 δ-amino ester derived 1f and ε-CF3 ε-amino ester derived 1g as the 2-azaallyl anion precursors, the corresponding quaternary δ-CF3 δ-amino acid 6b (45% yield, 93% ee) and ε-CF3 ε-amino acid derivative 6c (44% yield, 94% ee) were obtained in synthetically useful yields and excellent enantioselectivities along with the allylation products 7b (37% yield, 94% ee) and 7c (36% yield, 97% ee).
| a All reactions were carried out with 0.40 mmol 1, 0.44 mmol 2a, base (0.4 mmol) in 2 mL of DCE. Isolated yield. The ee value was determined by HPLC analysis. |
|---|
|
To further showcase the utility of this method, several synthetic transformations were conducted as shown in Scheme 2. Acidic hydrolysis of 3a and 4a afforded trifluoromethylated amino esters 8 and 9 in high yields with maintained enantioselectivities. I2-promoted intramolecular cyclization of compounds 8 and 9 provides a straightforward entry to highly functionalized pyrrolidines 10 and 11 in high yield with high diastereoselectivity control. Reduction of 8 and 9 with DIBAL-H followed by one-pot mesylation/intramolecular nucleophilic substitution produced enantioenriched aziridine 12 and azetidine 13 bearing a CF3-containing N-quaternary stereogenic center in good yields without loss of enantioselectivity (Scheme 2a). On the other hand, quaternary γ-CF3 γ-amino ester 6a and δ-CF3 δ-amino ester 6b could be directly transformed to γ-butyrolactam 14 and δ-valerolactam 15via acidic hydrolysis/lactamization. Furthermore, both branched allylation products 5a and 7a could be readily hydrolyzed under mild conditions, giving compound 16 in good yield with maintained stereoselectivity.
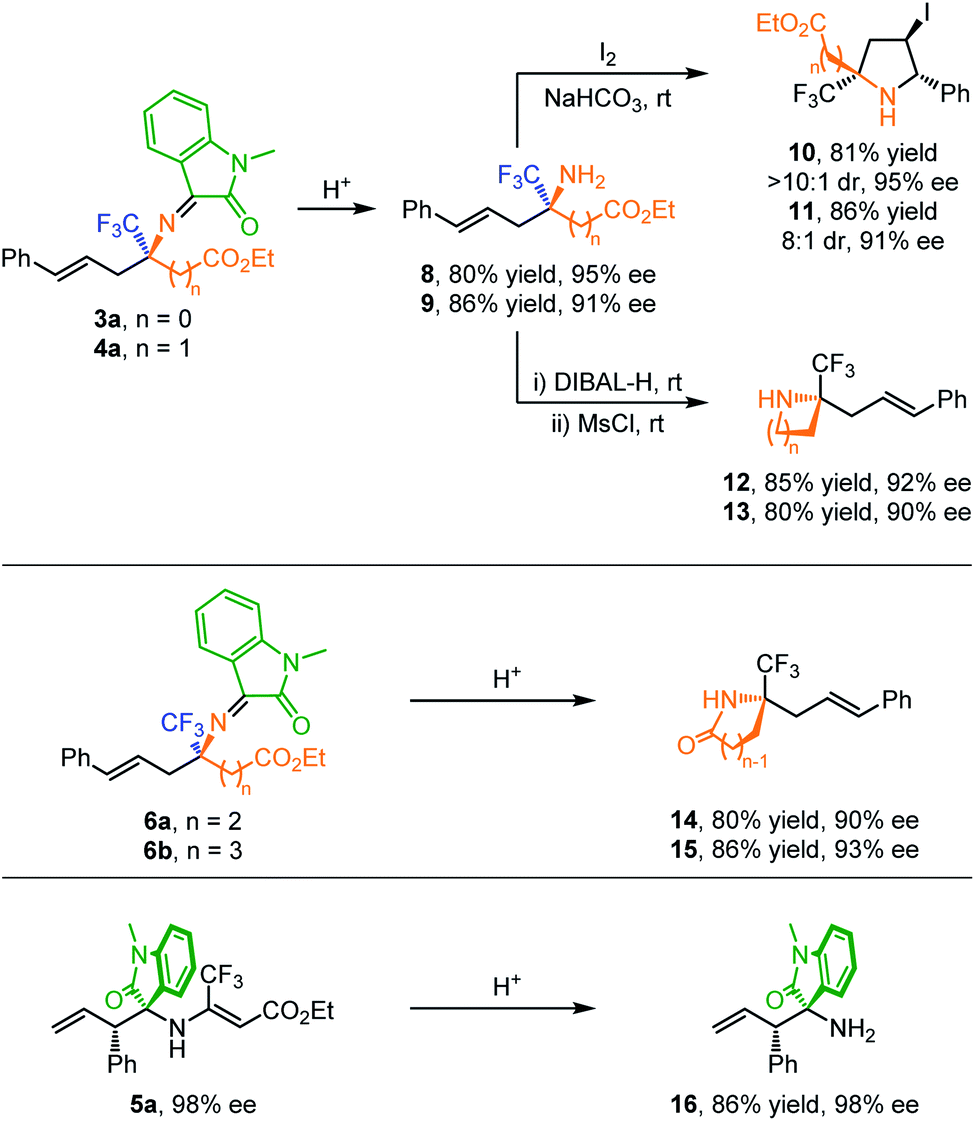 | ||
| Scheme 2 Synthetic transformation. | ||
Based on the experimental results and previous literature reports,11,13 a plausible mechanism was proposed to explain the stereochemistry outcome and the different reaction pathways (Scheme 3). The reaction between isatin-activated ketoimine ester (Z)-1c21 and 2a starts with the nucleophilic attack of either the Re- or Si-face of the 2-azaallyl carbanion via exclusive a'-regioselectivity of the azadienolate to the Re-face of the in situ formed π-allyl-iridium species,23 delivering diastereoisomeric branched allylation intermediates Int A and Int B. Driven by the steric congestion around the two adjacent stereogenic centers, Int A and Int B undergo a stereospecific aza-Cope rearrangement via highly ordered six-membered chair-like transition states TSA and TSB,24 respectively, in both of which the bulky CF3 group resides in the equatorial position, affording (R)-3a in good yield and excellent enantioselectivity. Similarly, the diastereomeric Int C and Int D are formed in the umpolung Ir-catalyzed allylation of 1d–g; however, the carbon atoms in the imine moiety of Int C/Int D are less electron-deficient compared with those in Int A/Int B, which would decelerate the subsequent 2-aza-Cope rearrangement. Int C could undergo further rearrangement to afford the corresponding amino acid derivatives 4a and 6a–cvia the energy-favored TSC, in which the bulky CF3 group resides in the equatorial position, while in TSD the bulky substituent (Ph) of the oxindole ring residing in the axial position would lead to an increasing steric congestion (with a larger A value)25 in comparison with TSC, no matter whether the CF3 or ester carbon chain resides in the axial position. Thus, Int D is incapable of undergoing further rearrangement at room temperature since the corresponding TSD is energetically and electronically disfavored. When isatin-activated β-CF3 β-amino ester derived 1d was employed as the reaction partner, the allylation intermediate Int D would readily convert into thermodynamically stable (S,S)-5a through imine/enamine isomerization.
 | ||
| Scheme 3 Proposed mechanism for Ir-catalyzed umpolung allylation/2-aza-Cope rearrangement of isatin-ketoimino esters 1c–g. | ||
Conclusions
In conclusion, we have developed a highly efficient Ir-catalyzed cascade allylation/2-aza-Cope rearrangement for the synthesis of quaternary trifluoromethylated α-amino acid derivatives in high yields with excellent enantioselectivities. The umpolung reactivity empowered by the activation of the isatin-ketoimine moiety is the key to success and obviates the intractable enantioselectivity control in previous Pd-catalyzed asymmetric linear α-allylation. In combination with quasi-parallel kinetic resolution or kinetic resolution, the generality of the method is further demonstrated by the first preparation of enantioenriched quaternary trifluoromethyl β-, γ-, δ- and ε-amino acids along with 3-amino oxindole in one-pot. Synthetic transformation of the obtained amino acid derivatives afforded a variety of biologically important compounds including trifluoromethyl-containing N-heterocycles and lactams. Further investigations on the mechanistic insights and synthetic applications of the current methodology are on-going in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (21525207, 21772147, 22071186) and Hubei Province NSF (2020CFA036). The Program of Introducing Talents of Discipline to Universities of China (111 Project) is also appreciated.Notes and references
- (a) J. Moschner, V. Stulberg, R. Fernandes, S. Huhmann, J. Leppkes and B. Koksch, Chem. Rev., 2019, 119, 10718 Search PubMed; (b) X.-L. Qiu and F.-L. Qing, Eur. J. Org. Chem., 2011, 2011, 3261 Search PubMed.
- (a) A. Giangaspero, L. Sandri and A. Tossi, Eur. J. Biochem., 2001, 268, 5589 Search PubMed; (b) M. R. Levengood, C. C. Kerwood, C. Chatterjee and W. A. van der Donk, ChemBioChem, 2009, 10, 911 Search PubMed; (c) J. Taira, Y. Kida, H. Yamaguchi, K. Kuwano, Y. Higashimoto and H. Kodama, J. Pept. Sci., 2010, 16, 607 Search PubMed; (d) A. A. Berger, J.-S. Völler, N. Budisa and B. Koksch, Acc. Chem. Res., 2017, 50, 2093 Search PubMed; (e) S. Huhmann and B. Koksch, Eur. J. Org. Chem., 2018, 2018, 3667 Search PubMed.
- (a) E. N. G. Marsh, Acc. Chem. Res., 2014, 47, 2878 Search PubMed; (b) M. Salwiczek, E. K. Nyakatura, U. I. M. Gerling, S. Ye and B. Koksch, Chem. Soc. Rev., 2012, 41, 2135 Search PubMed; (c) N. C. Yoder and K. Kumar, Chem. Soc. Rev., 2002, 31, 335 Search PubMed; (d) C. Jäckel and B. Koksch, Eur. J. Org. Chem., 2005, 2005, 4483 Search PubMed.
- (a) R. Smits, C. D. Cadicamo, K. Burger and B. Koksch, Chem. Soc. Rev., 2008, 37, 1727 Search PubMed; (b) J. Nie, H.-C. Guo, D. Cahard and J.-A. Ma, Chem. Rev., 2011, 111, 455 Search PubMed; (c) J. L. Aceña, A. E. Sorochinsky and V. A. Soloshonok, Synthesis, 2012, 44, 1591 Search PubMed.
- (a) D. Enders, K. Gottfried and G. Raabe, Adv. Synth. Catal., 2010, 352, 3147 Search PubMed; (b) G. Huang, J. Yang and X. Zhang, Chem. Commun., 2011, 47, 5587 Search PubMed; (c) R. Husmann, E. Sugiono, S. Mersmann, G. Raabe, M. Rueping and C. Bolm, Org. Lett., 2011, 13, 1044 Search PubMed; (d) Y.-L. Liu, T.-D. Shi, F. Zhou, X.-L. Zhao, X. Wang and J. Zhou, Org. Lett., 2011, 13, 3826 Search PubMed; (e) K. Morisaki, M. Sawa, J.-y. Nomaguchi, H. Morimoto, Y. Takeuchi, K. Mashima and T. Ohshima, Chem.–Eur. J., 2013, 19, 8417 Search PubMed; (f) H. Xie, A. Song, X. Song, X. Zhang and W. Wang, Tetrahedron Lett., 2013, 54, 1409 Search PubMed; (g) K. Morisaki, M. Sawa, R. Yonesaki, H. Morimoto, K. Mashima and T. Ohshima, J. Am. Chem. Soc., 2016, 138, 6194 Search PubMed; (h) Y.-L. Liu, X.-P. Yin and J. Zhou, Chin. J. Chem., 2018, 36, 321 Search PubMed; (i) U. Bhakta, P. V. Kattamuri, J. H. Siitonen, L. B. Alemany and L. Kürti, Org. Lett., 2019, 21, 9208 Search PubMed.
- (a) V. A. Soloshonok, A. G. Kirilenko, N. A. Fokina, V. P. Kukhar, S. V. Galushko, V. K. Švedas and G. Resnati, Tetrahedron: Asymmetry, 1994, 5, 1225 Search PubMed; (b) V. A. Soloshonok, A. G. Kirilenko, N. A. Fokina, I. P. Shishkina, S. V. Galushko, V. P. Kukhar, V. K. Švedas and E. V. Kozlova, Tetrahedron: Asymmetry, 1994, 5, 1119 Search PubMed; (c) V. A. Soloshonok and V. P. Kukhar, Tetrahedron, 1996, 52, 6953 Search PubMed; (d) V. Michaut, F. Metz, J.-M. Paris and J.-C. Plaquevent, J. Fluorine Chem., 2007, 128, 500 Search PubMed; (e) S. Fustero, C. del Pozo, S. Catalán, J. Alemán, A. Parra, V. Marcos and J. L. G. Ruano, Org. Lett., 2009, 11, 641 Search PubMed; (f) P. J. Duggan, M. Johnston and T. L. March, J. Org. Chem., 2010, 75, 7365 Search PubMed; (g) L. Brewitz, F. A. Arteaga, L. Yin, K. Alagiri, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2015, 137, 15929 Search PubMed; (h) Y.-Y. Peng, P. Liu, Z.-J. Liu, J.-T. Liu, H.-F. Mao and Y.-L. Yao, Tetrahedron, 2018, 74, 3074 Search PubMed; (i) M. R. Straub and V. B. Birman, Org. Lett., 2018, 20, 7550 Search PubMed.
- (a) N. Sewald, W. Hollweck, K. Mütze, C. Schierlinger, L. C. Seymour, K. Gaa, K. Burger, B. Koksch and H. D. Jakubke, Amino Acids, 1995, 8, 187 Search PubMed; (b) B. Koksch, N. Sewald, K. Burger and H. D. Jakubke, Amino Acids, 1996, 11, 425 Search PubMed; (c) P. Bravo, L. Bruché, C. Pesenti, F. Viani, A. Volonterio and M. Zanda, J. Fluorine Chem., 2001, 112, 153 Search PubMed; (d) N. Margiotta, P. Papadia, F. Lazzaro, M. Crucianelli, F. De Angelis, C. Pisano, L. Vesci and G. Natile, J. Med. Chem., 2005, 48, 7821 Search PubMed; (e) S. Rene and K. Beate, Curr. Top. Med. Chem., 2006, 6, 1483 Search PubMed.
- (a) H.-L. Teng, F.-L. Luo, H.-Y. Tao and C.-J. Wang, Org. Lett., 2011, 13, 5600 Search PubMed; (b) Z.-Y. Xue, Q.-H. Li, H.-Y. Tao and C.-J. Wang, J. Am. Chem. Soc., 2011, 133, 11757 Search PubMed; (c) H.-L. Teng, H. Huang and C.-J. Wang, Chem.–Eur. J., 2012, 18, 12614 Search PubMed; (d) L. Wei, S.-M. Xu, Q. Zhu, C. Che and C.-J. Wang, Angew. Chem., Int. Ed., 2017, 56, 12312 Search PubMed; (e) L. Wei, L. Xiao and C.-J. Wang, Adv. Synth. Catal., 2018, 360, 4715 Search PubMed; (f) L. Wei, Q. Zhu, S.-M. Xu, X. Chang and C.-J. Wang, J. Am. Chem. Soc., 2018, 140, 1508 Search PubMed; (g) H.-C. Liu, Y.-Z. Hu, Z.-F. Wang, H.-Y. Tao and C.-J. Wang, Chem.–Eur. J., 2019, 25, 8681 Search PubMed; (h) L. Wei, X. Chang and C.-J. Wang, Acc. Chem. Res., 2020, 53, 1084 Search PubMed.
- X.-S. Sun, Q. Ou-Yang, S.-M. Xu, X.-H. Wang, H.-Y. Tao, L. W. Chung and C.-J. Wang, Chem. Commun., 2020, 56, 3333 Search PubMed.
- (a) L. Wei, L. Xiao, Y. Hu, Z. Wang, H. Tao and C. Wang, Chin. J. Org. Chem., 2019, 39, 2119–2130 Search PubMed; (b) S. Tang, X. Zhang, J. Sun, D. Niu and J. J. Chruma, Chem. Rev., 2018, 118, 10393 Search PubMed.
- (a) C. Shen, R.-Q. Wang, L. Wei, Z.-F. Wang, H.-Y. Tao and C.-J. Wang, Org. Lett., 2019, 21, 6940 Search PubMed; (b) L.-M. Shi, X.-S. Sun, C. Shen, Z.-F. Wang, H.-Y. Tao and C.-J. Wang, Org. Lett., 2019, 21, 4842 Search PubMed; (c) Y. Wu, L. Hu and L. Deng, Nature, 2015, 523, 445 Search PubMed; (d) Y. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2014, 136, 4500 Search PubMed; (e) P. Chen, Z. Yue, J. Zhang, X. Lv, L. Wang and J. Zhang, Angew. Chem., Int. Ed., 2016, 55, 13316 Search PubMed; (f) Y.-L. Su, Y.-H. Li, Y.-G. Chen and Z.-Y. Han, Chem. Commun., 2017, 53, 1985 Search PubMed.
- Q. Cheng, H.-F. Tu, C. Zheng, J.-P. Qu, G. Helmchen and S.-L. You, Chem. Rev., 2019, 119, 1855 Search PubMed.
- (a) M. Kawatsura, H. Tsuji, K. Uchida and T. Itoh, Tetrahedron, 2011, 67, 7686 Search PubMed; (b) J. Liu, C.-G. Cao, H.-B. Sun, X. Zhang and D. Niu, J. Am. Chem. Soc., 2016, 138, 13103 Search PubMed; (c) W.-B. Liu, N. Okamoto, E. J. Alexy, A. Y. Hong, K. Tran and B. M. Stoltz, J. Am. Chem. Soc., 2016, 138, 5234 Search PubMed; (d) L. Wei, Q. Zhu, L. Xiao, H.-Y. Tao and C.-J. Wang, Nat. Commun., 2019, 10, 1594 Search PubMed; (e) L. Wei, L. Xiao, Z.-F. Wang, H.-Y. Tao and C.-J. Wang, Chin. J. Chem., 2020, 38, 82 Search PubMed; (f) C.-X. Zhuo and A. Fürstner, J. Am. Chem. Soc., 2018, 140, 10514 Search PubMed; (g) C.-G. Cao, B. He, Z. Fu and D. Niu, Org. Process Res. Dev., 2019, 23, 1758 Search PubMed; (h) Y. Wang, L.-F. Deng, X. Zhang and D. Niu, Org. Lett., 2019, 21, 6951 Search PubMed; (i) R.-Q. Wang, C. Shen, X. Cheng, Z.-F. Wang, H.-Y. Tao, X.-Q. Dong and C.-J. Wang, Chin. J. Chem., 2020, 38, 807 Search PubMed.
- (a) M. Winter, H. Kim and M. Waser, Eur. J. Org. Chem., 2019, 2019, 7122 Search PubMed; (b) For the early attempt with less than 10% ee on Pd-catalyzed allylation of N-PMP trifluoroalanine derivative, see: T. Konno, M. Kanda, T. Ishihara and H. Yamanaka, J. Fluorine Chem., 2005, 126, 1517 Search PubMed.
- (a) M. Hudlicky, J. Fluorine Chem., 1984, 25, 353 Search PubMed; (b) Y. Itoh, M. Yamanaka and K. Mikami, J. Am. Chem. Soc., 2004, 126, 13174 Search PubMed.
- J. F. Teichert and B. L. Feringa, Angew. Chem., Int. Ed., 2010, 49, 2486 Search PubMed.
- (a) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461 Search PubMed; (b) C. A. Kiener, C. Shu, C. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 14272 Search PubMed.
- Q. Yang, Y. Li, J.-D. Yang, Y. Liu, L. Zhang, S. Luo and J.-P. Cheng, Angew. Chem., Int. Ed. DOI:10.1002/anie.202008528.
- The predicted pKa values of 1a–1c in DMSO was obtained from, http://pka.luoszgroup.com/.
- X. Zhang, W.-B. Liu, Q. Cheng and S.-L. You, Organometallics, 2016, 35, 2467 Search PubMed.
- Crystallographic data have been deposited at the Cambridge Crystallographic Data Center (CCDC) as CCDC 2018214 (1c), 2018215 ((R)-3k) and 2018216 ((S,S)-5a).
- (a) Y. Nakao, B. K. S. Yeung, W. Y. Yoshida, P. J. Scheuer and M. Kelly-Borges, J. Am. Chem. Soc., 1995, 117, 8271 Search PubMed; (b) B. K. S. Yeung, Y. Nakao, R. B. Kinnel, J. R. Carney, W. Y. Yoshida, P. J. Scheuer and M. Kelly-Borges, J. Org. Chem., 1996, 61, 7168 Search PubMed; (c) Y. Nakao, J. Kuo, W. Y. Yoshida, M. Kelly and P. J. Scheuer, Org. Lett., 2003, 5, 1387 Search PubMed; (d) H. Takayama, I. Mori, M. Kitajima, N. Aimi and N. H. Lajis, Org. Lett., 2004, 6, 2945 Search PubMed; (e) G. Ding, L. Jiang, L. Guo, X. Chen, H. Zhang and Y. Che, J. Nat. Prod., 2008, 71, 1861 Search PubMed.
- M. Jäkel, J. Qu, T. Schnitzer and G. Helmchen, Chem.–Eur. J., 2013, 19, 16746 Search PubMed.
- H. E. Zimmerman and M. D. Traxler, J. Am. Chem. Soc., 1957, 79, 1920 Search PubMed.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry, Part A: Structure and Mechanisms, Springer, New York, 4th edn, 2000, p. 135 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2018214–2018216. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04685j |
| This journal is © The Royal Society of Chemistry 2020 |